 Debora Curci
Debora Curci Stefania Braidotti
Stefania Braidotti Natalia Maximova
Natalia Maximova
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pharmacol. , 23 October 2024
Sec. Obstetric and Pediatric Pharmacology
Volume 15 - 2024 | https://doi.org/10.3389/fphar.2024.1478381
This report describes a pediatric case of isolated agranulocytosis occurring months after hematopoietic stem cell transplantation (HSCT). Secondary cytopenia, or secondary transplant failure, affects 10%–25% of HSCT recipients, with potential triggers including viral infection, graft-versus-host disease (GVHD), sepsis, and certain medications. Viral reactivation was ruled out based on negative PCR results, while GVHD and sepsis were ruled out based on the patient’s clinical presentation. The patient, who received an HLA 10/10 unrelated donor T-cell transplant, underwent standard myeloablative conditioning to minimize the risk of graft rejection. However, agranulocytosis persisted even after discontinuation of myelotoxic drugs such as valganciclovir and ruxolitinib. Further investigation revealed that the patient had been taking febuxostat, which was subsequently discontinued, leading to a recovery of the neutrophil count. The European Medicines Agency lists agranulocytosis as a rare side effect of febuxostat. The effect of candidate genes and variants involved in febuxostat pharmacokinetics and pharmacodynamics was done using the Pharmacogenomics Knowledge Base (PharmGKB) to accurately evaluate an individual’s risk for neutropenia. This case suggests that genetic variants in renal transporters ABCG2 (exonic non-synonymous variant, rs2231137), SLC29A1 (rs747199 and rs628031), and ABCC4 (3′UTR SNP, rs3742106 and rs11568658) may contribute to drug-induced agranulocytosis. This finding underscores the importance of genetic profiling in the management of patients undergoing HSCT to prevent adverse drug reactions.
Asymptomatic hyperuricemia is not uncommon in hematopoietic stem cell transplantation (HSCT) recipients due to the consolidated use of immunosuppressive agents such as calcineurin inhibitors (Ben Salem et al., 2017). Traditional treatment with allopurinol is associated with complications and interactions that could be potentially serious, leading to lower efficacy if used in regular doses (Tayar et al., 2012).
Febuxostat, a xanthine oxidase inhibitor, reduces urate production and, therefore, reduces serum uric acid levels. It achieves this by inhibiting the conversion of hypoxanthine to xanthine and xanthine to uric acid. Unlike allopurinol, febuxostat is a non-purine selective inhibitor of the xanthine oxidase (XO) enzyme in that it works by inhibiting both the oxidized and reduced forms of XO without inhibiting the enzymes involved in purine or pyrimidine metabolism (Bridgeman and Chavez, 2015). Febuxostat lowers uric acid levels more effectively than allopurinol and serves as an alternative for patients who are intolerant to allopurinol (Becker et al., 2005).
Febuxostat undergoes hepatic metabolism in the cytochrome P450 (CYP) enzyme system into acyl-glucuronide metabolites. This metabolism occurs mainly through conjugation via uridine diphosphate glucuronosyltransferase (UGT) enzymes (UGT1A1, UGT1A3, UGT1A9, and UGT2B7), although a small portion is oxidized into active hydroxyl metabolites (67M-1, 67M-2, and 67M-4) by CYP1A2, CYP2C8, and CYP2C9. Drug interactions in vitro studies found that febuxostat had no measurable effect on the activities of the CYP1A2, CYP2C9, CYP2C19, and CYP2D6 isoenzymes (Mukoyoshi et al., 2008). Because XO is involved in the metabolism of azathioprine, mercaptopurine, and theophylline, inhibition of this enzyme can lead to toxicity due to increased drug levels. For this reason, febuxostat is not advised in patients treated with these drugs (Ernst and Fravel, 2009).
Febuxostat has not been reported to cause severe complications in pediatric patients, particularly hematological abnormalities. Still, we have reported one case of isolated severe neutropenia associated with the initiation of febuxostat therapy in a pediatric hematopoietic stem cell transplant recipient.
A 10-year-old Caucasian girl was admitted to our department with a high fever and petechial rash. One year before, for a history of fever, cytopenia, and hepatosplenomegaly associated with increased triglycerides and ferritin, sequencing analysis of the UNC13D gene evidenced a double-heterozygous mutation. Diagnosis of familial hemophagocytic lymphohistiocytosis type 3 (FLH3) was formalized. The patient underwent treatment according to the HLH-94 protocol with dexamethasone, etoposide, and ciclosporin, obtaining remission after 8 weeks of treatment.
Upon admission to our department, a physical examination revealed high fever, petechiae on the upper and lower limbs, and hepatosplenomegaly. The patient was found to have pancytopenia (neutrophils 430/mm3, platelets 13,000/mm3, and hemoglobin 8.1 g/dL), hyperferritinemia (ferritin 1,115 μg/L), and low natural killer (NK) cell activity. The FLH3 recurrence was confirmed. The patient was started on dexamethasone and ruxolitinib.
It’s known that HSCT is the only established curative treatment for patients with familial, relapsing, or severe and persistent disease (Janka and Lehmberg, 2013). An HLA-matched, AB0 mismatched unrelated donor was found on the international bone marrow donor registry. The conditioning regimen, which included treosulfan 14 g/m2/day for 3 days, fludarabine 160 mg/m2 total dose, melphalan 140 mg/m2, rituximab 375 mg/m2, and antithymocyte globulin was started 6 days before HSCT. Ruxolitinib was discontinued on the first day of conditioning because of possible adverse effects on engraftment and a lack of sufficient data on compatibility between ruxolitinib and drugs used during conditioning (Salit, 2022).
On day 0, they were infused with donor graft containing 11 × 106 CD34+/kg recipient body weight. The prophylaxis of graft-versus-host disease (GVHD) included tacrolimus and mycophenolate mofetil (MMF). Engraftment was achieved on day +11, and complete donor chimerism was reached on day +32. The early post-transplant period was complicated by severe cytokine release syndrome (CRS), which led to acute respiratory failure requiring continuous positive airway pressure (CPAP) and pulmonary hypertension. The IL-6 plasma levels reached 202.8 pg/mL (normal range ≤6.4 pg/mL). The CRS was treated with cortisone, tocilizumab, and continuous infusion of anakinra. Furthermore, a very high type 1 interferon signature (30.9, normal range ≤2.2) led us to restart treatment with ruxolitinib, which replaced the MMF. Furthermore, a high dose of labetalol has been used to correct severe refractory arterial hypertension. On day +12, cytomegalovirus (CMV) reactivation with a high blood viral load further complicated the clinical situation. The patient began antiviral treatment with foscarnet, which gradually decreased the viral load. The CMV clearance was obtained after 4 weeks of antiviral therapy. The foscarnet use caused a worsening of renal function, already compromised by prior treatment with cyclosporin. Foscarnet was replaced with valganciclovir under therapeutic drug monitoring (TDM), which was performed twice a week, maintaining the drug’s blood levels at the normal range’s lower-end cutoffs. Despite the change in antiviral therapy, the estimated glomerular filtration rate (eGFR) declined significantly, falling below 60 mL/min/1.73 m2. Taper the dose of tacrolimus was considered. Ruxolitinib with a low dose of tacrolimus was successfully used for GVHD prophylaxis.
In the following 2 months, the patient maintained mild renal dysfunction with persistent proteinuria and albuminuria (400 mg/g of creatinine and 130 mg/g of creatinine, respectively), elevated serum (3,790 ng/mL, normal range 1,010–1730 ng/mL) and urinary (2,657 ng/mL, normal range <300 ng/mL) β2-microglobulin, and an eGFR nailed between 75 and 88 mL/min/1.73 m2.
Due to the persistence of high serum uric acid levels (>9 mg/dL, normal range 2.5–6.0 mg/dL), the patient began treatment with allopurinol, which was interrupted after 2 weeks due to the appearance of a widespread skin rash. Because of the persistence of hyperuricemia, treatment with febuxostat 80 mg/day began. Serum uric acid levels dropped rapidly, and after the first week of treatment, the patient did not report any side effects from the new medication. Although the therapy was well tolerated, we documented the tendency towards moderate neutropenia (PMN ≤1,000/mm3). A complete blood count repeated 1 week later showed absolute agranulocytosis or grade 4 neutropenia categorized by Common Terminology Criteria for Adverse Events (CTCAE 5.0). No decrease in other blood cell counts was observed. The patient was in excellent condition, with no fever, systemic inflammation symptoms, or laboratory test abnormalities. Treatment with valganciclovir and ruxolitinib was promptly discontinued due to suspected pharmacological myelotoxicity. Furthermore, we tried to stimulate neutrophil production without responding with recombinant human granulocyte stimulating factor (G-CSF; filgrastim). A broad viral work-up was negative for CMV, Epstein-Barr virus (EBV), Adenovirus, Parvovirus, Herpes Simplex virus (HSV), human herpesvirus 6 (HHV-6) and 7 (HHV-7), and hepatitis viruses. The bone marrow smear showed no neutrophils, and the biopsy showed a normal number of megakaryocytes and no evidence of myelodysplasia. Hemophagocytic lymphohistiocytosis infiltration to bone marrow was not present. Peripheral blood full donor chimerism was reconfirmed. Additionally, autoimmune neutropenia (antineutrophil antibody, antinuclear antibody, anti-DNA, and extractable nuclear antigen were negative) and vitamin deficiencies were excluded.
Febuxostat was suspended on suspicion of drug-induced neutropenia. Five days later, the neutrophil count increased to 900/mm3; 2 weeks later, it was normal. Figure 1 shows the timeline graph of transplant-related events with corresponding treatments and absolute polymorphonuclear leukocyte (PMN) count trend.
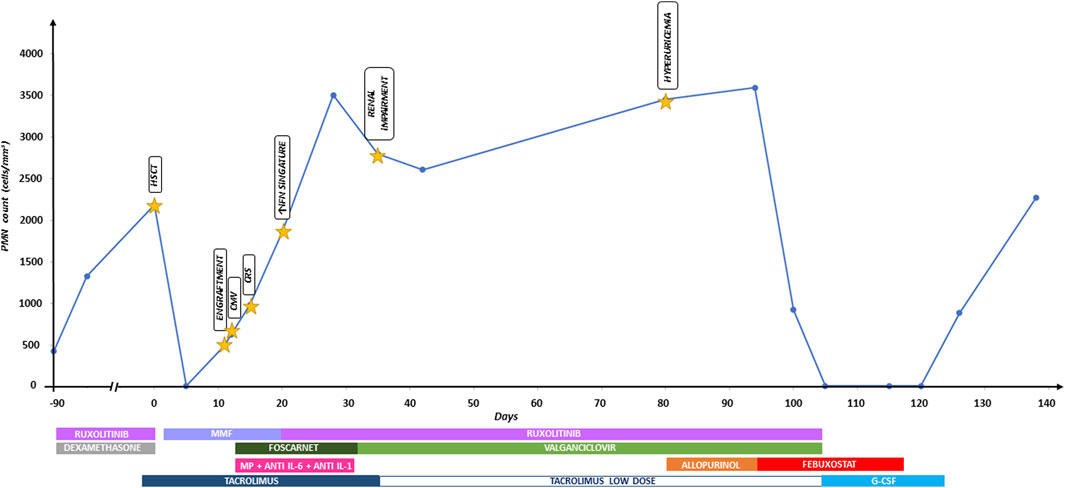
Figure 1. Timeline graph of transplant-related events with corresponding treatments and absolute polymorphonuclear leukocyte (PMN) count trend. Anti IL-6, tocilizumab; anti IL-1, anakinra; CMV, cytomegalovirus; CRS, cytokine release syndrome; G-CSF, granulocyte-colony stimulating factor; HSCT, hematopoietic stem cell transplantation; IFN, interferon; MMF, mycophenolate mofetil; MP, methylprednisolone.
The effect of candidate genes and variants involved in febuxostat pharmacokinetics and pharmacodynamics was done using the Pharmacogenomics Knowledge Base (PharmGKB) to evaluate an individual’s risk for neutropenia accurately. In particular, genetic variants in the renal transporters ABCG2 (exonic non-synonymous variant, rs2231137), ABCB1 (rs1128503, rs2229109, rs1045642 and rs2032582), ABCC4 (3′ UTR SNP, rs3742106 and rs11568658) and SLC29A1 (rs747199 and rs628031) was evaluated using Illumina genotyping arrays (Illumina Infinium HumanOmniExpressExome BeadChip). The array includes over 200,000 functional exonic markers, delivering unparalleled coverage of putative functional exonic variants. A total of 200 ng of gDNA (50 ng/μL) for each sample was processed according to Illumina’s Assay protocol. Normalization of raw image intensity data, genotype clustering, and individual sample genotype calls were performed using Illumina’s Genome Studio software. Allele detection and genotype calling were performed with Genome Studio software. The single-nucleotide polymorphism (SNP) of interest and the resulting patient genotypes are reported in Table 1. Figure 2 illustrates the mechanism of action of febuxostat and the drug’s interaction with the mentioned transporters.
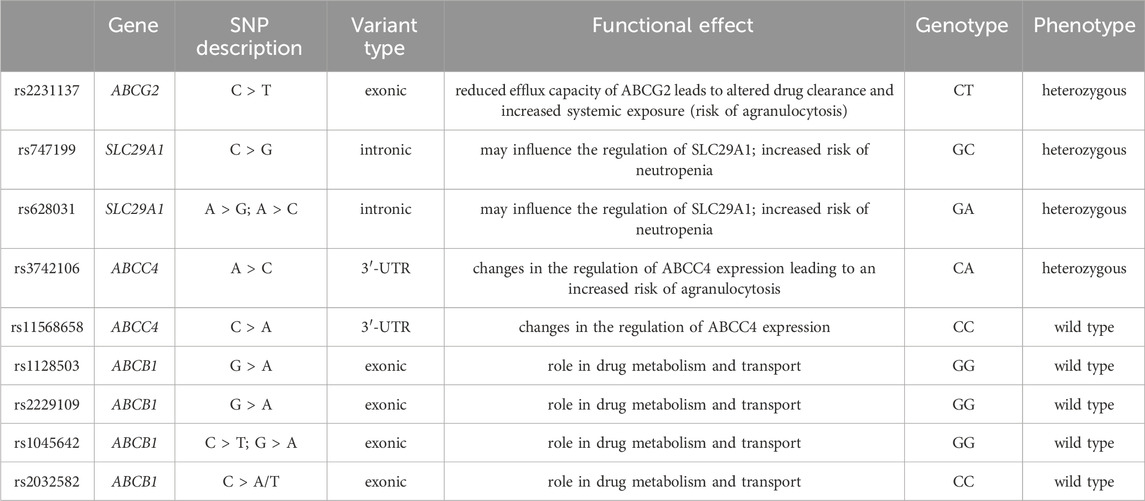
Table 1. Variants associated with febuxostat response according to PharmGKB and genotyping results in the case analyzed by Illumina Infinium HumanOmniExpressExome BeadChip.
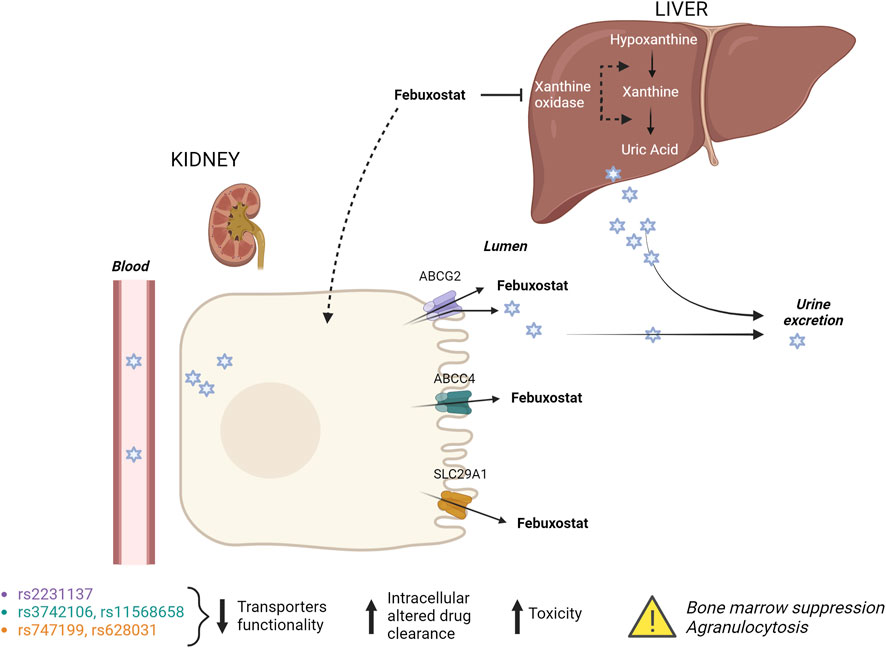
Figure 2. Febuxostat mechanism of action and its interaction with renal transporters. Febuxostat primarily inhibits xanthine oxidase in the liver. Variants in renal transporters may increase the risk of febuxostat-induced agranulocytosis.
After 1 year, the patient is doing well, takes no therapy, and leads an normal life. Neutrophil counts were monitored monthly for 6 months after the described episode and were constantly standard. Her kidney function has stabilized, maintaining serum uric acid, proteinuria, and albuminuria values slightly higher than expected, with eGFR returning to levels above 90 mL/min/1.73 m2.
In this report, we describe the case of a pediatric recipient who developed isolated agranulocytosis a few months after HSCT.
Secondary cytopenia, also known as secondary graft failure, is a relatively common complication after allogeneic HSCT, characterized by the loss of donor cells after initial engraftment (Bittencourt et al., 2005). From 10% to 25% of HSCT recipients can develop a transitory or permanent decrease in the absolute neutrophil count to less than 500 cells/mm3 (Bittencourt et al., 2002). Erythroid and megakaryocytic lineages may also be affected. Conditions associated with an increased occurrence of secondary graft failure including viral infections, especially CMV, HHV-6, and parvovirus, severe, treatment-refractory GVHD, sepsis, HLA disparity, reduced-intensity conditioning, low nucleated cell dose of the graft, T-cell depletion of the graft, allosensitization of the recipient, primary disease recurrence, and use of drugs inducing myelosuppression, such as ganciclovir, trimethoprim-sulfamethoxazole, mycophenolate mofetil (MMF) and ruxolitinib (Salzberger et al., 1997; Bittencourt et al., 2005; Nakamae et al., 2011; Locatelli et al., 2014; Imamura, 2021).
Viral infections account for a large part of post-transplant complications, with up to 90% of patients after allogeneic HSCT likely to experience reactivation of at least one virus (Hill et al., 2017; Düver et al., 2020). Viral infection, in particular CMV, can result in impaired graft function and, in the extreme cases, complete graft failure (Mayer et al., 1997; Dobonici et al., 1998; Steffens et al., 1998; Renzaho et al., 2020). Since viral reactivation could be the most likely cause in this child, we first performed a real-time polymerase chain reaction search for viruses of the Herpesviridae family, Parvovirus, and Adenovirus DNA, which was negative.
Bone marrow is an important target of GVHD, which damages not only stem cells but also the bone marrow niche (Müskens et al., 2021). Bone marrow aspirates from patients with GVHD show reduced numbers of progenitor cells with impaired proliferation in ex vivo colony-forming assays compared to patients without GVHD (Martínez-Jaramillo et al., 2001). Sepsis is an extreme example of inadequate host bone marrow response to severe infection. An excessive immune response with initial neutrophilia is followed by profound neutropenia, leukocyte anergy, and, consequently, an inability of the host to control the infection (Zhang et al., 2016).
GVHD was excluded from the causes because the patient had never developed any signs of either acute or chronic GVHD. Sepsis also did not fall within the criteria for differential diagnosis because the patient was in excellent general condition, was asymptomatic, and didn't have inflammation markers.
Other factors that increase the risk of graft failure are HLA-mismatched, T cell-depleted (TCD) or cord blood grafts and non-malignant hematological disease (Beatty et al., 1985; Gluckman et al., 1997; Remberger et al., 2007; Klein et al., 2023). Recipients from unrelated donors have a higher incidence of graft failure than recipients from HLA-identical donors. Comparing unrelated donors, HLA class I disparity was associated with an increased risk of graft failure (Mattsson et al., 2008). When total T-cell depletion was utilized to overcome the HLA barriers in haploidentical HSCT, there was a high rate of disease relapse and graft rejection. Studies of TCD graft rejection demonstrated that residual host-derived cytotoxic lymphocytes were activated and expanded in vivo, leading to donor graft rejection by targeting donor mismatched HLA molecules (Bierer et al., 1988). Similar results were obtained from the Spanish Group for Allogeneic Peripheral Blood Transplantation in CD34+ selected HSCT from HLA-identical siblings (Urbano-Ispizua et al., 2001). This study shows that the number of CD3+ cells in the graft with a threshold of 0.2 × 106/kg or less is the most critical factor in maintaining sustained engraftment in CD34+ selected HSCT from HLA-identical siblings.
Having received the T-cell repleted graft, which contained a very high number of stem cells, from an HLA 10/10 matched unrelated donor (MUD), our patient did not fall into the high-risk category for these factors.
Due to the low number of hematopoietic cells in a single harvest, allogeneic cord blood transplants have been employed mainly in treating children needing HSCT. However, in almost all published studies, the most important and limiting factor influencing cord blood transplant outcome resulted from the low cell dose infused, which was found to correlate with the rate of engraftment, speed of hematopoietic recovery, frequency of graft failure, and survival (Rubinstein et al., 1998; Rocha et al., 2001; Eapen et al., 2007; Kurtzberg et al., 2008). Allosensitization towards major HLA antigens or, less frequently, minor histocompatibility antigens that develop the heavily transfused recipients can contribute to the increased rejection rate in non-malignant diseases. In aplastic anemia, multiply transfused patients are more likely to develop graft rejection. Thus, HSCT performed at an early age is highly recommended to limit sensitization to histocompatibility antigens. As a consequence of disease status and numerous pretransplant transfusions, in analogy with aplastic anemia, the incidence of graft failure in thalassemia ranges from 8% to 12% according to the patient’s risk class (Bacigalupo et al., 2010). Our patient received only a few packed red blood cells and platelet units at the onset and the recurrence of hemophagocytic lymphohistiocytosis. Therefore, she wouldn't be at risk of allosensitization due to transfusions.
Other important factors for developing secondary graft failure are non-myeloablative (NMA) or reduced-intensity conditioning (RIC) and low-intensity pre-transplant immunosuppression. High-dose myeloablative radio and chemotherapy (MAC) are conventionally used as conditioning for HSCT, with advantages in transplant-related outcomes. MAC has a profound immunosuppressive effect on the host, limiting the ability to reject the graft (Petersen, 2007). RIC or NMA HSCT resulted in a three to four times increased risk of graft failure compared with MAC transplant (Olsson et al., 2013). Even an alloreactive immunological reaction mediated by residual host immunity persisting after the conditioning regimen can induce graft failure. Residual host T-cells are the most prominent effector cells mediating rejection (Masouridi-Levrat et al., 2016). T-cell-mediated graft rejection can occur in both HLA-mismatched and HLA-matched settings. In the latter case, rejection is due to responses directed against minor histocompatibility antigens (Kernan et al., 1987; Voogt et al., 1990).
Our patient received a standard myeloablative conditioning regimen associated with rabbit antithymocyte globulin (Thymoglobulin) to minimize the possibility of rejection.
Determining post-transplant chimerism is an essential aspect of post-transplant follow-up. It allows us to distinguish between graft failure, poor graft function, or primary disease recurrence (Delie et al., 2021). The peripheral blood chimerism determination showed that the patient has a stable chimera, confirming full donor chimerism.
Furthermore, a bone marrow biopsy excluded the primary disease recurrence. The hallmark of the FLH bone marrow picture is the presence of prominent hemophagocytosis with a decreased number of normal hematopoietic precursors (Henter et al., 1991). In our case, agranulocytosis was the only pathological finding. The bone marrow smear instead suggested probable drug toxicity. Valganciclovir and ruxolitinib were discontinued at the first evidence of neutropenia, as their myelotoxic effect is well known. Both drugs impair bone marrow cell proliferation, either by inhibiting DNA replication in bone marrow progenitor cells (valganciclovir) or disrupting cytokine-mediated survival signals (ruxolitinib), resulting in significant myelotoxicity (Salzberger et al., 1997; Locatelli et al., 2024). Even after 10 days of myelotoxic drug interruption and continuous G-CSF stimulation, agranulocytosis persisted.
The patient was still taking febuxostat, which replaced allopurinol due to its known side effects. Febuxostat therapy was discontinued, resulting in a rise in neutrophil count after 5 days.
The European Medicines Agency patient information leaflets on febuxostat report agranulocytosis as a rare event collected in the post-marketing experience. In the literature, we found only three reports of neutropenia/agranulocytosis following treatment with febuxostat. All three cases involved adult patients with chronic renal failure and other significant comorbidities (Kobayashi et al., 2013; Poh et al., 2017). A later published review reported no association between febuxostat treatment and agranulocytosis was confirmed (Jordan and Gresser, 2018).
The administration of febuxostat as an ABCG2 inhibitor may alter the pharmacokinetics and efficacy of ABCG2 substrate drugs. Patients with ABCG2 SNPs (decreased ABCG2 function) reportedly exhibit higher bioavailability of ABCG2 substrates (Yamasaki et al., 2008) than subjects with ABCG2 wild type (WT). Therefore, febuxostat could induce a similar effect on drug absorption. The genetic variant rs2231137 is known to reduce the efflux capacity of the ABCG2 transporter, leading to increased intracellular concentrations of substrates, among which febuxostat (Yamasaki et al., 2008). Higher intracellular drug concentrations may lead to increased toxicity, potentially affecting the bone marrow and leading to agranulocytosis. In addition, reduced ABCG2 function could lead to febuxostat accumulation in the kidneys, resulting in altered drug clearance and increased systemic exposure, further contributing to the risk of agranulocytosis.
For this reason, the presence of missense SNP in ABCG2, concomitant with other variants in renal drug transporters (SLC29A1 and ABCC4) in heterozygous, could affect the clinical drug efficacy in terms of risk of neutropenia. It is known that patients with rs11568658 in ABCC4 and kidney transplant may have an increased risk of neutropenia when treated with valganciclovir compared to patients with WT (Billat et al., 2016). For this reason, another genetic variant in the 3′ UTR of ABCC4 (rs3742106) may lead to changes in the regulation of ABCC4 expression, potentially reducing the efflux of febuxostat and its metabolites (Billat et al., 2016). Reduced ABCC4 function can result in higher intracellular concentrations of toxic metabolites, contributing to cellular toxicity in the bone marrow and ultimately leading to agranulocytosis. Additionally, impaired drug efflux could exacerbate febuxostat’s pharmacodynamic effects, increasing the risk of drug-induced bone marrow suppression. The variants rs747199 and rs628031 in SLC29A1, involved in the transport of nucleosides and some drugs across cell membranes could also affect the cellular uptake of febuxostat or its regular metabolism, leading to either increased exposure to toxic metabolites or an imbalance in cellular nucleotides, thereby contributing to bone marrow suppression and agranulocytosis. Lee et al. have demonstrated an association between neutropenia and genetic polymorphism in SLC29A1 (rs747199) in pediatric patients with inflammatory bowel diseases in treatment with thiopurine (Lee et al., 2015). In line with this result, our patient, heterozygous for variants in SLC29A1 (rs747199 and rs628031), may have developed neutropenia after including febuxostat in the therapeutic protocol.
The identified variants in ABCG2, SLC29A1, and ABCC4 may collectively contribute to an increased risk of febuxostat-induced agranulocytosis by altering the drug’s pharmacokinetics and pharmacodynamics through the following possible mechanisms: altering drug transport and clearance inducing higher systemic and intracellular levels of febuxostat; increasing exposure to toxic metabolites, leading to bone marrow suppression and disrupting normal cellular processes, which affect neutrophil production and survival. This case demonstrates that there exists an association between concomitant treatments (valganciclovir, tacrolimus, and febuxostat) and the onset of neutropenia. There are several limitations in this case. First of all, a single case limits the significance of these findings. Moreover, during treatment with febuxostat, the patient received other drugs, such as valganciclovir and ruxolitinib. The potential contributions of these additional drugs as the trigger of agranulocytosis due to febuxostat cannot be ruled out.
In conclusion, this case report suggests that the presence of ABCG2-SLC29A1-ABCC4 haplotypes may affect clinical outcomes, leading to neutropenia.
However, it is important to understand that after excluding febuxostat medication, resolution of neutropenia was observed, indicating that the identified haplotype could help clinicians choose the treatment option to avoid undesired effects. Complete blood count close monitoring should be recommended at the beginning of the treatment to monitor for side effects in individuals with renal failure or treatment-related frailty. In hospitals with the necessary tools, genetic tests to identify polymorphisms in candidate genes before starting febuxostat treatment could offer significant value for some patients.
The patient’s parents were pleased that the cause of the neutropenia was identified, and the complication resolved relatively quickly without subjecting the daughter to further treatments.
After stopping all medicine, they were grateful for the improvement in their daughter’s quality of life.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
DC: Data curation, Formal Analysis, Writing–review and editing. SB: Data curation, Formal Analysis, Writing–review and editing. NM: Investigation, Project administration, Supervision, Writing–original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Italian Ministry of Health, through the contribution given to the Institute for Maternal and Child Health IRCCS Burlo Garofolo, Trieste, Italy (grant number RC 44/24).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bacigalupo, A., Socie, G., Lanino, E., Prete, A., Locatelli, F., Locasciulli, A., et al. (2010). Fludarabine, cyclophosphamide, antithymocyte globulin, with or without low dose total body irradiation, for alternative donor transplants, in acquired severe aplastic anemia: a retrospective study from the EBMT-SAA Working Party. Haematologica 95, 976–982. doi:10.3324/haematol.2009.018267
Beatty, P. G., Clift, R. A., Mickelson, E. M., Nisperos, B. B., Flournoy, N., Martin, P. J., et al. (1985). Marrow transplantation from related donors other than HLA-identical siblings. N. Engl. J. Med. 313, 765–771. doi:10.1056/NEJM198509263131301
Becker, M. A., Schumacher, H. R., Wortmann, R. L., Macdonald, P. A., Eustace, D., Palo, W. A., et al. (2005). Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N. Engl. J. Med. 353, 2450–2461. doi:10.1056/NEJMoa050373
Ben Salem, C., Slim, R., Fathallah, N., and Hmouda, H. (2017). Drug-induced hyperuricaemia and gout. Rheumatol. Oxf. 56, 679–688. doi:10.1093/rheumatology/kew293
Bierer, B. E., Emerson, S. G., Antin, J., Maziarz, R., Rappeport, J. M., Smith, B. R., et al. (1988). Regulation of cytotoxic T lymphocyte-mediated graft rejection following bone marrow transplantation. Transplantation 46, 835–839. doi:10.1097/00007890-198812000-00009
Billat, P. A., Ossman, T., Saint-Marcoux, F., Essig, M., Rerolle, J. P., Kamar, N., et al. (2016). Multidrug resistance-associated protein 4 (MRP4) controls ganciclovir intracellular accumulation and contributes to ganciclovir-induced neutropenia in renal transplant patients. Pharmacol. Res. 111, 501–508. doi:10.1016/j.phrs.2016.07.012
Bittencourt, H., Rocha, V., Chevret, S., Socié, G., Espérou, H., Devergie, A., et al. (2002). Association of CD34 cell dose with hematopoietic recovery, infections, and other outcomes after HLA-identical sibling bone marrow transplantation. Blood 99, 2726–2733. doi:10.1182/blood.v99.8.2726
Bittencourt, H., Rocha, V., Filion, A., Ionescu, I., Herr, A. L., Garnier, F., et al. (2005). Granulocyte colony-stimulating factor for poor graft function after allogeneic stem cell transplantation: 3 days of G-CSF identifies long-term responders. Bone Marrow Transpl. 36, 431–435. doi:10.1038/sj.bmt.1705072
Bridgeman, M. B., and Chavez, B. (2015). Febuxostat for the treatment of gout. Expert Opin. Pharmacother. 16, 395–398. doi:10.1517/14656566.2015.985588
Delie, A., Verlinden, A., Beel, K., Deeren, D., Mazure, D., Baron, F., et al. (2021). Use of chimerism analysis after allogeneic stem cell transplantation: Belgian guidelines and review of the current literature. Acta Clin. Belg 76, 500–508. doi:10.1080/17843286.2020.1754635
Dobonici, M., Podlech, J., Steffens, H. P., Maiberger, S., and Reddehase, M. J. (1998). Evidence against a key role for transforming growth factor-beta1 in cytomegalovirus-induced bone marrow aplasia. J. Gen. Virol. 79 (Pt 4), 867–876. doi:10.1099/0022-1317-79-4-867
Düver, F., Weißbrich, B., Eyrich, M., Wölfl, M., Schlegel, P. G., and Wiegering, V. (2020). Viral reactivations following hematopoietic stem cell transplantation in pediatric patients - a single center 11-year analysis. PLoS One 15, e0228451. doi:10.1371/journal.pone.0228451
Eapen, M., Rubinstein, P., Zhang, M. J., Stevens, C., Kurtzberg, J., Scaradavou, A., et al. (2007). Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet 369, 1947–1954. doi:10.1016/S0140-6736(07)60915-5
Ernst, M. E., and Fravel, M. A. (2009). Febuxostat: a selective xanthine-oxidase/xanthine-dehydrogenase inhibitor for the management of hyperuricemia in adults with gout. Clin. Ther. 31, 2503–2518. doi:10.1016/j.clinthera.2009.11.033
Gluckman, E., Rocha, V., Boyer-Chammard, A., Locatelli, F., Arcese, W., Pasquini, R., et al. (1997). Outcome of cord-blood transplantation from related and unrelated donors. Eurocord transplant group and the European blood and marrow transplantation group. N. Engl. J. Med. 337, 373–381. doi:10.1056/NEJM199708073370602
Henter, J. I., Elinder, G., and Ost, A. (1991). Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL study group of the histiocyte society. Semin. Oncol. 18, 29–33.
Hill, J. A., Mayer, B. T., Xie, H., Leisenring, W. M., Huang, M. L., Stevens-Ayers, T., et al. (2017). The cumulative burden of double-stranded DNA virus detection after allogeneic HCT is associated with increased mortality. Blood 129, 2316–2325. doi:10.1182/blood-2016-10-748426
Imamura, M. (2021). Impaired hematopoiesis after allogeneic hematopoietic stem cell transplantation: its pathogenesis and potential treatments. Hemato 2, 43–63. doi:10.3390/hemato2010002
Janka, G. E., and Lehmberg, K. (2013). Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol. Am. Soc. Hematol. Educ. Program 2013, 605–611. doi:10.1182/asheducation-2013.1.605
Jordan, A., and Gresser, U. (2018). Side effects and interactions of the xanthine oxidase inhibitor febuxostat. Pharm. (Basel) 11, 51. doi:10.3390/ph11020051
Kernan, N. A., Flomenberg, N., Dupont, B., and O'reilly, R. J. (1987). GRAFT rejection in recipients of T-CELL-DEPLETED HLA-NONIDENTICAL marrow transplants for leukemia: identification of host-derived antidonor allocytotoxic T Lymphocytes1. Transplantation 43, 842–847. doi:10.1097/00007890-198743060-00014
Klein, O. R., Bonfim, C., Abraham, A., Ruggeri, A., Purtill, D., Cohen, S., et al. (2023). Transplant for non-malignant disorders: an International Society for Cell and Gene Therapy Stem Cell Engineering Committee report on the role of alternative donors, stem cell sources and graft engineering. Cytotherapy 25, 463–471. doi:10.1016/j.jcyt.2022.12.005
Kobayashi, S., Ogura, M., and Hosoya, T. (2013). Acute neutropenia associated with initiation of febuxostat therapy for hyperuricaemia in patients with chronic kidney disease. J. Clin. Pharm. Ther. 38, 258–261. doi:10.1111/jcpt.12057
Kurtzberg, J., Prasad, V. K., Carter, S. L., Wagner, J. E., Baxter-Lowe, L. A., Wall, D., et al. (2008). Results of the Cord Blood Transplantation Study (COBLT): clinical outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with hematologic malignancies. Blood 112, 4318–4327. doi:10.1182/blood-2007-06-098020
Lee, M. N., Kang, B., Choi, S. Y., Kim, M. J., Woo, S. Y., Kim, J. W., et al. (2015). Impact of genetic polymorphisms on 6-thioguanine nucleotide levels and toxicity in pediatric patients with IBD treated with azathioprine. Inflamm. Bowel Dis. 21, 2897–2908. doi:10.1097/MIB.0000000000000570
Locatelli, F., Antmen, B., Kang, H. J., Koh, K., Takahashi, Y., Kupesiz, A., et al. (2024). Ruxolitinib in treatment-naive or corticosteroid-refractory paediatric patients with chronic graft-versus-host disease (REACH5): interim analysis of a single-arm, multicentre, phase 2 study. Lancet Haematol. 11, e580–e592. doi:10.1016/S2352-3026(24)00174-1
Locatelli, F., Lucarelli, B., and Merli, P. (2014). Current and future approaches to treat graft failure after allogeneic hematopoietic stem cell transplantation. Expert Opin. Pharmacother. 15, 23–36. doi:10.1517/14656566.2014.852537
Martínez-Jaramillo, G., Gómez-Morales, E., Sánchez-Valle, E., and Mayani, H. (2001). Severe hematopoietic alterations in vitro, in bone marrow transplant recipients who develop graft-versus-host disease. J. Hematother Stem Cell Res. 10, 347–354. doi:10.1089/152581601750288957
Masouridi-Levrat, S., Simonetta, F., and Chalandon, Y. (2016). Immunological basis of bone marrow failure after allogeneic hematopoietic stem cell transplantation. Front. Immunol. 7, 362. doi:10.3389/fimmu.2016.00362
Mattsson, J., Ringdén, O., and Storb, R. (2008). Graft failure after allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transpl. 14, 165–170. doi:10.1016/j.bbmt.2007.10.025
Mayer, A., Podlech, J., Kurz, S., Steffens, H. P., Maiberger, S., Thalmeier, K., et al. (1997). Bone marrow failure by cytomegalovirus is associated with an in vivo deficiency in the expression of essential stromal hemopoietin genes. J. Virol. 71, 4589–4598. doi:10.1128/JVI.71.6.4589-4598.1997
Mukoyoshi, M., Nishimura, S., Hoshide, S., Umeda, S., Kanou, M., Taniguchi, K., et al. (2008). In vitro drug-drug interaction studies with febuxostat, a novel non-purine selective inhibitor of xanthine oxidase: plasma protein binding, identification of metabolic enzymes and cytochrome P450 inhibition. Xenobiotica 38, 496–510. doi:10.1080/00498250801956350
Müskens, K. F., Lindemans, C. A., and Belderbos, M. E. (2021). Hematopoietic dysfunction during graft-versus-host disease: a self-destructive process? Cells 10, 2051. doi:10.3390/cells10082051
Nakamae, H., Storer, B., Sandmaier, B. M., Maloney, D. G., Davis, C., Corey, L., et al. (2011). Cytopenias after day 28 in allogeneic hematopoietic cell transplantation: impact of recipient/donor factors, transplant conditions and myelotoxic drugs. Haematologica 96, 1838–1845. doi:10.3324/haematol.2011.044966
Olsson, R., Remberger, M., Schaffer, M., Berggren, D. M., Svahn, B. M., Mattsson, J., et al. (2013). Graft failure in the modern era of allogeneic hematopoietic SCT. Bone Marrow Transpl. 48, 537–543. doi:10.1038/bmt.2012.239
Petersen, S. L. (2007). Alloreactivity as therapeutic principle in the treatment of hematologic malignancies. Studies of clinical and immunologic aspects of allogeneic hematopoietic cell transplantation with nonmyeloablative conditioning. Dan. Med. Bull. 54, 112–139.
Poh, X. E., Lee, C. T., and Pei, S. N. (2017). Febuxostat-induced agranulocytosis in an end-stage renal disease patient: a case report. Med. Baltim. 96, e5863. doi:10.1097/MD.0000000000005863
Remberger, M., Watz, E., Ringdén, O., Mattsson, J., Shanwell, A., and Wikman, A. (2007). Major ABO blood group mismatch increases the risk for graft failure after unrelated donor hematopoietic stem cell transplantation. Biol. Blood Marrow Transpl. 13, 675–682. doi:10.1016/j.bbmt.2007.01.084
Renzaho, A., Podlech, J., Kühnapfel, B., Blaum, F., Reddehase, M. J., and Lemmermann, N. a.W. (2020). Cytomegalovirus-associated inhibition of hematopoiesis is preventable by cytoimmunotherapy with antiviral CD8 T cells. Front. Cell Infect. Microbiol. 10, 138. doi:10.3389/fcimb.2020.00138
Rocha, V., Cornish, J., Sievers, E. L., Filipovich, A., Locatelli, F., Peters, C., et al. (2001). Comparison of outcomes of unrelated bone marrow and umbilical cord blood transplants in children with acute leukemia. Blood 97, 2962–2971. doi:10.1182/blood.v97.10.2962
Rubinstein, P., Carrier, C., Scaradavou, A., Kurtzberg, J., Adamson, J., Migliaccio, A. R., et al. (1998). Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N. Engl. J. Med. 339, 1565–1577. doi:10.1056/NEJM199811263392201
Salit, R. B. (2022). The role of JAK inhibitors in hematopoietic cell transplantation. Bone Marrow Transpl. 57, 857–865. doi:10.1038/s41409-022-01649-y
Salzberger, B., Bowden, R. A., Hackman, R. C., Davis, C., and Boeckh, M. (1997). Neutropenia in allogeneic marrow transplant recipients receiving ganciclovir for prevention of cytomegalovirus disease: risk factors and outcome. Blood 90, 2502–2508. doi:10.1182/blood.v90.6.2502.2502_2502_2508
Steffens, H. P., Podlech, J., Kurz, S., Angele, P., Dreis, D., and Reddehase, M. J. (1998). Cytomegalovirus inhibits the engraftment of donor bone marrow cells by downregulation of hemopoietin gene expression in recipient stroma. J. Virol. 72, 5006–5015. doi:10.1128/JVI.72.6.5006-5015.1998
Tayar, J. H., Lopez-Olivo, M. A., and Suarez-Almazor, M. E. (2012). Febuxostat for treating chronic gout. Cochrane Database Syst. Rev. 11, Cd008653. doi:10.1002/14651858.CD008653.pub2
Urbano-Ispizua, A., Rozman, C., Pimentel, P., Solano, C., De La Rubia, J., Brunet, S., et al. (2001). The number of donor CD3(+) cells is the most important factor for graft failure after allogeneic transplantation of CD34(+) selected cells from peripheral blood from HLA-identical siblings. Blood 97, 383–387. doi:10.1182/blood.v97.2.383
Voogt, P. J., Fibbe, W. E., Marijt, W. A., Goulmy, E., Veenhof, W. F., Hamilton, M., et al. (1990). Rejection of bone-marrow graft by recipient-derived cytotoxic T lymphocytes against minor histocompatibility antigens. Lancet 335, 131–134. doi:10.1016/0140-6736(90)90003-n
Yamasaki, Y., Ieiri, I., Kusuhara, H., Sasaki, T., Kimura, M., Tabuchi, H., et al. (2008). Pharmacogenetic characterization of sulfasalazine disposition based on NAT2 and ABCG2 (BCRP) gene polymorphisms in humans. Clin. Pharmacol. Ther. 84, 95–103. doi:10.1038/sj.clpt.6100459
Keywords: hematopoietic stem cell recipient, pediatric, hyperuricemia, febuxostat, nonchemotherapy drug-induced agranulocytosis
Citation: Curci D, Braidotti S and Maximova N (2024) Febuxostat-induced agranulocytosis in a pediatric hematopoietic stem cell transplant recipient: Case Report and literature review. Front. Pharmacol. 15:1478381. doi: 10.3389/fphar.2024.1478381
Received: 09 August 2024; Accepted: 30 September 2024;
Published: 23 October 2024.
Edited by:
Margherita Neri, University of Ferrara, ItalyReviewed by:
Catherine M. T. Sherwin, University of Western Australia, AustraliaCopyright © 2024 Curci, Braidotti and Maximova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalia Maximova, bmF0YWxpYS5tYXhpbW92YUBidXJsby50cmllc3RlLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.