 Sameh Saber1*
Sameh Saber1* Rasha Abdelhady2,3
Rasha Abdelhady2,3 Mai A. Elhemely4*
Mai A. Elhemely4* Elsayed A. Elmorsy5,6*
Elsayed A. Elmorsy5,6* Rabab S. Hamad7,8*
Rabab S. Hamad7,8* Mustafa Ahmed Abdel-Reheim9,10*
Mustafa Ahmed Abdel-Reheim9,10* Attalla F. El-kott11,12
Attalla F. El-kott11,12 Mohammed A. AlShehri11
Mohammed A. AlShehri11 Kareem Morsy11,13Ali S. AlSheri11
Kareem Morsy11,13Ali S. AlSheri11 Mahmoud E. Youssef1
Mahmoud E. Youssef1- 1Department of Pharmacology, Faculty of Pharmacy, Delta University for Science and Technology, Gamasa, Egypt
- 2Pharmacology and Toxicology Department, Faculty of Pharmacy, Fayoum University, Fayoum, Egypt
- 3Pharmacology and Toxicology Department, Faculty of Pharmacy, Egyptian Chinese University, Cairo, Egypt
- 4School of Medical Sciences, Faculty of Biology, Medicine and Health, The University of Manchester, Manchester, United Kingdom
- 5Department of Pharmacology and Therapeutics, College of Medicine, Qassim University, Buraidah, Saudi Arabia
- 6Department of Clinical Pharmacology, Faculty of Medicine, Mansoura University, Mansoura, Egypt
- 7Biological Sciences Department, College of Science, King Faisal University, Al Ahsa, Saudi Arabia
- 8Central Laboratory, Theodor Bilharz Research Institute, Giza, Egypt
- 9Department of Pharmaceutical Sciences, College of Pharmacy, Shaqra University, Shaqra, Saudi Arabia
- 10Department of Pharmacology and Toxicology, Faculty of Pharmacy, Beni-Suef University, Beni Suef, Egypt
- 11Department of Biology, College of Science, King Khalid University, Abha, Saudi Arabia
- 12Department of Zoology, Faculty of Science, Damanhour University, Damanhour, Egypt
- 13Department of Zoology, Faculty of Science, Cairo University, Cairo, Egypt
Heat shock protein 90 (HSP90) is a pivotal molecular chaperone with multifaceted roles in cellular health and disease. Herein, we explore how HSP90 orchestrates cellular stress responses, particularly through its partnership with heat shock factor 1 (HSF-1). PU-H71, a selective inhibitor of HSP90, demonstrates significant potential in cancer therapy by targeting a wide array of oncogenic pathways. By inducing the degradation of multiple client proteins, PU-H71 disrupts critical signaling pathways such as MAPK, PI3K/Akt, JAK/STAT, EGFR, and mTOR, which are essential for cancer cell survival, proliferation, and metastasis. We examined its impact on combating triple-negative breast cancer and enhancing the effectiveness of carbon-ion beam therapy, offering new avenues for cancer treatment. Furthermore, the dual inhibition of HSP90A and HSP90B1 by PU-H71 proves highly effective in the context of myeloma, providing fresh hope for patients with this challenging malignancy. We delve into its potential to induce apoptosis in B-cell lymphomas that rely on Bcl6 for survival, highlighting its relevance in the realm of hematologic cancers. Shifting our focus to hepatocellular carcinoma, we explore innovative approaches to chemotherapy. Moreover, the current review elucidates the potential capacity of PU-H71 to suppress glial cell activation paving the way for developing novel therapeutic strategies for neuroinflammatory disorders. Additionally, the present report also suggests the promising role of PU-H71 in JAK2-dependent myeloproliferative neoplasms. Eventually, our report sheds more light on the multiple functions of HSP90 protein as well as the potential therapeutic benefit of its selective inhibitor PU-H71 in the context of an array of diseases, laying the foundations for the development of novel therapeutic approaches that could achieve better treatment outcomes.
1 Introduction
Heat shock proteins (HSPs) denote a class of highly conserved proteins playing a pivotal role in safeguarding the cells against several forms of stress. Notably, HSPs are central to a range of cellular functions including protein folding, unfolding, assembly, and disassembly. According to their molecular weight, HSPs are classified into main four subgroups, namely, HSP90, HSP70, HSP60, and the small HSP family (Table 1) (Hu et al., 2022). Conspicuously, HSP90 is a leading chaperone protein indispensable for correcting misfolded proteins. On the other hand, HSP70 functions by binding to partially unfolded protein fragments inhibiting their clumping. Meanwhile, HSP60 plays a fundamental role in the proper folding of proteins within specific cellular compartments (e.g., mitochondria). Lastly, the small HSP family, including HSP27, suppresses the aggregation of misfolded proteins (Navarro-Zaragoza et al., 2021). In times of stress, such as exposure to high temperatures, HSPs play a critical role in cell survival by latching onto and stabilizing damaged or partially unfolded proteins until the stressful conditions subside and normal cellular conditions are restored (Kakkar et al., 2014). This allows the proteins to undergo proper folding with the assistance of HSPs. The initial, inactivated state of the HSP90 chaperone cycle is denoted by the closed conformation. Upon binding of ATP, a structural alteration occurs in HSP90, causing it to transition into the open state. In this open state, HSP90 exhibits increased ATPase activity and becomes receptive to client proteins, which can then engage with it for protein folding (Figure 1) (Youssef et al., 2023). HSPs are classified according to their broad structural classes as described in Table 1.
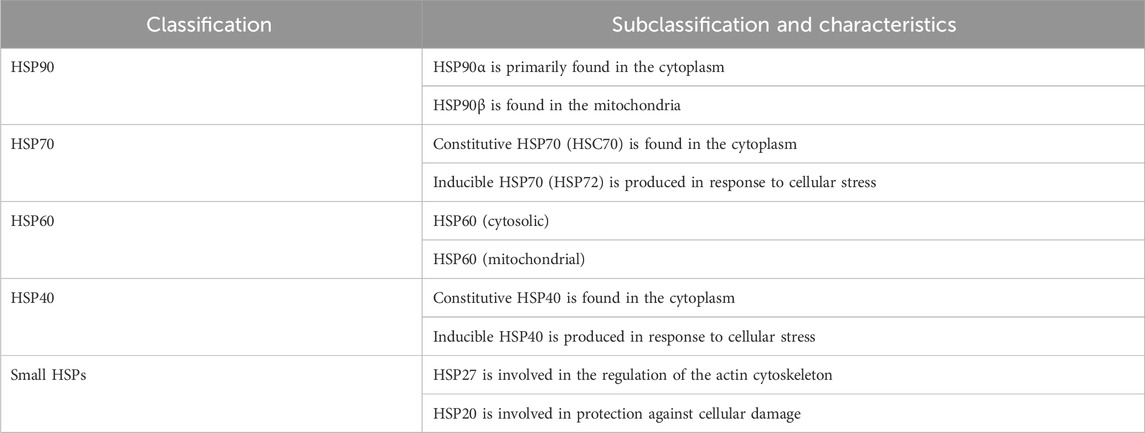
Table 1. Classification and characteristics of HSPs.
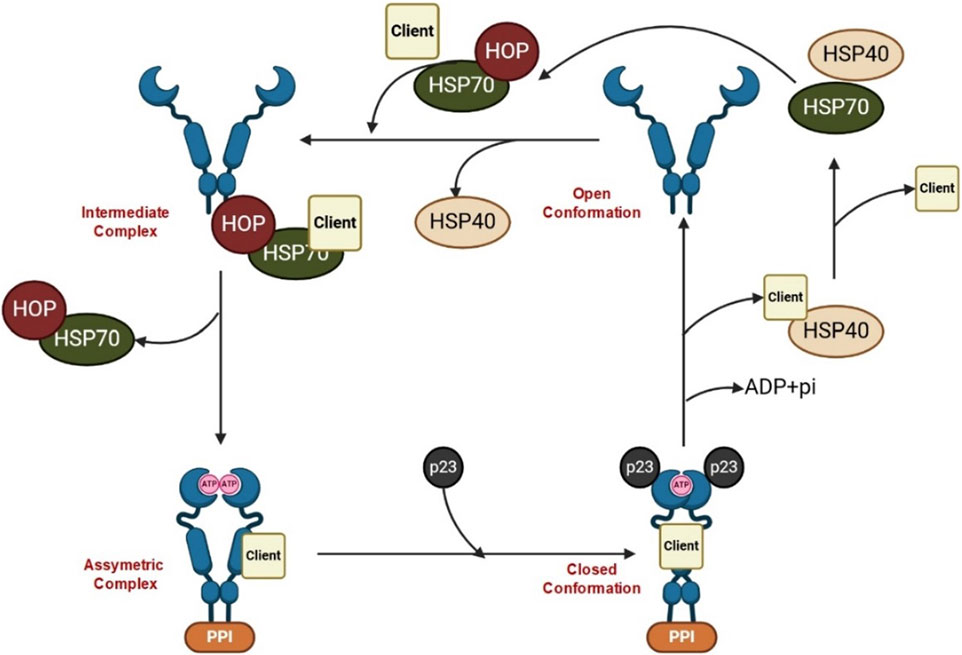
Figure 1. HSP90 cycle.
Inactive Hsp90 (open conformation) forms a dimer at its C terminus, leaving the N termini unassociated, while the Hsp70-captures client proteins and delivers them to Hsp90 with the help of co-chaperones. The co-chaperone Hop binds to both Hsp70 and Hsp90 through its TPR domains, acting as a scaffold to facilitate the transfer of the client protein from Hsp70 to Hsp90 (intermediate complex). Additional co-chaperones, including peptidyl-prolyl cis-trans isomerases (PPIase), associate with Hsp90, leading to the formation of an asymmetric complex. Subsequently, Hsp70 and Hop dissociate, and the co-chaperone p23 replaces them, forming the closed late conformation, where ATP hydrolysis occurs, resulting in the release of the client protein.
Heat shock protein 90 (HSP90) plays a crucial role as a key molecular helper, vital for safeguarding the functional stability and survival of cells when confronted with changing stress factors. Within the purview of HSPs, HSP90 has gained rising attention, fundamentally this could be ascribed to its crucial role in an array of normal physiological cellular processes as well as those correlated with disease progression. Through direct connections, HSP90 was reported to keep proteins associated with cell death in a state that is resistant to programmed cell death (Peng et al., 2022). The wide range of functions exerted by HSP90 is likely attributed to its ability to serve as a helper for numerous client proteins playing indispensable roles in health disorders such as cancer and neurodegenerative diseases (Sumi and Ghosh, 2022).
Targeting and inhibition of HSP90 were achieved by a class of drugs that have been classified as HSP90 inhibitors capable of destabilizing and degrading HSP90 client proteins, with subsequent suppression of multiple oncogenic signaling pathways simultaneously. This multi-target approach suggests that HSP90 inhibitors could be promising therapeutic agents for various neoplasms (Magwenyane et al., 2022). Notably, HSP90 inhibitors, such as geldanamycin derivatives (e.g., 17-AAG and 17-DMAG) and resorcinol-based compounds (e.g., ganetespib), displayed potent anti-cancer activity in preclinical trials and are currently undergoing clinical trials evaluating their safety profiles. The therapeutic benefit of HSP90 inhibitors could potentially exceed anticancer therapy. Markedly, ongoing studies are investigating their potential benefit in neurodegenerative disorders and other health conditions (Luo et al., 2010). HSP90 inhibitors are summarized in Table 2. Table 2 shows the classes of HSP90 inhibitors. The structural diversity of HSP90 inhibitors allows for the development of various drug candidates for cancer and other diseases.

Table 2. HSP90 inhibitors.
2 Role of HSP90 in tumorigenesis
HSP90 is a crucial molecular chaperone that plays a significant role in cancer biology by facilitating the proper folding, stabilization, and functional regulation of numerous proteins that are essential for tumorigenesis (Birbo et al., 2021). As a prominent member of the heat shock protein family, HSP90 assists in maintaining cellular proteostasis, particularly under the stress conditions often encountered in cancer cells. Its importance in cancer has led to extensive research focusing on HSP90 as a therapeutic target, given its involvement in various oncogenic processes.
HSP90 is known to interact with over 200 client proteins, many of which are involved in critical signaling pathways that control cell growth, survival, and metastasis (Kamal et al., 2004; Xu and Neckers, 2007; Sun et al., 2021). Among these client proteins are receptor tyrosine kinases such as human epidermal growth factor receptor 2 (HER2) and epidermal growth factor (EGFR) (Wang et al., 2016), which are essential for transmitting growth signals from the extracellular environment into the cell interior. HSP90 also interacts with key signaling molecules such as AKT and SRC, which are critical for cell survival and proliferation (Rong and Yang, 2018). HSP90 is also required for the stability and function of transcription factors such as hypoxia inducible factor (HIF-1) and p53, which are responsible for regulating hypoxia responses and controlling the cell cycle, respectively (Albadari et al., 2019; Shirai et al., 2021).
Furthermore, HSP90 stabilizes cell cycle regulators such as CDK4 and CDK6, which are required for cell cycle progression (Sager et al., 2022). In cancer cells, HSP90 protects mutated and overexpressed oncoproteins that would otherwise be degraded. This protective mechanism is especially important because cancer cells frequently experience high levels of cellular stress as a result of rapid proliferation and abnormal protein synthesis. As a result, HSP90 expression levels are typically two to ten times higher in cancer cells than in normal cells (Solárová et al., 2015; Birbo et al., 2021). This overexpression has been linked to a poor prognosis and increased resistance to various therapeutic interventions, highlighting the role of HSP90 in cancer progression.
Hanahan and Weinberg identified several hallmarks of cancer in which HSP90 plays a role. These characteristics include self-sufficiency in growth signals, apoptosis evasion, long-term angiogenesis, and increased tissue invasion and metastasis (Hanahan and Weinberg, 2011). HSP90 allows tumor cells to thrive even in adverse conditions such as nutrient deprivation or therapeutic stress by stabilizing oncogenic proteins involved in these processes (Jaeger and Whitesell, 2019). This ability to support malignant characteristics highlights the potential of targeting HSP90 in cancer therapy as a way to disrupt these critical pathways and induce tumor regression. Understanding the post-translational modifications (PTMs) of HSP90, such as phosphorylation, acetylation, and S-nitrosylation, is critical because they regulate chaperone activity and influence cancer development (Caruso Bavisotto et al., 2020).
3 Post-translational modifications of HSP90
3.1 Phosphorylation: key modulator of HSP90 function
Phosphorylation is one of the most studied HSP90 PTMs, and it has a significant impact on chaperone activity. Several kinases, such as SRC, BRAF, and casein kinase II (CK2), phosphorylate HSP90, affecting its ability to interact with client proteins (Bachman et al., 2018; Firnau and Brieger, 2022; Zhang et al., 2022). For example, SRC phosphorylates HSP90 at Tyr301, which is required for vascular endothelial growth factor receptor 2 (VEGFR2)-mediated angiogenesis, a critical process in tumor growth (Miyata et al., 2013). HSP90 modulates apoptosome formation via phosphorylation of Ser226 and Ser255, highlighting its role in apoptosis regulation (Mollapour and Neckers, 2012). In leukemia, reduced phosphorylation at these sites strengthens the interaction between HSP90 and APAF1, inhibiting cytochrome c-induced apoptosome assembly and contributing to chemoresistance (Ho et al., 2012).
Client kinases such as WEE1 phosphorylate HSP90, resulting in a feedback loop that improves its chaperoning ability for other kinases such as HER2 and CDK1 (Zhang et al., 2024). This interaction demonstrates the complexities of HSP90’s regulatory mechanisms in cancer. Interestingly, WEE1-mediated phosphorylation can reduce the efficacy of HSP90 inhibitors such as geldanamycin and 17-AAG, posing a challenge for therapeutic strategies. A better understanding of phosphorylation sites may lead to new approaches for sensitizing cancer cells to HSP90 inhibitors and overcoming resistance (Mollapour et al., 2010; Iwai et al., 2012; Lokeshwar, 2012).
3.2 Acetylation: impact on client protein stability
Acetylation of HSP90 is another important post-translational modification that can affect its chaperone activity. This modification frequently causes destabilization of client proteins, as seen when histone deacetylase (HDAC) inhibitors or HDAC6 silencing increase HSP90 acetylation levels (Liu P. et al., 2021). Acetylation of several lysine residues on HSP90 impairs its ability to bind client proteins and co-chaperones, including p23, resulting in decreased chaperone efficiency (Woodford et al., 2016; Liu et al., 2023). Importantly, the combination of HDAC inhibitors and HSP90 inhibitors has synergistic effects in certain cancers, particularly leukemia, while antagonistic effects are seen in some solid tumors (Krämer et al., 2014; Chen et al., 2020). These findings emphasize the tumor-specific nature of HSP90 acetylation and the complexities of using combination therapies.
3.3 S-nitrosylation: nitric oxide’s role in HSP90 inhibition
S-nitrosylation, or the modification of HSP90 by nitric oxide (NO) at Cys597, adds another layer of regulation by inhibiting the chaperone’s ATPase activity. This PTM impairs HSP90’s ability to stabilize client proteins, especially in endothelial cells where NO-mediated S-nitrosylation inhibits angiogenesis (Siragusa and Fleming, 2016; Rizza and Filomeni, 2020). Interestingly, exogenous NO can inhibit HSP90 in cancer cells, resulting in telomere shortening and reduced telomerase activity, both of which are required for cancer cell immortality (Trepel et al., 2010; Solárová et al., 2015). This finding lends credence to the theory that manipulating NO levels may have therapeutic potential in HSP90-driven cancers.
3.4 HSP90’s role in nuclear and chromatin events
Aside from its cytoplasmic functions, HSP90 influences several nuclear events, including the regulation of steroid hormone receptors, such as androgen and estrogen receptors. These receptors, which move between the cytoplasm and the nucleus, are essential in hormone-driven cancers such as breast and prostate cancer (Baker et al., 2018; Kaziales et al., 2020). HSP90 regulates their localization, stability, and transcriptional activity, influencing cancer progression.
HSP90 also interacts with key transcription factors such as heat shock transcription factor 1 (HSF1), which controls the expression of heat shock proteins in response to stress. Inhibition of HSP90 can prolong HSF1 activity, resulting in enhanced heat shock responses, which could be used in cancer therapy (Kijima et al., 2019; Cyran and Zhitkovich, 2022). Furthermore, HSP90 stabilizes the transcriptional repressor BCL6 in diffuse large B-cell lymphoma, HSP90 stabilizes the transcriptional repressor BCL6 in diffuse large B-cell lymphoma, which promotes tumorigenesis by inhibiting tumor suppressor genes such as TP53 (Xu-Monette et al., 2012). Targeting HSP90 in such situations may deactivate these genes and promote cancer cell apoptosis.
HSP90’s involvement in chromatin remodeling expands its role in cancer. The chaperone promotes the activity of chromatin-modifying enzymes, including the histone methyltransferase SMYD3, which is overexpressed in hepatocellular carcinoma and colorectal cancer. HSP90 increases SMYD3 activity, which contributes to the transcription of genes that promote cancer cell proliferation (Abu-Farha et al., 2011; Giakountis et al., 2017). Inhibiting HSP90 in these cancers can reduce SMYD3 activity, providing another therapeutic target.
4 HSP90 and heat shock factor 1 (HSF1)
Under stress, the heat shock response (HSR) is stimulated/triggered, leading to the upregulation of various cytoprotective HSPs. HSPs mediate cellular protective effects by alleviating the drastic consequences of aberrantly folded and distorted proteins, thus lessening their detrimental effects on the cell (Collier and Benesch, 2020). In mammals, cellular response to stress could result in destroying cellular proteins and it is principally regulated by the activation of HSF-1. Significantly, HSF-1 could regulate the expression of heat shock genes (Chang et al., 2021). Of note, HSF-1 activation involves multiple steps that start with oligomerization, then nuclear translocation and accumulation, and finally undergo post-translational modifications (Masser et al., 2020). HSP90 is implicated in controlling the activity of HSF. HSP90 forms a binding interaction with HSF-1, maintaining the monomeric form of the transcription factor under normal, non-stressed conditions. When the cell experiences stressors, such as exposure to elevated temperatures (heat shock), or when the function of HSP90 is disrupted, HSF-1 becomes detached from the HSP90 complex (Chin et al., 2023). This release leads to the activation of HSF-1 and its transformation into a trimeric form, and it then moves into the cell nucleus, where HSF-1 initiates an HSR, marked by the synthesis of HSPS such as HSP70 and HSP40 activator (Schmauder et al., 2022).
5 PU-H71
PU-H71 (Figure 2) is a potent small-molecule inhibitor that targets HSP90. HSP90 is frequently overexpressed in various cancers, contributing to the stabilization and activity of numerous proteins involved in tumor growth and survival, making it a critical therapeutic target (Mahajan et al., 2024; Rastogi et al., 2024).
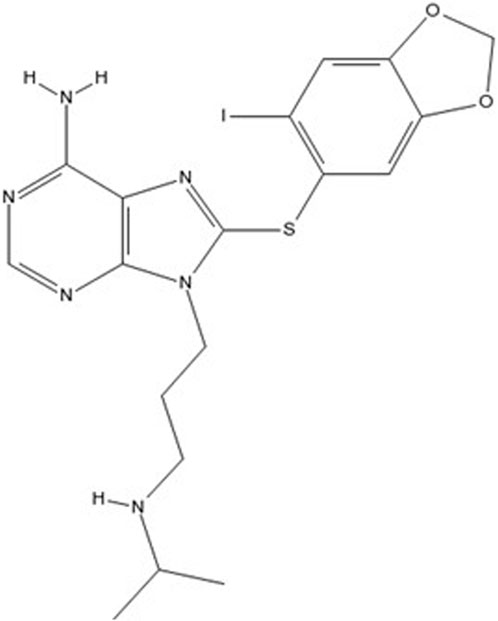
Figure 2. Chemical structure of PU-H71 (8-[(6-iodo-1,3-benzodioxol-5-yl)sulfanyl]-9-[3-(propan-2-ylamino)propyl]purin-6-amine).
PU-H71 binds with high affinity to the ATP-binding site of HSP90, inhibiting its chaperone activity. This binding disrupts the proper folding of client proteins, leading to their ubiquitination and subsequent proteasomal degradation (Schopf et al., 2017). The loss of these client proteins impairs several oncogenic signaling pathways, thereby inhibiting tumor cell proliferation and inducing apoptosis.
Preclinical studies have demonstrated that PU-H71 exhibits significant antitumor activity across a variety of cancer cell lines and xenograft models, particularly those dependent on HSP90 client proteins for growth and survival. These studies have highlighted the ability of PU-H71 to induce tumor regression and prolong survival without causing significant toxicity to normal tissues (Ambati et al., 2014; Trendowski, 2015; Martinet et al., 2022).
Clinical evaluations of PU-H71 have also shown promise. Phase I clinical trials have focused on determining the maximum tolerated dose, pharmacokinetics, and preliminary efficacy of PU-H71 in patients with advanced solid tumors and hematological malignancies. Results from these studies indicate that PU-H71 is well-tolerated and demonstrates antitumor activity, with manageable side effects predominantly involving gastrointestinal disturbances and reversible transaminase elevations. PU-H71 has been included in six clinical studies (NCT03166085; NCT03373877; NCT01581541), of which three (NCT03935555; NCT01393509; NCT01269593) are still active or recruiting, indicating that PU-H71 is the most promising purine-based inhibitor of HSP90 (Gerecitano et al., 2015; Speranza et al., 2018; Pemmaraju et al., 2019).
5.1 PU-H71: Discovery
PU-H71 was developed through a rational drug design approach aimed at creating more selective and potent inhibitors of HSP90, compared to previous generations. It is part of a class of compounds synthesized from a purine scaffold, designed to improve both the specificity and solubility of the drug. Earlier compounds like PU3 and PU24FCl, which also targeted HSP90, were optimized through structure-activity relationship studies, refining molecular structures to enhance binding affinity and therapeutic efficacy (Messaoudi et al., 2008). Through this process, PU-H71 emerged as one of the most effective inhibitors, with an IC50 value of approximately 50 nM for HSP90 in epichaperome-containing cells (Ambati et al., 2014).
5.2 Pharmacokinetics of PU-H71
The pharmacokinetics of PU-H71 were thoroughly investigated during clinical trials. One of the key findings was the compound’s mean terminal half-life of approximately 8.4 ± 3.6 h, indicating a relatively stable elimination profile across the range of doses tested (Speranza et al., 2018). Biodistribution studies utilizing positron emission tomography imaging revealed that PU-H71 selectively accumulates in tumor tissues that express epichaperomes, with a tumor retention rate observed in about 58% of patients (Dunphy et al., 2020). This tumor-selective accumulation is a promising feature that underscores the drug’s potential as a targeted therapy.
In terms of metabolism, in vitro studies revealed that PU-H71 undergoes limited biotransformation, with the primary metabolic pathways involving S-oxidation and O-demethylation (Speranza et al., 2018). Metabolites were present in plasma samples but accounted for less than 10% of the parent drug’s concentration, suggesting that metabolism has a minimal impact on the overall pharmacologic activity of PU-H71 (Speranza et al., 2018).
5.3 Development of PU-H71
Preclinical studies have demonstrated that PU-H71 exhibits significant antitumor activity across a variety of cancer cell lines and xenograft models, particularly those dependent on HSP90 client proteins for growth and survival. These studies have highlighted the ability of PU-H71 to induce tumor regression and prolong survival without causing significant toxicity to normal tissues (Ambati et al., 2014; Trendowski, 2015; Martinet et al., 2022).
Following successful preclinical testing, the first-in-human clinical trial of PU-H71 was initiated to evaluate its safety, tolerability, and pharmacokinetic profile. This phase I study involved patients with advanced solid tumors who had not responded to standard therapies. The trial followed a dose-escalation design, with doses ranging from 10 to 470 mg/m2, administered intravenously on days 1 and 8 of a 21-day treatment cycle (De Almeida et al., 2020). The results indicated that PU-H71 was well-tolerated at the tested doses, with no dose-limiting toxicities observed. However, some patients experienced grade 2 and 3 adverse events, which were closely monitored. The positive safety profile allowed for the continuation of the drug’s clinical evaluation in subsequent trials. PU-H71 has been included in six clinical studies (NCT03166085; NCT03373877; NCT01581541), of which three (NCT03935555; NCT01393509; NCT01269593) are still active or recruiting, indicating that PU-H71 is the most promising purine-based inhibitor of HSP90 (Gerecitano et al., 2015; Speranza et al., 2018; Pemmaraju et al., 2019).
6 Mechanism of action of PU-H71
PU-H71 demonstrates a high degree of selectivity for the ATP-binding sites of HSP90 that are specifically integrated into tumor epichaperome complexes (Dunphy et al., 2020). This selective binding is of paramount importance, as it significantly reduces the potential for off-target effects on healthy tissues where HSP90 does not exist in this altered form (Speranza et al., 2018; Li and Luo, 2023). The unique conformational landscape of HSP90 in tumors allows PU-H71 to engage with its ATP-binding sites effectively (Trendowski, 2015; Tramentozzi and Finotti, 2019). Upon binding, PU-H71 induces critical conformational changes in the HSP90 structure, which stabilizes client protein-HSP90 complexes. This stabilization is not merely a passive interaction; it actively facilitates the recruitment of these complexes to the proteasome, the cellular machinery responsible for degrading misfolded or unneeded proteins (Caldas-Lopes et al., 2009; Li et al., 2018). Consequently, this process leads to the degradation of oncogenic proteins that are essential for tumor cell survival and proliferation. By disrupting the stability of these proteins, PU-H71 effectively undermines the tumor’s ability to maintain its malignant characteristics, thereby presenting a targeted therapeutic strategy against cancer.
The epichaperome represents a highly interconnected network of chaperone proteins that plays a critical role in supporting various cellular processes associated with tumor biology, including proliferation, survival, and invasion (Hadizadeh Esfahani et al., 2018). Within this network, HSP90 acts as a central hub, facilitating the proper folding and function of numerous client proteins that are often dysregulated in cancer. When PU-H71 inhibits HSP90’s activity, it disrupts the integrity of this epichaperome network. The destabilization caused by PU-H71 leads to a significant reduction in the functionality of the epichaperome, impairing its ability to support tumor cell processes. As a result, tumor cells become more susceptible to stressors and less capable of maintaining their survival and proliferative advantages (Speranza et al., 2018; Jhaveri et al., 2020). Ultimately, this disruption contributes to increased tumor cell death, highlighting PU-H71’s potential as an effective therapeutic agent in targeting cancer through the modulation of chaperone networks. By interfering with the epichaperomes’ formation and function, PU-H71 not only targets individual oncogenic proteins but also undermines the broader chaperone-dependent mechanisms that tumors rely on for their growth and resilience.
7 Molecular mechanisms of PU-H71
Targeting HSP90 by PU-H71 disrupts multiple signaling pathways critical for cancer cell survival, proliferation, and metastasis. The compound exhibits a broad spectrum of anti-cancer activities by inducing the degradation of client proteins involved in key pathways such as Mitogen-Activated Protein Kinase (MAPK), Phosphoinositide 3-Kinase (PI3K)/protein kinase B (Akt), and Janus kinase (JAK)/signal transducers and activators of transcription (STAT) (Figure 3). Additionally, PU-H71 enhances the efficacy of other therapeutic agents and sensitizes cancer cells to treatments like radiation. This multi-faceted approach makes PU-H71 a promising candidate for cancer therapy, addressing the complexity and heterogeneity of tumor biology. Table 3 outlines the various mechanisms through which PU-H71 exerts its anti-cancer effects.
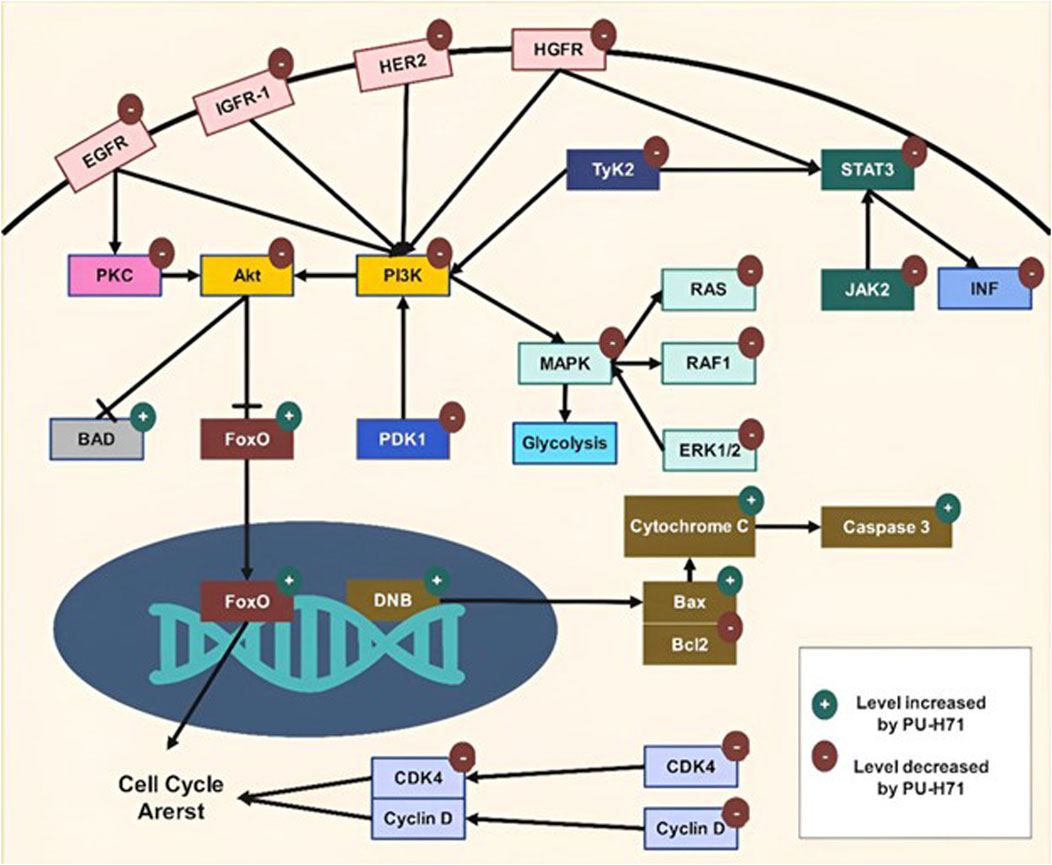
Figure 3. Molecular mechanisms of PU-H71.
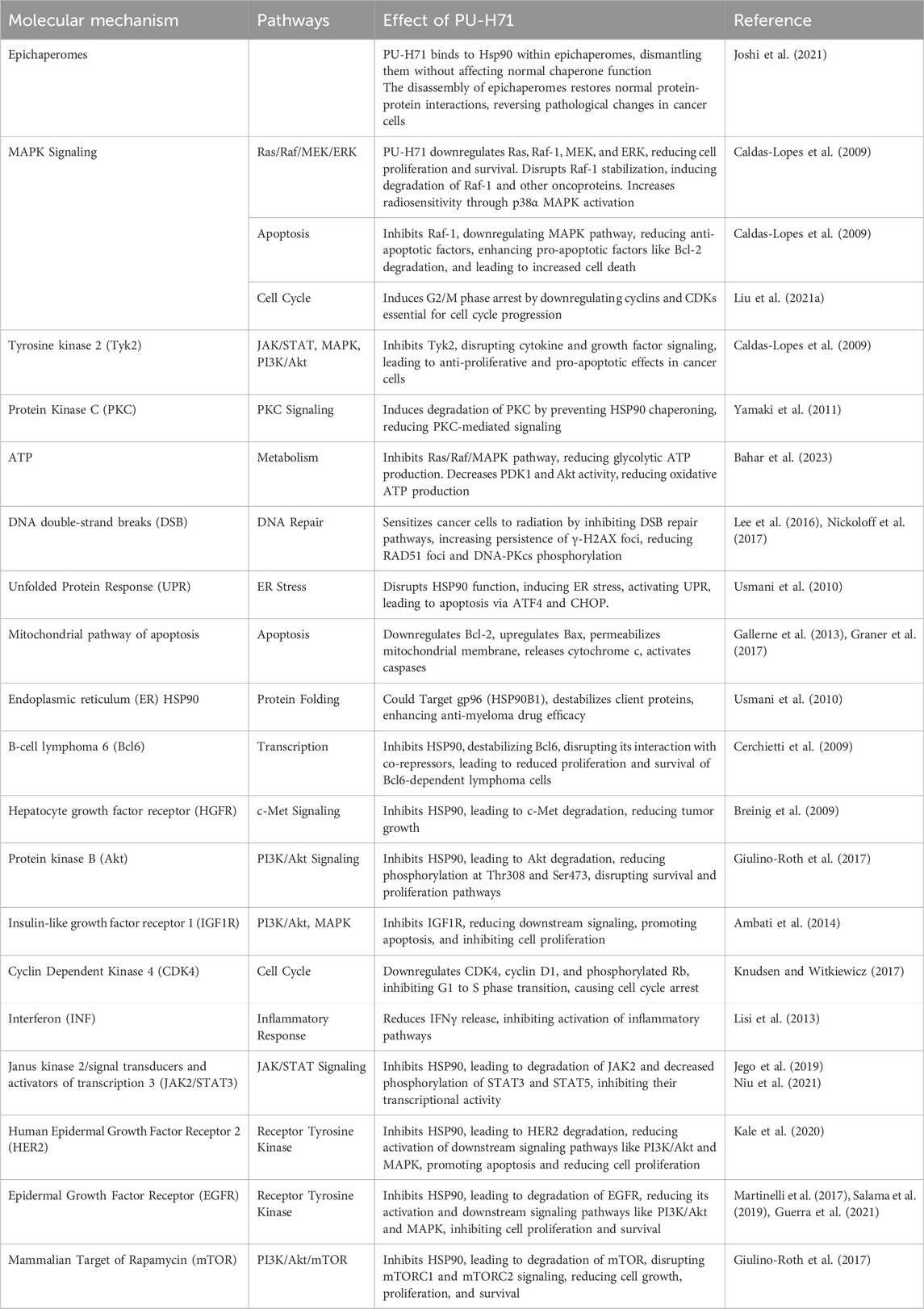
Table 3. Molecular mechanisms of PU-H71.
7.1 Epichaperomes
Epichaperomes are novel type of molecular structures formed by the assembly of chaperones and co-chaperones, representing a significant advance in our understanding of cellular stress responses, particularly in cancer and neurodegenerative disorders (Rodina et al., 2016). Unlike traditional chaperones, which help with protein folding and aggregation temporarily, epichaperomes form stable oligomeric complexes that remain in cells, fundamentally altering cellular proteostasis (Joshi et al., 2018; Yan et al., 2020; Ginsberg et al., 2021). These long-lived structures are critical in modulating protein-protein interactions (PPIs) within cells, especially under chronic stress conditions (Rodina et al., 2023; Chakrabarty et al., 2024). The formation of epichaperomes in diseased tissues provides new insights into disease mechanisms as well as potential therapeutic avenues, making them a promising intervention target.
Epichaperomes are derived from the chaperome, a network of over 300 chaperones and associated factors that regulate protein folding and quality control under normal conditions (Inda et al., 2020). However, epichaperomes differ from conventional chaperones in terms of stability and function. They serve as scaffolding platforms, reorganizing PPIs and altering cellular function in response to chronic stress, particularly in cancer cells (Ginsberg et al., 2023). Their specificity for diseased cells and tissues, as opposed to the widespread presence of traditional chaperones in healthy cells, makes them an appealing therapeutic target. In cancer, epichaperomes promote cell survival by supporting hyperconnected PPI networks, allowing malignant cells to survive in hostile microenvironments and evade therapeutic treatments (Digwal et al., 2022). Tumors with high epichaperomes expression often exhibit heightened vulnerability to specific inhibitors that disrupt these pathological structures.
A key therapeutic discovery is that small molecules, such as PU-H71, can selectively target epichaperomes and dismantle them without interfering with normal chaperone activity (Jhaveri et al., 2020). PU-H71 binds to Hsp90 within the epichaperome and induces conformational changes that cause the epichaperome to disassemble into its constituent chaperones. The disassembly of epichaperomes restores normal interactome structures, reversing pathological PPI changes. This therapeutic strategy has shown particular promise in cancers like Ras-driven pancreatic cancer, which has redundant signaling pathways that contribute to drug resistance (Joshi et al., 2021). In these cases, epichaperomes maintain interactome hyperconnectivity, which occurs when cancer cells rely on highly connected networks of PPIs to survive. Approximately 50%–60% of tumors express epichaperomes, with those displaying the highest levels of epichaperomes expression being characterized by extreme interactome hyperconnectivity and increased vulnerability to therapeutic interventions (Merugu et al., 2020). Furthermore, the breakdown of the epichaperome causes significant changes in cellular signaling pathways involved in survival and proliferation. For instance, decreased phosphorylation of key signaling proteins such as phospho-SYK and phospho-STAT5 has been documented, highlighting the downstream effects of epichaperome disruption on oncogenic signaling cascades (Roboz et al., 2018).
This approach exploits the vulnerability in cancer cells by pharmacologically manipulating epichaperome-mediated protein-protein interaction (PPI) networks. By forcing the interactome to reach maximum hyperconnectivity, cancer cells become more sensitive to existing therapies (Joshi et al., 2021). This strategy reactivates pre-existing protein pathways, preventing cancer cells from utilizing alternative signaling mechanisms to evade treatment. Unlike traditional therapies that target specific genetic mutations or protein aberrations, this method aims to control the entire interactome, thereby shutting down compensatory pathways that contribute to drug resistance (Rodina et al., 2016; Carter et al., 2023). Experiments in pancreatic cancer models, including cell lines, patient biospecimens, and xenografts, show that pharmacologically induced hyperconnectivity significantly improves the efficacy of current treatments (Joshi et al., 2021). By priming cancer cells with PU-H71 to induce a hyperconnected state, the efficacy of previously ineffective therapies was increased. This novel treatment paradigm represents a promising approach to overcoming drug resistance in cancers marked by high epichaperome expression and interactome plasticity (Joshi et al., 2021). PU-H71, by inducing hyperconnectivity, targets entire PPI networks rather than individual molecular nodes, resulting in a more comprehensive and effective therapeutic strategy.
In clinical settings, PU-H71 has shown promising results, especially in cancers with high epichaperome levels. Preclinical and early clinical trials have shown that patients with acute myeloid leukemia, which is characterized by elevated epichaperome expression, respond well to PU-H71 treatment, with some achieving long-term remission (Li and Adams, 2023; Seipel et al., 2023). These findings highlight the potential of PU-H71 as a therapeutic agent in cancers where the epichaperome plays an important role in disease progression.
7.2 Mitogen-activated protein kinase kinase (MAPK) signaling
PU-H71 treatment leads to a potent reduction in the proliferative, anti-apoptotic, and invasive potential of tumors. Among the pathways affected, PU-H71 induces downregulation of components of the Ras/Raf/MAPK pathway, contributing to its anti-proliferative effects (Pastena et al., 2024). In chronic myelogenous leukemia cells, PU-H71 was shown to identify the aberrant signalosome, with a focus on the Raf-MAPK pathway (Moulick et al., 2011). By inhibiting HSP90, PU-H71 destabilizes oncoproteins associated with the MAPK pathway, leading to their downregulation and impairment of downstream signaling (Seipel et al., 2023). PU-H71 has also been found to increase the radiosensitivity of breast cancer cells metastasized to visceral organs. The radiosensitizing effect of PU-H71 in combination with radiation therapy correlates with increased activation of p38α MAPK (García-Flores et al., 2023). Remarkably, recent studies have identified p38α MAPK as a potential tumor suppressor owing to its capacity to interfere with malignant cell transformation via regulating tissue homeostasis. This effect could be ascribed to modulating signals that balance cell proliferation and differentiation or induce apoptosis. As previously noted, PU-H71-mediated inhibition of HSP90 could improve the response of tumor cells to radiotherapy through modulation of p38 MAPK signaling.
Additionally, PU-H71 was reported to target MAPK pathways thus consequently decreasing tumor cell resistance to other anti-cancer treatment regimens enhancing their efficacy. Furthermore, combined administration of PU-H71 and the anti-cancer prodigiosin, displayed remarkable synergistic effects against malignant cells, that could be ascribed to targeting different aberrantly regulated pathways including MAPK (Anwar et al., 2020).
Moreover, PU-H71 was documented to disrupt the stabilization of v-Raf-1 (Raf-1) and other oncogenic proteins, leading to their degradation (Trendowski, 2015). It is noteworthy to highlight that, Raf-1 is a critical component of the MAPK signaling cascade aberrantly activated in cancer cells (Beeram et al., 2005).
Besides, PU-H71 anticancer properties could be due to the reported downregulation of the components of the Ras/Raf/MAPK pathway. The activation of such a pathway is initiated by Ras (a small GTPase) phosphorylation and activation by growth factor receptors. Subsequently, activated Ras interacts with Raf-1, leading to activation of the latter (Terrell and Morrison, 2019; Nolan et al., 2021; Ullah et al., 2022). Herein, the pathway activation could be disrupted by PU-H71 treatment through hampering Ras-Raf-1 interaction, thereby inhibiting Raf-1 activation and consequent downstream signaling through MEK and ERK, which are essential for cell proliferation and survival (Caldas-Lopes et al., 2009; Ambati et al., 2014).
Intriguingly, PU-H71 was reported to induce apoptosis in cancer cells that could be also ascribed to the observed Raf-1 inhibition. Also, MAPK pathway suppression following PU-H71 treatment resulted in a significant downregulation of anti-apoptotic factors that triggered the degradation of pro-survival proteins like B-cell lymphoma-2 (Bcl-2) (Gallerne et al., 2013). The restoration of balance between pro-apoptotic and anti-apoptotic signals leads to increased malignant cell death (Yamaki et al., 2011).
Anti-cancer effects of PU-H71 could result from its capacity to induce G2/M phase arrest interfering with cell cycle progression. This could be linked to the reported downregulation of cyclins and cyclin-dependent kinases (CDKs) that are necessary for cell cycle progression (Liu H. et al., 2021). In addition, Raf-1 inhibition with subsequent suppression of the MAPK pathway could probably contribute to the reported cell cycle arrest (Bahar et al., 2023).
7.3 Tyrosine kinase 2 (Tyk2)
Tyk2 belongs to the Janus kinase (JAK) family of tyrosine kinases, which are closely implicated in cytokine and growth factor signaling pathways playing an essential for cancer progression (Wöss et al., 2019). Tyk2 was reported to be aberrantly activated in different types of neoplasms (e.g., acute myeloid leukemia, and solid tumors including breast cancer) (Wöss et al., 2019). Aberrant activation of Tyk2 was documented to facilitate a range of abnormal characteristics of cancer cells including abnormal cellular growth and proliferation alongside invasion. Fundamentally, abnormal Tyk2 activation results in the activation of downstream signaling cascades, among these the signal transducers and activators of transcription (STAT), MAPK, and Phosphoinositide 3-kinase (PI3K)/Protein Kinase B (Akt) pathways were reported to be the most important. Meanwhile, suppression Tyk2 cascade was shown to interrupt such oncogenic signaling cascades, which could potentially contribute to its anti-tumor effects in preclinical cancer models (Caldas-Lopes et al., 2009; Kucine et al., 2015).
Remarkably, HSP90 is pivotal for Tyk2 stability and function (Akahane et al., 2016; Reynolds and Blagg, 2024), where HSP90 suppression following PU-H71 treatment has been reported to cause Tyk2 and inactivation of Tyk2, in various cancer models. In an earlier investigation on triple-negative breast cancer (TNBC) cells, PU-H71 exposure caused a remarkable downregulation of multiple oncogenic proteins, including components of the Ras/Raf/MAPK pathway. This could lead to further downregulation of Tyk2 (Ravandi et al., 2003; Dimri and Satyanarayana, 2020).
7.4 Protein kinase C (PKC)
HSP90 is important for the stability and function of many protein kinases like PKC (Kawano et al., 2021). PU-H71 binds to HSP90 and prevents it from chaperoning and stabilizing client proteins like PKC (Yamaki et al., 2011). Without the support of HSP90, PKC becomes misfolded and targeted for proteasomal degradation. This leads to a reduction in cellular PKC levels and inhibition of PKC-mediated signaling. The PKC family consists of 10 isozymes that regulate diverse cellular processes (Kawano et al., 2021). By inhibiting HSP90, PU-H71 is expected to affect multiple PKC isoforms simultaneously. However, the relative potency of PU-H71 against each PKC isoform has not been reported.
PU-H71 has been shown to downregulate components of the MAPK signaling pathway, which includes PKC. Specifically, PU-H71 induces the degradation of activated Akt, a key PKC substrate (Yamaki et al., 2011). The documented suppression of PKC signaling has markedly contributed to reported PU-H71 antitumor effects against TNBC. In addition to the reported effects on PKC, PU-H71 also has demonstrated suppression of activated glial cells in experimental autoimmune encephalomyelitis (EAE) and an animal model of multiple sclerosis (Lisi et al., 2013). Additionally, PU-H71’s anti-inflammatory properties could be mediated through the inhibition of PKC and other inflammatory signaling pathways.
7.5 Adenosin triphosphate (ATP)
Interestingly, PU-H71-mediated downregulation of Ras/Raf/MAPK pathway components was shown to (Bahar et al., 2023) decrease the rate of glycolytic ATP generation and consequently be implicated in regulating glycolysis and ATP production. Besides, HSP90 was reported be principally involved in regulating the 3-phosphoinositide-dependent protein kinase-1 (PDK1)-Akt signaling pathway (Basso et al., 2002; Xue et al., 2014; Bao et al., 2021). Markedly, HSP90 inhibition by PU-H71 could trigger PDK1 degradation, subsequently suppressing the Akt pathway that would then lead to a profound decline in both the activity of mitochondrial metabolism and oxidative ATP production (Mookerjee et al., 2017).
Moreover, PU-H71 was reported to promote apoptosis as well as degradation of oncogenic proteins that drive both uncontrolled cell proliferation (Speranza et al., 2018) this effect could be mainly mediated by reducing the demand for ATP by these cellular processes, plus decreasing overall ATP utilization.
7.6 DNA double-strand breaks (DSB)
Particularly, PU-H71 has a significant capability of sensitizing malignant cells to heavy ion radiation that could be owing to the observed inhibition of DSB repair pathways (Lee et al., 2016) as well as increasing the persistence of γ-H2AX foci that represent markers of unrepaired DSBs, in irradiated tumor cells (Lee et al., 2016). This finding suggests PU-H71-induced impairment of radiation-induced DSB repair mechanisms. Also, the displayed sensitization of malignant cells to radiation was attributed to the decreased formation of RAD51 foci as well as diminished phosphorylation of DNA-PKcs in irradiated tumor cells. Reportedly, both RAD51 and DNA-PKcs are DNA repair proteins central to homologous recombination and non-homologous end-joining DSB repair pathways, respectively. Moreover, RAD51 was reported to be upregulated in several neoplasms and it was responsible for chemoresistance, poor cancer prognosis, as well a higher incidence of metastasis (Nickoloff et al., 2017). Significantly, PU-H71 was shown to improve the efficacy of radiotherapy via suppressing radiation-induced DSB repair in irradiated tumor cells (Lee et al., 2016) eventually leading to increased cell death.
PU-H71-induced restraining of DSB repair could potentially be implicated in the observed sensitization of various cancer cell lines to heavy ion radiation, including, human lung cancer cells (Lee et al., 2016). As previously reported, PU-H71 promoted the cytotoxic properties of heavy ion radiation as reflected by both reduced clonogenic survival and increased cell death. Interestingly, the reported radio-sensitizing actions of PU-H71 were explicit to tumor cells and did not affect the survival of normal human fibroblasts (Li et al., 2016). Such precise sensitization, recommends PU-H71 as a novel therapeutic agent for enhancing the efficacy of heavy ion radiotherapy.
7.7 Unfolded protein response (UPR)
PU-H71 displayed complex and multiple effects on the UPR including several mechanisms eventually promoting malignant cell apoptosis. Firstly, it was noted that PU-H71 could generate Endoplasmic reticulum (ER) stress probably by suppressing HSP90 normal function. The documented inhibition of HSP90 function accelerates the compilation of both unfolded or misfolded proteins in the ER, triggering the UPR (Gallerne et al., 2013). Of note, UPR represents a cellular response to ER stress that objectively restores protein homeostasis through upregulating certain chaperones including Glucose-regulated protein 78 (GRP78), GRP94, and calreticulin. Outstandingly, PU-H71 triggers the UPR through the induction of the splicing of X-box binding protein 1 (XBP1) mRNA, defined as a key marker of UPR activation (Gallerne et al., 2013).
The main aims of UPR include restoration of protein homeostasis or induction of apoptosis if the stress persists. Apoptosis induction is potentially through activating the downstream transcription factors such as transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) promoting the expression of pro-apoptotic genes (Rah et al., 2016; Hu et al., 2019).
7.8 Mitochondrial pathway of apoptosis
One of the major actions of PU-H71 is the activation of the mitochondrial pathway of apoptosis via upregulation of pro-apoptotic proteins (e.g., Bcl-2-Associated X Protein (Bax)) alongside downregulation of anti-apoptotic proteins (Bcl-2) followed by enhanced permeabilization of the mitochondrial membrane with subsequent release of cytochrome c and caspases activation (Gallerne et al., 2013; Graner et al., 2017).
Mechanistically, the capacity of PU-H71 to trigger apoptosis was underlined by activating this mitochondrial pathway in multiple cancer cell lines such as melanoma, cervix, colon, liver, and lung cancer cells (Rastogi et al., 2024). Interestingly, previous findings displayed that PU-H71-induced apoptosis could potentially attenuate malignant cell resistance attributed to overexpression of the anti-apoptotic protein Bcl-2 (Gallerne et al., 2013). Previous findings also highlighted the remarkable contribution of the pro-apoptotic protein Bax to PU-H71-induced apoptosis where Bax-deficient cells were shown to resist PU-H71-elicited apoptosis (Kubra et al., 2020). Conversely, such resistance was partially overcome by combined administration of PU-H71 treatment with other anticancer therapies (e.g., cisplatin or melphalan) (Gallerne et al., 2013). In addition to apoptosis induction, PU-H71 was shown to cause cell cycle arrest at the G2-M phase that could be mediated through cyclin D1 downregulation (Ambati et al., 2014).
7.9 Endoplasmic reticulum (ER) HSP90
Over the past decade, gp96 or HSP90B1 also referred to as grp94, the ER HSP90 family member, has attracted distinct attention. This attention was due to gp96’s remarkable role in chaperoning antigenic peptides, enabling their presentation in the antigen-cross-presentation pathway (Marzec et al., 2012; Duan et al., 2021). Outstandingly, immunizations utilizing pure tumor-derived gp96-peptide complexes have displayed promising preclinical and clinical results in the treatment of multiple myeloma (Randazzo et al., 2012). In-vitro investigations demonstrated that bortezomib (disturbing protein anti-myeloma drug), rendered gp96 knockdown cells more susceptible. In addition, gp96 knockdown cells demonstrated a remarkable sensitivity to thalidomide alongside increased cell death induced by genotoxic drugs like melphalan and doxorubicin. These outcomes suggest that gp96 could potentially be involved in diminishing the efficacy of anti-myeloma therapies, meanwhile, gp96 knockdown augmented myeloma cells’ response to those agents (Usmani et al., 2010). Nevertheless, gp96 knockdown negatively affected the anti-myeloma activity of PU-H71 suggesting that the acute cytotoxic effect of HSP90 inhibitors is mediated through modulating both cytosolic HSP90 in addition to gp96 in the ER. PU-H71 displayed in vitro anti-myeloma efficacy as well. These findings highlight that HSP90 inhibitors, including PU-H71, target both cytosolic HSP90 and the ER-resident HSP gp96 to exert their anti-myeloma effects.
7.10 B-cell lymphoma 6 (Bcl6)
BCL6 is identified as a master transcription factor regulating T follicular helper cell proliferation, which is central to germinal center development and B cell proliferation. Remarkably, mutations in BCL6 could result in the development of B cell lymphomas that could be owing to unchecked B cell growth, therefor such mutations could be used as diagnostic markers for B cell lymphomas and other cancers (Mintz and Cyster, 2020).
Interestingly, HSP90 plays a pivotal role in the stability as well as the function of the transcription factor BCL6 (Hoter et al., 2018) Herein, PU-H71 binding to the N-terminal ATP/ADP pocket of HSP90 leads to a prominent inhibition of its ATPase activity plus a profound disruption of HSP90 chaperone function with consequent BCL6 destabilization and degradation (Sidera and Patsavoudi, 2014), reducing its activity. This effect was reflected by the reported decline in luciferase activity of BCL6 target promoters in cell lines following exposure to PU-H71.
The downregulation of BCL6 by PU-H71 is thought to be mediated through multiple mechanisms including, reduced stability and increased degradation of the BCL6 protein (Sidera and Patsavoudi, 2014); disruption of the interaction between BCL6 and its transcriptional co-repressors, such as Silencing Mediator of Retinoid and Thyroid hormone receptors (SMRT), Nuclear receptor corepressor (N-CoR), and BCL6 corepressor (BCoR) (Briones, 2010; Kuai et al., 2018; McLachlan et al., 2022); Interference with the autoregulatory loop that maintains high BCL6 expression in lymphoma cells (Yang and Green, 2019); and derepression of BCL6 target genes involved in cell cycle control, DNA repair, metabolism, and immune regulation. This results in the suppression of the proliferation and survival of BCL6-dependent lymphoma cells, making PU-H71 a promising therapeutic agent for BCL6-driven lymphomas.
7.11 Hepatocyte growth factor receptor (HGFR)
The hepatocyte growth factor receptor (HGFR), also known as c-Met, is a receptor tyrosine kinase that is primarily expressed on epithelial cells. Its only known high-affinity ligand is hepatocyte growth factor (HGF) (Fu et al., 2021). The binding of HGF to c-Met activates the receptor, leading to tyrosine phosphorylation and activation of downstream signaling pathways such as MAPK, STAT, and PI3K/Akt (Zhang et al., 2018). Deregulated c-Met signaling, due to factors like receptor overexpression, ligand overexpression, or activating mutations, is implicated in the development, progression, and metastasis of many human cancers. Therefore, c-Met represents an attractive target for anti-cancer therapies, and various agents targeting the HGF/c-Met pathway are under development, including small molecule inhibitors, antibodies, and decoy receptors (Matsumoto et al., 2017).
PU-H71 was found to potently increase the degradation of c-Met as a client protein to HSP90. Treatment with PU-H71 resulted in reduced c-Met protein levels and decreased phosphorylation/activation of c-Met in various cancer cell lines. The inhibition of c-Met by PU-H71 was associated with reduced tumor growth in preclinical cancer models, without causing significant toxicity (Speranza et al., 2018). The mechanism by which PU-H71 inhibits c-Met is through its ability to bind and inhibit the HSP90 chaperone. By disrupting the HSP90-c-Met interaction, PU-H71 leads to the destabilization and degradation of the c-Met receptor, thereby suppressing its oncogenic signaling (Giulino-Roth et al., 2017). This indirect targeting of c-Met via HSP90 inhibition represents a promising therapeutic approach for cancers driven by deregulated c-Met activity.
7.12 Protein kinase B (Akt)
HSP90 was found to have an important role in the PDK1-Akt signaling pathway through binding to PDK1. Meanwhile, proteasome-dependent degradation of PDK1 was demonstrated following HSP90 inhibition by PU-H71 (Yamaki et al., 2011). Akt is a client protein of HSP90 and requires HSP90 for its stability and activation. PU-H71 binds to the N-terminal ATP/ADP pocket of HSP90, inhibiting its ATPase activity. This leads to the destabilization and degradation of Akt by the proteasome (Giulino-Roth et al., 2017). Akt activation requires phosphorylation at two regulatory sites: Thr308 in the activation segment by PDK1 and Ser473 in the hydrophobic motif by mTORC2 (Baffi et al., 2021). The binding of PU-H71 to HSP90 inhibits the phosphorylation of Akt at these sites, preventing its activation (Giulino-Roth et al., 2017). The degradation of PDK1 by PU-H71 also reduces the phosphorylation of Akt at Thr308.
Active, phosphorylated Akt regulates a panel of proteins that control proliferation, growth, survival, and metabolism (Hoxhaj and Manning, 2020). Inhibition of Akt by PU-H71 disrupts these downstream pathways. For example, Akt phosphorylates and inactivates the Bcl-2-associated death promoter (proapoptotic protein Bad) (Trendowski, 2015; Seipel et al., 2023). Degradation of Akt by PU-H71 leads to dephosphorylation of Bad, allowing it to induce apoptosis. Additionally, Akt phosphorylates the forkhead box O (FoxO) transcription factors, inducing their nuclear exclusion and inhibition (Orea-Soufi et al., 2022). This effect was restrained by PU-H71 suppressing Akt activity, leading to nuclear localization of FoxO with subsequent upregulation of genes involved in cell cycle arrest and apoptosis.
7.13 Insulin-like growth factor receptor 1 (IGF1R)
Insulin-like growth factor receptor 1 (IGF1R), represents a receptor tyrosine kinase crucial for mediating the effects of insulin-like growth factor 1 (IGF-1) (Ambati et al., 2014). Upon binding to IGF1R, IGF-1 activates the receptor’s intrinsic kinase activity, leading to autophosphorylation and activation of downstream signaling cascades essential for promoting cell growth, proliferation, and survival, including PI3K/Akt and Ras/Raf/MEK/ERK (Tahimic et al., 2013).
Significantly, PU-H71 was reported to specifically target IGF1R inhibiting its phosphorylation and then consequently suppressing downstream signaling pathways that eventually led to decreased proliferation and apoptosis induction in malignant cells. Earlier investigations reported that PU-H71 could effectively decrease the viability of various cancer cell lines overexpressing IGF1R (Breinig et al., 2011; Terwisscha Van Scheltinga et al., 2014; Trendowski, 2015). Remarkably, suppression of IGF1R signaling following PU-H71 treatment led to a marked inhibition of other signaling trajectories pivotal for malignant cell survival leading to increased efficacy of chemotherapeutic agents potentially through induction of apoptosis (Bulatowicz and Wood, 2022).
Significantly, the PI3K/Akt pathway is a principal signaling cascade activated by IGF1R. Following IGF1R activation, PI3K is recruited and then activated, which in turn activates Akt. Of note, Akt promotes cell proliferation and survival through stimulating protein synthesis and inhibition of apoptotic pathways. PU-H71’s inhibition of IGF1R was reported to decrease Akt activation, thereby inducing apoptosis and inhibiting malignant cell proliferation (Gao et al., 2019).
Similar to the PI3K/Akt pathway, the Ras/Raf/MEK/ERK pathway was shown to be inhibited after IGF1R signaling suppression by PU-H71 leading to reduced cell proliferation and induction of apoptosis (Caldas-Lopes et al., 2009).
7.14 Cyclin-dependent kinase 4 (CDK4)
Cyclin Dependent Kinase 4 (CDK4) is defined as a serine/threonine kinase that plays a pivotal role in cell cycle progression. Initially, it complexes with cyclin D and phosphorylates the retinoblastoma protein (Rb) inducing the release of E2F transcription factors that in turn activate the expression of the genes required for the G1 to S phase transition (Pandey et al., 2019). Conversely, PU-H71 was reported to suppress CDK4, this effect could be mediated through either promoting degradation of HSP90 client proteins, including CDK4 (Serwetnyk and Blagg, 2021) or downregulating CDK4 (Knudsen and Witkiewicz, 2017).
A previous in-vitro study on breast cancer cells displayed that, PU-H71 attenuated CDK4 and induced G1 phase cell cycle arrest. Notably, PU-H71 exposure induced a profound suppression of the CDK4/cyclin D/Rb pathway through reducing protein expression of CDK4 and cyclin D1 with a subsequent decline in Rb phosphorylation (Pimienta et al., 2011). Additionally, PU-H71 demonstrated a prominent effect on other HSP90 client proteins central to abnormal cell proliferation, survival, and drug resistance, including epidermal growth factor receptor (EGFR), mammalian target of rapamycin (mTOR) and human epidermal growth factor receptor 2 (HER2) (Trendowski, 2015).
7.15 Interferon (INF)
The binding of PU-H71 to HSP90 will lead to the disruption of interferon (IFN) signaling. PU-H71 treatment reduces the release of IFNγ from activated T cells (Lisi et al., 2013). IFNγ is a key cytokine that mediates inflammatory and anti-viral responses. PU-H71 inhibits the activation of astrocytes by lipopolysaccharide and IFNγ, as measured by reduced nitric oxide synthase 2 (NOS2) expression and nitrite production where Activated astrocytes secrete inflammatory mediators like nitric oxide (Lisi et al., 2013).
The exact molecular mechanisms by which PU-H71 modulates interferon signaling are not fully elucidated. However, it likely involves the destabilization of key signaling proteins in the IFN pathway, such as JAK and STATs (Koppikar et al., 2012; Mbofung et al., 2017; Kuykendall et al., 2020). Degradation of these proteins by the proteasome upon HSP90 inhibition would impair IFN-induced gene transcription and the inflammatory response.
7.16 Janus kinase 2/signal transducers and activators of transcription 3 (JAK2/STAT3)
PU-H71 has shown promising antineoplastic effects in preclinical models of malignancies driven by aberrant JAK/STAT signaling (Kucine et al., 2015). PU-H71 can directly bind to oncogenic JAK2 mutants, such as JAK2V617F, which are common driver mutations in myeloproliferative neoplasms (MPNs) (Jego et al., 2019). This binding leads to the proteasomal degradation of mutant JAK2, abrogating its constitutive activation.
By binding to HSP90, PU-H71 disrupts the chaperone complex that maintains JAK2 in an active conformation. This results in a dose-dependent inhibition of JAK2 kinase activity and reduced phosphorylation of its downstream substrates (Rampal et al., 2014).
JAK2-mediated STAT3/STAT5 phosphorylation is a rate-limiting step in the activation of such oncogenic signaling cascade (Halim et al., 2020). Yet, STAT3/STAT5 phosphorylation was mitigated following PU-H71 exposure, which consequently prevented their dimerization, nuclear translocation, and transcriptional activity (Niu et al., 2021).
7.17 Human epidermal growth factor receptor 2 (HER2)
HER2 is a receptor tyrosine kinase that plays a crucial role in regulating the rate of cellular proliferation as well as cell survival. HER2 upregulation was reported to induce a remarkable increase in the rate of cell proliferation alongside apoptosis inhibition, contributing to the pathogenesis as well as the development of an array of malignancies, chiefly breast cancer (Hsu and Hung, 2016). Of note, approximately 20%–30% of diagnosed breast cancer cases demonstrated HER2 overexpression. Herein, HER2-positive tumors significantly demonstrated more aggressive behavior and poor prognosis compared to HER2-negative tumors (Cheng, 2024). Moreover, HER2 overexpression was documented in other malignancies (e.g., gastroesophageal adenocarcinomas, ovarian cancers, and endometrial cancers) (Iqbal and Iqbal, 2014).
HER2 activation involves its dimerization with other members of the ErbB family of receptors followed by provocation of downstream signaling axes, particularly, the PI3K/Akt and MAPK pathways (Elster et al., 2015) promoting cell proliferation, survival, and metastasis, highlighting HER2 as a significant druggable molecular target in our fight against cancer.
Conspicuously, HSP90 inhibition following PU-H71 treatment was shown to disrupt the chaperone’s interaction with HER2 stimulating its degradation and then reducing its levels in cancer cells (Li and Luo, 2023). Furthermore, PU-H71-mediated downregulation of HER2 expression was stated. Additionally, PU-H71 exposure displayed a significant impact on key oncogenic cascades indispensable for cancer cell survival and proliferation namely, ERK1/2 and Akt (Kale et al., 2020). The observed inhibition of these signaling cascades consequent to PU-H71 treatment could potentially induce significant cytotoxic effects in different molecular types of breast cancer, among which triple-negative breast cancer (TNBC) is the most important owing to the reported resistance to conventional therapies. This multimodal approach suggested that PU-H71 could be a promising candidate for the treatment of HER2-positive and other aggressive cancer types.
7.18 Epidermal growth factor receptor (EGFR)
EGFR is a key receptor tyrosine kinase, that belongs to the ErbB family that plays a pivotal role in regulating cellular proliferation as well as survival. EGFR is mostly overexpressed in a range of neoplasms (e.g., lung, brain and neck cancers) (Roskoski, 2014). Therefore, EGFR represents a potential druggable target for the development of novel targeted cancer therapies.
EGFR activation results in subsequent activation of other downstream cascades including PI3K/Akt pathway, Ras/Raf/MEK/ERK pathway, and Phospholipase C γ1 (PLCγ1)/PKC pathway (Martinelli et al., 2017; Salama et al., 2019; Guerra et al., 2021). Therefore, HSP90 inhibition by PU-H71 resulted in EGFR destabilization as well as degradation suppressing the activation of downstream cascades. This was shown to increase the sensitivity of cancer cells to conventional anti-cancer agents, especially, malignant cells harboring mutant forms of EGFR, which are often dependent on HSP90 for their stability (Lee et al., 2016).
7.19 Mammalian target of rapamycin (mTOR)
As previously stated, mTOR represents a key serine/threonine protein kinase central to regulating cell metabolism, growth, and survival in response to various environmental cues such as growth factors, nutrients, energy levels, and stress signals (Wu and Storey, 2021). In addition, The mTOR pathway was defined as a chief oncogenic pathway that contributed significantly to the pathogenesis of an array of malignancies as it was aberrantly hyperactivated by mutational activation of its upstream regulators (Popova and Jücker, 2021).
Notably, mTOR forms two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) both are key regulators of principal cellular processes. Significantly, mTORC1 is the primary regulator of protein synthesis, lipogenesis, and autophagy, while mTORC2 plays a remarkable role in regulating cell survival and cytoskeleton organization (Szwed et al., 2021). In cancer cells, constitutive activation of the mTOR pathway promotes several hallmarks of cancer, including increased protein synthesis, lipid metabolism, and inhibition of autophagy, which are vital for rapid cell growth and proliferation. Dysregulation of mTOR signaling has been implicated in various types of cancer, including lung, breast, liver, renal, pancreatic, and prostate cancers. PU-H71 was found to disrupt the mTOR pathway, leading to decreased protein synthesis and enhanced apoptosis in cancer cells (Giulino-Roth et al., 2017). This mechanism contributes to the therapeutic efficacy of PU-H71 in targeting mTOR-driven cancers.
PU-H71 exerts its effects through a range of molecular mechanisms targeting various pathways. It downregulates Ras, Raf-1, and ERK in MAPK signaling, reducing cell proliferation, enhancing radiosensitivity, and inducing G2/M phase arrest by disrupting cyclins and CDKs. PU-H71 inhibits Tyk2, impacting JAK/STAT, MAPK, and PI3K/Akt pathways to induce anti-proliferative and pro-apoptotic effects. It also induces PKC degradation by preventing HSP90 chaperoning. By inhibiting ATP production, PU-H71 disrupts glycolytic and oxidative metabolism. It activates mitochondrial apoptosis pathways, downregulating Bcl-2 and upregulating Bax, and targets ER HSP90 to destabilize client proteins. The drug affects HGFR by disrupting its stability and function, thereby reducing tumor growth. PU-H71 also inhibits Akt and IGF1R, affecting survival pathways and promoting apoptosis. It reduces inflammatory responses by decreasing IFNγ release and inhibits JAK2/STAT3 signaling. Finally, it targets HER2 and EGFR, reducing their activation and downstream signaling, thus inhibiting cell proliferation and promoting apoptosis.
8 Impact of PU-H71 on cancer
8.1 Impact on triple-negative breast cancer
Triple-negative breast cancer (TNBC) accounts for about 12% of all breast tumors and is more prevalent in premenopausal women of African and African-American heritage (Howard and Olopade, 2021). TNBCs are defined by their low degree of differentiation, and a significant majority of them are classified within the primary subtype of breast cancers based on gene manifestation analysis (Cserni et al., 2021; Fusco et al., 2022). In breast cancer, there is the existence of the molecular chaperone protein identified as HSP90. HSP90 plays a critical role in maintaining a substantial reservoir of active and properly folded oncoproteins. It has a distinct affinity for its activated form, serving as a safeguarding “biochemical buffer” that shields cancer-promoting oncogenes (Kunachowicz et al., 2024). TNBCs are distinguished by the absence of estrogen, progesterone, and HER2 receptor expression. Because there are no specific targets and targeted therapies available, as well as the diverse molecular makeup of TNBCs, the current treatment guidelines primarily rely on conventional chemotherapy. While this approach proves effective for certain patients, it leads to early relapse in a substantial portion of instances and does not offer a cure for individuals with metastatic TNBC (Medina et al., 2020; Vagia et al., 2020). The HSP90 inhibitor PU-H71 has demonstrated remarkable efficacy against these tumors. This agent has been shown to produce forcible and enduring anti-tumor impacts in TNBC xenografts, counting instances of full reaction and tumor reversion, free from inducing harm to the host (Anwar et al., 2020). Importantly, TNBC tumors have displayed continued responsiveness to PU-H71 retreatment for multiple cycles, extending over more than 5 months, with no signs of resistance or toxicity. Both in laboratory experiments (in vitro) and in living organisms (in vivo). PU-H71 has proven highly effective in consistently reducing and deactivating these proteins. It has been identified several mechanisms contributing to its anti-proliferative effects, including the suppression of factors in the Ras/Raf/MAPK pathway and the triggering of apoptosis by breaking down activated Akt and Bcl-xL. Additionally, PU-H71 inhibits various signaling pathways such as NF-κB, Akt, ERK2, Tyk2, and PKC, which collectively decrease the influential domination of TNBC (Caldas-Lopes et al., 2009; Ozgur and Tutar, 2014; He and Hu, 2018; Scott, 2018).
PU-H71’s cytotoxicity was evaluated in TNBC cell lines MDA-MB-468, MDA-MB-231, and HCC-1806 employing a test that measures ATP degrees (Caldas-Lopes et al., 2009). It had been noted that PU-H71 effectively prevented the expansion of these cells at concentrations known to bind to HSP90, indicating a correlation between its anti-tumor activity and its interaction with HSP90 (Chiosis et al., 2008). In an experiment conducted over a 48-h incubation period using an intensification of PU-H71 that was 5- to 10-fold more than its IC50 for expansion prevention (1 M), PU-H71 exhibited notable cytotoxicity (Anwar et al., 2020). Using PU-H71 as a data collection method offers preclinical evidence that achieving a comprehensive response is possible across molecularly diverse TNBC tumors using a single agent, an HSP90 inhibitor. The data collected with PU-H71 suggests that the variability in the effectiveness of other HSP90 inhibitors in TNBC tumors is not primarily due to a decrease in the number of HSP90-present tumors. Instead, it suggests that differences in their effectiveness may be attributed to factors unrelated to the target (HSP90), which remain undisturbed. In contrast to approaches that exclusively target specific proteins or signaling pathways within TNBC cells, PU-H71’s exceptional efficacy can be linked to its capacity to suppress HSP90, whether in laboratory settings (in vitro) or within living organisms (in vivo). HSP90 is a protein that forms complexes with numerous proteins pivotal for the cancerous characteristics of TNBC. PU-H71 induces concurrent instability and decrease of several crucial TNBC oncoproteins, a lot of which contribute to processes such as cell proliferation, anti-apoptotic capabilities, and metastasis.
8.2 Impact as cancer cell sensitizer for carbon-ion beam therapy
Carbon-ion radiotherapy (CIRT) has shown its efficacy in treating a range of tumor types, including lung cancer, prostate cancer, and bone/soft tissue cancer (Kamada et al., 2015). However, there is ongoing potential for further progress in the field of CIRT, especially in terms of effectively managing radioresistant tumors while minimizing adverse effects on healthy tissues. Radiosensitizers play a crucial role in enhancing radiotherapy by making tumor cells more responsive to radiation, enabling the achievement of similar therapeutic effects with lower radiation doses (Xie et al., 2019). While there is a wealth of research on radiosensitizers for X-ray-based therapies, there has been relatively limited investigation into radiosensitizers specifically designed for use with carbon-ion beams (Yanagi et al., 2009).
The prospect of targeting HSP90 for cancer treatment holds significant potential owing to its elevated expression in cancer cells in contrast to normal cells, and cancer cells exhibit a greater dependence on HSP90 for their survival (Jhaveri et al., 2012). Numerous inhibitors of HSP90 have been formulated and have demonstrated promising treatment efficacy; however, their therapeutic potential has been constrained due to associated toxicity. In several preclinical investigations, PU-H71 has demonstrated therapeutic efficacy in various preclinical models and is presently undergoing clinical trials. While CIRT has been effective in controlling tumors, there remains an opportunity for enhancement, particularly in terms of achieving tumor-specific radiosensitization (Li et al., 2016; Sokol and Durante, 2023).
There are currently two phase I clinical trials in progress, involving patients with solid tumors or lymphoma, So there is growing interest in PU-H71 as a promising novel drug (Li et al., 2016). PU-H71, regarded as one of the strong potential HSP90 inhibitors, is a resultant of PU-3 that has been created to possess both dispersibility and targeted binding affinity to the ATP-binding sites of HSP90 (Li et al., 2016). HSP90 operates through a sophisticated cycle regulation by ATP combination and dissociation. PU-H71 inhibits the function of HSP90 by preventing ATP from binding to it (Jhaveri et al., 2012). Inhibiting HSP90 represents an appealing approach to combined therapy, as there have been numerous reports indicating that HSP90 inhibitors can induce significant and lethal damage to tumor cells when used in binding with X-ray radiation therapy (Segawa et al., 2014). There have been only a limited number of studies examining the enhancing sensitivity of HSP90 inhibitors on high-LET C-ions, and the results of these studies remain a topic of debate and discussion (Musha et al., 2012). While PU-H71 when examined in isolation, there were no signs of toxicity to LM8 cells, it was demonstrated that after 24 h of incubation, PU-H71 significantly heightened the sensitivity of LM8 cells to both X-ray and C-ion exposure (Li et al., 2016). Due to the potent cell-killing capabilities of C-ions, making tumors more responsive to high-LET radiation, including C-ion therapy, through combination with other treatments, such as anti-cancer medications, can be particularly challenging (Li et al., 2016). Achieving successful combination therapy can indeed be complex. The potency of the standard treatment in a combination therapy approach should ideally surpass the effectiveness of each therapy component. Additionally, the side effects of the combined treatment must be less severe than the cumulative side effects regarding every specific therapy (Schirrmacher, 2019). Consequently, optimized doses for combinatorial treatment often should be administered at lower levels than the individual treatment doses to strike a balance between maximizing therapeutic benefit and minimizing adverse effects (Reece et al., 2007). PU-H71 has demonstrated the ability to sensitize LM8 cells at a dosage that doesn't disrupt cellular homeostasis. This suggests that PU-H71 holds noteworthy radiosensitizing qualities for CIRT (Li et al., 2015).
The potentially lethal impact of radiation on cells is primarily attributed to the induction of DNA double-strand breaks (DSBs) (Mladenov et al., 2013), HSP90 client proteins are prevalent in numerous DSB-associated proteins. Consequently, the focus was on exploring protein expression in the context of DSB repair and paying special attention to RAD51 and Ku70 levels (Pennisi et al., 2015; Khurana et al., 2016). These proteins play crucial roles in the primary cellular repair pathways, namely, homologous recombination and non-homologous end joining. Treatment with PU-H71 alone caused a reduction in RAD51 expression in LM8 cells (Ren et al., 2014). This suggests that PU-H71’s ability to enhance the sensitivity of C-ion therapy may also involve the non-homologous end-joining DSB repair pathway. Nevertheless, further investigation is necessary to fully elucidate the exact mechanism responsible for PU-H71’s radiosensitizing effect.
8.3 Impact on myeloma
In preclinical research, several HSP90 inhibitors have demonstrated effectiveness against myeloma, and at least three of these drugs have proceeded to Phase I/II clinical trials for the treatment of relapsed/refractory myeloma (Cavenagh et al., 2017). Preclinical studies utilizing PU-H71 in the treatment of diffuse large B-cell lymphoma and triple-negative breast cancer have indicated significant efficacy while causing minimal hematological or non-hematopoietic toxicity (Usmani et al., 2010). It is worth noting that the effectiveness of PU-H71 in treating multiple myeloma has not been thoroughly studied. However, in both IL-6 independent cell lines and IL-6 reliant cell line INA-6, PU-H71 has demonstrated comparable anti-myeloma efficacy (Usmani et al., 2010). Moreover, PU-H71 has exhibited in vitro synergy with other commonly used anti-myeloma medications. Additionally, PU-H71 has been shown to activate the UPR and induce apoptosis through the caspase-dependent pathway (Usmani et al., 2010). Over the past decade, there has been considerable research into the ER HSP90 family member known as gp96, or HSP90B1, also referred to as grp94. This research has been driven by gp96’s role in chaperoning antigenic peptides, enabling their presentation in the antigen-cross-presentation pathway (Marzec et al., 2012; Duan et al., 2021). Immunizations utilizing pure tumor-derived gp96-peptide complexes have demonstrated promising preclinical and clinical outcomes in the treatment of multiple myeloma (Qian et al., 2009). To assess the in vitro effectiveness of anti-myeloma drugs when gp96 was absent, researchers established a stable gp96 knockdown human myeloma cell line. The results aligned with expectations: bortezomib, known for disturbing protein balance, rendered gp96 knockdown cells more susceptible (Hua et al., 2013). Additionally, gp96 knockdown cells displayed heightened vulnerability to thalidomide, whose mechanism of action remains unidentified, as well as increased cell death induced by genotoxic drugs like melphalan and doxorubicin (Bagratuni et al., 2010; Hua et al., 2013). These findings imply that gp96 may play a role in reducing the efficacy of these anti-myeloma medications, and its absence amplified the sensitivity of myeloma cells to these agents (Usmani et al., 2010). However, the absence of gp96 had a significant negative impact on the anti-myeloma activity of PU-H71. This information indicates that HSP90 inhibitors affect not only cytosolic HSP90 but also gp96 in the ER, contributing to their acute cytotoxic effects (Hua et al., 2013). The novel purine-based HSP90 inhibitor, PU-H71, displayed in vitro anti-myeloma efficacy as well. These findings highlight that HSP90 inhibitors, including PU-H71, target both cytosolic HSP90 and the ER-resident HSP gp96 to exert their anti-myeloma effects. It is imperative to initiate a phase I clinical trial to assess the therapeutic efficacy of PU-H71 in individuals with refractory myeloma.
8.4 Effect on B-Cell lymphomas
The function of HSP90 and the impact of HSP90 inhibitors in the most popular kind of lymphoid malignancies, such as diffuse large B-cell lymphomas (DLBCL) that come from germinal center B-cells, remain largely unexplored and not well understood (Klein and Dalla-Favera, 2008). Bcl6, which is the most common oncogenic protein in DLBCL, has been identified as a client protein of HSP90 based on observations of how DLBCL cells respond to HSP90 inhibitors (Cerchietti et al., 2009; Calvo-Vidal et al., 2021). The interaction between HSP90 and Bcl6 influences the biology of normal germinal center B-cells as well as the pathophysiology of DLBCL to varying degrees (Ci et al., 2008). The functional and physical connection between HSP90 and Bcl6 may, in part, illustrate that treatment by directly controlling both Bcl6 mRNA and protein (Cerchietti et al., 2010). It is suggested that both mechanisms contribute to maintaining stable Bcl6 levels, although it is challenging to determine the precise extent to which HSP90 influences Bcl6 mRNA stability versus Bcl6 protein stability.
To establish germinal centers, where B-cells undergo affinity maturation, normal B-cells must undergo an upregulation process. HSP90 may play a role in supporting B-cell development by assisting in the accumulation of Bcl6 within germinal center B-cells, especially considering its localization in the nuclei of healthy Centro blasts (Hatzi and Melnick, 2014). These findings suggest that BCL6, which is part of a larger gene family subject to post-transcriptional regulation, could indeed function as a gene responsive to stress conditions (Cerchietti et al., 2009; Fernando et al., 2019; Liu et al., 2022). The fact that Bcl6 is a key target of HSP90 inhibitors in DLBCL aligns with the idea that it is essential for the survival of a subgroup of DLBCLs. This concept gains support from studies involving PU-H71, which showed that a version of Bcl6 resistant to degradation was enough to protect DLBCL cells (Culjkovic-Kraljacic et al., 2016). Studies like these emphasize the importance of discovering drugs that can enhance the effectiveness of conventional anti-lymphoma therapies while reducing their toxicity. It is noteworthy that HSP90 inhibitors have the propensity to accumulate preferentially within lymphomas for an extended period compared to normal tissues. In this regard, PU-H71 stands out as an exceptionally effective and fine-tolerated anti-lymphoma drug in vivo. Furthermore, finding that PU-H71 can play a role in the depression of Bcl6 target genes, that cause DNA damage checkpoints, raises the possibility that such medications could increase the cytotoxic activity of chemotherapy drugs. This effect was observed in various lymphoid cell lines, including DLBCLs (Zhao and Houry, 2005; Shinozaki et al., 2006).
8.5 Impact on human hepatocellular carcinoma
There has been a suggestion that HSP90 might be involved in the progression of hepatocellular carcinoma (HCC), potentially contributing to the hepatocarcinogenesis process (Mohammed et al., 2023a). Several factors that promote hepatocarcinogenesis, such as hepatocyte growth factor receptor, protein kinase B, IGF1R, cyclin-dependent kinase 4, and v-raf-1 murine leukemia viral oncogene homolog 1 exhibit abnormal expression and increased activity when influenced by HSP90. In general, HSP90 regulates the stability and activity of proteins represented as “client proteins,” (Saber et al., 2020; El-Kashef et al., 2022; Hasan Khudhair et al., 2022; Shaaban et al., 2022; Mohammed et al., 2023a; Mohammed et al., 2023b) and this is how it impacts protein homeostasis. Its activity is driven by adenosine triphosphate and involves dynamic interactions with co-chaperones (Workman et al., 2007; Pearl et al., 2008). The development of HCC is believed to be influenced by the upregulation of HSP90 in conjunction with increased CDK4 activity (Pascale et al., 2005). HCC typically leads to liver damage (Abdel-Ghany et al., 2015; Saber et al., 2018; Abdelhamid et al., 2022). Considering that geldanamycin analogs, which are HSP90 inhibitors, often cause dose-limiting hepatotoxicity, using HSP90 inhibition with these drugs may not be a suitable treatment option for this type of cancer (Pearl et al., 2008). Novel HSP90 inhibitors that do not contain the benzoquinone component found in geldanamycin, which is believed to contribute to hepatotoxicity, may offer potential prospects for HSP90 inhibition in the therapy of HCC in the future (Pearl et al., 2008). HCC is a major global health concern, with over 500,000 new cases diagnosed annually. It ranks as the sixth most common cancer worldwide (Abd El-Fattah et al., 2022; Nasr et al., 2023; Saber et al., 2023). Its incidence is on the rise globally (Nasr et al., 2023), and notably, it is the fastest-growing reason for cancer-related deaths among men in the United States (Bruix et al., 2004; El–Serag and Rudolph, 2007). Hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are the primary factors contributing to the development of HCC. However, almost any condition that results in cirrhosis, with alcoholism being a prominent example, can potentially lead to the development of hepatocarcinogenesis (Farazi and DePinho, 2006; Abdelhamid et al., 2021).
The prognosis for HCC remains unfavorable because it is often diagnosed at an advanced stage, and tumor recurrence is relatively common (Bruix et al., 2004). Worrisome increases in HCV infection rates are raising concerns about the possibility of an impending HCC pandemic. This underscores the critical and immediate need for effective treatments to address this growing public health issue (El–Serag and Rudolph, 2007). Inhibiting HSP90 simultaneously reduced various factors that promote hepatocarcinogenesis, suppressed the growth of HCC tumors, and induced apoptosis (Nouri-Vaskeh et al., 2020). The use of non-quinone compounds as HSP90 inhibitors appears to be a promising treatment strategy for HCC. In animal studies, the non-quinone HSP90 inhibitor, PU-H71, specifically accumulated in tumors, effectively inhibited the growth of HCC tumors, and was well-tolerated, suggesting a low risk of severe liver toxicity (Breinig et al., 2009). In in vivo experiments, the non-quinone HSP90 inhibitor PU-H71 demonstrated no adverse effects on liver function, and no toxicity was observed (Speranza et al., 2018). This finding underscores the growing evidence that non-quinone HSP90 inhibitors are well-tolerated in living organisms (Rodina et al., 2007; Chandarlapaty et al., 2008; Eccles et al., 2008). The quick removal of PU-H71 from the liver, combined with its effective retention in tumors, improves the advantageous therapeutic characteristics of non-quinone HSP90 inhibitors for treating HCC. Additionally, the fact that PU-H71 treatment did not substantially affect ERK, AKT, or CDK4 in mouse livers further supports this conclusion (Breinig et al., 2009). These findings build upon prior research involving a different non-quinone HSP90 inhibitor, emphasizing the potential advantages of such inhibitors for HCC therapy (Chandarlapaty et al., 2008).
8.6 Impact on glial cells
Geldanamycin (GA) and radicicol are natural compounds known for their ability to inhibit the N-terminal ATPase activity of HSP90. HSP90 primarily acts as a chaperone to oversee a set of proteins known as “client proteins” during the final stages of their maturation and folding, especially as they near their native, functional configuration. HSP90 is heavily involved in various signal transduction processes (Barginear et al., 2008). When compounds like radicicol inhibit the ATPase activity of HSP90, client proteins detach from the chaperone (Roe et al., 1999). Among these client proteins, transcription factors like HSF-1 can become activated, while others may either lose their function or be marked for degradation by the proteasome. The removal of HSF-1 from HSP90 facilitates its movement into the nucleus, where it initiates the heat shock response (HSR). The use of HSP90 inhibitor drugs to stimulate the HSR pharmacologically can efficiently inhibit the pro-inflammatory activation of glial cells and enhance clinical outcomes in conditions such as experimental autoimmune encephalomyelitis (EAE) (Lisi et al., 2013; Seasons et al., 2024). The anti-inflammatory impacts of the HSR appear to be conveyed through a decrease in the activation of the NF-κB transcription factor (Schroeder et al., 2024). Ansamycins, including GA, are indeed potent HSP90 inhibitors. Nevertheless, their practical use in clinical settings is restricted due to notable toxicity concerns and stability challenges (Chiosis et al., 2004). PU-H71 is recognized as a potent HSP90 inhibitor, and datasets related to PU-H71 display a strong correlation in terms of HSP90 inhibition. This suggests that these drugs share a similar molecular method of action. The HSP90 inhibitor PU-H71 has demonstrated beneficial effects in the context of neuroinflammatory diseases (Kim et al., 2009).
PU-H71 was found to inhibit the activation of astrocytes when stimulated by the bacterial endotoxin lipopolysaccharide (LPS). However, it did not have an inhibitory effect on the activation of inflammatory cytokines. Similarly, PU-H71, like 17-AAG (another HSP90 inhibitor), had relatively mild anti-inflammatory influences on microglial initiation. Conversely to ansamycins (like GA), PU-H71 had a restricted impact on the progress of EAE disease. Its effects included a reduction in peripheral T-cell triggering and IFN release (Lisi et al., 2013). These data suggest that PU-H71 shares some anti-inflammatory roles with ansamycins. However, they also indicate significant disparities in their action mechanisms between ansamycins and PU-H71 regarding the initiation of glial cells, which may explain the lower performance of PU-H71 in EAE. Both 17-AAG and PU-H71 target blocking the ATPase activity of HSP-90 by binding to its N-terminal ATP/ADP pocket. Notably, the chemical structure of PU-H71 is entirely dissimilar from that of 17-AAG. PU-H71 has a simple formulation and lacks the benzoquinone moiety found in 17-AAG (Trendowski, 2015).
The primary difference between PU-H71 and 17-AAG lies in the absence of a benzoquinone group, potentially leading to distinct pharmacological effects. Interestingly, the presence or absence of DT-diaphorase appears to play a significant role in determining the varied response to HSP90 inhibition between the two compounds, possibly serving as a resistance mechanism (Kelland et al., 1999). Additionally, the silencing of the NAD (P) H dehydrogenase quinone 1 (NQO1) gene due to hypermethylation limits the effectiveness of 17-AAG in Hep3B cells, but this limitation does not apply to PU-H71 (Tada et al., 2005; Broekgaarden et al., 2015). Consequently, the anti-tumor response to novel HSP90 inhibitors is greatly enhanced by the NQO1 status (Hadley and Hendricks, 2014). The restricted in vivo effectiveness of PU-H71 may be attributed to differences in its selectivity for particular cells and its ability to penetrate the blood-brain barrier (BBB). It is important to highlight that, under normal circumstances, PU-H71 has a very restricted capacity to penetrate the CNS (Lisi et al., 2013). The comparatively modest inhibitory impact of the drug noticed in vitro, coupled with the activation of glial cells using a combination of cytokines, may aid in understanding the drug’s restricted effects in MOG-induced EAE following treatment. Cytokines such as TNF, IFN, and IL-1, which are closely linked to neuroinflammatory processes, may play a substantial role in this particular context (Sosa and Forsthuber, 2011). Therefore, the use of a combination of these cytokines represents a more pertinent physiopathological stimulus compared to bacterial endotoxin when trying to replicate the conditions that glial cells encounter in neurodegenerative disorders such as multiple sclerosis (MS) within a living organism. In live animal experiments, drugs that exhibit substantial anti-inflammatory effects in laboratory settings and reduce the activity of NOS2 triggered by inflammatory stimuli like TII have been shown to alleviate the clinical symptoms of EAE, which serves as a model for MS (Lubina-Dąbrowska et al., 2017). LPS functions by binding to the Toll-like receptor 4 (TLR4) receptor, while cytokines interact with and activate various receptors through separate intracellular signaling pathways (Ciesielska et al., 2021; Youssef et al., 2021a; Youssef et al., 2021b; Afroz et al., 2022). The anti-inflammatory action of PU-H71 in glial cells appears to involve the inhibition of NF-κB activation, as suggested by in vitro studies using stably transfected C6 glioma cells. This mechanism may explain the observed effects in reducing nitrite levels in response to both LPS (LI) and a combination of cytokines (TII). PU-H71, when used at concentrations of 30 nM for LI-stimulated C6 cells and 50 nM for TII-stimulated C6 cells, was found to decrease NFκB promoter activity while simultaneously reducing the production of nitric oxide (NO) (Lisi et al., 2013).
8.7 Impact on myeloproliferative neoplasms
Myeloproliferative neoplasms (MPNs) are set of clonal hematological malignancies that encompass conditions such as polycythemia vera (PV), chronic myeloid leukemia (CML), primary myelofibrosis (PMF) and essential thrombocythemia (ET). These disorders involve the overproduction of certain blood cells in the bone marrow (Luque Paz et al., 2023; Dunn et al., 2024; Guo et al., 2024). Although it has been over three decades since the clonal origin of these conditions stemming from stem cells was established, the genetic basis of BCR-ABL-negative MPNs stayed enigmatic until multiple studies revealed a somatic activating mutation in the JAK2 kinase (JAK2V617F). This mutation is present in the overwhelming majority of PV patients and is observed in nearly 50% of patients with ET and PMF (James et al., 2005; Zhao et al., 2005). Follow-up investigations have uncovered somatic mutations in JAK2 exon 12 in instances of PV where the JAK2V617F mutation is absent. Furthermore, mutations in the thrombopoietin receptor, specifically MPLW515 L/K/A and MPLS505N, have been detected in a subset of patients with ET and PMF who do not possess the JAK2V617F mutation (Mario, 2007; Pietra et al., 2008). In vitro, the presence of JAK2/MPL mutations allows hematopoietic cells to proliferate even without the need for cytokines. This leads to increased activation of signaling pathways downstream of JAK2, including the STAT3/5, MAP kinase, and PI3K signaling pathways (Anand et al., 2011; Staerk and Constantinescu, 2012; Rao et al., 2019). Additionally, it is important to highlight that JAK2 or MPL mutations lead to a remarkably consistent pattern of myeloproliferation in vivo. This consistency is particularly notable in the context of conditions such as polycythemia (linked with JAK2V617F or JAK2 exon 12 mutations) and thrombocytosis/myelofibrosis (associated with JAK2V617F and MPLW515L mutations) (Wernig et al., 2006), This implies that the persistent activation of the JAK-STAT signaling pathway plays a crucial role in the progression of PV, ET, and PMF. PU-H71 demonstrates enhanced pharmacokinetic and pharmacodynamic characteristics, including its robust and sustained absorption by tumors. As a result, it leads to more efficient and enduring degradation of HSP90 client proteins (Jhaveri et al., 2020). Increasing PU-H71’s in vivo effectiveness doesn't appear to raise toxicity concerns. Extended treatment at effective doses has not shown significant hematopoietic or non-hematopoietic toxicities (Usmani et al., 2010; Levine et al., 2012).
Scientists used PU-H71 to assess HSP90 inhibition in JAK2-dependent cancers. PU-H71 demonstrated substantial anti-cancer effects in MPN cell lines, mouse models of MPN, and samples from MPN patients. PU-H71 treatment repressed cell proliferation in cells with JAK2/MPL mutations at doses related to JAK2 reduction and the suppression of downstream signaling pathways (Campbell and Green, 2006). Furthermore, giving PU-H71 to mice with JAK2V617F or MPLW515L mutations normalized blood parameters, reduced extramedullary hematopoiesis in both models, and decreased morbidity and mortality in the MPLW515L model, compared to mice receiving a control vehicle. Importantly, this therapeutic effect was achieved without causing hematopoietic or non-hematopoietic toxicities (Marubayashi et al., 2010). Additionally, PU-H71 treatment has been shown to inhibit abnormal erythrocytosis and megakaryopoiesis in the JAK2V617F and MPLW515L rodent models, while it does not impact normal erythrocytosis and megakaryopoiesis (Koppikar et al., 2010). These findings suggest that HSP90 inhibitor therapy with PU-H71 has a unique impact on the proliferation and signaling of the malignant clone. The specific effect of PU-H71 on JAK2/MPL mutant cells in vivo does not seem to be solely due to the mutant or activated JAK2’s increased reliance on the HSP90 chaperone complex (Marubayashi et al., 2010; Mullally, 2017).
Unlike previous experiments with a JAK2 inhibitor, PU-H71 treatment reduces the burden of mutant alleles in the MPLW515L murine MPN model. This strongly supports the idea of conducting clinical trials with PU-H71 for treating JAK2V617F/MPLW515L mutant MPN. In preclinical studies of PU-H71 and other HSP90 inhibitors, flow cytometric assays measuring JAK2 protein expression, phospho-STAT5 levels, and HSP70 induction could be utilized as pharmacodynamic assessments (Koppikar et al., 2010). PU-H71 and other HSP90 inhibitors affect numerous client proteins, likely contributing to their impact on myeloproliferation both in vitro and in vivo. However, evidence suggests that JAK2 is the primary target for HSP90 inhibitors in JAK2/MPL-mediated myeloproliferation (Weigert et al., 2012; Chakraborty et al., 2016; Hobbs et al., 2018). PU-H71 consistently resulted in JAK2 degradation and pathway inhibition in vitro and in vivo at comparable doses. Additionally, PU-H71 and two different JAK2 kinase inhibitors had a complementary, not synergistic, effect, implying a mutual mechanism of action in this situation (George et al., 2004). PU-H71 can serve as a valuable tool for identifying tumors depending on HSP90 chaperone proteins. This data could be integrated into genomic and proteomic databases to reveal new molecular targets across various human cancer types. The effectiveness of HSP90 inhibition by PU-H71 extends to a range of genetically diverse human cancers, highlighting the importance of conducting targeted drug trials with HSP90 inhibitors for treating MPNs and other malignancies (Gao et al., 2007; Hedvat et al., 2009).
9 The therapeutic potential of PU-H71 in non-tumor disorders
The reasoning behind the connection between protein aggregation and neurodegenerative conditions has been that blocking HSP90 leads to the activation of HSF-1, resulting in the production of HSP70, HSP40, and other chaperones, which in turn facilitate the disaggregation and breakdown of proteins. However, recent findings indicate that HSP90 has an added involvement in neurodegenerative processes. Specifically, HSP90 is involved in upholding the operational integrity of neuronal proteins that have an abnormal tendency, thereby permitting and sustaining the buildup of detrimental protein clumps (Waza et al., 2006).
Parkinson’s disease (PD), the most common neurodegenerative movement disorder, is marked by a complex interplay of pathological events (Westerlund et al., 2010). A substantial number of these events have been more recently associated with HSP90. Wang et al. have pointed out that Leucine-rich repeat kinase 2 (LRRK2), an enzyme whose altered forms already are found in both familial and sporadic cases of Parkinson’s disease, interacts with HSP90 in vivo to form a complex (Wang et al., 2008). The purine-based HSP90 inhibitor PU-H71 disrupted the interaction between HSP90 and LRRK2, resulting in the degradation of LRRK2 via the proteasome system. PU-H71 also decreased the neurotoxic effects induced by mutant LRRK2 and reversed the impairment in axon growth associated with the LRRK2 G2019S mutation in neurons (Wang et al., 2008). PARK6, a subtype of familial Parkinson’s disease inherited in an autosomal recessive fashion, is linked to a mutation that occurs in PTEN-induced kinase 1 (PINK1). PINK1 is a gene that codes for a potential mitochondrial serine/threonine kinase (Westerlund et al., 2010). The autosomal recessive inheritance pattern associated with this type of Parkinson’s disease indicates a strong connection between the disease’s development and the loss of PINK1 function. HSP90 always attaches to PINK1 to enhance its equilibrium. In cells exposed to the HSP90 inhibitor GM, the levels of PINK1 were significantly diminished through the ubiquitin-proteasome pathway (Moriwaki et al., 2008).
10 Conclusion
In this comprehensive review, we have explored the pivotal role of HSP90 as a key molecular player in maintaining cellular health and its intricate involvement in various disease contexts. We discussed how HSP90, in partnership with HSF-1, orchestrates stress responses, shedding light on its role in regulating the HSR. The therapeutic potential of PU-H71, a novel purine-based HSP90 inhibitor, has been a central focus of our investigation. We have elucidated its promise in addressing neurodegenerative disorders and its significant impact on triple-negative breast cancer, offering new avenues for cancer treatment. PU-H71’s ability to sensitize cancer cells for carbon-ion beam therapy suggests a novel approach to improving the efficacy of radiation therapy. Furthermore, the dual inhibition of HSP90 A and HSP90B1 by PU-H71 emerges as a potential breakthrough in myeloma treatment. Its capacity to induce apoptosis in B-cell lymphomas dependent on Bcl6 for survival underscores its relevance in hematologic cancers. Our exploration extends to HCC, where innovative chemotherapy approaches and PU-H71’s potential to inhibit the activation of glial cells hold promise for patients with hepatic and neuroinflammatory disorders. Finally, we emphasize PU-H71’s encouraging role in JAK2-dependent myeloproliferative neoplasms, highlighting its potential as a therapeutic avenue in hematology. Collectively, this review underscores the versatility of HSP90 and the immense therapeutic potential of PU-H71 in diverse disease scenarios, opening new doors to innovative treatments and improved patient outcomes in the realm of health and disease.
11 Authors’ perspectives and future considerations
From the author’s perspective, the multifaceted roles of HSP90 and the promising therapeutic potential of PU-H71 underscore the need for continued research and development in this field. Shedding more light on the underlying mechanisms of HSP90 and its inhibitors highlights them both as promising candidates for the development of novel targeted cancer therapies. Markedly, special attention should focus on optimizing precise drug delivery strategies to enhance the efficacy as well as selectivity of such novel therapeutic agents and to explore any potential combination therapies for maximum efficacy.
In addition, pondering the molecular pathways underlying PU-H71 actions is indispensable for tailoring novel therapeutic approaches to fulfilling specific patient needs. Further investigations on the long-term safety profiles of HSP90 inhibitors in addition to their potential for resistance mechanisms should be conducted to ensure their clinical success. The integration of pioneering technologies (e.g., precision medicine approaches and advanced imaging techniques) could potentially establish a platform for developing personalized as well as HSP90-targeted therapies.
Author contributions
SS: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. RA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. ME: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. EE: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. RH: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. MA-R: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. AE-K: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. MA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. KM: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. AA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. MY: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work under grant number RGP2/233/45. This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia (Grant No. KFU242193). The authors would like to thank the Deanship of Scientific Research at Shaqra University for supporting this work. The researchers would like to thank the Deanship of Graduate Studies and Scientific Research at Qassim University for financial support (QU-APC-2024-9/1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abd El-Fattah, E. E., Saber, S., Youssef, M. E., Eissa, H., El-Ahwany, E., Amin, N. A., et al. (2022). AKT-AMPKα-mTOR-dependent HIF-1α activation is a new therapeutic target for cancer treatment: a novel approach to repositioning the antidiabetic drug sitagliptin for the management of hepatocellular carcinoma. Front. Pharmacol. 12. doi:10.3389/fphar.2021.720173
Abdel-Ghany, R., Rabia, I., El-Ahwany, E., Saber, S., Gamal, R., Nagy, F., et al. (2015). Blockade of Pge2, Pgd2 receptors confers protection against prepatent schistosomiasis mansoni in mice. J. Egypt Soc. Parasitol. 45, 511–520. doi:10.12816/0017911
Abdelhamid, A. M., Saber, S., Youssef, M. E., Gaafar, A. G. A., Eissa, H., Abd-Eldayem, M. A., et al. (2022). Empagliflozin adjunct with metformin for the inhibition of hepatocellular carcinoma progression: emerging approach for new application. Biomed. and Pharmacother. 145, 112455. doi:10.1016/j.biopha.2021.112455
Abdelhamid, A. M., Youssef, M. E., Abd El-Fattah, E. E., Gobba, N. A., Gaafar, A. G. A., Girgis, S., et al. (2021). Blunting p38 MAPKα and ERK1/2 activities by empagliflozin enhances the antifibrotic effect of metformin and augments its AMPK-induced NF-κB inactivation in mice intoxicated with carbon tetrachloride. Life Sci. 286, 120070. doi:10.1016/j.lfs.2021.120070
Abu-Farha, M., Lanouette, S., Elisma, F., Tremblay, V., Butson, J., Figeys, D., et al. (2011). Proteomic analyses of the SMYD family interactomes identify HSP90 as a novel target for SMYD2. J. Mol. Cell Biol. 3, 301–308. doi:10.1093/jmcb/mjr025
Afroz, R., Tanvir, E. M., Tania, M., Fu, J., Kamal, M. A., and Khan, M. A. (2022). LPS/TLR4 pathways in breast cancer: insights into cell signalling. Curr. Med. Chem. 29, 2274–2289. doi:10.2174/0929867328666210811145043
Akahane, K., Sanda, T., Mansour, M. R., Radimerski, T., Deangelo, D. J., Weinstock, D. M., et al. (2016). HSP90 inhibition leads to degradation of the TYK2 kinase and apoptotic cell death in T-cell acute lymphoblastic leukemia. Leukemia 30, 219–228. doi:10.1038/leu.2015.222
Akram, A., Khalil, S., Halim, S. A., Younas, H., Iqbal, S., and Mehar, S. (2018). Therapeutic uses of HSP90 inhibitors in non-small cell lung carcinoma (NSCLC). Curr. Drug Metab. 19, 335–341. doi:10.2174/1389200219666180307122441
Albadari, N., Deng, S., and Li, W. (2019). The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 14, 667–682. doi:10.1080/17460441.2019.1613370
Ambati, S. R., Lopes, E. C., Kosugi, K., Mony, U., Zehir, A., Shah, S. K., et al. (2014). Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 8, 323–336. doi:10.1016/j.molonc.2013.12.005
Anand, S., Stedham, F., Beer, P., Gudgin, E., Ortmann, C. A., Bench, A., et al. (2011). Effects of the JAK2 mutation on the hematopoietic stem and progenitor compartment in human myeloproliferative neoplasms. Blood 118, 177–181. doi:10.1182/blood-2010-12-327593
Anwar, M. M., Shalaby, M., Embaby, A. M., Saeed, H., Agwa, M. M., and Hussein, A. (2020). Prodigiosin/PU-H71 as a novel potential combined therapy for triple negative breast cancer (TNBC): preclinical insights. Sci. Rep. 10, 14706. doi:10.1038/s41598-020-71157-w
Bachman, A. B., Keramisanou, D., Xu, W., Beebe, K., Moses, M. A., Vasantha Kumar, M. V., et al. (2018). Phosphorylation induced cochaperone unfolding promotes kinase recruitment and client class-specific Hsp90 phosphorylation. Nat. Commun. 9, 265. doi:10.1038/s41467-017-02711-w
Baffi, T. R., Lordén, G., Wozniak, J. M., Feichtner, A., Yeung, W., Kornev, A. P., et al. (2021). mTORC2 controls the activity of PKC and Akt by phosphorylating a conserved TOR interaction motif. Sci. Signal. 14, eabe4509. doi:10.1126/scisignal.abe4509
Bagratuni, T., Wu, P., Gonzalez De Castro, D., Davenport, E. L., Dickens, N. J., Walker, B. A., et al. (2010). XBP1s levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood 116, 250–253. doi:10.1182/blood-2010-01-263236
Bahar, M. E., Kim, H. J., and Kim, D. R. (2023). Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct. Target. Ther. 8, 455. doi:10.1038/s41392-023-01705-z
Baker, J. D., Ozsan, I., Rodriguez Ospina, S., Gulick, D., and Blair, L. J. (2018). Hsp90 heterocomplexes regulate steroid hormone receptors: from stress response to psychiatric disease. Int. J. Mol. Sci. 20, 79. doi:10.3390/ijms20010079
Bao, F., Hao, P., An, S., Yang, Y., Liu, Y., Hao, Q., et al. (2021). Akt scaffold proteins: the key to controlling specificity of Akt signaling. Am. J. Physiology-Cell Physiology 321, C429–C442. doi:10.1152/ajpcell.00146.2020
Barginear, M., Van Poznak, C., Rosen, N., Modi, S., Hudis, C., and Budman, D. (2008). The heat shock protein 90 chaperone complex: an evolving therapeutic target. Curr. cancer drug targets 8, 522–532. doi:10.2174/156800908785699379
Basso, A. D., Solit, D. B., Chiosis, G., Giri, B., Tsichlis, P., and Rosen, N. (2002). Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 277, 39858–39866. doi:10.1074/jbc.M206322200
Beeram, M., Patnaik, A., and Rowinsky, E. K. (2005). Raf: a strategic target for therapeutic development against cancer. J. Clin. Oncol. 23, 6771–6790. doi:10.1200/JCO.2005.08.036
Birbo, B., Madu, E. E., Madu, C. O., Jain, A., and Lu, Y. (2021). Role of HSP90 in cancer. Int. J. Mol. Sci. 22, 10317. doi:10.3390/ijms221910317
Breinig, M., Caldas-Lopes, E., Goeppert, B., Malz, M., Rieker, R., Bergmann, F., et al. (2009). Targeting heat shock protein 90 with non-quinone inhibitors: a novel chemotherapeutic approach in human hepatocellular carcinoma. Hepatology 50, 102–112. doi:10.1002/hep.22912
Breinig, M., Mayer, P., Harjung, A., Goeppert, B., Malz, M., Penzel, R., et al. (2011). Heat shock protein 90-sheltered overexpression of insulin-like growth factor 1 receptor contributes to malignancy of thymic epithelial tumors. Clin. Cancer Res. 17, 2237–2249. doi:10.1158/1078-0432.CCR-10-1689
Briones, J. (2010). Targeted therapy of BCL6-dependent diffuse large B-cell lymphomas by heat-shock protein 90 inhibition. Expert Rev. Hematol. 3, 157–159. doi:10.1586/ehm.10.11
Broekgaarden, M., Weijer, R., Van Gulik, T. M., Hamblin, M. R., and Heger, M. (2015). Tumor cell survival pathways activated by photodynamic therapy: a molecular basis for pharmacological inhibition strategies. Cancer Metastasis Rev. 34, 643–690. doi:10.1007/s10555-015-9588-7
Bruix, J., Boix, L., Sala, M., and Llovet, J. M. (2004). Focus on hepatocellular carcinoma. Cancer Cell 5, 215–219. doi:10.1016/s1535-6108(04)00058-3
Bulatowicz, J. J., and Wood, T. L. (2022). Activation versus inhibition of IGF1R: a dual role in breast tumorigenesis. Front. Endocrinol. (Lausanne) 13, 911079. doi:10.3389/fendo.2022.911079
Caldas-Lopes, E., Cerchietti, L., Ahn, J. H., Clement, C. C., Robles, A. I., Rodina, A., et al. (2009). Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. 106, 8368–8373. doi:10.1073/pnas.0903392106
Calvo-Vidal, M. N., Zamponi, N., Krumsiek, J., Stockslager, M. A., Revuelta, M. V., Phillip, J. M., et al. (2021). Oncogenic HSP90 facilitates metabolic alterations in aggressive B-cell lymphomas. Cancer Res. 81, 5202–5216. doi:10.1158/0008-5472.CAN-21-2734
Campbell, P. J., and Green, A. R. (2006). The myeloproliferative disorders. N. Engl. J. Med. 355, 2452–2466. doi:10.1056/NEJMra063728
Carter, B. Z., Mak, P. Y., Muftuoglu, M., Tao, W., Ke, B., Pei, J., et al. (2023). Epichaperome inhibition targets TP53-mutant AML and AML stem/progenitor cells. Blood 142, 1056–1070. doi:10.1182/blood.2022019047
Caruso Bavisotto, C., Alberti, G., Vitale, A. M., Paladino, L., Campanella, C., Rappa, F., et al. (2020). Hsp60 post-translational modifications: functional and pathological consequences. Front. Mol. Biosci. 7, 95. doi:10.3389/fmolb.2020.00095
Cavenagh, J., Oakervee, H., Baetiong-Caguioa, P., Davies, F., Gharibo, M., Rabin, N., et al. (2017). A phase I/II study of KW-2478, an Hsp90 inhibitor, in combination with bortezomib in patients with relapsed/refractory multiple myeloma. Br. J. Cancer 117, 1295–1302. doi:10.1038/bjc.2017.302
Cerchietti, L. C., Hatzi, K., Caldas-Lopes, E., Yang, S. N., Figueroa, M. E., Morin, R. D., et al. (2010). BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J. Clin. Investigation 120, 4569–4582. doi:10.1172/JCI42869
Cerchietti, L. C., Lopes, E. C., Yang, S. N., Hatzi, K., Bunting, K. L., Tsikitas, L. A., et al. (2009). A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6–dependent B cell lymphomas. Nat. Med. 15, 1369–1376. doi:10.1038/nm.2059
Chakrabarty, S., Wang, S., Roychowdhury, T., Ginsberg, S. D., and Chiosis, G. (2024). Introducing dysfunctional Protein-Protein Interactome (dfPPI) – a platform for systems-level protein-protein interaction (PPI) dysfunction investigation in disease. Curr. Opin. Struct. Biol. 88, 102886. doi:10.1016/j.sbi.2024.102886
Chakraborty, S. N., Leng, X., Perazzona, B., Sun, X., Lin, Y. H., and Arlinghaus, R. B. (2016). Combination of JAK2 and HSP90 inhibitors: an effective therapeutic option in drug-resistant chronic myelogenous leukemia. Genes Cancer 7, 201–208. doi:10.18632/genesandcancer.111
Chandarlapaty, S., Sawai, A., Ye, Q., Scott, A., Silinski, M., Huang, K., et al. (2008). SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase–dependent cancers. Clin. Cancer Res. 14, 240–248. doi:10.1158/1078-0432.CCR-07-1667
Chang, Y.-W., Wang, Y.-C., Zhang, X.-X., Iqbal, J., Lu, M.-X., and Du, Y.-Z. (2021). Transcriptional regulation of small heat shock protein genes by heat shock factor 1 (HSF1) in Liriomyza trifolii under heat stress. Cell Stress Chaperones 26, 835–843. doi:10.1007/s12192-021-01224-2
Chen, I.-C., Sethy, B., and Liou, J.-P. (2020). Recent update of HDAC inhibitors in lymphoma. Front. Cell Dev. Biol. 8, 576391. doi:10.3389/fcell.2020.576391
Chen, T. L., Harrington, B., Truxall, J., Wasmuth, R., Prouty, A., Sloan, S., et al. (2021). Preclinical evaluation of the Hsp90 inhibitor SNX-5422 in ibrutinib resistant CLL. J. Hematol. Oncol. 14, 36. doi:10.1186/s13045-021-01039-9
Cheng, X. (2024). A comprehensive review of HER2 in cancer biology and therapeutics. Genes 15, 903. doi:10.3390/genes15070903
Chin, Y., Gumilar, K. E., Li, X. G., Tjokroprawiro, B. A., Lu, C. H., Lu, J., et al. (2023). Targeting HSF1 for cancer treatment: mechanisms and inhibitor development. Theranostics 13, 2281–2300. doi:10.7150/thno.82431
Chiosis, G., Kang, Y., and Sun, W. (2008). Discovery and development of purine-scaffold Hsp90 inhibitors. Expert Opin. drug Discov. 3, 99–114. doi:10.1517/17460441.3.1.99
Chiosis, G., Vilenchik, M., Kim, J., and Solit, D. (2004). Hsp90: the vulnerable chaperone. Drug Discov. today 9, 881–888. doi:10.1016/S1359-6446(04)03245-3
Ci, W., Polo, J. M., and Melnick, A. (2008). B-cell lymphoma 6 and the molecular pathogenesis of diffuse large B-cell lymphoma. Curr. Opin. Hematol. 15, 381–390. doi:10.1097/MOH.0b013e328302c7df
Ciesielska, A., Matyjek, M., and Kwiatkowska, K. (2021). TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 78, 1233–1261. doi:10.1007/s00018-020-03656-y
Collier, M. P., and Benesch, J. L. P. (2020). Small heat-shock proteins and their role in mechanical stress. Cell Stress Chaperones 25, 601–613. doi:10.1007/s12192-020-01095-z
Cserni, G., Quinn, C. M., Foschini, M. P., Bianchi, S., Callagy, G., Chmielik, E., et al. (2021). Triple-negative breast cancer histological subtypes with a favourable prognosis. Cancers 13, 5694. doi:10.3390/cancers13225694
Culjkovic-Kraljacic, B., Fernando, T. M., Marullo, R., Calvo-Vidal, N., Verma, A., Yang, S., et al. (2016). Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood 127, 858–868. doi:10.1182/blood-2015-05-645069
Cyran, A. M., and Zhitkovich, A. (2022). Heat shock proteins and HSF1 in cancer. Front. Oncol. 12, 860320. doi:10.3389/fonc.2022.860320
De Almeida, S., Regimbeau, M., Jego, G., Garrido, C., Girodon, F., and Hermetet, F. (2020). Heat shock proteins and PD-1/PD-L1 as potential therapeutic targets in myeloproliferative neoplasms. Cancers 12, 2592. doi:10.3390/cancers12092592
Digwal, C. S., Sharma, S., Santhaseela, A. R., Ginsberg, S. D., and Chiosis, G. (2022). “Epichaperomes as a gateway to understanding, diagnosing, and treating disease through rebalancing protein–protein interaction networks,” in Protein homeostasis in drug discovery., 1–26.
Dimri, M., and Satyanarayana, A. (2020). Molecular signaling pathways and therapeutic targets in hepatocellular carcinoma. Cancers 12, 491. doi:10.3390/cancers12020491
Duan, X., Iwanowycz, S., Ngoi, S., Hill, M., Zhao, Q., and Liu, B. (2021). Molecular chaperone GRP94/GP96 in cancers: oncogenesis and therapeutic target. Front. Oncol. 11, 629846. doi:10.3389/fonc.2021.629846
Dunn, W. G., Mcloughlin, M. A., and Vassiliou, G. S. (2024). Clonal hematopoiesis and hematological malignancy. J. Clin. Investigation 134, e180065. doi:10.1172/JCI180065
Dunphy, M. P. S., Pressl, C., Pillarsetty, N., Grkovski, M., Modi, S., Jhaveri, K., et al. (2020). First-in-Human trial of epichaperome-targeted PET in patients with cancer. Clin. Cancer Res. 26, 5178–5187. doi:10.1158/1078-0432.CCR-19-3704
Eccles, S. A., Massey, A., Raynaud, F. I., Sharp, S. Y., Box, G., Valenti, M., et al. (2008). NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 68, 2850–2860. doi:10.1158/0008-5472.CAN-07-5256
El-Kashef, D. H., Youssef, M. E., Nasr, M., Alrouji, M., Alhajlah, S., Alomeir, O., et al. (2022). Pimitespib, an HSP90 inhibitor, augments nifuroxazide-induced disruption in the IL-6/STAT3/HIF-1α autocrine loop in rats with bleomycin-challenged lungs: evolutionary perspective in managing pulmonary fibrosis. Biomed. and Pharmacother. 153, 113487. doi:10.1016/j.biopha.2022.113487
El–Serag, H. B., and Rudolph, K. L. (2007). Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576. doi:10.1053/j.gastro.2007.04.061
Elster, N., Collins, D. M., Toomey, S., Crown, J., Eustace, A. J., and Hennessy, B. T. (2015). HER2-family signalling mechanisms, clinical implications and targeting in breast cancer. Breast Cancer Res. Treat. 149, 5–15. doi:10.1007/s10549-014-3250-x
Farazi, P. A., and Depinho, R. A. (2006). Hepatocellular carcinoma pathogenesis: from genes to environment. Nat. Rev. Cancer 6, 674–687. doi:10.1038/nrc1934
Fernando, T. M., Marullo, R., Pera Gresely, B., Phillip, J. M., Yang, S. N., Lundell-Smith, G., et al. (2019). BCL6 evolved to enable stress tolerance in vertebrates and is broadly required by cancer cells to adapt to stress. Cancer Discov. 9, 662–679. doi:10.1158/2159-8290.CD-17-1444
Firnau, M.-B., and Brieger, A. (2022). CK2 and the hallmarks of cancer. Biomedicines 10, 1987. doi:10.3390/biomedicines10081987
Fu, J., Su, X., Li, Z., Deng, L., Liu, X., Feng, X., et al. (2021). HGF/c-MET pathway in cancer: from molecular characterization to clinical evidence. Oncogene 40, 4625–4651. doi:10.1038/s41388-021-01863-w
Fusco, N., Sajjadi, E., Venetis, K., Ivanova, M., Andaloro, S., Guerini-Rocco, E., et al. (2022). Low-risk triple-negative breast cancers: clinico-pathological and molecular features. Crit. Rev. Oncology/Hematology 172, 103643. doi:10.1016/j.critrevonc.2022.103643
Gallerne, C., Prola, A., and Lemaire, C. (2013). Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1833, 1356–1366. doi:10.1016/j.bbamcr.2013.02.014
Gao, C., Peng, Y. N., Wang, H. Z., Fang, S. L., Zhang, M., Zhao, Q., et al. (2019). Inhibition of heat shock protein 90 as a novel platform for the treatment of cancer. Curr. Pharm. Des. 25, 849–855. doi:10.2174/1381612825666190503145944
Gao, S. P., Mark, K. G., Leslie, K., Pao, W., Motoi, N., Gerald, W. L., et al. (2007). Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. investigation 117, 3846–3856. doi:10.1172/JCI31871
García-Flores, N., Jiménez-Suárez, J., Garnés-García, C., Fernández-Aroca, D. M., Sabater, S., Andrés, I., et al. (2023). P38 MAPK and radiotherapy: foes or friends? Cancers (Basel) 15, 861. doi:10.3390/cancers15030861
George, P., Bali, P., Cohen, P., Tao, J., Guo, F., Sigua, C., et al. (2004). Cotreatment with 17-allylamino-demethoxygeldanamycin and FLT-3 kinase inhibitor PKC412 is highly effective against human acute myelogenous leukemia cells with mutant FLT-3. Cancer Res. 64, 3645–3652. doi:10.1158/0008-5472.CAN-04-0006
Gerecitano, J. F., Modi, S., Rampal, R., Drilon, A. E., Fury, M. G., Gounder, M. M., et al. (2015). Phase I trial of the HSP-90 inhibitor PU-H71. J. Clin. Oncol. 33, 2537. doi:10.1200/jco.2015.33.15_suppl.2537
Giakountis, A., Moulos, P., Sarris, M. E., Hatzis, P., and Talianidis, I. (2017). Smyd3-associated regulatory pathways in cancer. Seminars Cancer Biol. 42, 70–80. doi:10.1016/j.semcancer.2016.08.008
Ginsberg, S. D., Joshi, S., Sharma, S., Guzman, G., Wang, T., Arancio, O., et al. (2021). The penalty of stress - epichaperomes negatively reshaping the brain in neurodegenerative disorders. J. Neurochem. 159, 958–979. doi:10.1111/jnc.15525
Ginsberg, S. D., Sharma, S., Norton, L., and Chiosis, G. (2023). Targeting stressor-induced dysfunctions in protein–protein interaction networks via epichaperomes. Trends Pharmacol. Sci. 44, 20–33. doi:10.1016/j.tips.2022.10.006
Giulino-Roth, L., Van Besien, H. J., Dalton, T., Totonchy, J. E., Rodina, A., Taldone, T., et al. (2017). Inhibition of Hsp90 suppresses PI3K/AKT/mTOR signaling and has antitumor activity in burkitt lymphoma. Mol. Cancer Ther. 16, 1779–1790. doi:10.1158/1535-7163.MCT-16-0848
Graner, A. N., Hellwinkel, J. E., Lencioni, A. M., Madsen, H. J., Harland, T. A., Marchando, P., et al. (2017). HSP90 inhibitors in the context of heat shock and the unfolded protein response: effects on a primary canine pulmonary adenocarcinoma cell line. Int. J. Hyperth. 33, 303–317. doi:10.1080/02656736.2016.1256503
Guerra, F., Quintana, S., Giustina, S., Mendeluk, G., Jufe, L., Avagnina, M. A., et al. (2021). Investigation of EGFR/pi3k/Akt signaling pathway in seminomas. Biotech. and Histochem. 96, 125–137. doi:10.1080/10520295.2020.1776393
Guo, J., Walter, K., Quiros, P. M., Gu, M., Baxter, E. J., Danesh, J., et al. (2024). Inherited polygenic effects on common hematological traits influence clonal selection on JAK2V617F and the development of myeloproliferative neoplasms. Nat. Genet. 56, 273–280. doi:10.1038/s41588-023-01638-x
Hadizadeh Esfahani, A., Sverchkova, A., Saez-Rodriguez, J., Schuppert, A. A., and Brehme, M. (2018). A systematic atlas of chaperome deregulation topologies across the human cancer landscape. PLOS Comput. Biol. 14, e1005890. doi:10.1371/journal.pcbi.1005890
Hadley, K. E., and Hendricks, D. T. (2014). Use of NQO1 status as a selective biomarker for oesophageal squamous cell carcinomas with greater sensitivity to 17-AAG. BMC Cancer 14, 334. doi:10.1186/1471-2407-14-334
Halim, C. E., Deng, S., Ong, M. S., and Yap, C. T. (2020). Involvement of STAT5 in oncogenesis. Biomedicines 8, 316. doi:10.3390/biomedicines8090316
Hanahan, D., and Weinberg, R. a. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hasan Khudhair, D., Al-Gareeb, A. I., Al-Kuraishy, H. M., El-Kadem, A. H., Elekhnawy, E., Negm, W. A., et al. (2022). Combination of vitamin C and curcumin safeguards against methotrexate-induced acute liver injury in mice by synergistic antioxidant effects. Front. Med. 9, 866343. doi:10.3389/fmed.2022.866343
Hatzi, K., and Melnick, A. (2014). Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol. Med. 20, 343–352. doi:10.1016/j.molmed.2014.03.001
He, W., and Hu, H. (2018). BIIB021, an Hsp90 inhibitor: a promising therapeutic strategy for blood malignancies (Review). Oncol. Rep. 40, 3–15. doi:10.3892/or.2018.6422
Hedvat, M., Huszar, D., Herrmann, A., Gozgit, J. M., Schroeder, A., Sheehy, A., et al. (2009). The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 16, 487–497. doi:10.1016/j.ccr.2009.10.015
Ho, N., Li, A., Li, S., and Zhang, H. (2012). Heat shock protein 90 and role of its chemical inhibitors in treatment of hematologic malignancies. Pharmaceuticals 5, 779–801. doi:10.3390/ph5080779
Hobbs, G. S., Hanasoge Somasundara, A. V., Kleppe, M., Litvin, R., Arcila, M., Ahn, J., et al. (2018). Hsp90 inhibition disrupts JAK-STAT signaling and leads to reductions in splenomegaly in patients with myeloproliferative neoplasms. Haematologica 103, e5–e9. doi:10.3324/haematol.2017.177600
Hoter, A., El-Sabban, M. E., and Naim, H. Y. (2018). The HSP90 family: structure, regulation, function, and implications in health and disease. Int. J. Mol. Sci. 19, 2560. doi:10.3390/ijms19092560
Howard, F. M., and Olopade, O. I. (2021). Epidemiology of triple-negative breast cancer: a review. Cancer J. 27, 8–16. doi:10.1097/PPO.0000000000000500
Hoxhaj, G., and Manning, B. D. (2020). The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88. doi:10.1038/s41568-019-0216-7
Hsu, J. L., and Hung, M.-C. (2016). The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 35, 575–588. doi:10.1007/s10555-016-9649-6
Hu, C., Yang, J., Qi, Z., Wu, H., Wang, B., Zou, F., et al. (2020). Heat shock proteins: biological functions, pathological roles, and therapeutic opportunities. MedComm 3, e161. doi:10.1002/mco2.161
Hu, H., Tian, M., Ding, C., and Yu, S. (2019). The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 9, 3083. doi:10.3389/fimmu.2018.03083
Hua, Y., White-Gilbertson, S., Kellner, J., Rachidi, S., Usmani, S. Z., Chiosis, G., et al. (2013). Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clin. Cancer Res. 19, 6242–6251. doi:10.1158/1078-0432.CCR-13-2083
Inda, M. C., Joshi, S., Wang, T., Bolaender, A., Gandu, S., Koren Iii, J., et al. (2020). The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nat. Commun. 11, 319. doi:10.1038/s41467-019-14082-5
Iqbal, N., and Iqbal, N. (2014). Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol. Biol. Int. 2014, 852748. doi:10.1155/2014/852748
Iwai, A., Bourboulia, D., Mollapour, M., Jensen-Taubman, S., Lee, S., Donnelly, A. C., et al. (2012). Combined inhibition of Wee1 and Hsp90 activates intrinsic apoptosis in cancer cells. Cell Cycle 11, 3649–3655. doi:10.4161/cc.21926
Jaeger, A. M., and Whitesell, L. (2019). HSP90: enabler of cancer adaptation. Annu. Rev. Cancer Biol. 3, 275–297. doi:10.1146/annurev-cancerbio-030518-055533
James, C., Ugo, V., Le Couédic, J.-P., Staerk, J., Delhommeau, F., Lacout, C., et al. (2005). A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. nature 434, 1144–1148. doi:10.1038/nature03546
Jego, G., Hermetet, F., Girodon, F., and Garrido, C. (2019). Chaperoning STAT3/5 by heat shock proteins: interest of their targeting in cancer therapy. Cancers (Basel) 12, 21. doi:10.3390/cancers12010021
Jhaveri, K., Taldone, T., Modi, S., and Chiosis, G. (2012). Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochimica Biophysica Acta (BBA)-Molecular Cell Res. 1823, 742–755. doi:10.1016/j.bbamcr.2011.10.008
Jhaveri, K. L., Anjos, C. H. D., Taldone, T., Wang, R., Comen, E., Fornier, M., et al. (2020). “Measuring tumor epichaperome expression using [124I] PU-H71 positron emission tomography as a biomarker of response for PU-H71 plus nab-paclitaxel,” in HER2-Negative metastatic breast cancer (Alexandria, Virginia, United States: JCO Precision Oncology), 1414–1424.
Joshi, S., Gomes, E. D., Wang, T., Corben, A., Taldone, T., Gandu, S., et al. (2021). Pharmacologically controlling protein-protein interactions through epichaperomes for therapeutic vulnerability in cancer. Commun. Biol. 4, 1333. doi:10.1038/s42003-021-02842-3
Joshi, S., Wang, T., Araujo, T. L. S., Sharma, S., Brodsky, J. L., and Chiosis, G. (2018). Adapting to stress - chaperome networks in cancer. Nat. Rev. Cancer 18, 562–575. doi:10.1038/s41568-018-0020-9
Kakkar, V., Meister-Broekema, M., Minoia, M., Carra, S., and Kampinga, H. H. (2014). Barcoding heat shock proteins to human diseases: looking beyond the heat shock response. Dis. Models and Mech. 7, 421–434. doi:10.1242/dmm.014563
Kale, Ş., Korcum, A. F., Dündar, E., and Erin, N. (2020). HSP90 inhibitor PU-H71 increases radiosensitivity of breast cancer cells metastasized to visceral organs and alters the levels of inflammatory mediators. Naunyn-Schmiedeberg's Archives Pharmacol. 393, 253–262. doi:10.1007/s00210-019-01725-z
Kamada, T., Tsujii, H., Blakely, E. A., Debus, J., De Neve, W., Durante, M., et al. (2015). Carbon ion radiotherapy in Japan: an assessment of 20 years of clinical experience. Lancet Oncol. 16, e93–e100. doi:10.1016/S1470-2045(14)70412-7
Kamal, A., Boehm, M. F., and Burrows, F. J. (2004). Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol. Med. 10, 283–290. doi:10.1016/j.molmed.2004.04.006
Kawano, T., Inokuchi, J., Eto, M., Murata, M., and Kang, J. H. (2021). Activators and inhibitors of protein kinase C (PKC): their applications in clinical trials. Pharmaceutics 13, 1748. doi:10.3390/pharmaceutics13111748
Kaziales, A., Barkovits, K., Marcus, K., and Richter, K. (2020). Glucocorticoid receptor complexes form cooperatively with the Hsp90 co-chaperones Pp5 and FKBPs. Sci. Rep. 10, 10733. doi:10.1038/s41598-020-67645-8
Kelland, L. R., Sharp, S. Y., Rogers, P. M., Myers, T. G., and Workman, P. (1999). DT-diaphorase expression and tumor cell sensitivity to 17-Allylamino,17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. JNCI J. Natl. Cancer Inst. 91, 1940–1949. doi:10.1093/jnci/91.22.1940
Khurana, N., Laskar, S., Bhattacharyya, M. K., and Bhattacharyya, S. (2016). Hsp90 induces increased genomic instability toward DNA-damaging agents by tuning down RAD53 transcription. Mol. Biol. Cell 27, 2463–2478. doi:10.1091/mbc.E15-12-0867
Kijima, T., Prince, T., Neckers, L., Koga, F., and Fujii, Y. (2019). Heat shock factor 1 (HSF1)-targeted anticancer therapeutics: overview of current preclinical progress. Expert Opin. Ther. Targets 23, 369–377. doi:10.1080/14728222.2019.1602119
Kim, Y., Alarcon, S., Lee, S., Lee, M.-J., Giaccone, G., Neckers, L., et al. (2009). Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem. 9, 1479–1492. doi:10.2174/156802609789895728
Klein, U., and Dalla-Favera, R. (2008). Germinal centres: role in B-cell physiology and malignancy. Nat. Rev. Immunol. 8, 22–33. doi:10.1038/nri2217
Knudsen, E. S., and Witkiewicz, A. K. (2017). The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer 3, 39–55. doi:10.1016/j.trecan.2016.11.006
Koppikar, P., Abdel-Wahab, O., Hedvat, C., Marubayashi, S., Patel, J., Goel, A., et al. (2010). Efficacy of the JAK2 inhibitor INCB16562 in a murine model of MPLW515L-induced thrombocytosis and myelofibrosis. Blood, J. Am. Soc. Hematol. 115, 2919–2927. doi:10.1182/blood-2009-04-218842
Koppikar, P., Bhagwat, N., Kilpivaara, O., Manshouri, T., Adli, M., Hricik, T., et al. (2012). Heterodimeric JAK–STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 489, 155–159. doi:10.1038/nature11303
Krämer, O. H., Mahboobi, S., and Sellmer, A. (2014). Drugging the HDAC6–HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 35, 501–509. doi:10.1016/j.tips.2014.08.001
Kuai, Y., Gong, X., Ding, L., Li, F., Lei, L., Gong, Y., et al. (2018). Wilms’ tumor 1-associating protein plays an aggressive role in diffuse large B-cell lymphoma and forms a complex with BCL6 via Hsp90. Cell Commun. Signal. 16, 50. doi:10.1186/s12964-018-0258-6
Kubra, K.-T., Uddin, M. A., Akhter, M. S., and Barabutis, N. (2020). Hsp90 inhibitors induce the unfolded protein response in bovine and mice lung cells. Cell. Signal. 67, 109500. doi:10.1016/j.cellsig.2019.109500
Kucine, N., Marubayashi, S., Bhagwat, N., Papalexi, E., Koppikar, P., Sanchez Martin, M., et al. (2015). Tumor-specific HSP90 inhibition as a therapeutic approach in JAK-mutant acute lymphoblastic leukemias. Blood 126, 2479–2483. doi:10.1182/blood-2015-03-635821
Kunachowicz, D., Król-Kulikowska, M., Raczycka, W., Sleziak, J., Błażejewska, M., and Kulbacka, J. (2024). Heat shock proteins, a double-edged sword: significance in cancer progression, chemotherapy resistance and novel therapeutic perspectives. Cancers 16, 1500. doi:10.3390/cancers16081500
Kuykendall, A. T., Horvat, N. P., Pandey, G., Komrokji, R., and Reuther, G. W. (2020). Finding a jill for JAK: assessing past, present, and future JAK inhibitor combination approaches in myelofibrosis. Cancers 12, 2278. doi:10.3390/cancers12082278
Lee, Y., Li, H. K., Masaoka, A., Sunada, S., Hirakawa, H., Fujimori, A., et al. (2016). The purine scaffold Hsp90 inhibitor PU-H71 sensitizes cancer cells to heavy ion radiation by inhibiting DNA repair by homologous recombination and non-homologous end joining. Radiother. Oncol. 121, 162–168. doi:10.1016/j.radonc.2016.08.029
Levine, R. L., Koppikar, P., Marubayashi, S., Bhagwat, N., Taldone, T., Park, C. Y., et al. (2012). Combination therapy using JAK2 and HSP90 inhibitors increased efficacy in myelofibrosis in vivo. Blood 120, 805. doi:10.1182/blood.v120.21.805.805
Li, H. K., Matsumoto, Y., Furusawa, Y., and Kamada, T. (2016). PU-H71, a novel Hsp90 inhibitor, as a potential cancer-specific sensitizer to carbon-ion beam therapy. J. Radiat. Res. 57, 572–575. doi:10.1093/jrr/rrw054
Li, H. K., Morokoshi, Y., Daino, K., Furukawa, T., Kamada, T., Saga, T., et al. (2015). Transcriptomic signatures of auger electron radioimmunotherapy using nuclear targeting 111In-Trastuzumab for potential combination therapies. Cancer Biotherapy Radiopharm. 30, 349–358. doi:10.1089/cbr.2015.1882
Li, S., and Adams, P. D. (2023). Targeting the epichaperome to combat AML. Blood 142, 1031–1032. doi:10.1182/blood.2023021386
Li, T., Jiang, H.-L., Tong, Y.-G., and Lu, J.-J. (2018). Targeting the Hsp90-Cdc37-client protein interaction to disrupt Hsp90 chaperone machinery. J. Hematol. and Oncol. 11, 59. doi:10.1186/s13045-018-0602-8
Li, Z.-N., and Luo, Y. (2023). HSP90 inhibitors and cancer: prospects for use in targeted therapies (Review). Oncol. Rep. 49, 6. doi:10.3892/or.2022.8443
Lisi, L., Mcguire, S., Sharp, A., Chiosis, G., Navarra, P., Feinstein, D. L., et al. (2013). The novel HSP90 inhibitor, PU-H71, suppresses glial cell activation but weakly affects clinical signs of EAE. J. Neuroimmunol. 255, 1–7. doi:10.1016/j.jneuroim.2012.10.008
Liu, H., Lu, Z., Shi, X., Liu, L., Zhang, P., Golemis, E. A., et al. (2021a). HSP90 inhibition downregulates DNA replication and repair genes via E2F1 repression. J. Biol. Chem. 297, 100996. doi:10.1016/j.jbc.2021.100996
Liu, P., Xiao, J., Wang, Y., Song, X., Huang, L., Ren, Z., et al. (2021b). Posttranslational modification and beyond: interplay between histone deacetylase 6 and heat-shock protein 90. Mol. Med. 27, 110. doi:10.1186/s10020-021-00375-3
Liu, Y., Feng, J., Yuan, K., Wu, Z., Hu, L., Lu, Y., et al. (2022). The oncoprotein BCL6 enables solid tumor cells to evade genotoxic stress. eLife 11, e69255. doi:10.7554/eLife.69255
Liu, Y., Wang, X., Wang, J., Jin, Q., Cai, W., Pan, C., et al. (2023). Class Ⅱb histone deacetylase participates in postoperative cognitive dysfunction in elderly mice via HSP90/GR signaling pathway.
Lokeshwar, V. B. (2012). Wee1-Hsp90 inhibitor combination treatment. Cell Cycle 11, 3722. doi:10.4161/cc.22119
Lopes, E., Joungnam, K., Rondina, A., Moulick, K., and Chiosis, G. (2007). Targeting basal-like breast cancer via purine-scaffold Hsp90 inhibitors. Cancer Res. 67, 3964.
Lubina-Dąbrowska, N., Stepień, A., Sulkowski, G., Dąbrowska-Bouta, B., Langfort, J., and Chalimoniuk, M. (2017). Effects of IFN-β1a and IFN-β1b treatment on the expression of cytokines, inducible NOS (NOS type II), and myelin proteins in animal model of multiple sclerosis. Archivum Immunol. Ther. Exp. 65, 325–338. doi:10.1007/s00005-017-0458-6
Luo, W., Sun, W., Taldone, T., Rodina, A., and Chiosis, G. (2010). Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 5, 24. doi:10.1186/1750-1326-5-24
Luque Paz, D., Kralovics, R., and Skoda, R. C. (2023). Genetic basis and molecular profiling in myeloproliferative neoplasms. Blood 141, 1909–1921. doi:10.1182/blood.2022017578
Magwenyane, A. M., Ugbaja, S. C., Amoako, D. G., Somboro, A. M., Khan, R. B., and Kumalo, H. M. (2022). Heat shock protein 90 (HSP90) inhibitors as anticancer medicines: a review on the computer-aided drug discovery approaches over the past five years. Comput. Math. Methods Med. 2022, 2147763. doi:10.1155/2022/2147763
Mahajan, S., Grkovski, M., Staton, K. D., Ravassa, S., Domfe, K., Strauss, H. W., et al. (2024). Epichaperome-targeted myocardial imaging by 124I-PU-H71 PET. Clin. Transl. Imaging. doi:10.1007/s40336-024-00658-9
Mario, C. (2007). Somatic mutations of JAK2 exon 12 as a molecular basis of erythrocytosis. Haematologica 92, 1585–1589. doi:10.3324/haematol.11506
Martinelli, E., Morgillo, F., Troiani, T., and Ciardiello, F. (2017). Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat. Rev. 53, 61–69. doi:10.1016/j.ctrv.2016.12.001
Martinet, J., Sugita, M., Alhomoud, M., Abad, C., Chiosis, G., Roboz, G. J., et al. (2022). Impact of epichaperome inhibitor PU-H71 on anti-tumor T cell responses and its implications for immune and cellular therapy. Blood 140, 1171–1172. doi:10.1182/blood-2022-163563
Marubayashi, S., Koppikar, P., Taldone, T., Abdel-Wahab, O., West, N., Bhagwat, N., et al. (2010). HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Investigation 120, 3578–3593. doi:10.1172/JCI42442
Marzec, M., Eletto, D., and Argon, Y. (2012). GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1823, 774–787. doi:10.1016/j.bbamcr.2011.10.013
Masser, A. E., Ciccarelli, M., and Andréasson, C. (2020). Hsf1 on a leash – controlling the heat shock response by chaperone titration. Exp. Cell Res. 396, 112246. doi:10.1016/j.yexcr.2020.112246
Matsumoto, K., Umitsu, M., De Silva, D. M., Roy, A., and Bottaro, D. P. (2017). Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 108, 296–307. doi:10.1111/cas.13156
Mbofung, R. M., Mckenzie, J. A., Malu, S., Zhang, M., Peng, W., Liu, C., et al. (2017). HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes. Nat. Commun. 8, 451. doi:10.1038/s41467-017-00449-z
Mclachlan, T., Matthews, W. C., Jackson, E. R., Staudt, D. E., Douglas, A. M., Findlay, I. J., et al. (2022). B-Cell lymphoma 6 (BCL6): from master regulator of humoral immunity to oncogenic driver in pediatric cancers. Mol. Cancer Res. 20, 1711–1723. doi:10.1158/1541-7786.MCR-22-0567
Medina, M. A., Oza, G., Sharma, A., Arriaga, L. G., Hernández Hernández, J. M., Rotello, V. M., et al. (2020). Triple-negative breast cancer: a review of conventional and advanced therapeutic strategies. Int. J. Environ. Res. Public Health 17, 2078. doi:10.3390/ijerph17062078
Mellatyar, H., Talaei, S., Pilehvar-Soltanahmadi, Y., Barzegar, A., Akbarzadeh, A., Shahabi, A., et al. (2018). Targeted cancer therapy through 17-DMAG as an Hsp90 inhibitor: overview and current state of the art. Biomed. Pharmacother. 102, 608–617. doi:10.1016/j.biopha.2018.03.102
Merugu, S., Sharma, S., Kaner, J., Digwal, C., Sugita, M., Joshi, S., et al. (2020). “Chapter Thirteen - chemical probes and methods for single-cell detection and quantification of epichaperomes in hematologic malignancies,” in Methods in enzymology. Editor D. M. Chenoweth (Academic Press), 289–311.
Messaoudi, S., Peyrat, J. F., Brion, J. D., and Alami, M. (2008). Recent advances in Hsp90 inhibitors as antitumor agents. Anti-Cancer Agents Med. Chem. 8, 761–782. doi:10.2174/187152008785914824
Mintz, M. A., and Cyster, J. G. (2020). T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol. Rev. 296, 48–61. doi:10.1111/imr.12860
Miyata, Y. (2005). Hsp90 inhibitor geldanamycin and its derivatives as novel cancer chemotherapeutic agents. Curr. Pharm. Des. 11, 1131–1138. doi:10.2174/1381612053507585
Miyata, Y., Nakamoto, H., and Neckers, L. (2013). The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 19, 347–365. doi:10.2174/138161213804143725
Mladenov, E., Magin, S., Soni, A., and Iliakis, G. (2013). DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol. 3, 113. doi:10.3389/fonc.2013.00113
Modi, S., Stopeck, A., Linden, H., Solit, D., Chandarlapaty, S., Rosen, N., et al. (2011). HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin. Cancer Res. 17, 5132–5139. doi:10.1158/1078-0432.CCR-11-0072
Mohammed, O. A., Abdel-Reheim, M. A., Alamri, M. M. S., Alfaifi, J., Adam, M. I. E., Saleh, L. A., et al. (2023a). STA9090 as a potential therapeutic agent for liver fibrosis by modulating the HSP90/tβrii/proteasome interplay: novel insights from in vitro and in vivo investigations. Pharmaceuticals 16, 1080. doi:10.3390/ph16081080
Mohammed, O. A., Abdel-Reheim, M. A., Saleh, L. A., Alamri, M. M. S., Alfaifi, J., Adam, M. I. E., et al. (2023b). Alvespimycin exhibits potential anti-TGF-β signaling in the setting of a proteasome activator in rats with bleomycin-induced pulmonary fibrosis: a promising novel approach. Pharmaceuticals 16, 1123. doi:10.3390/ph16081123
Mollapour, M., and Neckers, L. (2012). Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1823, 648–655. doi:10.1016/j.bbamcr.2011.07.018
Mollapour, M., Tsutsumi, S., and Neckers, L. (2010). Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle 9, 2310–2316. doi:10.4161/cc.9.12.12054
Mookerjee, S. A., Gerencser, A. A., Nicholls, D. G., and Brand, M. D. (2017). Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 292, 7189–7207. doi:10.1074/jbc.M116.774471
Moriwaki, Y., Kim, Y.-J., Ido, Y., Misawa, H., Kawashima, K., Endo, S., et al. (2008). L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci. Res. 61, 43–48. doi:10.1016/j.neures.2008.01.006
Moulick, K., Ahn, J., Zong, H., Rodina, A., Cerchietti, L., Dagama, E., et al. (2011). Abstract 1263: affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 7, 818–826. doi:10.1038/nchembio.670
Mullally, A. (2017). MPN: next questions: potential new targets for MPN beside JAK2. Myeloma Leukemia 17, S99–S101. doi:10.1016/j.clml.2017.08.048
Musha, A., Yoshida, Y., Takahashi, T., Ando, K., Funayama, T., Kobayashi, Y., et al. (2012). Synergistic effect of heat shock protein 90 inhibitor, 17-allylamino-17-demethoxygeldanamycin and X-rays, but not carbon-ion beams, on lethality in human oral squamous cell carcinoma cells. J. Radiat. Res. 53, 545–550. doi:10.1093/jrr/rrs012
Nasr, M., Kira, A. Y., Saber, S., Essa, E. A., and El-Gizawy, S. A. (2023). Telmisartan-loaded lactosylated chitosan nanoparticles as a liver specific delivery system: synthesis, optimization and targeting efficiency. AAPS PharmSciTech 24, 144. doi:10.1208/s12249-023-02605-9
Navarro-Zaragoza, J., Cuenca-Bermejo, L., Almela, P., Laorden, M.-L., and Herrero, M.-T. (2021). Could small heat shock protein HSP27 Be a first-line target for preventing protein aggregation in Parkinson’s disease? Int. J. Mol. Sci. 22, 3038. doi:10.3390/ijms22063038
Nickoloff, J. A., Boss, M.-K., Allen, C. P., and Larue, S. M. (2017). Translational research in radiation-induced DNA damage signaling and repair. Transl. Cancer Res. 6, S875–S891. doi:10.21037/tcr.2017.06.02
Niu, M., Song, S., Su, Z., Wei, L., Li, L., Pu, W., et al. (2021). Inhibition of heat shock protein (HSP) 90 reverses signal transducer and activator of transcription (STAT) 3-mediated muscle wasting in cancer cachexia mice. Br. J. Pharmacol. 178, 4485–4500. doi:10.1111/bph.15625
Nolan, A. A., Aboud, N. K., Kolch, W., and Matallanas, D. (2021). Hidden targets in RAF signalling pathways to block oncogenic RAS signalling. Genes 12, 553. doi:10.3390/genes12040553
Nouri-Vaskeh, M., Alizadeh, L., Hajiasgharzadeh, K., Mokhtarzadeh, A., Halimi, M., and Baradaran, B. (2020). The role of HSP90 molecular chaperones in hepatocellular carcinoma. J. Cell. Physiology 235, 9110–9120. doi:10.1002/jcp.29776
Orea-Soufi, A., Paik, J., Bragança, J., Donlon, T. A., Willcox, B. J., and Link, W. (2022). FOXO transcription factors as therapeutic targets in human diseases. Trends Pharmacol. Sci. 43, 1070–1084. doi:10.1016/j.tips.2022.09.010
Ozgur, A., and Tutar, Y. (2014). Heat shock protein 90 inhibitors in Oncology. Curr. Proteomics 11, 2–16. doi:10.2174/1570164611666140415224635
Pandey, K., An, H. J., Kim, S. K., Lee, S. A., Kim, S., Lim, S. M., et al. (2019). Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: a review. Int. J. Cancer 145, 1179–1188. doi:10.1002/ijc.32020
Pascale, R. M., Simile, M. M., Calvisi, D. F., Frau, M., Muroni, M. R., Seddaiu, M. A., et al. (2005). Role of HSP90, CDC37, and CRM1 as modulators of P16INK4A activity in rat liver carcinogenesis and human liver cancer. Hepatology 42, 1310–1319. doi:10.1002/hep.20962
Pastena, P., Perera, H., Martinino, A., Kartsonis, W., and Giovinazzo, F. (2024). Unraveling biomarker signatures in triple-negative breast cancer: a systematic review for targeted approaches. Int. J. Mol. Sci. 25, 2559. doi:10.3390/ijms25052559
Pearl, L. H., Prodromou, C., and Workman, P. (2008). The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem. J. 410, 439–453. doi:10.1042/BJ20071640
Pemmaraju, N., Gundabolu, K., Pettit, K., Talpaz, M., Podoltsev, N. A., Schiller, G. J., et al. (2019). Phase 1b study of the epichaperome inhibitor PU-H71 administered orally with ruxolitinib continuation for the treatment of patients with myelofibrosis. Blood 134, 4178. doi:10.1182/blood-2019-130310
Peng, C., Zhao, F., Li, H., Li, L., Yang, Y., and Liu, F. (2022). HSP90 mediates the connection of multiple programmed cell death in diseases. Cell Death and Dis. 13, 929. doi:10.1038/s41419-022-05373-9
Pennisi, R., Ascenzi, P., and Di Masi, A. (2015). Hsp90: a new player in DNA repair? Biomolecules 5, 2589–2618. doi:10.3390/biom5042589
Pietra, D., Li, S., Brisci, A., Passamonti, F., Rumi, E., Theocharides, A., et al. (2008). Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood 111, 1686–1689. doi:10.1182/blood-2007-07-101576
Pimienta, G., Herbert, K. M., and Regan, L. (2011). A compound that inhibits the HOP–Hsp90 complex formation and has unique killing effects in breast cancer cell lines. Mol. Pharm. 8, 2252–2261. doi:10.1021/mp200346y
Popova, N. V., and Jücker, M. (2021). The role of mTOR signaling as a therapeutic target in cancer. Int. J. Mol. Sci. 22, 1743. doi:10.3390/ijms22041743
Qian, J., Hong, S., Wang, S., Zhang, L., Sun, L., Wang, M., et al. (2009). Myeloma cell line–derived, pooled heat shock proteins as a universal vaccine for immunotherapy of multiple myeloma. Blood, J. Am. Soc. Hematol. 114, 3880–3889. doi:10.1182/blood-2009-06-227355
Rah, B., Nayak, D., Rasool, R., Chakraborty, S., Katoch, A., Amin, H., et al. (2016). Reprogramming of molecular switching events in UPR driven ER stress: scope for development of anticancer therapeutics. Curr. Mol. Med. 16, 690–701. doi:10.2174/1566524016666160829152658
Rampal, R., Ahn, J., Abdel-Wahab, O., Nahas, M., Wang, K., Lipson, D., et al. (2014). Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. U. S. A. 111, E5401–E5410. doi:10.1073/pnas.1407792111
Randazzo, M., Terness, P., Opelz, G., and Kleist, C. (2012). Active-specific immunotherapy of human cancers with the heat shock protein Gp96—revisited. Int. J. Cancer 130, 2219–2231. doi:10.1002/ijc.27332
Rao, T. N., Hansen, N., Hilfiker, J., Rai, S., Majewska, J.-M., Leković, D., et al. (2019). JAK2-mutant hematopoietic cells display metabolic alterations that can be targeted to treat myeloproliferative neoplasms. Blood 134, 1832–1846. doi:10.1182/blood.2019000162
Rastogi, S., Joshi, A., Sato, N., Lee, S., Lee, M.-J., Trepel, J. B., et al. (2024). An update on the status of HSP90 inhibitors in cancer clinical trials. Cell Stress Chaperones 29, 519–539. doi:10.1016/j.cstres.2024.05.005
Ravandi, F., Talpaz, M., and Estrov, Z. (2003). Modulation of cellular signaling pathways: prospects for targeted therapy in hematological malignancies. Clin. Cancer Res. 9, 535–550.
Reece, C., Kumar, R., Nienow, D., and Nehra, A. (2007). Extending the rationale of combination therapy to unresponsive erectile dysfunction. Rev. Urology 9, 197–206.
Ren, J., Chu, Y., Ma, H., Zhang, Y., Zhang, X., Zhao, D., et al. (2014). Epigenetic interventions increase the radiation sensitivity of cancer cells. Curr. Pharm. Des. 20, 1857–1865. doi:10.2174/13816128113199990529
Reynolds, T. S., and Blagg, B. S. J. (2024). Extracellular heat shock protein 90 alpha (eHsp90α)'s role in cancer progression and the development of therapeutic strategies. Eur. J. Med. Chem. 277, 116736. doi:10.1016/j.ejmech.2024.116736
Rizza, S., and Filomeni, G. (2020). Exploiting S-nitrosylation for cancer therapy: facts and perspectives. Biochem. J. 477, 3649–3672. doi:10.1042/BCJ20200064
Roboz, G. J., Sugita, M., Mosquera, J. M., Wilkes, D. C., Nataraj, S., Jimenez-Flores, R. A., et al. (2018). Targeting the epichaperome as an effective precision medicine approach in a novel PML-SYK fusion acute myeloid leukemia. Blood 132, 1435. doi:10.1182/blood-2018-99-118764
Rodina, A., Vilenchik, M., Moulick, K., Aguirre, J., Kim, J., Chiang, A., et al. (2007). Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat. Chem. Biol. 3, 498–507. doi:10.1038/nchembio.2007.10
Rodina, A., Wang, T., Yan, P., Gomes, E. D., Dunphy, M. P. S., Pillarsetty, N., et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401. doi:10.1038/nature19807
Rodina, A., Xu, C., Digwal, C. S., Joshi, S., Patel, Y., Santhaseela, A. R., et al. (2023). Systems-level analyses of protein-protein interaction network dysfunctions via epichaperomics identify cancer-specific mechanisms of stress adaptation. Nat. Commun. 14, 3742. doi:10.1038/s41467-023-39241-7
Roe, S. M., Prodromou, C., O'brien, R., Ladbury, J. E., Piper, P. W., and Pearl, L. H. (1999). Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 42, 260–266. doi:10.1021/jm980403y
Rong, B., and Yang, S. (2018). Molecular mechanism and targeted therapy of Hsp90 involved in lung cancer: new discoveries and developments (Review). Int. J. Oncol. 52, 321–336. doi:10.3892/ijo.2017.4214
Roskoski, R. (2014). The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 79, 34–74. doi:10.1016/j.phrs.2013.11.002
Saber, S., Abd El-Kader, E. M., Sharaf, H., El-Shamy, R., El-Saeed, B., Mostafa, A., et al. (2020). Celastrol augments sensitivity of NLRP3 to CP-456773 by modulating HSP-90 and inducing autophagy in dextran sodium sulphate-induced colitis in rats. Toxicol. Appl. Pharmacol. 400, 115075. doi:10.1016/j.taap.2020.115075
Saber, S., Hasan, A. M., Mohammed, O. A., Saleh, L. A., Hashish, A. A., Alamri, M. M. S., et al. (2023). Ganetespib (STA-9090) augments sorafenib efficacy via necroptosis induction in hepatocellular carcinoma: implications from preclinical data for a novel therapeutic approach. Biomed. and Pharmacother. 164, 114918. doi:10.1016/j.biopha.2023.114918
Saber, S., Mahmoud, A., Helal, N., El-Ahwany, E., and Abdelghany, R. (2018). Liver protective effects of renin-angiotensin system inhibition have No survival benefits in hepatocellular carcinoma induced by repetitive administration of diethylnitrosamine in mice. Open Access Maced. J. Med. Sci. 6, 955–960. doi:10.3889/oamjms.2018.167
Sager, R. A., Backe, S. J., Ahanin, E., Smith, G., Nsouli, I., Woodford, M. R., et al. (2022). Therapeutic potential of CDK4/6 inhibitors in renal cell carcinoma. Nat. Rev. Urol. 19, 305–320. doi:10.1038/s41585-022-00571-8
Salama, M. F., Liu, M., Clarke, C. J., Espaillat, M. P., Haley, J. D., Jin, T., et al. (2019). PKCα is required for Akt-mTORC1 activation in non-small cell lung carcinoma (NSCLC) with EGFR mutation. Oncogene 38, 7311–7328. doi:10.1038/s41388-019-0950-z
Schirrmacher, V. (2019). From chemotherapy to biological therapy: a review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 54, 407–419. doi:10.3892/ijo.2018.4661
Schmauder, L., Sima, S., Hadj, A. B., Cesar, R., and Richter, K. (2022). Binding of the HSF-1 DNA-binding domain to multimeric C. elegans consensus HSEs is guided by cooperative interactions. Sci. Rep. 12, 8984. doi:10.1038/s41598-022-12736-x
Schopf, F. H., Biebl, M. M., and Buchner, J. (2017). The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360. doi:10.1038/nrm.2017.20
Schroeder, H. T., De Lemos Muller, C. H., Heck, T. G., Krause, M., and Homem De Bittencourt, P. I. (2024). Heat shock response during the resolution of inflammation and its progressive suppression in chronic-degenerative inflammatory diseases. Cell Stress Chaperones 29, 116–142. doi:10.1016/j.cstres.2024.01.002
Scott, L. M. (2018). “Inhibitors of the JAK/STAT pathway, with a focus on ruxolitinib and similar agents,” in Resistance of targeted therapies excluding antibodies for lymphomas. Editor A. J. M. Ferreri (Cham: Springer International Publishing), 107–134.
Seasons, G. M., Pellow, C., Kuipers, H. F., and Pike, G. B. (2024). Ultrasound and neuroinflammation: immune modulation via the heat shock response. Theranostics 14, 3150–3177. doi:10.7150/thno.96270
Segawa, T., Fujii, Y., Tanaka, A., Bando, S.-I., Okayasu, R., Ohnishi, K., et al. (2014). Radiosensitization of human lung cancer cells by the novel purine-scaffold Hsp90 inhibitor, PU-H71. Int. J. Mol. Med. 33, 559–564. doi:10.3892/ijmm.2013.1594
Seipel, K., Kohler, S., Bacher, U., and Pabst, T. (2023). HSP90 inhibitor PU-H71 in combination with BH3-mimetics in the treatment of acute myeloid leukemia. Curr. Issues Mol. Biol. 45, 7011–7026. doi:10.3390/cimb45090443
Serwetnyk, M. A., and Blagg, B. S. J. (2021). The disruption of protein−protein interactions with co-chaperones and client substrates as a strategy towards Hsp90 inhibition. Acta Pharm. Sin. B 11, 1446–1468. doi:10.1016/j.apsb.2020.11.015
Shaaban, A. A., Abdelhamid, A. M., Shaker, M. E., Cavalu, S., Maghiar, A. M., Alsayegh, A. A., et al. (2022). Combining the HSP90 inhibitor TAS-116 with metformin effectively degrades the NLRP3 and attenuates inflammasome activation in rats: a new management paradigm for ulcerative colitis. Biomed. and Pharmacother. 153, 113247. doi:10.1016/j.biopha.2022.113247
Shinozaki, F., Minami, M., Chiba, T., Suzuki, M., Yoshimatsu, K., Ichikawa, Y., et al. (2006). Depletion of hsp90beta induces multiple defects in B cell receptor signaling. J. Biol. Chem. 281, 16361–16369. doi:10.1074/jbc.M600891200
Shirai, Y., Chow, C. C. T., Kambe, G., Suwa, T., Kobayashi, M., Takahashi, I., et al. (2021). An overview of the recent development of anticancer agents targeting the HIF-1 transcription factor. Cancers 13, 2813. doi:10.3390/cancers13112813
Sidera, K., and Patsavoudi, E. (2014). HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat. Anti-Cancer Drug Discov. 9, 1–20. doi:10.2174/15748928113089990031
Siragusa, M., and Fleming, I. (2016). The eNOS signalosome and its link to endothelial dysfunction. Pflügers Archiv - Eur. J. Physiology 468, 1125–1137. doi:10.1007/s00424-016-1839-0
Smyth, T., Van Looy, T., Curry, J. E., Rodriguez-Lopez, A. M., Wozniak, A., Zhu, M., et al. (2012). The HSP90 inhibitor, AT13387, is effective against imatinib-sensitive and -resistant gastrointestinal stromal tumor models. Mol. Cancer Ther. 11, 1799–1808. doi:10.1158/1535-7163.MCT-11-1046
Sokol, O., and Durante, M. (2023). Carbon ions for hypoxic tumors: are we making the most of them? Cancers 15, 4494. doi:10.3390/cancers15184494
Solárová, Z., Mojžiš, J., and Solár, P. (2015). Hsp90 inhibitor as a sensitizer of cancer cells to different therapies (Review). Int. J. Oncol. 46, 907–926. doi:10.3892/ijo.2014.2791
Sosa, R. A., and Forsthuber, T. G. (2011). The critical role of antigen-presentation-induced cytokine crosstalk in the central nervous system in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Interferon and Cytokine Res. 31, 753–768. doi:10.1089/jir.2011.0052
Speranza, G., Anderson, L., Chen, A. P., Do, K., Eugeni, M., Weil, M., et al. (2018). First-in-human study of the epichaperome inhibitor PU-H71: clinical results and metabolic profile. Investig. New Drugs 36, 230–239. doi:10.1007/s10637-017-0495-3
Staerk, J., and Constantinescu, S. N. (2012). The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. Jakstat 1, 184–190. doi:10.4161/jkst.22071
Sumi, M. P., and Ghosh, A. (2022). Hsp90 in human diseases: molecular mechanisms to therapeutic approaches. Cells 11, 976. doi:10.3390/cells11060976
Sun, P., Wang, Y., Gao, T., Li, K., Zheng, D., Liu, A., et al. (2021). Hsp90 modulates human sperm capacitation via the Erk1/2 and p38 MAPK signaling pathways. Reproductive Biol. Endocrinol. 19, 39. doi:10.1186/s12958-021-00723-2
Szwed, A., Kim, E., and Jacinto, E. (2021). Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 101, 1371–1426. doi:10.1152/physrev.00026.2020
Tada, M., Yokosuka, O., Fukai, K., Chiba, T., Imazeki, F., Tokuhisa, T., et al. (2005). Hypermethylation of NAD(P)H: quinone oxidoreductase 1 (NQO1) gene in human hepatocellular carcinoma. J. Hepatol. 42, 511–519. doi:10.1016/j.jhep.2004.11.024
Tahimic, C. G. T., Wang, Y., and Bikle, D. D. (2013). Anabolic effects of IGF-1 signaling on the skeleton. Front. Endocrinol. 4, 6. doi:10.3389/fendo.2013.00006
Terrell, E. M., and Morrison, D. K. (2019). Ras-mediated activation of the raf family kinases. Cold Spring Harb. Perspect. Med. 9, a033746. doi:10.1101/cshperspect.a033746
Terwisscha Van Scheltinga, A. G. T., Berghuis, P., Nienhuis, H. H., Timmer-Bosscha, H., Pot, L., Gaykema, S. B. M., et al. (2014). Visualising dual downregulation of insulin-like growth factor receptor-1 and vascular endothelial growth factor-A by heat shock protein 90 inhibition effect in triple negative breast cancer. Eur. J. Cancer 50, 2508–2516. doi:10.1016/j.ejca.2014.06.008
Tramentozzi, E., and Finotti, P. (2019). Effects of purine-scaffold inhibitors on HUVECs: involvement of the purinergic pathway and interference with ATP. Implications for preventing the adverse effects of extracellular Grp94. Biochem. Biophysics Rep. 19, 100661. doi:10.1016/j.bbrep.2019.100661
Trendowski, M. (2015). PU-H71: an improvement on nature's solutions to oncogenic Hsp90 addiction. Pharmacol. Res. 99, 202–216. doi:10.1016/j.phrs.2015.06.007
Trepel, J., Mollapour, M., Giaccone, G., and Neckers, L. (2010). Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549. doi:10.1038/nrc2887
Ueno, T., Tsukuda, K., Toyooka, S., Ando, M., Takaoka, M., Soh, J., et al. (2012). Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer 76, 26–31. doi:10.1016/j.lungcan.2011.09.011
Ullah, R., Yin, Q., Snell, A. H., and Wan, L. (2022). RAF-MEK-ERK pathway in cancer evolution and treatment. Seminars Cancer Biol. 85, 123–154. doi:10.1016/j.semcancer.2021.05.010
Usmani, S. Z., Bona, R. D., Chiosis, G., and Li, Z. (2010). The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J. Hematol. and Oncol. 3, 40. doi:10.1186/1756-8722-3-40
Vagia, E., Mahalingam, D., and Cristofanilli, M. (2020). The landscape of targeted therapies in TNBC. Cancers 12, 916. doi:10.3390/cancers12040916
Wang, L., Xie, C., Greggio, E., Parisiadou, L., Shim, H., Sun, L., et al. (2008). The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J. Neurosci. 28, 3384–3391. doi:10.1523/JNEUROSCI.0185-08.2008
Wang, M., Shen, A., Zhang, C., Song, Z., Ai, J., Liu, H., et al. (2016). Development of heat shock protein (Hsp90) inhibitors to combat resistance to tyrosine kinase inhibitors through hsp90–kinase interactions. J. Med. Chem. 59, 5563–5586. doi:10.1021/acs.jmedchem.5b01106
Waza, M., Adachi, H., Katsuno, M., Minamiyama, M., Tanaka, F., Doyu, M., et al. (2006). Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J. Mol. Med. 84, 635–646. doi:10.1007/s00109-006-0066-0
Weigert, O., Lane, A. A., Bird, L., Kopp, N., Chapuy, B., Van Bodegom, D., et al. (2012). Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J. Exp. Med. 209, 259–273. doi:10.1084/jem.20111694
Wernig, G., Mercher, T., Okabe, R., Levine, R. L., Lee, B. H., and Gilliland, D. G. (2006). Expression of Jak2V617F causes a polycythemia vera–like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 107, 4274–4281. doi:10.1182/blood-2005-12-4824
Westerlund, M., Hoffer, B., and Olson, L. (2010). Parkinson's disease: exit toxins, enter genetics. Prog. Neurobiol. 90, 146–156. doi:10.1016/j.pneurobio.2009.11.001
Woodford, M. R., Dunn, D., Miller, J. B., Jamal, S., Neckers, L., and Mollapour, M. (2016). “Chapter two - impact of posttranslational modifications on the anticancer activity of Hsp90 inhibitors,” in Advances in cancer research. Editors J. Isaacs,, and L. Whitesell (Academic Press), 31–50.
Workman, P., Burrows, F., Neckers, L., and Rosen, N. (2007). Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N. Y. Acad. Sci. 1113, 202–216. doi:10.1196/annals.1391.012
Wöss, K., Simonović, N., Strobl, B., Macho-Maschler, S., and Müller, M. (2019). TYK2: an upstream kinase of STATs in cancer. Cancers 11, 1728. doi:10.3390/cancers11111728
Wu, C.-W., and Storey, K. B. (2021). mTOR signaling in metabolic stress adaptation. Biomolecules 11, 681. doi:10.3390/biom11050681
Wu, J., Wang, D., Nie, J., Zhang, D., Sun, L., Kan, S., et al. (2023). Design and synthesis of an Hsp90 and HDAC dual inhibitor as antitumor agent. Lett. Drug Des. and Discov. 20, 619–627. doi:10.2174/1570180819666220530145951
Xie, J., Gong, L., Zhu, S., Yong, Y., Gu, Z., and Zhao, Y. (2019). Emerging strategies of nanomaterial-mediated tumor radiosensitization. Adv. Mater. 31, 1802244. doi:10.1002/adma.201802244
Xu, W., and Neckers, L. (2007). Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin. Cancer Res. 13, 1625–1629. doi:10.1158/1078-0432.CCR-06-2966
Xue, N., Jin, J., Liu, D., Yan, R., Zhang, S., Yu, X., et al. (2014). Antiproliferative effect of HSP90 inhibitor Y306zh against pancreatic cancer is mediated by interruption of AKT and MAPK signaling pathways. Curr. Cancer Drug Targets 14, 671–683. doi:10.2174/1568009614666140908101523
Xu-Monette, Z. Y., Medeiros, L. J., Li, Y., Orlowski, R. Z., Andreeff, M., Bueso-Ramos, C. E., et al. (2012). Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood 119, 3668–3683. doi:10.1182/blood-2011-11-366062
Yamaki, H., Nakajima, M., Shimotohno, K. W., and Tanaka, N. (2011). Molecular basis for the actions of Hsp90 inhibitors and cancer therapy. J. Antibiotics 64, 635–644. doi:10.1038/ja.2011.60
Yan, P., Patel, H. J., Sharma, S., Corben, A., Wang, T., Panchal, P., et al. (2020). Molecular stressors engender protein connectivity dysfunction through aberrant N-glycosylation of a chaperone. Cell Rep. 31, 107840. doi:10.1016/j.celrep.2020.107840
Yanagi, T., Mizoe, J.-E., Hasegawa, A., Takagi, R., Bessho, H., Onda, T., et al. (2009). Mucosal malignant melanoma of the head and neck treated by carbon ion radiotherapy. Int. J. Radiat. Oncology* Biology* Phys. 74, 15–20. doi:10.1016/j.ijrobp.2008.07.056
Yang, H., and Green, M. R. (2019). Epigenetic programing of B-cell lymphoma by BCL6 and its genetic deregulation. Front. Cell Dev. Biol. 7, 272. doi:10.3389/fcell.2019.00272
Youssef, M. E., Cavalu, S., Hasan, A. M., Yahya, G., Abd-Eldayem, M. A., and Saber, S. (2023). Role of ganetespib, an HSP90 inhibitor, in cancer therapy: from molecular mechanisms to clinical practice. Int. J. Mol. Sci. 24, 5014. doi:10.3390/ijms24055014
Youssef, M. E., El-Mas, M. M., Abdelrazek, H. M., and El-Azab, M. F. (2021a). α7-nAChRs-mediated therapeutic angiogenesis accounts for the advantageous effect of low nicotine doses against myocardial infarction in rats. Eur. J. Pharmacol. 898, 173996. doi:10.1016/j.ejphar.2021.173996
Youssef, M. E., Moustafa, Y., and Abdelrazek, H. (2021b). Molecular mechanisms of α7-nAchR-mediated anti-inflammatory effects. Indian J. Physiology Pharmacol. 64, 158–173. doi:10.25259/ijpp_129_2020
Zhang, W., Li, Q., and Yin, R. (2024). Targeting WEE1 kinase in gynecological malignancies. Drug Des. Devel Ther. 18, 2449–2460. doi:10.2147/DDDT.S462056
Zhang, X., Li, S., Li, Z., Cheng, L., Liu, Z., and Wang, C. (2022). The therapeutic potential of targeting hsp90-cdc37 interactions in several diseases. Curr. Drug Targets 23, 1023–1038. doi:10.2174/1389450123666220408101544
Zhang, Y., Xia, M., Jin, K., Wang, S., Wei, H., Fan, C., et al. (2018). Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 17, 45. doi:10.1186/s12943-018-0796-y
Zhao, R., and Houry, W. A. (2005). Hsp90: a chaperone for protein folding and gene regulation. Biochem. Cell Biol. 83, 703–710. doi:10.1139/o05-158
Zhao, R., Xing, S., Li, Z., Fu, X., Li, Q., Krantz, S. B., et al. (2005). Identification of an acquired JAK2 mutation in polycythemia vera. J. Biol. Chem. 280, 22788–22792. doi:10.1074/jbc.C500138200
Glossary
17-AAG 17-Allylamino-17-demethoxygeldanamycin
17-DMAG 17-Dimethylaminoethylamino-17-demethoxygeldanamycin
Akt Protein Kinase B
ATF4 Activating Transcription Factor 4
ATP Adenosine Triphosphate
ATPase Adenosine Triphosphatase
Bax Bcl-2-Associated X Protein
Bcl-xL B-Cell Lymphoma-extra Large
Bcl6 B-Cell Lymphoma 6
Bcl-2 B-Cell Lymphoma 2
BCoR BCL6 Corepressor
C-ions Carbon Ions
CIRT Carbon-Ion Radiotherapy
CDK Cyclin-Dependent Kinase
CHOP C/EBP Homologous Protein
CK2 casein kinase II
DNA-PKcs DNA-Dependent Protein Kinase Catalytic Subunit
DSB DNA Double-Strand Break
DLBCL Diffuse Large B-Cell Lymphoma
ER Endoplasmic Reticulum
ERK Extracellular Signal-Regulated Kinase
FoxO Forkhead Box O
GRP78 Glucose-Regulated Protein 78
gp96 Glucose-Regulated Protein 94
HDAC histone deacetylase
HSP27 Heat Shock Protein 27
HSP40 Heat Shock Protein 40
HSP60 Heat Shock Protein 60
HSP70 Heat Shock Protein 70
HSP72 Inducible Heat Shock Protein 70
HSP90 Heat Shock Protein 90
HSP90α Heat Shock Protein 90 Alpha
HSP90β Heat Shock Protein 90 Beta
HSP90B1 Heat Shock Protein 90 Beta Family Member 1
HSC70 Constitutive Heat Shock Protein 70
HSF-1 Heat Shock transcription Factor 1
HSR Heat Shock Response
IC50 Half Maximal Inhibitory Concentration
IL-6 Interleukin 6
INF Interferon
IGF1R Insulin-Like Growth Factor 1 Receptor
JAK Janus Kinase
LET Linear Energy Transfer
MAPK Mitogen-Activated Protein Kinase
mTORC Mammalian Target of Rapamycin Complex
NF-κB Nuclear Factor kappa-light-chain-enhancer of activated B cells
NOS2 Nitric Oxide Synthase 2
PKC Protein Kinase C
PDK1 3-Phosphoinositide-Dependent Protein Kinase-1
PI3K Phosphoinositide 3-Kinase
PTMs post-translational modifications
RAD51 RAD51 Recombinase
Raf v-Raf-1 Murine Leukemia Viral Oncogene Homolog 1
Ras Rat Sarcoma (a family of related proteins)
SMRT Silencing Mediator for Retinoid and Thyroid Receptors
STAT Signal Transducers and Activators of Transcription
TNBC Triple-Negative Breast Cancer
Tyk2 Tyrosine Kinase 2
UPR Unfolded Protein Response
VEGFR2 vascular endothelial growth factor receptor 2
XBP1 X-Box Binding Protein 1
Keywords: Hsp90, PU-H71 (NSC 750424), tumorigenesis, apoptosis, oncogenic signals
Citation: Saber S, Abdelhady R, Elhemely MA, Elmorsy EA, Hamad RS, Abdel-Reheim MA, El-kott AF, AlShehri MA, Morsy K, AlSheri AS and Youssef ME (2024) PU-H71 (NSC 750424): a molecular masterpiece that targets HSP90 in cancer and beyond. Front. Pharmacol. 15:1475998. doi: 10.3389/fphar.2024.1475998
Received: 04 August 2024; Accepted: 22 October 2024;
Published: 05 November 2024.
Edited by:
Viviana di Giacomo, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Gabriela Chiosis, Memorial Sloan Kettering Cancer Center, United StatesErnest Duah, Sumitomo Dainippon Pharma Oncology, United States
Copyright © 2024 Saber, Abdelhady, Elhemely, Elmorsy, Hamad, Abdel-Reheim, El-kott, AlShehri, Morsy, AlSheri and Youssef. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sameh Saber, c2FtcGhhcm04MUBnbWFpbC5jbw==; Mai A. Elhemely, bWFpLmFsaUBtYW5jaGVzdGVyLmFjLnVrJiN4MDIwMGE7; Elsayed A. Elmorsy, YS5hbG1hcnNhQHF1LmVkdS5zYQ==; Rabab S. Hamad, cmhhbWFkQGtmdS5lZHUuc2E=; Mustafa Ahmed Abdel-Reheim, bS5haG1lZEBzdS5lZHUuc2E=