 Harriet Lampe
Harriet Lampe Laura Tam
Laura Tam Aaron R. Hansen
Aaron R. Hansen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 30 May 2024
Sec. Pharmacology of Anti-Cancer Drugs
Volume 15 - 2024 | https://doi.org/10.3389/fphar.2024.1399802
This article is part of the Research TopicGenomic Discoveries and Pharmaceutical Development in Urologic TumorsView all 17 articles
Metastatic castrate resistant prostate cancer (mCRPC) continues to have poor survival rates due to limited treatment options. Bi-specific T cell engagers (BiTEs) are a promising class of novel immunotherapies with demonstrated success in haematological malignancies and melanoma. BiTEs developed for tumour associated antigens in prostate cancer have entered clinical testing. These trials have been hampered by high rates of treatment related adverse events, minimal or transient anti-tumour efficacy and generation of high titres of anti-drug antibodies. This paper aims to analyse the challenges faced by the different BiTE therapy constructs and the mCRPC tumour microenvironment that result in therapeutic resistance and identify possible strategies to overcome these issues.
Globally, prostate cancer is the second most common cancer diagnosed amongst men and accounts for the fifth most common cause of malignancy-related deaths (Sung et al., 2021). Androgen deprivation therapy (ADT) has long remained the backbone of treatment of locally advanced or metastatic prostate cancer. The past 2 decades have seen multiple advancements in prostate cancer treatment, with demonstration of improved overall survival in metastatic castrate sensitive prostate cancer (mCSPC) with the introduction of androgen receptor signal inhibitor (ARSi) therapy, docetaxel chemotherapy and external radiation therapy, and in metastatic castrate resistant prostate cancer (mCRPC) with ARSis, a broader range of chemotherapy agents, and Radium-223 radionucleotide treatment (Petrylak et al., 2004; de Bono et al., 2011; Parker et al., 2013; Beer et al., 2014; James et al., 2016; Fizazi et al., 2017; Kyriakopoulos et al., 2018; Armstrong et al., 2019; Chi et al., 2019; Fizazi et al., 2022; Smith et al., 2022). The PSMA-targeted Lutetium-177 radionucleotide therapy has also shown survival benefit in mCRPC patients in the post-chemotherapy setting (Sartor et al., 2021). Additionally, for approximately 20% of men with mCRPC who harbour a homologous recombination defect (HRD), poly-ADP-ribose-polymerase (PARP) inhibitors such as olaparib and rucaparib have demonstrated better survival outcomes and are approved therapies in many countries (Hussain et al., 2020; Fizazi et al., 2023). Despite these significant advancements, overall survival rates amongst those with mCRPC remains low at only 34% at 5 years, thereby necessitating a need to develop better therapeutic options (SEER Explorer, 2023). BiTEs may represent a drug class that has the potential to improve clinical outcomes for prostate cancer patients and provide them with hope for a “brighter” future.
Immune checkpoint inhibitors (ICI) have revolutionised the management of a plethora of malignancies in the past decade (Robert, 2020). However, while their use in mCRPC has shown a marginal benefit in a small proportion of prostate cancer patients with mismatch repair (MMR) deficiency mutations, their use in the majority of mCRPC patients has been limited with a number of negative trials including KEYNOTE-641 and the KEYLYNK-010 trial (Kwon et al., 2014; Robinson et al., 2015; Kazandjian et al., 2016; Beer et al., 2017; Hansen et al., 2018; Antonarakis et al., 2019; Sharma et al., 2019). Earlier research in cancer vaccines have shown some success in the prostate cancer landscape. Sipuleucal-T, a dendritic cell-based vaccine which acts via a recombinant protein of granulocyte macrophage colony stimulating factor (GM-CSF) and prostatic acid phosphatase (PAP) to facilitate maturation of PAP-expressing antigen presentation cells, with subsequent T cell activation and PAP-expressing prostate cell killing, was approved by the US Food and Drug Administration (FDA) in 2010 for minimally symptomatic mCRPC. Although it demonstrated an overall survival (OS) benefit, particularly in a sub-population with reduced metastatic burden, and has acted as an important proof of concept for immunotherapies outside of ICIs in prostate cancer treatment, sipuleucal-T has failed to be incorporated into routine treatment for mCRPC due to doubts about limited clinical benefit (Kantoff et al., 2010). These modest results have driven research into other facets of immunotherapy such as T-cell engager (TCE) therapies, which aim to directly crosslink T-cells with tumour associated antigens (TAAs), thus driving localised cancer cell specific cytotoxicity, cytokine release, and downstream activation of a B-cell polyclonal humoral response (Zhou et al., 2021). TCE therapies include chimeric antigen-receptor-modified (CAR) T cells, in which patients’ own T-cells are genetically engineered ex vivo to express a chimeric T-cell receptor targeted at the desired TAA, and bi-specific T-cell engagers (BiTEs), antibody-based molecules offering a promising “off the shelf” option for achieving cross-linking of cancer and T-cells, which we shall examine in further detail.
BiTEs are monoclonal antibody (mAb) based molecules comprised of at least two conjoined antibody components which have respective specificity for a TAA of choice, and for an immune cell component, typically the conserved portion of the T-cell receptor, CD3, connected by a flexible linker moiety (Riethmüller, 2012). The mechanism of action of BiTEs involves activation of cytotoxic T-cells independent of the co-stimulation pathway, resulting in robust killing of cancer cells which has been demonstrated in vitro to have efficacy 100–10,000 fold greater than that of mAbs (Wolf et al., 2005).
BiTEs can be produced through chemical cross-linking of two antibody fragments, or via fusion of two different monoclonal antibody producing cell lines (so called “quadromas”) with subsequent purification of the desired protein outcome (Schaefer et al., 2011). Initial inefficiencies in BiTE production concerning high proportions of incorrect heterodimer products have been addressed with multiple strategies to improve correct heterodimerisation. These have included heavy chain “knobs in holes” alterations (Xu et al., 2015), “crossover” of light and heavy chain combinations within one Fab arm of a bispecific IgG antibody to reliably produce a single desired heterodimer (CrossMabs; (Klein et al., 2019)), and dual affinity re-targeting proteins (DARTs), which favour stable dimerization due to exchange of variable heavy and light chains between two scFv components (Johnson et al., 2010). While the original BiTE molecules comprised two single chain variable fragments (scFv) from two different mAbs connected by a flexible peptide linker, further drug development research has generated an array of structural variations conferring benefits of more efficient production, increased half-life and improved target binding (Suurs et al., 2019). Blinatumomab, an anti-CD3-CD19 BiTE, was the first in its class to be approved by the FDA in 2014 for the treatment of Philadelphia chromosome negative relapsed or refractory B-cell acute lymphoblastic leukaemia (Kantarjian et al., 2017). In solid tumours, tebentafusp, which targets gp100 and CD3, has demonstrated improved survival outcomes for patients with uveal melanoma (Nathan et al., 2021). While there are no other approved solid tumour BiTEs to date, work is currently underway in a variety of cancer histologies to further their development.
The use of BiTE therapies in prostate cancer has been trialled in several studies using different structures and TAA targets (See Table 1 for a complete list of finalised and ongoing trials). The first trial of a BiTE therapy in prostate cancer involved pasotuxizumab, an anti-CD3 and anti-prostate specific membrane antigen (PSMA) construct, administered either via subcutaneous injection or continuous intravenous infusion which produced a 54.9% reduction in PSA in the highest dose cohort, but was associated with 81% rate of Grade 3 or 4 treatment related adverse events (tr-AEs; (Hummel et al., 2021)). The trial was terminated in favour of acapatamab, an anti-CD3, anti-PSMA molecule with an IgG crystallisable fragment (Fc) to extend serum half-life. Unfortunately, while the PSA response of a 50% reduction (PSA50) was 34.3%, the rate of grade 3/4 tr-AEs was >50% and consequently acapatamab was not planned for further development (Ben et al., 2020). Subsequent studies have assessed alternative TAAs, including prostate stem cell antigen (PSCA), human kallikrein 2 (KLK2), delta-like protein 3 (DLL-3) and six transmembrane epithelial antigen of prostate 1 (STEAP-1). These studies have also explored a variety of alternative BiTE structures—addition of HLE or immune-interacting domains, incorporation of more complete antibody structures and differences in linker molecules. However, to date the vast majority have failed to move past Phase 1 clinical trials, displaying various combinations of prohibitive side effect profiles, limited anti-tumour efficacy and a high rate of anti-drug antibodies, amongst other issues (Simão et al., 2023). Given the increasing interest in BiTE therapies in prostate cancer, we will explore some of the factors impacting both safety and efficacy associated with this class of drugs.
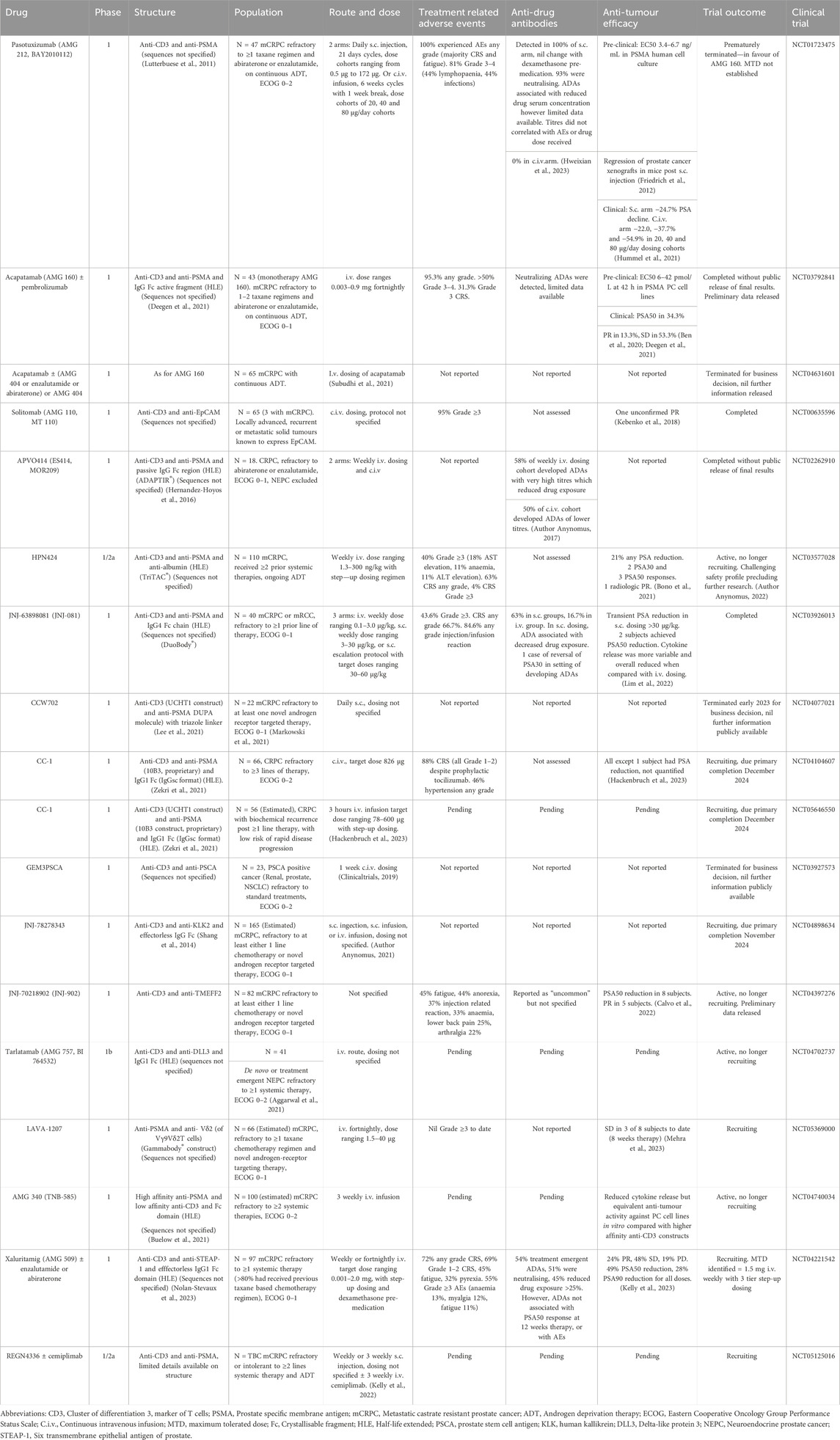
Table 1. Summary of clinical trials evaluating BiTE therapies in prostate cancer to date.
The original BiTE molecules were comprised of two single chain variable fragments (scFv) from two different mAbs connected by a flexible peptide linker (Ahmad et al., 2012). The small size of these molecules resulted in a short half-life with poor serum stability and rapid renal clearance, which necessitated administration via continuous intravenous infusion. This issue was subsequently addressed with the development of half-life extended (HLE) BiTE variants, which incorporated structures to increase molecular size and stability, enabling BiTEs to be formulated as intermittent infusions or subcutaneous injections, and consequently improving efficacy, convenience and cost of administration schedules. One method involved bonding together of two BiTEs to create a tetravalent “tandem diabody” (TandAbs; (Kipriyanov et al., 1999)). Alternative methods included addition of antibody heavy chain elements, either as part of a IgG-based structure, or as an isolated Fc domain to enable binding to the neonatal Fc receptor in recipient serum and thus reduce the rate of clearance (Brinkmann and Kontermann, 2017). Inclusion of an Fc region confers the ability to bind to the neonatal Fc receptor on innate immune cells, enhancing immune cell engagement. Other HLE strategies have included addition of an albumin receptor to improve serum stability, or molecular modification of heavy chains to increase half-life, such as with the XmAb technology (Zhukovsky et al., 2007). Overall, these customisations have created a myriad of BiTE structural variations which have facilitated administration via intermittent intravenous or subcutaneous routes, and also offer additional functional benefits.
The main adverse events of special interest with BiTEs are cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). CRS is a systemic inflammatory response syndrome that can produce symptoms ranging from fever to shock with multi-organ failure caused by high levels of cytokine release, particularly IFN-γ, IL-1 and IL-6 (Morris et al., 2022). CRS most commonly occurs hours to short days after the initial dose of TCE therapy, with some evidence showing reduced incidence and severity following pre-medication with dexamethasone and step-up dosing regimens (Shimabukuro-Vornhagen et al., 2018). Severe CRS can be treated with tocilizumab, an anti-IL-6 receptor mAb, without affecting therapeutic activity (Kauer et al., 2020). The occurrence of any-grade CRS in the studies on BiTE therapies in mCRPC to date ranges from 5% with JNJ-902% to 88% with CC-1, although it should be noted that there were no reports of associated mortality even with higher rates of CRS (Calvo et al., 2022; Heitmann et al., 2022).
ICANS pathophysiology remains uncertain, but patients often present with confusion, tremor or dizziness and can progress to seizures or irreversible encephalopathy over a course of days post treatment. Treatment consists of aggressive supportive therapy and corticosteroids. Tocilizumab is unable to cross the blood-brain barrier and has limited clinical utility in management. ICANS occurs infrequently with BiTE therapies in general and appears to be rarer in solid tumour BiTE studies (Siegler and Kenderian, 2020). There have been no formal diagnoses of ICANS in the studies of prostate cancer-targeted BiTEs to date, although seizures, a possible sign of ICANS, was reported in a single patient receiving HPN-424 (Bono et al., 2021).
CRS initially presented a clinical challenge to progression of BiTE therapies in solid tumours. However, with increased exposure to and recognition of the constellation of CRS in clinical settings, the improved availability of tocilizumab, and the growing evidence that even with high grade CRS, survival remains extremely high, this trAE should not be a major factor limiting use of BiTE therapies moving forward. Focus should instead be placed on reducing immunogenicity of the BiTE structure to curb hyperactive cytokine release where possible. Brandt et al. have proposed the “kill switch” mechanism for BiTE therapy similar to that adopted in experimental CAR T-cell therapies (Brandt et al., 2020). These mechanisms rely on the incorporation of an inducible self-destruct process into the original therapy which, when activated, results in the rapid removal of the treatment from the system. In CAR T-cell therapies, this has involved induction of the caspase-9 T-cell apoptosis pathway, or induction of T-cell transcription of viral components subsequently leading to T-cell apoptosis. While purely theoretical, inclusion of a cleavage site within the linker portion of a BiTE therapy could achieve similar effects.
A challenge in the adaptation of BiTEs from haematological to solid tumours has been the difficulty in finding solid organ TAAs which are highly specific to the desired tumour cell population, as many solid organ TAAs are also present at low levels in normal tissue. In this instance, BiTE activity poses a risk of so-called “on-target off-tumour” effects resulting from damage to these non-malignant tissues. These can be catastrophic, as demonstrated in trials of catumaxomab, an early example of a solid organ BiTE directed against epithelial cell adhesion molecule (EpCAM) and CD3 for EpCAM-positive advanced solid organ cancers causing malignant ascites, with one patient experiencing fatal acute fulminant liver failure (Linke et al., 2010).
PSMA, the most commonly targeted TAA in prostate cancer BiTE therapies to date, while being highly expressed in prostate cancer, also has high levels of expression within intestinal, liver, salivary glandular cells and proximal kidney tubules (Silver et al., 1997). There has been evidence of PSMA targeting leading to significant on-target off-tumour effects, for example, with the BiTE HPN424, which caused Grade 3–4 AST elevations in 18% patients, and ALT elevations in 11% (Bono et al., 2021). Although on-target off-tumour AEs appear to be relatively infrequent in trials of BiTEs targeting PSMA, research into more specific TAAs remains warranted to alleviate the risk of these outcomes.
Multiple alternative TAAs are under investigation as therapeutic targets in prostate cancer. One potential target proposed has been STEAP-1, which has some expression in lung tissue but otherwise has limited expression outside of the brain which is considered inaccessible to BiTE molecules (Xu et al., 2022). STEAP-1 has been targeted in a Phase 1 clinical trial using an agent called xaluritamig. Although 72% of patients experienced CRS, the vast majority (69%) were Grade 1–2 (Kelly et al., 2023). Another target being used in clinical trials is TMEFF2 which has expression limited to intestinal tissues, the male reproductive tissues and brain. JNJ-70218902 was designed to target TMEFF2 and entered phase I testing but limited available data exists regarding its efficacy and toxicity profile (Calvo et al., 2022). Kallikrein related peptidase-2 (KLK2) is a highly specific TAA with expression restricted to prostate tissue only and is the putative tumour target for JNJ-78278343. Both efficacy and safety data is still being awaited at this stage (Shang et al., 2014).
The current trials of BiTEs in prostate cancer treatment as described in Table 1 are all targeted at membrane bound extracellular proteins which can be readily accessed by nearby immune effector cells. However, these extracellular proteins represent only a fraction of potential tumour specific targets. Intracellular protein fragments expressed extracellularly bound to human leukocyte antigen (HLA) major histocompatibility complexes (MHC) through antigen presentation to immune cells offer a much more extensive suite of potential therapeutic targets not otherwise accessible to antibody based therapies (Trenevska et al., 2017). BiTEs designed to target these peptide-HLA complexes have been termed immune mobilising monoclonal T-cell receptors against cancer (ImmTACs). Tebentafusp, an approved BiTE therapy for uveal melanoma, is the first in class ImmTAC, designed to target a peptide-HLA combination of gp100:HLA-A*02:01 along with the CD3 component of the T-cell receptor (TCR, Nathan et al., 2021). Disadvantages of this therapy include restriction of eligibility of patients to those with matched HLA typing, challenges of identifying peptide:HLA combinations which are highly conserved across cancer cells and patient populations, and possibility of tumour evasion through downregulation of HLA cell surface expression (Trenevska et al., 2017; Maruta et al., 2019). Nevertheless, the development of ImmTACS represents a breakthrough in BiTE development, and further research into intracellular prostate cancer TAAs may reveal novel therapeutic targets with reduced on-target off-tumour effects.
In addition to targeting more tumour-specific TAAs, another approach to improve the localisation of BiTE activation to tumour tissue involves the incorporation of moieties targeting two different TAAs into a single BiTE construct. This has been explored pre-clinically in solid tumour cell lines with AMG-305, a dual targeted BiTE directed against P-cadherin and mesothelin which showed attenuated activity against cells expressing only one of these targets (Pham et al., 2023). Targeting of two separate TAAs may also offer a possible means of reducing immune escape through the common route of downregulated expression of a targeted TAA. However, once again, in the absence of TAAs which are more tightly limited to prostate cancer expression, these therapies continue to pose the risk of unwanted on-target off-tumour effects.
Another possible strategy to localise BiTE activation to tumour tissue is the incorporation of structural elements which prevent activity of the BiTE outside of conditions specific to the tumour microenvironment. One possible approach is masking of BiTE binding sites with structures designed to be cleaved away by tumour-resident proteases, hence restricting activity to tumour deposits (Geiger et al., 2020). Panchal et al. described generation of an EGFR-CD3 BiTE where the heavy and light variable chains of the anti-CD3 portion were separated by a linker degradable only by the tumour specific matrix metalloproteinase 9 (MMP9), permitting anti-CD3 activity only after MMP9 facilitated rearrangement of the molecule (Panchal et al., 2020). Another approach is addition of a second TAA target within a T-cell engager structure with the aim of improving tumour tissue specific cytotoxicity. A variation on this concept is the construction of “hemibodies,” two “half” antibodies each containing a different antigen binding scFv fragment fused to either a variable light or variable heavy chain of a CD3 antibody, designed to recombine to form a functional BiTE only in the presence of both TAAs. The proof-of-concept hemibody was targeted against HLA-A2 and CD45, and showed apoptosis restricted to dual positive tumour cells in animal models (Banaszek et al., 2019). To date these experiments have all been pre-clinical, and none have been targeted towards prostate cancer, however these results could be transferable to other BiTE structures in future (Panchal et al., 2020).
Drug related immunogenicity, or the response of a patient’s immune system to a drug, occurs via recognition of drug components as “non-self,” and is a key factor influencing the efficacy and adverse effects of BiTEs (Jawa et al., 2020). Protein sequences within the drug or drug excipients as part of the drug formulation are taken up by patients’ antigen presenting cells (APCs), with subsequent breakdown and expression of short protein epitopes on HLA surface molecules. T helper cell recognition of these epitopes as “non-self” will stimulate an immune response including a B cell humoral response with production of anti-drug antibodies (ADAs). ADAs can be broadly split into “neutralizing” antibodies which obstruct binding sites, and “non-neutralizing,” which bind epitopes which do not directly interfere with the drug’s action. ADAs have critical effects on a drug’s efficacy, pharmacokinetics, and adverse events through a wide array of mechanisms, including blocking or changing affinity of binding sites, prolongation or potentiation of drug clearance, aggregation of drug-antibody complexes and effects from inflammatory cytokine production (van Brummelen et al., 2016). Further complicating factors influencing the extent of immunogenicity to a drug include the route of administration, dose regimen, product storage, product purity, and the patient’s own immune system factors such as the presence of pre-existing cross-reacting ADAs and skew towards immunoregulatory or inflammatory immune responses. The desired alternative response is recognition of these epitopes as “self” by the immune system, with subsequent induction of immune tolerance via activation of T regulatory cells (Tregs).
The complex interactions between these therapies and the immune system are a fundamental part of the nature of T cell therapies. ADA quantification and their resultant physiological effects form the backbone of a multifaceted immune response assessment recommended by both the FDA and EMA for drug immunogenicity (Administration USFaD, 2010; Agency, 2017). It should be noted that current detection methods for ADAs are imperfect. A range of assays can be used to detect ADAs, with a wide variability in sensitivity and specificity. Furthermore, assays usually only detect one subclass of Fc immunoglobulin receptors, predominantly IgG, and may not detect drug-bound antibody, leading to under-reporting of ADAs (van Brummelen et al., 2016). Nevertheless, ADA analysis remains the most accurate method of assessing immunogenicity. Notably, these assessments are primarily conducted in or ex vivo in human clinical studies, given the limitations to mimick a nautral human immune response in animal or human cell lines.
Where data has been released for quantification of ADAs in clinical trials of BiTEs for prostate cancer, ADAs were frequently present in >50% subjects (See Table 1). This is an extremely high incidence compared to other biologic therapies on the market (van Brummelen et al., 2016). When compared with intravenous administration, subcutaneous dosing is associated with a high rate of neutralising antibodies, which has been ascribed to sequential antigen presentation from both APCs residing in the skin and then lymph node-resident APCs. These two “waves” of antigen presentation increase the formation of ADAs (Jarvi and Balu-Iyer, 2021). For instance, during testing of pasotuxizumab, induced ADAs were recorded in 100% subjects receiving s.c. dosing, with 93% of these being neutralizing, but none in those receiving the continuous i.v. dosing (Hweixian et al., 2023).
Further assessment of ADA subclasses and effects in BiTE therapies for mCRPC has been limited to studies in pasotuxizumab and xaluritamig (Hweixian et al., 2023; Kelly et al., 2023). Within this limited sample, it has been demonstrated that treatment emergent neutralizing ADAs can have either adverse (e.g., pasotuxizumab) or neutral (e.g., xaluritmag) clinical effects. (Hweixian et al., 2023; Kelly et al., 2023). For xaluritamag, 54% of subjects developed treatment emergent ADAs for an i.v. formulation, with 45% of subjects developing neutralising antibodies causing reduction in drug exposure by more than 25%. However, these ADAs were not associated with difference in PSA50 response at 12 weeks (Kelly et al., 2023). Conversely, non-neutralising ADAs can lead to formation of serum antibody complexes which are subsequently removed by the host immune system, which theoretically could affect drug efficacy despite their “benign” status, although this was not observed in the above two trials. Despite historical data in other fields showing an association between ADAs and certain AEs, the presence or titre of ADAs does not appear to correlate with the rate or severity of AEs in either population. Importantly, all ADAs, regardless of neutralising status, can dramatically alter pharmacokinetics of the drug by sustaining or expediting drug clearance, with likely construct specific effects on efficacy (Kelly et al., 2023). Unfortunately, a deeper analysis is limited by the restricted published data from clinical trials about ADAs and their effect on drug pharmacokinetics or pharmacodynamics.
The causes of the high rates of ADA formation in response to BiTE therapies are incompletely understood but derives at least partially from particularly immunogenic sequences within the drug’s structure, including effector Fv domains, Fc region, half-life extending domains, or peptide linkers. A number of methods can be used to determine the immunogenicity of these sequences, including screening of drug protein structure for known T helper or T regulatory binding epitopes; culturing of antigen presenting cells with ex vivo peripheral red blood cells with sequencing of epitopes expressed by APCs; or analysis of the binding sequences of extracted ADAs from trial subjects to match to a complementary epitope from the drug; with the latter detailed by Hweixian et al. for pasotuxizumab (van Brummelen et al., 2016; Hweixian et al., 2023). However, the protein sequences of the drug or ADAs detected have not been made available to the public domain for the majority of BiTEs trialled for prostate cancer so far. Release of this existing information, and wider testing of ADAs in ongoing and future trials would provide invaluable information in helping better understand the interplay between BiTEs and the humoral immune system. It could help to differentiate the concentrations or affinities at which ADAs become clinically significant, and to select drug components with less problematic immune responses. With this information, immune engineering of therapeutic protein structures could enable adjustments to homology to mask or remove immunogenic sequences, or addition of T regulatory epitopes promoting immune tolerance (van Brummelen et al., 2016).
Other factors known to contribute to immunogenicity include drug excipients, which can be comprised of contaminant proteins accidently purified with the therapeutic protein during production, or other components of the drug formulation such as trace heavy metals. Subcutaneous administration has been associated with increased ADAs over an i.v. route for previous biologics, as has re-exposure following a treatment free interval (Agency, 2017). Half-life extending domains, such as an Fc region, may also add to non-humoral immunogenicity through engagement of the immune system via the Fc receptor on APCs, neutrophils and NK cells. Although HLE molecules are attractive for the possibility they offer of a more convenient administration schedule, the added immunogenicity they seem to generate poses challenges, particularly to subcutaneous administration. Finally, there are patient specific factors at play, such as levels of pre-existing ADAs, and different HLA haplotypes influencing the particular epitopes that may produce a patient response (van Brummelen et al., 2016). Once again, limited testing or release of data concerning ADA quantity and sequences, and their association with patient HLA typing and drug excipients, prevents retrospective analysis of how to avoid highly immunogenic structures in the future. Greater transparency and dissemination of available data will assist in further targeted development of this class of agents.
Unfortunately, the majority of completed clinical trials of BiTEs against prostate cancer have demonstrated limited or inconsistent anti-tumour effects to date. There is a recurrent pattern of small percentages of study groups achieving PSA50, or radiologic partial response (PR) in clinical trials examining the use of HPN424, JNJ-081 and JNJ-902 (Bono et al., 2021; Calvo et al., 2022; Lim et al., 2022). Pasotuxizumab displayed slightly more promising results with a 19% PSA50 response and two long term PSA50 responders, however this trial was prematurely terminated in favour of acapatamab, which in turn delivered a PSA50 response in 34.3% of subjects (Ben et al., 2020; Hummel et al., 2021). However, the trial using acapatamab was also discontinued in the setting of a high incidence of trAEs. Xaluritamig has recently demonstrated the greatest anti-tumour efficacy in the field, with 49% of patients demonstrating a PSA50 response and 28% of patients also achieving a PSA90 reduction (Kelly et al., 2023).
Despite these promising results, when compared with haematological malignancies, immunotherapies in prostate cancer face multiple barriers to effectiveness, including intra- and inter-tumoral genotypic heterogeneity, and downregulation of TAA expression over time, leading to treatment escape or failure (Middelburg et al., 2021; Ge et al., 2022). They must also contend with a complex solid tumour microenvironment (TME), of which prostate cancer’s TME presents specific challenges.
The disappointing response of prostate cancer to ICIs has been predominantly attributed to its multifactorial “cold” or immunosuppressive tumour microenvironment (TME) in comparison to other common malignancies such as melanoma or lung. Metastatic CRPC TMEs are characterised by a dense stroma with relatively high proportions of cancer-associated fibroblasts (CAFs) and myeloid derived suppressor cells (MDSCs) which presents a physical barrier to anti-cancer therapies and results in low numbers of tumour infiltrating lymphocytes (TILs) and innate immune cells. Even within the limited TIL population there is a tendency towards an immunosuppressive phenotype, with a preponderance of Th2 and T regulatory (Treg) lymphocytes over Th1 counterparts, skewing against activation of CD8+ cytotoxic T cells and natural killer (NK) cells (Krueger et al., 2019). There is a similar overabundance of M2 anti-inflammatory macrophages instead of their inflammatory M1 counterparts. Metastatic CRPC TMEs are further masked from the immune system by a low tumour mutational burden with consequent reduced neoantigen expression as well as local overexpression of costimulatory molecules such as PD-1 and CTLA-4, which act as “self” markers, leading over time to an “exhausted” local immune cell phenotype (Gannon et al., 2009).
This immunosuppressive TME is theorised to limit the efficacy of the “bystander” effect of immunotherapies, in which successful cell dependent cytotoxicity creates a local environment of pro-inflammatory cytokines and subsequent upregulation of neoantigens and differentiation of inflammatory immune cell phenotypes, prompting cytotoxicity towards adjacent tumour cells (Ross et al., 2017). Notably, ADT, the cornerstone of prostate cancer treatment, is suspected to contribute to the immunosuppressive TME through increases in intratumoural Tregs, MDSCs and M2 macrophages, reduced markers of interaction between tumour cells and de-regulation of intratumoural Tregs (Pu et al., 2016; Qin et al., 2022). The immunosuppressive TME of prostate cancer is observed to become more extreme with progression to mCRPC, with bony metastases representing the most severe example of this phenotype (Jiao et al., 2019). Synchronous metastatic deposits of CRPC display significant genetic heterogeneity, further predisposing to immune escape and treatment resistance (Sun, 2021).
BiTEs were initially expected to provide a radical solution to many of the problems presented by “cold” TMEs, by localising T cell activation directly to malignant cells. Unfortunately, this has not eventuated, with BiTE therapies demonstrating limited anti-tumour activity in prostate cancer in studies to date. It is possible that the immunosuppressive TME limits the bystander effect of BiTEs to some extent, as it does for ICIs. It should also be noted that the recipients of these BiTEs comprise a heavily pre-treated and castrate-resistant group. In light of the mCRPC TME being highly immunosuppressive post ADT, ARSi and chemotherapy, it can be speculated that BiTEs may show greater efficacy in a less heavily pre-treated patient population, akin to the benefit seen in a similar sub-population with the sipuleucal-T vaccine.
There are a multitude of theories as to how to alter the TME pathophysiology to improve the action of BiTEs, predominantly based on improving either systemic or intratumoural immune effector cell populations and activity. A logical option is to trial a combination of immunotherapy with BiTE therapy, to simultaneously aim to reverse TIL anergy, prevent BiTE mediated upregulation of TIL PD-1 expression and tumour and stromal PD-L1 expression (Jiang et al., 2019; Belmontes et al., 2021). This strategy has been tested in prostate cancer BiTE trials (See Table 1), with combinations of anti-PSMA BiTEs with PD-1 inhibitors, namely, acapatamab and pembrolizumab, and REGN4336 and cemiplimab, but results from these combinations are not available (Ben et al., 2020; Kelly et al., 2022). Outside of prostate cancer, there has been great interest in this combination with multiple clinical trials underway (Belmontes et al., 2021). In solid tumours, preliminary phase 1 clinical trial data for a CEA and CD3 targeting BiTE showed increased disease response when paired with atezolizumab in the absence of increased toxicity (Tabernero et al., 2017). Alternative structural variations on the synergy of BiTEs and ICIs includes bifunctional checkpoint-inhibitory T-cell engagers (CiTEs), comprised of a BiTE crosslinked with a PD-L1 inhibitor to provide localised combination therapy and attempt to avoid systemic effects of ICIs (Herrmann et al., 2018).
An alternative experimental approach involves shifting focus to disruption of the stroma of the TME to permit increased TIL migration and improve BiTE intra-tumoural access. Brunker et al. developed a bi-specific antibody designed to crosslink the fibroblast activation protein (FAP) and the death receptor 5 (DR5), triggering the extrinsic apoptotic pathway for tumour cells, with successful cytotoxicity in FAP positive tumour stroma (Brünker et al., 2016). Another example of successful pre-clinical alteration of the structure of the TME has been the use of an oncolytic virus expressing a FAP-CD3 BiTE which successfully increased intra-tumoral accumulation of T cells and decreased FAP concentration, indicating fibroblast apoptosis in vivo testing (de Sostoa et al., 2019). The extracellular matrix could also be directly targeted with enzymes such as hyaluronidase to literally open a path for tumour infiltration by T cells (Eikenes et al., 2005). Yet further studies have targeted function and trafficking of MDSCs to reduce their immunosuppressive local effects (Middelburg et al., 2021). However, these approaches remain pre-clinical and have not been targeted to prostate cancer to date.
Rather than focusing on CD3 positive T-cells, some potential therapeutics instead aim to target alternative immune effector cell populations such as the pro-inflammatory natural killer (NK) cells. NK cells have been targeted via bi- and tri-specific NK-cell engagers (BiKEs and TriKEs) in clinical trials for haematological malignancies with promising results (Rothe et al., 2015; Vallera et al., 2016). γδ T-cells present a unique target in prostate cancer. Although they are relatively sparse in comparison to the more common αβ T-cells, they are particularly concentrated in prostate cancer tumours when compared with other solid organ tumours (Tosolini et al., 2017). Activation of intra-tumoral γδ T-cells could potentially kickstart immune cell activation within the prostate TME, and a clinical trial of LAVA-1207, a γδ T-cell-directed BiTE, is currently underway (Mehra et al., 2023).
A potential strategy for overcoming limited anti-tumour activity of current BiTEs would be to increase the binding affinity of the TAA or CD3 targeted structural components. Multiple antibody components targeting the same TAAs could be added to increase valency and target binding, as exemplified by the TandMab structure. Alternatively, reverse protein engineering could be utilised to design increased affinity of complementarity determining regions within the variable chains directed towards the relevant TAA. For example, Zekri et al. (2021) developed a proprietary PSMA binder, 10B3, which demonstrated increased reactivity against prostate cancer cells in vitro compared with a pre-existing J591 PSMA antibody. This improvement was attributed to alternative binding sites to PSMA recognised by the 10B3 molecule. However, it should be noted that protein engineering generally leads to deviation from native antibody structure and consequently higher risk of immunogenicity. Ultimately, while the expression of BiTE targets for prostate cancer remains non-specific to malignant tissue, attempts to increase TAA binding affinity risk increased rates of on-target off-tumour effects.
Metastatic CRPC is a prevalent disease which remains a challenging clinical entity to effectively treat, with limited response to the ICIs which have become cornerstones of treatment for multiple other cancers. Novel TCE therapy, particularly BiTE therapy, has been a promising area of immunotherapy development in the past decade with particular interest in their use in prostate cancer. Unfortunately, early investigations of various prostate cancer specific BiTE therapies have been limited by high incidences of intolerable adverse events and insufficient anti-tumour activity. Nevertheless, progress has been made in addressing these shortcomings through identifying a range of possible TAAs to exploit, extending the drug half-life allowing for more convenient administration schedules, and managing class specific AEs. Moreover, there is extensive research into multiple strategies as to how to overcome the challenges presented by the prostate cancer TME, the immunogenicity of BiTE constructs and focused targeting of BiTEs to tumour cells. With these developments and the possibilities of identification of more specific TAAs, improved affinity of BiTEs to TAAs, greater modelling and testing for immunogenicity, and modulation or mitigation of the anti-inflammatory mCRPC TME, it is reasonable to hope that BiTEs may provide a therapeutic benefit in the future for those afflicted with mCRPC.
HL: Writing–original draft, Writing–review and editing. LT: Supervision, Writing–review and editing. AH: Conceptualization, Supervision, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors would like to acknowledge the contributions of Gina Velli, of the Princess Alexandra Hospital Library and Knowledge Centre, who assisted with the initial literature search for this review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Administration USFaD (2010) Immunogenicity testing of therapeutic protein products - developing and validating assays for anti-drug antibody detection - guidelines for Industry.
Agency, E. M. (2017) Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins.
Aggarwal, R. R., Aparicio, A., Heidenreich, A., Sandhu, S. K., Zhang, Y., Salvati, M., et al. (2021). Phase 1b study of AMG 757, a half-life extended bispecific T-cell engager (HLE BiTEimmune-oncology therapy) targeting DLL3, in de novo or treatment emergent neuroendocrine prostate cancer (NEPC). J. Clin. Oncol. 39 (15), TPS5100. doi:10.1200/jco.2021.39.15_suppl.tps5100
Ahmad, Z. A., Yeap, S. K., Ali, A. M., Ho, W. Y., Alitheen, N. B., and Hamid, M. (2012). scFv antibody: principles and clinical application. Clin. Dev. Immunol. 2012, 980250. doi:10.1155/2012/980250
Antonarakis, E. S., Shaukat, F., Isaacsson Velho, P., Kaur, H., Shenderov, E., Pardoll, D. M., et al. (2019). Clinical features and therapeutic outcomes in men with advanced prostate cancer and DNA mismatch repair gene mutations. Eur. Urol. 75 (3), 378–382. doi:10.1016/j.eururo.2018.10.009
Armstrong, A. J., Szmulewitz, R. Z., Petrylak, D. P., Holzbeierlein, J., Villers, A., Azad, A., et al. (2019). ARCHES: a randomized, phase III study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J. Clin. Oncol. 37 (32), 2974–2986. doi:10.1200/JCO.19.00799
Author Anynomus (2017) Aptevo therapeutics and MorphoSys end joint development and - commercialization agreement for mor209/es414. Aptevo Therapeutics Inc. [press release].
Author Anynomus (2021). A study of JNJ-78278343, a T-cell-redirecting agent targeting human kallikrein 2 (KLK2), for advanced prostate cancer. Available at: https://clinicaltrials.gov/study/NCT04898634?tab=history.
Author Anynomus (2022) Harpoon therapeutics reports fourth quarter and full year 2021 financial results and provides corporate update<harpoon-therapeutics-reports-fourth-quarter-and.pdf>. Harpoon Therapeutics. [press release].
Banaszek, A., Bumm, T. G. P., Nowotny, B., Geis, M., Jacob, K., Wölfl, M., et al. (2019). On-target restoration of a split T cell-engaging antibody for precision immunotherapy. Nat. Commun. 10 (1), 5387. doi:10.1038/s41467-019-13196-0
Beer, T. M., Armstrong, A. J., Rathkopf, D. E., Loriot, Y., Sternberg, C. N., Higano, C. S., et al. (2014). Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 371 (5), 424–433. doi:10.1056/NEJMoa1405095
Beer, T. M., Kwon, E. D., Drake, C. G., Fizazi, K., Logothetis, C., Gravis, G., et al. (2017). Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J. Clin. Oncol. 35 (1), 40–47. doi:10.1200/JCO.2016.69.1584
Belmontes, B., Sawant, D. V., Zhong, W., Tan, H., Kaul, A., Aeffner, F., et al. (2021). Immunotherapy combinations overcome resistance to bispecific T cell engager treatment in T cell-cold solid tumors. Sci. Transl. Med. 13 (608), eabd1524. doi:10.1126/scitranslmed.abd1524
Ben, T., Horvath, L., Rettig, M., Fizazi, K., Lolkema, M. P., Dorff, T. B., et al. (2020). Phase I study of AMG 160, a half-life extended bispecific T-cell engager (HLE BiTE immune therapy) targeting prostate-specific membrane antigen, in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 38 (15), TPS5590. doi:10.1200/jco.2020.38.15_suppl.tps5590
Bono, J. S. D., Fong, L., Beer, T. M., Gao, X., Geynisman, D. M., Hab, I. I. I., et al. (2021). Results of an ongoing phase 1/2a dose escalation study of HPN424, a tri-specific half-life extended PSMA-targeting T-cell engager, in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 39 (15_Suppl. l), 5013. doi:10.1200/jco.2021.39.15_suppl.5013
Brandt, L. J. B., Barnkob, M. B., Michaels, Y. S., Heiselberg, J., and Barington, T. (2020). Emerging approaches for regulation and control of CAR T cells: a mini review. Front. Immunol. 11, 326. doi:10.3389/fimmu.2020.00326
Brinkmann, U., and Kontermann, R. E. (2017). The making of bispecific antibodies. MAbs 9 (2), 182–212. doi:10.1080/19420862.2016.1268307
Brünker, P., Wartha, K., Friess, T., Grau-Richards, S., Waldhauer, I., Koller, C. F., et al. (2016). RG7386, a novel tetravalent FAP-DR5 antibody, effectively triggers FAP-dependent, avidity-driven DR5 hyperclustering and tumor cell apoptosis. Mol. Cancer Ther. 15 (5), 946–957. doi:10.1158/1535-7163.MCT-15-0647
Buelow, B., Dalvi, P., Dang, K., Patel, A., Johal, K., Pham, D., et al. (2021). TNB585.001: a multicenter, phase 1, open-label, dose-escalation and expansion study of tnb-585, a bispecific T-cell engager targeting PSMA in subjects with metastatic castrate resistant prostate cancer. J. Clin. Oncol. 39 (15), TPS5092. doi:10.1200/jco.2021.39.15_suppl.tps5092
Calvo, E., Doger de Spéville, B., Carles Galceran, J., Peer, A., Sarid, D. L., Eigl, B. J., et al. (2022). 1367P JNJ-70218902 (JNJ-902), a TMEFF2 x CD3 bispecific antibody, in prostate cancer: initial results from a phase I dose escalation study. Ann. Oncol. 33, S1166. doi:10.1016/j.annonc.2022.07.1499
Chi, K. N., Agarwal, N., Bjartell, A., Chung, B. H., Pereira de Santana Gomes, A. J., Given, R., et al. (2019). Apalutamide for metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 381 (1), 13–24. doi:10.1056/NEJMoa1903307
Clinicaltrials (2019). “Study with bispecific antibody engaging T-cells,” in Patients with progressive cancer diseases with positive PSCA marker. Available at: https://clinicaltrials.gov/study/NCT03927573?tab=history.
de Bono, J. S., Logothetis, C. J., Molina, A., Fizazi, K., North, S., Chu, L., et al. (2011). Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 364 (21), 1995–2005. doi:10.1056/NEJMoa1014618
Deegen, P., Thomas, O., Nolan-Stevaux, O., Li, S., Wahl, J., Bogner, P., et al. (2021). The PSMA-targeting half-life extended BiTE therapy AMG 160 has potent antitumor activity in preclinical models of metastatic castration-resistant prostate cancer. Clin. CANCER Res. 27 (10), 2928–2937. doi:10.1158/1078-0432.CCR-20-3725
de Sostoa, J., Fajardo, C. A., Moreno, R., Ramos, M. D., Farrera-Sal, M., and Alemany, R. (2019). Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J. Immunother. Cancer 7 (1), 19. doi:10.1186/s40425-019-0505-4
Eikenes, L., Tari, M., Tufto, I., Bruland, O. S., and de Lange Davies, C. (2005). Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br. J. Cancer 93 (1), 81–88. doi:10.1038/sj.bjc.6602626
Fizazi, K., Foulon, S., Carles, J., Roubaud, G., McDermott, R., Fléchon, A., et al. (2022). Abiraterone plus prednisone added to androgen deprivation therapy and docetaxel in de novo metastatic castration-sensitive prostate cancer (PEACE-1): a multicentre, open-label, randomised, phase 3 study with a 2 × 2 factorial design. Lancet 399 (10336), 1695–1707. doi:10.1016/S0140-6736(22)00367-1
Fizazi, K., Piulats, J. M., Reaume, M. N., Ostler, P., McDermott, R., Gingerich, J. R., et al. (2023). Rucaparib or physician's choice in metastatic prostate cancer. N. Engl. J. Med. 388 (8), 719–732. doi:10.1056/NEJMoa2214676
Fizazi, K., Tran, N., Fein, L., Matsubara, N., Rodriguez-Antolin, A., Alekseev, B. Y., et al. (2017). Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 377 (4), 352–360. doi:10.1056/NEJMoa1704174
Friedrich, M., Raum, T., Lutterbuese, R., Voelkel, M., Deegen, P., Rau, D., et al. (2012). Regression of human prostate cancer xenografts in mice by AMG 212/bay2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol. CANCER Ther. 11 (12), 2664–2673. doi:10.1158/1535-7163.MCT-12-0042
Gannon, P. O., Poisson, A. O., Delvoye, N., Lapointe, R., Mes-Masson, A. M., and Saad, F. (2009). Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 348 (1-2), 9–17. doi:10.1016/j.jim.2009.06.004
Ge, R., Wang, Z., and Cheng, L. (2022). Tumor microenvironment heterogeneity an important mediator of prostate cancer progression and therapeutic resistance. npj Precis. Oncol. 6 (1), 31. doi:10.1038/s41698-022-00272-w
Geiger, M., Stubenrauch, K. G., Sam, J., Richter, W. F., Jordan, G., Eckmann, J., et al. (2020). Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat. Commun. 11 (1), 3196. doi:10.1038/s41467-020-16838-w
Hackenbruch, C., Heitmann, J. S., Walz, J. S., Federmann, B., Pflügler, M., Hadaschik, B. A., et al. (2023). ProSperA: phase I study to evaluate safety, tolerability and preliminary efficacy of a bispecific PSMAxCD3 antibody in men with biochemical recurrence of prostate cancer. J. Clin. Oncol. 41 (16_Suppl. l), TPS5114–TPS. doi:10.1200/jco.2023.41.16_suppl.tps5114
Hansen, A. R., Massard, C., Ott, P. A., Haas, N. B., Lopez, J. S., Ejadi, S., et al. (2018). Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann. Oncol. 29 (8), 1807–1813. doi:10.1093/annonc/mdy232
Heitmann, J. S., Walz, J. S., Pflügler, M., Marconato, M., Tegeler, C. M., Reusch, J., et al. (2022). Abstract CT141: CC-1, a bispecific PSMAxCD3 antibody for treatment of prostate carcinoma: results of the ongoing phase I dose escalation trial. Cancer Res. 82 (12), CT141. doi:10.1158/1538-7445.am2022-ct141
Hernandez-Hoyos, G., Sewell, T., Bader, R., Bannink, J., Chenault, R. A., Daugherty, M., et al. (2016). MOR209/ES414, a novel bispecific antibody targeting PSMA for the treatment of metastatic castration-resistant prostate cancer. Mol. Cancer Ther. 15 (9), 2155–2165. doi:10.1158/1535-7163.MCT-15-0242
Herrmann, M., Krupka, C., Deiser, K., Brauchle, B., Marcinek, A., Ogrinc Wagner, A., et al. (2018). Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 132 (23), 2484–2494. doi:10.1182/blood-2018-05-849802
Hummel, H.-D., Kufer, P., Grüllich, C., Seggewiss-Bernhardt, R., Deschler-Baier, B., Chatterjee, M., et al. (2021). Pasotuxizumab, a BiTE® immune therapy for castration-resistant prostate cancer: phase I, dose-escalation study findings. Immunotherapy 13 (2), 125–141. doi:10.2217/imt-2020-0256
Hussain, M., Mateo, J., Fizazi, K., Saad, F., Shore, N., Sandhu, S., et al. (2020). Survival with olaparib in metastatic castration-resistant prostate cancer. N. Engl. J. Med. 383 (24), 2345–2357. doi:10.1056/NEJMoa2022485
Hweixian, L. P., Kelly, H., Nathaniel, T., Bin, W., Christian, B., Christian, W., et al. (2023). Characterization and root cause analysis of immunogenicity to pasotuxizumab (AMG 212), a prostate-specific membrane antigen-targeting bispecific T-cell engager therapy. Front. Immunol. 14, 1261070. doi:10.3389/fimmu.2023.1261070
James, N. D., Sydes, M. R., Clarke, N. W., Mason, M. D., Dearnaley, D. P., Spears, M. R., et al. (2016). Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 387 (10024), 1163–1177. doi:10.1016/S0140-6736(15)01037-5
Jarvi, N. L., and Balu-Iyer, S. V. (2021). Immunogenicity challenges associated with subcutaneous delivery of therapeutic proteins. BioDrugs 35 (2), 125–146. doi:10.1007/s40259-020-00465-4
Jawa, V., Terry, F., Gokemeijer, J., Mitra-Kaushik, S., Roberts, B. J., Tourdot, S., et al. (2020). T-cell dependent immunogenicity of protein therapeutics pre-clinical assessment and mitigation–updated consensus and review 2020. Front. Immunol. 11, 1301. doi:10.3389/fimmu.2020.01301
Jiang, X., Wang, J., Deng, X., Xiong, F., Ge, J., Xiang, B., et al. (2019). Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 18 (1), 10. doi:10.1186/s12943-018-0928-4
Jiao, S., Subudhi, S. K., Aparicio, A., Ge, Z., Guan, B., Miura, Y., et al. (2019). Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell 179 (5), 1177–1190. doi:10.1016/j.cell.2019.10.029
Johnson, S., Burke, S., Huang, L., Gorlatov, S., Li, H., Wang, W., et al. (2010). Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J. Mol. Biol. 399 (3), 436–449. doi:10.1016/j.jmb.2010.04.001
Kantarjian, H., Stein, A., Gokbuget, N., Fielding, A. K., Schuh, A. C., Ribera, J. M., et al. (2017). Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 376 (9), 836–847. doi:10.1056/NEJMoa1609783
Kantoff, P. W., Higano, C. S., Shore, N. D., Berger, E. R., Small, E. J., Penson, D. F., et al. (2010). Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 363 (5), 411–422. doi:10.1056/NEJMoa1001294
Kauer, J., Horner, S., Osburg, L., Muller, S., Marklin, M., Heitmann, J. S., et al. (2020). Tocilizumab, but not dexamethasone, prevents CRS without affecting antitumor activity of bispecific antibodies. J. Immunother. Cancer 8 (1), e000621. doi:10.1136/jitc-2020-000621
Kazandjian, D., Suzman, D. L., Blumenthal, G., Mushti, S., He, K., Libeg, M., et al. (2016). FDA approval summary: nivolumab for the treatment of metastatic non-small cell lung cancer with progression on or after platinum-based chemotherapy. Oncologist 21 (5), 634–642. doi:10.1634/theoncologist.2015-0507
Kebenko, M., Goebeler, M. E., Wolf, M., Hasenburg, A., Seggewiss-Bernhardt, R., Ritter, B., et al. (2018). A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology 7 (8), e1450710. doi:10.1080/2162402X.2018.1450710
Kelly, W. K., Danila, D. C., Lin, C. C., Lee, J. L., Matsubara, N., Ward, P. J., et al. (2023). Xaluritamig, a STEAP1 x CD3 XmAb 2+1 immune therapy for metastatic castration-resistant prostate cancer: results from dose exploration in a first-in-human study. Cancer Discov. OF1-OF14. doi:10.1158/2159-8290.CD-23-0964
Kelly, W. K., Thanigaimani, P., Sun, F., Seebach, F. A., Lowy, I., Sandigursky, S., et al. (2022). A phase 1/2 study of REGN4336, a PSMAxCD3 bispecific antibody, alone and in combination with cemiplimab in patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 40 (16_Suppl. l), TPS5105–TPS. doi:10.1200/jco.2022.40.16_suppl.tps5105
Kipriyanov, S. M., Moldenhauer, G., Schuhmacher, J., Cochlovius, B., Von der Lieth, C. W., Matys, E. R., et al. (1999). Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 293 (1), 41–56. doi:10.1006/jmbi.1999.3156
Klein, C., Schaefer, W., Regula, J. T., Dumontet, C., Brinkmann, U., Bacac, M., et al. (2019). Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. 154, 21–31. doi:10.1016/j.ymeth.2018.11.008
Krueger, T. E., Thorek, D. L. J., Meeker, A. K., Isaacs, J. T., and Brennen, W. N. (2019). Tumor-infiltrating mesenchymal stem cells: drivers of the immunosuppressive tumor microenvironment in prostate cancer? Prostate 79 (3), 320–330. doi:10.1002/pros.23738
Kwon, E. D., Drake, C. G., Scher, H. I., Fizazi, K., Bossi, A., van den Eertwegh, A. J. M., et al. (2014). Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 15 (7), 700–712. doi:10.1016/S1470-2045(14)70189-5
Kyriakopoulos, C. E., Chen, Y. H., Carducci, M. A., Liu, G., Jarrard, D. F., Hahn, N. M., et al. (2018). Chemohormonal therapy in metastatic hormone-sensitive prostate cancer: long-term survival analysis of the randomized phase III E3805 CHAARTED trial. J. Clin. Oncol. 36 (11), 1080–1087. doi:10.1200/JCO.2017.75.3657
Lee, S. C., Ma, J. S. Y., Kim, M. S., Laborda, E., Choi, S. H., Hampton, E. N., et al. (2021). A PSMA-targeted bispecific antibody for prostate cancer driven by a small-molecule targeting ligand. Sci. Adv. 7 (33), eabi8193. doi:10.1126/sciadv.abi8193
Lim, E. A., Schweizer, M. T., Chi, K. N., Aggarwal, R. R., Agarwal, N., Gulley, J. L., et al. (2022). Safety and preliminary clinical activity of JNJ-63898081 (JNJ-081), a PSMA and CD3 bispecific antibody, for the treatment of metastatic castrate-resistant prostate cancer (mCRPC). J. Clin. Oncol. 40 (6_Suppl. l), 279. doi:10.1200/jco.2022.40.6_suppl.279
Linke, R., Klein, A., and Seimetz, D. (2010). Catumaxomab: clinical development and future directions. MAbs 2 (2), 129–136. doi:10.4161/mabs.2.2.11221
Lutterbuese, R., Friedrich, M., Kischel, R., Rau, D., Hoffmann, P., Ebert, E., et al. (2011). Abstract 4561: preclinical characterization of MT112/BAY 2010112, a novel PSMA/CD3-bispecific BiTE antibody for the treatment of prostate cancer. Cancer Res. 71 (8), 4561. doi:10.1158/1538-7445.am2011-4561
Markowski, M. C., Kilari, D., Eisenberger, M. A., McKay, R. R., Dreicer, R., Trikha, M., et al. (2021). Phase I study of CCW702, a bispecific small molecule-antibody conjugate targeting PSMA and CD3 in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 39 (15_Suppl. l), TPS5094–TPS. doi:10.1200/jco.2021.39.15_suppl.tps5094
Maruta, M., Ochi, T., Tanimoto, K., Asai, H., Saitou, T., Fujiwara, H., et al. (2019). Direct comparison of target-reactivity and cross-reactivity induced by CAR- and BiTE-redirected T cells for the development of antibody-based T-cell therapy. Sci. Rep. 9 (1), 13293. doi:10.1038/s41598-019-49834-2
Mehra, N., Robbrecht, D., Voortman, J., Parren, P. W., Macia, S., Veeneman, J., et al. (2023). Early dose escalation of LAVA-1207, a novel bispecific gamma-delta T-cell engager (Gammabody), in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 41 (6_Suppl. l), 153. doi:10.1200/jco.2023.41.6_suppl.153
Middelburg, J., Kemper, K., Engelberts, P., Labrijn, A. F., Schuurman, J., and van Hall, T. (2021). Overcoming challenges for CD3-bispecific antibody therapy in solid tumors. Cancers (Basel) 13 (2), 287. doi:10.3390/cancers13020287
Morris, E. C., Neelapu, S. S., Giavridis, T., and Sadelain, M. (2022). Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 22 (2), 85–96. doi:10.1038/s41577-021-00547-6
Nathan, P., Hassel, J. C., Rutkowski, P., Baurain, J. F., Butler, M. O., Schlaak, M., et al. (2021). Overall survival benefit with tebentafusp in metastatic uveal melanoma. N. Engl. J. Med. 385 (13), 1196–1206. doi:10.1056/NEJMoa2103485
Nolan-Stevaux, O., Li, C., Liang, L., Zhan, J., Estrada, J., Osgood, T., et al. (2023). AMG 509 (xaluritamig), an anti-STEAP1 XmAb 2+1 T-cell redirecting immune therapy with avidity-dependent activity against prostate cancer. Cancer Discov. 14, 90–103. doi:10.1158/2159-8290.CD-23-0984
Panchal, A., Seto, P., Wall, R., Hillier, B. J., Zhu, Y., Krakow, J., et al. (2020). COBRA™: a highly potent conditionally active T cell engager engineered for the treatment of solid tumors. MAbs 12 (1), 1792130. doi:10.1080/19420862.2020.1792130
Parker, C., Nilsson, S., Heinrich, D., Helle, S. I., O'Sullivan, J. M., Fosså, S. D., et al. (2013). Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 369 (3), 213–223. doi:10.1056/NEJMoa1213755
Petrylak, D. P., Tangen, C. M., Hussain, M. H. A., Lara, P. N., Jones, J. A., Taplin, M. E., et al. (2004). Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 351 (15), 1513–1520. doi:10.1056/NEJMoa041318
Pham, E., Lutterbuese, P., Deegen, P., Mariano, N., Matthes, K., Wahl, J., et al. (2023). Abstract ND06: AMG 305, a dual targeting BiTE®molecule with selective activity for solid tumors that co-express CDH3 and MSLN. Cancer Res. 83 (7_Suppl. ment), ND06–ND. doi:10.1158/1538-7445.am2023-nd06
Pu, Y., Xu, M., Liang, Y., Yang, K., Guo, Y., Yang, X., et al. (2016). Androgen receptor antagonists compromise T cell response against prostate cancer leading to early tumor relapse. Sci. Transl. Med. 8 (333), 333ra47–ra47. doi:10.1126/scitranslmed.aad5659
Qin, C., Wang, J., Du, Y., and Xu, T. (2022). Immunosuppressive environment in response to androgen deprivation treatment in prostate cancer. Front. Endocrinol. 13, 1055826. doi:10.3389/fendo.2022.1055826
Riethmüller, G. (2012). Symmetry breaking: bispecific antibodies, the beginnings, and 50 years on. Cancer Immun. 12, 12.
Robert, C. (2020). A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 11 (1), 3801. doi:10.1038/s41467-020-17670-y
Robinson, D., Van Allen, E. M., Wu, Y. M., Schultz, N., Lonigro, R. J., Mosquera, J. M., et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 162 (2), 454. doi:10.1016/j.cell.2015.06.053
Ross, S. L., Sherman, M., McElroy, P. L., Lofgren, J. A., Moody, G., Baeuerle, P. A., et al. (2017). Bispecific T cell engager (BiTE®) antibody constructs can mediate bystander tumor cell killing. PLoS One 12 (8), e0183390. doi:10.1371/journal.pone.0183390
Rothe, A., Sasse, S., Topp, M. S., Eichenauer, D. A., Hummel, H., Reiners, K. S., et al. (2015). A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 125 (26), 4024–4031. doi:10.1182/blood-2014-12-614636
Sartor, O., de Bono, J., Chi, K. N., Fizazi, K., Herrmann, K., Rahbar, K., et al. (2021). Lutetium-177–PSMA-617 for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 385 (12), 1091–1103. doi:10.1056/NEJMoa2107322
Schaefer, W., Regula, J. T., Bähner, M., Schanzer, J., Croasdale, R., Dürr, H., et al. (2011). Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. U. S. A. 108 (27), 11187–11192. doi:10.1073/pnas.1019002108
SEER Explorer (2023) An interactive website for SEER cancer statistics: surveillance Research Program. National Cancer Institute. [updated 16/11/2023. SEER Incidence Data, November 2022 Submission (1975-2020), SEER 22 registries (excluding Illinois and Massachusetts) Available at: https://seer.cancer.gov/statistics-network/explorer/.
Shang, Z., Niu, Y., Cai, Q., Chen, J., Tian, J., Yeh, S., et al. (2014). Human kallikrein 2 (KLK2) promotes prostate cancer cell growth via function as a modulator to promote the ARA70-enhanced androgen receptor transactivation. Tumor Biol. 35, 1881–1890. doi:10.1007/s13277-013-1253-6
Sharma, P., Pachynski, R. K., Narayan, V., Flechon, A., Gravis, G., Galsky, M. D., et al. (2019). Initial results from a phase II study of nivolumab (NIVO) plus ipilimumab (IPI) for the treatment of metastatic castration-resistant prostate cancer (mCRPC; CheckMate 650). J. Clin. Oncol. 37 (7_Suppl. l), 142. doi:10.1200/jco.2019.37.7_suppl.142
Shimabukuro-Vornhagen, A., Gödel, P., Subklewe, M., Stemmler, H. J., Schlößer, H. A., Schlaak, M., et al. (2018). Cytokine release syndrome. J. Immunother. Cancer 6 (1), 56. doi:10.1186/s40425-018-0343-9
Siegler, E. L., and Kenderian, S. S. (2020). Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front. Immunol. 11, 1973. doi:10.3389/fimmu.2020.01973
Silver, D. A., Pellicer, I., Fair, W. R., Heston, W. D., and Cordon-Cardo, C. (1997). Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 3 (1), 81–85.
Simão, D. C., Zarrabi, K. K., Mendes, J. L., Luz, R., Garcia, J. A., Kelly, W. K., et al. (2023). Bispecific T-cell engagers therapies in solid tumors: focusing on prostate cancer. Cancers 15 (5), 1412. doi:10.3390/cancers15051412
Smith, M. R., Hussain, M., Saad, F., Fizazi, K., Sternberg, C. N., Crawford, E. D., et al. (2022). Darolutamide and survival in metastatic, hormone-sensitive prostate cancer. N. Engl. J. Med. 386 (12), 1132–1142. doi:10.1056/NEJMoa2119115
Subudhi, S. K., Siddiqui, B. A., Maly, J. J., Nandagopal, L., Lam, E. T., Whang, Y. E., et al. (2021). Safety and efficacy of AMG 160, a half-life extended BiTE immune therapy targeting prostate-specific membrane antigen (PSMA), and other therapies for metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 39 (15), TPS5088. doi:10.1200/jco.2021.39.15_suppl.tps5088
Sun, B. L. (2021). Immunotherapy in treatment of metastatic prostate cancer: an approach to circumvent immunosuppressive tumor microenvironment. Prostate 81 (15), 1125–1134. doi:10.1002/pros.24213
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Suurs, F. V., Lub-de Hooge, M. N., de Vries, E. G. E., and de Groot, D. J. A. (2019). A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 201, 103–119. doi:10.1016/j.pharmthera.2019.04.006
Tabernero, J., Melero, I., Ros, W., Argiles, G., Marabelle, A., Rodriguez-Ruiz, M. E., et al. (2017). Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 35 (15_Suppl. l), 3002. doi:10.1200/jco.2017.35.15_suppl.3002
Tosolini, M., Pont, F., Poupot, M., Vergez, F., Nicolau-Travers, M. L., Vermijlen, D., et al. (2017). Assessment of tumor-infiltrating TCRVγ9Vδ2 γδ lymphocyte abundance by deconvolution of human cancers microarrays. Oncoimmunology 6 (3), e1284723. doi:10.1080/2162402X.2017.1284723
Trenevska, I., Li, D., and Banham, A. H. (2017). Therapeutic antibodies against intracellular tumor antigens. Front. Immunol. 8, 1001. doi:10.3389/fimmu.2017.01001
Vallera, D. A., Felices, M., McElmurry, R., McCullar, V., Zhou, X., Schmohl, J. U., et al. (2016). IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin. Cancer Res. 22 (14), 3440–3450. doi:10.1158/1078-0432.CCR-15-2710
van Brummelen, E. M., Ros, W., Wolbink, G., Beijnen, J. H., and Schellens, J. H. (2016). Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist 21 (10), 1260–1268. doi:10.1634/theoncologist.2016-0061
Wolf, E., Hofmeister, R., Kufer, P., Schlereth, B., and Baeuerle, P. A. (2005). BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov. Today 10 (18), 1237–1244. doi:10.1016/S1359-6446(05)03554-3
Xu, M., Evans, L., Bizzaro, C. L., Quaglia, F., Verrillo, C. E., Li, L., et al. (2022). STEAP1-4 (Six-Transmembrane epithelial antigen of the prostate 1-4) and their clinical implications for prostate cancer. Cancers (Basel) 14 (16), 4034. doi:10.3390/cancers14164034
Xu, Y., Lee, J., Tran, C., Heibeck, T. H., Wang, W. D., Yang, J., et al. (2015). Production of bispecific antibodies in "knobs-into-holes" using a cell-free expression system. MAbs 7 (1), 231–242. doi:10.4161/19420862.2015.989013
Zekri, L., Vogt, F., Osburg, L., Müller, S., Kauer, J., Manz, T., et al. (2021). An IgG-based bispecific antibody for improved dual targeting in PSMA-positive cancer. EMBO Mol. Med. 13 (2), e11902. doi:10.15252/emmm.201911902
Zhou, S., Liu, M., Ren, F., Meng, X., and Yu, J. (2021). The landscape of bispecific T cell engager in cancer treatment. Biomark. Res. 9 (1), 38. doi:10.1186/s40364-021-00294-9
Keywords: prostate cancer, metastatic castrate resistant prostate cancer, novel immunotherapies, bi-specific T-cell engager therapy, T-cell engager therapy, bi-specific antibody therapy, BiTE
Citation: Lampe H, Tam L and Hansen AR (2024) Bi-specific T-cell engagers (BiTEs) in prostate cancer and strategies to enhance development: hope for a BiTE-r future. Front. Pharmacol. 15:1399802. doi: 10.3389/fphar.2024.1399802
Received: 12 March 2024; Accepted: 13 May 2024;
Published: 30 May 2024.
Edited by:
Huan Yang, Huazhong University of Science and Technology, ChinaReviewed by:
Jagpreet Singh Nanda, Cedars Sinai Medical Center, United StatesCopyright © 2024 Lampe, Tam and Hansen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron R. Hansen, YWFyb24uci5oYW5zZW5AaGVhbHRoLnFsZC5nb3YuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.