
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 27 February 2024
Sec. Neuropharmacology
Volume 15 - 2024 | https://doi.org/10.3389/fphar.2024.1363212
 Qing-Wei Ma1†Rui-Ting Han1†Zi-Jie Wu1Jun-Jie Zhou1Meng-Ting Chen1Xiang-Zhi Zhang1
Qing-Wei Ma1†Rui-Ting Han1†Zi-Jie Wu1Jun-Jie Zhou1Meng-Ting Chen1Xiang-Zhi Zhang1 Wen-Zhe Ma2
Wen-Zhe Ma2 Na Feng1*
Na Feng1*Both continuous oxidative stress and poly (ADP-ribose) polymerase 1 (PARP-1) activation occur in neurodegenerative diseases such as Parkinson’s disease. PARP-1 inhibition can reverse mitochondrial damage and has a neuroprotective effect. In a previous study, we synthesized melatonin derivative 6a (MD6a) and reported that it has excellent antioxidant activity and significantly reduces α-synuclein aggregation in Caenorhabditis elegans; however, the underlying mechanism is largely unknown. In the present study, we revealed that MD6a is a potential PARP-1 inhibitor, leading to mammalian targe of rapamycin/heat shock factor 1 signaling downregulation and reducing heat shock protein 4 and 6 expression, thus helping to maintain protein homeostasis and improve mitochondrial function. Together, these findings suggest that MD6a might be a viable candidate for the prevention and treatment of Parkinson’s disease.
Parkinson’s disease (PD) is a common neurological degenerative disease in middle-aged and older adults; most patients develop the disease after the age of 60. Its clinical manifestations are motor dysfunction, reduced cognitive function, and depression (Jankovic, 2007), which can seriously affect patients’ quality of life. In recent years, mitochondrial dysfunction has been considered as a crucial defect occurring in the early PD pathogenesis for the loss of dopaminergic neurons (Subramaniam and Chesselet, 2013), which is closely related to aging (Chandra et al., 2017), environmental exposure (Huang et al., 2022), and genetic factors (Ye et al., 2023). Mitochondrial dysfunction and damage—including mitochondrial DNA mutations, mitochondrial electron transport chain (ETC) dysfunction, and mitochondrial reactive oxygen species (ROS) production increase—are widespread in patients with PD. This dysfunction leads to reduced energy production in the mitochondria and increased oxidative stress, which can subsequently cause cell damage and neuronal degeneration (Prasuhn et al., 2020). Studies have shown that more than 90% of sporadic PD cases are caused by mitochondrial dysfunction, which compromise the power source of nerve cells. This aggravates the accumulation of damaged mitochondria, which fails to produce enough energy for the cell, and causes the gradual death of neurons, and eventually leading to the development of PD (Magalhaes et al., 2021).
Poly (ADP-ribose) polymerase 1 (PARP-1) is a DNA repair enzyme that is mainly found in the nuclei of eukaryotes (Bai, 2015; Ray Chaudhuri and Nussenzweig, 2017). Under normal physiological conditions, PARP-1 is as a sensor of unligated Okazaki fragments during DNA replication and assists their repair (Hanzlikova et al., 2018). In pathological states, a large amount of DNA is damaged and PARP-1 becomes overactivated. This inhibits mitochondrial ETC activity, thus resulting in mitochondrial energy metabolism disorders, chromatin agglutination, and eventually programmed cell death activated by PARP-1 (known as parthanatos) (Wang et al., 2009; Wang et al., 2011; Wang et al., 2016). Continuous oxidative stress and PARP-1 activation are present in neurodegenerative diseases such as PD, and PARP-1-mediated parthanatos is one of the main forms of neuronal death in this disease (Martire et al., 2015). Inhibiting PARP-1 activation can reverse mitochondrial damage and reduce genetic defects of mitochondrial metabolism, which has a neuroprotective effect (Sun et al., 2021).
Melatonin is a natural antioxidant in the human body. A relatively high concentration of melatonin is maintained in neuronal mitochondria (Reiter et al., 2017); this mitochondria-targeted melatonin removes free radicals generated by oxidative phosphorylation via various antioxidant pathways, protects mitochondrial complexes I and IV, and increases adenosine triphosphate (ATP) synthesis (Díaz-Casado et al., 2017). Moreover, studies have reported that melatonin has a neuroprotective effect in PD. Melatonin can reduce nitrite release from astrocytes (Gonzalez, 2020), decrease the neurotoxic effects of nitric oxide (López et al., 2017), reduce dopaminergic neuron apoptosis, and prevent neuroinflammation. It can also inhibit the formation of Lewy bodies by affecting the expression and aggregation of α-synuclein (α-syn), or just directly binding to α-syn, thus improving the dopaminergic system (Sae-Ung et al., 2011).
In a previous study, we synthesized the C7-substituted melatonin derivative 6a (MD6a) using a C/H functionalization reaction. We revealed that MD6a has good antioxidant activity and can reduce ROS levels in wild-type N2 and NL5901, with an optimal concentration of 10 μM (He et al., 2022). We utilized C. elegans (Caenorhabditis elegans) as a model studying for the PD pathology because it has several advantages, including a short life cycle, easy to maintain and eight DAergic neurons containing conserved DAergic pathway and genes with human orthologs. Moreover, its PD-like phenotypes can be easily generated and analyzed in laboratory (Brunetti et al., 2020; Caldwell et al., 2020). We found that MD6a significantly reduces α-syn aggregation in NL5901 nematodes and has neuroprotective effects against 6-hydroxydopamine (6-OHDA)-induced dopaminergic neuron damage. These results suggest that MD6a may be a potential treatment for PD. However, the mechanism and targets underlying the neuroprotective effects of MD6a in PD remain largely unknown. In this study, we demonstrated that MD6a acts as a PARP-1 inhibitor to reduce α-syn aggregation and enhance mitochondrial function in a C. elegans PD model.
The wild type worm (Bristol N2) and transgenic line of NL5901 (pkIs2386, unc-54p::alphasynuclein::YFP) were obtained from the Caenorhabditis Genetics Center (CGC). Worms were grown on the nematode growth medium (NGM) agar plate and fed with Escherichia coli OP50 at 20°C (Brenner, 1974). Synchronization of nematode culture was achieved using treatment with sodium hypochlorite and 1 M NaOH (1:1). The synchronized eggs were cultured in M9 buffer for 24 h at 20°C to hatch L1 larvae. Then, the L1 stage worms were transferred to NGM plate.
RNAi feeding experiments were performed on synchronized L1 to L4 larvae at 20°C. E. coli HT115 (DE3) containing empty vector (pL4440) or target genes strains were cultured overnight in LB medium containing 100 μg/mL ampicillin and 12.5 μg/mL tetracycline at 37°C. Then spread to NGM plates containing 25 μg/mL carbenicillin and 1 mM isopropyl 1-thio-b-β-galactopyranoside (IPTG) at 37°C overnight. The synchronized NL5901 worms were transferred to NGM plates seeded with RNAi bacteria and allowed to grow until mature. RNAi efficiency was evaluated prior to starting the experiment.
Effect of MD6a on the α-syn aggregation was evaluated using NL5901 strain. Briefly, age-synchronized worms were washed three times with M9 buffer to get rid of remaining bacteria and mounted onto 2% agarose pads. Then, worms were immobilized with 20 mM sodium azide. To monitor the α-syn aggregation, YFP protein was visualized and photographed with a fluorescence microscope (Olympus BX63). The fluorescence intensity was quantified using ImageJ.
Briefly, the collected worms added extract liquid were grinded by freeze-thawing with liquid nitrogen. Centrifugation was performed at 4°C for 10 min at 12,000 rmp and 30 µL of the supernatant was taken and then assayed with the ATP Content Assay Kit (Solarbio). And protein quantification was performed with BCA Protein Assay kit (Biosharp).
Oxygen consumption rates were measured using the Oxytherm (Hansatech, United Kingdom), a Clark-type oxygen electrode as described previously (Schulz et al., 2007). The collected nematodes were resuspended in 1 mL of M9 and transferred to the chamber. Oxygen concentration was monitored with a Clark electrode in a closed chamber for 10 min. The nematodes were subsequently collected. Protein concentration was measured using the BCA Protein Assay kit.
At least 15 worms on day 0 of adulthood were randomly selected and transferred to a glass slide containing 10 µL M9 buffer. After allowing the worms to acclimate to the liquid medium for 30 s, their movement was continuously recorded by a microscope (MZ62) for 10 s.
To determine mitochondrial ROS levels, nematodes were stained with MitoSOX™ Red for 20 min. Then, the worms were anaesthetised with 20 mM sodium azide solution and mounted on microscopic slides. The fluorescence was examined under a fluorescence microscope (Olympus BX63). The Fluorescence intensity was quantified using ImageJ.
Nematodes were cultured to the L3 stage placed into 100 nM TMRE dishes stained for 24 h, and worms were transferred to plates without dye for 1 h prior to imaging to clear the gut of residual dye (Berry et al., 2023). Nematodes were mounted on 2% agarose pads under 20 mM sodium azide anesthesia. Changes in mitochondrial membrane potential fluorescence intensity were observed using a confocal (Leica TCS SP8). Fluorescence intensity was quantified using ImageJ.
Nematodes were treated with MD6a to incubate the L4 stage, and total RNA was extracted from nematodes of each treatment group using TRIzol reagent (Invitrogen), as previously described (Lapierre et al., 2013). The RNA of worms was reverse transcribed into cDNA by using PrimeScript™ RT reagent Kit with gDNA Eraser. Real-time PCR was performed according to the primers designed in Table 1.
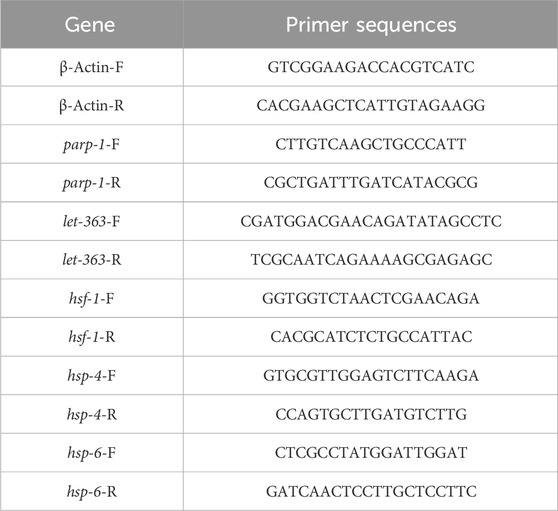
TABLE 1. Primers used for qPCR.
The accumulation and aggregation of α-syn is known as one of the pathological features in PD patients. Previous studies have demonstrated that pathological α-syn can activate PARP-1, thus leading to the loss of dopaminergic neurons in PD (Kam et al., 2018). We used the C. elegans strain NL590, which expresses human α-syn fused with YFP under an unc-54 promoter, to elucidate the effects of MD6a on α-syn aggregation. The parp-1 mRNA levels in wild-type N2 and NL5901 worms were examined by qPCR. The result showed parp-1 expression in NL5901 nematodes was significantly higher than that in N2 worms, treatment with 10 μM MD6a significantly reduced parp-1 mRNA levels in NL5901 nematodes (Figure 1A). To confirm the role of parp-1 in the neuroprotective effects of MD6a, we genetically knocked down parp-1 expression using RNAi. Reduced α-syn accumulation was observed in NL5901 worms treated with both 10 μM MD6a and parp-1 RNAi (Figures 1B,C). However, under parp-1 RNAi, 10 μM MD6a failed to further reduce α-syn aggregation in NL5901 nematodes compared with parp-1 RNAi group (Figures 1B,C). Together, these results indicate that the MD6a-induced reduction of α-syn aggregation in C. elegans is dependent on parp-1.
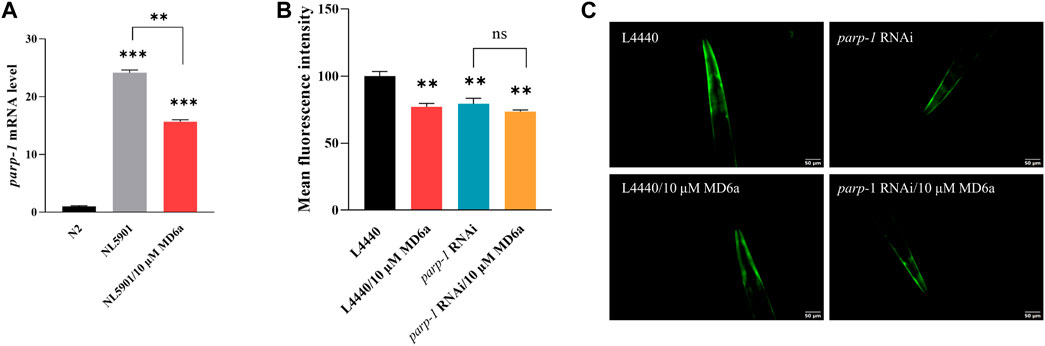
FIGURE 1. The MD6a-mediated reduction of α-syn in Caenorhabditis elegans is dependent on parp-1. (A) qPCR measurement of parp-1 expression in N2, NL5901 and 10 µM MD6a-treated NL5901 worms. (B, C) Quantification (B) and fluorescence images (C) of α-syn levels in NL5901 worms treated with 10 μM MD6a and parp-1 RNAi. Differences were analyzed using the t-test; *p < 0.05, **p < 0.01, NS (no significance), compared with the control group.
Mitochondrial dysfunction is widely considered as a main contributor to neurodegeneration in PD (Park et al., 2018). The complex interplay between mitochondrial dynamics and bioenergetics is especially important for neuronal function. In general, neurons have high energy demands that require many functional mitochondria. The activation of pathological parp-1 causes mitochondrial dysfunction, leading to ATP depletion and mitochondrial membrane potential decline (Cipriani et al., 2005; Luo and Kraus, 2011). To elucidate whether MD6a acts as a PARP-1 inhibitor to contribute to protecting mitochondrial function in PD, we measured the oxygen consumption rate (OCR), ATP levels, movement ability, mitochondrial ROS production and mitochondrial membrane potential. The results demonstrated that MD6a treatment significantly affected mitochondrial biogenesis in NL5901 worms, with markedly increased OCR levels (Figure 2A), ATP production (Figure 2B), movement ability (Figure 2C) and mitochondrial membrane potential (Figures 2D,E), while observably decreased mitochondrial ROS levels (Figures 2F,G). Furthermore, parp-1 RNAi could mimic all these beneficial effects of MD6a which suggested that MD6a improve mitochondrial dysfunction through parp-1. However, MD6a could not further increased OCR, ATP, movement, mitochondrial membrane potential levels (Figures 2A–E), and decreased mitochondrial ROS levels (Figures 2F,G) under parp-1 RNAi. Taken together, these results indicate that MD6a significantly enhances mitochondrial function in the PD nematode model through parp-1.
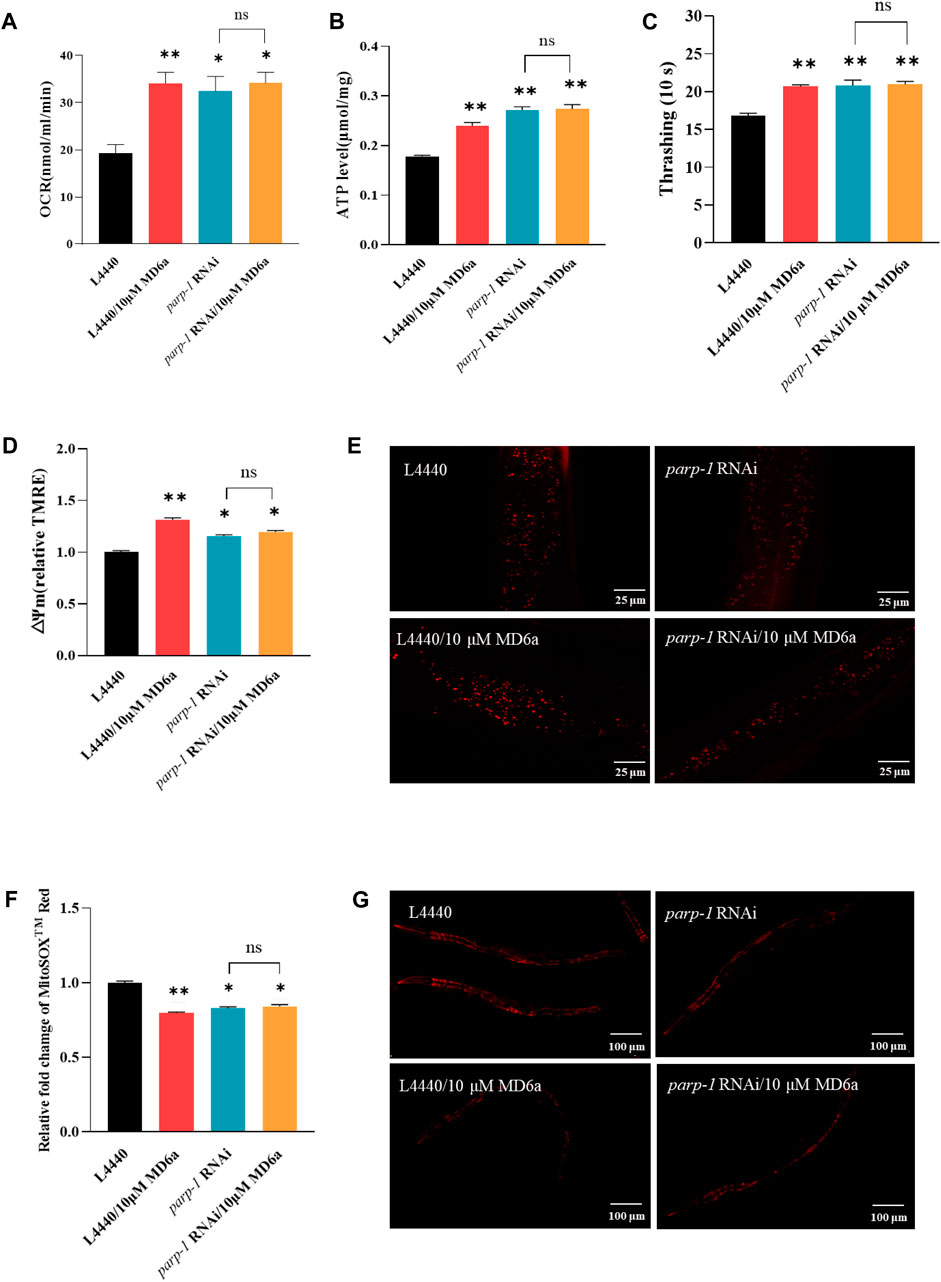
FIGURE 2. MD6a-mediated protection of mitochondrial function in Caenorhabditis elegans is dependent on parp-1. (A) Rate of oxygen consumption in NL5901 worms treated with 10 μM MD6a and parp-1 RNAi. (B) ATP levels in NL5901 worms treated with 10 μM MD6a and parp-1 RNAi. (C) Movement ability in NL5901 worms treated with 10 μM MD6a and parp-1 RNAi. (D, E) Quantification (D) and fluorescence images (E) of mitochondrial membrane potential levels in NL5901 worms treated with 10 μM MD6a and parp-1 RNAi. (F, G) Quantification (F) and fluorescence images (G) of mitochondrial ROS levels in NL5901 worms treated with 10 μM MD6a and parp-1 RNAi. Differences were analyzed using the t-test; *p < 0.05, **p < 0.01, NS (no significance), compared with the control group.
The mammalian target of rapamycin (mTOR) signaling pathway plays an important homeostatic function in the regulation of energy metabolism, cell survival, senescence, and neurodegeneration (Wullschleger et al., 2006). Moreover, increasing evidence indicates that mTOR is critical for the pathogenesis of PD. A previous study reported that mTOR protein expression levels are increased in the temporal cortex of patients with α-syn accumulation (Crews et al., 2010). We explored the effects of MD6a on the TOR signaling pathway in C. elegans, and revealed that the expression of let-363, an ortholog of TORC1, was downregulated by 10 μM MD6a (Figure 3A). Furthermore, although 10 μM MD6a did not decrease let-363 expression in parp-1 RNAi-treated worms (Figure 3A), it decreased parp-1 expression in let-363 RNAi-treated worms (Figure 3B). These results suggest that the MD6a-mediated regulation of let-363 is dependent on parp-1. Additionally, the fluorescence intensity of α-syn was reduced by let-363 RNAi treatment in NL5901 nematodes; however, 10 μM MD6a did not reduce α-syn fluorescence intensity in let-363 RNAi-treated nematodes compared with let-363 RNAi group (Figure 3D). These findings indicate that MD6a downregulates let-363 in a parp-1-dependent manner to reduce α-syn aggregation.
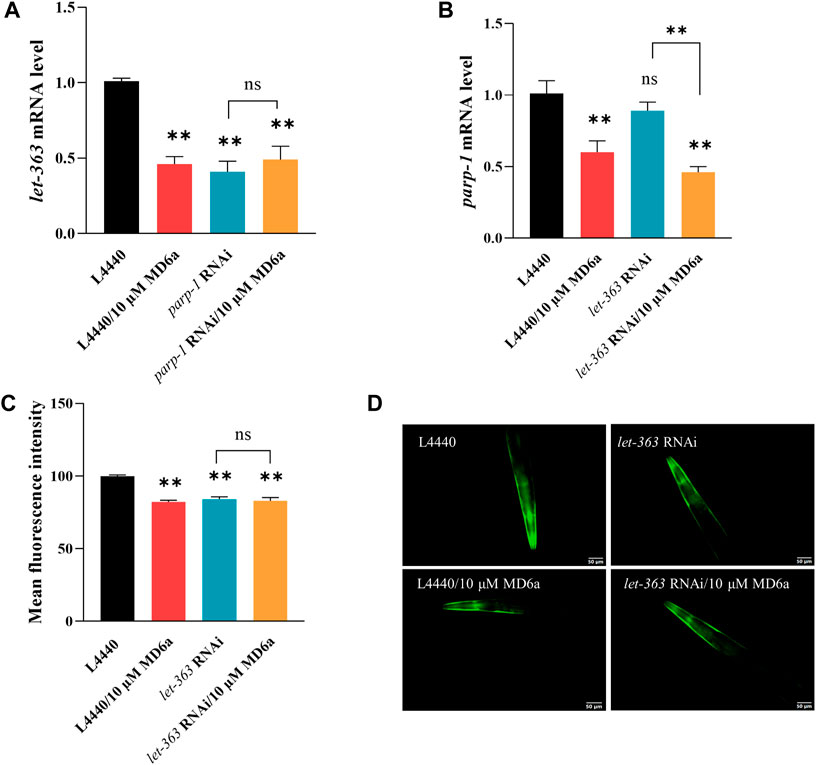
FIGURE 3. MD6a affects let-363 to exert neuroprotective effects in PD. (A) qPCR measurement of let-363 expression in NL5901 worms treated with 10 µM MD6a and parp-1 RNAi. (B) qPCR measurement of parp-1 expression in NL5901 worms treated with 10 µM MD6a and let-363 RNAi. (C, D) Quantification (C) and fluorescence images (D) of α-syn levels in NL5901 worms treated with 10 μM MD6a and let-363 RNAi. Results are presented as the mean ± SEM of three independent experiments performed in triplicate. Differences were analyzed using the t-test; *p < 0.05, **p < 0.01, NS (no significance), compared with the control group.
Previous studies have identified a number of transcription factors downstream of TOR, including DAF-16/FOXO, SKN-1/NRF, HSF-1/HSFs, PHA-4/FOXA, HLH-30/TFEB, and RPC-1/POL III (Blackwell et al., 2019). In this study, we found hsf-1 mRNA levels were downregulated by treatment with 10 μM MD6a, and parp-1 RNAi blocked this MD6a-mediated reduction (Figure 4A). Previous reports have demonstrated that α-syn aggregation is related to dysfunction of the protein degradation pathway, including of heat shock proteins (HSPs) (Jones et al., 2014). HSP-4 and HSP-6 are involved in regulating intracellular protein degradation processes by binding to damaged or obsolete proteins and removing unstable proteins via endoplasmic reticulum-associated protein degradation (ERAD) (Shin et al., 2022) and mitochondria-associated degradation pathway (Rolland et al., 2019). We thus evaluated the mRNA levels of hsp-4 and hsp-6, which are associated with HSPs in NL5901 worms. The results revealed that MD6a significantly reduced degradation-regulated genes in NL5901 nematodes (Figures 4B,C). Furthermore, parp-1 RNAi reduced the expression levels of hsp-4 and hsp-6 in NL5901 worms. However, 10 μM MD6a treatment could not further reduce the mRNA levels of hsp-4 and hsp-6 under parp-1 RNAi (Figures 4B,C). Together, these results indicate that MD6a inhibits PARP-1 downregulation of the let-363/hsf-1 signaling pathway by targeting the reduction of hsp-4 and hsp-6 mRNA levels.
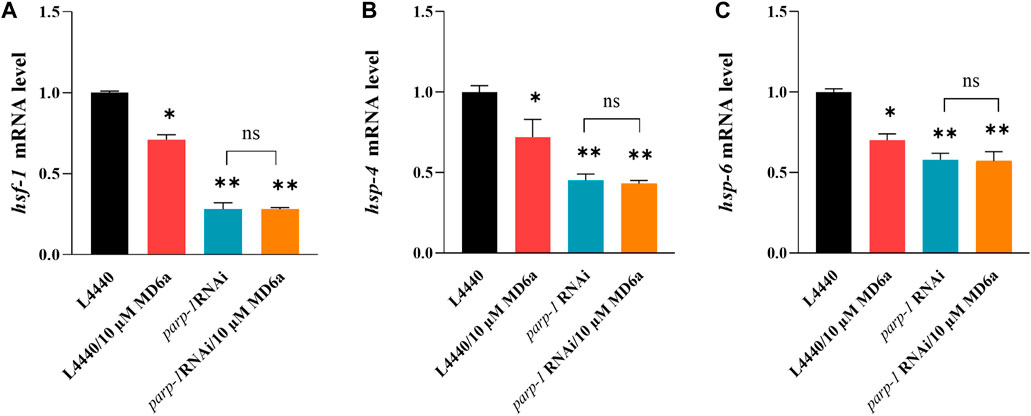
FIGURE 4. MD6a mediates hsf-1 to exert neuroprotective effects in PD. (A) qPCR measurement of hsf-1 expression in NL5901 worms treated with 10 µM MD6a and parp-1 RNAi. (B) qPCR measurement of hsp-4 expression in NL5901 worms treated with 10 µM MD6a and parp-1 RNAi. (C) qPCR measurement of hsp-6 expression in NL5901 worms treated with 10 µM MD6a and parp-1 RNAi. Results are presented as the mean ± SEM of three independent experiments performed in triplicate. Differences were analyzed using the t-test; *p < 0.05, **p < 0.01, NS (no significance), compared with the control group.
HSF-1 is a heat shock transcription factor that regulates the synthesis of heat shock proteins to assist cells cope with proteotoxic stress (Kyriakou et al., 2022). It is also a key effector of longevity signaling (Lu et al., 2020). Recent studies have reported that HSF-1 plays crucial role in the pathogenesis of PD (Govindan et al., 2018; Zheng et al., 2023). Hyperactivation of HSF-1 was associated with the onset of PD, and the elevated activity of HSF-1 and increased expression levels of heat shock proteins in patients with PD lead to increased sensitivity of neurons to proteotoxic stress, which accelerate the progression of the disease (Kim et al., 2016). Although the exact relationship between HSF-1 and PD remains to be elucidated in further studies, the current findings suggest that HSF-1 may play an important role in the pathogenesis and progression of PD by regulating the synthesis of heat shock proteins, which affect neuronal survival and function.
Mitochondrial dysfunction and protein homeostasis imbalances are two essential factors in the pathogenesis of Parkinson’s, which are closely linked (Chiti and Dobson, 2017; Malpartida et al., 2020). It has been found that misfolded proteins in the cytoplasm are recruited to mitochondria via chaperone proteins and degraded by mitochondrial proteases, which facilitate the cell maintain protein homeostasis (Li et al., 2019). However, excessive accumulation of mitochondria unfolded proteins response (UPRMT) can compromise mitochondrial integrity and accelerate the symptom of PD. In C. elegans, α-syn and PD-associated disease variants can not only induce the UPRMT, but also dysregulate the UPRMT synergistically potentiate dopaminergic neurotoxicity (Martinez et al., 2017). Various studies revealed that persistent endoplasmic reticulum (ER) stress has been linked to neurodegenerative diseases, such as PD. Anesthesia-induced neurotoxicity is related to ER stress, which is attenuated by HSP-4 downregulation (Shin et al., 2022). HSF-1 decreases the expression of HSP-4 and HSP-6, which contributes to the maintenance of ER and mitochondrial homeostasis, reduces ER stress and mitochondrial damage, and thus protects neurological function.
In the present study, we found that MD6a acts as a PARP-1 inhibitor to reduce α-syn aggregation and enhance mitochondrial function in C. elegans through TOR/HSF-1 signaling. MD6a inhibits PARP-1 to downregulate let-363/hsf-1 signaling by targeting hsp-4 and hsp-6, thus improving mitochondrial function and maintaining protein homeostasis in PD (Figure 5). Together, our findings indicate that MD6a may serve as a potential PARP-1 inhibitor for the prevention and treatment of PD.
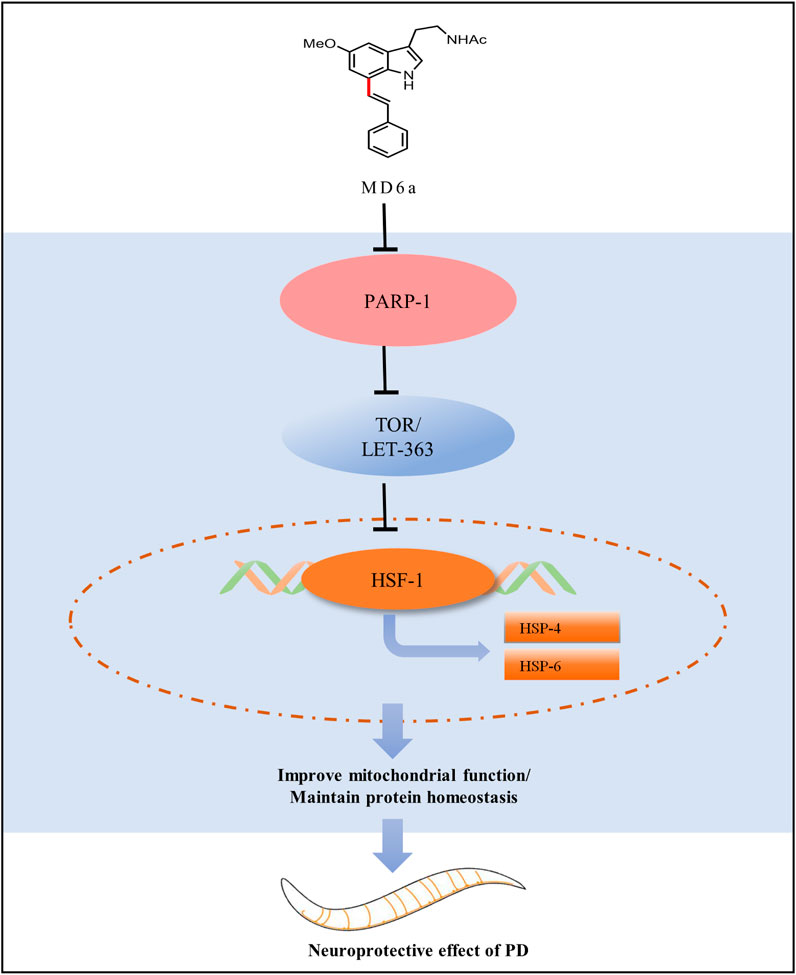
FIGURE 5. Working model for the complete pathway through which MD6a may exert a neuroprotective role in PD.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The manuscript presents research on animals that do not require ethical approval for their study.
NF: Conceptualization, Resources, Supervision, Writing–original draft, Writing–review and editing. Q-WM: Formal Analysis, Investigation, Writing–original draft. R-TH: Conceptualization, Formal Analysis, Investigation, Methodology, Writing–original draft. Z-JW: Data curation, Software, Validation, Writing–review and editing. J-JZ: Methodology, Writing–review and editing. M-TC: Methodology, Writing–review and editing. X-ZZ: Funding acquisition, Resources, Supervision, Writing–review and editing. W-ZM: Funding acquisition, Methodology, Resources, Supervision, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by NSFC (22101213) and the Hong Kong–Macao Joint Research and Development Fund of Wuyi University (2019WGALH12).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bai, P. (2015). Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol. cell 58, 947–958. doi:10.1016/j.molcel.2015.01.034
Berry, B. J., Vodičková, A., Müller-Eigner, A., Meng, C., Ludwig, C., Kaeberlein, M., et al. (2023). Optogenetic rejuvenation of mitochondrial membrane potential extends C. elegans lifespan. Nat. aging 3, 157–161. doi:10.1038/s43587-022-00340-7
Blackwell, T. K., Sewell, A. K., Wu, Z., and Han, M. (2019). TOR signaling in Caenorhabditis elegans development, metabolism, and aging. Genetics 213, 329–360. doi:10.1534/genetics.119.302504
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. doi:10.1093/genetics/77.1.71
Brunetti, G., Di Rosa, G., Scuto, M., Leri, M., Stefani, M., Schmitz-Linneweber, C., et al. (2020). Healthspan maintenance and prevention of Parkinson's-like phenotypes with hydroxytyrosol and oleuropein aglycone in C. elegans. Int. J. Mol. Sci. 21 (7), 2588. doi:10.3390/ijms21072588
Caldwell, K. A., Willicott, C. W., and Caldwell, G. A. (2020). Modeling neurodegeneration in Caenorhabditis elegans. Dis. models Mech. 13 (10), dmm046110. doi:10.1242/dmm.046110
Chandra, G., Shenoi, R. A., Anand, R., Rajamma, U., and Mohanakumar, K. P. (2017). Reinforcing mitochondrial functions in aging brain: an insight into Parkinson's disease therapeutics. J. Chem. Neuroanat. 95, 29–42. doi:10.1016/j.jchemneu.2017.12.004
Chiti, F., and Dobson, C. M. (2017). Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. doi:10.1146/annurev-biochem-061516-045115
Cipriani, G., Rapizzi, E., Vannacci, A., Rizzuto, R., Moroni, F., and Chiarugi, A. (2005). Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J. Biol. Chem. 280, 17227–17234. doi:10.1074/jbc.M414526200
Crews, L., Spencer, B., Desplats, P., Patrick, C., Paulino, A., Rockenstein, E., et al. (2010). Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PloS one 5, e9313. doi:10.1371/journal.pone.0009313
Díaz-Casado, M. E., Rusanova, I., Aranda, P., Fernández-Ortiz, M., Sayed, R. K. A., Fernández-Gil, B. I., et al. (2017). In vivo determination of mitochondrial respiration in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated zebrafish reveals the efficacy of melatonin in restoring mitochondrial normalcy. Zebrafish 15, 15–26. doi:10.1089/zeb.2017.1479
Gonzalez, A. (2020). Antioxidants and neuron-astrocyte interplay in brain physiology: melatonin, a neighbor to rely on. Neurochem. Res. 46, 34–50. doi:10.1007/s11064-020-02972-w
Govindan, S., Amirthalingam, M., Duraisamy, K., Govindhan, T., Sundararaj, N., and Palanisamy, S. (2018). Phytochemicals-induced hormesis protects Caenorhabditis elegans against α-synuclein protein aggregation and stress through modulating HSF-1 and SKN-1/Nrf2 signaling pathways. Biomed. Pharmacother. 102, 812–822. doi:10.1016/j.biopha.2018.03.128
Hanzlikova, H., Kalasova, I., Demin, A. A., Pennicott, L. E., Cihlarova, Z., and Caldecott, K. W. (2018). The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol. cell 71, 319–331. doi:10.1016/j.molcel.2018.06.004
He, L., Du, J. J., Zhou, J. J., Chen, M. T., Luo, L., Li, B. Q., et al. (2022). Synthesis of melatonin derivatives and the neuroprotective effects on Parkinson's disease models of Caenorhabditis elegans. Front. Chem. 10, 918116. doi:10.3389/fchem.2022.918116
Huang, M., Bargues-Carot, A., Riaz, Z., Wickham, H., Zenitsky, G., Jin, H., et al. (2022). Impact of environmental risk factors on mitochondrial dysfunction, neuroinflammation, protein misfolding, and oxidative stress in the etiopathogenesis of Parkinson's disease. Int. J. Mol. Sci. 23, 10808. doi:10.3390/ijms231810808
Jankovic, J. (2007). Parkinson's disease: clinical features and diagnosis. J. neurology, Neurosurg. psychiatry 79 (4), 368–376. doi:10.1136/jnnp.2007.131045
Jones, D. R., Moussaud, S., and McLean, P. (2014). Targeting heat shock proteins to modulate α-synuclein toxicity. Ther. Adv. neurological Disord. 7, 33–51. doi:10.1177/1756285613493469
Kam, T. I., Mao, X., Park, H., Chou, S. C., Karuppagounder, S. S., Umanah, G. E., et al. (2018). Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson's disease. Science 362, eaat8407. doi:10.1126/science.aat8407
Kim, E., Wang, B., Sastry, N., Masliah, E., Nelson, P. T., Cai, H., et al. (2016). NEDD4-mediated HSF1 degradation underlies α-synucleinopathy. Hum. Mol. Genet. 25 (2), 211–222. doi:10.1093/hmg/ddv445
Kyriakou, E., Taouktsi, E., and Syntichaki, P. (2022). The thermal stress coping network of the nematode Caenorhabditis elegans. Int. J. Mol. Sci. 23 (23), 14907. doi:10.3390/ijms232314907
Lapierre, L. R., De Magalhaes Filho, C. D., McQuary, P. R., Chu, C. C., Visvikis, O., Chang, J. T., et al. (2013). The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 4, 2267. doi:10.1038/ncomms3267
Li, Y., Xue, Y., Xu, X., Wang, G., Liu, Y., Wu, H., et al. (2019). A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 38 (3), e98786. doi:10.15252/embj.201798786
López, A., Ortiz, F., Doerrier, C., Venegas, C., Fernández-Ortiz, M., Aranda, P., et al. (2017). Mitochondrial impairment and melatonin protection in parkinsonian mice do not depend of inducible or neuronal nitric oxide synthases. PloS one 12, e0183090. doi:10.1371/journal.pone.0183090
Lu, M., Tan, L., Zhou, X. G., Yang, Z. L., Zhu, Q., Chen, J. N., et al. (2020). Secoisolariciresinol diglucoside delays the progression of aging-related diseases and extends the lifespan of Caenorhabditis elegans via DAF-16 and HSF-1. Oxidative Med. Cell. Longev. 2020, 1293935. doi:10.1155/2020/1293935
Luo, X., and Kraus, W. L. (2011). A one and a two expanding roles for poly(ADP-ribose) polymerases in metabolism. Cell metab. 13, 353–355. doi:10.1016/j.cmet.2011.03.011
Magalhaes, J., Tresse, E., Ejlerskov, P., Hu, E., Liu, Y., Marin, A., et al. (2021). PIAS2-mediated blockade of IFN-β signaling: a basis for sporadic Parkinson disease dementia. Mol. psychiatry 26, 6083–6099. doi:10.1038/s41380-021-01207-w
Malpartida, A. B., Williamson, M., Narendra, D. P., Wade-Martins, R., and Ryan, B. J. (2020). Mitochondrial dysfunction and mitophagy in Parkinson's disease: from mechanism to therapy. Trends Biochem. Sci. 46 (4), 329–343. doi:10.1016/j.tibs.2020.11.007
Martinez, B. A., Petersen, D. A., Gaeta, A. L., Stanley, S. P., Caldwell, G. A., and Caldwell, K. A. (2017). Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans models of Parkinson's disease. J. Neurosci. official J. Soc. Neurosci. 37 (46), 11085–11100. doi:10.1523/JNEUROSCI.1294-17.2017
Martire, S., Mosca, L., and d'Erme, M. (2015). PARP-1 involvement in neurodegeneration: a focus on Alzheimer's and Parkinson's diseases. Mech. ageing Dev. 146-148, 53–64. doi:10.1016/j.mad.2015.04.001
Park, J. S., Davis, R. L., and Sue, C. M. (2018). Mitochondrial dysfunction in Parkinson's disease: New mechanistic insights and therapeutic perspectives. Curr. neurology Neurosci. Rep. 18 (5), 21. doi:10.1007/s11910-018-0829-3
Prasuhn, J., Davis, R. L., and Kumar, K. R. (2020). Targeting mitochondrial impairment in Parkinson's disease: challenges and opportunities. Front. cell Dev. Biol. 8, 615461. doi:10.3389/fcell.2020.615461
Ray Chaudhuri, A., and Nussenzweig, A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. cell Biol. 18, 610–621. doi:10.1038/nrm.2017.53
Reiter, R. J., Rosales-Corral, S., Tan, D. X., Jou, M. J., Galano, A., and Xu, B. (2017). Melatonin as a mitochondria-targeted antioxidant: one of evolution's best ideas. Cell. Mol. life Sci. CMLS 74, 3863–3881. doi:10.1007/s00018-017-2609-7
Rolland, S. G., Schneid, S., Schwarz, M., Rackles, E., Fischer, C., Haeussler, S., et al. (2019). Compromised mitochondrial protein import acts as a signal for UPRmt. Cell Rep. 28 (7), 1659–1669. doi:10.1016/j.celrep.2019.07.049
Sae-Ung, K., Uéda, K., Govitrapong, P., and Phansuwan-Pujito, P. (2011). Melatonin reduces the expression of alpha-synuclein in the dopamine containing neuronal regions of amphetamine-treated postnatal rats. J. pineal Res. 52, 128–137. doi:10.1111/j.1600-079X.2011.00927.x
Schulz, T. J., Zarse, K., Voigt, A., Urban, N., Birringer, M., and Ristow, M. (2007). Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell metab. 6 (4), 280–293. doi:10.1016/j.cmet.2007.08.011
Shin, H. J., Koo, B. W., Yoon, J., Kim, H., Do, S. H., and Na, H. S. (2022). Melatonin reduces the endoplasmic reticulum stress and polyubiquitinated protein accumulation induced by repeated anesthesia exposure in Caenorhabditis elegans. Sci. Rep. 12 (1), 5783. doi:10.1038/s41598-022-09853-y
Subramaniam, S. R., and Chesselet, M. F. (2013). Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Prog. Neurobiol. 106-107, 17–32. doi:10.1016/j.pneurobio.2013.04.004
Sun, J., Liu, J., Gao, C., Zheng, J., Zhang, J., Ding, Y., et al. (2021). Targeted delivery of PARP inhibitors to neuronal mitochondria via biomimetic engineered nanosystems in a mouse model of traumatic brain injury. Acta biomater. 140, 573–585. doi:10.1016/j.actbio.2021.12.023
Wang, Y., An, R., Umanah, G. K., Park, H., Nambiar, K., Eacker, S. M., et al. (2016). A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354, aad6872. doi:10.1126/science.aad6872
Wang, Y., Dawson, V. L., and Dawson, T. M. (2009). Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp. Neurol. 218, 193–220 2. doi:10.1016/j.expneurol.2009.03.020
Wang, Y., Kim, N. S., Haince, J. F., Kang, H. C., David, K. K., Andrabi, S. A., et al. (2011). Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 4, ra20. doi:10.1126/scisignal.2000902
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. doi:10.1016/j.cell.2006.01.016
Ye, H., Robak, L. A., Yu, M., Cykowski, M., and Shulman, J. M. (2023). Genetics and pathogenesis of Parkinson's syndrome. Annu. Rev. pathology 18, 95–121. doi:10.1146/annurev-pathmechdis-031521-034145
Keywords: melatonin derivative, Parkinson’s disease, PARP-1, mTOR, mitochondrial dysfunction
Citation: Ma Q-W, Han R-T, Wu Z-J, Zhou J-J, Chen M-T, Zhang X-Z, Ma W-Z and Feng N (2024) Melatonin derivative 6a as a PARP-1 inhibitor for the treatment of Parkinson’s disease. Front. Pharmacol. 15:1363212. doi: 10.3389/fphar.2024.1363212
Received: 30 December 2023; Accepted: 12 February 2024;
Published: 27 February 2024.
Edited by:
Xi Zheng, The State University of New Jersey, United StatesReviewed by:
Wu Zhengrong, Lanzhou University, ChinaCopyright © 2024 Ma, Han, Wu, Zhou, Chen, Zhang, Ma and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na Feng, d3l1Y2hlbWZuQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.