
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 11 January 2024
Sec. Experimental Pharmacology and Drug Discovery
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1328950
This article is part of the Research TopicExperimental and computational aspects of bioactive proteins from animal venoms: An insight into pharmacological properties and drug discoveryView all 10 articles
 Rachel H. Clare1,2*†
Rachel H. Clare1,2*† Charlotte A. Dawson1,2†Adam Westhorpe1,2
Charlotte A. Dawson1,2†Adam Westhorpe1,2 Laura-Oana Albulescu1,2
Laura-Oana Albulescu1,2 Christopher M. Woodley3
Christopher M. Woodley3 Nada Mosallam3
Nada Mosallam3 Daniel J. W. Chong3
Daniel J. W. Chong3 Jeroen Kool4
Jeroen Kool4 Neil G. Berry3Paul M. O’Neill3
Neil G. Berry3Paul M. O’Neill3 Nicholas R. Casewell1,2*
Nicholas R. Casewell1,2*Snakebite envenoming results in ∼100,000 deaths per year, with close to four times as many victims left with life-long sequelae. Current antivenom therapies have several limitations including high cost, variable cross-snake species efficacy and a requirement for intravenous administration in a clinical setting. Next-generation snakebite therapies are being widely investigated with the aim to improve cost, efficacy, and safety. In recent years several small molecule drugs have shown considerable promise for snakebite indication, with oral bioavailability particularly promising for community delivery rapidly after a snakebite. However, only two such drugs have entered clinical development for snakebite. To offset the risk of attrition during clinical trials and to better explore the chemical space for small molecule venom toxin inhibitors, here we describe the first high throughput drug screen against snake venom metalloproteinases (SVMPs)—a pathogenic toxin family responsible for causing haemorrhage and coagulopathy. Following validation of a 384-well fluorescent enzymatic assay, we screened a repurposed drug library of 3,547 compounds against five geographically distinct and toxin variable snake venoms. Our drug screen resulted in the identification of 14 compounds with pan-species inhibitory activity. Following secondary potency testing, four SVMP inhibitors were identified with nanomolar EC50s comparable to the previously identified matrix metalloproteinase inhibitor marimastat and superior to the metal chelator dimercaprol, doubling the current global portfolio of SVMP inhibitors. Following analysis of their chemical structure and ADME properties, two hit-to-lead compounds were identified. These clear starting points for the initiation of medicinal chemistry campaigns provide the basis for the first ever designer snakebite specific small molecules.
Snakebite envenoming is responsible for perhaps as many as 138,000 deaths annually, while the extensive morbidity burden of sequalae is estimated to represent around 6 million disability adjusted life years (DALYs) (Gutiérrez et al., 2017). Snakebite predominately affects rural populations of the tropics, and consequently the World Health Organization (WHO) recently classified snakebite envenoming as a priority neglected tropical disease (NTD) (Chippaux, 2017). Snakebite can result in catastrophic healthcare expenditure, as well as subsequent loss of livelihood, due to the loss or incapacitation of the primary earner in a household (Herzel et al., 2018; Harrison et al., 2019). In addition, the psychological impact of surviving a snakebite and related socio-economic implications (e.g., social stigma, ostracization and loss of time in education) can cause substantial long-term impacts on individuals and their families (Williams et al., 2011a; Harrison et al., 2019; Kasturiratne et al., 2021; Oliveira et al., 2022).
One of the major challenges that snakebite victims face is restricted access to effective treatment, since current antivenom therapies are often unavailable in the remote rural and indigenous communities that are most vulnerable to snakebite (Gutiérrez et al., 2017; Longbottom et al., 2018; Harrison et al., 2019; Beck et al., 2022). This challenge with antivenom availability is largely attributed to the need for cold chain storage and high costs associated with provision (Williams et al., 2011b). However, despite antivenom currently being the only approved therapeutic for the treatment of snakebite, these animal-derived polyclonal antibody-based therapies have several other limitations associated with them. Antivenom can only be given in a healthcare setting due to the high risk of adverse reactions and the need for intravenous administration (Williams et al., 2011a; de Silva et al., 2016) and their mode of production, i.e., raising animal polyclonal antibodies against a specific venom or venoms, results in products that are restricted in terms of the snake species they are effective against (Harrison et al., 2017; Laxme et al., 2019; Ainsworth et al., 2020).
Snake venoms are a complex mix of several protein families, peptides, and small molecules, which have evolved over millions of years to facilitate prey capture (Tasoulis and Isbister, 2017; Tasoulis et al., 2021). Snake venoms often display compositional variation, and while this is most commonly observed between snake species, there are also several examples of extensive intraspecific and ontogenetic venom variation reported in the literature (Chippaux et al., 1991; Currier et al., 2010; Casewell et al., 2020; 2014; Amazonas et al., 2018; Laxme et al., 2019; Oliveira et al., 2019). Venom variation results in variation in toxin functionality, with associated human snakebite pathologies being equally diverse; the most prominent being neurotoxicity (neuromuscular paralysis), cytotoxicity (local swelling, blistering and tissue necrosis) and haemotoxicity (haemorrhage and coagulopathy) (Warrell, 2010; Gutiérrez et al., 2017; Slagboom et al., 2017; Longbottom et al., 2018). Of the many venom components found across medically important venomous snakes (i.e., families Viperidae and Elapidae), four toxin families are considered amongst the most important therapeutic targets due to their role contributing to these pathologies: three finger toxins (3FTx), snake venom serine proteases (SVSP), phospholipases A2 (PLA2) and snake venom metalloproteinases (SVMP) (Ferraz et al., 2019).
Snake venom metalloproteinases (SVMPs) are zinc-dependent endoproteolytic toxins that evolved from ADAM genes (Moura-da-Silva et al., 1996; Casewell et al., 2012). These toxins are found in the venoms of almost all medically important snakes, though they are most abundant in viperids, where they can account for up to 74% of total venom composition (average, 36%) (Tasoulis and Isbister, 2017; Dawson et al., 2021). SVMPs can be broadly divided into three classes based on their tertiary structure (P-I, P-II, and P-III), with each being progressively larger and more structurally complex due to the presence of additional domains (Fox and Serrano, 2008; 2005). Haemorrhage is the most pathologically serious complication resulting from SVMP activity and can occur both systemically and locally. The degradation of collagen and other key extracellular matrix proteins by SVMPs weakens the vascular endothelium, with the impact of haemodynamic forces on this weakened endothelium then enabling blood to escape from the vessels (Gutiérrez et al., 2016; Asega et al., 2020). In addition, several SVMPs also target different points in the coagulation cascade, particularly the common pathway components, fibrinogen, prothrombin and factor X (Kini and Koh, 2016; Asega et al., 2020). These key clotting factors are then consumed, either by their activation or degradation, ultimately leading to the depletion of fibrinogen and, in severe cases, venom induced consumption coagulopathy (Maduwage and Isbister, 2014; Kini and Koh, 2016).
Recent innovations in snakebite therapeutics have centred around the use of monoclonal antibodies or small molecule toxin inhibitors (i.e., drugs), with the latter focusing on their potential application against PLA2 and SVMP rich venoms (Lewin et al., 2016; Knudsen and Laustsen, 2018; Albulescu et al., 2020b; Clare et al., 2021; Hall et al., 2022; Menzies et al., 2022). So far, two groups of molecules with differing modes of action have been found to inhibit SVMP activity. The first are metal chelators that bind Zn2+; the leading candidate from this class is 2, 3-Dimercapto-1-propanesulfonic acid (DMPS: Unithiol), a drug licensed for the treatment of heavy metal poisoning, marketed under the name Dimaval®, and available in both oral and parenteral forms (Drisko, 2018). DMPS exhibits efficacy in in vivo models of snakebite envenoming against venoms rich in SVMP toxins (Albulescu et al., 2020a; Menzies et al., 2022), and recently completed Phase I dose optimisation studies for snakebite indication (Abouyannis et al., 2022). The second drug group are the matrix metalloproteinase inhibitors (MMPi); these compounds include a zinc-binding motif and directly interact with the active site of these enzymes (Gutiérrez et al., 2021). Marimastat has emerged as an exciting early candidate from this group; initially developed as an anti-cancer drug, marimastat passed into Phase III clinical trials, where ultimately development was halted due to lack of efficacy (Winer et al., 2018). Marimastat exhibits broad and potent inhibition of SVMP toxins in vitro (Xie et al., 2020; Menzies et al., 2022) and exerts promising preclinical efficacy against the local and systemic haemotoxic effects of several snake venoms in small animal models (Albulescu et al., 2020b; Hall et al., 2022; Menzies et al., 2022). Like DMPS, marimastat exhibits desirable oral bioavailability (Millar et al., 1998), which is an exciting characteristic for future snakebite therapeutics, as drugs could potentially be rapidly administered in rural communities soon after a bite. Reducing current delays to snakebite treatment is likely to substantially improve patient outcomes (Gutiérrez et al., 2017). One of the other major potential benefits of drugs like DMPS and marimastat is the opportunity to prioritise those with existing clinical data or regulatory approvals, which could result in major reductions in the time and costs associated with translating drugs to market (Ashburn and Thor, 2004).
Although both DMPS and the PLA2 inhibiting drug varespladib have moved into clinical development for snakebite indication (Abouyannis et al., 2022; Carter et al., 2022), the snakebite drug portfolio remains extremely limited, with attrition a major risk. There is a strong need to develop a broad portfolio of potential snakebite drugs in the early hit discovery stage to allow the selection of candidates with the highest chance of success, and to provide multiple back-up compounds should a primary hit series fail during hit-to-lead optimisation.
Consequently, in this study we performed a repurposed drug screen to identify novel SVMP toxin-inhibiting drugs. We first report on the validation of a high-throughput primary screen capable of assessing SVMP inhibition and processing ∼3,500 compounds per day. Next, we performed a drug discovery campaign by screening a commercial human pharmacopoeia library consisting of 3,547 post-Phase I drugs, with the aim of identifying hits for direct repurposing or as starting points for future medicinal chemistry campaigns. The results of our screening campaign, the first of its kind applied in the context of snakebite, yielded four novel compounds with promise for downstream development.
Venoms were extracted from specimens of Echis ocellatus (likely Echis romani following recent taxonomic reclassification (Trape, 2018), and awaiting molecular confirmation) (Nigeria), Crotalus atrox (United States), and Bitis arietans (Nigeria) maintained within the herpetarium at the Centre for Snakebite Research and Interventions (CSRI) at the Liverpool School of Tropical Medicine (LSTM). Crude venoms were pooled by species and lyophilised for long term storage at 2°C–8°C. Venoms from Calloselasma rhodostoma (Thailand) and Bothrops jararaca (Brazil) were sourced from the historical collection of lyophilised venoms at LSTM. Venoms were reconstituted to 10 mg/mL in sterile Phosphate Buffered Saline (PBS) (Cat no: 20012027, ThermoFisher) prior to use.
Several matrix metalloproteinase inhibitors were commercially sourced, namely, marimastat ((2S,3R)-N4-[(1S)-2,2-Dimethyl-1-[(methylamino)carbonyl]propyl]-N1,2-dihydroxy-3-(2-methylpropyl)butanediamide, ≥99%, Cat no: 2631, Tocris Bioscience), prinomastat hydrochloride (AG3340 hydrochloride, ≥98%, Cat no: HY-12170A, MedChemExpress), tanomastat (4′-chloro-γ-oxo-αS-[(henylthiol)methyl]-[1,1′-biphenyl]-4-butanoic acid, ≥98%, Cat no: 19258, Cayman chemicals), batimastat ((2R,3S)-N4-Hydroxy-N1-[(1S)-2-(methylamino)-2-oxo-1-(phenylmethyl)ethyl]-2-(2-methylpropyl)-3-[(2-thienylthio)methyl]butanediamide, ≥98%, Cat no: SML0041, Sigma) and doxycycline (6-Desoxy-5-hydroxytetracycline hydrochloride hemihydrate hemiethanolate, ≥93.5%, Cat no: D9891, Sigma). The metal chelators dimercaprol (2,3-dimercapto-1-propanol, ≥98% iodometric, Cat no: 64046, Sigma) and DMPS (2,3-dimercapto-1-propane-sulfonic acid sodium salt monohydrate, 95%, Cat no: H56578, Alfa Aesar) were also commercially sourced. Small molecules with alternative toxin targets were tested as negative drug controls; varespladib (2-[[3-(2-Amino-2-oxoacetyl)-2-ethyl-1-(phenylmethyl)-1H-indol-4-yl]oxy]-acetic acid, ≥98%, Cat no: SML1100, Sigma) and nafamostat mesylate ((6-Carbamimidoylnaphthalen-2-yl)4-(diaminomethylideneamino)benzoate methanesulfonic acid, > 97%, Cat no: ab141432, Abcam).
For the primary high-throughput screen (HTS) a bespoke repurposed drug library (a combination of the HY-L035 and HY-L026 libraries with overlaps removed) was purchased from MedChemExpress. The library was curated by MedChemExpress to contain a diverse chemical panel of 3,547 post-Phase I and approved drugs that have completed preclinical and clinical studies for a wide range of diseases, as well as having well-characterised bioactivity, safety, and bioavailability properties, making them suitable for drug repurposing (Supplementary Datasheet).
Drugs were either supplied at 10 mM or reconstituted in dimethyl sulfoxide (DMSO) (≥99.7%, Cat no: D2650, Sigma) to the same concentration and stored at −20°C. Daughter plates were created with 40 µL per well of 1 mM stocks on V-bottomed 384-well plates (Cat no: 781280, Greiner) to allow the creation of assay-ready plates using a VIAFLO 384 electronic pipette (Integra), excluding columns 1 and 24. Daughter plates and assay-ready plates were stored at −20°C, with the latter used within a month of creation. For SVMP assay-ready plates, 0.91 µL of each drug from the respective daughter plate was plated, such that in the final assay volume of 91 μL, a final concentration of 10 µM was reached. Controls were processed in a similar manner into columns 1 and 24 of each plate; DMSO, PBS, 10 µM marimastat, and 50 nM marimastat, with each condition repeated 4 times within each control column. In secondary screening, dose response curves were created, with a top final assay concentration of 10 µM and reaching sub-nanomolar concentrations (concentration range was dependent on specific plate layout), with each concentration tested in duplicate. For DMPS and dimercaprol, dose response curves started at a concentration of 160 µM to account for reduced inhibitory potency compared to other compounds, with each concentration tested in duplicate.
The SVMP activity of crude venom in the presence of inhibitors or vehicle control (DMSO) was measured using a quenched fluorogenic substrate (substrate for MMPs, ADAMs and Cathepsins, Cat no: ES010, Bio-Techne). The substrate was diluted in reaction buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5) and used at a final assay concentration of 7.5 µM (supplied as a 6.2 mM stock). Reactions consisted of 1 µg of venom in 15 µL PBS co-incubated with 0.91 µL of inhibitor at a concentration of 1 mM. The 384-well plate (Cat no: 781101, Greiner) was briefly centrifuged in a Platefuge (Benchmark Scientific) and incubated at 37°C for 25 min. Following incubation, the plate was acclimatised to room temperature for 5 min, before the addition of freshly prepared fluorogenic substrate (75 µL of 9.2 µM). The plates were read on a CLARIOstar microplate reader (BMG Labtech), at an excitation wavelength of 320–10 nm and emission wavelength of 420–10 nm with 10 flashes per well at 25°C, at a pre-defined end read time. The end-read times were selected independently for each venom, as the plateau point for the venom + vehicle control (maximum fluorescence) identified in an initial 1-h kinetic read.
The SVMP activity for the end-read data was calculated for each test condition as a percentage of the mean of the venom + vehicle wells (100% activity), with a baseline of the marimastat 10 µM controls representing 0% activity (Supplementary Equation S1). For the dose response screens, absolute half maximal effective concentration (EC50) values were calculated from the percentage inhibition values by fitting a nonlinear regression curve for the normalised response (variable slope) for each compound using GraphPad Prism 9.0 (GraphPad Software, San Diego, United States).
In addition, these controls were used to calculate the Z prime (Z′) as a quality control measure for each screening plate (Supplementary Equation S2).
The repurposed HTS library was screened for common Pan-Assay Interference compoundS (PAINS) substructures (Baell and Holloway, 2010) using the in-built PAINS filter in the RDKIT cheminformatics package version 2022.03.5 as implemented in Python (Landrum, 2006). Tree Manifold Approximation Projection (TMAP) visualisation was performed using the TMAP python module version 1.0.6. (Probst and Reymond, 2020). ADME properties of hit molecules and radar plots were obtained using the SwissADME web server (Daina et al., 2017). Compounds were hand-picked based on their predicted ADME properties and potential further optimisation.
The SVMP assay implemented here has previously been used for quantifying venom activity and toxin inhibition and is based on the adapted use of a commercial assay designed for human MMPs (Albulescu et al., 2020b; Hou et al., 2021; Menzies et al., 2022; Nguyen et al., 2022). The fluorescence-quenched peptide substrate measures the activity of peptidases such as SVMPs which cleave the amide bond between the fluorescent group and the quencher group, resulting in an increase in fluorescence. When metalloproteinase activity is inhibited, the quencher is undisturbed, resulting in only background fluorescence. To increase the throughput of this assay, we implemented the use of robotics and associated adaptation of the method described by Albulescu et al., 2020b, including reducing the substrate concentration from 10 μM to 7.5 µM to minimise the cost of completing a high-throughput screen. The 384-well assay was semi-automated by utilising a VIAFLO 384-channel pipette which allowed single-step addition of both the venom and substrate to the assay-ready drug plates. This two-step protocol, in conjunction with assay ready drug plates, could be utilised to test a panel of venoms in a single batch run. The capacity to create assay-ready plates was provided by the VIAFLO 384-channel pipette’s ability to stamp out multiple plates from manually created master drug plates, enabling creation of either single-dose primary screening or downstream dose-response plates. The finalised high-throughput-compatible method is demonstrated in Figure 1.
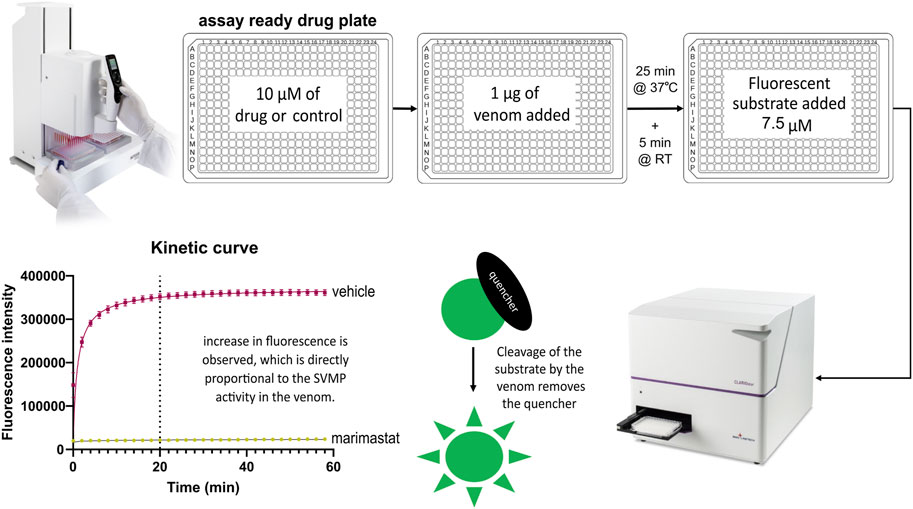
FIGURE 1. High-throughput SVMP screening workflow. This three-step screen is completed within 2 hours. Master drug plates are created with selected drugs of interest and control samples including negative vehicle controls (DMSO) and positive controls (the SVMP inhibiting MMPi marimastat). Using the VIAFLO 384 handheld electronic pipette (Integra), assay-ready drug plates are created by adding 0.91 µL of compound from a master plate. To initiate the assay, 1 µg of venom is added and the plate is incubated for 25 min at 37°C, followed by a 5 min acclimatisation to room temperature (RT). The fluorescence-quenched peptide substrate is added to all wells at a final concentration of 7.5 µM with a final assay volume of 91 µL including 10 µM of drug. Following an incubation time selected based on the plateau point previously determined by a kinetic read (e.g., 20 min for Echis ocellatus in this figure) the plate is read with triplicate endreads. Active drugs inhibit the ability of the venom to cleave the quencher off the fluorescent substrate, resulting in a low fluorescent read out, whereas inactive drugs will give a high fluorescence read due to uninhibited venom activity.
Through the adaptation to using end-point measurements rather than the original 1-h kinetic measurement, the time required to read a single plate was reduced to less than 100 s. The endpoint was selected as the plateau point of substrate cleavage, measured in triplicate. This endpoint was dependent on the SVMP activity profile of the individual venoms selected; for example, the endpoint for E. ocellatus venom was selected as 20 min post substrate addition (drug vehicle control, DMSO in Figure 2A). This provided the capacity to stagger multiple plate batches when working with large drug libraries, thus increasing the throughput capacity of the assay from a maximum of seven plates/day for a kinetic read to a maximum of 21 plates/day with this endpoint read, whilst maintaining a strong assay window.
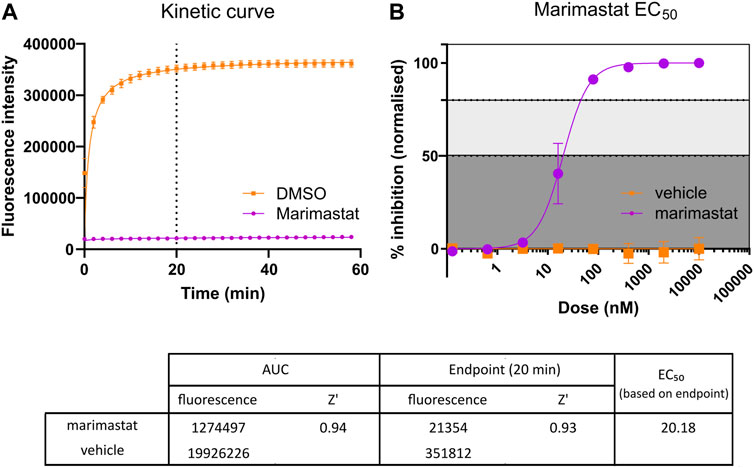
FIGURE 2. Comparison of kinetic Area Under Curve (AUC) measurement and endpoint measurement. (A) Kinetic curve measurement of positive (10 µM marimastat + venom) and negative (DMSO vehicle + venom) controls. Following the protocol outlined in Figure 1, kinetic measurement was taken for 60 min, and the AUC measurement calculated (n = 96). Dotted line indicates timepoint at which comparative endpoint reads were taken (20 min). (B) EC50 curve generated from positive and negative controls ran as a dose response plate created, starting at 10 µM and diluting 1:5 down to 128 pM. Raw data from the 20-min timepoint was normalised to a percentage inhibition based on the positive (100%) and negative (0%) controls and plotted in a dose-dependent manner (n = 2). The table demonstrates the raw values and Z′ values for both methods of response calculation. Z′ values were calculated by comparison of positive and negative raw values across the respective test plates. Z′ values range from 0 to 1, with values closer to 1 indicating clearly defined positive and negative controls, with little to no overlap in values between controls. The EC50 of marimastat was calculated through nonlinear trendline fit ([inhibitor] vs. normalized response—Variable slope) using Graphpad Prism version 9.
The aim of this screening campaign was to identify novel molecules with activity at <10 µM in the primary screen and, ideally, exhibiting low nanomolar activity EC50 in secondary screening comparable to marimastat’s EC50 demonstrated in Figure 2B (Clare et al., 2019). Based on this, positive (10 µM marimastat) and negative (vehicle) controls were included on all screening plates as a quality control assessment. For E. ocellatus there was a 16-fold assay window between the fluorescence of the controls at the selected 20-min endpoint (351,812 and 21,354 for vehicle control and marimastat, respectively) (Figure 2A). Furthermore, to align with industry practice for drug discovery campaigns, the controls were used to calculate Z′ for each plate to monitor the assay window and variation in the data as an additional quality control step (Zhang et al., 1999). The Z′ is indicative of how discrete each control is; the closer the Z′ value is to 1, the less likely there is to be overlap between the positive and negative control values. Therefore, the Z′ of 0.93 emphasises the robust nature of this readout due to a combination of large assay window and minor variation in the data from 96 samples per control (Figure 2A).
To ensure that the optimised methodology was robust for HTS, we assessed intra- and inter-plate signal uniformity across triplicate 384-well plates containing either the negative control (vehicle control; plates 1–3) or the positive control (10 µM marimastat; plates 4–6). As seen in Supplementary Figure S1A, there was signal uniformity both horizontally and vertically across each plate (coefficient of variation [CV%] between 2.42 and 3.93), though there was a degree of inter-plate variance in raw signal values between repeated experiments, as best evidenced by reduced signal in plate 1 compared to plates 2 and 3 (mean 270,148 vs. 360,521 and 391,484 fluorescence AU, respectively). However, the assay window was still greater than 11-fold between the lowest vehicle control signal (plate 1) and the highest marimastat signal (plate 5). Normalising the data to percentage of inhibition based on the positive and negative controls, with 10 µM marimastat representing 100% inhibition, corrected this variance (Supplementary Figure S1B) and allowed for inter-plate consistency across a HTS for identifying drug hits.
Following validation of positive and negative controls, we then determined whether there was any inter-well interference when control wells were interleaved; as this is a fluorescence-based assay, there is potential for a high-signal well to bleed over into low-signal wells and be erroneously detected. Consequently, full rows of wells in duplicate (i.e., rows A + B, C + D, etc.) were plated containing PBS, DMSO, or marimastat at either 10 µM or a suboptimal 50 nM concentration (Supplementary Figure S2). Minimal variance was observed between the positive (raw signal range from 275,763–409,939 across all plates) and negative (raw signal range from 18,037–27,605 across all plates) controls, confirmed by a CV% from the raw data of 3.24% or lower across all conditions from all plates, and resulting in Z′ of 0.89–0.94. Although there is variation in the 50 nM control (percentage inhibition ranges from 21.7% to 78.7%), the condition is clearly discrete from the positive and negative readings. In addition, no spill-over into the previously defined strong hit classification was detected (>80% inhibition) (Supplementary Figure S2).
As a validation screen, we tested a panel of drugs known to inhibit the activity of snake venom toxins. These included metal chelators (DMPS and dimercaprol) and a broad range of MMPis (marimastat, prinomastat, batimastat, tanomastat and doxycycline). In addition, we included the drugs varespladib and nafamostat which are known to target other snake venom toxins (PLA2 and SVSP, respectively), but not SVMPs, as negative controls. The observed EC50s were as expected based on prior experience, with marimastat, prinomastat and batimastat all having strong SVMP inhibitory activity into the low nanomolar range while the metal chelators, which are known to have lower potency in this assay, displayed EC50s in the low micromolar range (Figure 3). As expected, the negative control drugs exhibited no activity at the top 10 µM dose tested, and therefore EC50 curves were not created (Figure 3).
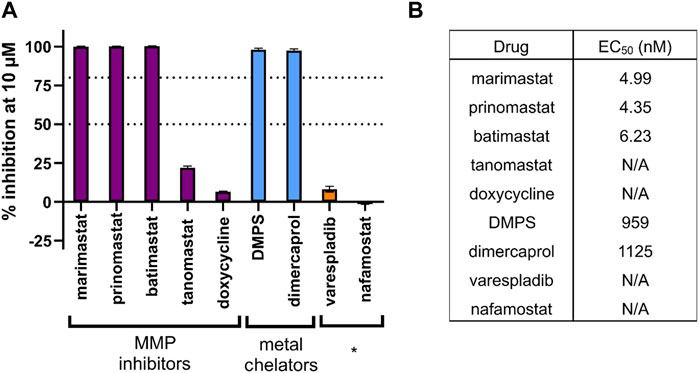
FIGURE 3. The inhibitory potency of various drugs against the SVMP activity of E. ocellatus venom using the validated HTS assay. (A) Bar graph demonstrating the percentage inhibition (when normalised to positive and negative controls) of compounds tested through the screening method. These include chemical groups broadly known to inhibit SVMPs, namely, matrix metalloproteinase inhibitors (MMPi) and metal chelators. * indicates known inhibitors of other snake venom toxin families (PLA2 and serine proteases) used as negative controls. Dotted lines are present at the cutoff values for mediocre (50% inhibition) and strong (80% inhibition) hit identification. (B) EC50 data calculated for each compound from the 20-min endpoint read data. Compounds with an EC50 displaying N/A did not cross the 50% inhibition threshold at any dose and therefore EC50s could not be calculated.
The strong, consistent Z′ results and clear signal windows from these validation experiments provided confidence to expand this approach to four other medically important viper venoms. These came from four distinct viper genera (family Viperidae) found on four different continents, and were selected to account for well-known variations in venom composition among snakes (Tasoulis and Isbister, 2017; Casewell et al., 2020), including variable SVMP content (Table 1). All venoms were initially measured using the original kinetic read method of the assay, and their fluorescence profile examined to determine the plateau point of their respective fluorescence signals (Figure 4A). Upon interrogation of these profiles, the following time points were selected for endpoint read data collection: Echis ocellatus and Calloselasma rhodostoma, 20 min; Crotalus atrox and Bothrops jararaca, 30 min; Bitis arietans, 40 min.

TABLE 1. Details of the venoms tested in the HTS and statistics on the resulting primary screen.
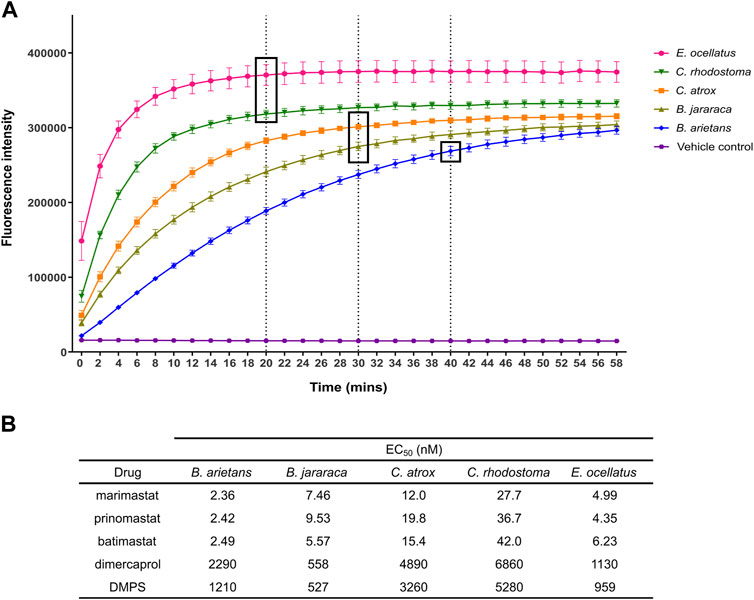
FIGURE 4. Identification of endpoint read times for each venom in the panel, and example EC50s for each venom from the panel of known inhibitors. (A) Kinetic read of venom-only activity in the SVMP assay to identify endpoint read times. Timepoint selection was based on the time at which quencher cleaving had predominantly ceased, as indicated by a plateau in fluorescence values. Venoms with greater SVMP activity generally demonstrated earlier plateau (E. ocellatus, C. rhodostoma) when compared to other venoms. Dotted lines and boxes indicate selected timepoint for the respective venoms. (B) Table showing EC50 values of SVMP inhibitors from the known inhibitors when screened against the panel of five viper venoms as a dilution series.
The same inhibitors that were used to validate the initial experiments with E. ocellatus venom were then applied to these additional venoms (Figure 4B). The same potency differences between the metal chelators and the MMPis were observed with the four new venoms with EC50s of >500 nM maintained for the metal chelators compared to <50 nM for the MMPis. This potency difference between the two drug classes is likely due to their distinct mechanism of action (Albulescu et al., 2020b; Xie et al., 2020). There were also differences between drugs of the same class; across all venoms, DMPS produced lower EC50 values relative to dimercaprol (DMPS = 527–5,284 nM range across all venoms, average 2,248 nM; dimercaprol = 558–6,860 nM, average 3,144 nM). Between the MMPis, minimal difference was observed between the drugs for each of the venoms tested, with all EC50 values lower than 50 nM against all venoms. However, for all drug classes, the EC50s were higher (∼2–20 fold) against C. rhodostoma and C. atrox venoms compared to those obtained for B. arietans, B. jararaca and E. ocellatus venom.
Following the full validation of the assay, we next embarked on a HTS of a repurposed drug library consisting of 3,547 post-Phase I compounds. Screening of the 11-plate library at 10 µM against a single venom was possible in approximately 2 hours, depending on the venom being used for screening (20-min plateau time required 2 hours, 40-min plateau time required 2 hours and 20 min). This throughput meant that the full screen of the library against all five venoms was completed within 5 days. Consistent Z′ of between 0.71–0.96 across all 11 screening plates for each venom (total of 55 × 384 well plates) indicated that the robustness of the assay was retained during the screening campaign (Table 1).
Normalised screening results were characterised into three discrete groups; non-hits (≤50% inhibition compared to control), mediocre hits (50%–80% inhibition), and strong hits (>80% inhibition). The hit rate for strong hits (>80% inhibition) ranged from 0.5% to 1.0% depending on the venom screened, with the highest rate of strong hits observed against B. jararaca venom (35 hits, 1.0%) (Table 1). The lowest hit rates were observed against C. rhodostoma and C. atrox venoms (17 and 19 hits respectively; both ∼0.5%). A greater disparity was observed in the number of mediocre hits recovered (>50% < 80% inhibition), with the highest number of hits observed against B. jararaca venom (37 hits, 1.0%), followed by B. arietans (22 hits, 0.6%). Far fewer mediocre hits were observed against the remaining venoms (E. ocellatus, 4; C. atrox, 5; C. rhodostoma, 5), each representing only 0.1% of the library. It is worth noting that this order of hit rate correlates with the rank ordering of EC50 potencies described earlier for known SVMP inhibitors, with C. rhodostoma and C. atrox venom seemingly being more challenging to inhibit.
To prioritise hits for progression into secondary EC50 screening, recovered hits were assessed for their consistent activity across all five venoms (Figure 5). In total, we had 109 compounds that were either a mediocre or strong hit in at least one venom, but of these, only 10 were strong hits in all five venoms. A further four were strong hits in at least three venoms and mediocre in the additional two venoms. These 14 lead hits are presented in Figure 5A. There were a further six compounds with strong activity in two or three venoms, but lacking activity (<50% inhibition) in the additional venoms (apart from ethylenediamine tetracetic acid (EDTA), which displayed strong activity in two venoms and mediocre in a third) (Figure 5B). Of the remaining 89 compounds, inhibitory activity varied between venoms, with 32 compounds (36%) identified as strong hits against one venom (mainly B. jararaca) but lacking activity in the remaining four venoms. This emphasises the value in testing several venoms to maximise the chance of identifying pan-species inhibitory activities.
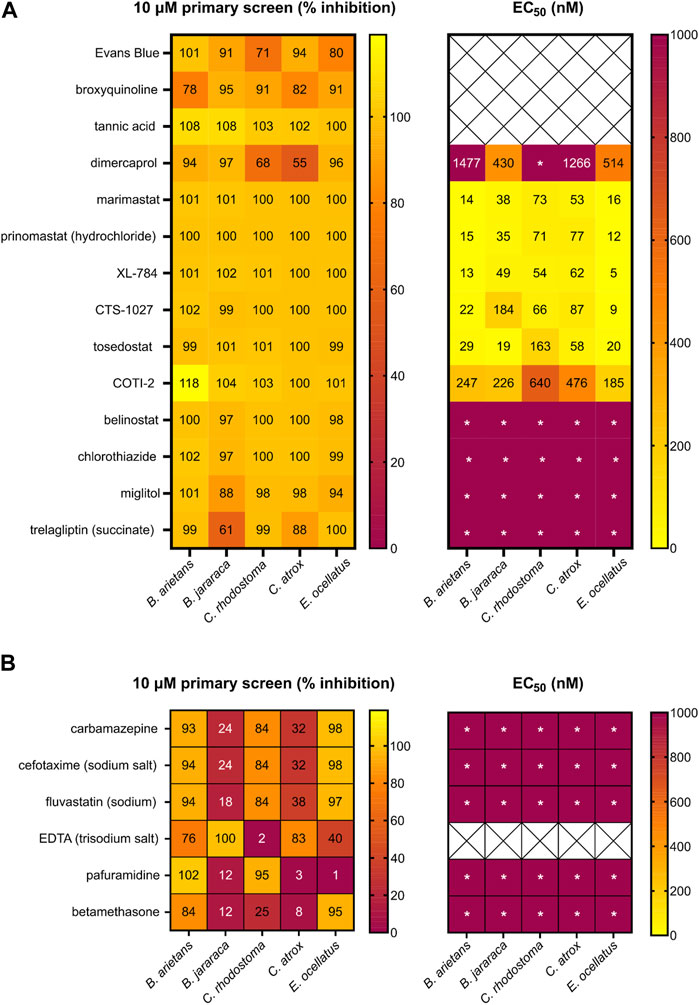
FIGURE 5. Heatmap of the hits from the primary and secondary EC50 screen of the repurposed compound library. Left panels present the primary screen at 10 µM and right panels present the secondary dose response screen. (A) Percentage inhibition of 14 compounds identified as strong hits in three or more venoms during the primary screening of the repurposed library (left) and EC50 data of 11 compounds progressed into the secondary screen (right). (B) Percentage inhibition for six strong hits in the primary screen against at least two venoms (left) and EC50 (right). Wells demonstrating * indicate an EC50 was not calculated due to low activity at the highest doses.
Of the 14 hits with pan-species activity (Figure 5A), all except three were progressed into secondary EC50 testing. Evans Blue and tannic acid were not considered further due to their high molecular weights coupled with known polypharmacology, while broxyquinoline was considered an intractable starting point due to previously reported toxic side effects (Strandvik and Zetterström, 1968; Baumgartner et al., 1979). The remaining 11 pan-species hits included the known and previously described SVMP inhibitors dimercaprol, marimastat and prinomastat (Rucavado et al., 2000; Ainsworth et al., 2018; Albulescu et al., 2020b; Chowdhury et al., 2021). These were progressed as additional controls in the EC50 screen and exhibited inhibitory potencies as anticipated (i.e., Figure 5A in comparison with Figure 4B).
The remaining eight novel hits fell into three broad inhibitory categories when tested against the five venoms in this secondary screen (Figure 5A). Three drugs (XL-784, CTS-1027 and tosedostat) had comparable EC50 activity (5–184 nM) to marimastat and prinomastat across all venoms. COTI-2 was slightly less active, producing EC50s that were approximately 10-fold lower, though these were still superior (>2 fold) to dimercaprol across all venoms (EC50s, 185–640 nM). The four remaining drugs (belinostat, chlorothiazide, miglitol and trelagliptin) were less active than dimercaprol, with EC50s greater than 1 µM against each of the venoms.
Although discovery of drug compounds with pan-species activity was the overarching goal of this project, hits with strong activity against two or more venoms were also progressed to EC50 testing (Figure 5B) to ensure valuable hits had not been excluded (e.g., false negatives against certain venoms). We still excluded EDTA since it has been investigated previously as a potential snakebite therapy and was found to be inferior to other metal chelators (DMPS and dimercaprol), whilst having poor therapeutic characteristics; high affinity for calcium, poor safety profile, and requirement for slow intravenous administration (Flora and Pachauri, 2010; Ainsworth et al., 2018; Albulescu et al., 2020a). Of the remaining six hits in this category (Figure 5B), when tested in the EC50 screen, all displayed poor activity against all venoms (EC50s > 1 µM), thus further validating the focus on hits with pan-species activity in the primary screen.
To scrutinise this repurposed library from a structural activity relationship (SAR) perspective, we used Tree Manifold Approximation Projection (TMAP) to visualise the HTS chemical space explored and to identify trends among closely related scaffolds (Probst and Reymond, 2020). Briefly, TMAP displays chemical space as a tree manifold displaying similar groups of compounds as branches while the distance between each compound represents similarity–we would therefore expect similar compounds to exist within the same sub-branches. TMAP representation of our dataset showed that the majority of the mediocre hits detected were structurally distinct from other hits (Figure 6). However, for the lead compounds, which included the four novel hits with nanomolar EC50s (XL-784, CTS-1027, COTI-2 and tosedostat) and the known SVMP inhibitors (marimastat, prinomastat and dimercaprol), this approach allowed identification of two distinct groups: i) CTS-1027, XL-784 and prinomastat—all lipophilic non-peptidomimetic hydroxamate MMPis (Johnson et al., 2006; Kahraman et al., 2009) and ii) marimastat and tosedostat—an established peptidomimetic SVMP inhibitor and a structurally similar compound, respectively. Dimercaprol and COTI-2 are visibly isolated from the other hits in separate branches and exist as singletons. This reduces confidence in the latter compounds as starting points for optimisation due to lack of evidence of an SVMP-inhibitory scaffold, with their closest neighbours in the HTS showing no SVMP-inhibitory activity. This hypothesis is further supported by the poorer performance of COTI-2 and dimercaprol in secondary EC50 screens compared to other hits. The structures of the lead compounds are presented in Figure 7.
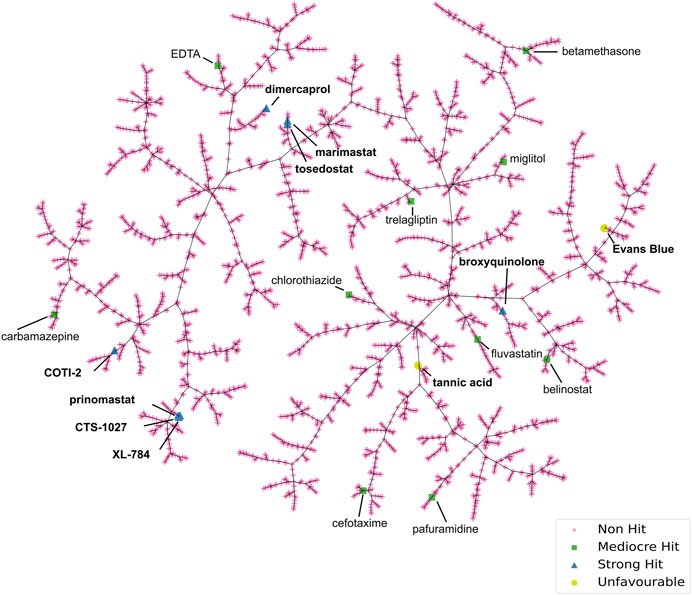
FIGURE 6. Tree Manifold Approximation Projection (TMAP) representation of the chemical space covered by the HTS library. Non-hits are shown as pink crosses, mediocre hits are shown as green squares, and strong hits are shown as blue triangles. Unfavourable hits are shown as yellow circles.
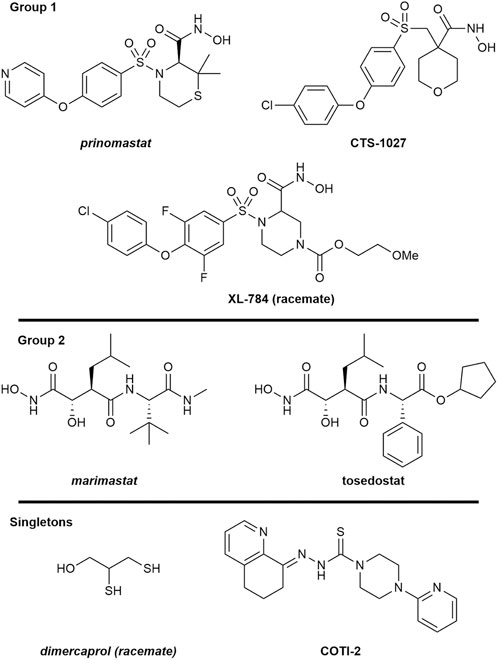
FIGURE 7. Structures of the seven lead SVMP compounds including three known inhibitors (italics) and four novel hits with nanomolar EC50s grouped by structural similarity determined by TMAP. Structures are drawn by ChemDraw based on SMILES provided by the source of the drug screening library.
We used SwissADME to evaluate the predicted physicochemical properties and drug-likeness of the seven lead compounds (Figure 7) using their validated machine-learning models (Daina et al., 2017). Predicted values indicating lipophilicity (XLOGP3), molecular weight (MW), polarity (topological polar surface area, TPSA), non-planarity (fraction of carbons in the sp3 hybridisation state, Fsp3), molecular flexibility (number of rotatable bonds, RotB) and solubility are provided in Table 2, alongside radar plots for visualisation in Figure 8.
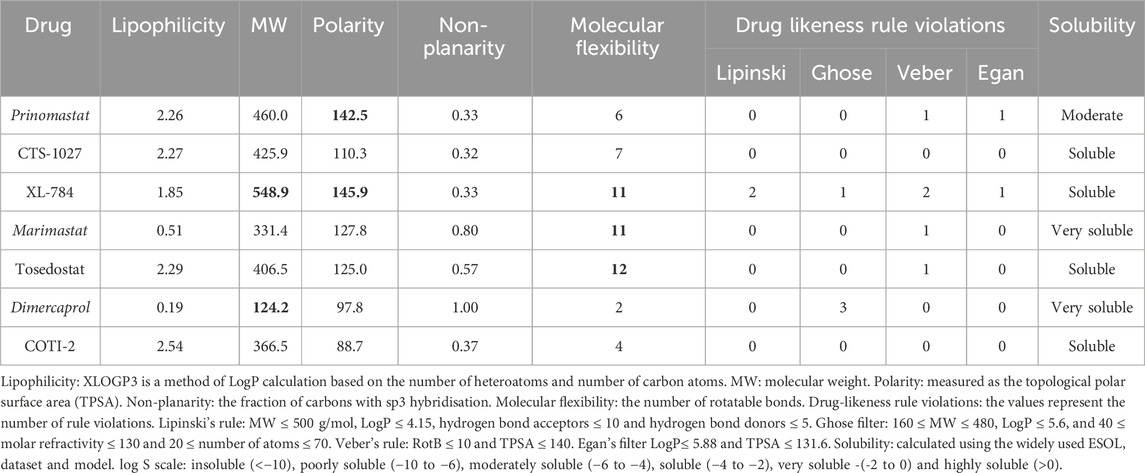
TABLE 2. Selected SwissADME predicted properties of the seven lead SVMP compounds including three known inhibitors (italics) and four novel inhibitors. Properties of concern based on violations of drug-likeness rules are indicated in bold.
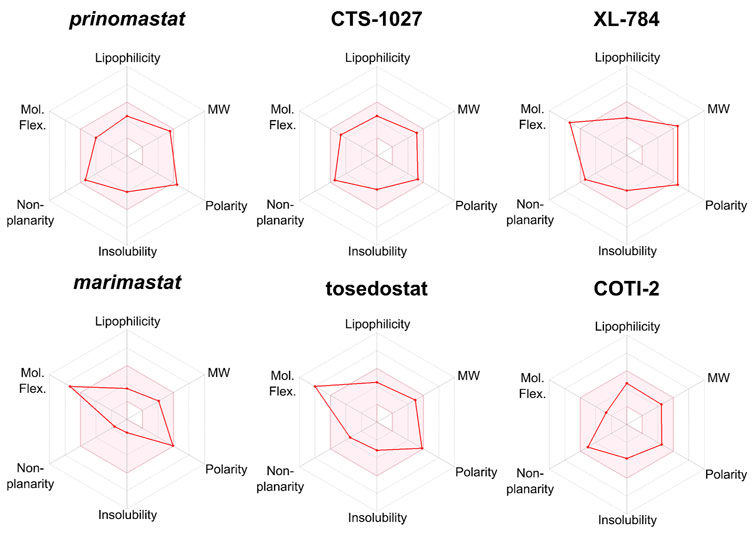
FIGURE 8. Radar plots generated by SwissADME for lead compounds of interest. The shaded area represents the ideal range for oral bioavailability. Thresholds: Lipophilicity: XLOGP3 between −0.7 and 5.0, Molecular Weight (MW): 150 < MW < 500 g/mol; Polarity: 20 < TPSA 130 Å2, Insolubility: −log S < 6, non-planarity: fraction of carbons in the sp3 hybridization not less than 0.25, and Molecular Flexibility (Mol.Flex.): no more than 9 rotatable bonds.
SwissADME assesses drug-likeness through design rules as determined by Lipinski, Ghose, Veber, and Egan (Ghose et al., 1999; Egan et al., 2000; Lipinski et al., 2001; Veber et al., 2002). These rules use the predicted ADME properties and defined thresholds to determine whether a compound violates a rule. The number of violations of these rules is summarised in Table 2. All compounds possess no greater than 2 violations to any rule with the exception of dimercaprol which possesses three Ghose violations due to molecular weight < 160 Da, molar refractivity < 40 and total number of atoms < 20 (Ghose et al., 1999). CTS-1027 and COTI-2 have no violations across all 4 rules, whilst marimastat and tosedostat have a single violation in Veber’s rule due to having the number of rotatable bonds as > 9 (molecular flexibility). Prinomastat has one violation in both Veber and Ghose design rules due to its polarity (TPSA = 142.5 Å2), though considering the thresholds of these rules (TPSA > 140 Å2 and 131.6 Å2, respectively) these violations are minor. XL-784 is the only compound to have violations across all four rules. This is due to its high molecular weight and number of hydrogen bond donors (Lipinski and Ghose), as well as its polarity and flexibility (Veber and Egan).
Outside the parameters used to determine rule violations, SwissADME also profiles solubility by three methods; an implementation of the ESOL method (Delaney, 2004), a model adapted from (Ali et al., 2012), and a method developed by SILICOS-IT (Daina et al., 2017). Low solubility of molecules may cause issues with absorption. No hit compounds were found to be poorly soluble (Table 2) supporting the insight from design rules that the majority of hits are unlikely to experience issues with absorption. Overall, assessment of predicted ADME properties suggests that the novel hits CTS-1027, COTI-2 and tosedostat are the most promising hits for optimisation, while XL-784 would require more extensive optimisation of its ADME properties.
SVMPs are a major snake venom toxin family responsible for causing substantial pathology in snakebite patients, while conventional antivenom therapies are often associated with a high incidence of adverse effects, as well as financial and logistical constraints. Consequently, efforts are being made to find alternative therapies that could overcome these existing barriers for victims of snakebite. Through validated miniaturisation of an existing fluorometric assay and the application of liquid handling instruments, we have developed a high-throughput screening platform with the capacity to screen ∼7,000 individual compounds against a venom of interest in a single day. Utilising this HTS platform, we screened 3,547 post-Phase I compounds in singleton at 10 µM against five medically important viper venoms of diverse taxonomy, geographic location and SVMP content. The robust nature of this HTS is quantified by consistently high Z′ across assay plates (0.71–0.96), providing confidence in the results obtained. Out of the 0.4% hit rate from the primary screen (14 strong pan-species inhibitors), after secondary testing and assessment of physiochemical properties, four discrete clusters of structurally similar strong (nM EC50s) hit compounds were identified (Figures 6, 7). Three hit compounds (XL-784, CTS-1027 and tosedostat) shared structural similarities with MMP inhibitors previously shown to also inhibit SVMPs (marimastat and prinomastat (Howes et al., 2007; Arias et al., 2017; Albulescu et al., 2020b; Menzies et al., 2022)). XL-784, first prepared by Exelixis, and CTS-1027 (also known as RS-130830), originally prepared by Roche Bio-Science, are both known MMP inhibitors, while both tosedostat and COTI-2 have unknown MMP inhibitory activity. Due to the less favourable EC50 performance in the secondary screen, COTI-2 was deprioritised as a backup compound.
Despite promising inhibitory potencies and ADME properties, tosedostat retains a major limitation as a future SVMP inhibiting drug. Tosedostat is a pro-drug that is metabolised into its active form, which is an aminopeptidase inhibitor investigated for cancer indications. This transformation involves cleavage of a labile ester linkage by serum esterases to reveal a carboxylic acid necessary for its aminopeptidase inhibitory function (Krige et al., 2008). As our HTS was not carried out under the appropriate metabolic conditions to generate tosedostat’s active metabolite, it can be assumed that the anti-SVMP activity of this drug is mediated by its ester form. For our purposes, hydrolysis to the free acid would add unnecessary polar functionality which may have negative implications on absorption, metabolism, and overall exposure at the target SVMPs in vivo. For these reasons, we have deprioritised tosedostat for onward development.
The ADME properties described in Table 2 and Figures 7, 8 suggest that XL-784 is perhaps less attractive due to its high molecular weight, polarity, molecular flexibility, and number of hydrogen bond acceptors—all of which are associated with poor bioavailability, and XL-784 also has a reported low aqueous solubility of 20 μg/mL (Williams et al., 2011b). For these reasons, analogues of XL-784 with improved pharmacokinetic properties are being evaluated in our medicinal chemistry program. CTS-1027 and prinomastat provide the best medicinal chemistry starting points for future SVMP inhibitor discovery due to their desirable physicochemical and structural properties and minor violations of drug-likeness rules. However, it should be noted that CTS-1027 and prinomastat were flagged in SwissADME as having toxicity and pharmacokinetic (PK) concerns. Prinomastat previously progressed into Phase III trials where musculoskeletal toxicity was observed in a significant proportion of patients that received a treatment course of 15 mg bi-daily dosing for 3–36 months, as well as an increased risk of venous thromboembolic disease in cancer patients receiving combination chemotherapy (Bissett et al., 2005). It is anticipated that these side effects were due to inhibition of MMP-1. Consequently, a more selective MMP/SVMP inhibitor will be pursued in our drug discovery campaign to avoid joint toxicities. CTS-1027 previously entered Phase II clinical trials for the treatment of hepatitis C, and a range of serious adverse events were associated with the administration of 30 mg per day for up to 24 weeks (e.g., single incidences of anaemia, hematemesis, upper GI haemorrhage, chest pain, staphylococcal abscess, prostate cancer and chronic obstructive pulmonary disease) (Conatus Pharmaceuticals Inc, 2012). It should be noted that these toxicity concerns were associated with a long-term treatment course and high drug dosage, whereas projected snakebite therapy would likely only involve short course dosing for perhaps up to 72 h. Additionally, these side effects are reversible following treatment termination, thereby avoiding permanent pathologies (Scatena, 2000).
The HTS assay described here identifies compounds that directly inhibit the enzymatic activity of SVMP toxins (Neumann et al., 2004; Gutiérrez et al., 2021). One limitation of this assay is that compounds that inhibit SVMP activity through metal chelation will not be observed as highly potent in our HTS and thus may not be selected as hits for progression. This is important to note as the metal chelator dimercaprol has been shown to successfully inhibit SVMP activity in vivo, with the derivative DMPS currently being planned for progression into Phase II clinical trials for snakebite (Abouyannis et al., 2022). Therefore, screening to identify compounds with similar mode of action would require an alternative screening model that better represents the phenotypic nature of snakebite envenoming. Another caveat to consider is that fluorescence-based drug screens are vulnerable to interference from compounds that are either fluorescent quenchers (false positives) or auto-fluorescent (false negatives) (Yang et al., 2020). Diversity based screening libraries often contain compounds with several aromatic rings in their structure which are inherently fluorescent in the lower-blue range of the spectrum (excitation ∼ 350 nm and emission of 450–495 nm) (Thorne et al., 2010). The emission wavelength of this assay is 410–420 nm, therefore such false negatives are likely to be minimal. Chemical scrutiny of hits prior to chemical selection can be used to identify quenching moieties and deprioritised for onward progression, while false negatives with auto-fluorescent characteristics could be progressed into secondary assays if they possessed promising structural similarities to known inhibitors or demonstrate inhibitory activity in orthogonal, non-fluorescent assays.
Prior to the initiation of medicinal chemistry campaigns around the two hit-to-lead compounds described here (CTS-1027 and prinomastat), traction along a defined drug discovery pipeline would be essential, inclusive of activity confirmation in additional in vitro assays and critical evaluation PK/PD properties prior to progression into in vivo testing. Although we do not anticipate dropouts in in vivo experiments (MMPis such as marimastat and metal chelators such as DMPS have previously displayed activity in murine envenoming models (Albulescu et al., 2020a; 2020b; Menzies et al., 2022)), discrepancies between in vitro enzymatic screening and results from in vivo testing could occur due to the increased degree of complexity of venom pathology in a biological system rather than an enzymatic in vitro setting. For example, SVMPs directly target the coagulation cascade through the activation of factor X and/or prothrombin, which are effects that this current enzymatic HTS platform cannot determine. Marimastat and prinomastat are known to inhibit SVMP activation of the clotting cascade, so a further set of experiments to confirm that hits identified in this assay show similar inhibition of coagulopathy would be beneficial as a tertiary screen (Mackessy and Stephen, 2009; Cabral-Pacheco et al., 2020). In addition to tertiary in vitro testing, the pharmacokinetic and pharmacodynamic (PK/PD) properties of the lead compounds need to be considered to allow for rational dose design prior to progression into preclinical in vivo models. One benefit of screening repurposed compound libraries, such as in this HTS study, is that many of the compounds involved will have already been subjected to human safety and efficacy testing, which will provide data of value for considering human dose prediction.
As described above, we have identified two novel lead candidates which are poised for progression along our drug discovery pipeline (tertiary screening and PK/PD evaluation inclusive of informed dose selection). Prinomastat and CTS-1027 are viable as parent compounds for the establishment of medicinal chemistry campaigns to develop the first designer toxin-specific small molecule for the treatment of snakebite. To our knowledge, this is the first time a validated, scalable HTS platform has been demonstrated for the investigation of small molecule inhibitors against medically relevant snake venom toxins. We hope that this work will help to overcome barriers of throughput and accelerate the future identification of alternative snakebite therapeutics. In addition to small molecules, this platform is amenable to use to discover monoclonal antibody-based therapeutics (Knudsen and Laustsen, 2018; Laustsen, 2018; Harrison and Gutiérrez, 2019; Knudsen et al., 2019), as well as for high-throughput profiling of SVMP activity from a diversity of venoms (e.g., macroevolutionary or population-level analyses). The platform can also be utilised for libraries of any size; smaller, rationally selected libraries can be screened rapidly (e.g., analogues from a medicinal chemistry campaign) and progressed directly to secondary EC50 screening, whilst the scalability of this platform makes it equally applicable to larger, more diverse libraries such as the ReFRAME collection (12,000 small molecules which have reached clinical development or undergone significant preclinical development (Janes et al., 2018)) and AstraZeneca’s open innovation programme (AstraZeneca, 2022). Furthermore, it is feasible this HTS could be scaled up further based on the precedence for ultra-HTS discovery programmes which use the same fluorescent substrate (Madoux et al., 2016; Hou et al., 2021).
Our ongoing work aims to develop and validate a drug discovery pipeline from primary in vitro HTS through to preclinical testing, paving the way for the discovery and development of novel snakebite therapeutics against pathogenically important snake venom toxins. The automated set up of this HTS platform has also been applied in parallel to other toxin families, such as the PLA2s (Albulescu et al., 2020b). Collectively, these studies have the potential to vastly broaden the portfolio of preclinical stage small molecules for snakebite with options for novel drug combinations directed at distinct snake venom components. Following the long-term goal of successful progression through snakebite targeted clinical development regulatory approved oral drugs that can be rapidly delivered in the community soon after a bite to alleviate the severe, often life-threatening effects would provide a paradigm shift in snakebite envenoming therapeutics (Clare et al., 2021; Gutiérrez et al., 2021; Abouyannis et al., 2022; Carter et al., 2022).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
RC: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Writing–original draft. CD: Data curation, Formal Analysis, Methodology, Validation, Writing–original draft. AW: Data curation, Formal Analysis, Methodology, Validation, Writing–original draft. L-OA: Methodology, Validation, Writing–review and editing. CW: Data curation, Formal Analysis, Methodology, Writing–original draft. NM: Data curation, Methodology, Writing–original draft. DC: Data curation, Methodology, Writing–original draft. JK: Funding acquisition, Writing–review and editing, Conceptualization. NB: Funding acquisition, Writing–review and editing, Conceptualization, Methodology. PO’N: Funding acquisition, Writing–review and editing, Conceptualization, Methodology. NC: Funding acquisition, Conceptualization, Methodology, Writing–original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by: i) A Director’s Catalyst Fund from LSTM [supported by Wellcome Institutional Strategic Support Fund 3 (204806/Z/16/Z) and LSTM Internal Funding] awarded to RC, ii) a UK Medical Research Council-funded Confidence in Concept Award (MC_PC_15040) to NC, and iii) a Wellcome Trust grant awarded to JK, NB, PO’N and NC (221712/Z/20/Z). This research was funded in part by the Wellcome Trust. For the purpose of open access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
We thank Paul Rowley and Edouard Crittenden for maintenance and husbandry of the snake collection and provision of venom samples at LSTM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1328950/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Full-plate control data separated by column and row to demonstrate inter- and intra-plate consistency. (A) Graphs demonstrating the raw data from three independently-ran control plates, containing either full maximum signal (vehicle: DMSO) or full baseline signal (10 µM marimastat), with values split by row (left) or column (right). Consistently high vehicle values and consistently baseline marimastat values can be seen irrespective of where on the plate they are located. (B) Normalised data from the same experiments as shown in (A). Normalisation of this data reduces inter-plate variation, as can be seen with plate 1 compared to plates 2 and 3 in (A), without affecting the separation of the positive and negative control signal windows.
SUPPLEMENTARY FIGURE S2 | Interleaved control data separated by column and row to demonstrate inter- and intra-plate consistency. Graphs demonstrating the normalised results from three independently-ran control plates, containing alternating rows of maximum signal (vehicle: DMSO), baseline signal (10 µM marimastat), venom vehicle (PBS) or a suboptimal dose of marimastat (50 nM). Values ordered by row (top) or column (bottom). As with Supplementary Figure S1, consistency can be seen in matched conditions regardless of the location of the well on the plate, as well as between independently repeated plates. The suboptimal dose of marimastat selected demonstrates that it inhibits at around 50% that of the baseline concentration.
SUPPLEMENTARY DATASHEET S1 | Repurposed Drug library—The details provided by MedChemExpress of the bespoke 3,547 repurposed drug library created for snakebite drug discovery screening are displayed. This is inclusive of chemical structural data through simplified molecular-input line-entry system (SMILES) keys (column K) and CAS number.
Abouyannis, M., FitzGerald, R., Ngama, M., Mwangudzah, H., Nyambura, Y. K., Ngome, S., et al. (2022). TRUE-1: trial of Repurposed Unithiol for snakebite Envenoming phase 1 (safety, tolerability, pharmacokinetics and pharmacodynamics in healthy Kenyan adults). Wellcome Open Res. 7, 90. doi:10.12688/wellcomeopenres.17682.1
Ainsworth, S., Menzies, S. K., Casewell, N. R., and Harrison, R. A. (2020). An analysis of preclinical efficacy testing of antivenoms for sub-Saharan Africa: inadequate independent scrutiny and poor-quality reporting are barriers to improving snakebite treatment and management. PLoS Negl. Trop. Dis. 14, e0008579. doi:10.1371/journal.pntd.0008579
Ainsworth, S., Slagboom, J., Alomran, N., Pla, D., Alhamdi, Y., King, S. I., et al. (2018). The paraspecific neutralisation of snake venom induced coagulopathy by antivenoms. Commun. Biol. 1, 34. doi:10.1038/s42003-018-0039-1
Albulescu, L.-O., Hale, M. S., Ainsworth, S., Alsolaiss, J., Crittenden, E., Calvete, J. J., et al. (2020a). Preclinical validation of a repurposed metal chelator as an early-intervention therapeutic for hemotoxic snakebite. Sci. Transl. Med. 12, eaay8314. doi:10.1126/scitranslmed.aay8314
Albulescu, L.-O., Xie, C., Ainsworth, S., Alsolaiss, J., Crittenden, E., Dawson, C. A., et al. (2020b). A therapeutic combination of two small molecule toxin inhibitors provides broad preclinical efficacy against viper snakebite. Nat. Commun. 11, 6094. doi:10.1038/s41467-020-19981-6
Ali, J., Camilleri, P., Brown, M. B., Hutt, A. J., and Kirton, S. B. (2012). Revisiting the general solubility equation: in silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 52, 420–428. doi:10.1021/ci200387c
Amazonas, D. R., Portes-Junior, J. A., Nishiyama-, M. Y., Nicolau, C. A., Chalkidis, H. M., Mourão, R. H. V., et al. (2018). Molecular mechanisms underlying intraspecific variation in snake venom. J. Proteomics 181, 60–72. doi:10.1016/j.jprot.2018.03.032
Arias, A. S., Rucavado, A., and Gutiérrez, J. M. (2017). Peptidomimetic hydroxamate metalloproteinase inhibitors abrogate local and systemic toxicity induced by Echis ocellatus (saw-scaled) snake venom. Toxicon 132, 40–49. doi:10.1016/j.toxicon.2017.04.001
Asega, A. F., Menezes, M. C., Trevisan-Silva, D., Cajado-Carvalho, D., Bertholim, L., Oliveira, A. K., et al. (2020). Cleavage of proteoglycans, plasma proteins and the platelet-derived growth factor receptor in the hemorrhagic process induced by snake venom metalloproteinases. Sci. Rep. 10, 12912. doi:10.1038/s41598-020-69396-y
Ashburn, T. T., and Thor, K. B. (2004). Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 3, 673–683. doi:10.1038/nrd1468
AstraZeneca (2022). AstraZeneca open innovation. Available at: https://openinnovation.astrazeneca.com.
Baell, J. B., and Holloway, G. A. (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740. doi:10.1021/jm901137j
Baumgartner, G., Gawel, M. J., Kaeser, H. E., Pallis, C. A., Rose, F. C., Schaumburg, H. H., et al. (1979). Neurotoxicity of halogenated hydroxyquinolines: clinical analysis of cases reported outside Japan. J. Neurol. Neurosurg. Psychiatry 42, 1073–1083. doi:10.1136/jnnp.42.12.1073
Beck, T. P., Tupetz, A., Farias, A. S., Silva-Neto, A., Rocha, T., Smith, E. R., et al. (2022). Mapping of clinical management resources for snakebites and other animal envenomings in the Brazilian Amazon. Toxicon X 16, 100137. doi:10.1016/j.toxcx.2022.100137
Bissett, D., O’Byrne, K. J., von Pawel, J., Gatzemeier, U., Price, A., Nicolson, M., et al. (2005). Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 23, 842–849. doi:10.1200/JCO.2005.03.170
Cabral-Pacheco, G. A., Garza-Veloz, I., Castruita-De la Rosa, C., Ramirez-Acuña, J. M., Perez-Romero, B. A., Guerrero-Rodriguez, J. F., et al. (2020). The roles of matrix metalloproteinases and their inhibitors in human diseases. Int. J. Mol. Sci. 21, 9739. doi:10.3390/ijms21249739
Calvete, J. J., Fasoli, E., Sanz, L., Boschetti, E., and Righetti, P. G. (2009). Exploring the venom proteome of the western diamondback rattlesnake, Crotalus atrox, via snake venomics and combinatorial peptide ligand library approaches. J. Proteome Res. 8, 3055–3067. doi:10.1021/pr900249q
Carter, R. W., Gerardo, C. J., Samuel, S. P., Kumar, S., Kotehal, S. D., Mukherjee, P. P., et al. (2022). The BRAVO clinical study protocol: oral varespladib for inhibition of secretory phospholipase A2 in the treatment of snakebite envenoming. Toxins 15, 22. doi:10.3390/toxins15010022
Casewell, N. R., Huttley, G. A., and Wüster, W. (2012). Dynamic evolution of venom proteins in squamate reptiles. Nat. Commun. 3, 1066. doi:10.1038/ncomms2065
Casewell, N. R., Jackson, T. N. W., Laustsen, A. H., and Sunagar, K. (2020). Causes and consequences of snake venom variation. Trends Pharmacol. Sci. 41, 570–581. doi:10.1016/j.tips.2020.05.006
Casewell, N. R., Wagstaff, S. C., Wüster, W., Cook, D. A. N., Bolton, F. M. S., King, S. I., et al. (2014). Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. Proc. Natl. Acad. Sci. U. S. A. 111, 9205–9210. doi:10.1073/pnas.1405484111
Chippaux, J.-P. (2017). Snakebite envenomation turns again into a neglected tropical disease. J. Venom. Anim. Toxins Trop. Dis. 23, 38. doi:10.1186/s40409-017-0127-6
Chippaux, J. P., Williams, V., and White, J. (1991). Snake venom variability: methods of study, results and interpretation. Toxicon Off. J. Int. Soc. Toxinology 29, 1279–1303. doi:10.1016/0041-0101(91)90116-9
Chowdhury, A., Zdenek, C. N., Lewin, M. R., Carter, R., Jagar, T., Ostanek, E., et al. (2021). Venom-induced blood disturbances by palearctic viperid snakes, and their relative neutralization by antivenoms and enzyme-inhibitors. Front. Immunol. 12, 688802. doi:10.3389/fimmu.2021.688802
Clare, R. H., Clark, R., Bardelle, C., Harper, P., Collier, M., Johnston, K. L., et al. (2019). Development of a high-throughput cytometric screen to identify anti-wolbachia compounds: the power of public–private partnership. SLAS Discov. Adv. Sci. Drug Discov. 24, 537–547. doi:10.1177/2472555219838341
Clare, R. H., Hall, S. R., Patel, R. N., and Casewell, N. R. (2021). Small molecule drug discovery for neglected tropical snakebite. Trends Pharmacol. Sci. 42, 340–353. doi:10.1016/j.tips.2021.02.005
Conatus Pharmaceuticals Inc. (2012). A trial of CTS-1027 in interferon-naive hepatitis C patients (clinical trial registration No. NCT00925990). Available at: https://clinicaltrials.gov/study/NCT00925990.
Currier, R. B., Harrison, R. A., Rowley, P. D., Laing, G. D., and Wagstaff, S. C. (2010). Intra-specific variation in venom of the African Puff Adder (Bitis arietans): differential expression and activity of snake venom metalloproteinases (SVMPs). Toxicon Off. J. Int. Soc. Toxinology 55, 864–873. doi:10.1016/j.toxicon.2009.12.009
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717. doi:10.1038/srep42717
Dawson, C. A., Ainsworth, S., Albulescu, L.-O., and Casewell, N. R. (2021). “Snake venom metalloproteinases,” in Handbook of venoms and toxins of reptiles (USA: CRC Press), 363–379.
Delaney, J. S. (2004). ESOL: estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 44, 1000–1005. doi:10.1021/ci034243x
de Silva, H. A., Ryan, N. M., and de Silva, H. J. (2016). Adverse reactions to snake antivenom, and their prevention and treatment. Br. J. Clin. Pharmacol. 81, 446–452. doi:10.1111/bcp.12739
Dingwoke, E. J., Adamude, F. A., Mohamed, G., Klein, A., Salihu, A., Abubakar, M. S., et al. (2021). Venom proteomic analysis of medically important Nigerian viper Echis ocellatus and Bitis arietans snake species. Biochem. Biophys. Rep. 28, 101164. doi:10.1016/j.bbrep.2021.101164
Drisko, J. A. (2018). “Chapter 107 - chelation therapy,” in Integrative medicine Editor D. Rakel Fourth Edition (Germany: Elsevier), 1004–1015.e3. doi:10.1016/B978-0-323-35868-2.00107-9
Egan, W. J., Merz, K. M., and Baldwin, J. J. (2000). Prediction of drug absorption using multivariate statistics. J. Med. Chem. 43, 3867–3877. doi:10.1021/jm000292e
Ferraz, C. R., Arrahman, A., Xie, C., Casewell, N. R., Lewis, R. J., Kool, J., et al. (2019). Multifunctional toxins in snake venoms and therapeutic implications: from pain to hemorrhage and necrosis. Front. Ecol. Evol. 7. doi:10.3389/fevo.2019.00218
Flora, S. J. S., and Pachauri, V. (2010). Chelation in metal intoxication. Int. J. Environ. Res. Public. Health 7, 2745–2788. doi:10.3390/ijerph7072745
Fox, J. W., and Serrano, S. M. T. (2005). Structural considerations of the snake venom metalloproteinases, key members of the M12 reprolysin family of metalloproteinases. Toxicon Off. J. Int. Soc. Toxinology 45, 969–985. doi:10.1016/j.toxicon.2005.02.012
Fox, J. W., and Serrano, S. M. T. (2008). Insights into and speculations about snake venom metalloproteinase (SVMP) synthesis, folding and disulfide bond formation and their contribution to venom complexity. FEBS J. 275, 3016–3030. doi:10.1111/j.1742-4658.2008.06466.x
Ghose, A. K., Viswanadhan, V. N., and Wendoloski, J. J. (1999). A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1, 55–68. doi:10.1021/cc9800071
Gonçalves-Machado, L., Pla, D., Sanz, L., Jorge, R. J. B., Leitão-De-Araújo, M., Alves, M. L. M., et al. (2016). Combined venomics, venom gland transcriptomics, bioactivities, and antivenomics of two Bothrops jararaca populations from geographic isolated regions within the Brazilian Atlantic rainforest. J. Proteomics, Proteomics Evol. Ecol. 135, 73–89. doi:10.1016/j.jprot.2015.04.029
Gutiérrez, J. M., Albulescu, L.-O., Clare, R. H., Casewell, N. R., Abd El-Aziz, T. M., Escalante, T., et al. (2021). The search for natural and synthetic inhibitors that would complement antivenoms as therapeutics for snakebite envenoming. Toxins 13, 451. doi:10.3390/toxins13070451
Gutiérrez, J. M., Calvete, J. J., Habib, A. G., Harrison, R. A., Williams, D. J., and Warrell, D. A. (2017). Snakebite envenoming. Nat. Rev. Dis. Primer 3, 17063–17121. doi:10.1038/nrdp.2017.63
Gutiérrez, J. M., Escalante, T., Rucavado, A., and Herrera, C. (2016). Hemorrhage caused by snake venom metalloproteinases: a journey of discovery and understanding. Toxins 8, 93. doi:10.3390/toxins8040093
Hall, S. R., Rasmussen, S. A., Crittenden, E., Dawson, C. A., Bartlett, K. E., Westhorpe, A. P., et al. 2022. Repurposed drugs and their combinations prevent morbidity-inducing dermonecrosis caused by diverse cytotoxic snake venoms. doi:10.1101/2022.05.20.492855
Harrison, R. A., Casewell, N. R., Ainsworth, S. A., and Lalloo, D. G. (2019). The time is now: a call for action to translate recent momentum on tackling tropical snakebite into sustained benefit for victims. Trans. R. Soc. Trop. Med. Hyg. 113, 835–838. doi:10.1093/trstmh/try134
Harrison, R. A., Gutiérrez, J. M., Suntravat, M., Rodriguez-Acosta, A., Galan, J. A., and Salazar, E. (2019). The neutralization efficacy of expired polyvalent antivenoms: an alternative option. Toxicon Off. J. Int. Soc. Toxinology 168, 32–39. doi:10.1016/j.toxicon.2019.06.216
Harrison, R. A., Oluoch, G. O., Ainsworth, S., Alsolaiss, J., Bolton, F., Arias, A.-S., et al. (2017). Preclinical antivenom-efficacy testing reveals potentially disturbing deficiencies of snakebite treatment capability in East Africa. PLoS Negl. Trop. Dis. 11, e0005969. doi:10.1371/journal.pntd.0005969
Herzel, B. J., Samuel, S. P., Bulfone, T. C., Raj, C. S., Lewin, M., and Kahn, J. G. (2018). Snakebite: an exploratory cost-effectiveness analysis of adjunct treatment strategies. Am. J. Trop. Med. Hyg. 99, 404–412. doi:10.4269/ajtmh.17-0922
Hou, S., Diez, J., Wang, C., Becker-Pauly, C., Fields, G. B., Bannister, T., et al. (2021). Discovery and optimization of selective inhibitors of meprin α (Part I). Pharmaceuticals 14, 203. doi:10.3390/ph14030203
Howes, J.-M., Theakston, R. D. G., and Laing, G. D. (2007). Neutralization of the haemorrhagic activities of viperine snake venoms and venom metalloproteinases using synthetic peptide inhibitors and chelators. Toxicon 49, 734–739. doi:10.1016/j.toxicon.2006.11.020
Janes, J., Young, M. E., Chen, E., Rogers, N. H., Burgstaller-Muehlbacher, S., Hughes, L. D., et al. (2018). The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. 115, 10750–10755. doi:10.1073/pnas.1810137115
Johnson, J. L., Fritsche-Danielson, R., Behrendt, M., Westin-Eriksson, A., Wennbo, H., Herslof, M., et al. (2006). Effect of broad-spectrum matrix metalloproteinase inhibition on atherosclerotic plaque stability. Cardiovasc. Res. 71, 586–595. doi:10.1016/j.cardiores.2006.05.009
Kahraman, A., Bronk, S. F., Cazanave, S., Werneburg, N. W., Mott, J. L., Contreras, P. C., et al. (2009). Matrix metalloproteinase inhibitor, CTS-1027, attenuates liver injury and fibrosis in the bile duct-ligated mouse. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 39, 805–813. doi:10.1111/j.1872-034X.2009.00541.x
Kasturiratne, A., Lalloo, D. G., and Janaka de Silva, H. (2021). Chronic health effects and cost of snakebite. Toxicon X 9, 100074. –10. doi:10.1016/j.toxcx.2021.100074
Kini, R. M., and Koh, C. Y. (2016). Metalloproteases affecting blood coagulation, fibrinolysis and platelet aggregation from snake venoms: definition and nomenclature of interaction sites. Toxins 8, 284. doi:10.3390/toxins8100284
Knudsen, C., and Laustsen, A. H. (2018). Recent advances in next generation snakebite antivenoms. Trop. Med. Infect. Dis. 3, 42. doi:10.3390/tropicalmed3020042
Knudsen, C., Ledsgaard, L., Dehli, R. I., Ahmadi, S., Sørensen, C. V., and Laustsen, A. H. (2019). Engineering and design considerations for next-generation snakebite antivenoms. Toxicon 167, 67–75. doi:10.1016/j.toxicon.2019.06.005
Krige, D., Needham, L. A., Bawden, L. J., Flores, N., Farmer, H., Miles, L. E. C., et al. (2008). CHR-2797: an antiproliferative aminopeptidase inhibitor that leads to amino acid deprivation in human leukemic cells. Cancer Res. 68, 6669–6679. doi:10.1158/0008-5472.CAN-07-6627
Laustsen, A. H. (2018). Toxin-centric development approach for next-generation antivenoms. Toxicon 150, 195–197. doi:10.1016/j.toxicon.2018.05.021
Laxme, R. R. S., Khochare, S., Souza, H.F. de, Ahuja, B., Suranse, V., Martin, G., et al. (2019). Beyond the ‘big four’: venom profiling of the medically important yet neglected Indian snakes reveals disturbing antivenom deficiencies. PLoS Negl. Trop. Dis. 13, e0007899. doi:10.1371/journal.pntd.0007899
Lewin, M., Samuel, S., Merkel, J., and Bickler, P. (2016). Varespladib (LY315920) appears to Be a potent, broad-spectrum, inhibitor of snake venom phospholipase A2 and a possible pre-referral treatment for envenomation. Toxins 8, 248. doi:10.3390/toxins8090248
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26. doi:10.1016/S0169-409X(00)00129-0
Longbottom, J., Shearer, F. M., Devine, M., Alcoba, G., Chappuis, F., Weiss, D. J., et al. (2018). Vulnerability to snakebite envenoming: a global mapping of hotspots. Lancet 392, 673–684. doi:10.1016/S0140-6736(18)31224-8
Mackessy S. P. M., and Stephen P. (2009). Handbook of venoms and toxins of reptiles (Boca Raton: CRC Press). doi:10.1201/9781420008661
Madoux, F., Dreymuller, D., Pettiloud, J.-P., Santos, R., Becker-Pauly, C., Ludwig, A., et al. (2016). Discovery of an enzyme and substrate selective inhibitor of ADAM10 using an exosite-binding glycosylated substrate. Sci. Rep. 6, 11. doi:10.1038/s41598-016-0013-4
Maduwage, K., and Isbister, G. K. (2014). Current treatment for venom-induced consumption coagulopathy resulting from snakebite. PLoS Negl. Trop. Dis. 8, e3220. doi:10.1371/journal.pntd.0003220
Menzies, S. K., Clare, R. H., Xie, C., Westhorpe, A., Hall, S. R., Edge, R. J., et al. (2022). In vitro and in vivo preclinical venom inhibition assays identify metalloproteinase inhibiting drugs as potential future treatments for snakebite envenoming by Dispholidus typus. Toxicon X 14, 100118. doi:10.1016/j.toxcx.2022.100118
Millar, A. W., Brown, P. D., Moore, J., Galloway, W. A., Cornish, A. G., Lenehan, T. J., et al. (1998). Results of single and repeat dose studies of the oral matrix metalloproteinase inhibitor marimastat in healthy male volunteers. Br. J. Clin. Pharmacol. 45, 21–26. doi:10.1046/j.1365-2125.1998.00639.x
Moura-da-Silva, A. M., Theakston, R. D., and Crampton, J. M. (1996). Evolution of disintegrin cysteine-rich and mammalian matrix-degrading metalloproteinases: gene duplication and divergence of a common ancestor rather than convergent evolution. J. Mol. Evol. 43, 263–269. doi:10.1007/BF02338834
Neumann, U., Kubota, H., Frei, K., Ganu, V., and Leppert, D. (2004). Characterization of Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, a fluorogenic substrate with increased specificity constants for collagenases and tumor necrosis factor converting enzyme. Anal. Biochem. 328, 166–173. doi:10.1016/j.ab.2003.12.035
Nguyen, G. T. T., O’Brien, C., Wouters, Y., Seneci, L., Gallissà-Calzado, A., Campos-Pinto, I., et al. (2022). High-throughput proteomics and in vitro functional characterization of the 26 medically most important elapids and vipers from sub-Saharan Africa. GigaScience 11, giac121. doi:10.1093/gigascience/giac121
Oliveira, I. S., Ananias, C. B., Medeiros, J. M., Franco, M. V. S., Ferreira, I. G., Cerni, F. A., et al. (2022). Medical management after lancehead snakebite in north amazon: a case report of long-term disability. Toxins 14, 494. doi:10.3390/toxins14070494
Oliveira, I.S. de, Cardoso, I. A., Bordon, K. de C. F., Carone, S. E. I., Boldrini-França, J., Pucca, M. B., et al. (2019). Global proteomic and functional analysis of Crotalus durissus collilineatus individual venom variation and its impact on envenoming. J. Proteomics, V. ICAP Proteogenomics 191, 153–165. doi:10.1016/j.jprot.2018.02.020
Probst, D., and Reymond, J.-L. (2020). Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminformatics 12, 12. doi:10.1186/s13321-020-0416-x
Rucavado, A., Escalante, T., Franceschi, A., Chaves, F., León, G., Cury, Y., et al. (2000). Inhibition of local hemorrhage and dermonecrosis induced by Bothrops asper snake venom: effectiveness of early in situ administration of the peptidomimetic metalloproteinase inhibitor batimastat and the chelating agent CaNa2EDTA. Am. J. Trop. Med. Hyg. 63, 313–319. doi:10.4269/ajtmh.2000.63.313
Scatena, R. (2000). Prinomastat, a hydroxamate-based matrix metalloproteinase inhibitor. A novel pharmacological approach for tissue remodelling-related diseases. Expert Opin. Investig. Drugs 9, 2159–2165. doi:10.1517/13543784.9.9.2159
Slagboom, J., Kool, J., Harrison, R. A., and Casewell, N. R. (2017). Haemotoxic snake venoms: their functional activity, impact on snakebite victims and pharmaceutical promise. Br. J. Haematol. 177, 947–959. doi:10.1111/bjh.14591
Strandvik, B., and Zetterström, R. (1968). Amaurosis after broxyquinoline. Lancet Issue 7548 291, 922–923. Originally published as Volume 1. doi:10.1016/S0140-6736(68)90280-8
Tang, E. L. H., Tan, C. H., Fung, S. Y., and Tan, N. H. (2016). Venomics of Calloselasma rhodostoma, the Malayan pit viper: a complex toxin arsenal unraveled. J. Proteomics 148, 44–56. doi:10.1016/j.jprot.2016.07.006
Tasoulis, T., and Isbister, G. K. (2017). A review and database of snake venom proteomes. Toxins 9, 290. doi:10.3390/toxins9090290
Tasoulis, T., Pukala, T. L., and Isbister, G. K. (2021). Investigating toxin diversity and abundance in snake venom proteomes. Front. Pharmacol. 12, 768015. doi:10.3389/fphar.2021.768015
Thorne, N., Auld, D. S., and Inglese, J. (2010). Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 14, 315–324. doi:10.1016/j.cbpa.2010.03.020
Trape, J.-F. (2018). Partition d’Echis ocellatus Stemmler, 1970 (Squamata, Viperidae), avec la description d’une espèce nouvelle. Bull. Société Herpétologique Fr. 167, 13–34.
Veber, D. F., Johnson, S. R., Cheng, H.-Y., Smith, B. R., Ward, K. W., and Kopple, K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623. doi:10.1021/jm020017n
Williams, D. J., Gutiérrez, J.-M., Calvete, J. J., Wüster, W., Ratanabanangkoon, K., Paiva, O., et al. (2011a). Ending the drought: new strategies for improving the flow of affordable, effective antivenoms in Asia and Africa. J. Proteomics 74, 1735–1767. doi:10.1016/j.jprot.2011.05.027
Williams, J. M., Zhang, J., North, P., Lacy, S., Yakes, M., Dahly-Vernon, A., et al. (2011b). Evaluation of metalloprotease inhibitors on hypertension and diabetic nephropathy. Am. J. Physiol.-Ren. Physiol. 300, F983–F998. doi:10.1152/ajprenal.00262.2010
Winer, A., Adams, S., and Mignatti, P. (2018). Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future Successes. Mol. Cancer Ther. 17, 1147–1155. doi:10.1158/1535-7163.MCT-17-0646
Xie, C., Albulescu, L., Bittenbinder, M. A., Somsen, G., Vonk, F., Casewell, N. R., et al. (2020). Neutralizing effects of small molecule inhibitors and metal chelators on coagulopathic Viperinae snake venom toxins (preprint). Pharmacol. Toxicol. doi:10.1101/2020.06.02.129643
Yang, Z.-Y., He, J.-H., Lu, A.-P., Hou, T.-J., and Cao, D.-S. (2020). Frequent hitters: nuisance artifacts in high-throughput screening. Drug Discov. Today 25, 657–667. doi:10.1016/j.drudis.2020.01.014
Keywords: venom, toxin, snakebite envenoming, SVMP, small molecule drugs, neglected tropical diseases
Citation: Clare RH, Dawson CA, Westhorpe A, Albulescu L-O, Woodley CM, Mosallam N, Chong DJW, Kool J, Berry NG, O’Neill PM and Casewell NR (2024) Snakebite drug discovery: high-throughput screening to identify novel snake venom metalloproteinase toxin inhibitors. Front. Pharmacol. 14:1328950. doi: 10.3389/fphar.2023.1328950
Received: 27 October 2023; Accepted: 21 December 2023;
Published: 11 January 2024.
Edited by:
Michelle Yap, Monash University Malaysia, MalaysiaReviewed by:
Manuela Berto Pucca, Sao Paulo State University, BrazilCopyright © 2024 Clare, Dawson, Westhorpe, Albulescu, Woodley, Mosallam, Chong, Kool, Berry, O’Neill and Casewell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel H. Clare, cmFjaGVsLmNsYXJlQGxzdG1lZC5hYy51aw==; Nicholas R. Casewell, TmljaG9sYXMuQ2FzZXdlbGxAbHN0bWVkLmFjLnVr
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.