 Marco Echeverria-Villalobos1*
Marco Echeverria-Villalobos1* Victor Tortorici2,3
Victor Tortorici2,3 Beatriz E. Brito4
Beatriz E. Brito4 David Ryskamp5
David Ryskamp5 Alberto Uribe1
Alberto Uribe1 Tristan Weaver1
Tristan Weaver1- 1Anesthesiology Department, The Ohio State University Wexner Medical Center, Columbus, OH, United States
- 2Neuroscience Laboratory, Faculty of Science, Department of Behavioral Sciences, Universidad Metropolitana, Caracas, Venezuela
- 3Neurophysiology Laboratory, Center of Biophysics and Biochemistry, Venezuelan Institute for Scientific Research (IVIC), Caracas, Venezuela
- 4Immunopathology Laboratory, Center of Experimental Medicine, Venezuelan Institute for Scientific Research (IVIC), Caracas, Venezuela
- 5College of Medicine, The Ohio State University, Columbus, OH, United States
Current evidence suggests that activation of glial and immune cells leads to increased production of proinflammatory mediators, creating a neuroinflammatory state. Neuroinflammation has been proven to be a fundamental mechanism in the genesis of acute pain and its transition to neuropathic and chronic pain. A noxious event that stimulates peripheral afferent nerve fibers may also activate pronociceptive receptors situated at the dorsal root ganglion and dorsal horn of the spinal cord, as well as peripheral glial cells, setting off the so-called peripheral sensitization and spreading neuroinflammation to the brain. Once activated, microglia produce cytokines, chemokines, and neuropeptides that can increase the sensitivity and firing properties of second-order neurons, upregulating the signaling of nociceptive information to the cerebral cortex. This process, known as central sensitization, is crucial for chronification of acute pain. Immune-neuronal interactions are also implicated in the lesser-known complex regulatory relationship between pain and opioids. Current evidence suggests that activated immune and glial cells can alter neuronal function, induce, and maintain pathological pain, and disrupt the analgesic effects of opioid drugs by contributing to the development of tolerance and dependence, even causing paradoxical hyperalgesia. Such alterations may occur when the neuronal environment is impacted by trauma, inflammation, and immune-derived molecules, or when opioids induce proinflammatory glial activation. Hence, understanding these intricate interactions may help in managing pain signaling and opioid efficacy beyond the classical pharmacological approach.
1 Introduction
According to the International Association for the Study of Pain (IASP), chronic pain persists or recurs for over 3 months and is a leading source of human suffering and disability (Treede et al., 2019). A cornucopia of etiological factors has been implicated in the genesis of chronic pain. It may be the primary symptom of a non-specific underlying disease (i.e., fibromyalgia), a sign of a progressive local/systemic disorder (i.e., rheumatic inflammatory diseases, Diabetes Mellitus), or secondary to direct nerve injury (Vergne-Salle and Bertin, 2021). More recently, a new pain mechanism identified as “nociplastic pain” has been proposed. It occurs in individuals with an abnormally elevated nociception sufficient to activate peripheral nociceptors without clear evidence of tissue damage, neural injury, or disease (Kosek et al., 2016). Although central sensitization is not included in the definition of nociplastic pain, patients with this condition frequently refer to signs of central sensitization, suggesting that nociceptive and neuroplastic pain may coexist (Kosek et al., 2016). Unlike acute pain, which functions as a defensive mechanism, chronic pain is considered maladaptive because it does not provide additional protective or recuperative benefits (Walters, 2019). Peripheral sensitization is the most common mechanism of pain initiation; nonetheless, the chronicity of pain is mainly determined by central sensitization. The neuroinflammatory response also gives rise to structural and functional maladaptive changes in the peripheral and central somatosensory pathways, modifying the pain-signaling process. The existing evidence suggests that activation of glial cells and other non-neuronal cells, such as immune cells (neutrophils, macrophages, T-cells, and mast cells), leads to increased production and release of proinflammatory mediators that play a crucial role in developing and maintaining neuropathic and chronic pain (Hains et al., 2006; Chiu et al., 2012; Du et al., 2023).
The main goal of this article is to review the current evidence on the molecular mechanisms that support the role of neuroinflammation as an important etiopathogenic factor in the transition from acute postoperative pain to chronic postoperative pain (CPSP). Similarly, the neuroimmune interactions associated with the decrease in the analgesic effect of opioid drugs are considered, including those involved in the development of opioid-induced inflammation, opioid tolerance, and paradoxical hyperalgesia.
2 The leading role of the immune response and glial activation in the transition from acute to neuropathic and chronic pain
Acute pain after surgery is almost a sine qua non condition; however, this kind of pain is ineffectively treated. It transitions to a state of chronic pain, often with neuropathic characteristics, that is refractory to treatment with opioids (Haroutiunian et al., 2013; Honkanen et al., 2021). The incidence of pain persisting beyond the period of wound healing after surgery is approximately 10%–85%, depending on the type of surgery. This has been typified as CPSP, with 5%–10% of cases rating the pain as severe, affecting patients quality of life and creating a significant economic burden for the health systems (Kehlet et al., 2006; Johansen et al., 2012; Jin et al., 2021; Jin et al., 2023). The most recent revision of the International Classification of Diseases (ICD-11) defines CPSP as “pain developing or increasing in intensity after a surgical procedure, in the area of the surgery, persisting beyond the healing process (i.e., at least 3 months) and not better explained by another cause such as infection, malignancy or a pre-existing pain condition”. Similarly, the IASP defines it as “chronic pain that develops or increases in intensity after a surgical procedure or a tissue injury and persists beyond the healing process, i.e., at least 3 months after the surgery or tissue trauma” (Schug et al., 2019).
Currently, overwhelming evidence supports the fundamental role of neuroinflammation in pain; however, its etiopathogenic mechanisms are not yet fully elucidated. A neuroinflammatory state is characterized by the activation of glial cells, generation of proinflammatory mediators (cytokines and chemokines, among others), and changes in the vasculature, resulting in increased permeability and leukocytic infiltration, as well as alterations in gene expression (Ji et al., 2006; Ji et al., 2018; Donnelly et al., 2020). Increasing evidence shows that glial tissue activation (including microglia, astrocytes, oligodendrocytes, satellite glial cells, and ependymal cells) plays a pivotal role in developing peripheral and central sensitization (Figure 1).
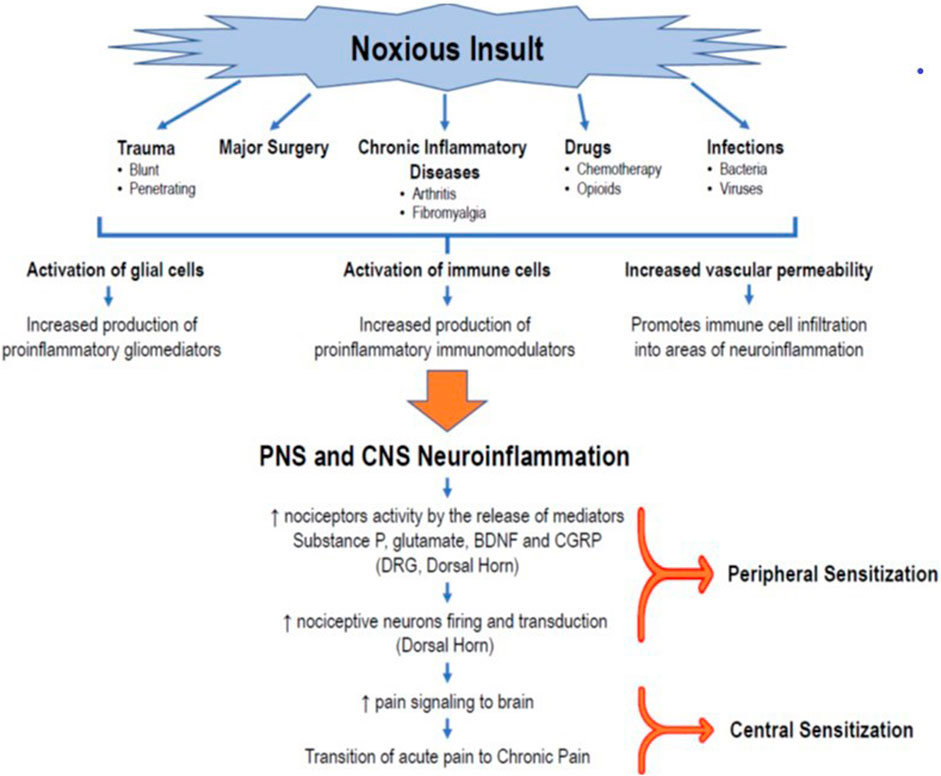
FIGURE 1. Role of neuroinflammation in the transition to CPSP. The occurrence of a noxious insult, such as trauma, major surgery, chronic inflammatory diseases, drugs (chemotherapy, opioids), or infections, triggers an initial immunoinflammatory response that, in turn, promotes the activation of glial cells (microglia, astrocytes, oligodendrocytes) in the peripheral sensory system, creating a state of peripheral neuroinflammation. At the same time, the production and release of inflammatory mediators produce changes in vascular permeability that facilitate the infiltration of immune cells that activate the glial cells of the CNS, generating the release of more proinflammatory mediators and a process of neuroinflammation in the CNS. This milieu of cytokines, chemokines, neuropeptides (substance P, CGRP), and neurotrophic factors leads to changes in the plasticity of second-order neurons in the posterior horn of the spinal cord. This results in an exaggerated firing activity and amplification of nociceptive signaling in the DRG and the posterior horn of the spinal cord, establishing the peripheral sensitization phenomenon. The transmission of painful signals to the brain, together with the process of neuroinflammation, keeps the nociceptor neurons of the brain’s pain centers stimulated, giving rise to a state of central desensitization, and promoting the transition from acute pain to neuropathic or chronic pain. BDNF = brain-derived neurotrophic factor; CGRP = calcitonin gene-related peptide; CNS = central nervous system, DRG = dorsal root ganglion.
2.1 Role of immune cells
In the perioperative period, harmful events, such as surgical incisions and organ manipulation, cause tissue and nerve damage, which triggers the activation of immune cells, glial cells, and nociceptive neurons. These cells then set forth the release of pro-inflammatory mediators causing a pervasive state of inflammation in the peripheral and central nervous system. If the inflammation is not adequately resolved and central sensitization is established, the nociceptive state may progress from acute postoperative pain to CPSP once (Ji et al., 2006; Ji et al., 2013; Ji et al., 2016; Pinho-Ribeiro et al., 2017; Ji et al., 2018; Ji, et al., 2019). Additionally, recent evidence from preclinical studies shows that epidermic resident cells, keratocytes, and dendritic cells (DCs) play an active role in inflammatory pain arising from surgical tissue injury as well as in the development of neuropathic pain and CPSP (Manjavachi et al., 2014; Hesselink et al., 2016; Guo et al., 2020; Silva et al., 2022). Keratocytes lie close to sensory afferent nerves and when stimulated by surgical incision initiate a nociceptive response mediated by the production and release of various neuroactivator agents such as cytokines (TNF-α and IL-1β), calcitonin gene-related peptide receptor (CGRP), acetylcholine, adenosine triphosphate (ATP), nerve growth factors, neuropeptides, and other neurotransmitters contributing to peripheral neuroinflammation (Guo et al., 2020). Studies in animal models have shown that activated keratocytes can also elicit afferent firing and sensitization, resulting in postsurgical incisional hypersensitivity (Hesselink et al., 2016; Ritter-Jones et al., 2016; Guo et al., 2020; Silva et al., 2022). DCs and Langerhans cells are another group of skin-resident cells that become upregulated after tissue injury and can directly activate peripheral nociceptive neurons (nociceptors), releasing chemokines CCL17 and CCL22 through the shared receptor CCR4, contributing to neuroinflammation and postoperative pain (Silva et al., 2022).
Activated immune cells (macrophages, neutrophils, T-cells, and mast cells) also produce and release pro-inflammatory mediators that interact with peripheral nerve terminals and their somas located in the dorsal root ganglia (DRG), which relay in laminae I and II of the dorsal horn of the spinal cord (Pinho-Ribeiro et al., 2017). DRG macrophages are critical contributors to the initiation and persistence of neuropathic pain in male and female mice, without the sexual dimorphism reported in microglial cells (Yu et al., 2020). It has been recently demonstrated that immediately after a nerve injury, there is a significant proliferation of resident macrophages in the DRG ipsilateral to the nerve lesion, which produces proinflammatory cytokines such as TNF-α, IL-1β, and HMGB1 (Hu and McLachlan, 2003; Guimarães et al., 2023). The proximity of macrophages to the cell bodies of primary nociceptors in the DRG facilitates the activation of primary sensory neurons by the proinflammatory cytokines (Chen et al., 2018)”.
Macrophages are tissue immune cells that are stimulated early when nerve injury occurs. Until recently, macrophages were thought to be two polarized forms of immune cells (M1 and M2) with distinct phenotypic qualities and functional properties. Accordingly, M1 activity inhibited cell proliferation and caused tissue damage, whereas M2 activity promoted cell proliferation and tissue repair. However, recent evidence has shown that the expression of specific polarization markers for M1/M2 phenotypes in vitro observations can be influenced not only by a specific stimulus but even by a specific stimulation time sequence (Purcu Duygu et al., 2022).
In vivo, the situation is even more complex, as there is a wide range of different macrophages depending on the conditions of the microenvironment (Strizova et al., 2023). Moreover, most surface markers identified on macrophages generated in vitro do not translate to the situation in vivo (Orecchioni et al., 2020). These facts suggest that macrophages exhibit phenotypic plasticity and can adopt different activation states in response to different conditions. Therefore, the current phenotypic status encompasses an expanded spectrum of possibilities that includes multiple subsets of macrophages identified by their different activation states, functional properties, and surface marker expression. To further complicate matters, certain subsets of macrophages may not be completely distinct from one another and may share common functional activities.
These dualistic implications also apply to microglia, the macrophages of the brain, whose phenotype is also influenced by the central nervous system microenvironment. The repertoire of microglial states and functions goes beyond the dichotomy of “resting vs activated,” “M1 vs M2,” or “good vs bad microglia,” as demonstrated by the impressive work of Paolicelli et al. (2022), a group of multidisciplinary experts who advance the understanding of microglial states as a dynamic concept, emphasize the importance of considering microglial function, and provide a new conceptual framework for this type of dichotomy.
Therefore, the large phenotypic heterogeneity exhibited by macrophages might lead to oversimplification in light of recent observations. In the transitional phase, it might be appropriate, albeit risky, to describe macrophages as M1-like and M2-like to define their different roles rather than using the conventional M1 and M2 nomenclature (Strizova et al., 2023). However, the latter would be very helpful when referring to previous work reporting on the dichotomy.
It is normally accepted that M1-like macrophages liberate proinflammatory cytokines (TNFα, IL-1β, IL-6), chemokines (CCL2, CCL3, CCL4), and nitric oxide (NO). They also respond to damage-associated molecular patterns (DAMPs), and pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS). This results in further recruitment and activation of other immune cells (neutrophils, monocytes, mast cells, T-cells) and unmyelinated afferents, causing the sensitization of primary nociceptive neurons at the DRG (Nicol et al., 1997; Oh et al., 2001; Obreja et al., 2002; Zhang and An, 2007). In a rodent model of arthritis, a proinflammatory macrophage phenotype similar to the M1-like subtype was found in the rat DRG, and activation of these macrophages stimulated DRG neurons to release calcitonin gene-related peptide (CGRP), which plays an active role in the persistence of the pain state (Massier et al., 2015). Conversely, M2-like macrophages promote analgesia in response to IL-4, which stimulates the production of high levels of anti-inflammatory cytokines, such as IL-10 (Celik et al., 2020). IL-4 has also been shown to induce the M2-like subtype to produce endogenous opioid peptides that bind to peripheral opioid receptors, further contributing to pain relief by deactivating neuropathy-triggered mechanical hypersensitivity (Pannell et al., 2016; Celik et al., 2020). Furthermore, the action of the cytokine IL-4 also promotes the transcription of M2-like associated genes while reducing the transcription of M1-like associated genes (Kiguchi et al., 2015).
Neutrophils are polymorphonuclear leukocytes representing the first-line cells in the innate immune response to tissue damage or infections. However, their role in neuroimmune interactions has only recently been studied in greater detail (Chavan et al., 2017; Pavlov et al., 2018). Neutrophils are almost always absent around the intact nerves. However, when tissue damage occurs, they become activated by locally produced mediators such as proinflammatory cytokines, growth factors, leukotrienes B4 (LB4), DAMPs, and prostanoids such as prostaglandin E2 (PGE2). That milieu of mediators enhances neutrophil migration toward the sites of inflammation. Subsequent neutrophil infiltration of afferent nerve endings activates and sensitizes the terminals of the peripheral nociceptors and increases vascular permeability (Kalaczkowska and Kubes, 2013). Once activated, neutrophils continue releasing mediators, contributing to nociceptive amplification (Levine et al., 1984; Grace et al., 2014). Several studies have demonstrated that endoneural neutrophil invasion of peripheral nerves and DRGs occurs after induced chronic constriction injury of peripheral nerves. This neutrophil infiltration is correlated with an increase in the production of monocyte chemoattractant protein-1 (MCP-1, also known as CCL2) and neutrophil-derived elastase. Both are regarded as essential mediators in the development of hyperalgesia in animal models of neuropathic pain (Perkins and Tracey, 2000; Morin et al., 2007; Bali and Kuner, 2017). Contrarily, some researchers have questioned the role of cytokines in mechanical hyperalgesia while assigning a more important role to the release of bradykinin, PGE2, and sympathomimetic amines such as adrenaline, epinephrine, and norepinephrine (Green, 1974; Cunha et al., 2008). Neutrophils also participate in the process of neuroinflammation and post-incisional hypersensitivity through the production of other mediators such as metalloproteases, reactive oxygen species (ROS), hydrogen, and endothelins, which further increase nociceptors excitability (Steen et al., 1992; Wang et al., 2004; Woo et al., 2004; Mujenda et al., 2007). In the CNS, neutrophil infiltration occurs as a result of changes in the permeability of the blood-brain barrier (BBB) secondary to the neuroinflammation process, and the chemoattractant effect of chemokines CXCL2 and CXCL6, released by meningeal mast cells and astrocytes. This neutrophilic invasion causes demyelination and axonal damage (Simmons et al., 2014; Pierson et al., 2018).
T lymphocytes (T-cells) are part of the adaptive immune system. Studies have shown that mechanical allodynia after nerve injury is associated with infiltration of the DRG by T-cells (Vicuña et al., 2015). However, preclinical studies in mice demonstrated that the active involvement of T-cells in the pathogenesis of neuropathic pain depends on the subtype of T-cells and the mouses gender (Sorge et al., 2015). Interestingly, T-cells also actively participate in the pain resolution process. After a period of hypernociception, the PNS and CNS can enter into a state of apparent deactivation (remission), also called “latent sensitization” (Marvizon et al., 2015). In this state, any noxious or stressful event leading to the reactivation of nociceptors, including the use of naloxone, will trigger a prolonged hyperalgesic response with changes in the expression of pain-related genes (Parada et al., 2003; Rivat et al., 2007; Price and Ray, 2019). T-cells and macrophages generate endogenous opioid peptides such as β-endorphins and enkephalins, which play a key role in preventing the reappearance of pain during latent sensitization (Stein et al., 1990; Ritter-Jones et al., 2016). Likewise, the anti-inflammatory cytokine IL-4, produced by mast cells, granulocytes, and T helper 2 (Th2) cells, exerts a modulatory effect on proinflammatory mediators and induces M2-like macrophages to release opioid peptides that attenuate pain (Celik et al., 2020).
Mast cells are located very close to nociceptive neurons; once activated, they release neuroactive proinflammatory cytokines (TNF-α, IL-1β, IL-6), chemokines, histamine, bradykinin, proteases, nerve growth factor (NGF), and substance P (SP). This release of proinflammatory cytokines generates the initial nociceptive signaling that modulates thermal and mechanical sensitivity through specific receptors (Chatterjee and Martinov, 2015). Crosstalk between mast cells and nociceptive neurons is established through their common receptor NK1. In addition, receptors located in the mast cells (S1P1, S1P2, and CRHR) interact with cytokines, chemokines, and NGF released by nociceptive neurons, sustaining the hyperactivation of nociceptors in the DRG and spinal dorsal horn and causing neurogenic inflammation (Kempuraj et al., 2019). Several authors have reported that substance P released from primary afferent nerve terminals not only interacts with its canonical receptor NK1 but can also activate the mast cell-specific receptor Mrgprb2 to promote the release of proinflammatory cytokines and chemokines, as well as facilitate the migration of immune cells to nociceptors (McNeil et al., 2015; Green et al., 2019). Additionally, NGF released by mast cells plays a primary role in peripheral sensitization by allowing the phosphorylation of transient receptor potential vanilloid 1 (TRPV1), which is a determinant factor in pain transduction. At the same time, TNF-α, IL-1β, and IL-6 also activate TRPV1 and other channel receptors like transient receptor potential ankyrin 1 (TRPA1), and sodium channels Nav1.7, Nav1.8, Nav1.9 in the nociceptor. Activation of these targets further contributes to peripheral sensitization and enhances signaling between nociceptor cell bodies in the DRG and the cerebral cortex where it will be processed as a pain sensation, subsequently resulting in central sensitization (Basbaum et al., 2009; Ji, et al., 2016; Pinho-Ribeiro et al., 2017; Locke et al., 2020).
Another determinant factor in the central sensitization process is the activation of Toll-Like Receptors (TLRs) in neurons and glial cells (Lacagnina et al., 2018; Zhang et al., 2020; Liu et al., 2022). Increased TLR signaling in macrophages, glial cells, and sensory neurons, accompanied by decreased action potential thresholds in nociceptive neurons, leads to increased nociceptive excitability and firing. The resulting exaggerated nociceptive response is a critical factor in the transition to persistent pain (Pinho-Ribeiro et al., 2017; Lacagnina et al., 2018; Locke et al., 2020). Furthermore, disruption of the BBB during the neuroinflammatory response allows for massive entry of immune cells into the CNS. The subsequent direct activation of nociceptive neurons and glial cells along the cerebral pathways in the brain worsens neuroinflammation and central sensitization, which perpetuates pain signaling, and facilitates the transition to chronic pain (Patel et al., 2015; Kempuraj et al., 2017; Gupta and IkkaHarvima, 2018; Mastrangelo et al., 2018).
In a rodent model, a proinflammatory phenotype similar to M1 in activated macrophages in the DRG was demonstrated, promoting the production of CGRP, which plays an active role in the persistence of the pain state (Massier et al., 2015).
2.2 Glial cell activation is essential in the transition from acute to chronic pain
Based on emerging evidence, the activation of glial cells is a phenomenon closely related to the origin and persistence of neuropathic and chronic pain, therefore it might be considered a “gliopathy” (Ji, Berta, and Nedergaard, 2013). Under normal conditions, the glial cells, especially microglia, provide microenvironmental conditions that promote neuronal development, synaptic pruning, and circuit formation, as well as the modulation of synaptic connectivity, neurotransmission, and neuroplasticity (Um, 2017). Microglial cells in the CNS emulate the phagocytic function of peripheral macrophages, helping in clearing the neural environment of damaged cells, microbial agents, and debris (Hanisch and Kettenmann, 2007). Astrocytes provide structural and metabolic support to glutamatergic synaptic transmission, regulating the extracellular concentration of glutamate (Vandenberg and Ryan, 2013; Ji et al., 2019). At the same time, oligodendrocytes speed up the synaptic transmission of the electrical impulse and are actively involved in pain in various ways. The production of IL-33 by oligodendrocytes in the spinal cord mediates the activation of microglial ST2 receptors in a mouse model of neuropathic pain (Malta et al., 2019). Glial cells also release anti-inflammatory cytokines that play an active role in repairing neurotoxic damage caused by neuroinflammation (Tiwari et al., 2014). Both in the DRG and in the trigeminal ganglion, satellite glial cells (SGCs) are located near the nuclei of nociceptive neurons, creating a neural structure unique to the PNS. Under normal conditions, this neuron-SGC coupling helps to maintain neuronal homeostasis, particularly by protecting axonic insulation and the integrity of the neural soma. Recent preclinical studies showed that SGCs have a key role in the neural repair process (Xiao et al., 2015; Hanani and Spray, 2020; Gazerani, 2021). Activation of SGCs after nerve damage leads to changes in K+ channels and increased release of cytokines and ATP (Mujenda et al., 2007). Immediately following nerve injury, there is an upregulation of the ATP receptor subtype P2X4 in spinal microglia, which influences microglial signaling to promote mechanical allodynia (Tsuda et al., 2003).
This functional relationship between neurons and SGCs contributes to the development of neuronal hyperactivity, and both peripheral, as well as the chronification of pain (Mujenda et al., 2007). After a peripheral nerve lesion, the degeneration produced by TNF-α and IL-1β release from the Schwann cells (SCs) significantly contributes to the progression to neuropathic pain (Fang et al., 2023).
Shortly after nerve damage, signs of microgliosis appear in the ipsilateral dorsal horn of the spinal cord within 2–3 days. It reaches peak levels in 4–7 days, before progressively declining weeks to months after the nerve lesion (Kohno et al., 2018). Spinal microglia activation originates from proinflammatory cytokines, chemokines, extracellular proteases, purines, excitatory neurotransmitters, and neuropeptides released by macrophages, natural killer cells (NK), and T-cells (Grace et al., 2014). T Helper 1 cells (Th1) also produce interferons (IFNs) that react with type 1FN receptors (IFNR) expressed by microglia, astrocytes, and neurons (Tan et al., 2021). IFNs vary in their effect when interacting with IFNR. IFNγ exhibits proinflammatory actions that activate glial cells and nociceptive neurons, contributing to the development of pain. IFNα and IFNβ promote the inactivation of microglia and astrocytes, as well as the inhibition of synaptic transmission in the spinal cord. In effect, they encourage restoration of the peripheral and central sensitization processes, leading to the resolution of neuropathic or chronic pain (Tsuda et al., 2009; Tan et al., 2021).
The complex array of inflammatory glial and immune mediators also includes other signaling molecules originating from the damaged nerve tissue, such as DAMPs and PAMPs (including LPS) (Ji et al., 2019; Gong et al., 2020; Jurga et al., 2020). Neural damage results in the overexpression of peripheral and central neurotrophic factors generated by neural damage, such as NGF, brain-derived neurotrophic factor (BDNF), and glial cell line-derived neurotrophic factor (GDNF) (Salio et al., 2014), neurotransmitters and neuropeptides (substance P, glutamate, CGRP) (Gwak et al., 2017). Additionally, activated astrocytes located in the presynaptic sensory neurons of the DRG and dorsal horns generate chemokines that intervene in microglial and oligodendrocyte activation (Chen et al., 2014; Ji et al., 2018; Ji et al., 2019; Liu et al., 2019; Malta et al., 2019). Upregulation of ATP receptor subtype P2X2 in spinal microglia immediately after nerve injury is also involved in microglial signaling that leads to mechanical allodynia (Tsuda et al., 2003). As mentioned before, once the microglial cells are activated, a highly dynamic process begins that leads to the production of multivariate morphological, metabolic, and functional states (Paolicelli et al., 2022) with a unique role in maintaining or repairing the neuroinflammatory state. These include the classically activated proinflammatory M1-like phenotype, and the intermittently activated M2-like phenotype microglia with anti-inflammatory and reparative effects (Willemen et al., 2014; Chen et al., 2018). M2-like phenotype microglia produce inflammation, cytotoxicity, and vascular changes leading to increased BBB permeability, and prolongation of the immune response (Jurga et al., 2020).
The synthesis and release of all these inflammation by-products in the PNS and CNS significantly affect synaptic transmission and interneuronal networking excitability, influencing the initiation and maintenance of pain (Coull et al., 2005; Ji et al., 2006; Pezet and McMahon, 2006; Kawasaki et al., 2008; Vezzani and Viviani, 2015). The resulting neuroinflammatory state creates an exaggerated afferent input that promotes changes in plasticity in nociceptive neurons and synaptic transmission at the spinal cord level, increasing hyperexcitability of nociceptors, enhancement of signaling transduction to the brain and facilitating the development of central sensitization, which perpetuates the pain state and the transition to neuropathic or chronic pain (von Banchet et al., 2009).
2.3 The resolution phase of the neuroinflammatory damage
After peripheral nerve injury, transected axons elicit degeneration of the distal nerve endings and in the axotomized nociceptors bodies in the DRG. The post-injury neuroinflammation state that occurs can originate deleterious consequences (neuropathic and/or chronic pain) and beneficial effects triggering reparative processes in the injured peripheral nerve as well as in the DRGs nociceptive neurons. The axonal regeneration and neuron functional recovery is not an autonomous process and depends on immune cells, especially macrophages, and glial cells (Schwann cells). On day 3 after nerve injury, macrophages accumulate, most at the distal end and less proximal to the lesion (Taskinen and Roytta, 1997). Almost at the same time (day 4), macrophage accumulation can be detected at the DRG (Lu and Richardson, 1993). This noxious event activates chemokines CCL2 in the nerve injury region, Schwann cells, and in the DRG, which binds to CCR2, G-protein-coupled receptors located in the macrophages, attracting them to the distal nerve cell body area as well as to the DRG (Ransohoff, 2009). Macrophages and Schwann cells facilitate the removal of axon debris, myelin phagocytosis, and the clearance of molecules from degenerating axons that inhibit neural regeneration and axonal outgrowth in the nerve endings and the DRG (Zigmond and Echevarria, 2019). Post-traumatic neural regeneration is almost an exclusive property of the PNS, however, not all injured axons achieve complete regeneration (Gordon et al., 2009) since the neural repair mechanism in the PNS is not only slow but often incomplete. Resident macrophages are cells found in peripheral nerves and ganglia while infiltrating macrophages access the neural tissue after a nerve injury or infection, outnumbering the resident macrophages. Although resident macrophages are the “first responder cells” after nerve injury, later, infiltrating macrophages outnumber them (Mueller et al., 2003). Unlike peripheral axonal damage or transection, there is no macrophage accumulation with crushing damage to the dorsal root neurons, in which regeneration of centrally projecting axons does not occur (Kwon et al., 2013).
During the resolution phase that follows the initial post-injury activation, M1-like macrophages can transition into M2-like macrophages (Van der Bossche et al., 2016). Several recent studies showed that infiltrating macrophages attracted to the nerve injury and to the DRG express M2-like subtype which not only release antiinflammatory cytokines (IL-4, IL-10, IL-13, and TGF-β) depending upon the pathologic state (Willemen et al., 2014). M2-like macrophages also release other mediators that promote repair of the axonal and neuron damage such as neurotrophic factors, growth factors, colony-stimulating 1 (CSF-1), and progranulin (Kwon et al., 2013; Martinez and Gordon, 2014; Wynn and Vannella, 2016; Jurga et al., 2020). Oncomodulin, secreted by macrophages and granulocytes, promotes neuron outgrowth in axotomized sensory neurons in the DRG (DeFrancesco-Lisowitz et al., 2015; Kwon et al., 2013). In vitro studies demonstrated that cytokine IL-1β secreted by macrophages stimulates the production of NGF and other neurotrophins such as BNDF, NT3, and NT4/5, which enhances regeneration at the distal nerve stump (Barrette et al., 2008). A recent study conducted by Feng et al. reported that the self-renovation of resident macrophages in the DRG is a contributing factor to axonal regeneration very similar to the self-renewal of glial cells in the brain (Feng et al., 2023).
Microglial polarization into the M2-like phenotype is also stimulated by the presence of IL-4 and IL-13 cytokines produced by T-cells (Th2) (). After activation, the M2-like microglial phenotype releases antiinflammatory cytokines (IL-10 and TGFβ), (Saijo et al., 2013; Jurga et al., 2020).
M2-like phenotype microglia plays a decisive role in repairing neural damage by inhibiting neuroinflammation, restoring neuronal homeostasis, removing cellular debris through phagocytosis, and protecting the extracellular matrix (Jurga et al., 2020).
Other components involved in the resolution phase of neuroinflammation are resolvins and protectins, which are a group of molecules derived from omega-3 fatty acids. Resolvins and protectins represent one part of a biochemical arsenal that functions to restore homeostasis once the initial inflammatory response is over (Sommer and Frank, 2011). New data suggest these substances might have future applications as analgesic drugs that reduce inflammatory pain by blocking TRP channels and NMDA receptors in somatosensory neurons located in the dorsal horn of the spinal cord (Ji et al., 2011; Sommer and Frank, 2011). Additionally, resolvins and protectines exhibit a neuromodulator-like profile that affects only pathological pain sensations, but not normal sensations evoked by painful stimuli (Ji et al., 2016; Roh et al., 2020).
The transition from acute to chronic pain can start shortly after the event that triggered the onset of the acute pain (2–3 weeks) (Price and Ray, 2019). Effective modulation of the neuroinflammation and initiation of the restorative process requires a balanced expression of M1-like and M2-like microglial phenotypes. A recent preclinical study by Li et al. in rats revealed that activated microglia mediate the transformation of spinal cord astrocytes predominantly into the A1 phenotype, which promotes neuroinflammation and neurotoxicity, and favors the appearance of CPSP, and to a lesser extent into the A2 phenotype, which provides neuroprotection and restoration (Li et al., 2020). The existing preclinical and clinical evidence suggests that after this transitional process, the maintenance of the chronic pain state is mainly attributed to central sensitization; however, recent clinical studies using neuroimaging have shown that peripheral nerve blocks in patients with neuropathic pain provide pain relief. This suggests that sustained primary afferent output from nociceptors and altered neuroplasticity, characterized by hyperexcitability and over-signaling in the synaptic relays at the spinal cord play a crucial role in the development of chronic and neuropathic pain after surgery (Haroutiunian et al., 2013; Haroutiunian et al., 2014; Vaso et al., 2014; Ratte and Prescott, 2016).
3 Importance of the enteric nervous system in acute and chronic pain
Increasing evidence supports the role of continuous functional interdependence of the microbiota, enteric nervous system (ENS), PNS, and CNS. The so-called microbiota-gut-brain axis is composed of the colonic myenteric plexuses, the dorsal root ganglion, the nucleus solitary tract (NST), and the periaqueductal grey. It constitutes an important factor in the pathogenesis of acute visceral, nociplastic, neuropathic, and chronic pain; however, most studies remain in the realm of research in animal models of pain (Harotounian et al., 2014; Vaso et al., 2014; Ratte and Prescott, 2016). The intrinsic structure of the ENS consists of a neural network of resident neurons and glial cells located in the intestinal mucosa and the inner muscularis propria. The ENS integrates and transduces immune, inflammatory, and neuroendocrine signals that reach the brain tissue through the vagus nerve and the BBB, producing alterations in its permeability and allowing the translocation of immune and inflammatory mediators to the brain tissue. Current evidence also suggests that enteric neuronal plasticity and glial activation are fundamental players in the development of neuropathic and chronic pain (Morales-Soto and Gulbransen, 2019). The alleged mechanisms proposed to activate enteric neurons and glial cells include the production of immune and inflammatory mediators and neuromodulatory by-products of bacterial metabolism (Grubišić and Gulbransen, 2017). Enteric neuronal plasticity is directly involved in sensitizing visceral afferent sensory nerve fibers, augmenting neural sensitivity, and increasing firing patterns in the peripheral pain neural networking, spinal cord, and brain, which are all regulated by glial cell hyperactivity (Grubišić and Gulbransen, 2017; Morales-Soto and Gulbransen, 2019). Subtle differences in microbiota composition, amino acid levels, and neurotransmitters have been detected in individuals suffering from several chronic pain syndromes, such as fibromyalgia (Clos-Garcia et al., 2019; Ji et al., 2019; Minerbi et al., 2019; Bistoletti et al., 2020). Qualitative and quantitative alterations in the microbiota, or dysbiosis, stimulate the production and release of proinflammatory substances by enteric immune and glial cells. Some noxious events, including surgical trauma and bowel surgery, produce dysbiosis and activation of IL-1 receptor type I (IL-1R1), which induces a reactive phenotype of enteric glial cells (EGCs) called enteric gliosis (Schneider et al., 2021). Once activated, EGCs release cytokines, chemokines, and colony-stimulating factors 1 and 3 (CSF1, CSF3) which are key elements modulating macrophage activation (Grubišić and Gulbransen, 2017; Schneider et al., 2022). The bidirectional communication between EGCs and macrophages is crucial to enteric neuroinflammation and visceral afferent nerve hypersensitivity (Schneider et al., 2022). Vagal afferent neurons (VAN) are the enteric primary nociceptive neurons that integrate gut signaling and connect VAN via the vagus nerve to the NST in the brainstem. In this way, dysbiosis modulates inflammation in the afferent vagal nerve and the transmission of noxious information from the gut to the brain (Kim et al., 2020). The visceral nociceptor neurons and VAN hyperactivity also induce nociceptor sensitivity in the PNS, which is substantive in generating chronic visceral pain and transitioning from acute to chronic pain. Schneider et al. recently identified another potential pathway for enteric gliosis and neuroinflammation following intestinal surgery. According to them, surgical trauma triggers ATP release that binds to purinergic receptors (P2X receptors), drives enteric gliosis and intestinal inflammation, and is mainly responsible for visceral hypersensitivity and abdominal pain (Schneider et al., 2021) Although not wholly studied, enteric gliosis and gliotransmitter release have been suggested to be closely associated with the activation of enteric nociceptive neurons and TRVP1 sensory neurons in the mesenteric plexus, contributing to the development of prolonged visceral pain (Figure 2). However, the mechanisms involved in the changes in sensitivity in the PNS and CNS resulting from the interaction of gliotransmitters with enteric nociceptors remain largely unknown (Xu et al., 2008; Morales-Soto and Gulbransen, 2019). After the neuroinflammatory response accompanying the initial neural injury, the healing process begins and promotes tissue restoration, neutrophil apoptosis, and scarification. However, nociceptor activation and the release of immunoinflammatory mediators may persist elevated for weeks or months, leading to the transition to chronic or neuropathic pain (Chavan et al., 2017).
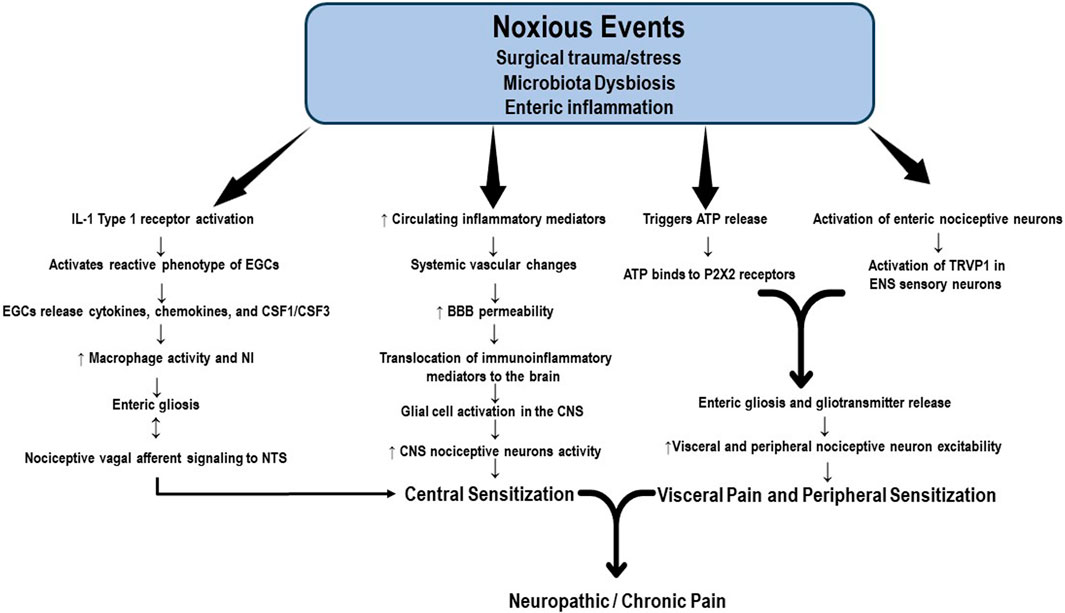
FIGURE 2. Potential mechanisms of enteric gliosis. Noxious events, including surgical trauma and bowel surgery, produce enteric dysbiosis and activation of the IL-1 receptor, which induces a reactive phenotype of enteric glial cells (EGCs), a process called enteric gliosis. EGCs release cytokines, chemokines, and colony-stimulating factors 1 and 3 (CSF1, CSF3), key elements modulating macrophage activation. The crosstalk between EGCs and macrophages is crucial to enteric neuroinflammation and visceral afferent nerve hyperactivity. Vagal afferent neurons (VAN) are the primary enteric nociceptive neurons that integrate gut signaling and connect VAN via the vagus nerve to the NST in the brainstem. Enteric dysbiosis modulates inflammation in the afferent vagal nerve and the transmission of noxious information from the gut to the brain, contributing to central sensitization. The visceral nociceptive neurons and VAN hyperactivity also induce nociceptor sensitivity in the peripheral nervous system. Surgical trauma triggers the release of ATP, which ultimately binds to purinergic receptors (P2XR). This signaling pathway represents another factor responsible for the visceral hypersensitivity and chronic abdominal pain associated with enteric gliosis. Gliotransmitter release activates the mesenteric plexus enteric nociceptive neurons and TRVP1 sensory neurons. Nociceptor activation and the release of immunoinflammatory mediators may persistently elevate for weeks or months, leading to the transition to chronic or neuropathic pain. BBB: brain blood barrier; CSF1: colony-stimulating factor 1; CSF3: colony-stimulating factor 3; EGCs: enteric glial cells; NST: nucleus solitary tract; P2X2: purinergic 2 × 2 receptor; TRVP1: transient receptor potential vanilloid receptor.
4 Neuroinflammation associated with opioids
The belief that opioids exert their effects solely by binding to their receptors does not adequately support the pharmacological basis of their use as pain control agents in clinical practice. The appearance of undesirable effects that compromise analgesic efficacy, such as hyperalgesia, allodynia, tolerance, and increased opportunistic infections, particularly in prolonged use scenarios, suggests the involvement of other mechanisms. This section considers some aspects of the immune response as elements of the opioid-analgesic equation. It is important to note that interactions between the nervous and immune systems are not exclusively limited to inflammatory conditions. Communication between these systems also occurs as a consequence of signaling crosstalk between immunocompetent cells, glia, endothelial cells, and neuronal cells in peripheral and central locations, and ultimately alters neuronal function (Watkins et al., 2007; Shah and Choi, 2017; Morioka et al., 2019). It is also important to recognize that pain processing is not simply the result of signals traveling from a damaged zone to the brain cortex but the confluence of multiple dynamic influences that can enhance or suppress nociceptive messages (Milligan and Watkins, 2009). Before unraveling the complex relationship between opioids and neuroinflammation, the beneficial analgesic actions of opioids must be separated from the detrimental interactions that underlie their undesirable effects. Additionally, it must be remembered that drugs currently used to treat clinical pain conditions were developed to target neurons before the discovery of these complex interactions that compromise opioid efficacy (Watkins et al., 2007).
The seminal work of Goldstein et al. brought to the forefront that opioids could also nonselective bind to non-opioid receptors (Goldstein et al., 1971). Years after, Wybran et al. reported that morphine possessed an immunosuppressive effect that could be modulated by naloxone, a classic antagonist of opioid activity (Wybran et al., 1979). Later, Watkins et al. published an elegant paper reporting that morphine administration caused glial activation in the spinal cord. As a result, it led to the release of substances that antagonize opioid action while causing pro-inflammatory effects and inducing a central immune response (Watkins et al., 2005). The participation of non-neuronal cells in these mechanisms further complicates the pharmacology of opioids.
Research aimed at clarifying this less-known opioid action identified TLR4 as a critical element of this “atypical” signaling (Figure 3). This transmembrane receptor belongs to the Toll-like receptor (TLR) family, which activates the innate immune response by recognizing PAMPs (from viruses, bacteria, protozoa, and fungi) or DAMPs as a consequence of tissue damage or cell death (Moresco et al., 2011; Zhang et al., 2020). In an interesting study, Grace et al. suggested that morphine administration leads to the persistent release of DAMPs with actions mediated by TLR4 and the purinergic receptor P2X7R (Grace et al., 2016).
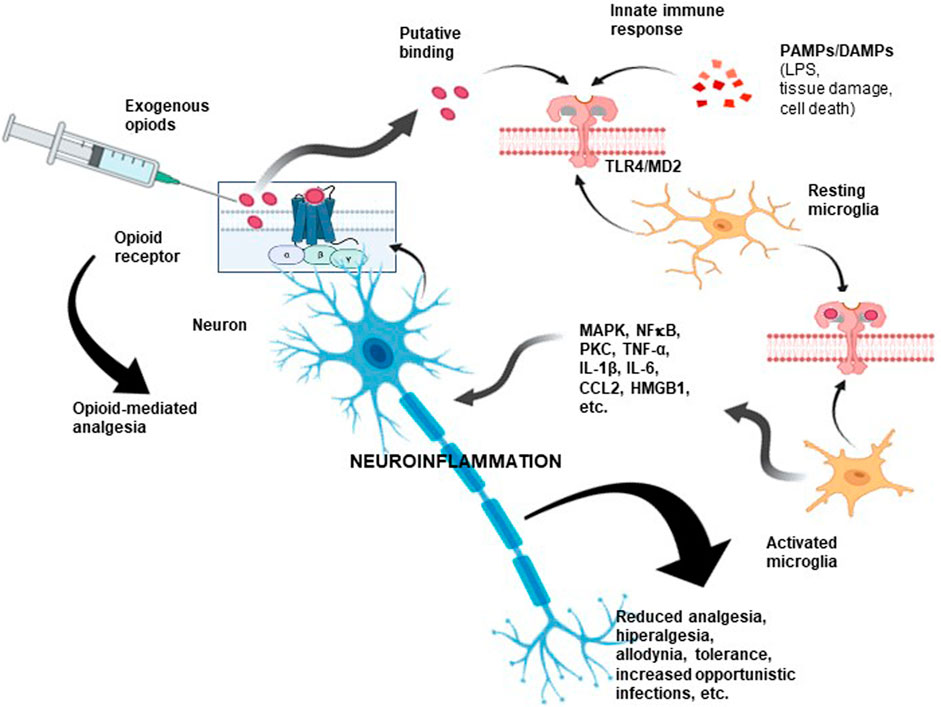
FIGURE 3. Opioid-associated neuroinflammation. As a consequence of opioid administration, proinflammatory scenarios become activated via TLR4/MD-2. As shown, opioids could also bind nonselectively to a non-opioid receptor due to interactions with critical sites shared by LPS, some PAMPs, and DAMPs, inducing downstream signaling. These interactions may compromise analgesic efficacy and cause unwanted opioid side effects. Hence, an opioid could be considered a proinflammatory molecule. This type of neuroimmune interaction could partly explain the unwanted side effects observed after prolonged administration of opioids, including the development of tolerance, allodynia, paradoxical hyperalgesic effects, and even the induction of a proinflammatory response. TLR4: toll-like receptor 4; MD-2: myeloid differentiation factor 2; PAMPs: pathogen-associated molecular pattern molecules; DAMPs: damage-associated molecular pattern molecules.
LPS, a component of the cell wall of Gram-negative bacteria, is a classic exogenous TLR4 agonist. However, a comprehensive review by Zhang et al. showed evidence that opioid receptor agonists directly activate TLR4, even in the absence of LPS (Zhang et al., 2020). Of note, this property is also shared with endogenous opioids, which, by the way, can be derived from immune cells. Unlike the interaction between opioid and opioid receptors, the binding of opioids to TLR4 is not stereoselective. Interestingly, morphine-3-glucuronic acid (M3G), an inactive metabolite without affinity for opioid receptors, also activates TLR4 (Wu et al., 2007; Hutchinson et al., 2010).
The high mobility group box 1 protein (HMGB1), an endogenous TLR4 agonist, is an alarmin that acts in synergy with endogenous and exogenous danger signals to promote inflammation. HMGB1 is secreted not only by reactive immunocompetent cells but also by neurons and glial cells (Takeuchi and Akira, 2010; Shah and Choi, 2017). It is important to note that TLR4 has been associated with morphine-induced HMGB1 production (Qian et al., 2020) and that HMGB1 is associated with abnormal pain processing and development of tolerance, hyperalgesia, and allodynia (Zhang et al., 2020).
Myeloid differentiation factor 2 (MD2), is an essential component of the TLR4 signaling receptor complex that recognizes and initiates an innate immune response to bacterial LPS. Opioid binding to MD2 activates signaling processes that lead to increased expression of the nuclear factor kappa-light-chain enhancer of activated B cells (NF-kB) and the production of pro-inflammatory cytokines such as TNF-a, IL-1β, and IL-6 (Takeuchi and Akira, 2010; Liu et al., 2022). Furthermore, opioid and TLR4 signaling activates the mitogen-activated protein kinase (MAPK) pathway, leading to neuroinflammation (Zhang et al., 2020). These pieces of evidence suggest that opioids may function as proinflammatory-like cytokines causing neuroinflammatory and immunosuppressive effects. It also highlights potential intracellular crosstalk mechanisms between immune cells, glial products, and neurons (Wang et al., 2012; Grace et al., 2014; Grace et al., 2015).
As suggested above, the interaction between opioids and the TLR4 receptor may also explain the hyperalgesia, allodynia, and tolerance observed after morphine administration, which has been associated with opioid-induced proinflammatory glial activation (Hutchinson et al., 2010). Evidence indicates that opioid-induced immune effects could explain the drugs decreased efficacy, including the critical role that non-neuronal immunocompetent cells, such as astrocytes and microglia, could play (Shah and Choi, 2017). As discussed earlier, the endogenous TLR4 agonist HMGB1 can translocate from the nucleus to the cytoplasm or the extracellular space. The binding of transmembrane TLR4 initiates the innate immune response, which activates NF-κB and mediates the transcription of pro-inflammatory cytokines in macrophages, monocytes, and glial cells. HMGB1 binds to several receptor systems, including TLR2, TLR4-5, and the receptor for advanced glycosylation end products (RAGE). All these receptors are implicated in the antianalgesic effects of opioids (He et al., 2013; Das et al., 2016; Morioka et al., 2019). Prior work has demonstrated that morphine administration increases HMGB1 expression and release. Additionally, it has been shown to promote mechanical allodynia, which may be sustained for months after the discontinuation of morphine treatment. In this setting, investigators noted an increase in IL-1β release from activated spinal microglia and the NOD-like receptor protein 3 (NLRP3) inflammasome (Grace et al., 2016), a cytosolic multiprotein oligomer of the innate immune system responsible for activating inflammatory responses. Interestingly, this multiprotein complex activates IL-1β through proteolytic cleavage by caspase-1. TLR4 has also been associated with opioid-induced hyperalgesia via protein kinase C (Araldi et al., 2019). It should be noted that the involvement of TLR4 in the antianalgesic effects of opioids has also been demonstrated in TLR4 mutant and knockout mice (Mattioli et al., 2014), suggesting that perhaps anti-opioid activity does not depend exclusively on TLR4 but can be complemented and facilitated by this receptor.
As we already know, pain induced by a stimulus or pathological damage sets in motion multiple interactions in the neuronal environment. The appropriate knowledge of these multipartite interactions might help avoid vicious cycles that cause pain persistence and counteract the analgesic effect of opioids. Such interactions may also be triggered by opioid administration alone, disrupting the innate response to infections and tissue damage. Therefore, we must expand our knowledge of analgesic pharmacology and the related neuroimmune responses. This might help us to understand that the apparent pronociceptive incongruities observed after sustained opioid administration have a reason to exist. These discrepancies become an interesting paradox when selecting the most appropriate analgesic for pain management. Fortunately, the contributions of recognized research teams are opening new avenues for deciphering these entangled concepts. They are also expanding frontiers in pain management. Specifically, they beckon the future development of therapeutic agents that target immune-neuronal-glial interactions during pain processing without inhibiting these interactions restorative and protective properties. Additionally, they reveal opportunities to design new analgesic molecules that may avoid non-neuronal sites while retaining their original neuronal binding properties. Perhaps the solution to these problems lies in maintaining an adequate homeostatic balance among the cellular constituents that inhabit the same neighborhood in which neurons exist. Most undesirable effects of opioids are not evident under basal conditions in glial and immunocompetent cells.
5 Summary and conclusion
This narrative review was undertaken to address the complex cellular mechanisms involved in the genesis of neuroinflammation as a fundamental factor contributing to the progression and chronicity of pain as well as the immunoinflammatory response resulting from the activation of glial cells by opioids, which aggravates the neuroinflammatory process and causes the development of tolerance, dependence, or paradoxical hyperalgesia that interferes with the analgesic effects of opioid drugs. We also sought to identify the most recent evidence supporting the usefulness and effectiveness of alternative non-pharmacological interventions, such as regulation of the microbiota-gut-brain axis, which can also modulate glial cell activation and create more effective multimodal synergistic therapies for patients with chronic pain syndromes, improving their clinical and functional outcomes.
Author contributions
ME-V: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing. VT: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing–original draft, Writing–review and editing. BEB: Conceptualization, Formal Analysis, Investigation, Methodology, Validation, Writing–original draft, Writing–review and editing. DR: Formal Analysis, Investigation, Writing–original draft, Writing–review and editing. AU: Conceptualization, Formal Analysis, Methodology, Resources, Validation, Writing–review and editing. TW: Conceptualization, Formal Analysis, Resources, Supervision, Validation, Writing–review and editing.
Funding
The author(s) declare no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors acknowledge McKenna Carr, BSc, from the Department of Anesthesiology, The Ohio State University, Wexner Medical Center, Columbus, OH, for her contribution during the final editing process.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Araldi, D., Oliver, B., Green, P. G., and Levine, J. D. (2019). Role of nociceptor toll-like receptor 4 (TLR4) in opioid-induced hyperalgesia and hyperalgesic priming. J. Neurosci. 39, 6414–6424. doi:10.1523/JNEUROSCI.0966-19.2019
Bali, K. K., and Kuner, R. (2017). Therapeutic potential for leukocyte elastase in chronic pain states harboring a neuropathic component. Pain 158, 2243–2258. doi:10.1097/j.pain.0000000000001032
Barrette, B., Hebert, M. A., Filali, M., Lafortune, K., Vallieres, N., Gowing, G., et al. (2008). Requirement of myeloid cells for axon regeneration. J. Neurosci. 28, 9363–9376. doi:10.1523/JNEUROSCI.1447-08.2008
Basbaum, A. I., Bautista, D. M., Grégory, S, and Julius, D. (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. doi:10.1016/j.cell.2009.09.028
Bistoletti, M., Bosi, A., Banfi, D., Giaroni, C., and Andreina, B. (2020). The microbiota-gut-brain axis: focus on the fundamental communication pathways. Prog. Mol. Biol. Transl. Sci. 176, 43–110. doi:10.1016/bs.pmbts.2020.08.012
Celik, M. Ö., Labuz, D., Keye, J., Glauben, R., and Machelska, H. (2020). IL-4 induces M2 macrophages to produce sustained analgesia via opioids, JCI insight 5, e133093. doi:10.1172/jci.insight.133093
Chatterjee, D., and Martinov, T. (2015). Mast cells: versatile gatekeepers of pain. Mol. Immunol. 63, 38–44. doi:10.1016/j.molimm.2014.03.001
Chavan, S. S., Pavlov, V. A., and Tracey, K. J. (2017). Mechanisms and therapeutic relevance of neuro-immune communication. Immunity 46, 927–942. doi:10.1016/j.immuni.2017.06.008
Chen, G., Chul-Kyu, P., Xie, R.-G., Berta, T., Nedergaard, M., and Ji, R.-R. (2014). Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 137, 2193–2209. doi:10.1093/brain/awu140
Chen, G., Zhang, Y.-Q., J Qadri, Y., Serhan, C. N., and Ji, R.-R. (2018). Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron 100, 1292–1311. doi:10.1016/j.neuron.2018.11.009
Chiu, I. M., Hehn, C. A. V., and Woolf, C. J. (2012). Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat. Neurosci. 15, 1063–1067. doi:10.1038/nn.3144
Clos-Garcia, M., Andrés-Marin, N., Fernández-Eulate, G., Abecia, L., JoséLavín, L., Sebastiaan van Liempd, , et al. (2019). Gut microbiome and serum metabolome analyses identify molecular biomarkers and altered glutamate metabolism in fibromyalgia. EBioMedicine 46, 499–511. doi:10.1016/j.ebiom.2019.07.031
Coull, J. A. M., Beggs, S., Boudreau, D., Boivin, D., Tsuda, M., Inoue, K., et al. (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. doi:10.1038/nature04223
Cunha, T. M., Verri, W. A., Schivo, I. R., Napimoga, M. H., Parada, C. A., Poole, S., et al. (2008). Crucial role of neutrophils in the development of mechanical inflammatory hypernociception. J. Leucocyte Biol. 83, 824–832. doi:10.1189/jlb.0907654
Das, N., Dewan, V., Grace, P. M., Gunn, R. J., Tamura, R., Tzarum, N., et al. (2016). HMGB1 activates proinflammatory signaling via TLR5 leading to allodynia. Cell Rep. 17, 1128–1140. doi:10.1016/j.celrep.2016.09.076
DeFrancesco-Lisowitz, A., Lindborg, J. A., Niemi, J. P., and Zigmond, R. E. (2015). The neuroimmunology of degeneration and regeneration in the peripheral nervous system. Neuroscience 302, 174–203. doi:10.1016/j.neuroscience.2014.09.027
Donnelly, C. R., Andriessen, A. S., Chen, G., Wang, K., Jiang, C., Maixner, W., et al. (2020). Central nervous system targets: glial cell mechanisms in chronic pain. Neurotherapeutics 17, 846–860. doi:10.1007/s13311-020-00905-7
Du, J., Yi, M., Xi, D., Wang, S., Liu, B., Shao, X., et al. (2023). Satellite glial cells drive the transition from acute to chronic pain in a rat model of hyperalgesic priming. Front. Mol. Neurosci. 16, 1089162. doi:10.3389/fnmol.2023.1089162
Fang, X.-X., Zhai, M.-N., Zhu, M., Cheng, He, Wang, H., Wang, J., et al. (2023). Inflammation in pathogenesis of chronic pain: foe and friend. Mol. Pain 19, 17448069231178176. doi:10.1177/17448069231178176
Feng, R., Muraleedharan Saraswathy, V., Mokalled, M. H., and Cavalli, V. (2023). Self-renewing macrophages in dorsal root ganglia contribute to promote nerve regeneration. Proc. Natl. Acad. Sci. 120 (7), e2215906120. doi:10.1073/pnas.2215906120
Gazerani, P. (2021). Satellite glial cells in pain research: a targeted viewpoint of potential and future directions. Front. Pain Res. 2, 646068. doi:10.3389/fpain.2021.646068
Goldstein, A., LouiseLowney, I., and Pal, B. K. (1971). Stereospecific and nonspecific interactions of the morphine congener levorphanol in subcellular fractions of mouse brain. Proc. Natl. Acad. Sci. 68, 1742–1747. doi:10.1073/pnas.68.8.1742
Gong, T., Liu, L., Jiang, W., and Zhou, R. (2020). DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 20, 95–112. doi:10.1038/s41577-019-0215-7
Gordon, T., Chan, K. M., Sulaiman, O. A., Udina, E., Amirjani, N., and Brushart, T. M. (2009). Accelerating axon growth to overcome limitations in functional recovery after peripheral nerve injury. Neurosurgery 65, A132–A144. doi:10.1227/01.NEU.0000335650.09473.D3
Grace, P. M., Hutchinson, M. R., Maier, S. F., and Watkins, L. R. (2014). Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 14, 217–231. doi:10.1038/nri3621
Grace, P. M., Keith, A. S., Galer, E. L., Urban, D. J., Wang, X., Baratta, M. V., et al. (2016). Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. 113, E3441–E3450. doi:10.1073/pnas.1602070113
Grace, P. M., Maier, S. F., and Watkins, L. R. (2015). Opioid-induced central immune signaling: implications for opioid analgesia. J. Head Face Pain 55, 475–489. doi:10.1111/head.12552
Green, D. P., Limjunyawong, N., Gour, N., Pundir, P., and Dong, X. (2019). A mast-cell-specific receptor mediates neurogenic inflammation and pain. Neuron 101, 412–420. doi:10.1016/j.neuron.2019.01.012
Green, K. L. (1974). Mechanism of the pro-inflammatory activity of sympathomimetic amines in thermic oedema of the rat paw. Br. J. Pharmacol. 50, 243–251. doi:10.1111/j.1476-5381.1974.tb08568.x
Grubišić, V., and Gulbransen, B. D. (2017). Enteric glia: the most alimentary of all glia. J. Physiology 595, 557–570. doi:10.1113/JP271021
Guimarães, R. M., Aníbal-Silva, C. E., Davoli-Ferreira, M., Gomes, F. I. F., Mendes, A., Cavallini, M. C. M., et al. (2023). Neuron-associated macrophage proliferation in the sensory ganglia is associated with peripheral nerve injury-induced neuropathic pain involving CX3CR1 signaling. Elife 12, e78515. doi:10.7554/eLife.78515
Guo, R., Hao, J., Ma, D., Li, H., Liao, K., and Wang, Y. (2020). Persistent proliferation of keratinocytes and prolonged expression of pronociceptive inflammatory mediators might be associated with the postoperative pain in KK mice. Mol. Pain 16, 1744806920927284. doi:10.1177/1744806920927284
Gupta, K., and IlkkaHarvima, T. (2018). Mast cell-neural interactions contribute to pain and itch. Immunol. Rev. 282, 168–187. doi:10.1111/imr.12622
Gwak, Y. S., Claire, E., and Woo Leem, J. (2017). Neuronal-glial interactions maintain chronic neuropathic pain after spinal cord injury, Neural Plast. 2017, 2480689. doi:10.1155/2017/2480689
Hains, , Bryan, C., and Waxman, S. G. (2006). Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J. Neurosci. 26, 4308–4317. doi:10.1523/JNEUROSCI.0003-06.2006
Hanani, M., and Spray, D. C. (2020). Emerging importance of satellite glia in nervous system function and dysfunction. Nat. Rev. Neurosci. 21, 485–498. doi:10.1038/s41583-020-0333-z
Hanisch, U.-K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi:10.1038/nn1997
Haroutiunian, S., Nikolajsen, L., Nanna Brix, F., and Troels Staehelin, J. (2013). The neuropathic component in persistent postsurgical pain: a systematic literature review. Pain 154, 95–102. doi:10.1016/j.pain.2012.09.010
Haroutounian, S., Nikolajsen, L., Bendtsen, T. F., Finnerup, N. B., Kristensen, A. D., JørgenHasselstrøm, B., et al. (2014). Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 155, 1272–1279. doi:10.1016/j.pain.2014.03.022
He, Z., Guo, Q., Xiao, M., He, C., and Zou, W. (2013). Intrathecal lentivirus-mediated transfer of interleukin-10 attenuates chronic constriction injury-induced neuropathic pain through modulation of spinal high-mobility group box 1 in rats. Pain Physician 16, E615–E625. doi:10.36076/ppj.2013/16/e615
Hesselink, K., Jan, M., Kopsky, D. J., and ArunBhaskar, K. (2016). Skin matters! The role of keratinocytes in nociception: a rational argument for the development of topical analgesics. J. Pain Res. 10, 1–8. doi:10.2147/JPR.S122765
Honkanen, N., Mustonen, L., Kalso, E., Meretoja, T., and Hanna, H. (2021). Breast reconstruction after breast cancer surgery–persistent pain and quality of life 1–8 years after breast reconstruction. Scand. J. Pain 21, 522–529. doi:10.1515/sjpain-2021-0026
Hu, P., and McLachlan, E. M. (2003). Distinct functional types of macrophages in dorsal root ganglia and spinal nerves proximal to sciatic and spinal nerve transections in the rat. Exp. Neurol. 184, 590–605. doi:10.1016/S0014-4886(03)00307-8
Hutchinson, M. R., Zhang, Y., Shridhar, M., Evans, J. H., Buchanan, M. M., Zhao, T. X., et al. (2010). Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain, Behav. Immun. 24, 83–95. doi:10.1016/j.bbi.2009.08.004
Ji, R.-R., Alexander, C., and Zhang, Y.-Q. (2016). Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577. doi:10.1126/science.aaf8924
Ji, R.-R., Berta, T., and Nedergaard, M. (2013). Glia and pain: is chronic pain a gliopathy? Pain 154, S10–S28. doi:10.1016/j.pain.2013.06.022
Ji, R.-R., Donnelly, C. R., and Nedergaard, M. (2019). Astrocytes in chronic pain and itch. Nat. Rev. Neurosci. 20, 667–685. doi:10.1038/s41583-019-0218-1
Ji, R.-R., Kawasaki, Y., Zhuang, Z.-Ye, Wen, Y.-R., and Decosterd, I. (2006). Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2, 259–269. doi:10.1017/S1740925X07000403
Ji, R.-R., Nackley, A., Huh, Y., Terrando, N., and Maixner, W. (2018). Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 129, 343–366. doi:10.1097/ALN.0000000000002130
Ji, R.-R., Xu, Z.-Z., Gary, S., and Serhan, C. N. (2011). Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 34, 599–609. doi:10.1016/j.tins.2011.08.005
Jin, J., Chen, Q., Su, M., Du, X., Zhang, D., and Qin, P. (2021). Prevalence and predictors of chronic postsurgical pain after colorectal surgery: a prospective study. Colorectal Dis. 23, 1878–1889. doi:10.1111/codi.15640
Jin, J., Zhang, T., Xiong, X., Chen, H., Jiang, Y., and He, S. (2023). A prospective study of chronic postsurgical pain in elderly patients: incidence, characteristics, and risk factors. BMC Geriatr. 23, 289–300. doi:10.1186/s12877-023-04006-w
Johansen, A., Romundstad, L., Nielsen, C. S., Schirmer, H., and Audun, S. (2012). Persistent postsurgical pain in a general population: prevalence and predictors in the Tromsø study. Pain 153, 1390–1396. doi:10.1016/j.pain.2012.02.018
Jurga, A. M., Paleczna, M., and Kuter, K. Z. (2020). Overview of general and discriminating markers of differential microglia phenotypes. Front. Cell. Neurosci. 14, 198. doi:10.3389/fncel.2020.00198
Kalaczkowska, E., and Kubes, P. (2013). Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175. doi:10.1038/nri3399
Kawasaki, Y., Zhang, L., Cheng, J.-K., and Ji, R.-R. (2008). Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 28, 5189–5194. doi:10.1523/JNEUROSCI.3338-07.2008
Kehlet, H., Jensen, T. S., and Woolf, C. J. (2006). Persistent postsurgical pain: risk factors and prevention. Lancet 367, 1618–1625. doi:10.1016/S0140-6736(06)68700-X
Kempuraj, D., Mentor, S., Thangavel, R., Ahmed, M. E., Govindhasamy Pushpavathi, S., Raikwar, S. P., et al. (2019). Mast cells in stress, pain, blood-brain barrier, neuroinflammation, and Alzheimers disease. Front. Cell. Neurosci. 54, 54. doi:10.3389/fncel.2019.00054
Kempuraj, D., Thangavel, R., Selvakumar, G. P., Zaheer, S., Ahmed, M. E., Raikwar, S. P., et al. (2017). Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation and neurodegeneration. Front. Cell. Neurosci. 11, 216. doi:10.3389/fncel.2017.00216
Kiguchi, N., Kobayashi, Y., Saika, F., Sakaguchi, H., Maeda, T., and Kishioka, S. (2015). Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain 156, 684–693. doi:10.1097/j.pain.0000000000000097
Kim, J. S., Kirkland, R. A., Lee, S. H., Cawthon, C. R., Rzepka, K. W., Minaya, D. M., et al. (2020). Gut microbiota composition modulates inflammation and structure of the vagal afferent pathway. Physiology Behav. 225, 113082. doi:10.1016/j.physbeh.2020.113082
Kohno, K., Kitano, J., Kohro, Y., Tozaki-Saitoh, H., Inoue, K., and Tsuda, M. (2018). Temporal kinetics of microgliosis in the spinal dorsal horn after peripheral nerve injury in rodents. Biol. Pharm. Bull. 41, 1096–1102. doi:10.1248/bpb.b18-00278
Kosek, E., Cohen, M., Baron, R., Gebhart, G. F., Juan-Antonio Mico, , Rice, A. S. C., et al. (2016). Do we need a third mechanistic descriptor for chronic pain states? Pain 157, 1382–1386. doi:10.1097/j.pain.0000000000000507
Kwon, M. J., Kim, J., Shin, H., Jeong, S. R., Kang, Y. M., Choi, J. Y., et al. (2013). Contribution of macrophages to enhanced regenerative capacity of dorsal root ganglia sensory neurons by conditioning injury. J. Neurosci. 33, 15095–15108. doi:10.1523/JNEUROSCI.0278-13.2013
Lacagnina, M. J., Watkins, L. R., and Grace, P. M. (2018). Toll-like receptors and their role in persistent pain. Pharmacol. Ther. 184, 145–158. doi:10.1016/j.pharmthera.2017.10.006
Levine, J. D., Lau, W., Kwiat, G., and Goetzl, E. J. (1984). Leukotriene B4 produces hyperalgesia that is dependent on polymorphonuclear leukocytes. Science 225, 743–745. doi:10.1126/science.6087456
Li, T., Liu, T., Chen, X., Li, Li, Feng, M., Zhang, Y., et al. (2020). Microglia induce the transformation of A1/A2 reactive astrocytes via the CXCR7/PI3K/Akt pathway in chronic post-surgical pain. J. Neuroinflammation 17, 211–215. doi:10.1186/s12974-020-01891-5
Liu, X., Yang, W., Zhu, C., Sun, S., Wu, S., Wang, L., et al. (2022). Toll-like receptors and their role in neuropathic pain and migraine. Mol. Brain 15, 73–79. doi:10.1186/s13041-022-00960-5
Liu, Z.-Y., Song, Z.-W., Guo, S.-Wu, He, J.-S., Wang, S.-Yu, Zhu, J.-G., et al. (2019). CXCL12/CXCR4 signaling contributes to neuropathic pain via central sensitization mechanisms in a rat spinal nerve ligation model. CNS Neurosci. Ther. 25, 922–936. doi:10.1111/cns.13128
Locke, S., Yousefpour, N., Mannarino, M., Xing, S., Yashmin, F., Bourassa, V., et al. (2020). Peripheral and central nervous system alterations in a rat model of inflammatory arthritis. Pain 161, 1483–1496. doi:10.1097/j.pain.0000000000001837
Lu, X., and Richardson, P. M. (1993). Responses of macrophages in rat dorsal root ganglia following peripheral nerve injury. J. Neurocytol. 22, 334–341. doi:10.1007/BF01195557
Malta, I., Moraes, T., Rodrigues, G., Franco, P., and Galdino, G. (2019). The role of oligodendrocytes in chronic pain: cellular and molecular mechanisms. J. Physiol. Pharmacol. 70, 299–309. doi:10.26402/jpp.2019.5.02
Manjavachi, M. N., Costa, R., Nara Lins, Q., and JoãoCalixto, B. (2014). The role of keratinocyte-derived chemokine (KC) on hyperalgesia caused by peripheral nerve injury in mice. Neuropharmacology 79, 17–27. doi:10.1016/j.neuropharm.2013.10.026
Martinez, F. O., and Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. 6, 13. doi:10.12703/P6-13
Marvizon, J. C., Walwyn, W., Minasyan, A., Chen, W., and Taylor, B. K. (2015). Latent sensitization: a model for stress-sensitive chronic pain. Curr. Protoc. Neurosci. 71, 1–9. doi:10.1002/0471142301.ns0950s71
Massier, J., Eitner, A., Gisela Segond von, B., and Schaible, H.-G. (2015). Effects of differently activated rodent macrophages on sensory neurons: implications for arthritis pain. Arthritis & Rheumatology 67, 2263–2272. doi:10.1002/art.39134
Mastrangelo, F., Ronconi, G., Kritas, S., Conti, P., and Tettamanti, L. (2018). New concepts in neuroinflammation: mast cells pro-inflammatory and anti-inflammatory cytokine mediators. J. Biol. Regul. Homeost. Agents 32, 449–454.
Mattioli, T. A., Leduc-Pessah, H., Skelhorne-Gross, G., Nicol, C. J. B., Milne, B., Tuan, T., et al. (2014). Toll-like receptor 4 mutant and null mice retain morphine-induced tolerance, hyperalgesia, and physical dependence. PLoS One 9, e97361. doi:10.1371/journal.pone.0097361
McNeil, B. D., Pundir, P., Meeker, S., Han, L., BradleyUndem, J., Kulka, M., et al. (2015). Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 519, 237–241. doi:10.1038/nature14022
Milligan, E. D., and Watkins, L. R. (2009). Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 10, 23–36. doi:10.1038/nrn2533
Minerbi, A., Gonzalez, E., Brereton, N. J. B., Abraham, A., Dewar, K., Fitzcharles, M.-A., et al. (2019). Altered microbiome composition in individuals with fibromyalgia. Pain 160, 2589–2602. doi:10.1097/j.pain.0000000000001640
Morales-Soto, W., and Gulbransen, B. D. (2019). Enteric glia: a new player in abdominal pain. Cell. Mol. Gastroenterology Hepatology 7, 433–445. doi:10.1016/j.jcmgh.2018.11.005
Moresco, E. M. Y., LaVine, D., and Beutler, B. (2011). Toll-like receptors. Curr. Biol. 21, R488–R493. doi:10.1016/j.cub.2011.05.039
Morin, N., Owolabi, S. A., Harty, M. W., Papa, E. F., Tracy, T. F., Shaw, S. K., et al. (2007). Neutrophils invade lumbar dorsal root ganglia after chronic constriction injury of the sciatic nerve. J. Neuroimmunol. 184, 164–171. doi:10.1016/j.jneuroim.2006.12.009
Morioka, N., Miyauchi, K., Miyashita, K., Kochi, T., Zhang, F. F., Nakamura, Y., et al. (2019). Spinal high-mobility group box-1 induces long-lasting mechanical hypersensitivity through the toll-like receptor 4 and upregulation of interleukin-1β in activated astrocytes. J. Neurochem. 150, 738–758. doi:10.1111/jnc.14812
Mueller, M., Leonhard, C., Wacker, K., Ringelstein, E. B., Okabe, M., Hickey, W. F., et al. (2003). Macrophage response to peripheral nerve injury: the quantitative contribution of resident and hematogenous macrophages. Lab. Invest. 83, 175–185. doi:10.1097/01.lab.0000056993.28149.bf
Mujenda, F. H., Duarte, A. M., Reilly, E. K., and Strichartz, G. R. (2007). Cutaneous endothelin-A receptors elevate post-incisional pain. PAIN 133, 161–173. doi:10.1016/j.pain.2007.03.021
Nicol, G. D., Lopshire, J. C., and Pafford, C. M. (1997). Tumor necrosis factor enhances the capsaicin sensitivity of rat sensory neurons. J. Neurosci. 17, 975–982. doi:10.1523/JNEUROSCI.17-03-00975.1997
Obreja, O., ParvinderRathee, K., KathrinLips, S., Distler, C., and Kress, M. (2002). IL-1 beta potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB J. 16, 1497–1503. doi:10.1096/fj.02-0101com
Oh, S. B., Tran, P. B., Gillard, S. E., Hurley, R. W., Hammond, D. L., and Miller, R. J. (2001). Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J. Neurosci. 21, 5027–5035. doi:10.1523/JNEUROSCI.21-14-05027.2001
Orecchioni, M., Yanal, G., Akula Bala, P., and Ley, K. (2019). Macrophage polarization: different gene signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively activated macrophages. Front. Immunol. 10, 1–14. doi:10.3389/fimmu.2019.01084
Pannell, M., Labuz, D., Celik, M. Ö., Keye, J., Batra, A., Siegmund, B., et al. (2016). Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J. Neuroinflammation 13, 262–317. doi:10.1186/s12974-016-0735-z
Paolicelli, R. C., Sierra, A., Stevens, B., Tremblay, M. E., Aguzzi, A., Ajami, B., et al. (2022). Microglia states and nomenclature: a field at its crossroads. Neuron 110 (21), 3458–3483. doi:10.1016/j.neuron.2022.10.020
Parada, C. A., Yeh, J. J., Joseph, E. K., and Levine, J. D. (2003). Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur. J. Neurosci. 17, 1847–1852. doi:10.1046/j.1460-9568.2003.02626.x
Patel, J. P., and Frey, B. N. (2015). Disruption in the blood-brain barrier: the missing link between brain and body inflammation in bipolar disorder?, Neural Plast. 2015, 708306. doi:10.1155/2015/708306
Pavlov, V. A., Chavan, S. S., and Tracey, K. J. (2018). Molecular and functional neuroscience in immunity. Annu. Rev. Immunol. 36, 783–812. doi:10.1146/annurev-immunol-042617-053158
Perkins, N. M., and Tracey, D. J. (2000). Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience 101, 745–757. doi:10.1016/S0306-4522(00)00396-1
Pezet, S., and McMahon, S. B. (2006). Neurotrophins: mediators and modulators of pain. Annu. Rev. Neurosci. 29, 507–538. doi:10.1146/annurev.neuro.29.051605.112929
Pierson, E. R., Wagner, C. A., and JoanGoverman, M. (2018). The contribution of neutrophils to CNS autoimmunity. Clin. Immunol. 189, 23–28. doi:10.1016/j.clim.2016.06.017
Pinho-Ribeiro, F. A., Verri, W. A., and Chiu, I. M. (2017). Nociceptor sensory neuron–immune interactions in pain and inflammation. Trends Immunol. 38, 5–19. doi:10.1016/j.it.2016.10.001
Price, T. J., and Ray, P. R. (2019). Recent advances toward understanding the mysteries of the acute to chronic pain transition. Curr. Opin. Physiology 11, 42–50. doi:10.1016/j.cophys.2019.05.015
Purcu Duygu, U., Asli, K., Yonca, E., Yavuz, D., Asli, S., Gerhard, W., et al. (2022). Effect of stimulation time on the expression of human macrophage polarization markers. PLoS ONE 17, e0265196. doi:10.1371/journal.pone.0265196
Qian, J., Zhu, Y., Bai, L., Gao, Y., Jiang, M., Xing, F., et al. (2020). Chronic morphine-mediated upregulation of high mobility group box 1 in the spinal cord contributes to analgesic tolerance and hyperalgesia in rats. Neurotherapeutics 17, 722–742. doi:10.1007/s13311-019-00800-w
Ransohoff, R. M. (2009). Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity 31, 711–721. doi:10.1016/j.immuni.2009.09.010
Ratte, S., and Prescott, S. A. (2016). Afferent hyperexcitability in neuropathic pain and the inconvenient truth about its degeneracy. Curr. Opin. Neurobiol. 36, 31–37. doi:10.1016/j.conb.2015.08.007
Ritter-Jones, M., Najjar, S., and Albers, K. M. (2016). Keratinocytes as modulators of sensory afferent firing. Pain 157, 786–787. doi:10.1097/j.pain.0000000000000490
Rivat, C., Laboureyras, E., Laulin, J.-P., Le Roy, C., Richebé, P., and Guy, S. (2007). Non-nociceptive environmental stress induces hyperalgesia, not analgesia, in pain and opioid-experienced rats. Neuropsychopharmacology 32, 2217–2228. doi:10.1038/sj.npp.1301340
Roh, J., Go, E. J., Park, J.-W., Kim, Y.Ho, and Chul-Kyu, P. (2020). Resolvins: potent pain inhibiting lipid mediators via transient receptor potential regulation. Front. Cell Dev. Biol. 8, 584206. doi:10.3389/fcell.2020.584206
Saijo, K., Crotti, A., and Glass, C. K. (2013). Regulation of microglia activation and deactivation by nuclear receptors. Glia 61, 104–111. doi:10.1002/glia.22423
Salio, C., Ferrini, F., Muthuraju, S., and Merighi, A. (2014). Presynaptic modulation of spinal nociceptive transmission by glial cell line-derived neurotrophic factor (GDNF). J. Neurosci. 34, 13819–13833. doi:10.1523/JNEUROSCI.0808-14.2014
Schneider, R., Leven, P., Mallesh, S., Breßer, M., Schneider, L., Mazzotta, E., et al. (2022). IL-1-dependent enteric gliosis guides intestinal inflammation and dysmotility and modulates macrophage function. Commun. Biol. 5, 811–816. doi:10.1038/s42003-022-03772-4
Schneider, R., Leven, P., Tim, G., Kuzmanov, I., Lysson, M., Schneiker, B., et al. (2021). A novel P2X2-dependent purinergic mechanism of enteric gliosis in intestinal inflammation. EMBO Mol. Med. 13, e12724. doi:10.15252/emmm.202012724
Schug, S. A., Lavandhomme, P., Barke, A., Korwisi, B., Rief, W., Treede, R.-D., et al. (2019). The IASP classification of chronic pain for ICD-11: chronic postsurgical or posttraumatic pain. Pain 160, 45–52. doi:10.1097/j.pain.0000000000001413
Shah, M., and Choi, S. (2017). Toll-like receptor-dependent negative effects of opioids: a battle between analgesia and hyperalgesia. Front. Immunol. 8, 642. doi:10.3389/fimmu.2017.00642
Silva, R., Iftinca, M., Gomes, F. I. F., Segal, J. P., Smith, O. M. A., Bannerman, C. A., et al. (2022). Skin-resident dendritic cells mediate postoperative pain via CCR4 on sensory neurons. Proc. Natl. Acad. Sci. 119, e2118238119. doi:10.1073/pnas.2118238119
Simmons, S. B., Denny, L., and JoanGoverman, M. (2014). Cytokine-regulated neutrophil recruitment is required for brain but not spinal cord inflammation during experimental autoimmune encephalomyelitis. J. Immunol. 193, 555–563. doi:10.4049/jimmunol.1400807
Sommer, C., and Frank, B. (2011). Resolvins and inflammatory pain. F1000 Med. Rep. 3, 19. doi:10.3410/M3-19
Sorge, R. E., JosianeMapplebeck, C. S., Rosen, S., Beggs, S., Taves, S., JessicaAlexander, K., et al. (2015). Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 18, 1081–1083. doi:10.1038/nn.4053
Steen, K. H., Reeh, P. W., Anton, F., and Ho, H. (1992). Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J. Neurosci. 12, 86–95. doi:10.1523/JNEUROSCI.12-01-00086.1992
Stein, C., Hassan, A. H., Przewłocki, R., Gramsch, C., Peter, K., and Herz, A. (1990). Opioids from immunocytes interact with receptors on sensory nerves to inhibit nociception in inflammation. Proc. Natl. Acad. Sci. 87, 5935–5939. doi:10.1073/pnas.87.15.5935
Strizova, Z., Iva, B., Robin, B., Rene, N., Eva, C., Lily, K. F., et al. (2023). M1/M2 macrophages and their overlaps - myth or reality? Clin. Sci. (Lond) 137 (15), 1067–1093. doi:10.1042/CS20220531
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi:10.1016/j.cell.2010.01.022
Tan, P.-H., Ji, J., Yeh, C.-C., and Ji, R.-R. (2021). Interferons in pain and infections: emerging roles in neuro-immune and neuro-glial interactions. Front. Immunol. 12, 783725. doi:10.3389/fimmu.2021.783725
Taskinen, H. S., and Roytta, M. (1997). The dynamics of macrophage recruitment after nerve transection. Acta Neuropathol. 93, 252–259. doi:10.1007/s004010050611
Tiwari, V., Guan, Y., and Raja, S. N. (2014). Modulating the delicate glial–neuronal interactions in neuropathic pain: promises and potential caveats. Neurosci. Biobehav. Rev. 45, 19–27. doi:10.1016/j.neubiorev.2014.05.002
Treede, R.-D., Rief, W., Barke, A., Aziz, Q., Bennett, M. I., Benoliel, R., et al. (2019). Chronic pain as a symptom or a disease: the IASP classification of chronic pain for the international classification of diseases (ICD-11). Pain 160, 19–27. doi:10.1097/j.pain.0000000000001384
Tsuda, M., Masuda, T., Kitano, J., Shimoyama, H., Tozaki-Saitoh, H., and Inoue, K. (2009). IFN-γ receptor signaling mediates spinal microglia activation driving neuropathic pain. Proc. Natl. Acad. Sci. 106, 8032–8037. doi:10.1073/pnas.0810420106
Tsuda, M., Shigemoto-Mogami, Y., Koizumi, S., Mizokoshi, A., Kohsaka, S., Salter, M. W., et al. (2003). P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424, 778–783. doi:10.1038/nature01786
Um, Ji W. (2017). Roles of glial cells in sculpting inhibitory synapses and neural circuits. Front. Mol. Neurosci. 10, 381. doi:10.3389/fnmol.2017.00381
Vandenberg, R. J., and Ryan, R. M. (2013). Mechanisms of glutamate transport. Physiol. Rev. 93, 1621–1657. doi:10.1152/physrev.00007.2013
Van den Bossche, J., Baardman, J., Otto, N. A., van der Velden, S., Neele, A. E., van den Berg, S. M., et al. (2016). Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 17, 684–696. doi:10.1016/j.celrep.2016.09.008
Vaso, A., Haim-Moshe, A., Gjika, A., Zahaj, S., Zhurda, T., Vyshka, G., et al. (2014). Peripheral nervous system origin of phantom limb pain. Pain 155, 1384–1391. doi:10.1016/j.pain.2014.04.018
Vergne-Salle, P., and Bertin, P. (2021). Chronic pain and neuroinflammation. Jt. Bone Spine 88, 105222. doi:10.1016/j.jbspin.2021.105222
Vezzani, A., and Viviani, B. (2015). Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 96, 70–82. doi:10.1016/j.neuropharm.2014.10.027
Vicuña, L., Strochlic, D. E., Latremoliere, A., Kumar Bali, K., Simonetti, M., Husainie, D., et al. (2015). The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell–derived leukocyte elastase. Nat. Med. 21, 518–523. doi:10.1038/nm.3852
von Banchet, , Segond, G., Boettger, M. K., Fischer, N., Gajda, M., Bräuer, R., et al. (2009). Experimental arthritis causes tumor necrosis factor-alpha-dependent infiltration of macrophages into rat dorsal root ganglia which correlates with pain-related behavior. Pain 145, 151–159. doi:10.1016/j.pain.2009.06.002
Walters, E. T. (2019). Adaptive mechanisms driving maladaptive pain: how chronic ongoing activity in primary nociceptors can enhance evolutionary fitness after severe injury. Philosophical Trans. R. Soc. B 374, 20190277. doi:10.1098/rstb.2019.0277
Wang, X., Loram, L. C., Ramos, K., J de Jesus, A., Jacob, T., Cheng, K., et al. (2012). Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. 109, 6325–6330. doi:10.1073/pnas.1200130109
Wang, Z.-Q., Frank, P., Cuzzocrea, S., Galen, K., Lightfoot, R., Masini, E., et al. (2004). A newly identified role for superoxide in inflammatory pain. J. Pharmacol. Exp. Ther. 309, 869–878. doi:10.1124/jpet.103.064154
Watkins, L. R., Hutchinson, M. R., Johnston, I. N., and Maier, S. F. (2005). Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 28, 661–669. doi:10.1016/j.tins.2005.10.001
Watkins, L. R., Hutchinson, M. R., Milligan, E. D., and Maier, S. F. (2007). “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res. Rev. 56, 148–169. doi:10.1016/j.brainresrev.2007.06.006
Willemen, H. L. D. M., Eijkelkamp, N., Wang, H., Mack, M., Zijlstra, J., Heijnen, C. J., et al. (2014). Monocytes/macrophages control resolution of transient inflammatory pain. J. Pain 15, 496–506. doi:10.1016/j.jpain.2014.01.491
Woo, Y. C., Park, S. S., Subieta, A. R., and Brennan, T. J. (2004). Changes in tissue pH and temperature after incision indicate acidosis may contribute to postoperative pain. J. Am. Soc. Anesthesiol. 101, 468–475. doi:10.1097/00000542-200408000-00029
Wu, H.-en, Hong, J.-S., and Tseng, L. F. (2007). Stereoselective action of (+)-morphine over (−)-morphine in attenuating the (−)-morphine-produced antinociception via the naloxone-sensitive sigma receptor in the mouse. Eur. J. Pharmacol. 571, 145–151. doi:10.1016/j.ejphar.2007.06.012
Wybran, J., Appelboom, T., Famaey, J.-P., and Govaerts, A. (1979). Suggestive evidence for receptors for morphine and methionine-enkephalin on normal human blood T lymphocytes. J. Immunol. 123, 1068–1070. doi:10.4049/jimmunol.123.3.1068
Wynn, T. A., and Vannella, K. M. (2016). Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462. doi:10.1016/j.immuni.2016.02.015
Xiao, Y., Faucherre, A.le, Pola-Morell, L., Heddleston, J. M., Liu, T.-Li, Chew, T.-L., et al. (2015). High-resolution live imaging reveals axon-glia interactions during peripheral nerve injury and repair in zebrafish. Dis. Models Mech. 8, 553–564. doi:10.1242/dmm.018184
Xu, G.-Y., Shenoy, M., Winston, J. H., Mittal, S., and Jay Pasricha, P. (2008). P2X receptor-mediated visceral hyperalgesia in a rat model of chronic visceral hypersensitivity. Gut 57, 1230–1237. doi:10.1136/gut.2007.134221
Yu, X., Liu, H., Hamel, K. A., Morvan, M. G., Yu, S., Leff, J., et al. (2020). Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain.Nat. Commun. 11.264. doi:10.1038/s41467-019-13839-2
Zhang, J.-M., and An, J. (2007). Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 45, 27–37. doi:10.1097/AIA.0b013e318034194e
Zhang, P., Yang, M., Chen, C., Liu, L., Wei, X., and Zeng, Si (2020). Toll-like receptor 4 (TLR4)/opioid receptor pathway crosstalk and impact on opioid analgesia, immune function, and gastrointestinal motility. Front. Immunol. 11, 1455. doi:10.3389/fimmu.2020.01455
Keywords: acute pain, chronic pain, neuroinflammation, dysbiosis, gliosis, inflammatory mediators, opioids
Citation: Echeverria-Villalobos M, Tortorici V, Brito BE, Ryskamp D, Uribe A and Weaver T (2023) The role of neuroinflammation in the transition of acute to chronic pain and the opioid-induced hyperalgesia and tolerance. Front. Pharmacol. 14:1297931. doi: 10.3389/fphar.2023.1297931
Received: 20 September 2023; Accepted: 04 December 2023;
Published: 15 December 2023.
Edited by:
Nicolau Beckmann, Novartis Institutes for BioMedical Research, SwitzerlandReviewed by:
Giulia Magni, University of Milan, ItalyFrancisco Isaac Fernandes Gomes, University of São Paulo, Brazil
Copyright © 2023 Echeverria-Villalobos, Tortorici, Brito, Ryskamp, Uribe and Weaver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Echeverria-Villalobos, TWFyY28uRWNoZXZlcnJpYVZpbGxhbG9ib3NAb3N1bWMuZWR1