 Helen M. Reid1Mark Maginn1C. Michael Perkins1Eamon P. Mulvaney1Malcolm Boyce2Takahiro Yamamoto2B. Therese Kinsella1*
Helen M. Reid1Mark Maginn1C. Michael Perkins1Eamon P. Mulvaney1Malcolm Boyce2Takahiro Yamamoto2B. Therese Kinsella1*- 1ATXA Therapeutics Limited, UCD Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Belfield, Dublin, Ireland
- 2Hammersmith Medicines Research, London, United Kingdom
Background: The thromboxane receptor (TP) antagonist NTP42 is in clinical development for treatment of cardiopulmonary diseases, such as pulmonary arterial hypertension. In this randomized, placebo-controlled Phase I clinical trial, NTP42, administered as the oral formulation NTP42:KVA4, was evaluated for safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) in healthy males.
Methods: The first-in-human trial had three Parts: A, single ascending dose (SAD) study with seven groups given 0.25–243 mg NTP42:KVA4 or placebo; B, food effect study where one SAD group (9 mg) was also given NTP42:KVA4 or placebo after a high-fat breakfast; C, multiple ascending dose study with three groups given 15–135 mg NTP42:KVA4 or placebo once-daily for 7 days.
Results: Seventy-nine volunteers participated. No serious adverse events occurred, where any drug- or placebo-related adverse events were mild to moderate, with no correlation to NTP42:KVA4 dose. NTP42 was rapidly absorbed, yielding dose proportional increases in exposure after single and repeat dosing. PK confirmed that, with a clearance (T1/2) of 18.7 h, NTP42:KVA4 is suited to once-daily dosing, can be taken with or without food, and does not accumulate on repeat dosing. At doses ≥1 mg, NTP42 led to complete and sustained inhibition of thromboxane-, but not ADP-, induced platelet aggregation ex vivo, with direct correlation between NTP42 exposure and duration of PD effects.
Conclusion: Orally administered NTP42:KVA4 was well tolerated, with favorable PK/PD profiles and evidence of specific TP target engagement. These findings support continued clinical development of NTP42:KVA4 for cardiopulmonary or other relevant diseases with unmet needs.
Clinical Trial Registration: clinicaltrials.gov, identifier NCT04919863.
Introduction
The prostanoid thromboxane (TX) A2 and the free-radical derived isoprostane 8-iso-prostaglandin (PG) F2α signal through the thromboxane receptor (TP) to regulate multiple processes including platelet aggregation, constriction, and proliferation of vascular and pulmonary smooth muscle. They also mediate potent pro-inflammatory, pro-mitogenic, and pro-fibrotic effects within the lungs, heart, and wider cardiovascular system. Imbalances in the levels of TXA2, 8-iso-PGF2α, or the TP have been implicated in a range of cardiopulmonary diseases (Davi et al., 2012; Capra et al., 2013; Capra et al., 2014; Fontana et al., 2014; Al-Naamani et al., 2016; West et al., 2016; Badimon et al., 2021; Ashton, 2023), and aberrant TXA2/TP signaling contributes to cardiac dysfunction in multiple preclinical disease-models (Hoffmann et al., 1993; Nakamura et al., 1996; Francois et al., 2004; Wacker et al., 2006; Francois et al., 2008; Wacker et al., 2009; West et al., 2016; West et al., 2019).
NTP42 is a novel TP antagonist in clinical development for the treatment of cardiopulmonary diseases such as pulmonary arterial hypertension (PAH) (Mulvaney et al., 2020a; Mulvaney et al., 2020b; Mulvaney et al., 2022). NTP42 inhibits both TXA2-and 8-iso-PGF2α-induced signaling, being highly specific for the human TP, acting as neither a TP agonist, or agonist or antagonist of other prostanoid receptors (Mulvaney et al., 2020a).
PAH is a rare disease characterized by structural and functional changes in the pulmonary vasculature resulting in elevated pulmonary vascular resistance (PVR), right ventricular (RV) hypertrophy and, ultimately, RV failure and death (Vonk Noordegraaf and Galie, 2011; Naeije and Manes, 2014; Ryan and Archer, 2014). Preclinical efficacy data shows that NTP42 can attenuate key PAH disease hallmarks, many of which are also found in related cardiopulmonary conditions (Mulvaney et al., 2020a; Mulvaney et al., 2020b; Mulvaney et al., 2022). In the monocrotaline (MCT)-and Sugen/Hypoxia-induced PAH models, the active pharmaceutical ingredient (API) NTP42 attenuated pulmonary pathologies and consequent RV effects (Mulvaney et al., 2020a; Mulvaney et al., 2020b). Moreover, NTP42:KVA4, an oral formulation of NTP42 developed as an investigational medicinal product (IMP) for clinical use, was evaluated in both the MCT-PAH and pulmonary artery banding (PAB) model of RV overload (Mulvaney et al., 2022). In the MCT-PAH model, efficacy of NTP42:KVA4 was consistent with previous findings using the NTP42 API. In the PAB model, NTP42:KVA4 promoted a beneficial pattern of cardiac hypertrophy, resulting in significantly improved cardiac function. Moreover, findings in the PAB model pointed to a direct cardioprotective effect for NTP42:KVA4 as a component of its clinical potential, not only in PAH, but also in more prevalent cardiac conditions involving RV dysfunction.
In a Phase II clinical trial in PAH patients that evaluated Terbogrel, which also targets the TXA2/TP pathway, the development of acute leg pain in participants led to the study being prematurely terminated during enrolment (Langleben et al., 2002). Notably, Terbogrel acts as both a TP antagonist and TXA2 synthase (TXS) inhibitor (Langleben et al., 2002). As subsequently reported, leg pain occurred due to Terbogrel’s inhibition of TXS which, while reducing TXA2 generation, resulted in a shift towards synthesis of prostacyclin, a potent pain inducer (Berkenkopf and Weichman, 1988; Shindo et al., 1991; Murata et al., 1997; Langleben et al., 2002; Popp et al., 2009). In contrast to Terbogrel, NTP42 is a highly selective TP antagonist and does not act as a TXS inhibitor (Mulvaney et al., 2020a). Thus, NTP42 will not lead to increased prostacyclin synthesis or to pain induction associated with dual TP antagonists and TXS inhibitors, such as Terbogrel (Kinsella and Reid, 2018; Kinsella and Reid, 2019; Mulvaney et al., 2020a).
The objective of this study was to evaluate the safety, tolerability, pharmacokinetic (PK) and pharmacodynamic (PD) properties of the selective TP antagonist NTP42, delivered orally as NTP42:KVA4, in healthy adult volunteers.
Material and methods
Study design and subjects
This first-in-human Phase I, single-center, double-blinded, randomized, placebo-controlled trial aimed to assess the safety and tolerability of NTP42:KVA4 in healthy male volunteers. It also aimed to evaluate the PK profile and PD effect of NTP42, after single and repeated doses of NTP42:KVA4.
Eligible subjects were aged 18–55 years, with a body mass index of 18–30 kg/m2, and with no clinically significant medical conditions. All subjects provided written informed consent. The trial was conducted in 3 parts: Part A, a single ascending dose (SAD) study; Part B, a food effect study, and Part C, a multiple ascending dose (MAD) study. In Part A, fifty-five fasted subjects were randomly assigned to receive a single oral dose of placebo or NTP42:KVA4 (0.25–243 mg). The 0.25 mg starting dose was selected based on a review of preclinical toxicokinetic data and predicted to have no pharmacological activity. For Part B, one group from Part A (9 mg NTP42:KVA4) received a second dose of NTP42:KVA4 or placebo after consuming a high fat breakfast. In Part C, twenty-four fasted subjects were randomly assigned to three increasing dose Groups to receive repeat, once daily oral NTP42:KVA4 (15–135 mg) or matching placebo for seven consecutive days.
At an initial screening visit, prospective trial participants were assessed for eligibility, including a full physical exam. Those eligible with no clinically significant electrocardiogram (ECG), vital signs or laboratory test (hematology, coagulation, biochemistry, and urinalysis) findings were admitted to the clinical site and randomized on Day −1 for all parts and dose groups. As the Phase I trial was conducted during the height of the Coronavirus 2 (Covid) pandemic (24th May 2021 through 13th January 2022), specific screening and other safety measures were introduced, both at admission and throughout all aspects of the trial.
For each dose group in Part A, a sentinel cohort of two subjects received either NTP42:KVA4 or placebo (1:1 randomization) before the remaining subjects were dosed. Eligible subjects were confined to the clinical site from the day before dosing (Day −1), administered a single dose of NTP42:KVA4 or placebo on Day 1, and discharged from the clinic on the morning of Day 3. Subjects returned to the clinical site for a follow-up review at 7–10 days after their final dose for end-of-study safety assessments (including physical examination, hematology, coagulation, biochemistry, urinalysis, and ECG). For Groups 4, 5, 6, and 7 (9, 27, 81, and 243 mg NTP42:KVA4 doses), PK plasma collection and PD assessment was also performed at the follow-up visit.
Part C subjects were confined to the clinical site for the duration of the in-house period from Day −1, administered once daily oral doses of NTP42:KVA4 or placebo from Day 1 to Day 7, discharged from the clinic on the morning of Day 9, and returned to the clinic 7–10 days after their last dose for a follow-up review and end-of-study safety assessments (including physical examination, hematology, coagulation, biochemistry, urinalysis, ECG, PK plasma collection and PD assessment).
The study was conducted by Hammersmith Medicines Research (London, United Kingdom) in compliance with Good Clinical Practice (GCP), which has its origins in the Declaration of Helsinki. A Safety Review Group (SRG) was established to determine and approve the dose levels for all parts of the trial. The SRG reviewed blinded safety, tolerability, and anonymized PK and PD data for each previous dose group before proceeding with the next dose escalation or before progressing from Part A to Parts B or C.
Study drug
NTP42 (API; N-(tert-butylcarbamoyl)-5-cyano-2-((4'-(trifluoromethoxy) -[1,1′-biphenyl]-3-yl)oxy)benzenesulfonamide) is formulated with the excipient Kollidon® VA 64 at a ratio of 1:4 (drug substance: polymer) for oral administration where the IMP is referred to as NTP42:KVA4. The matching placebo for NTP42:KVA4 was prepared as a fill of the corresponding weight of the excipient Kollidon® VA 64 only. NTP42:KVA4 and its matching placebo were manufactured and dispensed in accordance with current good manufacturing practices (cGMP) with the required doses given as an oral suspension (NTP42:KVA4) or in solution (placebo) in water. NTP42:KVA4 and the placebo were given to subjects in a blinded manner.
Safety and tolerability
Safety assessments, including physical examination, vital signs, 12-lead ECG, laboratory safety tests (hematology, coagulation, clinical chemistry, and urinalysis), for all parts of the study were performed at screening, pre-dose, following dosing up to 48 h after the last dose, and at follow-up.
Pharmacokinetics
In Parts A and B, PK parameters for NTP42 were assessed following collection of serial blood samples at pre-dose and frequently up to 48 h after each dose and at follow-up. In Part C, samples were taken before and frequently up to 18 h after the first dose (Day 1); before and 12 h after the second dose (Day 2); before each dose on Days 3–6; before and frequently up to 48 h after the last dose and at follow-up. PK analyses were performed at Analytical Services International (London, United Kingdom) using liquid chromatography/tandem mass spectrometry (LC-MS/MS) and a validated method for the quantification of NTP42 in human plasma. Noncompartmental PK analyses following the linear trapezoidal linear interpolation calculation method were performed using Phoenix WinNonlin (Version 8.3).
Statistical methods
This study was an exploratory trial and there were no null hypotheses to be tested. All descriptive statistical analyses were performed using Statistical Analysis Software (SAS; Version 9.4).
Pharmacodynamics
In Part A, blood was taken for assessment of TXA2 (U46619)- or, as controls, vehicle- and ADP-induced platelet aggregation before and frequently up to 48 h after dosing on Day 1 and at follow-up. In Part C, samples were taken before and frequently up to 12 h after the first dose (Day 1); before each dose on Days 2–6; before and frequently up to 48 h after the last dose (Day 7), and at follow-up. There was no PD assessment undertaken for Part B.
Blood was collected using 3.2% sodium citrate monovettes and centrifuged at 150 g for 15 min at room temperature to prepare platelet rich plasma (PRP). Platelet poor plasma (PPP) was obtained by re-centrifugation of the blood samples at 2,500 × g for 20 min at room temperature. A platelet count was performed and the PRP adjusted to 250 × 109 platelets/L by diluting PRP with autologous PPP.
Platelet aggregation assays were performed on the Helena AggRAM aggregometer, using the internally validated method developed prior to Trial commencement. In these assays, PRP samples were maintained at 37 °C and stirred throughout analyses. PRP samples from each individual were treated with either the TP agonist U46619 (1.5 µM in duplicate for all dose groups) or, as controls, saline or adenosine diphosphate (10 µM ADP; an agonist for the purinergic P2Y1/P2Y12 receptors expressed in platelets). Platelet aggregation was monitored for 6 min following stimulation.
Results
Subject disposition
A total of 79 subjects were enrolled, 55 in the SAD study (Part A) and 24 in the MAD study (Part C). In Part B, 8 subjects who had received a single dose of IMP or placebo in Part A received a second dose after consuming a high-fat breakfast. While no subjects withdrew from the trial, six subjects from Part A were not available to participate in Part B as originally intended. Therefore, to ensure a cross-over design, six replacements were recruited. While the mean age (39 years) of the subjects participating in Part B, including these replacements, was higher than in Part A (32 years), the range of 22–51 years in Part B was in line with that of all other groups. Subject demographics and key baseline parameters (e.g., vital signs and clinical laboratory data) are summarized in Table 1 and in Supplementary Table S1.
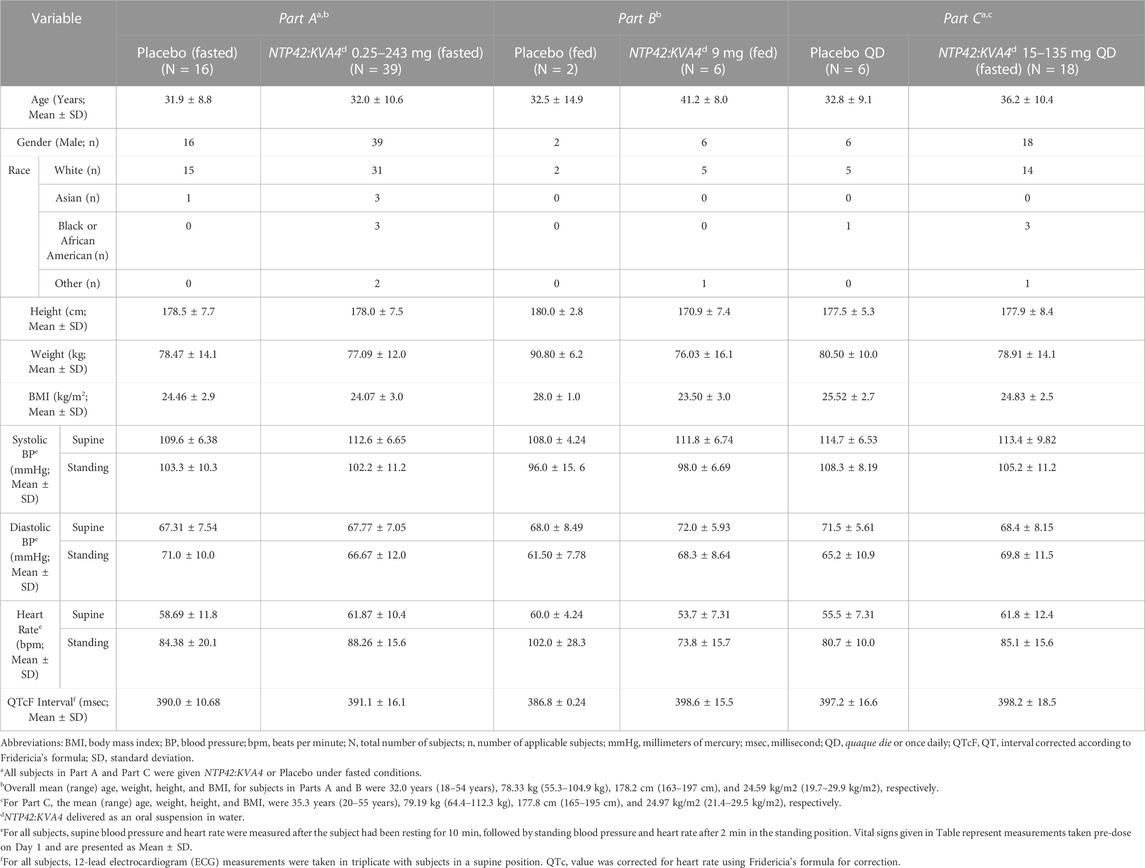
TABLE 1. Summary of demographic details of trial participants.
Safety and tolerability
All subjects completed the trial with no serious adverse events (SAEs), other significant adverse events (AEs) or AEs leading to subject withdrawal. As summarized in Table 2, all AEs observed after NTP42:KVA4 or placebo dosing (i.e., treatment-emergent AEs, TEAEs) were classed as mild or moderate in severity.

TABLE 2. Summary of possible drug- and placebo-treatment-emergent adverse events.
In Part A, TEAEs determined to be possibly related to the drug or placebo were reported following 0.25, 1, 3, and 27 mg NTP42:KVA4 and placebo, but not 9, 81, or 243 mg NTP42:KVA4 (Table 3). The most frequently observed drug- or placebo-related TEAE was dizziness, followed by orthostatic hypotension (Table 3). There was no correlation between NTP42:KVA4 dose and overall TEAE incidence or intensity. In Part B, no TEAEs were reported after dosing of NTP42:KVA4 or placebo in the fed state (Tables 2, 3). In Part C, while TEAEs were reported for placebo and 15, 45, and 135 mg NTP42:KVA4, of these, only 2 subjects dosed with 45 mg NTP42:KVA4 had a TEAE considered as possibly NTP42-related (Tables 2, 4).
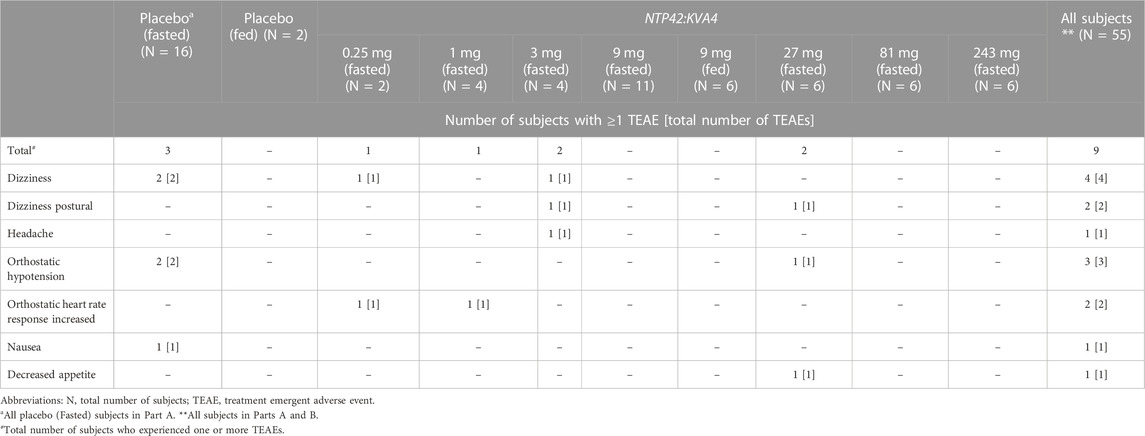
TABLE 3. Likely drug- and placebo-related treatment-emergent adverse events in parts a and B.
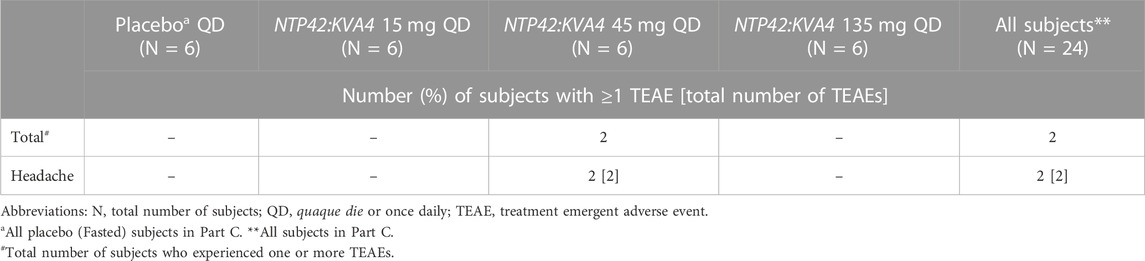
TABLE 4. Likely drug- and placebo-related treatment-emergent adverse events in part C.
For Parts A-C, there were no clinically relevant changes in physical examination findings, laboratory parameters (Supplementary Table S2), or ECG. Vital signs were within normal limits or, if outside reference ranges, not deemed clinically significant (Supplementary Table S3). Minor, non-significant decreases in systolic blood pressure on standing were the most frequent observation in the NTP42:KVA4-or placebo-dose groups, followed by an increase in heart rate on standing.
Pharmacokinetics
In Part A, NTP42 was detected in plasma from all subjects receiving NTP42:KVA4 (Figure 1A; Supplementary Figure S1A). NTP42 was rapidly absorbed, being first detected 30 min or 10 min after single doses of 0.25 mg or ≥1 mg, respectively, with mean peak concentrations observed 2–4 h post-dose (Cmax; Table 5). Generally, mean Cmax values increased proportionally with dose, e.g., with 27-fold dose increase (9–243 mg), Cmax increased 28-fold. Clearance of NTP42 was similar across all dose groups showing a gradual decline over 24–48 h with terminal elimination (T1/2) from 15 to 23 h following doses ≥1 mg NTP42:KVA4. Total NTP42 exposure (area under the curve, AUC) increased less than dose proportionally with NTP42:KVA4 dose between 1 and 243 mg (156-fold following a 243-fold dose increase; Table 5).
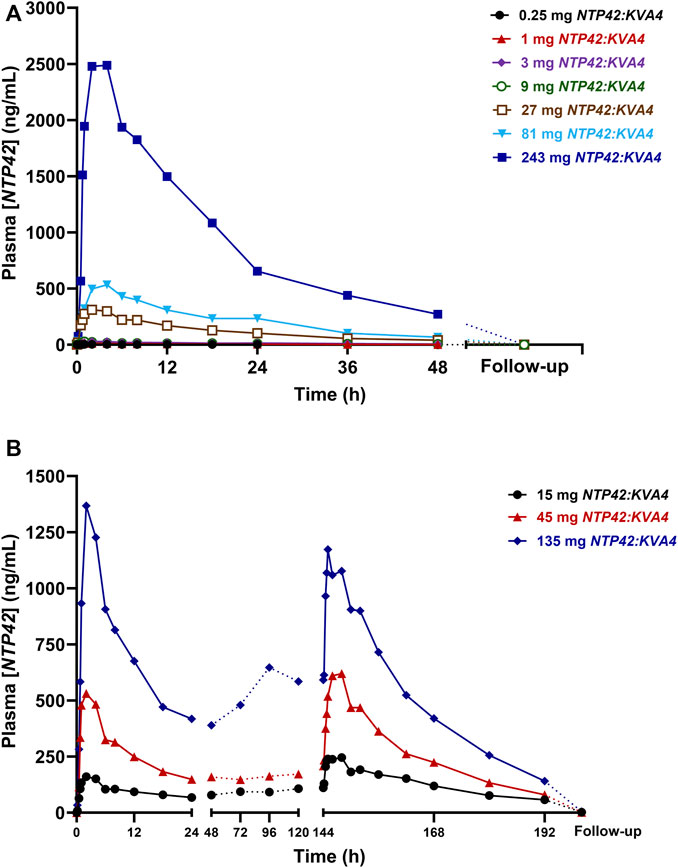
FIGURE 1. Mean NTP42 Plasma Concentrations Following Single and Repeat Oral Doses of NTP42:KVA4 in Fasting Healthy Male Subjects (Parts A and C) (A,B) Serial blood samples were collected pre-dose, frequently up to 48 h after the last dose and at follow-up (some 7–10 days after last NTP42:KVA4 dose) from subjects given either single oral doses of placebo or 0.25 mg–243 mg NTP42:KVA4 (Panel A) or repeat single daily oral doses of placebo or 15 mg–135 mg NTP42:KVA4 for 7 days (Panel B). Data is presented as mean changes in concentrations of NTP42 in plasma (Plasma [NTP42], ng/mL) from pre-dose to follow-up (some 7–10 days after the last NTP42:KVA4 dose) as determined by liquid-chromatography/tandem mass spectrometry (LC-MS/MS) and a validated method for the quantification of NTP42 in human plasma. The limit for quantitation (LoQ) for the bioanalytical assay was 1 ng/mL.
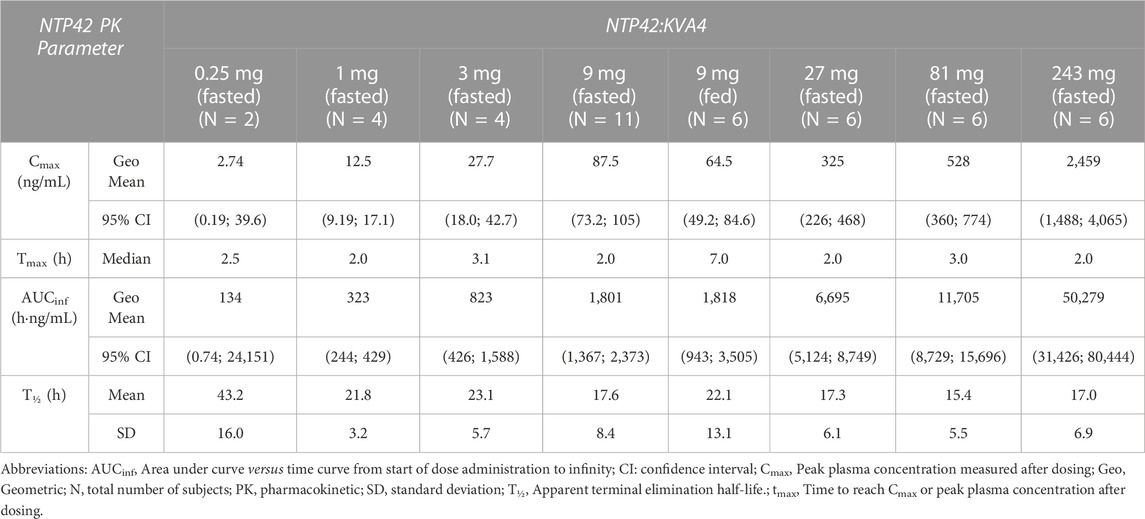
TABLE 5. Summary of NTP42 plasma pharmacokinetics following single doses of NTP42:KVA4 in fasting healthy male subjects (parts A and B).
In Part B subjects, after consuming a high fat breakfast, absorption was slightly delayed with peak NTP42 plasma concentrations observed 4–8 h after NTP42:KVA4 dosing and the Cmax decreased (87.5 ng/mL in fasted subjects, 64.5 ng/mL in fed state, Tables 5, 6). However, there was no overall difference in the AUC in the fed-compared to the fasted-states (Table 5; geometric mean AUCinf after 9 mg NTP42:KVA4, 1,801 and 1,818 h∙ng/mL in fasted and fed states, respectively). ANOVA analysis confirmed that while food reduced Cmax, it had no effect on the exposure of NTP42 (AUCinf; Table 6).
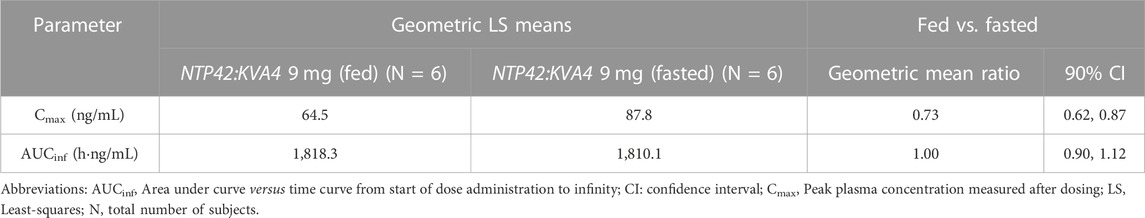
TABLE 6. Effect of food on NTP42 plasma pharmacokinetic parameters after 9 mg NTP42:KVA4 given in fed and fasted states.
In Part C, NTP42 was detected in plasma from all subjects receiving repeated doses of NTP42:KVA4 (15–135 mg), being first detected at 10 min after initial dosing (Figure 1B; Supplementary Figure S1B). Peak NTP42 concentrations were observed at 2 h post-dose after the first dose (Day 1) and at 4 h post-dose on Day 7 (Table 7). Cmax increased close to dose proportionally on Day 1, while on Day 7, following repeat dosing, Cmax increased less than dose proportionally (4.5-fold increase in Cmax with 9-fold dose increase). Mean T1/2 values ranged from 14.9 to 26.1 h on Day 1 and 19.6–23.1 h on Day 7 (Table 7). Total exposure (AUC) after single and repeat dosing increased less than dose proportionally in Part C, i.e., AUCτ increased 7.0-fold on Day 1 and 4.24-fold on Day 7 with a 9-fold dose increase.
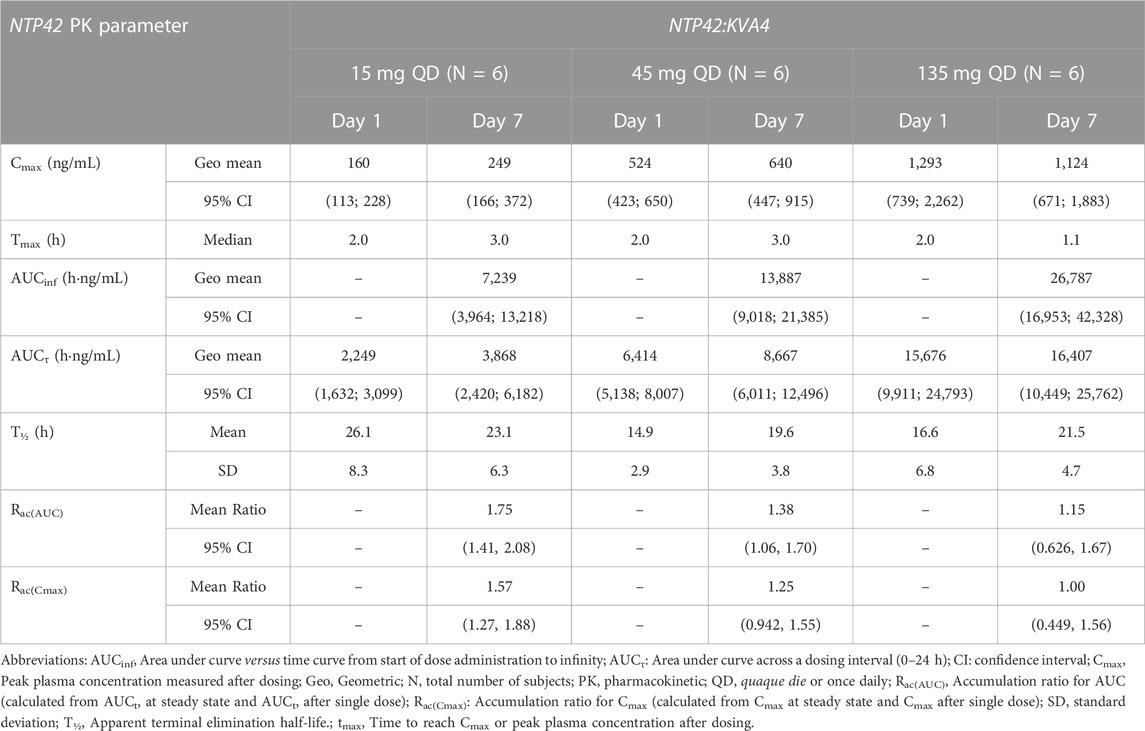
TABLE 7. Summary of NTP42 plasma pharmacokinetics following repeat doses of NTP42:KVA4 in fasting healthy male subjects (part C).
In all Part C subjects, NTP42 was quantifiable pre-dose on Days 2–7 with mean Ctrough indicating that steady-state exposure was attained between Days 3–6 (Figure 1B). Moreover, mean Day 1–7 accumulation ratios of NTP42 for AUCτ and Cmax were higher for the lowest dose tested (15 mg) and decreased as doses increased (Table 7). Mean ratios for AUCτ and Cmax of 1.15 and 1.00, respectively, in subjects receiving repeated dosing of 135 mg NTP42:KVA4 suggests little to no accumulation of NTP42 at the highest dose level.
Pharmacodynamics
As a PD read-out and surrogate marker of clinical efficacy for TP antagonists (Guth et al., 2004; Gaussem et al., 2005; Sakariassen et al., 2009; Fiessinger et al., 2010; Lesault et al., 2011), the ability of NTP42 to inhibit thromboxane (TX)A2- but not adenosine diphosphate (ADP)-induced platelet aggregation ex vivo was assessed after single and repeat dosing of NTP42:KVA4. As food consumption is known to interfere with ex vivo platelet assays, aggregations were not performed for Part B.
No platelet aggregation was induced by the drug vehicle (saline), either at baseline (pre-dose) and after single or repeat dosing of NTP42:KVA4 at all doses tested (data not shown). At pre-dose, both platelet agonists, U46619 and ADP, induced strong platelet aggregation responses in all subjects (Figure 2; Supplementary Figure S2).
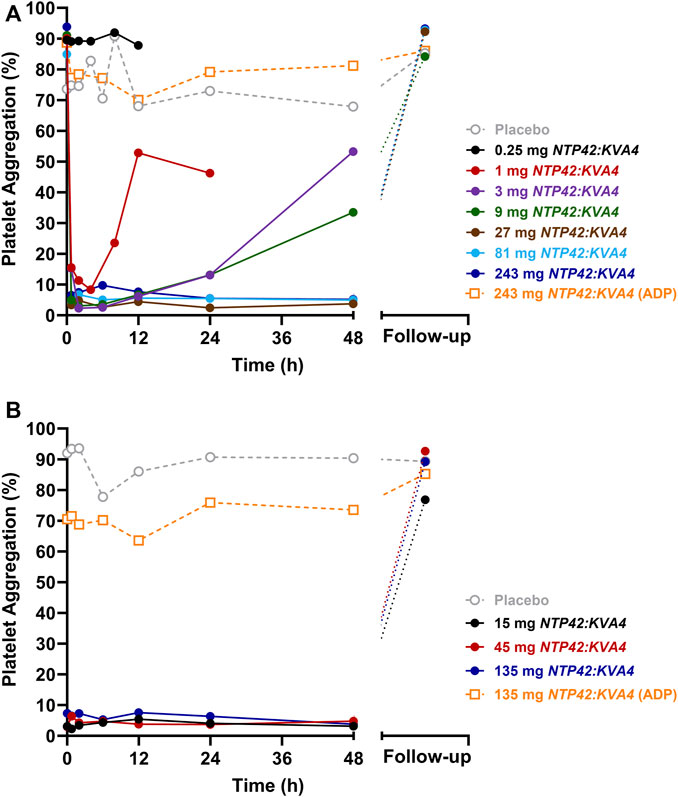
FIGURE 2. Effect of Single and Repeat Oral Dosing of NTP42:KVA4 on Human Platelet Aggregation Following U46619 and ADP Stimulation (A) Aggregation of platelets obtained from subjects given single oral doses of placebo (grey dashed line) or 0.25 mg–243 mg NTP42:KVA4 (solid-colored lines) following stimulation with 1.5 µM U46619 or 10 µM ADP (243 mg dose group only; orange dashed line). (B) Aggregation of platelets obtained from subjects given repeat oral doses of placebo (grey dashed line) or 15 mg–135 mg NTP42:KVA4 (solid-colored lines) following stimulation with 1.5 µM U46619 or 10 µM ADP (135 mg dose group only; orange dashed line). In both panels, data are presented as mean changes in maximal platelet aggregation (Platelet Aggregation; %) from pre-dose to follow-up (some 7–10 days after the last NTP42:KVA4 dose) as determined using the Helena AggRAM aggregometer.
After single oral dosing at 0.25 mg NTP42:KVA4, no inhibition of U46619-induced platelet aggregation was observed, consistent with the lack of a pharmacological effect of NTP42 at this low dose (Figure 2A; black line). Following single dosing of ≥1 mg NTP42:KVA4, dose-dependent inhibition of U46619-induced platelet aggregation was observed with complete inhibition (≥90%) following ≥3 mg NTP42:KVA4 that was sustained for up to 24 h (Figure 2A; Supplementary Figure S2A). Aggregation responses returned to baseline in a dose- and time-dependent manner, with all NTP42:KVA4 doses (1–243 mg) back at pre-dose baseline levels at the follow-up assessment (7–10 days after dosing). No inhibition of U46619-induced platelet aggregation was observed following placebo (Figure 2A; dashed grey line).
In Part C, complete inhibition of U46619-induced platelet aggregation occurred at all doses tested (Figure 2B; Supplementary Figure S2B), which was sustained from 45 min after the first dose on Day 1 until at least 48 h after the last dose on Day 7. Aggregation responses returned to baseline at follow-up for most individuals with only 2 subjects in the 15 mg dose group showing 18% and 50% inhibition of aggregation remaining at this visit. There were no underlying characteristics in these two subjects compared to other subjects in the same group or, indeed, in those dosed with 45 mg or 135 mg NTP42:KVA4 that might explain the more prolonged inhibition of U46619-induced platelet aggregation.
In both Parts A and C, a slight non-significant decrease (≤10%) in mean ADP-induced platelet aggregation from baseline was observed after single and repeat dosing at all NTP42:KVA4 or placebo doses (Figure 2; orange dashed lines). The greatest decreases (≥20%) in ADP-induced responses were observed at 12 h post-dose in all subjects receiving NTP42:KVA4. However, a similar decrease in ADP-induced aggregation was also observed in subjects receiving the placebo. Overall, there was no substantive inhibition of ADP-induced platelet aggregation after single or repeat oral NTP42:KVA4 dosing, confirming the specificity of NTP42 for TXA2/TP-, and not ADP/(P2Y1/P2Y12)-, receptor responses.
Discussion
This study is the first clinical evaluation of NTP42, a novel TP antagonist in development as a treatment for cardiopulmonary diseases such as PAH and other related conditions. NTP42, administered as the IMP NTP42:KVA4, was found to be safe and well-tolerated after single oral dosing up to 243 mg and repeat once-daily oral dosing up to 135 mg.
In all parts of the trial, there were no significant AEs or SAEs that either stopped dose escalation or resulted in subject withdrawal. Any drug- or placebo-related TEAEs were considered mild or moderate in severity, with no SAEs and no overall correlation in incidence or severity of the TEAE to NTP42:KVA4 dose. Also, there was no correlation between TEAEs, either incidence or severity, with age, ethnicity, or other clinical characteristics of trial participants. The most common drug- or placebo-related TEAEs were dizziness, orthostatic hypotension, and headache. It was notable that in Part B, following food consumption, no TEAEs were observed in any subject. Thus, it is suggested that the TEAEs observed in Parts A and C may be procedural as subjects were fasted and supine for long periods before orthostatic assessments. Overall, there were no clinically significant changes in vital signs, ECGs, or safety laboratory tests at any of doses tested, including no changes in coagulation parameters such as activated partial thromboplastin time (aPTT) and international normalized ratio (INR).
NTP42 was rapidly absorbed after single and repeat oral dosing of NTP42:KVA4, with dose proportional increases in exposure and favorable clearance rates indicating that NTP42:KVA4 may be suited for use as a once-a-day oral medication. The data also suggests that NTP42:KVA4 may be taken with or without food, another key benefit for patient compliance. Moreover, there was excellent correlation between NTP42 plasma levels and PD effect. At doses ≥1 mg NTP42:KVA4, complete and sustained inhibition of U46619-, but not ADP-, induced aggregation was observed, confirming selective TP-target engagement. The duration of inhibition of U46619-induced aggregation was dependent on the plasma levels of NTP42 after dosing, where U46619-induced platelet responses returned to baseline (pre-dose) levels in a dose-dependent manner.
Inhibition of TP-mediated platelet aggregation has been used to determine doses of TP antagonists to be tested for efficacy in early-stage clinical trials in multiple disease indications (Guth et al., 2004; Gaussem et al., 2005; Sakariassen et al., 2009; Fiessinger et al., 2010; Lesault et al., 2011; Richardson et al., 2013). While inhibition of TP-mediated platelet aggregation is not a regulatory-approved marker of efficacy in PAH patients per se, there is ample evidence for PAH being, in part, a prothrombotic state caused by the dysregulation of platelet activation, coagulation, fibrinolysis, and endothelial cell interaction (Rabinovitch, 2012). For example, Zhu et al. outlined a key role for platelet-derived transforming growth factor (TGF)-β in the metabolic reprogramming of pulmonary arterial smooth muscle cells and subsequent pulmonary vascular remodeling (Zhu et al., 2022). In that study, therapies that reduce platelet TGF-β release have strong potential to translate into clinically efficacious treatments for PAH (Zhu et al., 2022). Notably, Sotatercept, in clinical development for PAH, acts as a ligand trap for members of the TGF-β superfamily (Yung et al., 2020; Humbert et al., 2021; Hoeper et al., 2023). In Phase III trials, benefits for Sotatercept were seen across key clinical parameters, including the 6-min walk distance (6MWD) test, the primary clinical endpoint (Hoeper et al., 2023). However, using a multicomponent improvement (MCI) index, a composite measure based on pre-specified improvements in functional class (FC), 6MWD and NT-proBNP, only 40% of patients responded to Sotatercept delivered in combination with background PAH therapy (Hoeper et al., 2023). This latter finding demonstrates that there remains a clear need for novel therapies for PAH, in particular those with the potential to address all the clinical hallmarks of the disease, to lessen premature mortality while robustly improving patient quality-of-life.
Notwithstanding the potential benefits of TP antagonism acting through platelet-derived mechanisms in cardiopulmonary diseases such as PAH, or in other conditions, extensive data from preclinical models has shown NTP42’s efficacy in treating diverse pulmonary and cardiac pathologies (Mulvaney et al., 2020a; Mulvaney et al., 2020b; Mulvaney et al., 2022). With benefits in alleviating excessive vasoconstriction, pulmonary artery remodeling, as well as pulmonary inflammation and fibrosis in the preclinical setting, NTP42 is anticipated to lead to a reduction in PVR, the regulatory-approved marker of hemodynamic efficacy in PAH. Moreover, our preclinical efficacy data using the PAB model demonstrates that NTP42 may have a direct cardioprotective benefit, highly distinct from other PAH standard-of-care (SoC) therapies or indeed pipeline compounds (Mulvaney et al., 2022). These findings are relevant when considering that, in up to a third of patients with reduced PVR on PAH SoC therapy, RV function continues to decrease, with poorer survival outcomes (van de Veerdonk et al., 2011). Thus, the ideal PAH therapy should not only target pulmonary vasoconstriction and vascular remodeling but should also demonstrate direct cardioprotective effects and promote beneficial adaptation and maintenance or improvement in RV function.
Phase I clinical trials, by their nature, being typically conducted at a single clinical site, may suffer from a lack of ethnic diversity in the volunteer cohort recruited. As this FIH Phase I trial was conducted in healthy male, mostly white, Caucasian volunteers, it is acknowledged that the safety and tolerability findings and PK/PD profiles reported herein may differ in other diverse ethnic groups, females, and/or diseased patient cohorts. The safety, tolerability, PK/PD, and clinical efficacy profiles of NTP42 will be investigated in Phase II/III trials in relevant patient cohorts.
In conclusion, this study showed that NTP42:KVA4 is safe and well tolerated, with favorable PK/PD characteristics and clear evidence of specific TP target engagement. These findings, combined with compelling preclinical efficacy data (Mulvaney et al., 2020a; Mulvaney et al., 2020b; Mulvaney et al., 2022), support the continued development of NTP42:KVA4 for the treatment of cardiopulmonary diseases, such as PAH, or other conditions involving aberrant TXA2/TP signaling.
Data availability statement
The datasets presented in this article are not readily available because trial data may only be made available under the terms of a Confidential Disclosure Agreement (CDA). Requests to access the datasets should be directed to therese.kinsella@atxatherapeutics.com.
Ethics statement
The studies involving humans were approved by the United Kingdom NHS Health Research Authority (HRA), North East—York. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
HR: Conceptualization, Data curation, Formal Analysis, Methodology, Project administration, Validation, Visualization, Writing–original draft, Writing–review and editing. MM: Formal Analysis, Validation, Writing–review and editing. CP: Formal Analysis, Methodology, Supervision, Validation, Writing–review and editing. EM: Conceptualization, Methodology, Project administration, Validation, Visualization, Writing–original draft, Writing–review and editing. MB: Investigation, Methodology, Resources, Supervision, Writing–review and editing. TY: Investigation, Methodology, Resources, Supervision, Writing–review and editing. BK: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This Phase I clinical trial was sponsored and financially supported in full by ATXA Therapeutics Limited (ATXA). The company was the recipient of a grant from the European Commission Horizon 2020 SME Instrument Program (Project Number: 822258) which helped to fund the earlier preclinical and drug development work. These funding sources supported ATXA Therapeutics’ research and development program, and the individual funding bodies did not play a role in the study design, data collection, data analysis, data interpretation or in writing the manuscript.
Acknowledgments
We thank staff at Hammersmith Medicines Research, including Matilde Luce, Norena Fernandes, Steluta Bolboceanu, Maliha Ahmed, Nathalie Hopchet and relevant teams. We also thank Analytical Services International and Niche Science and Technology.
Conflict of interest
HR, EM, and BK were employees of ATXA Therapeutics Limited at the time of conducting the work reported in this manuscript. BK is also the founder and a member of the board of directors of ATXA Therapeutics Limited. The clinical trial, performed at Hammersmith Medicines Research, was sponsored by ATXA Therapeutics Ltd. ATXA received grant funding from the European Commission Horizon 2020 SME Instrument Program (Project Number: 822258) and, since 2023, is a recipient of European Commission funding through European Innovation Council Accelerator (Project Number: 190143893) program.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1296188/full#supplementary-material
References
Al-Naamani, N., Palevsky, H. I., Lederer, D. J., Horn, E. M., Mathai, S. C., Roberts, K. E., et al. (2016). Prognostic significance of biomarkers in pulmonary arterial hypertension. Ann. Am. Thorac. Soc. 13 (1), 25–30. doi:10.1513/AnnalsATS.201508-543OC
Ashton, A. W. (2023). Preparing to strike: acute events in signaling by the serpentine receptor for thromboxane a(2). Pharmacol. Ther. 248, 108478. Epub 20230613. doi:10.1016/j.pharmthera.2023.108478
Badimon, L., Vilahur, G., Rocca, B., and Patrono, C. (2021). The key contribution of platelet and vascular arachidonic acid metabolism to the pathophysiology of atherothrombosis. Cardiovasc Res. 117 (9), 2001–2015. doi:10.1093/cvr/cvab003
Berkenkopf, J. W., and Weichman, B. M. (1988). Production of prostacyclin in mice following intraperitoneal injection of acetic acid, phenylbenzoquinone and zymosan: its role in the writhing response. Prostaglandins 36 (5), 693–709. doi:10.1016/0090-6980(88)90014-7
Capra, V., Back, M., Angiolillo, D. J., Cattaneo, M., and Sakariassen, K. S. (2014). Impact of vascular thromboxane prostanoid receptor activation on hemostasis, thrombosis, oxidative stress, and inflammation. J. Thromb. Haemost. 12 (2), 126–137. doi:10.1111/jth.12472
Capra, V., Busnelli, M., Perenna, A., Ambrosio, M., Accomazzo, M. R., Gales, C., et al. (2013). Full and partial agonists of thromboxane prostanoid receptor unveil fine tuning of receptor superactive conformation and G protein activation. PloS one 8 (3), e60475. Epub 2013/04/05. doi:10.1371/journal.pone.0060475
Davi, G., Santilli, F., and Vazzana, N. (2012). Thromboxane receptors antagonists and/or synthase inhibitors. Handb. Exp. Pharmacol. (210), 261–286. doi:10.1007/978-3-642-29423-5_11
Fiessinger, J. N., Bounameaux, H., Cairols, M. A., Clement, D. L., Coccheri, S., Fletcher, J. P., et al. (2010). Thromboxane antagonism with terutroban in peripheral arterial disease: the taipad study. J. Thromb. Haemost. 8 (11), 2369–2376. doi:10.1111/j.1538-7836.2010.04020.x
Fontana, P., Zufferey, A., Daali, Y., and Reny, J. L. (2014). Antiplatelet therapy: targeting the Txa2 pathway. J. Cardiovasc Transl. Res. 7 (1), 29–38. Epub 20131219. doi:10.1007/s12265-013-9529-1
Francois, H., Athirakul, K., Mao, L., Rockman, H., and Coffman, T. M. (2004). Role for thromboxane receptors in angiotensin-ii-induced hypertension. Hypertension 43 (2), 364–369. Epub 2004/01/14. doi:10.1161/01.HYP.0000112225.27560.24
Francois, H., Makhanova, N., Ruiz, P., Ellison, J., Mao, L., Rockman, H. A., et al. (2008). A role for the thromboxane receptor in L-name hypertension. Am. J. Physiol. Ren. Physiol. 295 (4), F1096–F1102. Epub 2008/08/08. doi:10.1152/ajprenal.00369.2007
Gaussem, P., Reny, J. L., Thalamas, C., Chatelain, N., Kroumova, M., Jude, B., et al. (2005). The specific thromboxane receptor antagonist S18886: pharmacokinetic and pharmacodynamic studies. J. Thromb. Haemost. 3 (7), 1437–1445. doi:10.1111/j.1538-7836.2005.01468.x
Guth, B. D., Narjes, H., Schubert, H. D., Tanswell, P., Riedel, A., and Nehmiz, G. (2004). Pharmacokinetics and pharmacodynamics of Terbogrel, a combined thromboxane A2 receptor and synthase inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 58 (1), 40–51. Epub 2004/06/23. doi:10.1111/j.1365-2125.2004.02083.x
Hoeper, M. M., Badesch, D. B., Ghofrani, H. A., Gibbs, J. S. R., Gomberg-Maitland, M., McLaughlin, V. V., et al. (2023). Phase 3 trial of Sotatercept for treatment of pulmonary arterial hypertension. N. Engl. J. Med. 388, 1478–1490. Epub 20230306. doi:10.1056/NEJMoa2213558
Hoffmann, P., Heinroth-Hoffmann, I., and Toraason, M. (1993). Alterations by a thromboxane A2 analog (U46619) of calcium dynamics in isolated rat cardiomyocytes. J. Pharmacol. Exp. Ther. 264 (1), 336–344. Epub 1993/01/01. https://jpet.aspetjournals.org/content/264/1/336.
Humbert, M., McLaughlin, V., Gibbs, J. S. R., Gomberg-Maitland, M., Hoeper, M. M., Preston, I. R., et al. (2021). Sotatercept for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 384 (13), 1204–1215. doi:10.1056/NEJMoa2024277
Kinsella, B. T., and Reid, H. M. (2018). Thromboxane receptor antagonists. US PTO (patent No. 9,932,304. Fully Granted April 3, 2018).
Kinsella, B. T., and Reid, H. M. (2019). Thromboxane receptor antagonists. US PTO (patent No. 10,357,504. Fully Granted July 23, 2019).
Langleben, D., Christman, B. W., Barst, R. J., Dias, V. C., Galie, N., Higenbottam, T. W., et al. (2002). Effects of the thromboxane synthetase inhibitor and receptor antagonist Terbogrel in patients with primary pulmonary hypertension. Am. heart J. 143 (5), E4. Epub 2002/06/01. doi:10.1067/mhj.2002.121806
Lesault, P. F., Boyer, L., Pelle, G., Covali-Noroc, A., Rideau, D., Akakpo, S., et al. (2011). Daily administration of the tp receptor antagonist terutroban improved endothelial function in high-cardiovascular-risk patients with atherosclerosis. Br. J. Clin. Pharmacol. 71 (6), 844–851. doi:10.1111/j.1365-2125.2010.03858.x
Mulvaney, E. P., Reid, H. M., Bialesova, L., Bouchard, A., Salvail, D., and Kinsella, B. T. (2020a). Ntp42, a novel antagonist of the thromboxane receptor, attenuates experimentally induced pulmonary arterial hypertension. BMC Pulm. Med. 20 (1), 85. Epub 2020/04/08. doi:10.1186/s12890-020-1113-2
Mulvaney, E. P., Reid, H. M., Bialesova, L., Mendes-Ferreira, P., Adao, R., Bras-Silva, C., et al. (2020b). Efficacy of the thromboxane receptor antagonist Ntp42 alone, or in combination with sildenafil, in the sugen/hypoxia-induced model of pulmonary arterial hypertension. Eur. J. Pharmacol. 889, 173658. Epub 2020/10/31. doi:10.1016/j.ejphar.2020.173658
Mulvaney, E. P., Renzo, F., Adao, R., Dupre, E., Bialesova, L., Salvatore, V., et al. (2022). The thromboxane receptor antagonist Ntp42 promotes beneficial adaptation and preserves cardiac function in experimental models of right heart overload. Front. Cardiovasc Med. 9, 1063967. Epub 20221214. doi:10.3389/fcvm.2022.1063967
Murata, T., Ushikubi, F., Matsuoka, T., Hirata, M., Yamasaki, A., Sugimoto, Y., et al. (1997). Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 388 (6643), 678–682. doi:10.1038/41780
Naeije, R., and Manes, A. (2014). The right ventricle in pulmonary arterial hypertension. Eur. Respir. Rev. 23 (134), 476–487. Epub 2014/12/03. doi:10.1183/09059180.00007414
Nakamura, F., Minshall, R. D., Le Breton, G. C., and Rabito, S. F. (1996). Thromboxane A2 mediates the stimulation of inositol 1,4,5-trisphosphate production and intracellular calcium mobilization by bradykinin in neonatal rat ventricular cardiomyocytes. Hypertension 28 (3), 444–449. Epub 1996/09/01. doi:10.1161/01.hyp.28.3.444
Popp, L., Haussler, A., Olliges, A., Nusing, R., Narumiya, S., Geisslinger, G., et al. (2009). Comparison of nociceptive behavior in prostaglandin E, F, D, prostacyclin and thromboxane receptor knockout mice. Eur. J. Pain 13 (7), 691–703. Epub 20081019. doi:10.1016/j.ejpain.2008.09.001
Rabinovitch, M. (2012). Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest 122 (12), 4306–4313. doi:10.1172/JCI60658
Richardson, A., Sakariassen, K. S., Meyer, J. P., Alberts, P., and Sorensen, A. S. (2013). Single ascending oral dose pharmacokinetics and pharmacodynamics study of ev-077: the specific inhibitor of prostanoid- and isoprostane-induced cellular activation. Eur. J. Clin. Pharmacol. 69 (3), 459–465. Epub 2012/07/21. doi:10.1007/s00228-012-1348-9
Ryan, J. J., and Archer, S. L. (2014). The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 115 (1), 176–188. Epub 2014/06/22. doi:10.1161/CIRCRESAHA.113.301129
Sakariassen, K. S., Alberts, P., Fontana, P., Mann, J., Bounameaux, H., and Sorensen, A. S. (2009). Effect of pharmaceutical interventions targeting thromboxane receptors and thromboxane synthase in cardiovascular and renal diseases. Future Cardiol. 5 (5), 479–493. doi:10.2217/fca.09.33
Shindo, H., Tawata, M., Aida, K., and Onaya, T. (1991). Clinical efficacy of a stable prostacyclin analog, iloprost, in diabetic neuropathy. Prostaglandins 41 (1), 85–96. doi:10.1016/0090-6980(91)90108-r
van de Veerdonk, M. C., Kind, T., Marcus, J. T., Mauritz, G. J., Heymans, M. W., Bogaard, H. J., et al. (2011). Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J. Am. Coll. Cardiol. 58 (24), 2511–2519. Epub 2011/12/03. doi:10.1016/j.jacc.2011.06.068
Vonk Noordegraaf, A., and Galie, N. (2011). The role of the right ventricle in pulmonary arterial hypertension. Eur. Respir. Rev. 20 (122), 243–253. doi:10.1183/09059180.00006511
Wacker, M. J., Best, S. R., Kosloski, L. M., Stachura, C. J., Smoot, R. L., Porter, C. B., et al. (2006). Thromboxane A2-induced arrhythmias in the anesthetized rabbit. Am. J. Physiol. Heart Circ. Physiol. 290 (4), H1353–H1361. Epub 2005/12/13. doi:10.1152/ajpheart.00930.2005
Wacker, M. J., Kosloski, L. M., Gilbert, W. J., Touchberry, C. D., Moore, D. S., Kelly, J. K., et al. (2009). Inhibition of thromboxane A2-induced arrhythmias and intracellular calcium changes in cardiac myocytes by blockade of the inositol trisphosphate pathway. J. Pharmacol. Exp. Ther. 331 (3), 917–924. Epub 2009/09/11. doi:10.1124/jpet.109.157677
West, J. D., Galindo, C. L., Kim, K., Shin, J. J., Atkinson, J. B., Macias-Perez, I., et al. (2019). Antagonism of the thromboxane-prostanoid receptor as a potential therapy for cardiomyopathy of muscular dystrophy. J. Am. Heart Assoc. 8 (21), e011902. Epub 2019/10/31. doi:10.1161/JAHA.118.011902
West, J. D., Voss, B. M., Pavliv, L., de Caestecker, M., Hemnes, A. R., and Carrier, E. J. (2016). Antagonism of the thromboxane-prostanoid receptor is cardioprotective against right ventricular pressure overload. Pulm. Circ. 6 (2), 211–223. Epub 2016/06/03. doi:10.1086/686140
Yung, L. M., Yang, P., Joshi, S., Augur, Z. M., Kim, S. S. J., Bocobo, G. A., et al. (2020). Actriia-fc rebalances activin/gdf versus bmp signaling in pulmonary hypertension. Sci. Transl. Med. 12 (543), eaaz5660. Epub 2020/05/15. doi:10.1126/scitranslmed.aaz5660
Zhu, Y., Shu, D., Gong, X., Lu, M., Feng, Q., Zeng, X. B., et al. (2022). Platelet-derived TGF (transforming growth factor)-β1 enhances the aerobic glycolysis of pulmonary arterial smooth muscle cells by PKM2 (pyruvate kinase muscle isoform 2) upregulation. Hypertension 79 (5), 932–945. Epub 20220302. doi:10.1161/HYPERTENSIONAHA.121.18684
Keywords: thromboxane, safety, thromboxane receptor, NTP42, thromboxane receptor antagonist
Citation: Reid HM, Maginn M, Perkins CM, Mulvaney EP, Boyce M, Yamamoto T and Kinsella BT (2023) Evaluation of NTP42, a novel thromboxane receptor antagonist, in a first-in-human phase I clinical trial. Front. Pharmacol. 14:1296188. doi: 10.3389/fphar.2023.1296188
Received: 22 September 2023; Accepted: 04 December 2023;
Published: 21 December 2023.
Edited by:
Emanuela Ricciotti, University of Pennsylvania, United StatesReviewed by:
Erica J. Carrier, Vanderbilt University Medical Center, United StatesAnthony Ashton, The University of Sydney, Australia
Copyright © 2023 Reid, Maginn, Perkins, Mulvaney, Boyce, Yamamoto and Kinsella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: B. Therese Kinsella, therese.kinsella@atxatherapeutics.com