
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 14 September 2023
Sec. Pharmacogenetics and Pharmacogenomics
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1248898
This article is part of the Research TopicIs there still room for Pharmacogenetics in 2023? The cases of Infectious Diseases and OncologyView all 5 articles
 Georgia Ragia1,2
Georgia Ragia1,2 Anthi Maslarinou1,2
Anthi Maslarinou1,2 Natalia Atzemian1,2
Natalia Atzemian1,2 Eirini Biziota3Triantafyllia Koukaki3
Eirini Biziota3Triantafyllia Koukaki3 Charalampia Ioannou1Ioanna Balgkouranidou3
Charalampia Ioannou1Ioanna Balgkouranidou3 George Kolios1,2Stylianos Kakolyris3Nikolaos Xenidis3†Kyriakos Amarantidis3
George Kolios1,2Stylianos Kakolyris3Nikolaos Xenidis3†Kyriakos Amarantidis3 Vangelis G. Manolopoulos1,2,4*
Vangelis G. Manolopoulos1,2,4*Introduction: Dihydropyrimidine dehydrogenase (DPD), encoded by DPYD gene, is the rate-limiting enzyme responsible for fluoropyrimidine (FP) catabolism. DPYD gene variants seriously affect DPD activity and are well validated predictors of FP-associated toxicity. DPYD variants rs3918290, rs55886062, rs67376798, and rs75017182 are currently included in FP genetic-based dosing guidelines and are recommended for genotyping by the European Medicines Agency (EMA) before treatment initiation. In Greece, however, no data exist on DPYD genotyping. The aim of the present study was to analyze prevalence of DPYD rs3918290, rs55886062, rs67376798, rs75017182, and, additionally, rs1801160 variants, and assess their association with FP-induced toxicity in Greek cancer patients.
Methods: Study group consisted of 313 FP-treated cancer patients. DPYD genotyping was conducted on QuantStudio ™ 12K Flex Real-Time PCR System (ThermoFisher Scientific) using the TaqMan® assays C__30633851_20 (rs3918290), C__11985548_10 (rs55886062), C__27530948_10 (rs67376798), C_104846637_10 (rs75017182) and C__11372171_10 (rs1801160).
Results: Any grade toxicity (1-4) was recorded in 208 patients (66.5%). Out of them, 25 patients (12%) experienced grade 3-4 toxicity. DPYD EMA recommended variants were detected in 9 patients (2.9%), all experiencing toxicity (p = 0.031, 100% specificity). This frequency was found increased in grade 3-4 toxicity cases (12%, p = 0.004, 97.9% specificity). DPYD deficiency increased the odds of grade 3-4 toxicity (OR: 6.493, p = 0.014) and of grade 1-4 gastrointestinal (OR: 13.990, p = 0.014), neurological (OR: 4.134, p = 0.040) and nutrition/metabolism (OR: 4.821, p = 0.035) toxicities. FP dose intensity was significantly reduced in DPYD deficient patients (β = −0.060, p <0.001). DPYD rs1801160 variant was not associated with FP-induced toxicity or dose intensity. Triple interaction of DPYD*TYMS*MTHFR was associated with grade 3-4 toxicity (OR: 3.725, p = 0.007).
Conclusion: Our findings confirm the clinical validity of DPYD reduced function alleles as risk factors for development of FP-associated toxicity in the Greek population. Pre-treatment DPYD genotyping should be implemented in clinical practice and guide FP dosing. DPYD*gene interactions merit further investigation as to their potential to increase the prognostic value of DPYD genotyping and improve safety of FP-based chemotherapy.
The fluoropyrimidines (FPs) 5-fluorouracil (5-FU) and its oral prodrug capecitabine (CAP) constitute an efficient treatment of solid tumors. Despite their well-documented efficacy, toxicity is a serious drawback of FP therapy (Maslarinou et al., 2023). Toxicity rate in FP-treated patients has been reported to range between 10%–40%. Additionally, death rates due to FP-induced toxicity are relatively high. Per year, in France and the United States, respectively, approximately 150 and 1,300 FP-related deaths occur (Barin-Le Guellec et al., 2020; Deac et al., 2020).
Pharmacogenomics can be applied in FP therapy via the well-documented association of DPYD variations with FP-induced severe toxicity and dose requirements. DPYD encodes for dihydropyrimidine dehydrogenase (DPD), the rate limiting enzyme of 5-FU activation. To date, DPYD constitutes one of the most well characterized genetic loci. More than 80 DPYD variants have been identified and their functional effect on DPD activity has been characterized (Amstutz et al., 2018). Among them, DPYD*2A (rs3918290) and *13 (rs55886062) are the most common deleterious alleles leading to complete DPD loss of function (activity value 0), whereas c.2846T>A (rs67376798), and c.1129–5923C>G (rs75017182, HapB3) lead to decreased DPD activity (activity value 0.5). The released guidelines by Clinical Pharmacogenetics Implementation Consortium (CPIC) (Amstutz et al., 2018) and the Dutch Pharmacogenetics Working Group (DPWG) (Lunenburg et al., 2020) recommend FP dose reduction in DPYD defective patients ranging from 25% to 50% (activity score 0.5-1.5) or avoidance of FP use (activity score 0) (Amstutz et al., 2018; Lunenburg et al., 2020).
The benefits of DPYD genotyping towards reduction of FP-induced toxicity have been shown in a plethora of studies [reviewed in (Maslarinou et al., 2023)] with an established cost-effectiveness (Brooks et al., 2022; van der Wouden et al., 2022). Overall, results show that DPYD rs3918290, rs55886062, rs67376798, and rs75017182 variations are associated with increased toxicity risk and that this risk can be reduced when dose is genotype-adjusted without confronting therapeutic response (Maslarinou et al., 2023). Since April 2020, EMA recommends that “Patients should be tested for the lack of the enzyme DPD before starting cancer treatment with fluorouracil or with the related medicines, capecitabine and tegafur,” and that either phenotyping or genotyping can be used (EMA, 2020). Upfront DPYD genotyping of the four DPYD variants has been endorsed by several countries in Europe, including Germany, Austria, the Netherlands, Switzerland, the United Kingdom, Spain, and Italy (Martens et al., 2019; Wörmann et al., 2020; Begré et al., 2022; Etienne-Grimaldi et al., 2022; García-Alfonso et al., 2022; Wang et al., 2022; White et al., 2022; Bignucolo et al., 2023). In Italy, additionally to the four EMA recommended DPYD variants, rs1801160 (c.2194G>A, p.Val732Ile, DPYD*6) is also included in their ethnic dosing guidelines (Bignucolo et al., 2023). DPYD*6 is assigned by CPIC as a normal function allele, however, results of a recent meta-analysis show that DPYD*6 allele carriers have a 1.73-fold increased overall toxicity risk (Kim et al., 2022), therefore, in addition to the already known DPYD variants, *6 allele could be evaluated pre-emptively to reduce the risk of FP-induced toxicity (Del Re et al., 2019).
Despite the undisputable significance of DPYD variations on FP dosing decisions, for several countries, including Greece, data on the prevalence of DPYD variants and on their ethnicity-specific effect on FP-induced toxicity is still lacking. To fill this gap, the aim of the present study was to characterize the frequency of DPYD *2A, *13, c.2846T>A, HapB3, and *6 alleles in Greek cancer patients treated with the FPs 5-FU or CAP and to further analyze their association with FP-induced (severe) toxicity and dose intensity. Additionally, we have recently proposed the concept of a polygenic FP dosing algorithm (Maslarinou et al., 2023). Towards this direction, we have re-analyzed previous genetic associations found in the study population (Ioannou et al., 2021; Ioannou et al., 2022) in view of the presence of DPYD variations.
Patient cohort has been described previously in detail (Ioannou et al., 2021; Ioannou et al., 2022). In brief, a total of 313 unrelated cancer patients (160 males and 153 females; mean age 64.2 years ± 10.6, range 34–88) treated with 5-FU or CAP in monotherapy or in combination with other antineoplastic drugs were included in the study. All patients were inpatients of the Department of Oncology of the Academic General Hospital of Alexandroupolis in Greece and were retrospectively enrolled from February 2018 to December 2019.
For each patient demographic and clinical data were recorded, including scheme dosage, duration of treatment and administration route, toxicity incidence and its effect on treatment. Toxicity was recorded by grade according to the common terminology criteria for adverse events v5.0 (National Cancer Institute (NCI), 2017). All patients were closely monitored by the same team of oncologists who were responsible for clinical decisions on chemotherapeutic regimen, dosages, timeline of drug administration and therapy discontinuation.
Informed written consent was obtained from each patient. The protocol of the study was approved by the Scientific Council and the Ethics Committee of the Academic General Hospital of Alexandroupolis (Greece) and was conducted according to the Declaration of Helsinki.
Approximately 3 mL blood was collected in EDTA tubes. Genomic DNA was extracted from peripheral whole blood using Gentra Puregene Blood Kit (QiagenR/Qiagen, MD, United States) or MagCore Automated Nucleic Acid Extractor (RBC Bioscience, New Taipei City, Taiwan), according to the instructions of the manufacturer, and stored at −20°C until use. DNA purity and quantity were assessed by UV-Vis Spectrophotometer Q5000 (Quawell) and Qubit 4 fluorometer (ThermoFisher Scientific), respectively.
DPYD genotyping was conducted in 96-well plates on QuantStudio™ 12K Flex Real-Time PCR System (ThermoFisher Scientific) using the pre-designed TaqMan® allelic discrimination assays C__30633851_20 (rs3918290), C__11985548_10 (rs55886062), C__27530948_10 (rs67376798), C_104846637_10 (rs75017182) and C__11372171_10 (rs1801160) (ThermoFisher Scientific).
Each reaction was carried out in 10 μL of a total reaction volume containing 5 µL of TaqMan Genotyping Master Mix, 0.5 μL of 20x TaqMan assay and 4.5 μL (approximately 20 ng) of genomic DNA. PCR conditions for rs3918290, rs55886062, rs67376798, and rs1801160 were the following: 60 °C for 30 s (pre-read stage), 95 °C for 10 min, 50 cycles of 95 °C for 15 s and 60 °C for 1:30 min (PCR stage), and a final post-read stage at 60 °C for 30 s. For rs75017182, PCR conditions were identical except for 40 cycles of 95 °C for 15 s and 60 °C for 1 min in PCR stage. Non template controls were included in triplicates in each reaction. In 10% of randomly selected samples, genotyping was replicated by an independent researcher, with a 100% accordance in results.
For genotype calling, QuantStudio 12K Flex Software v1.5 was used. In the presence of at least one variant allele allelic discrimination plots were automatically generated, whereas in the absence of variations, genotype calling was based on multicomponent plots for each sample.
Shapiro–Wilk test was used to assess normality of continuous variables. Continuous variables are expressed as mean ± standard deviation (SD) in the case of normal distribution, otherwise they are expressed as median (25th, 75th percentiles). Based on normality, parametric (independent t-test or one-way ANOVA) and non-parametric (Mann–Whitney test or Kruskal–Wallis test) tests were used to compare continuous variables between two or more groups, as appropriate. Chi-square (χ2)-test or Fisher’s exact test was used for categorical data comparisons depending on number of observations. Allele frequencies were estimated by the gene counting method. Departure from Hardy–Weinberg equilibrium was estimated by an exact two-sided probability test using the formula provided by Weir (1996). True/False positive and negative cases were calculated to express sensitivity, specificity, positive (PPV) and negative (NPV) predictive values of DPYD genotyping in FP-induced any grade and grade 3-4 toxicity.
Logistic regression analysis with a stepwise forward selection procedure adjusted for sex, age, and weight was performed, with toxicity and severe toxicity incidence as the dependent variable and DPYD genotype or DPYD*other genes interaction as independent variable, to calculate odds ratio (OR). To estimate the likelihood of FP dose intensity associated with DPYD polymorphisms, beta coefficient (β) with 95% C.I. were calculated with a multivariable linear regression analysis with a stepwise forward selection procedure with FP dose intensity as a dependent variable and sex, age, weight, and DPYD deficiency as independent variables. A p-value less than 0.05 was considered statistically significant. The Statistical Package for Social Sciences for Windows, Version 27.0 (IBM Corp., NY, United States) was used for all analyses.
Patient demographic and clinical characteristics are presented in Table 1. The complete profile of patient clinicopathological data in pooled sample as well as stratified by sex has been previously described in detail (Ioannou et al., 2021; Ioannou et al., 2022). In terms of toxicity, FP-induced any grade (1-4) toxicity prevalence was 66.5% (208 patients) and the majority of adverse events (AEs) were gastrointestinal (37.1%). A total of 26 cases of grade 3-4 toxicity were recorded in 25 patients (12% prevalence within grade 1-4 toxicity). For a total of 79 patients, toxicity led to reduction of the administered dosage (n = 44), and/or delayed drug administration or therapy discontinuation (n = 59). Such adjustments were made in different chemotherapy cycles; n = 21 in cycle 1, n = 25 in cycle 2, n = 14 in cycle 3, n = 12 in cycle 4, n = 9 in cycle 5, n = 22 in cycle 6 and above.
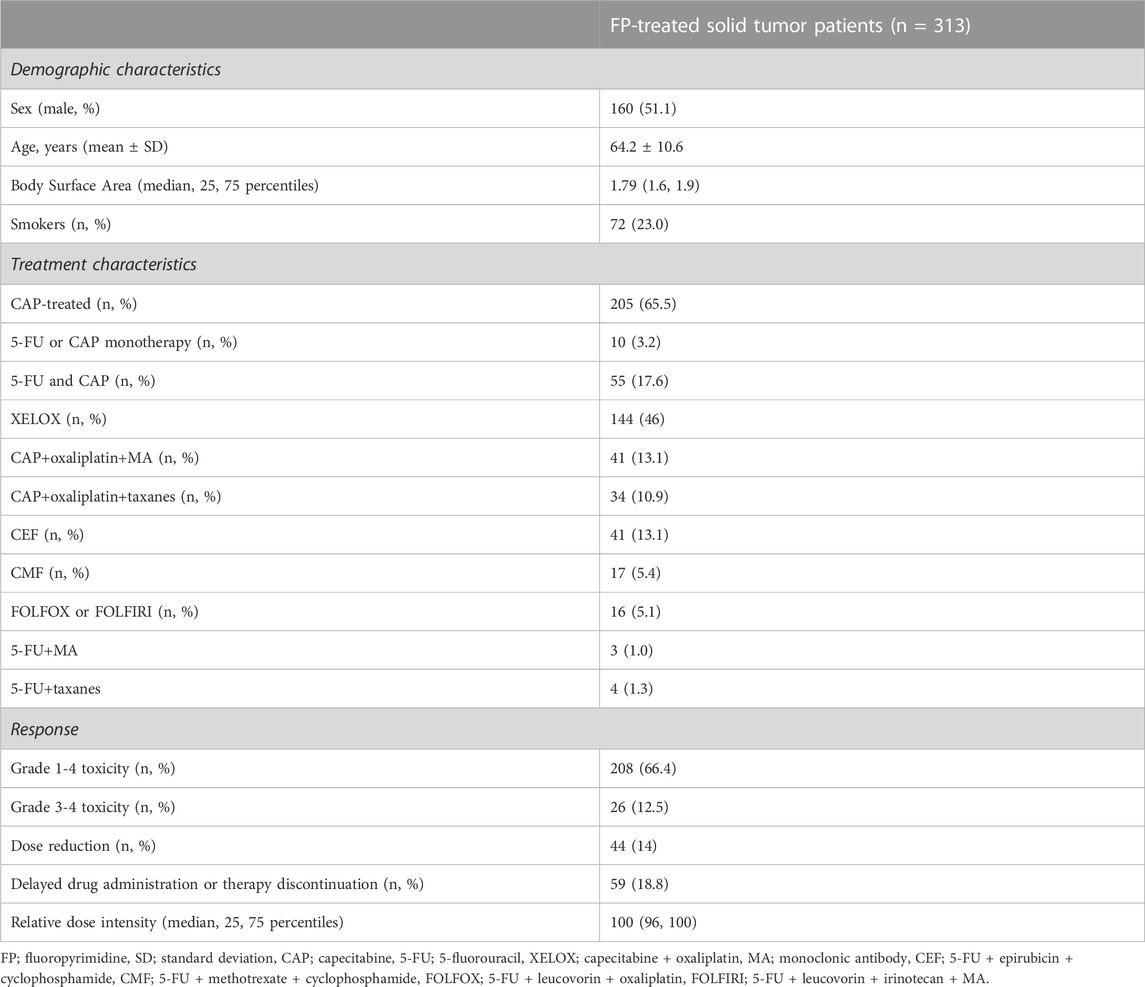
TABLE 1. Main characteristics of patient cohort.
Among the four EMA-recommended DPYD variants, we have found 9 heterozygous carriers (2.9% prevalence); one patient carrying *2A deleterious allele (0.3%), and 8 patients carrying the reduced function rs75017182 allele (2.6%). Both DPYD *13 and rs67376798 alleles were absent from the studied population (95% C.I. 0.0001%–0.6%). DPYD rs1801160 polymorphism was more common in our population (42 heterozygous patients, 13.4%, and 1 homozygous patient, 0.3%) (Table 2).
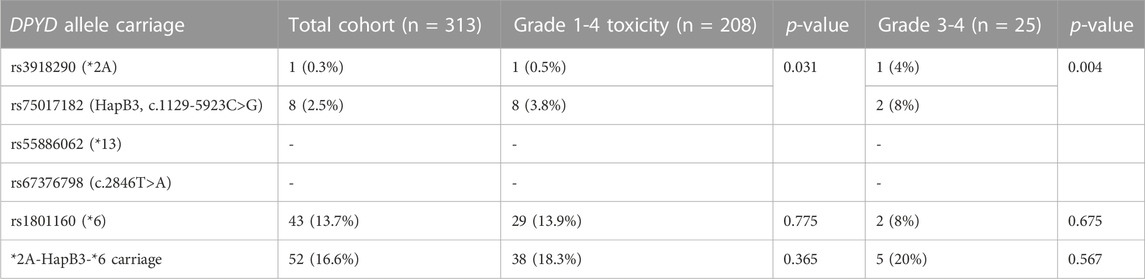
TABLE 2. Prevalence of DPYD variants in total patient cohort and stratified per toxicity grade.
All 9 patients carrying DPYD *2A or rs75017182 alleles experienced toxicity (p = 0.031). For grade 1-4 toxicity, PPV was 100%, NPV was 34.5%, with a 100% specificity and 4.3% sensitivity. DPYD deficiency increased odds of any grade gastrointestinal (OR: 13.99, 95% C.I. 1.71-114.22, p = 0.014), neurological (OR: 4.13, 95% C.I. 1.07-16.04, p = 0.040) and nutrition/metabolism (OR: 4.82, 95% C.I. 1.12-20.73, p = 0.035) toxicities.
In grade 3-4 toxicity cases prevalence of DPYD *2A or rs75017182 alleles was higher (3 out of 25 patients, 12.0%, p = 0.028) (PPV 33.3%, NPV 92.8%, specificity 97.9%, sensitivity 12%). DPYD deficiency increased odds of grade 3-4 toxicity in any system (OR: 6.49, 95% C.I. 1.45-29.06, p = 0.014). For gastrointestinal toxicity, DPYD defective variants significantly increased the odds of grade 3-4 toxicity (OR: 45.94, 95% C.I. 4.69-449.95, p = 0.001). Carriers of DPYD defective variants did not present with grade 3-4 toxicity in other systems.
In a model adjusted for sex, age, and weight, FP dose intensity was significantly reduced in DPYD deficient patients (β = −0.060, 95% C.I. −0.085, −0.035, p <0.001).
DPYD rs1801160 variant was not associated independently with FP-induced toxicity (p = 0.78 for any grade toxicity, p = 0.68 for grade 3-4 toxicity) or after adjusting for DPYD *2A and rs75017182 alleles (p = 0.77 for any grade toxicity, p = 0.51 for grade 3-4 toxicity). DPYD rs1801160 had no effect on dose intensity (p = 0.37).
Eight patients carrying DPYD *2A and rs75017182 variants, were treated with CAP-based combination chemotherapeutic scheme; 5 patients were on XELOX, and 3 patients on CAP+MA, whereas one patient was treated both with CAP or 5-FU based scheme (CAP or 5-FU +taxanes). The majority of patients (n = 7, 77.8%) had colorectal cancer. Eight out of nine DPYD variant carriers developed gastrointestinal toxicity. Oncologists proceeded to dose or therapy adjustment in 4 patients (5% of patients with dose or therapy adjustment). Dose was reduced only in the DPYD*2A carrier at the second cycle of chemotherapy due to grade 3-4 toxicity, as it is described in more detail in Section 3.5. For two patients who developed grade 1-2 toxicity, scheduled chemotherapy administration was delayed at cycle 4, whereas for the last carrier of DPYD variation, chemotherapeutic scheme was changed after first cycle.
In our study, one patient was carrier of the deleterious DPYD*2A allele having, thus, severely reduced DPD activity (activity score 1). This is the case of a 63 years old male colorectal cancer patient with pulmonary metastasis, treated with CAP at an initial daily dose of 3,500 mg. At the second cycle of chemotherapy, patient developed grade 1-2 diarrhea and CAP dose was reduced to 3,000 mg. At the third cycle, due to persisting diarrhea (grade 4) CAP dose was further reduced at 2,000 mg. At this dose, reduced by 43% of initially prescribed, patient did not experience any toxicity. According to CPIC dosing guidelines, the recommended starting dose for DPYD*2A heterozygous carriers is half of the standard dose. Prospective genotyping of this patient and subsequent DPYD-genotype CAP dose adjustment would have probably resulted in diarrhea avoidance and safe CAP treatment.
In previous studies in the same patient cohort published by our team, we have found that TYMS-TSER 2R/2R genotype was associated with FP dose reduction due to toxicity in female patients (Ioannou et al., 2021) and that MTHFR 665C>T polymorphism increased both need for FP dose reduction (OR 5.05) and percentage of dose reduction (β = 3.318) again in female patients (Ioannou et al., 2022). We have herein further analyzed the interaction of DPYD*TYMS*MTHFR towards a polygenic prediction of FP-induced toxicity (Table 3). Neither TYMS nor MTHFR were independently associated in regression analysis with grade 3-4 toxicity in the pooled sample, however, DPYD*TYMS TSER 2R/2R and DPYD*MTHFR 665T+ interactions were associated with increased odds of severe toxicity (OR: 2.89, 95% C.I. 1.23-6.84, p = 0.015 and OR: 6.18, 95% C.I. 1.97-19.42, p = 0.002, respectively). Additionally, triple gene interaction DPYD*TYMS*MTHFR was associated with grade 3-4 toxicity (OR: 3.73, 95% C.I. 1.43-9.71, p = 0.007). No gene interactions were found for dose intensity (data not shown).
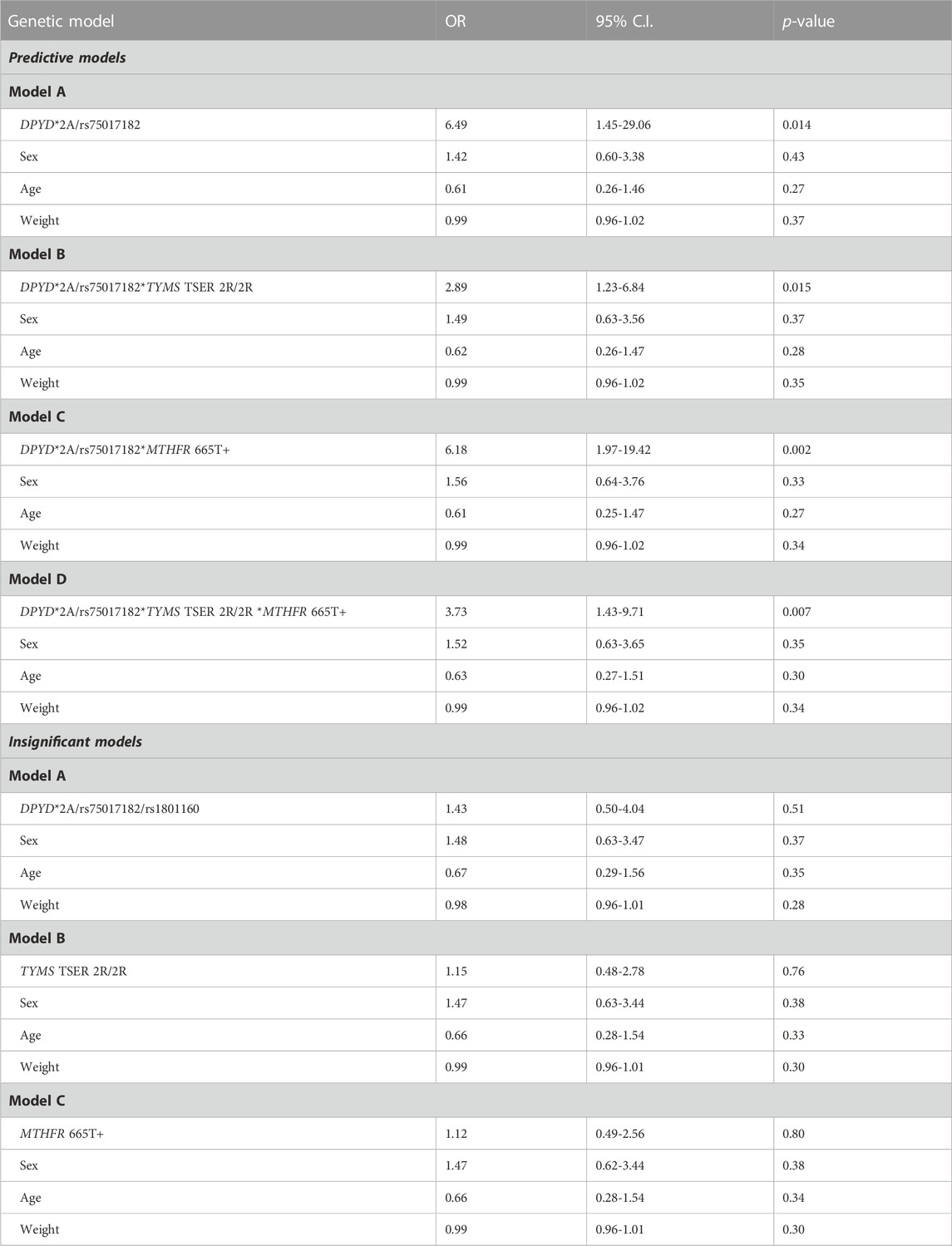
TABLE 3. Logistic regression analysis adjusted for sex, age and weight to estimate the odds of grade 3-4 toxicity in different genetic models.
In the present study we have genotyped a patient cohort consisting of 313 Greek cancer patients treated with the FPs 5-FU or CAP for five DPYD SNPs, namely, the EMA-recommended *2A, rs75017182, *13 and rs67376798, and, additionally, rs1801160. We have estimated the frequency of the four EMA-recommended polymorphism carriers at 2.9% and have found significant associations of DPYD variations with any grade FP-induced toxicity, severe toxicity and dose intensity. Analysis of DPYD rs1801160 SNP, did not improve the prediction models, albeit this SNP was higher in frequency (13.7% carriers). We have further applied a polygenic FP-induced toxicity prediction model in the study population and have found significant interactions among DPYD, TYMS, and MTHFR genes associated with grade 3-4 toxicity.
DPYD genotyping of *2A, rs75017182, *13, and rs67376798 constitutes the cornerstone of FP pharmacogenomics. Genotype-based FP dose reduction guided by these variations is currently implemented in several European countries prior to FP therapy initiation. However, to date, for Greece, data on DPYD variant frequency and on their effect on FP response in terms of toxicity is missing. In the studied population, we have detected heterozygosity of *2A and rs 75017182 variant alleles in nine patients. The DPYD deficiency frequency of 2.9% found in our study is consistent with the frequency of lower than 5% reported in European populations (Innocenti et al., 2020). For *13 and rs67376798 polymorphisms that were absent from the study population, the reported frequency in European populations ranges from 0% to 0.5%. DPYD rs1801160 polymorphism was more common in our population with an estimated allele frequency of 7% (13.7% carriers), in accordance with the frequency of approximately 5% reported in European populations (Kim et al., 2022).
DPYD*2A and rs75017182 polymorphisms were strongly associated with any grade toxicity; all patients carrying DPYD*2A and rs75017182 experienced FP-induced adverse events. In terms of severe (grade 3-4) toxicity, frequency of DPYD*2A and rs75017182 variants was increased (3 out of the 9 patients carrying these variants experienced severe toxicity). Both in any grade and in grade 3-4 toxicity, DPYD genotyping shows high specificity (100% and 97.9%, respectively). Sensitivity, however, is rather low ranging from 4.3% for any grade to 12% for grade 3-4 toxicity. Indeed, as described in other studies, the combined sensitivity of the major DPYD variants to predict 5-FU related toxicity is relatively low (5.3%), whereas specificity is above 99% (Lee et al., 2014). Therefore, in our study we confirm that in the Greek population DPYD genotyping of the EMA recommended polymorphisms has a prognostic value for FP-induced toxicity and can be used to select the optimal FP dose.
Additionally to the EMA-recommended DPYD polymorphisms, we have further analyzed DPYD rs1801160 variation. In our population, despite the rather high frequency of rs1801160 polymorphism, we found no association with FP-induced toxicity endpoints. DPYD rs1801160 variant is classified as a normal function allele, however, in different studies it was shown that after correcting for the four EMA-recommended alleles, rs1801160 increases risk of FP-induced toxicity (Del Re et al., 2019; Kim et al., 2022). The frequency of rs1801160 in Greek patient population is similar to the one reported in Europeans, however, adding rs1801160 in our prediction models decreased both PPV (from 100% to 72% for any grade toxicity) and specificity (from 100% to 86.7%). Our study could detect an effect of the magnitude reported in the meta-analysis by (Kim et al., 2022), if present in our population, with a power >75%. DPYD rs1801160 has not been associated straight forward with FP-induced toxicity in several studies (Toffoli et al., 2015; Varma et al., 2019), there are, however, reports showing that this variant allele may induce neutropenia risk (Ruzzo et al., 2017). In the recent meta-analysis by (Kim et al., 2022), rs1801160 shows an increased odds of neutropenia (OR 1.87) which is slightly increased to the odds of overall toxicity (OR 1.72). Therefore, in our study the lack of association could be hindered by the lack of records on levels of neutrophils. It should also be acknowledged that, in addition to DPYD genotype, other factors, notably microRNAs (Deac et al., 2021), have been shown to be predictive factors for FP toxicity; miR-27A is such a case. Interestingly, miR-27A rs895819 polymorphism was associated with FP-toxicity in DPYD variant allele carriers (Meulendijks et al., 2016). Thus, it cannot be excluded that DPYD rs1801160 effect is minor per se, but it can be masked by the presence of other variants. Such findings should be independently tested in different ethnic populations in order to establish the best fit genetic model to accurately predict patients who are at increased risk for FP-induced toxicity. Otherwise, implementing multiple variants without proven efficacy in a specific population endangers overestimating toxicity risk and underdosing of patients. It should be acknowledged that ethnicity is a significant contributor that should be taken into account as an adjusting factor for current genotype-based dosing algorithms. This has been shown for different pharmacogenomic based dosing algorithms, with CYP2C9/VKORC1 pharmacogenomic dosing algorithm for vitamin K antagonists standing as a gold example (Ragia et al., 2017). Specifically for the FPs and DPYD variations, differences have been reported in Asian populations in whom DPYD variations have only a minor role in FP-related toxicity (Kanai et al., 2022).
DPYD undisputedly drives personalization of FP-dosing. However, a main limitation of DPYD polymorphisms is their rather low frequency resulting in low sensitivity. This fact highlights the need both to identify additional DPYD deleterious variants and to incorporate additional genes in dose prediction guidelines. DPYD is a well characterized genetic locus. It is known that additional, albeit rare variants, exist that may significantly increase FP-induced toxicity risk. Rare DPYD variants could contribute in predicting a larger fraction of DPD deficiencies and partially address the missing heritability and improve prediction of FP-induced toxicity (De Luca et al., 2022; De Mattia et al., 2022; Lešnjaković et al., 2023). It is expected, however, that even if additional rare variants are incorporated into DPYD dosing guidelines, sensitivity will remain low. We have recently discussed in detail that, beyond DPYD, several genes are involved FP-induced toxicity and that strong evidence exists that we are gradually moving from single DPYD based FP-dosing to a multigenic dosing approach (Maslarinou et al., 2023). Towards this direction, we have re-analyzed in our study population the association of TYMS and MTHFR gene polymorphisms in interaction with DPYD polymorphisms. We had previously shown that TYMS and MTHFR variations affect FP-response in a sex specific manner (Ioannou et al., 2021; Ioannou et al., 2022). In these studies, both TYMS and MTHFR were not independently associated with FP-induced toxicity. Interaction analyses show that, after correcting for DPYD, TYMS and MTHFR variations secondary increase FP-induced toxicity risk. Interestingly, the triple gene interaction DPYD*TYMS*MTHFR confers approximately a 4-fold increased odds of grade 3-4 toxicity, irrespectively of sex, while both 95% C.I. and p-value were improved compared to DPYD alone. TYMS and MTHFR encode for proteins that are not involved in 5-FU breakdown; these are thymidylate synthase, a target of FPs, and methylene tetrahydrofolate reductase that interferes with FP activity via thymidylate synthase inhibition (Maslarinou et al., 2023). The DPYD*TYMS*MTHFR interaction, thus, may be suggestive for additive effects of FP pharmacokinetic and pharmacodynamic variant factors and needs further investigation in larger patient cohorts. Ultimately, multi pharmaco-omics approach can and will be used in oncology (Ragia and Manolopoulos, 2022). It should be acknowledged that both machine learning and artificial intelligence models should be employed to classify the variants associated with FP-induced toxicity and construct tools for the clinical application of multigenic interactions (Shrestha et al., 2018; Maslarinou et al., 2023).
Our study has several strengths. Patient population is well characterized in terms of toxicity incidence. Additionally, all patients were closely monitored by the same small group of oncologists reducing thus variability in clinical decisions. All patients were previously genotyped for TYMS and MTHFR polymorphisms allowing therefore generation of gene interaction analyses. DPYD genotyping was performed by use of validated pre-designed TaqMan assays and 100% accordance in genotyping call was obtained by independent researchers. We should also acknowledge that unavoidable limitations exist in the study design. This is a retrospective study, therefore, the benefits of prospective DPYD genotyping in reducing FP-induced toxicity incidence cannot be assessed. Rare variants of DPYD that can shed additional light into toxicity mechanisms were not sequenced in our population. Additionally, in interaction analyses, other genes showing association with FP dosing requirements and toxicity incidence, such as ENOSF1, were not included. Patient chemotherapeutic schemes are FP-based, albeit it cannot be excluded that toxicity was induced by other agents. In our cohort, all DPYD variant carriers were treated with FP-based combination chemotherapeutic schemes; pharmacogenomic markers of other chemotherapeutic agents were not analyzed.
In conclusion, our study shows that approximately 3% of the Greek cancer patient population have reduced DPYD activity. We confirm the DPYD clinical implications in terms of FP-induced toxicity. DPYD *2A, rs75017182, *13 and rs67376798 based dosing can be applied in the Greek cancer population prior of FP therapy initiation. Towards a polygenic FP-dosing algorithm, interaction of DPYD*TYMS*MTHFR needs further validation in different populations.
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by the Scientific Council and Ethics Committee of the Academic General Hospital of Alexandroupolis (Greece). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
GR: study conception and design, experimental procedures, analysis and interpretation of results, manuscript preparation and editing. AM: experimental procedures. NA: experimental procedures. EB: patient enrollment, clinical examination, clinical decisions, sample acquisition, data collection. TK: patient enrollment, clinical examination, clinical decisions, sample acquisition, data collection. CI: data collection, experimental procedures. IB: sample acquisition, data collection. GK: manuscript review and editing. SK: patient enrollment, clinical examination, clinical decisions, sample acquisition, data collection. NX: patient enrollment, clinical examination, clinical decisions, sample acquisition, data collection. KA: patient enrollment, clinical examination, clinical decisions, sample acquisition, data collection. VGM: study conception and design, funding acquisition, interpretation of results, manuscript review and editing, and final manuscript editing. All authors contributed to the article and approved the submitted version.
Financial support for project IMPReS (MIS 5047189) was provided to VGM by the Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) co-financed by Greece and the European Union (European Regional Development Fund).
This paper is dedicated to the memory of Professor Nikolaos Xenidis (1966–2022).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Amstutz, U., Henricks, L. M., Offer, S. M., Barbarino, J., Schellens, J. H. M., Swen, J. J., et al. (2018). Clinical pharmacogenetics implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin. Pharmacol. Ther. 103 (2), 210–216. doi:10.1002/cpt.911
Barin-Le Guellec, C., Lafay-Chebassier, C., Ingrand, I., Tournamille, J. F., Boudet, A., Lanoue, M. C., et al. (2020). Toxicities associated with chemotherapy regimens containing a fluoropyrimidine: A real-life evaluation in France. Eur. J. Cancer 124, 37–46. doi:10.1016/j.ejca.2019.09.028
Begré, U. B. M., Jörger, M., Aebi, S., Amstutz, U., and Largiadèr, C. R. (2022). Clinical implementation of DPYD pharmacogenetic testing to prevent early-onset fluoropyrimidine-related toxicity in cancer patients in Switzerland. Front. Pharmacol. 13, 885259. doi:10.3389/fphar.2022.885259
Bignucolo, A., De Mattia, E., Roncato, R., Peruzzi, E., Scarabel, L., D'Andrea, M., et al. (2023). Ten-year experience with pharmacogenetic testing for DPYD in a national cancer center in Italy: Lessons learned on the path to implementation. Front. Pharmacol. 14, 1199462. doi:10.3389/fphar.2023.1199462
Brooks, G. A., Tapp, S., Daly, A. T., Busam, J. A., and Tosteson, A. N. A. (2022). Cost-effectiveness of DPYD genotyping prior to fluoropyrimidine-based adjuvant chemotherapy for colon cancer. Clin. Colorectal Cancer 21 (3), e189–e195. doi:10.1016/j.clcc.2022.05.001
De Luca, O., Salerno, G., De Bernardini, D., Torre, M. S., Simmaco, M., Lionetto, L., et al. (2022). Predicting dihydropyrimidine dehydrogenase deficiency and related 5-fluorouracil toxicity: Opportunities and challenges of DPYD exon sequencing and the role of phenotyping assays. Int. J. Mol. Sci. 23 (22), 13923. doi:10.3390/ijms232213923
De Mattia, E., Silvestri, M., Polesel, J., Ecca, F., Mezzalira, S., Scarabel, L., et al. (2022). Rare genetic variant burden in DPYD predicts severe fluoropyrimidine-related toxicity risk. Biomed. Pharmacother. 154, 113644. doi:10.1016/j.biopha.2022.113644
Deac, A. L., Burz, C. C., Militaru, C., Bocșan, I. C., Pop, R. M., Achimaș-Cadariu, P., et al. (2021). Role of microRNAs in fluoropyrimidine-related toxicity: what we know. Eur. Rev. Med. Pharmacol. Sci. 25 (8), 3306–3315. doi:10.26355/eurrev_202104_25742
Deac, A. L., Burz, C. C., Bocse, H. F., Bocsan, I. C., and Buzoianu, A. D. (2020). A review on the importance of genotyping and phenotyping in fluoropyrimidine treatment. Med. Pharm. Rep. 93 (3), 223–230. doi:10.15386/mpr-1564
Del Re, M., Cinieri, S., Michelucci, A., Salvadori, S., Loupakis, F., Schirripa, M., et al. (2019). DPYD*6 plays an important role in fluoropyrimidine toxicity in addition to DPYD*2A and c.2846A>T: a comprehensive analysis in 1254 patients. Pharmacogenomics J. 19 (6), 556–563. doi:10.1038/s41397-019-0077-1
EMA (2020). EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine. Amsterdam, Netherlands: European Medicines Agency.
Etienne-Grimaldi, M. C., Pallet, N., Boige, V., Ciccolini, J., Chouchana, L., Barin-Le Guellec, C., et al. (2022). Current diagnostic and clinical issues of screening for dihydropyrimidine dehydrogenase deficiency. Eur. J. Cancer 181, 3–17. doi:10.1016/j.ejca.2022.11.028
García-Alfonso, P., Saiz-Rodríguez, M., Mondéjar, R., Salazar, J., Páez, D., Borobia, A. M., et al. (2022). Consensus of experts from the Spanish Pharmacogenetics and Pharmacogenomics Society and the Spanish Society of Medical Oncology for the genotyping of DPYD in cancer patients who are candidates for treatment with fluoropyrimidines. Clin. Transl. Oncol. 24 (3), 483–494. doi:10.1007/s12094-021-02708-4
Innocenti, F., Mills, S. C., Sanoff, H., Ciccolini, J., Lenz, H. J., and Milano, G. (2020). All you need to know about DPYD genetic testing for patients treated with fluorouracil and capecitabine: A practitioner-friendly guide. JCO Oncol. Pract. 16 (12), 793–798. doi:10.1200/op.20.00553
Ioannou, C., Ragia, G., Balgkouranidou, I., Xenidis, N., Amarantidis, K., Koukaki, T., et al. (2021). Gender-dependent association of TYMS-TSER polymorphism with 5-fluorouracil or capecitabine-based chemotherapy toxicity. Pharmacogenomics 22 (11), 669–680. doi:10.2217/pgs-2021-0031
Ioannou, C., Ragia, G., Balgkouranidou, I., Xenidis, N., Amarantidis, K., Koukaki, T., et al. (2022). MTHFR c.665C>T guided fluoropyrimidine therapy in cancer: gender-dependent effect on dose requirements. Drug Metab. Pers. Ther. 37 (3), 323–327. doi:10.1515/dmpt-2021-0219
Kanai, M., Kawaguchi, T., Kotaka, M., Manaka, D., Hasegawa, J., Takagane, A., et al. (2022). Poor association between dihydropyrimidine dehydrogenase (DPYD) genotype and fluoropyrimidine-induced toxicity in an Asian population. Cancer Med. 12 (7), 7808–7814. doi:10.1002/cam4.5541
Kim, W., Cho, Y. A., Kim, D. C., and Lee, K. E. (2022). Elevated risk of fluoropyrimidine-associated toxicity in European patients with DPYD genetic polymorphism: A systematic review and meta-analysis. J. Pers. Med. 12 (2), 225. doi:10.3390/jpm12020225
Lee, A. M., Shi, Q., Pavey, E., Alberts, S. R., Sargent, D. J., Sinicrope, F. A., et al. (2014). DPYD variants as predictors of 5-fluorouracil toxicity in adjuvant colon cancer treatment (NCCTG N0147). J. Natl. Cancer Inst. 106 (12), dju298. doi:10.1093/jnci/dju298
Lešnjaković, L., Ganoci, L., Bilić, I., Šimičević, L., Mucalo, I., Pleština, S., et al. (2023). DPYD genotyping and predicting fluoropyrimidine toxicity: where do we stand? Pharmacogenomics 24 (2), 93–106. doi:10.2217/pgs-2022-0135
Lunenburg, C., van der Wouden, C. H., Nijenhuis, M., Crommentuijn-van Rhenen, M. H., de Boer-Veger, N. J., Buunk, A. M., et al. (2020). Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet. 28 (4), 508–517. doi:10.1038/s41431-019-0540-0
Martens, F. K., Huntjens, D. W., Rigter, T., Bartels, M., Bet, P. M., and Cornel, M. C. (2019). DPD testing before treatment with fluoropyrimidines in the Amsterdam UMCs: An evaluation of current pharmacogenetic practice. Front. Pharmacol. 10, 1609. doi:10.3389/fphar.2019.01609
Maslarinou, A., Manolopoulos, V. G., and Ragia, G. (2023). Pharmacogenomic-guided dosing of fluoropyrimidines beyond DPYD: time for a polygenic algorithm?. Front. Pharmacol. 14, 1184523. doi:10.3389/fphar.2023.1184523
Meulendijks, D., Henricks, L. M., Amstutz, U., Froehlich, T. K., Largiadèr, C. R., Beijnen, J. H., et al. (2016). Rs895819 in MIR27A improves the predictive value of DPYD variants to identify patients at risk of severe fluoropyrimidine-associated toxicity. Int. J. Cancer 138 (11), 2752–2761. doi:10.1002/ijc.30014
National Cancer Institute (NCI) (2017). Common Terminology Criteria for Adverse Events (CTCAE) v5.0. Available at: https://ctep.cancer.gov/protocolDevelopment/electronicapplications/ctc.htm#ctc50.
Ragia, G., Kolovou, V., Kolovou, G., Konstantinides, S., Maltezos, E., Tavridou, A., et al. (2017). A novel acenocoumarol pharmacogenomic dosing algorithm for the Greek population of EU-PACT trial. Pharmacogenomics 18 (1), 23–34. doi:10.2217/pgs-2016-0126
Ragia, G., and Manolopoulos, V. G. (2022). The revolution of pharmaco-omics: ready to open new avenues in materializing precision medicine? Pharmacogenomics 23 (16), 869–872. doi:10.2217/pgs-2022-0145
Ruzzo, A., Graziano, F., Galli, F., Galli, F., Rulli, E., Lonardi, S., et al. (2017). Dihydropyrimidine dehydrogenase pharmacogenetics for predicting fluoropyrimidine-related toxicity in the randomised, phase III adjuvant TOSCA trial in high-risk colon cancer patients. Br. J. Cancer 117 (9), 1269–1277. doi:10.1038/bjc.2017.289
Shrestha, S., Zhang, C., Jerde, C. R., Nie, Q., Li, H., Offer, S. M., et al. (2018). Gene-specific variant classifier (DPYD-Varifier) to identify deleterious alleles of dihydropyrimidine dehydrogenase. Clin. Pharmacol. Ther. 104 (4), 709–718. doi:10.1002/cpt.1020
Toffoli, G., Giodini, L., Buonadonna, A., Berretta, M., De Paoli, A., Scalone, S., et al. (2015). Clinical validity of a DPYD-based pharmacogenetic test to predict severe toxicity to fluoropyrimidines. Int. J. Cancer 137 (12), 2971–2980. doi:10.1002/ijc.29654
van der Wouden, C. H., Marck, H., Guchelaar, H. J., Swen, J. J., and van den Hout, W. B. (2022). Cost-effectiveness of pharmacogenomics-guided prescribing to prevent gene-drug-related deaths: A decision-analytic model. Front. Pharmacol. 13, 918493. doi:10.3389/fphar.2022.918493
Varma, K. A., Jayanthi, M., Dubashi, B., and Shewade, D. G. (2019). Influence of DPYD*9A, DPYD*6 and GSTP1 ile105val genetic polymorphisms on capecitabine and oxaliplatin (CAPOX) associated toxicities in colorectal cancer (CRC) patients. Asian Pac J. Cancer Prev. 20 (10), 3093–3100. doi:10.31557/apjcp.2019.20.10.3093
Wang, L., Howlett, S., and Essapen, S. (2022). Treating patients with dihydropyrimidine dehydrogenase (DPD) deficiency with fluoropyrimidine chemotherapy since the onset of routine prospective testing-The experience of a large oncology center in the United Kingdom. Semin. Oncol. 49 (2), 170–177. doi:10.1053/j.seminoncol.2021.11.004
Weir, B. S. (1996). “Disequilibrium,” in Genetic Data Analysis II. Massachusetts, United States: Sinauer Associates Inc., 91–139.
White, C., Scott, R. J., Paul, C., Ziolkowski, A., Mossman, D., Fox, S. B., et al. (2022). Dihydropyrimidine dehydrogenase deficiency and implementation of upfront DPYD genotyping. Clin. Pharmacol. Ther. 112 (4), 791–802. doi:10.1002/cpt.2667
Keywords: DPYD, pharmacogenomics, fluoropyrimidines, 5-fluοrouracil, capecitabine, clinical implementation, polygenic algorithm, DPYD*TYMS*MTHFR interaction
Citation: Ragia G, Maslarinou A, Atzemian N, Biziota E, Koukaki T, Ioannou C, Balgkouranidou I, Kolios G, Kakolyris S, Xenidis N, Amarantidis K and Manolopoulos VG (2023) Implementing pharmacogenetic testing in fluoropyrimidine-treated cancer patients: DPYD genotyping to guide chemotherapy dosing in Greece. Front. Pharmacol. 14:1248898. doi: 10.3389/fphar.2023.1248898
Received: 27 June 2023; Accepted: 30 August 2023;
Published: 14 September 2023.
Edited by:
Jessica Cusato, University of Turin, ItalyReviewed by:
Ursula Amstutz, University of Bern, SwitzerlandCopyright © 2023 Ragia, Maslarinou, Atzemian, Biziota, Koukaki, Ioannou, Balgkouranidou, Kolios, Kakolyris, Xenidis, Amarantidis and Manolopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vangelis G. Manolopoulos, ZW1hbm9sb3BAbWVkLmR1dGguZ3I=
†Deceased
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.