 Lixiang Feng
Lixiang Feng Xingyu Chen1
Xingyu Chen1 Yujing Huang
Yujing Huang Na Xie
Na Xie- 1West China School of Basic Medical Sciences and Forensic Medicine, Sichuan University, and State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, and Collaborative Innovation Center for Biotherapy, Chengdu, China
- 2Hainan Cancer Clinical Medical Center of the First Affiliated Hospital, Key Laboratory of Tropical Cardiovascular Diseases Research of Hainan Province and Key Laboratory of Emergency and Trauma of Ministry of Education, Hainan Medical University, Haikou, China
- 3Department of Pathology, Hainan Women and Children Medical Center, Hainan Medical University, Haikou, China
Immune cells are essential for initiating and developing the fibrotic process by releasing cytokines and growth factors that activate fibroblasts and promote extracellular matrix deposition. Immunometabolism describes how metabolic alterations affect the function of immune cells and how inflammation and immune responses regulate systemic metabolism. The disturbed immune cell function and their interactions with other cells in the tissue microenvironment lead to the origin and advancement of fibrosis. Understanding the dysregulated metabolic alterations and interactions between fibroblasts and the immune cells is critical for providing new therapeutic targets for fibrosis. This review provides an overview of recent advances in the pathophysiology of fibrosis from the immunometabolism aspect, highlighting the altered metabolic pathways in critical immune cell populations and the impact of inflammation on fibroblast metabolism during the development of fibrosis. We also discuss how this knowledge could be leveraged to develop novel therapeutic strategies for treating fibrotic diseases.
1 Introduction
Fibrosis in various organisms, such as idiopathic pulmonary fibrosis (IPF), hepatic fibrosis, nephrogenic fibrosis, cardiac fibrosis and systemic sclerosis (SSc), might result in organ failure and death if left unchecked. In response to harmful irritants, including pathogens, toxins, oxidative stress, and autoimmune reactions, fibroblasts of multiple origins, such as mesenchymal cells, resident fibroblasts, and trans-differentiation of other cell types, launch the injury healing reactions via remodeling the extracellular environment to repair tissue integrity (Wynn and Ramalingam, 2012). Normally, the fibrotic program is turned off when tissue healing occurs. However, a chronic inflammatory milieu and dysregulated tissue healing caused by persistent tissue injury result in fibrosis by inducing fibroblast activation and excessive extracellular matrix (ECM) protein deposition (Meng et al., 2016). Specifically, sustained TGF-β and pro-inflammatory cytokines promote the differentiation of fibroblasts into myofibroblasts, which are responsible of secreting collagen and other pro-fibrotic factors (Darby and Hewitson, 2007; Travers et al., 2016). In fibrosis, excessively enhanced ECM production causes an increase in organ stiffness and tissue thickening, ultimately leading to organ dysfunction (Herrera et al., 2018).
Activation of immune cells, including macrophages, neutrophils, B cells and T cells, is crucial for tissue damage repair (Arora et al., 2018; Franco et al., 2020). Recent studies indicate that immune cells undergo cellular metabolic reprogramming during the development of various profibrotic diseases, suggesting a crucial role of immunometabolism in fibrosis (Srivastava et al., 2018; Cho et al., 2020). Compared to the homeostatic immune cells that rely on the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) for efficient energy production, immune cell activation requires increased glycolysis to supply energy and metabolic intermediates (Lam et al., 2016). Additionally, glutamine metabolism and fatty acid oxidation (FAO) can fuel the TCA cycle to support the energy generation in mitochondria. These metabolic alterations play a vital role in regulating immune cell differentiation and the production of inflammatory factors, promoting inflammation (Xu et al., 2021; Pérez and Rius-Pérez, 2022). From the immunometabolism aspect, the aberrant metabolic reprogramming alters the immune cell function, leading to a chronic inflammatory environment and contributing to fibrosis development (Xu et al., 2021; Pérez and Rius-Pérez, 2022). Additionally, the inflammation and immune responses regulate fibroblast metabolism to increase ECM deposition, further aggravating fibrosis (Zhu et al., 2019). Therefore, targeting the abnormal metabolic signaling molecules may provide a practical pathway to slow fibrosis progression.
This review discusses the crucial role of immunometabolism in fibrotic processes and the clinical applications of intervening with metabolic pathways for treating fibrotic diseases.
2 Inflammation-driven metabolic reprogramming in fibrotic fibroblasts
Increasing evidence showed that metabolic reprogramming in activated fibroblasts is an essential feature of fibrosis and its associated diseases (Wang et al., 2019; Wang Q. et al., 2021; Ning et al., 2023). Chronic inflammation induces the metabolic alterations in fibrotic fibroblasts, including glycolysis, amino acid metabolism, fatty acid oxidation (FAO), and fatty acid synthesis (FAS). These metabolic alterations have been implicated in forming ECM and inflammatory environments (Figure 1). Additionally, TGF-β and its associated signaling pathways have been approved to be a key regulator for metabolic reprogramming-driven fibrosis (Jiang et al., 2015; Xie et al., 2015; Wang F. et al., 2023) (Figure 2).
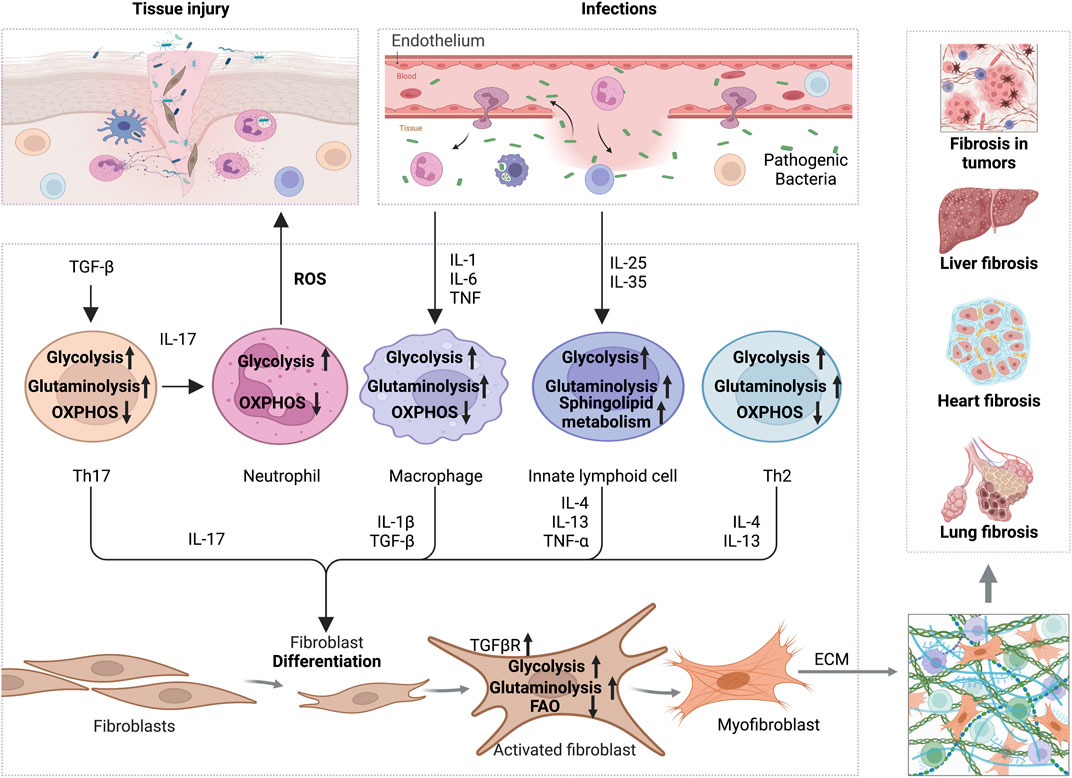
FIGURE 1. The interplay of inflammation with metabolic reprogramming in the profibrotic environment. TGF-β boosts glycolysis and glutaminolysis in Th17 to promote IL-17 release, which increases ROS secretion by neutrophils and thus promotes tissue injury. On the other hand, increased TGF-βR expression on the surface of fibroblasts promotes glycolysis and glutaminolysis to support ECM production. In addition, pro-inflammatory factors, like IL-1, IL-6, and TNF-α secreted by infected or damaged tissues, enhance the glycolysis and glutaminolysis in macrophages, thus increasing the expression of IL-1β and TGF-β to activate the fibrotic response. Cytokines, including IL-25 and IL-35, increase the glycolysis, sphingolipid metabolism and glutaminolysis in innate lymphocytes, which promote the secretion of IL-4, IL-13 and TNF-α, and activation of myofibroblasts. Th2 cells also secrete IL-4 and IL-13 in a glycolysis and glutaminolysis dependent manner to exert profibrotic effect. Ultimately, the activated myofibroblasts contribute to ECM formation and promote fibrosis in various tissues, including liver, heart, lung and even tumors. Abbreviations: ECM, extracellular matrix; FAO, fatty acid oxidation; IL-1, interleukin-1; IL-4, interleukin-4; IL-6, interleukin-6; IL-10, interleukin-10; IL-13, interleukin-17; IL-17, interleukin-17; IL-25, interleukin-25; IL-35, interleukin-35; ROS, reactive oxygen species; TNF-α, tumor necrosis factor alpha; TGF-β, transforming growth factor-beta; TGFβR,TGFβ receptor.
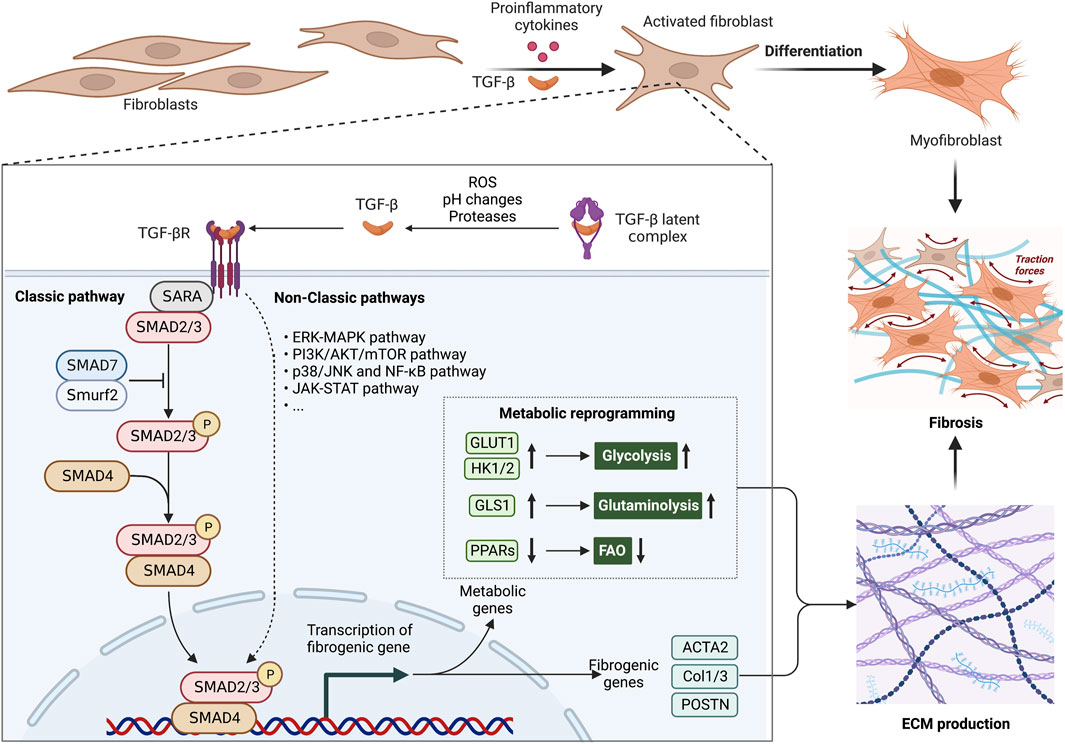
FIGURE 2. Metabolic alterations are involved in the profibrotic role of TGF-β1 signaling pathway. The aberrant TGF-β1 signaling pathway is an essential pathogenic mechanism in fibrosis. The TGF-β latent complex is activated by proteases, ROS and pH changes in the microenvironment. Activated TGF-β induces the differentiation of fibroblasts into myofibroblasts, by inducing metabolic enzymes in glycolysis and glutaminolysis, as well as inhibiting PPAR signaling-mediated fatty acid oxidation (FAO). These metabolic alterations offer ATP and biosynthetic intermediates for more ECM production. In fibrosis, abnormally ECM production causes an increase in organ stiffness, tissue thickening and lack of mobility, ultimately leading to organ dysfunction. Abbreviations: ERK, extracellular signal-regulated kinase; FAO, fatty acid oxidation; JNK, Jun N-terminal kinase; JAK, Janus kinase; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PI3K, phosphoinositide 3-kinase; ROS, reactive oxygen species; SMAD, suppressor of mothers against decapentaplegic; Smurf2, SMAD ubiquitination regulatory factor 2; STAT, signal transducer and activator of transcription; TGF-β, transforming growth factor-β; TGFβR,TGFβ receptor.
2.1 Increased glycolysis in fibrosis
Microenvironmental factors, such as inflammatory factors, lactate and hypoxia, induce the glycolytic reprogramming of fibroblasts during fibrosis. Pro-inflammatory factors released by immune cells, such as PDGF-BB, IL-12, IL-1β and MIP-1β, promote the glycolytic process in fibroblasts. The metabolic switch from OXPHOS to glycolysis can offer more ATP and biosynthetic intermediates, which act as the building blocks for the excessive production of ECM (Kruse and Bornstein, 1975; Im et al., 1976; Xie et al., 2015).
The accumulating lactate in the microenvironment will also boost the glycolysis in stroma cells, promoting ECM deposition and fibrotic process. Activated immune cells with an increased glycolysis release large amounts of lactate, which induces the HIF-1-mediated expression of glycolytic genes, including GLUT1, LDH, and hexokinase (HK), in fibroblasts (Kozlov et al., 2020). Similarly, the elevated level of lactate in the lung tissue of IFP patients due to hypoxia increased the glycolysis inmesenchymal progenitor cells (MPCs) through the HIF-1α pathway. This metabolic reprogramming promotes the differentiation of MPCs into fibroblasts, which further contributes to the exacerbation of lung fibrosis (Yang et al., 2023). While pharmacological and siRNA-mediated suppression of LDH, an enzyme that catalyzes the conversion of pyruvate to lactate, in lung fibroblasts impeded the transition of fibroblast to myofibroblast (Kottmann et al., 2015). Lactate also promotes the migration and accumulation of T cells at the injury sites to create a chronic inflammation environment, which further activate fibroblast to synthesis ECM (Haas et al., 2015). Consistently, higher levels of lactate in the blister fluid of SSc patients was proven to increase collagen production (Taki et al., 2020).
Additionally, increased glycolysis may also fuel the TCA cycle to produce more mitochondrial metabolic intermediates that drive fibrotic progression. Rats with carbon tetrachloride (CCl4)-induced liver fibrosis had higher levels of the TCA cycle metabolites in their serum (Chang et al., 2017). Higher levels of succinate in TGF-β-stimulated lung fibroblasts stabilize HIF-1α to induce glycolysis and decrease FAO, thereby increasing the production of ECM (Xie et al., 2015; Wang Z. et al., 2021). The early intermediates in the TCA cycle, like α-ketoglutarate (α-KG), are crucial precursors for collagen synthesis. Increased glycolysis might promote fibrosis by increasing the α-KG production from the TCA cycle to support the collagen synthesis (Henderson and O'Reilly, 2021). Additionally, the NADH and FADH2 derived from the increased TCA cycle can then supply electrons to the electron transport chain (ETC) and drive OXPHOS, resulting in an elevated production of reactive oxygen species (ROS). ROS can induce TGF-β gene expression in various cell types, including macrophages and fibroblasts (Pociask et al., 2004; Jobling et al., 2006; Liu and Gaston Pravia, 2010). It has been shown that ROS convert inactive latent TGF-β to active formation by oxidizing latency association protein (LAP) (Pociask et al., 2004). Through activating TGF-β pathway, H2O2 drives epithelial mesenchymal transition (EMT) in lung epithelial cells to promote lung fibrosis (Kume et al., 2023).
2.2 Elevated glutaminolysis
In addition to glycolysis, glutaminolysis can also fuel the TCA cycle by glutamate dehydrogenase (GDH)-catalyzed conversion of glutamate into α-KG. Glutaminolysis plays a crucial role in amino acid synthesis, lipid metabolism, and energy production, providing energy and materials for ECM synthesis in fibroblasts. Glutaminolysis enhances glycolysis via the α-KG/mTOR/HIF-1α pathway, which plays an important role in fibrosis. Therefore, elevated glutaminolysis is also viewed as a crucial factor in the development of fibrosis (Gibb et al., 2022).
Inflammation at the site of injury and infection increases glutaminolysis in fibroblasts, providing energy and resources for ECM synthesis and secretion, which in turn promotes fibrosis (Henderson et al., 2020; Qiu et al., 2021). By activating SMAD3 and p38-MAPK signaling pathways, TGF-β1 increases the glutaminase 1 (GLS1) expression and glutaminolysis in myofibroblasts and cardiac fibroblasts (Bernard et al., 2018; Henderson et al., 2020; Chattopadhyaya et al., 2022). Inhibiting glutaminolysis results in a reduce in α-KG and less ECM production in fibroblasts (Henderson et al., 2020). Fibrosis development is slowed by inhibiting the enzymatic activity of GDH with epigallocatechin-3-gallate (EGCG) both in vitro and in vivo (Karsdal et al., 2020; Yin et al., 2022). Moreover, the transamination of glutamine to glutamate at the first stage of glutaminolysis provides nitrogens for amino acid synthesis and subsequent collagen production (Karsdal et al., 2020). α-KG also increases collagen translation and stability through activating mTOR signaling pathway and promoting collagen proline hydroxylation (Shi et al., 2022). As a result of mTORC1 activation, there is an increase in nutritional absorption, which in turn promotes glutaminolysis by increasing glutamine availability. Additionally, mTORC1 phosphorylates LARP6 to increase collagen expression in fibrosis (Zhang and Stefanovic, 2017).
Furthermore, the TCA cycle intermediates serve as cofactors for epigenetic regulatory enzymes in fibroblasts, such as histone demethylases or histone acetylases. α-KG is a cofactor for Jumonji C domain-containing demethylase 3 (JMJD3), which specifically regulates gene expression by demethylating the trimethylation on histone H3 lysine 27 (H3K27me3) (Tsukada et al., 2006). Increased α-KG derived from glutaminolysis enhances the JMJD3 activity, promoting fibrosis by inducing histone demethylation in particular regulatory regions (Bai et al., 2019; Jia et al., 2019). For example, JMJD3 decreases the accumulation of H3K27me3 at the FRA2 promoter, leading to fibroblast activation in SSc (Bergmann et al., 2018). In a mouse model of obstructive nephropathy, JMJD3 is markedly increased and activated in the kidney with the development of renal fibrosis. JMJD3 activates myeloid fibroblasts by increasing the H3K27 dimethylation in the obstructed nephropathy (An et al., 2023). However, some studies suggest a protective effect of JMJD3 in fibrosis. JMJD3 protects against renal fibrosis by inhibiting fibrotic signals, including TGF-β and Notch signaling (Yu et al., 2021). Consistently, inhibition of JMJD3 in rat hepatic stellate cells has been found to increase the levels of H3K27me3 and fibrosis markers (Jiang et al., 2021). More detailed studies are needed to elucidate whether α-KG regulate the role of JMJD3 in fibrosis in order to provide more strategies for the treatment of fibrosis. Besides, α-KG also modulate histone methylation dynamics of H3K9 and H3K4 mediated by the lysine specific demethylase 1 (LSD1). LSD1 epigenetically activates TGF-β1/Smad3 signaling, which contributes to pulmonary myofibroblast differentiation and fibrosis (Pan et al., 2020; Dong et al., 2022). Additionally, citrate derived from TCA cycle can be converted to acetyl-CoA, which is the substrate for histone acetylation and plays an important role in fibrosis (Wellen et al., 2009).
2.3 Reduced lipid metabolism
Fatty acid (FA) metabolism also provides energy to assist cells in adapting to environmental changes (Bartlett and Eaton, 2004; de Carvalho and Caramujo, 2018). In response to inflammation caused by tissue injury and infection, the aberrant FAO and consequent lipid synthesis impair ECM breakdown, contributing to the pathogenesis of fibrotic diseases (Chu et al., 2019; Zhao et al., 2019). It has been found that long-chain fatty acid transporter cluster of differentiation 36 (CD36) interacts with the elements of ECM, suggesting an interplay of fatty acid metabolism and ECM production (Febbraio et al., 2001; Susztak et al., 2005). In rheumatoid arthritis (RA), CD36 expression in fibroblasts is increased by macrophage-derived inflammatory mediators, including interleukin-1β (IL-1β), IL-6 and interferon gamma (IFN-γ) (Ahmed et al., 2008; Komatsu and Takayanagi, 2012). The binding of CD36 to thrombospondin 1 (TSP-1) inhibits the breakdown of ECM (Brown and Ahmed, 2019). Another crosstalk mechanism of lipid metabolism and ECM homeostasis is the TGF-β1/SMAD3-mediated repression of PGC1α or PPARs. The increased MiR-27a in diabetic nephropathy as a result of thylakoid cell damage activates TGF-β/SMAD3 signaling and inhibits PPARγ. The decreased levels of PPARγ result in FAO inhibition and lipid accumulation by reducing the expression of carnitine palmitoyl transferase 1 (CPT1) and acyl-CoA oxidase (ACOX) in fibroblasts, leading to renal tubular interstitial fibrosis (Wang Q. et al., 2018; Zhao et al., 2019). In patients with renal fibrosis, the absence of FAO raises CD36 expression in renal tubular epithelial cells, which in turn increases lipid uptake and intracellular lipid storage, promoting the synthesis and secretion of ECM. The lipotoxicity of excessive lipid accumulation also promotes the development of fibrosis (Herman-Edelstein et al., 2014; Yang et al., 2017). Additionally, fatty acid uptake is also regulated by caveolin-1, a principal component of caveolae, which modulate the surface availability of CD36. In response to bleomycin (BLM), CdCl2, TGF-β and irradiation, the caveolin-1 in fibroblasts is reduced, which concomitantly decreases the surface localization of CD36. Then, the CD36-mediated uptake of fatty acids is diminished, driving a metabolic switch from OXPHOS to aerobic glycolysis in fibroblasts (Ring et al., 2006; Wang et al., 2006; Del Galdo et al., 2008; Castello-Cros et al., 2011).
The disturbed FA metabolism also contributes to malfunction of epithelial cells, which is crucial for the development of pulmonary fibrosis. Alveolar type 2 cells have the most active lipid metabolism among lung epithelial cells, rendering them frequently prone to injury (Barkauskas et al., 2013). Disturbed FA biosynthesis and composition will lead to endoplasmic reticulum (ER) stress by regulating the transportation of folded proteins in lipid droplets. The persistent ER stress due to reduced lipid synthesis can cause damage to lung epithelial cells (Velázquez et al., 2016). These damaged lung epithelial cells create an inflammatory environment and the metabolic reprogramming of fibroblasts, leading to lung fibrosis (Katzen and Beers, 2020). According to the aforementioned evidence, a promising approach for treatment of fibrosis involves targeting lipid metabolic activities, such as FAO and lipid synthesis.
2.4 TGF-β mediates the crosstalk between inflammation and metabolic alteration
The profibrotic cytokine TGF-β is a significant part of complicated molecular mechanisms behind fibrosis (Hu et al., 2018). TGF-β mediates the crosstalk between inflammation and metabolic alteration in fibroblasts (Figure 2), which contributes to enhanced ECM synthesis and subsequent fibrosis (Yin et al., 2019; Henderson et al., 2020; Gao and Chen, 2022). During injury tissue repair and infections, activated macrophages and T cells release large amounts of TGF-β (Nakazaki et al., 2021; Zhao et al., 2023). Th17 cells increase the production of TGF-β in response to pro-inflammatory cytokine IL-17A (Lei et al., 2016). Moreover, TGF-β is a key component of senescence-associated secretory phenotype (SASP) factors derived from senescent cells. Cellular senescence is involved in lung fibrosis by releasing SASP factors and recruiting a large number of inflammatory cells into tissue and organs (Goliwas and Deshane, 2020). In addition, senescent cells secrete numerous pro-inflammatory cytokines which may drive the metabolic changes of fibroblasts and promote the synthesis of ECM proteins, contributing to the development of fibrosis (Jun and Lau, 2017; Wiley et al., 2019; Zhang et al., 2019; Blokland et al., 2020). Metabolic reprogramming like increased glycolytic pathway and mitochondrial dysfunction increase reactive oxygen species (ROS) production in senescent fibroblasts, which trigger the secretion of TGF-β (Hassona et al., 2013; Correia-Melo et al., 2016; Wiley et al., 2016). TGF-β/SMAD signaling pathway induces the senescence of adjacent cells in a paracrine manner by upregulating the cell cycle inhibitors p21, p27 and p15 (Muñoz-Espín and Serrano, 2014). In response to TGF-β, TGF-βRI induces the formation and nucleus translocation of SMAD 2/3/4 complex, which activates the transcription of profibrotic and metabolic genes while suppresses antifibrotic targets (Budi et al., 2017). Particularly, TGF-β signaling promotes cell proliferation, fibroblast-to-myofibroblast differentiation, and ECM synthesis, as well as reduces matrix protease secretion in the context of fibroblasts (Chen et al., 2018; Eser and Jänne, 2018). Interestingly, in mouse and human fibrotic lung tissue, cadherin-11 ensures continuous activation of myofibroblasts by linking TGF-β-producing macrophages and TGF-β-activated myofibroblasts (Lodyga et al., 2019).
Besides regulating inflammation, TGF-β induces the metabolic reprogramming of fibroblasts, resulting in an increased glycolysis and glutamine catabolism, as well as enhanced ECM synthesis (Yin et al., 2019; Henderson et al., 2020; Gao and Chen, 2022). TGF-β induces glycolysis and glutaminolysis to boost collagen production in fibroblasts. The expression of glycolytic transporter GLUT1 and metabolic enzyme HK1/2 is increased by TGF-β signaling to promote glycolysis in fibroblasts (Masumi et al., 1993; Yin et al., 2019; Chen et al., 2022). Fibroblasts in IPF have increased HK2, an enzyme that produces glucose 6-phosphate during the initial phase of glycolysis, which increased collagen expression through YAP (Yin et al., 2019). It has also been shown that GLUT1 and HK1/2 are upregulated in keloid fibroblasts, promoting the collagen synthesis and fibrosis development (Vinaik et al., 2020; Wang Q. et al., 2021). TGF-β also induces the glycolytic reprogramming by HIF-1 in hepatic stellate cells (HSCs), which plays an important role in increasing collagen production and finally hepatic fibrosis (Hanna et al., 2013; Mohammad Omar et al., 2022). Additionally, TGF-β1 upregulates glutaminase (GLS) expression in myofibroblasts, thereby increasing glutaminolysis to provide energy for the synthesis of ECM protein (Bernard et al., 2018).
3 Immunometabolism controls immune cells in fibrosis
Immunometabolism is essential in controlling the immune cell function and activity during fibrotic process (Figure 3). In this section, we review recent advances in the mechanisms underlying metabolic regulation of immune cells, including macrophages, T cells, B cells and neutrophils, as well their role in fibrotic diseases.
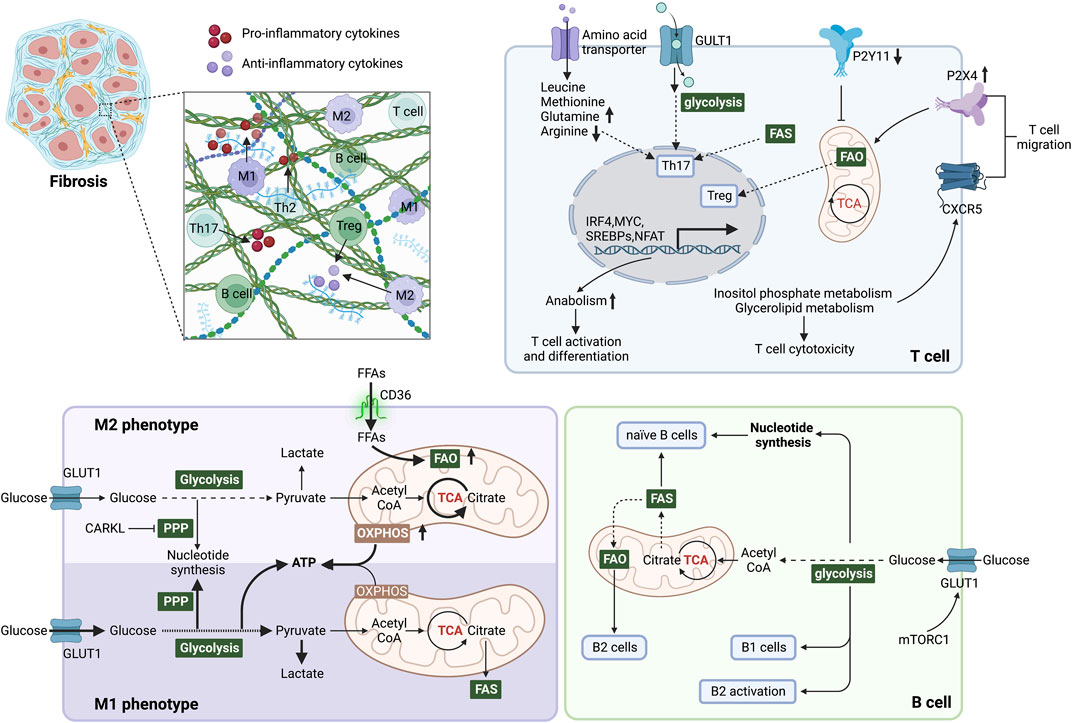
FIGURE 3. Immunometabolism determines immune cell function and inflammatory microenvironment during fibrosis. In M1 phenotype macrophages, glycolysis is the primary energy supply mode, elevating lactate production and inhibiting OXPHOS in mitochondria. In addition, the pentose phosphate pathway (PPP) is enhanced to generate NADPH, promoting the synthesis and release of inflammatory factors. Elevated FAO and OXPHOS promote macrophage polarization to the M2 phenotype. CARKL inhibits PPP and converts macrophages to the M2 phenotype. Glycolysis, glutaminolysis and lipid synthesis are required for Th17 cell differentiation and function, but in contrast, Treg cells perform catabolism and enhanced FAO to induce mitochondrial oxidative metabolism. Purinergic receptors influence mitochondrial metabolism by sensing local ATP content, which affects T cell migration. Mitochondrial metabolism is blocked by P2Y11 and activated by P2X4. Inositol phosphate metabolism and glycerolipid metabolism promote the migration of T cells by stabilizing the follicle homing receptor CXCR5 on their surface. Naive B cells mainly obtain energy and nucleotide biosynthesis through the TCA cycle and OXPHOS. The survival and function of B1 cells are dependent on glycolysis, whereas splenic B2 cells rely on FAO. During B2 cell activation, elevated GLUT1 expression induced by c-Myc and mTORC1 increases glucose uptake and shifts FAO to glycolysis. After metabolic reprogramming, immune cells regulate the microenvironment by secreting pro/anti-inflammatory factors, thereby affecting fibrosis. Abbreviations: ATP, adenosine triphosphate; CARKL, carbohydrate kinase-like protein; c-Myc, proto-oncogene Myc; FAS, fatty acid synthesis; FFAs, free fatty acids; FAO, fatty acid oxidation; GLUT1, glucose transporter 1; mTORC1, mTOR complex 1; mTOR: mammalian target of rapamycin; PPP, pentose phosphate pathway; TCA, tricarboxylic acid.
3.1 Macrophages
Under normal conditions, the inactive macrophages in a quiescent state, called naive macrophages or M0 macrophages, are supplied with ATP mainly by aerobic oxidation of glucose and β-oxidation of fatty acids (Rőszer, 2015; Ramond et al., 2019). In respond to stimulation, metabolic reprogramming drives polarization of macrophages into either pro-inflammatory M1 or anti-inflammatory M2 phenotype, thereby performing a distinct role in fibrosis (Wynn and Barron, 2010; Malyshev and Malyshev, 2015).
M1 macrophages undergo a shift from OXPHOS to glycolysis in response to stimulus (Figure 4), like interferon-γ (IFN-γ) secreted by Th1 lymphocytes, exogenous lipopolysaccharide (LPS), exogenous DNA and viral infection. This metabolic reprogramming induces secretion of various cytokines to promote inflammation (Orecchioni et al., 2019; Kang and Kumanogoh, 2020; Lin et al., 2021). M1 macrophages increase the GLUT1 expression for glucose uptake, which supports the enhanced aerobic glycolysis for rapid ATP production (Freemerman et al., 2014). However, the TCA cycle is blocked at the citrate and succinate production steps due to the reduced levels of isocitrate dehydrogenase (IDH) and succinate dehydrogenase (SDH) during M1 polarization (Jha et al., 2015; Corcoran and O'Neill, 2016). The accumulation of cellular lactate, citrate and succinate then regulate their energy metabolism by increasing the levels of intracellular PGE2, ROS, and NO production (El Kasmi and Stenmark, 2015; Corcoran and O'Neill, 2016). Succinate supports the, ETC and ROS generation (El Kasmi and Stenmark, 2015). Succinate or ROS can maintain the stability of HIF-1α, which enhances glycolysis by increasing GLUT1-mediated glucose uptake and LDH-dependent lactate production, driving M1 macrophage polarization (Wang T. et al., 2017; Liu et al., 2017). Iron metabolism also regulates the polarization of M1 macrophages. Excess iron frequently activates M1 polarization and induces the expression of inflammatory factors, such as TNF-α and IL-12 (Sindrilaru et al., 2011). Notably, M1 instead of M2 polarization occurs almost immediately after upregulation of transferrin receptor 1 (TfR1), a membrane receptor that uptake of circulating transferrin-bound iron, and downregulation of ferroportin, the only known iron exporter (DeRosa and Leftin, 2021; Ho et al., 2022).
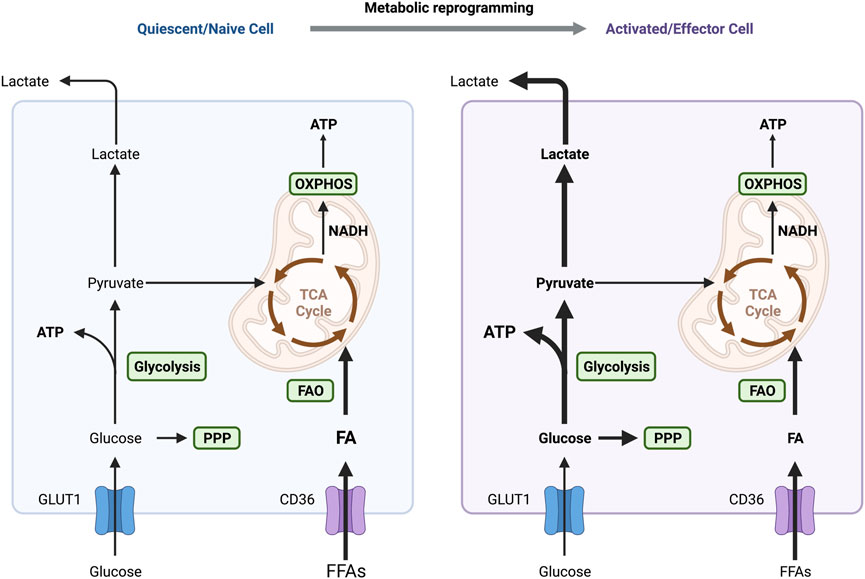
FIGURE 4. Different metabolic patterns in quiescent and activated immune cells. OXPHOS and FAO are the primary sources of energy in naive and quiescent macrophages and T cells to maintain regular cellular life activities. One of the primary variables in the activation of these immune cells is the metabolic switch between glycolysis and OXPHOS. IFN-γ and LPS-activated macrophages exhibit an increase in aerobic glycolysis, which supplies the energy needed for M1 macrophages to polarize and exhibit pro-inflammatory actions quickly. Similarly, energy supply mode in T cells switches to aerobic glycolysis in response to antigenic stimulation, activating and transforming them into effector cells. Abbreviations: ATP, adenosine triphosphate; FAO, fatty acid oxidation; OXPHOS, oxidative phosphorylation; PPP, pentose phosphate pathway; TCA, tricarboxylic acid.
The metabolic reprogramming is crucial for the production of inflammatory factors. ROS can enhance the DNA binding ability of p65 and increase the NF-κB-mediated expression of inflammatory factors, such as IL-1β and IL-6, which contribute to the polarization of macrophages toward the M1 phenotype (Genard et al., 2017). HIF-1α also promotes the metabolic reprogramming of M1 macrophage polarization and subsequent inflammatory reactions (Corcoran and O'Neill, 2016). In the process of M1 macrophage polarization, HIF-1α upregulate the expression of IL-1β (Tannahill et al., 2013). Additionally, pentose phosphate pathway (PPP) is a critical pathway of high flux glycolysis supporting inflammatory macrophage biosynthesis. The elevated NADPH generation from PPP supports fatty acid synthesis for the production of inflammatory mediators, such as leukotrienes and prostaglandins (Forrester et al., 2020). The main function of classically activated M1 macrophages is to combat acutely harmful infections by producing pro-inflammatory cytokines, including IL-1β, IL-2, IL-6, IL-12, IL-23, TNF-α, and chemokines like nitric oxide (NO) and ROS. However, the excessive amount of pro-inflammatory cytokines secreted by M1 macrophage promotes fibrotic disease during chronical tissue repair (Wick et al., 2010; Xu et al., 2012). For example, TGF-β and pro-inflammatory cytokines like IL-1β can induce fibroblast activation and differentiation into myofibroblasts, leading to overproduction of fibrogenic genes and ECM (Gieling et al., 2009; Gasse et al., 2011; Upagupta et al., 2018; Lv S. L. et al., 2021).
M0 macrophages polarize into the M2 phenotype, which facilitate the healing process after tissue damage in exposure to IL-4, IL-10, IL-13, TGF-β, and glucocorticoids (Stein et al., 1992; Rőszer, 2015; Ishida et al., 2023). The energy of anti-inflammatory M2 macrophages is mainly provided by OXPHOS and FAO (Liu et al., 2017; Van den Bossche et al., 2017). In response to IL-4 and IL-13, STAT6 together with peroxisome proliferator activated receptor-γ coactivator-1β (PGC-1β) stimulates PPARγ signaling pathway and FAO, which polarizes the macrophages toward the M2 phenotype (Olefsky and Glass, 2010). Increased CD36 expression and enhanced FAO in M2 macrophages produce more acetyl CoA, which can enter the TCA cycle to ensure the production of ATP from OXPHOS (Chowdhury et al., 2019; Wilson et al., 2019). In addition, IL-6, IL-13 and IL-14 stimulate HIF-2α to drive metabolic reprogramming and polarization of macrophages to M2 phenotype (Schultze et al., 2015). HIF-2α can subsequently activate PPAR-γ to induce the expression of arginase-1 (Arg-1), which catalyzes the urea cycle and increases NO production (Wang Q. et al., 2018). In terms of iron metabolism, in contrast to M1 macrophages, the expression of iron metabolism-related proteins, such as ferritin and TfR1, is decreased during the polarization of M2 macrophages (DeRosa and Leftin, 2021). The uptake of ω-6 and ω-3 unsaturated fatty acids facilities the synthesis of pro-resolving mediators, such as maresins, protectins, lipoxins, and resolvins (Serhan and Levy, 2018). They bind to ALX/FPR2, GPR18, and chemerine of macrophages, promoting the transition of macrophages into M2 phenotypes while inhibiting the production of pro-inflammatory molecules (Serhan, 2014; Chiang and Serhan, 2017). Pro-resolving chemicals produced by M2 macrophages, including TGF-β and IL-10, inhibit inflammation by enhancing tissue repair and remodeling (Cui et al., 2023).
Metabolic reprogramming-mediated macrophage polarization promotes fibrotic diseases. It has been shown that M2 macrophages ensure profibrotic function in multiple ways. Anti-inflammatory M2 macrophages promote the progression of pulmonary fibrosis by secreting platelet-derived growth factor (PDGF), IL-1β, and TGF-β (Goda et al., 2020). M2 macrophages promote ECM production by recruiting fibroblasts, inducing EMT and increasing L-arginine metabolism for collagen synthesis (He et al., 2019; Wang et al., 2020). Notably, macrophages play a dual role in liver fibrosis, i.e., M1 macrophages promote liver fibrosis while M2 macrophages attenuate liver fibrosis (Kisseleva and Brenner, 2021; Wang Z. et al., 2023). The metabolic reprogramming is required for M1 macrophages to produce inflammatory mediators, which induce the differentiation of HSCs into myofibroblasts for excessive ECM generation (Bility et al., 2014; Xie et al., 2021). In addition, during the inflammatory phase, M1 microphages increase the production of tissue inhibitor of matrix metalloproteinase (TIMP) to inhibit ECM degradation (Mahalanobish et al., 2020). However, M2 macrophages degrade ECM during the anti-inflammatory phase by secreting MMPs (i.e., MMP1, MMP12, MMP9), thus promoting fibrosis resolution (Ramachandran et al., 2012; Zhao et al., 2022). Oxidative stress is a key event in pulmonary fibrosis due to inflammation. ROS promote the conversion of macrophages from the M1 phenotype to the M2 phenotype, thereby contributing to the development of pulmonary fibrosis (Kurundkar and Thannickal, 2016). NADPH oxidases (NOXs)-derived ROS are responsible for monocyte-to-macrophage differentiation by activating MAPK/JNK and ERK signaling, which affecting M2 macrophage polarization (Xu et al., 2016). Mitochondrial ROS in M1 macrophages induce STAT6-dependent JMJD3 expression, which increases the transcriptional activation of M2 genes (He et al., 2016).
3.2 T cell
Infiltrated T cells participate in wound healing and tissue repair by promoting the activation of HCSs, formation of myofibroblasts and inflammation (Pellicoro et al., 2014; Koyama and Brenner, 2017; Rao et al., 2021). Metabolic reprogramming is required to meet the growth, proliferation and effector functions of T cells during fibrotic responses (Figure 4).
To maintain aerobic glycolysis, T cells increase GLUT1 expression and the glucose uptake through the CD28-dependent PI3K/Akt signaling pathway (Frauwirth et al., 2002). It has been found that mTOR-activated HIF-1α increased the expression of glycolytic enzymes, including HK and phosphofructokinase and pyruvate dehydrogenase kinase (PDK) in T cells (Düvel et al., 2010; Waickman and Powell, 2012). The mTORC1 signaling pathway in T cells also promotes fibroblast activation, contributing to the development of renal interstitial fibrosis (Chen et al., 2016). Naive T cells from RA patients have impaired glycolytic flux as a result of elevated glucose-6-phosphate dehydrogenase (G6PD) expression. Overexpression of G6PD shunts glucose into PPP for NADPH production, which eliminates the ROS and promotes the differentiation of T cells to pro-inflammatory Th1 and Th17 cells in RA (Yang et al., 2016). Alterations in the PPP pathway also render T cells sensitive to necrosis and significantly promote inflammatory responses in patients with systemic lupus erythematosus (Gergely et al., 2002). Upon T cell activation, enhanced aerobic glycolysis leads to activation of JAK-STAT1 pathway and secretion of IFN-γ, which further increases the classical IFN-stimulated gene expression and functions as a crude fibrogenic agent in fibrosis development (Villarino et al., 2017).
Amino acid metabolism, including glutamine, arginine, serine and glycine, is critical in T cell proliferation and differentiation (Wang et al., 2011; Kishton et al., 2016; Ma et al., 2017). Amino acids serve as the basic structure for protein synthesis, one source for providing energy and upstream regulators of mTORC1 (Rebsamen et al., 2015; Huang et al., 2021). It has recently been found that amino acids are converted into α-ketoglutarate, which can fuel the TCA cycle to produce energy and metabolites required for T cell activation (Newsholme et al., 1985; Wang and Green, 2012). Amino acids serve as upstream regulators of mTORC1 to modify intracellular ATP levels and energy metabolism (Shi et al., 2019; Huang et al., 2021). The disrupted amino acid metabolism exacerbates CCl4-induced liver fibrosis by promoting the release of fibrotic mediators from T cells, including TGF-β, IL-4, IL-6 and IL-17 (Yoshizaki et al., 2010; Shan et al., 2016). T cell receptor signaling can increase the expression and cell membrane localization of amino acid transporter (Nakaya et al., 2014). SLC7A5, a transporter for branched neutral amino acids such as leucine and phenylalanine, is highly increased in activated CD8+ T cells in response to antigen stimulus (Sinclair et al., 2013). CD4+ T cells are unable to proliferate and differentiate into Th1 and Th 17 cells in SLC7A5-deficient mice (Nakaya et al., 2014).
Iron homeostasis is also critical in the activation of T cells and in the regulation of their functions (Bonaccorsi-Riani et al., 2015). Reduced numbers of mature T cells were found in H-ferritin knockout mice, indicating that ferritin-stored iron is important for the survival of T lymphocytes (Vanoaica et al., 2014). During early T cell activation, mediated by IL-2, T cells take up iron via TfR1 to assist T cell activation (Macedo et al., 2004). Notably, iron inhibits Th1 cell differentiation, IFN-γ expression via upregulating of T cell immunoglobulin and mucin containing protein-3 (TIM-3) (Pfeifhofer-Obermair et al., 2021). Interestingly, some iron oxide nanoparticle-based adjuvants can instead promote the induction of Th1 and Th17 activation and immune responses (Neto et al., 2018)]. Similarly, iron has been shown to inhibit Th2 cell differentiation and immune function (Wang S. et al., 2021). TfR1 expression is higher in Treg than in CD4+ T cells, which leads to more iron translocation into Treg cells, and this in turn mediates Treg death (Feng et al., 2021).
Compared to Th1 and Th17 cells with high levels of glycolysis and low levels of FAO, Tregs with a low level of GLUT1 have a higher rate of lipid oxidation to fuel the mitochondrial, ETC for proliferation and differentiation (Michalek et al., 2011). Of note, FAO-driven Tregs are essential in regulating the development of fibrosis. In a diabetic mouse model, the ketogenic diet (KD), a very low-carbohydrate, high-fat diet, inhibits the mitochondrial respiration, FA synthesis and FAO in Tregs (Tao et al., 2021). This metabolic alteration impairs the Treg expansion and function, promoting cardiac fibroblast activation and interstitial fibrosis (Tao et al., 2021). Similarly, depletion of Tregs cells increases Th1/Th2 cytokine and collagen deposition, contributing to the diphtheria toxin-induced lung fibrosis (Moyé et al., 2020).
3.3 B cell
The altered immunometabolism in B-cell is essential in fibrotic diseases such as the autoimmune disease systemic lupus erythematosus (SLE) (Oleinika et al., 2019; Lee et al., 2021). FAO is the primary energy supply mode in inactivated germinal center B cells (Weisel et al., 2020). Multiple metabolic pathways are activated to provide materials and energy for the proliferation and differentiation of B cells following antigen recognition (Woodland et al., 2008; Caro-Maldonado et al., 2014; Cyster and Allen, 2019). In T cell-dependent (TD) B cell activation, IL-4/STAT6 axis induces the expression of GLUT1 to increase glycolysis (Dufort et al., 2007; Cyster and Allen, 2019). T cell-independent (TI) antigen stimulates TLR4-PI3K-AKT signaling pathway, which also promotes the GLUT1-mediating glucose uptake and subsequent glycolysis (Caro-Maldonado et al., 2014; Jayachandran et al., 2018). Moreover, HIF-1α is activated by ERK-STAT3 and NF-kB pathways following BCR and TLR stimulation (Meng et al., 2018). HIF-1α together with c-Myc induces the expression of GLUT1 and glutamine transporter, increasing glycolysis and glutaminolysis (Caro-Maldonado et al., 2014). Additionally, ATP-citrate lyase (ACLY) is increased by LPS to promote fatty acid synthesis, including FFAs, neutral and acidic phospholipids and cholesterol, supporting the proliferation and differentiation of B cells (Dufort et al., 2014). Furthermore, it has been recently found that iron is a key element in promoting B cell proliferation and antibody production after TD or TI stimulation (Jiang et al., 2019). Iron promotes B-cell proliferation by inducing histone 3 lysine 9 demethylation at the cyclin E1 promoter, thereby increase cyclin E1 expression (Jiang et al., 2019). Similarly, lactoferrin (LF), a pleiotropic iron-binding glycoprotein, stimulates TGF-β signaling to produce IgA and IgG2b in B cell (Jang Y. S. et al., 2015). B cells quickly die following a mitochondrial reprogramming initiated by BCR stimulation alone. However, mitochondrial dysfunction is prevented by TLRs signaling or CD40 co-stimulation (Akkaya et al., 2018). Upon co-stimulation by IL-4 and CD40L, the metabolic stress due to elevated glycolysis and mitochondrial biosynthesis leads to ROS accumulation, which could be counteracted by GSK3 (Jellusova et al., 2017) (Figure 5). The production of ROS by the mitochondria following metabolic reprogramming is related to mitochondrial mass and membrane potential (Echtay et al., 2002; Jang K. J. et al., 2015). Low mitochondrial mass and membrane potential carry out plasma cell differentiation (PCD) in B cells, and high mitochondrial mass and membrane potential drive class switching recombination (CSR) (Jang K. J. et al., 2015) (Figure 5). CSR is a process of molecular rearrangement in which a group of downstream CH genes exchange genes encoding the heavy chain constant region CH in B cells (Chi et al., 2020). Therefore, alterations of CH exon clusters result in production of IgG, IgA, or IgE, in which the antigen-binding variable region is unaltered (He et al., 2016).
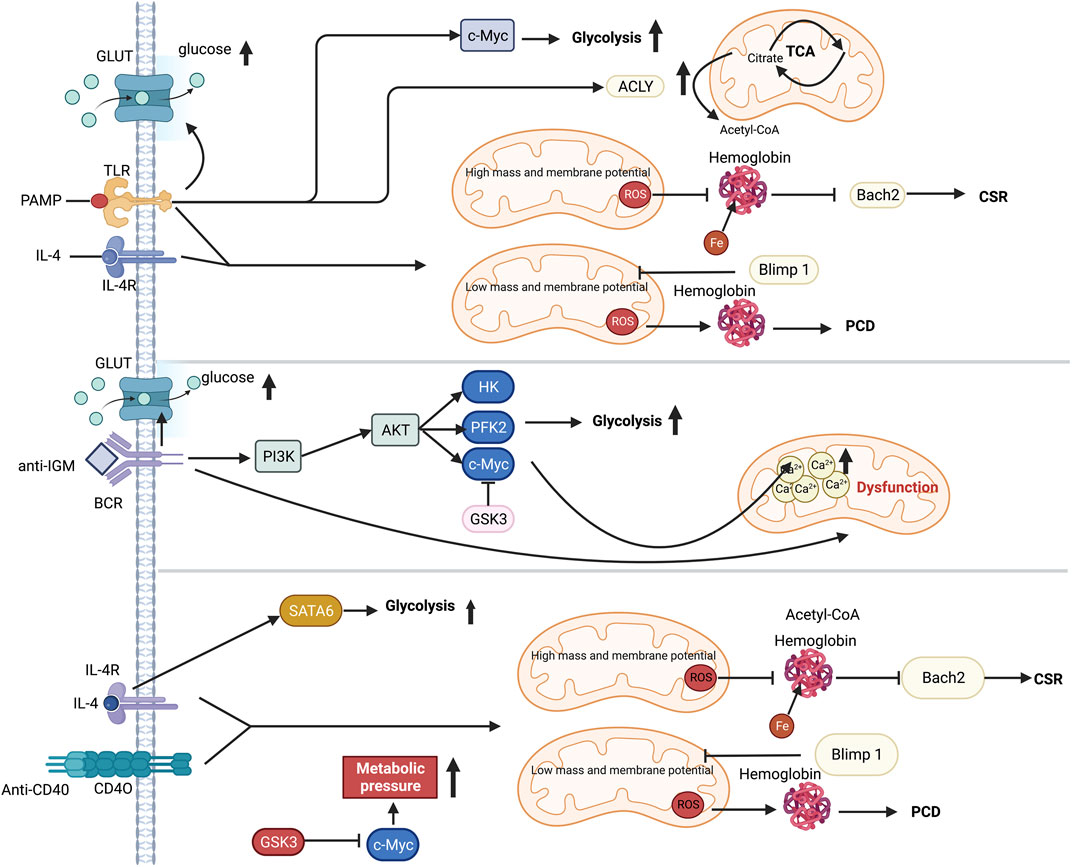
FIGURE 5. Metabolic reprogramming in B cell following TI or TD stimulation. Selected metabolic process is induced by TI or TD stimulation. While PI3K and GSK3 coordination is necessary for anti-IgM-induced metabolic change, T cell dependent antigen (TD-Ag) results in an increase in c-Myc-dependent glycolysis. The cytokine IL-4 can increase glycolysis by activating STAT6. Mitochondria perform a significant role in distorting the fates of B cells after antigen stimulation. Co-stimulation signal is essential for preserving B cells since excessive mitochondrial respiration during single-BCR stimulation results in Ca2+ buildup and ultimately leads to death. The production of mitochondrial ROS, which in turn regulates either Blimp1 or Barch2, two crucial transcriptional regulators of plasma development and differentiation, has an impact on hemoglobin stability. Abbreviations: ACLY, ATP-citrate lyase; ATP, adenosine triphosphate; AKT, protein kinase b; BCR, B cell receptor; CSR, class-switch recombination; c-Myc, proto-oncogene Myc; GLUT, glucose transporter; GSK3, Glycogen synthase kinase-3; HK, hexokinase; IL-4, interleukin-4; IL-4R, interleukin-4 receptor; IGM, immunoglobulin M; PCD, plasma cell differentiation; PAMP, pathogen-associated molecular patterns; PFK2, phosphofructokinase 2; PI3K, phosphatidylinositol 3-kinase; STAT6, signal transducer and activator of transcription 6; TLR, toll like receptor.
Metabolic switch is essential for plasma cell differentiation and antibody production. Plasmablasts consistently express Blimp1, XBP-1 and IRF, which promote the differentiation of mature B cells into plasma cells and B memory cells, as well as induce antibody production (Aronov and Tirosh, 2016; Tellier et al., 2016; Cyster and Allen, 2019). It has been found that plasma cells in the intestine had increased glycolysis and OXPHOS compared to neoplastic B cells (Kunisawa, 2017). The antibody production in plasma cells requires increased protein synthesis, elevated number of ER and related organelles to provide more anabolic raw materials (Manz and Radbruch, 2002). ACLY serves as a link between glucose metabolism and lipid synthesis, which enhances ER activity depending on lipid synthesis (van Anken et al., 2003). Activated ACLY induces the glucose incorporation into biosynthesis of lipids, such as phosphatidylcholine (PC) and ceramide (CM), which are required for ER expansion in B lymphocytes. Inhibiting ACLY-mediated glucose-dependent de novo lipogenesis will impair the plasma cell proliferation and differentiation, as indicated by reduced differentiation markers, CD138 and Blimp-1 in response to LPS stimulation (Dufort et al., 2014). Vitamin B1 is an essential coenzyme for the maintenance of pyruvate dehydrogenase (PDH) activity in the TCA cycle and is crucial in the synthesis and secretion of antibodies by plasma cells (Purroy et al., 2020). Long-lived plasma cells and short-lived plasma cells have different metabolic patterns (Tarlinton et al., 2008). Long-lived plasma cells exhibit higher levels of autophagosomes and surface amino acid transport protein CD98 (Lam and Bhattacharya, 2018). Long-lived plasma cells use glucose for protein glycosylation modification in the absence of long-term antigenic stimulation and metabolic stress. After being re-stimulated with pathogen antigens and autoantigen, long-lived plasma cells take up large amounts of glucose for pyruvate-dependent respiration (McHeyzer-Williams and McHeyzer-Williams, 2005; Jang et al., 2016; Zeng et al., 2020). Mice lacking mitochondrial pyruvate carrier 2 (Mpc2), a protein that transports pyruvate from matrix to mitochondria, have a shorter plasma cell lifetime and release less antigen-specific antibodies (Bertolotti et al., 2010; Lightman et al., 2019).
Metabolic reprogramming of B cells is associated with the development of fibrotic diseases. Disrupted formation of autoantibodies serves as a primary pathogenic factor for SLE, a systemic autoimmune disease (Spada et al., 2015). It has been observed that overactivation of PI3K/AKT/mTOR signaling pathway and metabolic reprogramming occur in B cells of SLE patients (Wu et al., 2007; Jones et al., 2016). Inhibition of mTORC1 dramatically reduces autoantibody production, which improves the SLE in mice (Jones et al., 2016; Geier and Perl, 2021). When mTORC1 is suppressed, the expression of immunoglobulin-binding protein (BiP) and other proteins necessary for protein synthesis is decreased, leading to reduced antibody production (Jones et al., 2016). In addition to harming joints, numerous other organs and tissues are also affected by the chronic inflammatory autoimmune illness known as RA (Yang et al., 2019). Similarly, EZH2-driven plasmablast differentiation and generation of autoantibody in RA patients are caused by overactivation of mTORC1 and methionine-dependent spleen tyrosine kinase (Syk) pathways (Zhang et al., 2020). In addition, disrupted lipid or/and glucose metabolism in B cells is frequently observed in metabolic illnesses, eventually leading to fibrosis (Brittain et al., 2016). For example, B cells in obese individuals produce pro-inflammatory factors, like IL-2, IFN-γ, TNF-α and IL-10, compared to wasting individuals. These cytokines make adipose and other tissues susceptible to chronic tissue inflammation in obesity, which can lead to systemic metabolic disorders linked to fibrosis (DeFuria et al., 2013; McLaughlin et al., 2017; Wijngaarden et al., 2022). On the other hand, B cells polarize into regulatory B cells that constitutively secrete IL-10 to control ongoing chronic inflammation in the adipose tissue milieu, which is mediated by saturated FFAs derived from adipocytes (Eguchi et al., 2012).
4 Targeting metabolic dysregulation for fibrosis therapy
The metabolic pathways, including glycolysis, FAS, FAO, are the main targets for fibrosis prevention and treatment. Inhibition of key enzymes and regulatory signaling pathways in glycolytic process has been shown to effectively prevent fibrosis by reducing pro-inflammatory immune cell activation and ECM production from fibroblasts. In addition, numerous clinical or preclinical studies have shown that enhancing FAS and FAO by inducing PPAR activity promotes the anti-inflammatory and anti-fibrotic cell phenotype, providing a viable strategy for antifibrotic therapy. On this occasion, we summarize the strategies for targeting metabolic changes in immune and stroma cells for antifibrotic therapy (Table 1).

TABLE 1. Targeting metabolic dysfunction for antifibrosis.
Given the important role of glucose metabolism in producing ECM, targeting glycolysis for antifibrotic therapy has yielded some positive results. For example, inhibiting glycolysis by 2-DG, an inhibitor of hexokinase2 (HK2), or 3PO, a small-molecule inhibitor of PFKFB3, prevents the conversion of lung fibroblasts to myofibroblasts and reduces collagen synthesis (Xie et al., 2015; Hu et al., 2020). Inhibition of glycolysis by 2-DG and shikonin attenuates the extent of renal fibrosis in mice. Impaired glycolysis in renal fibroblasts results in reduced expression of fibrosis markers, including α-SMA and fibronectin, elevated intracellular pH and decreased lactate accumulation (Ding et al., 2017). In addition, plant-derived triterpene celastrol inhibits glycolysis in macrophages by changing the conformation of pyruvate kinase isozymes M2 (PKM2) and inhibiting PKM1. This metabolic alteration polarizes pro-inflammatory M1 macrophages to anti-inflammatory M2 phenotypes, reducing inflammation and fibrosis in the liver (Fan et al., 2022). Repressing glycolysis and ROS production in CD4+ T cells by PGK1 inhibitor NG52 impairs the differentiation of Th17, Th1 and Treg cells, reducing inflammatory injury and subsequent myocardial fibrosis in mice and patients with myocarditis (Lu et al., 2023). D-mannose, a monosaccharide, retards the progression of ulcerative colitis in mice by blocking gluconeogenesis in macrophages and thereby inhibiting succinate-mediated activation of HIF-1α (Torretta et al., 2020).
Antifibrosis research has made potential strides by modifying fatty acid metabolism. Increasing the expression of CPT1 and peroxisomal ACOX1 with C75, a synthetic CPT1 activator, maintains the renal cell viability and inhibits renal fibrosis (Kang et al., 2015). Similarly, C75 reduces the production of collagen, fibronectin and α-SMA by inhibiting FAS, which attenuates the bleomycin-induced pulmonary fibrosis in mice (Jung et al., 2018). Astragaloside IV alleviates palmitate-mediated fibrosis in human glomerular mesangial cells by suppressing CD36 expression (Su et al., 2019). The receptors for FFAs, including GPR40 and GPR84, can also be promising targets for alleviating fibrosis (Gagnon et al., 2018). It has been found that GPR40 inhibits fibrosis, while GPR84 promotes it in a mouse model of renal fibrosis with unilateral ureteral obstruction. TGF-β promotes fibrosis by decreasing GPR40 expression while increasing GPR84 expression (Gagnon et al., 2018). Activating GPR40 with PBI-4050 also inhibits adenine-induced kidney fibrosis in mice (Thibodeau et al., 2019).
The PPAR signaling pathway is crucial for lipid metabolism, including fatty acid uptake and oxidation (Wei et al., 2010; Lakshmi et al., 2017; Cui et al., 2023). PPAR signaling-regulated FAO can promote cell-mediated ECM degradation, which is countered by profibrotic TGF-β (Zhao et al., 2019). As expected, a number of drugs that activate the PPAR signaling pathway exhibit beneficial effect on fibrosis. In a phase 2 clinical trial, the PPARα/δ dual agonist elafibranor improves lipid metabolism and reduces inflammation, which delays liver fibrosis in non-alcoholic steatohepatitis (NASH) patients (Ratziu et al., 2016). Dioscin, a steroidal saponin obtained from the roots of Dioscorea nipponica Makino, can reduce the severity of dextran sulfate sodium (DSS)-induced colitis in mice. This is achieved by increasing the mTORC2/PPARγ-mediated FAO for M2 polarization and decreasing the mTORC1/HIF-1-regulated glycolysis for M1 polarization (Wu et al., 2021). In NASH model mice, the PPARα/γ dual agonist saroglitazar and the PPARα/γ/δ triple agonist lanifibranor are effective in protecting the liver from metabolic disorders and fibrosis (Wettstein et al., 2017; Jain et al., 2018). Apigenin, a flavonoid derived from fruits and vegetables, alleviates inflammation by polarizing M1 macrophages to M2 macrophages (Feng et al., 2016). Apigenin inhibits the NF-κB activation by blocking PPARγ-mediated nuclear translocation of p65, promoting macrophage M2 polarization (Feng et al., 2016). Moreover, apigenin has also been shown to inhibit the expression of SREBP-1c and Fas proteins in alcohol-induced liver injury (Feng et al., 2016; Wang F. et al., 2017). However, one of the major issues with the current application of PPAR agonists for treating fibrosis is the incomplete comprehension of the interactions between PPAR isomers. For example, PPARα agonists can increase serum FFA levels, while the PPARγ agonists can decrease serum FFA levels in a rat model of NASH. Therefore, a better understanding of the interactions between these isoforms is needed to optimize the therapeutic efficacy of targeting PPAR pathway in fibrosis.
Boosting FAO is a strategy for polarizing macrophages toward anti-inflammatory M2 phenotype and preventing fibrosis. Spermidine, a natural polyamine, enhances FAO and OXPHOS fluxes by activating AMPK and HIF-1α during M2 macrophage polarization, which could effectively prevent the progression of colitis to fibrosis (Liu et al., 2020). Didymin, a dietary citrus flavonoid, prevents the progression of ulcerative colitis to fibrosis by enhancing 4 hydroxypropyl-CoA dehydrogenase beta-mediated FAO in macrophages (Lv Q. et al., 2021). Pirfenidone-increased FAO polarizes macrophages to the M2 phenotype and attenuates steatohepatitis in a mouse model (Chen et al., 2019). Fenofibrate attenuates liver fibrosis in mice by activating PPARα and enhancing FAO in macrophages, which drives their polarization to M2 macrophages (Lakhia et al., 2018). MCC950 enhances FAO by inducing CPT1β and CD36 expression in macrophages, and inhibiting GLUT4 to reduce glucose utilization. These metabolic alterations induce the infiltration of M2 macrophages into the sites of cardiac inflammation, reducing myocardial inflammatory injury and preventing myocardial fibrosis (Wang et al., 2022).
Hormone therapy, one of the significant treatments for fibrosis, also plays an important role in regulating cellular respiration and oxidative catabolism. Thyroid hormones are crosstalked with multiple metabolic pathways by regulating PPARs, mTOR and AMPK signaling, which regulate cellular energy metabolism. It has been observed that using thyroid hormones can improve mitochondrial function by increasing PGC1α and PINK1 activity in a mouse bleomycin-induced pulmonary fibrosis model (Wang K. et al., 2018; Yu et al., 2018). The enhanced mitochondrial biosynthetic function and cellular respiration in response to thyroid hormones ultimately alleviate pulmonary fibrosis. Additionally, thyroid hormone can increase miR34a expression to inhibit TGF-β-induced EMT in renal fibrosis (Lu et al., 2013). Thyroid hormone has also been found to bind to thyroid nuclear receptors, which inhibits TGF-β-SMAD signaling and reduces skin fibrosis (Alonso-Merino et al., 2016).
5 Conclusion and prospects
Fibrosis is a sequelae of tissue damage which is a potential risk factor for organ dysfunction and even individual death. Although fibrosis poses a great risk to human health, there is still no effective means to prevent and treat fibrosis. The key process leading to tissue fibrosis is the infiltration of immune cells, which create a chronic inflammatory environment at the injury site. Immune cells must undergo metabolic reprogramming to provide sufficient energy and metabolites for their activation and cytokine production, which is well known as immunometabolism. In recent years, an increasing number of studies have shown that immunometabolism contributes to the development and progression of fibrosis. Altered glycolysis, amino acid metabolism, and lipid metabolism induce the formation of inflammatory environment and production of ECM during fibrosis and its related diseases. Notably, these metabolic alterations are also involved in the profibrotic role of TGF-β1 signaling pathway. Insights into the roles of immunometabolism in fibrosis reveal opportunities for potential therapeutic treatment. In particular, the metabolic changes that precede phenotypic alterations in immune cells can be targeted to improve immune cell function, alleviating the progression of inflammation and fibrosis. While many antifibrotic drugs targeting metabolic dysregulation have shown powerful effects in experimental animal models, more clinical studies are needed to test the therapeutic efficacy and adverse effects of these drugs on fibrotic diseases. Moreover, further research is still required to elucidate how the interplay of inflammation and metabolic changes promotes fibrosis, what is the difference in the immunometabolism during physiological tissue repair and pathophysiological fibrotic process, and what is the contributing metabolic pattern in specific tissue, like liver and lung, for fibrotic process, hoping to develop more effective and safer antifibrotic drugs.
Author contributions
LF, XC, YH, and XZ wrote the manuscript and made the figures, LF organized the tables, NX and SZ conceived, designed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from National Key R&D Program of China (2020YFA0509400), Guangdong Basic and Applied Basic Research Foundation (2019B030302012), the National Natural Science Foundation of China (81821002, 82130082, 82341004, 81972665), 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD22007 and ZYJC21004), the specific research fund of The Innovation Platform for Academicians of Hainan Province (YSPTZX202208), and Hainan Province Clinical Medical Center (QWYH202175).
Acknowledgments
BioRender was used to create the figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, S., Marotte, H., Kwan, K., Ruth, J. H., Campbell, P. L., Rabquer, B. J., et al. (2008). Epigallocatechin-3-gallate inhibits IL-6 synthesis and suppresses transsignaling by enhancing soluble gp130 production. Proc. Natl. Acad. Sci. U. S. A. 105 (38), 14692–14697. doi:10.1073/pnas.0802675105
Akkaya, M., Traba, J., Roesler, A. S., Miozzo, P., Akkaya, B., Theall, B. P., et al. (2018). Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat. Immunol. 19 (8), 871–884. doi:10.1038/s41590-018-0156-5
Alonso-Merino, E., Martín Orozco, R., Ruíz-Llorente, L., Martínez-Iglesias, O. A., Velasco-Martín, J. P., Montero-Pedrazuela, A., et al. (2016). Thyroid hormones inhibit TGF-β signaling and attenuate fibrotic responses. Proc. Natl. Acad. Sci. U. S. A. 113 (24), E3451–E3460. doi:10.1073/pnas.1506113113
An, C., Jiao, B., Du, H., Tran, M., Song, B., Wang, P., et al. (2023). Jumonji domain-containing protein-3 (JMJD3) promotes myeloid fibroblast activation and macrophage polarization in kidney fibrosis. Br. J. Pharmacol. doi:10.1111/bph.16096
Aronov, M., and Tirosh, B. (2016). Metabolic control of plasma cell differentiation- what we know and what we don't know. J. Clin. Immunol. 36 (Suppl. 1), 12–17. doi:10.1007/s10875-016-0246-9
Arora, S., Dev, K., Agarwal, B., Das, P., and Syed, M. A. (2018). Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 223 (4-5), 383–396. doi:10.1016/j.imbio.2017.11.001
Bai, L., Bernard, K., Tang, X., Hu, M., Horowitz, J. C., Thannickal, V. J., et al. (2019). Glutaminolysis epigenetically regulates antiapoptotic gene expression in idiopathic pulmonary fibrosis fibroblasts. Am. J. Respir. Cell Mol. Biol. 60 (1), 49–57. doi:10.1165/rcmb.2018-0180OC
Barkauskas, C. E., Cronce, M. J., Rackley, C. R., Bowie, E. J., Keene, D. R., Stripp, B. R., et al. (2013). Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest. 123 (7), 3025–3036. doi:10.1172/jci68782
Bartlett, K., and Eaton, S. (2004). Mitochondrial beta-oxidation. Eur. J. Biochem. 271 (3), 462–469. doi:10.1046/j.1432-1033.2003.03947.x
Bergmann, C., Brandt, A., Merlevede, B., Hallenberger, L., Dees, C., Wohlfahrt, T., et al. (2018). The histone demethylase Jumonji domain-containing protein 3 (JMJD3) regulates fibroblast activation in systemic sclerosis. Ann. Rheum. Dis. 77 (1), 150–158. doi:10.1136/annrheumdis-2017-211501
Bernard, K., Logsdon, N. J., Benavides, G. A., Sanders, Y., Zhang, J., Darley-Usmar, V. M., et al. (2018). Glutaminolysis is required for transforming growth factor-β1-induced myofibroblast differentiation and activation. J. Biol. Chem. 293 (4), 1218–1228. doi:10.1074/jbc.RA117.000444
Bertolotti, M., Yim, S. H., Garcia-Manteiga, J. M., Masciarelli, S., Kim, Y. J., Kang, M. H., et al. (2010). B- to plasma-cell terminal differentiation entails oxidative stress and profound reshaping of the antioxidant responses. Antioxid. Redox Signal 13 (8), 1133–1144. doi:10.1089/ars.2009.3079
Bility, M. T., Cheng, L., Zhang, Z., Luan, Y., Li, F., Chi, L., et al. (2014). Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: Induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 10 (3), e1004032. doi:10.1371/journal.ppat.1004032
Blokland, K. E. C., Waters, D. W., Schuliga, M., Read, J., Pouwels, S. D., Grainge, C. L., et al. (2020). Senescence of IPF lung fibroblasts disrupt alveolar epithelial cell proliferation and promote migration in wound healing. Pharmaceutics 12 (4), 389. doi:10.3390/pharmaceutics12040389
Bonaccorsi-Riani, E., Danger, R., Lozano, J. J., Martinez-Picola, M., Kodela, E., Mas-Malavila, R., et al. (2015). Iron deficiency impairs intra-hepatic lymphocyte mediated immune response. PLoS One 10 (8), e0136106. doi:10.1371/journal.pone.0136106
Brittain, E. L., Talati, M., Fessel, J. P., Zhu, H., Penner, N., Calcutt, M. W., et al. (2016). Fatty acid metabolic defects and right ventricular lipotoxicity in human pulmonary arterial hypertension. Circulation 133 (20), 1936–1944. doi:10.1161/circulationaha.115.019351
Brown, M., and Ahmed, S. (2019). Emerging role of proprotein convertase subtilisin/kexin type-9 (PCSK-9) in inflammation and diseases. Toxicol. Appl. Pharmacol. 370, 170–177. doi:10.1016/j.taap.2019.03.018
Budi, E. H., Duan, D., and Derynck, R. (2017). Transforming growth factor-β receptors and smads: Regulatory complexity and functional versatility. Trends Cell Biol. 27 (9), 658–672. doi:10.1016/j.tcb.2017.04.005
Burgess, H. A., Daugherty, L. E., Thatcher, T. H., Lakatos, H. F., Ray, D. M., Redonnet, M., et al. (2005). PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: Implications for therapy of lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 288 (6), L1146–L1153. doi:10.1152/ajplung.00383.2004
Calvier, L., Chouvarine, P., Legchenko, E., Hoffmann, N., Geldner, J., Borchert, P., et al. (2017). PPARγ links BMP2 and TGFβ1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab. 25 (5), 1118–1134. doi:10.1016/j.cmet.2017.03.011
Caro-Maldonado, A., Wang, R., Nichols, A. G., Kuraoka, M., Milasta, S., Sun, L. D., et al. (2014). Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol. 192 (8), 3626–3636. doi:10.4049/jimmunol.1302062
Castello-Cros, R., Whitaker-Menezes, D., Molchansky, A., Purkins, G., Soslowsky, L. J., Beason, D. P., et al. (2011). Scleroderma-like properties of skin from caveolin-1-deficient mice: Implications for new treatment strategies in patients with fibrosis and systemic sclerosis. Cell Cycle 10 (13), 2140–2150. doi:10.4161/cc.10.13.16227
Chang, H., Meng, H. Y., Liu, S. M., Wang, Y., Yang, X. X., Lu, F., et al. (2017). Identification of key metabolic changes during liver fibrosis progression in rats using a urine and serum metabolomics approach. Sci. Rep. 7 (1), 11433. doi:10.1038/s41598-017-11759-z
Chattopadhyaya, S., Nagalingam, R. S., Ledingham, D. A., Moffatt, T. L., Al-Hattab, D. S., Narhan, P., et al. (2022). Regulation of cardiac fibroblast GLS1 expression by scleraxis. Cells 11 (9), 1471. doi:10.3390/cells11091471
Chen, G., Dong, Z., Liu, H., Liu, Y., Duan, S., Liu, Y., et al. (2016). mTOR signaling regulates protective activity of transferred CD4+Foxp3+ T cells in repair of acute kidney injury. J. Immunol. 197 (10), 3917–3926. doi:10.4049/jimmunol.1601251
Chen, G., Ni, Y., Nagata, N., Xu, L., Zhuge, F., Nagashimada, M., et al. (2019). Pirfenidone prevents and reverses hepatic insulin resistance and steatohepatitis by polarizing M2 macrophages. Lab. Invest. 99 (9), 1335–1348. doi:10.1038/s41374-019-0255-4
Chen, L., Yang, T., Lu, D. W., Zhao, H., Feng, Y. L., Chen, H., et al. (2018). Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 101, 670–681. doi:10.1016/j.biopha.2018.02.090
Chen, Q. T., Zhang, Z. Y., Huang, Q. L., Chen, H. Z., Hong, W. B., Lin, T., et al. (2022). HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat. Metab. 4 (10), 1306–1321. doi:10.1038/s42255-022-00642-5
Cheng, D., Xu, Q., Wang, Y., Li, G., Sun, W., Ma, D., et al. (2021). Metformin attenuates silica-induced pulmonary fibrosis via AMPK signaling. J. Transl. Med. 19 (1), 349. doi:10.1186/s12967-021-03036-5
Chi, X., Li, Y., and Qiu, X. (2020). V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: Mechanism and regulation. Immunology 160 (3), 233–247. doi:10.1111/imm.13176
Chiang, N., and Serhan, C. N. (2017). Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Asp. Med. 58, 114–129. doi:10.1016/j.mam.2017.03.005
Cho, S. J., Moon, J. S., Nikahira, K., Yun, H. S., Harris, R., Hong, K. S., et al. (2020). GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax 75 (3), 227–236. doi:10.1136/thoraxjnl-2019-213571
Chowdhury, D., Alrefai, H., Landero Figueroa, J. A., Candor, K., Porollo, A., Fecher, R., et al. (2019). Metallothionein 3 controls the phenotype and metabolic programming of alternatively activated macrophages. Cell Rep. 27 (13), 3873–3886. doi:10.1016/j.celrep.2019.05.093
Chu, S. G., Villalba, J. A., Liang, X., Xiong, K., Tsoyi, K., Ith, B., et al. (2019). Palmitic acid-rich high-fat diet exacerbates experimental pulmonary fibrosis by modulating endoplasmic reticulum stress. Am. J. Respir. Cell Mol. Biol. 61 (6), 737–746. doi:10.1165/rcmb.2018-0324OC
Corcoran, S. E., and O'Neill, L. A. (2016). HIF1α and metabolic reprogramming in inflammation. J. Clin. Invest. 126 (10), 3699–3707. doi:10.1172/jci84431
Correia-Melo, C., Marques, F. D., Anderson, R., Hewitt, G., Hewitt, R., Cole, J., et al. (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. Embo J. 35 (7), 724–742. doi:10.15252/embj.201592862
Cui, X., Zhao, H., Wei, S., Du, Q., Dong, K., Yan, Y., et al. (2023). Hepatocellular carcinoma-derived FOXO1 inhibits tumor progression by suppressing IL-6 secretion from macrophages. Neoplasia 40, 100900. doi:10.1016/j.neo.2023.100900
Cyster, J. G., and Allen, C. D. C. (2019). B cell responses: Cell interaction dynamics and decisions. Cell 177 (3), 524–540. doi:10.1016/j.cell.2019.03.016
Darby, I. A., and Hewitson, T. D. (2007). Fibroblast differentiation in wound healing and fibrosis. Int. Rev. Cytol. 257, 143–179. doi:10.1016/s0074-7696(07)57004-x
de Carvalho, C., and Caramujo, M. J. (2018). The various roles of fatty acids. Molecules 23 (10), 2583. doi:10.3390/molecules23102583
DeFuria, J., Belkina, A. C., Jagannathan-Bogdan, M., Snyder-Cappione, J., Carr, J. D., Nersesova, Y. R., et al. (2013). B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. U. S. A. 110 (13), 5133–5138. doi:10.1073/pnas.1215840110
Del Galdo, F., Lisanti, M. P., and Jimenez, S. A. (2008). Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr. Opin. Rheumatol. 20 (6), 713–719. doi:10.1097/bor.0b013e3283103d27
DeRosa, A., and Leftin, A. (2021). The iron curtain: Macrophages at the interface of systemic and microenvironmental iron metabolism and immune response in cancer. Front. Immunol. 12, 614294. doi:10.3389/fimmu.2021.614294
Ding, H., Jiang, L., Xu, J., Bai, F., Zhou, Y., Yuan, Q., et al. (2017). Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Ren. Physiol. 313 (3), F561–f575. doi:10.1152/ajprenal.00036.2017
Dong, L., Yu, L., and Zhong, J. (2022). Histone lysine-specific demethylase 1 induced renal fibrosis via decreasing sirtuin 3 expression and activating TGF-β1/Smad3 pathway in diabetic nephropathy. Diabetol. Metab. Syndr. 14 (1), 2. doi:10.1186/s13098-021-00771-z
Dufort, F. J., Bleiman, B. F., Gumina, M. R., Blair, D., Wagner, D. J., Roberts, M. F., et al. (2007). Cutting edge: IL-4-mediated protection of primary B lymphocytes from apoptosis via stat6-dependent regulation of glycolytic metabolism. J. Immunol. 179 (8), 4953–4957. doi:10.4049/jimmunol.179.8.4953
Dufort, F. J., Gumina, M. R., Ta, N. L., Tao, Y., Heyse, S. A., Scott, D. A., et al. (2014). Glucose-dependent de novo lipogenesis in B lymphocytes: A requirement for atp-citrate lyase in lipopolysaccharide-induced differentiation. J. Biol. Chem. 289 (10), 7011–7024. doi:10.1074/jbc.M114.551051
Düvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39 (2), 171–183. doi:10.1016/j.molcel.2010.06.022
Echtay, K. S., Murphy, M. P., Smith, R. A., Talbot, D. A., and Brand, M. D. (2002). Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J. Biol. Chem. 277 (49), 47129–47135. doi:10.1074/jbc.M208262200
Eguchi, K., Manabe, I., Oishi-Tanaka, Y., Ohsugi, M., Kono, N., Ogata, F., et al. (2012). Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 15 (4), 518–533. doi:10.1016/j.cmet.2012.01.023
El Kasmi, K. C., and Stenmark, K. R. (2015). Contribution of metabolic reprogramming to macrophage plasticity and function. Semin. Immunol. 27 (4), 267–275. doi:10.1016/j.smim.2015.09.001
Eser, P., and Jänne, P. A. (2018). TGFβ pathway inhibition in the treatment of non-small cell lung cancer. Pharmacol. Ther. 184, 112–130. doi:10.1016/j.pharmthera.2017.11.004
Fan, N., Zhang, X., Zhao, W., Zhao, J., Luo, D., Sun, Y., et al. (2022). Covalent inhibition of pyruvate kinase M2 reprograms metabolic and inflammatory pathways in hepatic macrophages against non-alcoholic fatty liver disease. Int. J. Biol. Sci. 18 (14), 5260–5275. doi:10.7150/ijbs.73890
Febbraio, M., Hajjar, D. P., and Silverstein, R. L. (2001). CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Invest. 108 (6), 785–791. doi:10.1172/jci14006
Feng, P., Yang, Q., Luo, L., Sun, Y., Lv, W., Wan, S., et al. (2021). The kinase PDK1 regulates regulatory T cell survival via controlling redox homeostasis. Theranostics 11 (19), 9503–9518. doi:10.7150/thno.63992
Feng, X., Weng, D., Zhou, F., Owen, Y. D., Qin, H., Zhao, J., et al. (2016). Activation of PPARγ by a natural flavonoid modulator, apigenin ameliorates obesity-related inflammation via regulation of macrophage polarization. EBioMedicine 9, 61–76. doi:10.1016/j.ebiom.2016.06.017
Forrester, J. V., Kuffova, L., and Delibegovic, M. (2020). The role of inflammation in diabetic retinopathy. Front. Immunol. 11, 583687. doi:10.3389/fimmu.2020.583687
Franco, F., Jaccard, A., Romero, P., Yu, Y. R., and Ho, P. C. (2020). Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2 (10), 1001–1012. doi:10.1038/s42255-020-00280-9
Frauwirth, K. A., Riley, J. L., Harris, M. H., Parry, R. V., Rathmell, J. C., Plas, D. R., et al. (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity 16 (6), 769–777. doi:10.1016/s1074-7613(02)00323-0
Freemerman, A. J., Johnson, A. R., Sacks, G. N., Milner, J. J., Kirk, E. L., Troester, M. A., et al. (2014). Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 289 (11), 7884–7896. doi:10.1074/jbc.M113.522037
Gagnon, L., Leduc, M., Thibodeau, J. F., Zhang, M. Z., Grouix, B., Sarra-Bournet, F., et al. (2018). A newly discovered antifibrotic pathway regulated by two fatty acid receptors: GPR40 and GPR84. Am. J. Pathol. 188 (5), 1132–1148. doi:10.1016/j.ajpath.2018.01.009
Gao, Z., and Chen, X. (2022). Fatty acid β-oxidation in kidney diseases: Perspectives on pathophysiological mechanisms and therapeutic opportunities. Front. Pharmacol. 13, 805281. doi:10.3389/fphar.2022.805281
Gasse, P., Riteau, N., Vacher, R., Michel, M. L., Fautrel, A., di Padova, F., et al. (2011). IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS One 6 (8), e23185. doi:10.1371/journal.pone.0023185
Geier, C., and Perl, A. (2021). Therapeutic mTOR blockade in systemic autoimmunity: Implications for antiviral immunity and extension of lifespan. Autoimmun. Rev. 20 (12), 102984. doi:10.1016/j.autrev.2021.102984
Genard, G., Lucas, S., and Michiels, C. (2017). Reprogramming of tumor-associated macrophages with anticancer therapies: Radiotherapy versus chemo- and immunotherapies. Front. Immunol. 8, 828. doi:10.3389/fimmu.2017.00828
Gergely, P., Grossman, C., Niland, B., Puskas, F., Neupane, H., Allam, F., et al. (2002). Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 46 (1), 175–190. doi:10.1002/1529-0131(200201)46:1<175::Aid-art10015>3.0.Co;2-h
Gibb, A. A., Murray, E. K., Huynh, A. T., Gaspar, R. B., Ploesch, T. L., Bedi, K., et al. (2022). Glutaminolysis is essential for myofibroblast persistence and in vivo targeting reverses fibrosis and cardiac dysfunction in heart failure. Circulation 145 (21), 1625–1628. doi:10.1161/circulationaha.121.057879
Gieling, R. G., Wallace, K., and Han, Y. P. (2009). Interleukin-1 participates in the progression from liver injury to fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 296 (6), G1324–G1331. doi:10.1152/ajpgi.90564.2008
Goda, C., Balli, D., Black, M., Milewski, D., Le, T., Ustiyan, V., et al. (2020). Loss of FOXM1 in macrophages promotes pulmonary fibrosis by activating p38 MAPK signaling pathway. PLoS Genet. 16 (4), e1008692. doi:10.1371/journal.pgen.1008692
Goliwas, K. F., and Deshane, J. S. (2020). Extracellular vesicles: Bidirectional accelerators of cellular senescence in fibrosis? Am. J. Respir. Cell Mol. Biol. 63 (5), 547–548. doi:10.1165/rcmb.2020-0313ED
Goodwin, J., Choi, H., Hsieh, M. H., Neugent, M. L., Ahn, J. M., Hayenga, H. N., et al. (2018). Targeting hypoxia-inducible factor-1α/pyruvate dehydrogenase kinase 1 Axis by dichloroacetate suppresses bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 58 (2), 216–231. doi:10.1165/rcmb.2016-0186OC
Haas, R., Smith, J., Rocher-Ros, V., Nadkarni, S., Montero-Melendez, T., D'Acquisto, F., et al. (2015). Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 13 (7), e1002202. doi:10.1371/journal.pbio.1002202
Han, Y. C., Tang, S. Q., Liu, Y. T., Li, A. M., Zhan, M., Yang, M., et al. (2021). AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 12 (10), 925. doi:10.1038/s41419-021-04184-8
Hanna, C., Hubchak, S. C., Liang, X., Rozen-Zvi, B., Schumacker, P. T., Hayashida, T., et al. (2013). Hypoxia-inducible factor-2α and TGF-β signaling interact to promote normoxic glomerular fibrogenesis. Am. J. Physiol. Ren. Physiol. 305 (9), F1323–F1331. doi:10.1152/ajprenal.00155.2013
Hassona, Y., Cirillo, N., Lim, K. P., Herman, A., Mellone, M., Thomas, G. J., et al. (2013). Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-β. Carcinogenesis 34 (6), 1286–1295. doi:10.1093/carcin/bgt035
He, C., Larson-Casey, J. L., Gu, L., Ryan, A. J., Murthy, S., and Carter, A. B. (2016). Cu,Zn-Superoxide dismutase-mediated redox regulation of Jumonji domain containing 3 modulates macrophage polarization and pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 55 (1), 58–71. doi:10.1165/rcmb.2015-0183OC
He, S., Chen, D., Hu, M., Zhang, L., Liu, C., Traini, D., et al. (2019). Bronchial epithelial cell extracellular vesicles ameliorate epithelial-mesenchymal transition in COPD pathogenesis by alleviating M2 macrophage polarization. Nanomedicine 18, 259–271. doi:10.1016/j.nano.2019.03.010
Henderson, J., Duffy, L., Stratton, R., Ford, D., and O'Reilly, S. (2020). Metabolic reprogramming of glycolysis and glutamine metabolism are key events in myofibroblast transition in systemic sclerosis pathogenesis. J. Cell Mol. Med. 24 (23), 14026–14038. doi:10.1111/jcmm.16013
Henderson, J., and O'Reilly, S. (2021). The emerging role of metabolism in fibrosis. Trends Endocrinol. Metab. 32 (8), 639–653. doi:10.1016/j.tem.2021.05.003
Herman-Edelstein, M., Scherzer, P., Tobar, A., Levi, M., and Gafter, U. (2014). Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 55 (3), 561–572. doi:10.1194/jlr.P040501
Herrera, J., Henke, C. A., and Bitterman, P. B. (2018). Extracellular matrix as a driver of progressive fibrosis. J. Clin. Invest. 128 (1), 45–53. doi:10.1172/jci93557
Ho, T., Nichols, M., Nair, G., Radford, K., Kjarsgaard, M., Huang, C., et al. (2022). Iron in airway macrophages and infective exacerbations of chronic obstructive pulmonary disease. Respir. Res. 23 (1), 8. doi:10.1186/s12931-022-01929-7
Hu, H. H., Chen, D. Q., Wang, Y. N., Feng, Y. L., Cao, G., Vaziri, N. D., et al. (2018). New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 292, 76–83. doi:10.1016/j.cbi.2018.07.008
Hu, X., Xu, Q., Wan, H., Hu, Y., Xing, S., Yang, H., et al. (2020). PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab. Invest. 100 (6), 801–811. doi:10.1038/s41374-020-0404-9
Huang, H., Zhou, P., Wei, J., Long, L., Shi, H., Dhungana, Y., et al. (2021). In vivo CRISPR screening reveals nutrient signaling processes underpinning CD8(+) T cell fate decisions. Cell 184 (5), 1245–1261.e21. doi:10.1016/j.cell.2021.02.021
Im, M. J., Freshwater, M. F., and Hoopes, J. E. (1976). Enzyme activities in granulation tissue: Energy for collagen synthesis. J. Surg. Res. 20 (2), 121–125. doi:10.1016/0022-4804(76)90108-6
Ishida, K., Nagatake, T., Saika, A., Kawai, S., Node, E., Hosomi, K., et al. (2023). Induction of unique macrophage subset by simultaneous stimulation with LPS and IL-4. Front. Immunol. 14, 1111729. doi:10.3389/fimmu.2023.1111729
Jain, M. R., Giri, S. R., Bhoi, B., Trivedi, C., Rath, A., Rathod, R., et al. (2018). Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 38 (6), 1084–1094. doi:10.1111/liv.13634
Jang, E., Cho, W. S., Oh, Y. K., Cho, M. L., Kim, J. M., Paik, D. J., et al. (2016). Splenic long-lived plasma cells promote the development of follicular helper T cells during autoimmune responses. J. Immunol. 196 (3), 1026–1035. doi:10.4049/jimmunol.1401059
Jang, K. J., Mano, H., Aoki, K., Hayashi, T., Muto, A., Nambu, Y., et al. (2015a). Mitochondrial function provides instructive signals for activation-induced B-cell fates. Nat. Commun. 6, 6750. doi:10.1038/ncomms7750
Jang, Y. S., Seo, G. Y., Lee, J. M., Seo, H. Y., Han, H. J., Kim, S. J., et al. (2015b). Lactoferrin causes IgA and IgG2b isotype switching through betaglycan binding and activation of canonical TGF-β signaling. Mucosal Immunol. 8 (4), 906–917. doi:10.1038/mi.2014.121
Jayachandran, N., Mejia, E. M., Sheikholeslami, K., Sher, A. A., Hou, S., Hatch, G. M., et al. (2018). TAPP adaptors control B cell metabolism by modulating the phosphatidylinositol 3-kinase signaling pathway: A novel regulatory circuit preventing autoimmunity. J. Immunol. 201 (2), 406–416. doi:10.4049/jimmunol.1701440
Jellusova, J., Cato, M. H., Apgar, J. R., Ramezani-Rad, P., Leung, C. R., Chen, C., et al. (2017). Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 18 (3), 303–312. doi:10.1038/ni.3664
Jha, A. K., Huang, S. C., Sergushichev, A., Lampropoulou, V., Ivanova, Y., Loginicheva, E., et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42 (3), 419–430. doi:10.1016/j.immuni.2015.02.005
Jia, Y., Reddy, M. A., Das, S., Oh, H. J., Abdollahi, M., Yuan, H., et al. (2019). Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-β1-induced gene expression in mesangial cells and diabetic kidney. J. Biol. Chem. 294 (34), 12695–12707. doi:10.1074/jbc.RA119.007575
Jiang, L., Xiao, L., Sugiura, H., Huang, X., Ali, A., Kuro-o, M., et al. (2015). Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 34 (30), 3908–3916. doi:10.1038/onc.2014.321
Jiang, Y., Li, C., Wu, Q., An, P., Huang, L., Wang, J., et al. (2019). Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nat. Commun. 10 (1), 2935. doi:10.1038/s41467-019-11002-5
Jiang, Y., Xiang, C., Zhong, F., Zhang, Y., Wang, L., Zhao, Y., et al. (2021). Histone H3K27 methyltransferase EZH2 and demethylase JMJD3 regulate hepatic stellate cells activation and liver fibrosis. Theranostics 11 (1), 361–378. doi:10.7150/thno.46360
Jobling, M. F., Mott, J. D., Finnegan, M. T., Jurukovski, V., Erickson, A. C., Walian, P. J., et al. (2006). Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 166 (6), 839–848. doi:10.1667/rr0695.1
Jones, D. D., Gaudette, B. T., Wilmore, J. R., Chernova, I., Bortnick, A., Weiss, B. M., et al. (2016). mTOR has distinct functions in generating versus sustaining humoral immunity. J. Clin. Invest. 126 (11), 4250–4261. doi:10.1172/jci86504
Jun, J. I., and Lau, L. F. (2017). CCN2 induces cellular senescence in fibroblasts. J. Cell Commun. Signal 11 (1), 15–23. doi:10.1007/s12079-016-0359-1
Jung, M. Y., Kang, J. H., Hernandez, D. M., Yin, X., Andrianifahanana, M., Wang, Y., et al. (2018). Fatty acid synthase is required for profibrotic TGF-β signaling. Faseb J. 32 (7), 3803–3815. doi:10.1096/fj.201701187R
Kang, H. M., Ahn, S. H., Choi, P., Ko, Y. A., Han, S. H., Chinga, F., et al. (2015). Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21 (1), 37–46. doi:10.1038/nm.3762
Kang, S., and Kumanogoh, A. (2020). The spectrum of macrophage activation by immunometabolism. Int. Immunol. 32 (7), 467–473. doi:10.1093/intimm/dxaa017
Karsdal, M. A., Daniels, S. J., Holm Nielsen, S., Bager, C., Rasmussen, D. G. K., Loomba, R., et al. (2020). Collagen biology and non-invasive biomarkers of liver fibrosis. Liver Int. 40 (4), 736–750. doi:10.1111/liv.14390
Katzen, J., and Beers, M. F. (2020). Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Invest. 130 (10), 5088–5099. doi:10.1172/jci139519
Khalil, N., Manganas, H., Ryerson, C. J., Shapera, S., Cantin, A. M., Hernandez, P., et al. (2019). Phase 2 clinical trial of PBI-4050 in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 53 (3), 1800663. doi:10.1183/13993003.00663-2018
Kim, D. H., Choi, H. I., Park, J. S., Kim, C. S., Bae, E. H., Ma, S. K., et al. (2019). Src-mediated crosstalk between FXR and YAP protects against renal fibrosis. Faseb J. 33 (10), 11109–11122. doi:10.1096/fj.201900325R
Kishton, R. J., Sukumar, M., and Restifo, N. P. (2016). Arginine arms T cells to thrive and survive. Cell Metab. 24 (5), 647–648. doi:10.1016/j.cmet.2016.10.019
Kisseleva, T., and Brenner, D. (2021). Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 18 (3), 151–166. doi:10.1038/s41575-020-00372-7
Komatsu, N., and Takayanagi, H. (2012). Inflammation and bone destruction in arthritis: Synergistic activity of immune and mesenchymal cells in joints. Front. Immunol. 3, 77. doi:10.3389/fimmu.2012.00077
Kottmann, R. M., Trawick, E., Judge, J. L., Wahl, L. A., Epa, A. P., Owens, K. M., et al. (2015). Pharmacologic inhibition of lactate production prevents myofibroblast differentiation. Am. J. Physiol. Lung Cell Mol. Physiol. 309 (11), L1305–L1312. doi:10.1152/ajplung.00058.2015
Koyama, Y., and Brenner, D. A. (2017). Liver inflammation and fibrosis. J. Clin. Invest. 127 (1), 55–64. doi:10.1172/jci88881
Kozlov, A. M., Lone, A., Betts, D. H., and Cumming, R. C. (2020). Lactate preconditioning promotes a HIF-1α-mediated metabolic shift from OXPHOS to glycolysis in normal human diploid fibroblasts. Sci. Rep. 10 (1), 8388. doi:10.1038/s41598-020-65193-9
Kruse, N. J., and Bornstein, P. (1975). The metabolic requirements for transcellular movement and secretion of collagen. J. Biol. Chem. 250 (13), 4841–4847. doi:10.1016/s0021-9258(19)41245-3
Kume, H., Yamada, R., Sato, Y., and Togawa, R. (2023). Airway smooth muscle regulated by oxidative stress in COPD. Antioxidants (Basel) 12 (1), 142. doi:10.3390/antiox12010142
Kunisawa, J. (2017). Metabolic changes during B cell differentiation for the production of intestinal IgA antibody. Cell Mol. Life Sci. 74 (8), 1503–1509. doi:10.1007/s00018-016-2414-8
Kurundkar, A., and Thannickal, V. J. (2016). Redox mechanisms in age-related lung fibrosis. Redox Biol. 9, 67–76. doi:10.1016/j.redox.2016.06.005
Lakhia, R., Yheskel, M., Flaten, A., Quittner-Strom, E. B., Holland, W. L., and Patel, V. (2018). PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am. J. Physiol. Ren. Physiol. 314 (1), F122–f131. doi:10.1152/ajprenal.00352.2017
Lakshmi, S. P., Reddy, A. T., and Reddy, R. C. (2017). Transforming growth factor β suppresses peroxisome proliferator-activated receptor γ expression via both SMAD binding and novel TGF-β inhibitory elements. Biochem. J. 474 (9), 1531–1546. doi:10.1042/bcj20160943
Lam, W. Y., Becker, A. M., Kennerly, K. M., Wong, R., Curtis, J. D., Llufrio, E. M., et al. (2016). Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity 45 (1), 60–73. doi:10.1016/j.immuni.2016.06.011
Lam, W. Y., and Bhattacharya, D. (2018). Metabolic links between plasma cell survival, secretion, and stress. Trends Immunol. 39 (1), 19–27. doi:10.1016/j.it.2017.08.007
Lee, D. S. W., Rojas, O. L., and Gommerman, J. L. (2021). B cell depletion therapies in autoimmune disease: Advances and mechanistic insights. Nat. Rev. Drug Discov. 20 (3), 179–199. doi:10.1038/s41573-020-00092-2
Lei, L., Zhao, C., Qin, F., He, Z. Y., Wang, X., and Zhong, X. N. (2016). Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin. Exp. Rheumatol. 34 (5), 14–22.
Li, L., Luo, W., Qian, Y., Zhu, W., Qian, J., Li, J., et al. (2019). Luteolin protects against diabetic cardiomyopathy by inhibiting NF-κB-mediated inflammation and activating the Nrf2-mediated antioxidant responses. Phytomedicine 59, 152774. doi:10.1016/j.phymed.2018.11.034
Lightman, S. M., Utley, A., and Lee, K. P. (2019). Survival of long-lived plasma cells (LLPC): Piecing together the puzzle. Front. Immunol. 10, 965. doi:10.3389/fimmu.2019.00965
Lin, P., Ji, H. H., Li, Y. J., and Guo, S. D. (2021). Macrophage plasticity and atherosclerosis therapy. Front. Mol. Biosci. 8, 679797. doi:10.3389/fmolb.2021.679797
Liu, H., Takagaki, Y., Kumagai, A., Kanasaki, K., and Koya, D. (2021). The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1α accumulation. J. Diabetes Investig. 12 (5), 697–709. doi:10.1111/jdi.13478
Liu, P. S., Wang, H., Li, X., Chao, T., Teav, T., Christen, S., et al. (2017). α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18 (9), 985–994. doi:10.1038/ni.3796
Liu, R., Li, X., Ma, H., Yang, Q., Shang, Q., Song, L., et al. (2020). Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent AMPK activation, Hif-1α upregulation and autophagy. Free Radic. Biol. Med. 161, 339–350. doi:10.1016/j.freeradbiomed.2020.10.029
Liu, R. M., and Gaston Pravia, K. A. (2010). Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic. Biol. Med. 48 (1), 1–15. doi:10.1016/j.freeradbiomed.2009.09.026
Lodyga, M., Cambridge, E., Karvonen, H. M., Pakshir, P., Wu, B., Boo, S., et al. (2019). Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci. Signal 12 (564), eaao3469. doi:10.1126/scisignal.aao3469
Lu, Y., Zhao, N., Wu, Y., Yang, S., Wu, Q., Dong, Q., et al. (2023). Inhibition of phosphoglycerate kinase 1 attenuates autoimmune myocarditis by reprogramming CD4+ T cell metabolism. Cardiovasc Res. 119, 1377–1389. doi:10.1093/cvr/cvad029
Lu, Y., Zhou, Y., and Tian, W. (2013). Combining Hi-C data with phylogenetic correlation to predict the target genes of distal regulatory elements in human genome. Nucleic Acids Res. 41 (22), 10391–10402. doi:10.1093/nar/gkt785
Lv, Q., Xing, Y., Liu, Y., Chen, Q., Xu, J., Hu, L., et al. (2021a). Didymin switches M1-like toward M2-like macrophage to ameliorate ulcerative colitis via fatty acid oxidation. Pharmacol. Res. 169, 105613. doi:10.1016/j.phrs.2021.105613
Lv, S. L., Zeng, Z. F., Gan, W. Q., Wang, W. Q., Li, T. G., Hou, Y. F., et al. (2021b). Lp-PLA2 inhibition prevents Ang II-induced cardiac inflammation and fibrosis by blocking macrophage NLRP3 inflammasome activation. Acta Pharmacol. Sin. 42 (12), 2016–2032. doi:10.1038/s41401-021-00703-7
Ma, E. H., Bantug, G., Griss, T., Condotta, S., Johnson, R. M., Samborska, B., et al. (2017). Serine is an essential metabolite for effector T cell expansion. Cell Metab. 25 (2), 345–357. doi:10.1016/j.cmet.2016.12.011
Macedo, M. F., de Sousa, M., Ned, R. M., Mascarenhas, C., Andrews, N. C., and Correia-Neves, M. (2004). Transferrin is required for early T-cell differentiation. Immunology 112 (4), 543–549. doi:10.1111/j.1365-2567.2004.01915.x
Mahalanobish, S., Saha, S., Dutta, S., and Sil, P. C. (2020). Matrix metalloproteinase: An upcoming therapeutic approach for idiopathic pulmonary fibrosis. Pharmacol. Res. 152, 104591. doi:10.1016/j.phrs.2019.104591
Malyshev, I., and Malyshev, Y. (2015). Current concept and update of the macrophage plasticity concept: Intracellular mechanisms of reprogramming and M3 macrophage "switch" phenotype. Biomed. Res. Int. 2015, 341308. doi:10.1155/2015/341308
Manz, R. A., and Radbruch, A. (2002). Plasma cells for a lifetime? Eur. J. Immunol. 32 (4), 923–927. doi:10.1002/1521-4141(200204)32:4<923::AID-IMMU923>3.0.CO;2-1
Masumi, A., Akamatsu, Y., and Kitagawa, T. (1993). Modulation of the synthesis and glycosylation of the glucose transporter protein by transforming growth factor-beta 1 in Swiss 3T3 fibroblasts. Biochim. Biophys. Acta 1145 (2), 227–234. doi:10.1016/0005-2736(93)90293-9
McHeyzer-Williams, L. J., and McHeyzer-Williams, M. G. (2005). Antigen-specific memory B cell development. Annu. Rev. Immunol. 23, 487–513. doi:10.1146/annurev.immunol.23.021704.115732
McLaughlin, T., Ackerman, S. E., Shen, L., and Engleman, E. (2017). Role of innate and adaptive immunity in obesity-associated metabolic disease. J. Clin. Invest. 127 (1), 5–13. doi:10.1172/jci88876
Meng, X., Grötsch, B., Luo, Y., Knaup, K. X., Wiesener, M. S., Chen, X. X., et al. (2018). Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 9 (1), 251. doi:10.1038/s41467-017-02683-x
Meng, X. M., Nikolic-Paterson, D. J., and Lan, H. Y. (2016). TGF-Β: The master regulator of fibrosis. Nat. Rev. Nephrol. 12 (6), 325–338. doi:10.1038/nrneph.2016.48
Michalek, R. D., Gerriets, V. A., Jacobs, S. R., Macintyre, A. N., MacIver, N. J., Mason, E. F., et al. (2011). Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186 (6), 3299–3303. doi:10.4049/jimmunol.1003613
Mohammad Omar, J., Hai, Y., and Jin, S. (2022). Hypoxia-induced factor and its role in liver fibrosis. PeerJ 10, e14299. doi:10.7717/peerj.14299
Moyé, S., Bormann, T., Maus, R., Sparwasser, T., Sandrock, I., Prinz, I., et al. (2020). Regulatory T cells limit pneumococcus-induced exacerbation of lung fibrosis in mice. J. Immunol. 204 (9), 2429–2438. doi:10.4049/jimmunol.1900980
Muñoz-Espín, D., and Serrano, M. (2014). Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 15 (7), 482–496. doi:10.1038/nrm3823
Nakaya, M., Xiao, Y., Zhou, X., Chang, J. H., Chang, M., Cheng, X., et al. (2014). Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40 (5), 692–705. doi:10.1016/j.immuni.2014.04.007
Nakazaki, M., Morita, T., Lankford, K. L., Askenase, P. W., and Kocsis, J. D. (2021). Small extracellular vesicles released by infused mesenchymal stromal cells target M2 macrophages and promote TGF-β upregulation, microvascular stabilization and functional recovery in a rodent model of severe spinal cord injury. J. Extracell. Vesicles 10 (11), e12137. doi:10.1002/jev2.12137
Neto, L. M. M., Zufelato, N., de Sousa-Júnior, A. A., Trentini, M. M., da Costa, A. C., Bakuzis, A. F., et al. (2018). Specific T cell induction using iron oxide based nanoparticles as subunit vaccine adjuvant. Hum. Vaccin Immunother. 14 (11), 2786–2801. doi:10.1080/21645515.2018.1489192
Newsholme, E. A., Crabtree, B., and Ardawi, M. S. (1985). Glutamine metabolism in lymphocytes: Its biochemical, physiological and clinical importance. Q. J. Exp. Physiol. 70 (4), 473–489. doi:10.1113/expphysiol.1985.sp002935
Ning, L., Shishi, Z., Bo, W., and Huiqing, L. (2023). Targeting immunometabolism against acute lung injury. Clin. Immunol. 249, 109289. doi:10.1016/j.clim.2023.109289
Olefsky, J. M., and Glass, C. K. (2010). Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 72, 219–246. doi:10.1146/annurev-physiol-021909-135846
Oleinika, K., Mauri, C., and Salama, A. D. (2019). Effector and regulatory B cells in immune-mediated kidney disease. Nat. Rev. Nephrol. 15 (1), 11–26. doi:10.1038/s41581-018-0074-7
Orecchioni, M., Ghosheh, Y., Pramod, A. B., and Ley, K. (2019). Macrophage polarization: Different gene signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively activated macrophages. Front. Immunol. 10, 1084. doi:10.3389/fimmu.2019.01084
Oruqaj, G., Karnati, S., Vijayan, V., Kotarkonda, L. K., Boateng, E., Zhang, W., et al. (2015). Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-β signaling. Proc. Natl. Acad. Sci. U. S. A. 112 (16), E2048–E2057. doi:10.1073/pnas.1415111112
Pan, X., Li, J., Tu, X., Wu, C., Liu, H., Luo, Y., et al. (2020). Lysine-specific demethylase-1 regulates fibroblast activation in pulmonary fibrosis via TGF-β1/Smad3 pathway. Pharmacol. Res. 152, 104592. doi:10.1016/j.phrs.2019.104592
Pellicoro, A., Ramachandran, P., Iredale, J. P., and Fallowfield, J. A. (2014). Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 14 (3), 181–194. doi:10.1038/nri3623
Pérez, S., and Rius-Pérez, S. (2022). Macrophage polarization and reprogramming in acute inflammation: A redox perspective. Antioxidants (Basel) 11 (7), 1394. doi:10.3390/antiox11071394
Pfeifhofer-Obermair, C., Tymoszuk, P., Nairz, M., Schroll, A., Klais, G., Demetz, E., et al. (2021). Regulation of Th1 T cell differentiation by iron via upregulation of T cell immunoglobulin and mucin containing protein-3 (TIM-3). Front. Immunol. 12, 637809. doi:10.3389/fimmu.2021.637809
Pociask, D. A., Sime, P. J., and Brody, A. R. (2004). Asbestos-derived reactive oxygen species activate TGF-beta1. Lab. Invest. 84 (8), 1013–1023. doi:10.1038/labinvest.3700109
Purroy, R., Medina-Carbonero, M., Ros, J., and Tamarit, J. (2020). Frataxin-deficient cardiomyocytes present an altered thiol-redox state which targets actin and pyruvate dehydrogenase. Redox Biol. 32, 101520. doi:10.1016/j.redox.2020.101520
Qiu, J., Wu, B., Goodman, S. B., Berry, G. J., Goronzy, J. J., and Weyand, C. M. (2021). Metabolic control of autoimmunity and tissue inflammation in rheumatoid arthritis. Front. Immunol. 12, 652771. doi:10.3389/fimmu.2021.652771
Ramachandran, P., Pellicoro, A., Vernon, M. A., Boulter, L., Aucott, R. L., Ali, A., et al. (2012). Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. U. S. A. 109 (46), E3186–E3195. doi:10.1073/pnas.1119964109
Ramond, E., Jamet, A., Coureuil, M., and Charbit, A. (2019). Pivotal role of mitochondria in macrophage response to bacterial pathogens. Front. Immunol. 10, 2461. doi:10.3389/fimmu.2019.02461
Rao, M., Wang, X., Guo, G., Wang, L., Chen, S., Yin, P., et al. (2021). Resolving the intertwining of inflammation and fibrosis in human heart failure at single-cell level. Basic Res. Cardiol. 116 (1), 55. doi:10.1007/s00395-021-00897-1
Ratziu, V., Harrison, S. A., Francque, S., Bedossa, P., Lehert, P., Serfaty, L., et al. (2016). Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150 (5), 1147–1159.e5. doi:10.1053/j.gastro.2016.01.038
Rebsamen, M., Pochini, L., Stasyk, T., de Araújo, M. E., Galluccio, M., Kandasamy, R. K., et al. (2015). SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519 (7544), 477–481. doi:10.1038/nature14107
Ring, A., Le Lay, S., Pohl, J., Verkade, P., and Stremmel, W. (2006). Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochim. Biophys. Acta 1761 (4), 416–423. doi:10.1016/j.bbalip.2006.03.016
Rőszer, T. (2015). Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 816460. doi:10.1155/2015/816460
Routh, R. E., Johnson, J. H., and McCarthy, K. J. (2002). Troglitazone suppresses the secretion of type I collagen by mesangial cells in vitro. Kidney Int. 61 (4), 1365–1376. doi:10.1046/j.1523-1755.2002.00277.x
Schultze, J. L., Schmieder, A., and Goerdt, S. (2015). Macrophage activation in human diseases. Semin. Immunol. 27 (4), 249–256. doi:10.1016/j.smim.2015.07.003
Serhan, C. N., and Levy, B. D. (2018). Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 128 (7), 2657–2669. doi:10.1172/jci97943
Serhan, C. N. (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature 510 (7503), 92–101. doi:10.1038/nature13479
Shan, L., Ding, Y., Fu, Y., Zhou, L., Dong, X., Chen, S., et al. (2016). mTOR overactivation in mesenchymal cells aggravates CCl(4)- induced liver fibrosis. Sci. Rep. 6, 36037. doi:10.1038/srep36037
Shi, H., Chapman, N. M., Wen, J., Guy, C., Long, L., Dhungana, Y., et al. (2019). Amino acids license kinase mTORC1 activity and Treg cell function via small G proteins rag and rheb. Immunity 51 (6), 1012–1027. doi:10.1016/j.immuni.2019.10.001
Shi, R., Xu, M., Ye, H., Gao, S., Li, J., Li, H., et al. (2022). Cycloheximide promotes type I collagen maturation mainly via collagen prolyl 4-hydroxylase subunit α2. Acta Biochim. Biophys. Sin. (Shanghai) 54 (12), 1832–1840. doi:10.3724/abbs.2022191
Sinclair, L. V., Rolf, J., Emslie, E., Shi, Y. B., Taylor, P. M., and Cantrell, D. A. (2013). Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14 (5), 500–508. doi:10.1038/ni.2556
Sindrilaru, A., Peters, T., Wieschalka, S., Baican, C., Baican, A., Peter, H., et al. (2011). An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Invest. 121 (3), 985–997. doi:10.1172/jci44490
Spada, R., Rojas, J. M., and Barber, D. F. (2015). Recent findings on the role of natural killer cells in the pathogenesis of systemic lupus erythematosus. J. Leukoc. Biol. 98 (4), 479–487. doi:10.1189/jlb.4RU0315-081RR
Srivastava, S. P., Li, J., Kitada, M., Fujita, H., Yamada, Y., Goodwin, J. E., et al. (2018). SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis. 9 (10), 997. doi:10.1038/s41419-018-1057-0
Stein, M., Keshav, S., Harris, N., and Gordon, S. (1992). Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 176 (1), 287–292. doi:10.1084/jem.176.1.287
Steneberg, P., Lindahl, E., Dahl, U., Lidh, E., Straseviciene, J., Backlund, F., et al. (2018). PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight 3 (12), e99114. doi:10.1172/jci.insight.99114
Su, Y., Chen, Q., Ma, K., Ju, Y., Ji, T., Wang, Z., et al. (2019). Astragaloside IV inhibits palmitate-mediated oxidative stress and fibrosis in human glomerular mesangial cells via downregulation of CD36 expression. Pharmacol. Rep. 71 (2), 319–329. doi:10.1016/j.pharep.2018.12.008
Susztak, K., Ciccone, E., McCue, P., Sharma, K., and Böttinger, E. P. (2005). Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2 (2), e45. doi:10.1371/journal.pmed.0020045
Takano, R., Yoshida, M., Inoue, M., Honda, T., Nakashima, R., Matsumoto, K., et al. (2015). Discovery of DS-1558: A potent and orally bioavailable GPR40 agonist. ACS Med. Chem. Lett. 6 (3), 266–270. doi:10.1021/ml500391n
Taki, Z., Gostjeva, E., Thilly, W., Yaseen, B., Lopez, H., Mirza, M., et al. (2020). Pathogenic activation of mesenchymal stem cells is induced by the disease microenvironment in systemic sclerosis. Arthritis Rheumatol. 72 (8), 1361–1374. doi:10.1002/art.41267
Tannahill, G. M., Curtis, A. M., Adamik, J., Palsson-McDermott, E. M., McGettrick, A. F., Goel, G., et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496 (7444), 238–242. doi:10.1038/nature11986
Tao, J., Chen, H., Wang, Y. J., Qiu, J. X., Meng, Q. Q., Zou, R. J., et al. (2021). Ketogenic diet suppressed T-regulatory cells and promoted cardiac fibrosis via reducing mitochondria-associated membranes and inhibiting mitochondrial function. Oxid. Med. Cell Longev. 2021, 5512322. doi:10.1155/2021/5512322
Tarlinton, D., Radbruch, A., Hiepe, F., and Dörner, T. (2008). Plasma cell differentiation and survival. Curr. Opin. Immunol. 20 (2), 162–169. doi:10.1016/j.coi.2008.03.016
Tellier, J., Shi, W., Minnich, M., Liao, Y., Crawford, S., Smyth, G. K., et al. (2016). Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat. Immunol. 17 (3), 323–330. doi:10.1038/ni.3348
Thibodeau, J. F., Simard, J. C., Holterman, C. E., Blais, A., Cloutier, M. P., Medeiros, T., et al. (2019). PBI-4050 via GPR40 activation improves adenine-induced kidney injury in mice. Clin. Sci. (Lond) 133 (14), 1587–1602. doi:10.1042/cs20190479
Tian, J., Zhang, M., Suo, M., Liu, D., Wang, X., Liu, M., et al. (2021). Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J. Cell Mol. Med. 25 (16), 7642–7659. doi:10.1111/jcmm.16601
Torretta, S., Scagliola, A., Ricci, L., Mainini, F., Di Marco, S., Cuccovillo, I., et al. (2020). D-mannose suppresses macrophage IL-1β production. Nat. Commun. 11 (1), 6343. doi:10.1038/s41467-020-20164-6
Travers, J. G., Kamal, F. A., Robbins, J., Yutzey, K. E., and Blaxall, B. C. (2016). Cardiac fibrosis: The fibroblast awakens. Circ. Res. 118 (6), 1021–1040. doi:10.1161/circresaha.115.306565
Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., et al. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439 (7078), 811–816. doi:10.1038/nature04433
Upagupta, C., Shimbori, C., Alsilmi, R., and Kolb, M. (2018). Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 27 (148), 180033. doi:10.1183/16000617.0033-2018
van Anken, E., Romijn, E. P., Maggioni, C., Mezghrani, A., Sitia, R., Braakman, I., et al. (2003). Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity 18 (2), 243–253. doi:10.1016/s1074-7613(03)00024-4
Van den Bossche, J., O'Neill, L. A., and Menon, D. (2017). Macrophage immunometabolism: Where are we (going)? Trends Immunol. 38 (6), 395–406. doi:10.1016/j.it.2017.03.001
Vanoaica, L., Richman, L., Jaworski, M., Darshan, D., Luther, S. A., and Kühn, L. C. (2014). Conditional deletion of ferritin h in mice reduces B and T lymphocyte populations. PLoS One 9 (2), e89270. doi:10.1371/journal.pone.0089270
Velázquez, A. P., Tatsuta, T., Ghillebert, R., Drescher, I., and Graef, M. (2016). Lipid droplet-mediated ER homeostasis regulates autophagy and cell survival during starvation. J. Cell Biol. 212 (6), 621–631. doi:10.1083/jcb.201508102
Villarino, A. V., Kanno, Y., and O'Shea, J. J. (2017). Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 18 (4), 374–384. doi:10.1038/ni.3691
Vinaik, R., Barayan, D., Auger, C., Abdullahi, A., and Jeschke, M. G. (2020). Regulation of glycolysis and the Warburg effect in wound healing. JCI Insight 5 (17), e138949. doi:10.1172/jci.insight.138949
Waickman, A. T., and Powell, J. D. (2012). mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 249 (1), 43–58. doi:10.1111/j.1600-065X.2012.01152.x
Wang, F., Liu, J. C., Zhou, R. J., Zhao, X., Liu, M., Ye, H., et al. (2017a). Apigenin protects against alcohol-induced liver injury in mice by regulating hepatic CYP2E1-mediated oxidative stress and PPARα-mediated lipogenic gene expression. Chem. Biol. Interact. 275, 171–177. doi:10.1016/j.cbi.2017.08.006
Wang, F., Yin, X., Fan, Y. M., Zhang, X., Ma, C., Jia, K., et al. (2023a). Upregulation of glycolytic enzyme PFKFB3 by deubiquitinase OTUD4 promotes cardiac fibrosis post myocardial infarction. J. Mol. Med. Berl. 101 (6), 743–756. doi:10.1007/s00109-023-02323-6
Wang, J., Xu, L., Xiang, Z., Ren, Y., Zheng, X., Zhao, Q., et al. (2020). Microcystin-LR ameliorates pulmonary fibrosis via modulating CD206(+) M2-like macrophage polarization. Cell Death Dis. 11 (2), 136. doi:10.1038/s41419-020-2329-z
Wang, K., Chen, Y., Zhang, P., Lin, P., Xie, N., and Wu, M. (2019). Protective features of autophagy in pulmonary infection and inflammatory diseases. Cells 8 (2), 123. doi:10.3390/cells8020123
Wang, K., Zhang, T., Lei, Y., Li, X., Jiang, J., Lan, J., et al. (2018a). Identification of ANXA2 (annexin A2) as a specific bleomycin target to induce pulmonary fibrosis by impeding TFEB-mediated autophagic flux. Autophagy 14 (2), 269–282. doi:10.1080/15548627.2017.1409405
Wang, M., Zhao, M., Yu, J., Xu, Y., Zhang, J., Liu, J., et al. (2022). MCC950, a selective NLRP3 inhibitor, attenuates adverse cardiac remodeling following heart failure through improving the cardiometabolic dysfunction in obese mice. Front. Cardiovasc Med. 9, 727474. doi:10.3389/fcvm.2022.727474
Wang, Q., He, Z., Huang, M., Liu, T., Wang, Y., Xu, H., et al. (2018b). Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat. Commun. 9 (1), 559. doi:10.1038/s41467-018-03050-0
Wang, Q., Wang, P., Qin, Z., Yang, X., Pan, B., Nie, F., et al. (2021a). Altered glucose metabolism and cell function in keloid fibroblasts under hypoxia. Redox Biol. 38, 101815. doi:10.1016/j.redox.2020.101815
Wang, R., Dillon, C. P., Shi, L. Z., Milasta, S., Carter, R., Finkelstein, D., et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35 (6), 871–882. doi:10.1016/j.immuni.2011.09.021
Wang, R., and Green, D. R. (2012). Metabolic checkpoints in activated T cells. Nat. Immunol. 13 (10), 907–915. doi:10.1038/ni.2386
Wang, S., Cheng, M., Peng, P., Lou, Y., Zhang, A., and Liu, P. (2021b). Iron released after cryo-thermal therapy induced M1 macrophage polarization, promoting the differentiation of CD4(+) T cells into CTLs. Int. J. Mol. Sci. 22 (13), 7010. doi:10.3390/ijms22137010
Wang, T., Liu, H., Lian, G., Zhang, S. Y., Wang, X., and Jiang, C. (2017b). HIF1α-Induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediat. Inflamm. 2017, 9029327. doi:10.1155/2017/9029327
Wang, X. M., Zhang, Y., Kim, H. P., Zhou, Z., Feghali-Bostwick, C. A., Liu, F., et al. (2006). Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 203 (13), 2895–2906. doi:10.1084/jem.20061536
Wang, Z., Chen, L., Huang, Y., Luo, M., Wang, H., Jiang, Z., et al. (2021c). Pharmaceutical targeting of succinate dehydrogenase in fibroblasts controls bleomycin-induced lung fibrosis. Redox Biol. 46, 102082. doi:10.1016/j.redox.2021.102082
Wang, Z., Du, K., Jin, N., Tang, B., and Zhang, W. (2023b). Macrophage in liver fibrosis: Identities and mechanisms. Int. Immunopharmacol. 120, 110357. doi:10.1016/j.intimp.2023.110357
Wei, J., Ghosh, A. K., Sargent, J. L., Komura, K., Wu, M., Huang, Q. Q., et al. (2010). PPARγ downregulation by TGFß in fibroblast and impaired expression and function in systemic sclerosis: A novel mechanism for progressive fibrogenesis. PLoS One 5 (11), e13778. doi:10.1371/journal.pone.0013778
Weisel, F. J., Mullett, S. J., Elsner, R. A., Menk, A. V., Trivedi, N., Luo, W., et al. (2020). Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat. Immunol. 21 (3), 331–342. doi:10.1038/s41590-020-0598-4
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R., and Thompson, C. B. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324 (5930), 1076–1080. doi:10.1126/science.1164097
Wettstein, G., Luccarini, J. M., Poekes, L., Faye, P., Kupkowski, F., Adarbes, V., et al. (2017). The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 1 (6), 524–537. doi:10.1002/hep4.1057
Wick, G., Backovic, A., Rabensteiner, E., Plank, N., Schwentner, C., and Sgonc, R. (2010). The immunology of fibrosis: Innate and adaptive responses. Trends Immunol. 31 (3), 110–119. doi:10.1016/j.it.2009.12.001
Wijngaarden, L. H., Taselaar, A. E., Nuijten, F., van der Harst, E., Klaassen, R. A., Kuijper, T. M., et al. (2022). T and B Cell composition and cytokine producing capacity before and after bariatric surgery. Front. Immunol. 13, 888278. doi:10.3389/fimmu.2022.888278
Wiley, C. D., Brumwell, A. N., Davis, S. S., Jackson, J. R., Valdovinos, A., Calhoun, C., et al. (2019). Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 4 (24), e130056. doi:10.1172/jci.insight.130056
Wiley, C. D., Velarde, M. C., Lecot, P., Liu, S., Sarnoski, E. A., Freund, A., et al. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23 (2), 303–314. doi:10.1016/j.cmet.2015.11.011
Wilson, J. L., Mayr, H. K., and Weichhart, T. (2019). Metabolic programming of macrophages: Implications in the pathogenesis of granulomatous disease. Front. Immunol. 10, 2265. doi:10.3389/fimmu.2019.02265
Woodland, R. T., Fox, C. J., Schmidt, M. R., Hammerman, P. S., Opferman, J. T., Korsmeyer, S. J., et al. (2008). Multiple signaling pathways promote B lymphocyte stimulator dependent B-cell growth and survival. Blood 111 (2), 750–760. doi:10.1182/blood-2007-03-077222
Wu, M. M., Wang, Q. M., Huang, B. Y., Mai, C. T., Wang, C. L., Wang, T. T., et al. (2021). Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol. Res. 172, 105796. doi:10.1016/j.phrs.2021.105796
Wu, T., Qin, X., Kurepa, Z., Kumar, K. R., Liu, K., Kanta, H., et al. (2007). Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus. J. Clin. Invest. 117 (8), 2186–2196. doi:10.1172/jci30398
Wynn, T. A., and Barron, L. (2010). Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 30 (3), 245–257. doi:10.1055/s-0030-1255354
Wynn, T. A., and Ramalingam, T. R. (2012). Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 18 (7), 1028–1040. doi:10.1038/nm.2807
Xiang, J., Yang, G., Ma, C., Wei, L., Wu, H., Zhang, W., et al. (2021). Tectorigenin alleviates intrahepatic cholestasis by inhibiting hepatic inflammation and bile accumulation via activation of PPARγ. Br. J. Pharmacol. 178 (12), 2443–2460. doi:10.1111/bph.15429
Xie, N., Tan, Z., Banerjee, S., Cui, H., Ge, J., Liu, R. M., et al. (2015). Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am. J. Respir. Crit. Care Med. 192 (12), 1462–1474. doi:10.1164/rccm.201504-0780OC
Xie, X., Lv, H., Liu, C., Su, X., Yu, Z., Song, S., et al. (2021). HBeAg mediates inflammatory functions of macrophages by TLR2 contributing to hepatic fibrosis. BMC Med. 19 (1), 247. doi:10.1186/s12916-021-02085-3
Xu, K., Yin, N., Peng, M., Stamatiades, E. G., Shyu, A., Li, P., et al. (2021). Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science 371 (6527), 405–410. doi:10.1126/science.abb2683
Xu, Q., Choksi, S., Qu, J., Jang, J., Choe, M., Banfi, B., et al. (2016). NADPH oxidases are essential for macrophage differentiation. J. Biol. Chem. 291 (38), 20030–20041. doi:10.1074/jbc.M116.731216
Xu, R., Zhang, Z., and Wang, F. S. (2012). Liver fibrosis: Mechanisms of immune-mediated liver injury. Cell Mol. Immunol. 9 (4), 296–301. doi:10.1038/cmi.2011.53
Yang, L., Gilbertsen, A., Xia, H., Benyumov, A., Smith, K., Herrera, J., et al. (2023). Hypoxia enhances IPF mesenchymal progenitor cell fibrogenicity via the lactate/GPR81/HIF1α pathway. JCI Insight 8 (4), e163820. doi:10.1172/jci.insight.163820
Yang, P., Qian, F. Y., Zhang, M. F., Xu, A. L., Wang, X., Jiang, B. P., et al. (2019). Th17 cell pathogenicity and plasticity in rheumatoid arthritis. J. Leukoc. Biol. 106 (6), 1233–1240. doi:10.1002/jlb.4ru0619-197r
Yang, X., Okamura, D. M., Lu, X., Chen, Y., Moorhead, J., Varghese, Z., et al. (2017). CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 13 (12), 769–781. doi:10.1038/nrneph.2017.126
Yang, Z., Shen, Y., Oishi, H., Matteson, E. L., Tian, L., Goronzy, J. J., et al. (2016). Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci. Transl. Med. 8 (331), 331ra38. doi:10.1126/scitranslmed.aad7151
Yin, X., Choudhury, M., Kang, J. H., Schaefbauer, K. J., Jung, M. Y., Andrianifahanana, M., et al. (2019). Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci. Signal 12 (612), eaax4067. doi:10.1126/scisignal.aax4067
Yin, X., Peng, J., Gu, L., Liu, Y., Li, X., Wu, J., et al. (2022). Targeting glutamine metabolism in hepatic stellate cells alleviates liver fibrosis. Cell Death Dis. 13 (11), 955. doi:10.1038/s41419-022-05409-0
Yoshizaki, A., Yanaba, K., Yoshizaki, A., Iwata, Y., Komura, K., Ogawa, F., et al. (2010). Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 62 (8), 2476–2487. doi:10.1002/art.27498
Yu, C., Xiong, C., Tang, J., Hou, X., Liu, N., Bayliss, G., et al. (2021). Histone demethylase JMJD3 protects against renal fibrosis by suppressing TGFβ and Notch signaling and preserving PTEN expression. Theranostics 11 (6), 2706–2721. doi:10.7150/thno.48679
Yu, G., Tzouvelekis, A., Wang, R., Herazo-Maya, J. D., Ibarra, G. H., Srivastava, A., et al. (2018). Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 24 (1), 39–49. doi:10.1038/nm.4447
Zeng, Q. H., Wei, Y., Lao, X. M., Chen, D. P., Huang, C. X., Lin, Q. Y., et al. (2020). B cells polarize pathogenic inflammatory T helper subsets through ICOSL-dependent glycolysis. Sci. Adv. 6 (37), eabb6296. doi:10.1126/sciadv.abb6296
Zhang, L. M., Zhang, J., Zhang, Y., Fei, C., Wang, L., Yi, Z. W., et al. (2019). Interleukin-18 promotes fibroblast senescence in pulmonary fibrosis through down-regulating Klotho expression. Biomed. Pharmacother. 113, 108756. doi:10.1016/j.biopha.2019.108756
Zhang, M., Iwata, S., Hajime, M., Ohkubo, N., Todoroki, Y., Miyata, H., et al. (2020). Methionine commits cells to differentiate into plasmablasts through epigenetic regulation of BTB and CNC homolog 2 by the methyltransferase EZH2. Arthritis Rheumatol. 72 (7), 1143–1153. doi:10.1002/art.41208
Zhang, Y., and Stefanovic, B. (2017). mTORC1 phosphorylates LARP6 to stimulate type I collagen expression. Sci. Rep. 7, 41173. doi:10.1038/srep41173
Zhao, W., Xu, D., Hong, W., Zhang, L., Wu, Q., Gao, M., et al. (2022). Grossamide attenuates inflammation by balancing macrophage polarization through metabolic reprogramming of macrophages in mice. Int. Immunopharmacol. 112, 109190. doi:10.1016/j.intimp.2022.109190
Zhao, X., Psarianos, P., Ghoraie, L. S., Yip, K., Goldstein, D., Gilbert, R., et al. (2019). Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat. Metab. 1 (1), 147–157. doi:10.1038/s42255-018-0008-5
Zhao, X., Yang, W., Yu, T., Yu, Y., Cui, X., Zhou, Z., et al. (2023). Th17 cell-derived amphiregulin promotes colitis-associated intestinal fibrosis through activation of mTOR and MEK in intestinal myofibroblasts. Gastroenterology 164 (1), 89–102. doi:10.1053/j.gastro.2022.09.006
Zhu, H., Chen, W., Liu, D., and Luo, H. (2019). The role of metabolism in the pathogenesis of systemic sclerosis. Metabolism 93, 44–51. doi:10.1016/j.metabol.2018.12.004
Glossary

Keywords: immunometabolism, fibrosis, inflammation, therapies, fibroblast
Citation: Feng L, Chen X, Huang Y, Zhang X, Zheng S and Xie N (2023) Immunometabolism changes in fibrosis: from mechanisms to therapeutic strategies. Front. Pharmacol. 14:1243675. doi: 10.3389/fphar.2023.1243675
Received: 21 June 2023; Accepted: 17 July 2023;
Published: 27 July 2023.
Edited by:
Chao He, University of Alabama at Birmingham, United StatesReviewed by:
Francisco O. Silva, University of Texas Southwestern Medical Center, United StatesQianjiang Hu, University of Pittsburgh, United States
Jyotsana Pandey, University of Alabama at Birmingham, United States
Copyright © 2023 Feng, Chen, Huang, Zhang, Zheng and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na Xie, bmF4aWVAc2N1LmVkdS5jbg==; Shaojiang Zheng, emhlbmdzajIwMDhAMTYzLmNvbQ==