 Samet Poyraz
Samet Poyraz H. Ali Döndaş
H. Ali Döndaş Naciye Yaktubay Döndaş
Naciye Yaktubay Döndaş José M. Sansano
José M. Sansano
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 06 September 2023
Sec. Pharmacology of Infectious Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1239658
To overcome numerous health disorders, heterocyclic structures of synthetic or natural origin are utilized, and notably, the emergence of various side effects of existing drugs used for treatment or the resistance of disease-causing microorganisms renders drugs ineffective. Therefore, the discovery of potential therapeutic agents that utilize different modes of action is of utmost significance to circumvent these constraints. Pyrrolidines, pyrrolidine-alkaloids, and pyrrolidine-based hybrid molecules are present in many natural products and pharmacologically important agents. Their key roles in pharmacotherapy make them a versatile scaffold for designing and developing novel biologically active compounds and drug candidates. This review aims to provide an overview of recent advancements (especially during 2015–2023) in the exploration of pyrrolidine derivatives, emphasizing their significance as fundamental components of the skeletal structure. In contrast to previous reviews that have predominantly focused on a singular biological activity associated with these molecules, this review consolidates findings from various investigations encompassing a wide range of important activities (antimicrobial, antiviral, anticancer, anti-inflammatory, anticonvulsant, cholinesterase inhibition, and carbonic anhydrase inhibition) exhibited by pyrrolidine derivatives. This study is also anticipated to serve as a valuable resource for drug research and development endeavors, offering significant insights and guidance.
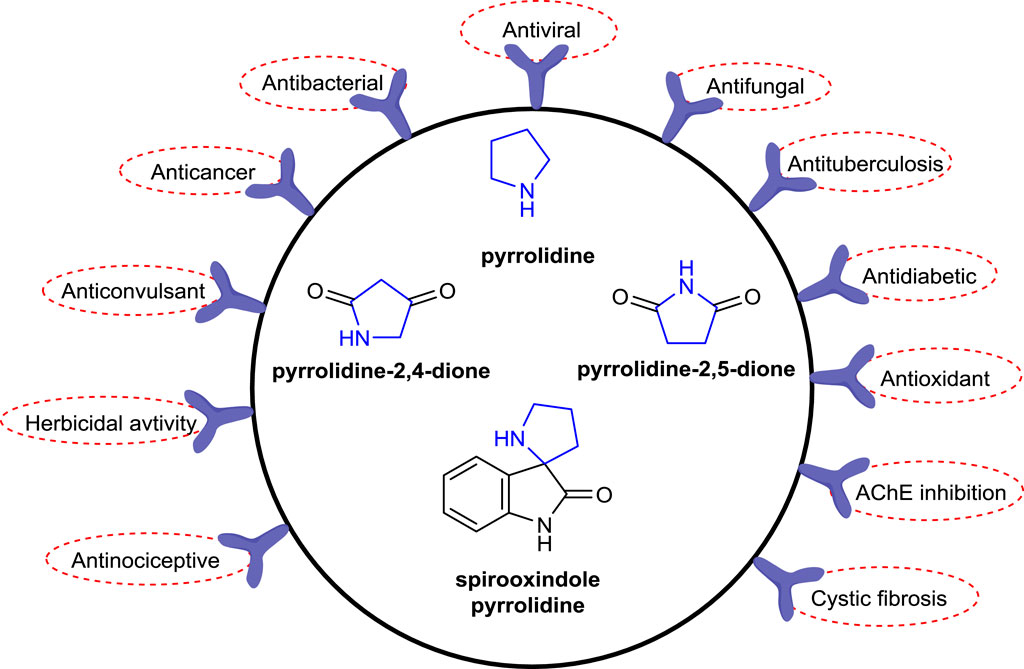
The pyrrolidine ring, also known as tetrahydropyrrole, which is one of the important heterocyclic compounds containing five-membered nitrogen atoms, is the core structure of numerous biologically and pharmacologically active molecules, as well as many alkaloids and bioactive compounds (Bhat and Tilve, 2014; Li Petri et al., 2021). Compounds bearing pyrrolidine scaffolds continue to be utilized as intermediates in drug research and development (R&D) studies for the development of molecules that could be new drug candidates (Jeelan Basha et al., 2022; Bhat et al., 2023a). Some pyrrolidine derivatives are known to be employed as pharmacophore groups, with some having antibacterial (Huang et al., 2015), antifungal (Lukowska-Chojnacka et al., 2019), antiviral (Moni et al., 2015), antimalarial (Meyers et al., 2019), antitumoral (Li et al., 2020), anti-inflammatory (Jan et al., 2020), anticonvulsant (Góra et al., 2020), and antioxidant (Hussain et al., 2019) activities, while others have diverse enzyme inhibitory effects (Zhou et al., 2019; Salve and Jadhav, 2021; Toumi et al., 2021; Poyraz et al., 2023a). In addition, several compounds with essential bioactive characteristics have been reported in the literature that comprise fused or spiropyrrolidine rings (Łowicki and Przybylski, 2022). The well-known drugs with a pyrrolidine ring in their structural skeleton (Figure 1) include clemastine 1 (antihistaminic), procyclidine 2, glycopyrronium 3 (anticholinergic), aniracetam 4 (anti-Alzheimer), clindamycin 5, anisomycin 6 (antibacterial), captopril 7, enalapril 8, bepridil 9 (antihypertensive), rolipram 10 (antidepressant), and ethosuximide 11 (antiepileptic) (https://go.drugbank.com/categories/DBCAT000663). Furthermore, the therapeutic molecules with a pyrrolidine ring in their structures such as daridorexant 12 (insomnia), pacritinib 13 (JAK-2 inhibitor), and futibatinib 14 (FGFR-4 inhibitor) were approved by the FDA in 2022 (Benedetto Tiz et al., 2022) (Figure 2).

FIGURE 1. Pyrrolidine-based marketed drugs.
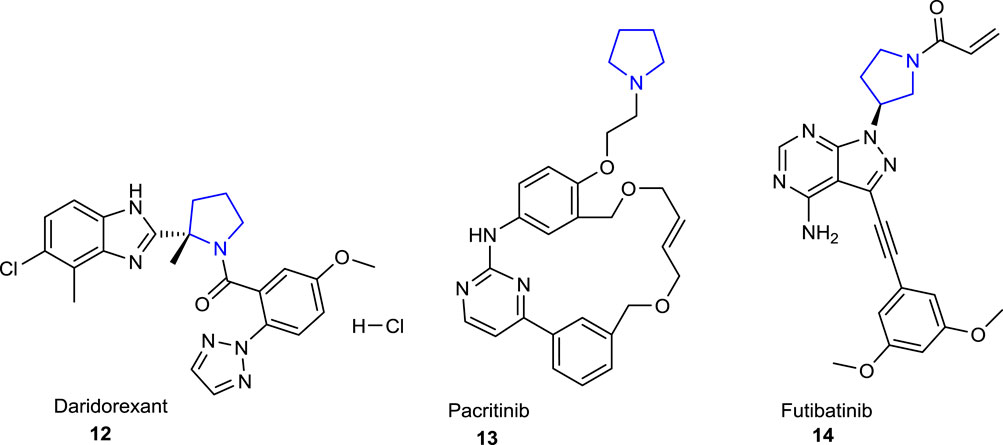
FIGURE 2. FDA-approved pyrrolidine-containing drugs in 2022.
The molecular diversity and complexity of such pyrrolidine-based molecules allow us to design and develop more active and less toxic drug candidates by taking into account the elucidation of structure–activity relationship (SAR) and quantitative structure–activity relationship (QSAR) in the synthetic pathway (Vitaku et al., 2014; Bhat et al., 2023a; Bhat et al., 2023b). So, in this review, the most relevant pharmaceutical uses of pyrrolidine frameworks are detailed.
Our research group designed the synthesis of significant hybrid compound series containing cyclic or bicyclic pyrrolidine rings and reported their bioactive properties. The pyrrolidine hybrids were incorporated with important pharmacophore moieties such as N-benzoylthiourea, thiohydantoin, thiazole, imidazole, and indole (Belveren et al., 2017; Poyraz et al., 2017; Poyraz et al., 2018; Belveren et al., 2019; Poyraz et al., 2023b).
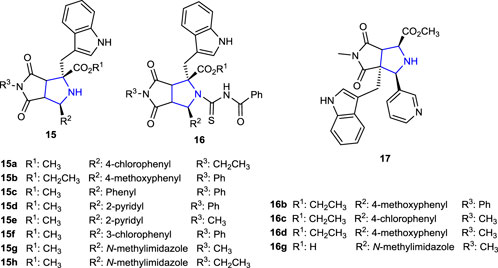
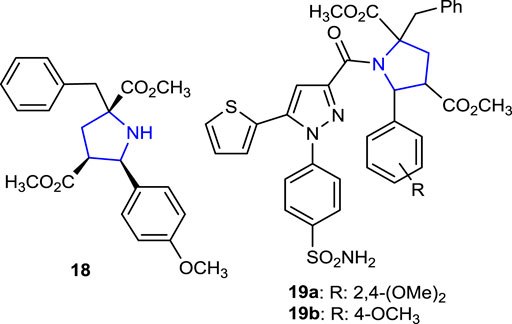
Recently, our research group designed N-benzoylthiourea-pyrrolidine carboxylic acid derivative compounds carrying a series of imidazole rings as cholinesterase inhibitors, and their acetylcholinesterase (AChE) and Butyrylcholinesterase (BuChE) inhibition properties were compared with those of the reference tacrine. The most active compounds were found to be phenyl- and methyl-substituted 15g (IC50: 0.029 µM), 15h (IC50: 0.041 µM), and 16g (IC50: 0.087 µM) with an indole ring in their structures (Poyraz et al., 2023a). Moreover, as part of our ongoing research work related to pyrroline-based potential bioactive molecules, we recently reported the design and synthesis of novel pyrrolidine-based benzenesulfonamide derivatives as carbonic anhydrase/acetylcholinesterase inhibitors, together with their antimicrobial properties. Thus, compounds 19a and 19b bearing 2,4-dimethoxyphenyl and 4-methoxyphenyl substituents were the most promising AChE inhibitor candidates with the Ki values of 22.34 ± 4.53 nM and 27.21 ± 3.96 nM, respectively, compared to tacrine inhibition. Compound 18 without a sulfonamide moiety showed significant inhibition of the hCAI enzyme, with a Ki value of 17.61 ± 3.58 nM, approximately 10 times greater than that of the reference AZA (164.22 ± 14.13 nM), and for the hCAII enzyme, with a Ki value of 5.14 ± 0.61 nM, approximately 26 times greater than that of the reference AZA (132.53 ± 7.44 nM) (Poyraz et al., 2023a).
The pyrrolidine core is part of the general polycyclic framework 20, a potential bioactive compound for the treatment of cystic fibrosis (CF). In this sense, more sophisticated analogs 21 containing this polycycle 20 can be classified as promising drugs for this purpose. Some studies regarding herbicide and pesticide activities, as well as surveys for plant growth control, are under investigation (Belabbes et al., 2023).
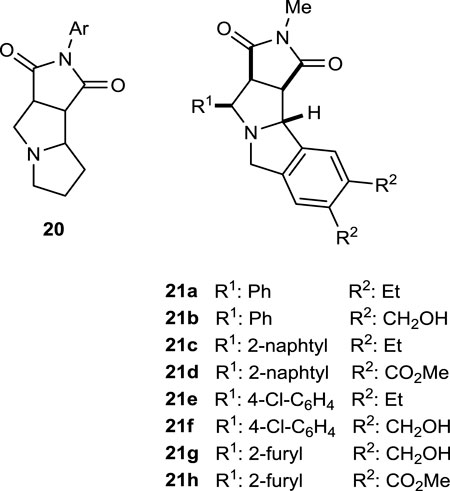
Frejat et al. (2022) synthesized 1,2,4-oxadiazole pyrrolidine derivatives and tested their antibacterial activity against different bacterial strains by inhibiting DNA gyrase and topoisomerase IV which are important targets for antibacterial agents. Compounds 22a (180 ± 20 nM), 22b (210 ± 20 nM), 22c (120 ± 10 nM), 22d (250 ± 20 nM), and 22e (270 ± 20 nM) inhibited E. coli DNA gyrase at concentrations similar to the standard novobiocin (IC50 = 170 nM). Notably, the 4-chlorophenyl group is present in the structures of three (22b–d) of these five active molecules. Likewise, compounds 22a, 22b, 22d, and 22e showed activity close to that of novobiocin (IC50 = 11 μM and 27 µM, respectively) against E. coli and S. aureus topoisomerase IV, while compound 22c (IC50 = 3.07 µM and 8.2 µM, respectively), containing 4-chlorophenyl substituents on the pyrrolidine ring and 5-hydroxymethyl furan moiety on the 1,2,4 oxadiazole group, was more active than the reference. Compound 22c could be considered the lead compound for future research.
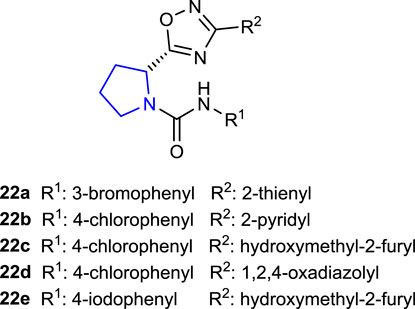
Salve and Jadhav (2021) synthesized novel pyrrolidine sulfonamide derivatives as DPP-IV inhibitors and compared their in vitro antidiabetic effects with those of vildagliptin as a control. Compounds 23a–d showed 56.32%, 44.29%, 49.62%, and 66.32% inhibition, respectively. Compound 23d with the 4-trifluorophenyl substitution on the 1,2,4-oxadiazole group exhibited the best inhibition (IC50: 11.32 ± 1.59 μM) against the DPP-IV enzyme among these compounds.
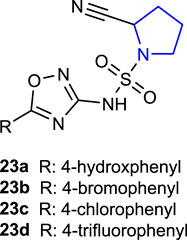
Sirin et al. (2021) synthesized the pyrrolidine-substituted 3-amido-9-ethylcarbazole derivative and performed an activity screening. Compound 24 showed an antiproliferative effect against HT-29 and SH-SY5Y cells, acetylcholinesterase inhibition activity, and antioxidant activity.
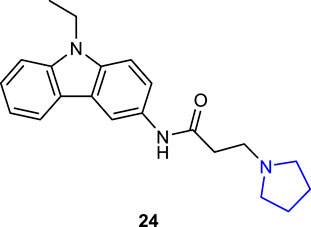
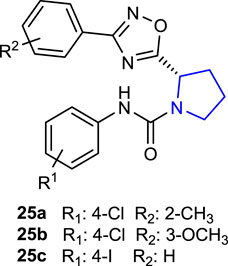
Ruan et al. (2020) designed and synthesized pyrrolidine oxadiazoles, which could be used as new anthelmintic drug candidates due to their resistance to over-used anthelmintic drugs against parasitic roundworms and reported their activity against the worm Haemonchus contortus. Compounds 25a (2-CH3) and 25b (3-OCH3) were discovered to be promising candidates against xL3 motility (IC50 = 0.78 µM for each), whereas compound 25c (4-I) was identified as an efficient inhibitor of L4 development (IC50 = 3.2 µM). Moreover, these compounds exhibited good selectivity against mammalian epithelial cells with limited inhibition of cell proliferation at 50 mM.
Martinez-Bailen et al. (2020) synthesized multimeric pyrrolidine iminosugars as inhibitors of important therapeutic enzymes, human β-glucocerebrosidase (GCase) and α-galactosidase A (α-Gal A), and tested them as a multivalent enzyme activity enhancer for Fabry disease. One of the nonavalent compounds showed 375-fold higher inhibition (0.20 µM) of the α-Gal A enzyme than the monovalent reference. In R301G fibroblasts from Fabry disease patients, the compound that increases the activity of the misfolded enzyme by 5.2-fold at 2.5 µM could be a candidate for a polyvalent α-Gal A activity enhancer.
Li et al. (2020) designed and synthesized pyrrolidine-containing derivatives as antagonists of the chemokine receptor CXCR4, which is responsible for essential activities in HIV infection, inflammation/autoimmune disorders, and cancer metastasis, and investigated their in vivo anticancer metastatic potential. Compound 26, which contains pyridine, piperazine, and pyrimidine, together with the pyrrolidine ring, was identified as a CXCR4 antagonist with strong binding affinity to the CXCR4 receptor (IC50 = 79 nM) and a capability to inhibit CXCL12-induced cytosolic calcium flux (IC50 = 0.25 nM). In addition, to its favorable in vitro safety profile, compound 7 also demonstrated significantly enhanced metabolic stability in human and rat liver microsomes, as well as remarkable efficacy in a mouse cancer metastasis model. According to the experimental findings, compound 7 appeared to be promising in the development of CXCR4 antagonists.
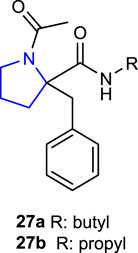
Sreekanth and Jha (2020) synthesized 1-acetyl-2-benzylpyrrolidine-2-carboxylic acid and its derivatives and evaluated their antimicrobial activities. Antibacterial activity studies were performed against some Gram-negative (E. coli and P. aeruginosa) and Gram-positive (S. aureus and B. subtilis) bacterial strains and compared with the reference antibiotic chloramphenicol. Compound 27a, containing a butyl substituent, had the same effect (MIC: 16 μg/mL) against the Gram-positive bacteria S. aureus and B. subtilis using chloramphenicol as the reference, whereas compound 27b, containing a propyl substituent, had the same effect (MIC: 16 μg/mL) against S. aureus bacterial strains using chloramphenicol as the reference. Antifungal activity studies were conducted against the Candida albicans strain using ketoconazole as a reference. Compound 27a (MIC: 32 μg/mL), which is also the most active compound against the C. albicans strain, could be considered a promising molecule for further studies.
The pyrrolidine derivatives 15c–f, bearing an indole moiety at the R2 position and several aromatic substituents at the R3 position in their structure, and compound 17, obtained by ring arrangement exhibited twice the activity (MIC: 62.5 μg/mL) of the reference ampicillin against the A. baumannii strain (Belveren et al., 2019).
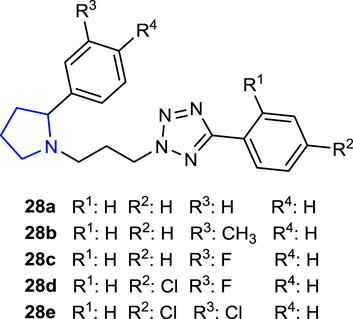
Since tetrazole derivatives are already known to have powerful antifungal properties, Lukowska-Chojnacka et al. (2019) synthesized tetrazoles bearing a pyrrolidine ring and tested them as prospective antifungal agents against C. albicans. While compound 28b (-CH3 substituted) was determined to be the most effective antifungal agent, randomly selected compounds 28a, 28d (-Cl, -F), and 28e (-Cl, -Cl) exhibited little to no toxicity against the Vero cell line and Galleria mellonella. In the invertebrate model of disseminated candidiasis, compounds 28b and 28c (-F) showed the highest effectiveness against biofilm formation in vitro and also exhibited activity in vivo. Mitochondrial damage (XTT assay) and decreased adherence to the TR-146 cell line at 46.05 µM demonstrated necrotic cell death via fungal membrane interaction of compound 28b.
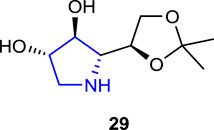
Guazzelli et al. (2019) synthesized polyhydroxylated pyrrolidine derivatives containing D-gluco- and D-galacto- units, which have the potential to inhibit glycosidase and aldose reductase enzymes and play an important role in the treatment of diabetes, a multifactorial disorder, and assessed their inhibitory properties. Among the synthesized pyrrolidine derivatives, compound 29 (57% inhibition) showed the best inhibitory property against ALR2 on an in vitro model of diabetic retinopathy. Furthermore, compound 29 decreased cell death processes in the photoreceptor-like 661w hyperglycemic cell line and restored physiological levels of oxidative stress, decreasing ALR2-associated ROS production.
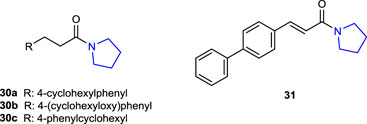
Zhou et al. (2019) synthesized a series of pyrrolidine hybrid molecules by changing the substituents at the terminal phenyl groups of pyrrolidine amide derivatives and tested their inhibitory properties against N-acylethanolamine acid amidase (NAAA), one of the key enzymes involved in the degradation of fatty acid ethanolamides (FAEs), specifically palmitoylethanolamide (PEA). Compounds 30a (4-cyclohexylphenyl; IC50: 0.5 ± 0.1 μM), 30b (4-(cyclohexyloxy)phenyl); IC50: 0.7 ± 0.2 μM), 30c (4-phenylcyclohexyl; IC50: 0.48 ± 0.11 μM), and 31 (4-phenylphenyl; IC50: 1.5 ± 0.22 μM) displayed potent inhibitory activity toward NAAA but had no such effect toward FAAH. Compound 31 showed good selectivity (IC50 for NAAA: 1.5 ± 0.22 μM and for FAAH: 550 ± 70 μM) with the strongest inhibition by reversible and competitive mechanisms against the NAAA enzyme as well as high in vitro anti-inflammatory activity in the lipopolysaccharide (LPS)-induced acute lung injury (ALI) model.
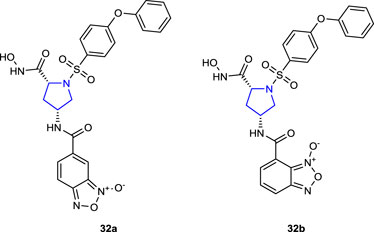
Zhang et al. (2018) designed and synthesized a series of benzofuroxane pyrrolidine hydroxamates as matrix metalloproteinase (MMP) inhibitors with nitric oxide-releasing activity and evaluated their antiproliferative activity. Among the target compounds, compounds 32a (IC50 = 102 ± 31.4 and 162 ± 10.5 nM) and 32b (IC50 = 182 ± 25.2 and 242 ± 29.2 nM) with 4-phenoxyphenylsulfonyl groups in their structures inhibited MMP-2 and MMP-9 enzymes better than control LY52 (IC50 = 266 ± 19.2 and 360 ± 30.1 nM). Meta-substituted benzofuroxane molecules exhibited more inhibitory activity than the ortho-substituted molecules. Furthermore, compounds were tested against A549, ES-2, HeLa, K562, MCF-7, and MDA-MB-231 cancer cells, and the findings were compared to those of the positive control LY52. Compound 32a, which demonstrated the highest enzyme activity, also demonstrated an antiproliferative effect against HeLa cells, roughly 60 times greater than that of the control LY52 with an IC50 value of 3.82 ± 0.11 µM. All compounds, but 32a (34.43 μM/L) in particular, released moderate quantities of NO.
In another study, while N-benzoylthiourea-pyrrolidine derivatives containing N-phenyl 16b and N-methyl 16d groups were found to be four times more active against the A. baumannii strain and twice more active against A. hydrophila than the reference. Compound 16c, containing the 4-chlorophenyl group, showed better antituberculosis (anti-TB) activity than the reference ethambutol. Compounds 15a and 15b, which are among the pyrrolidine derivatives used in the synthesis of the corresponding compounds, also showed four times higher activity (MIC: 31.25 μg/mL) against A. baumannii than the reference ampicillin (MIC: 125 μg/mL) (Poyraz et al., 2018).
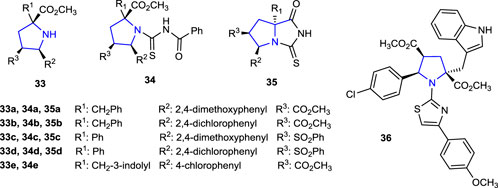
The anti-TB activities of the thiohydantoin-pyrrolidine derivatives 35a–d and the compounds 33a–d and 34a–d, used in the synthesis to access these molecules 35, were tested against Mycobacterium tuberculosis using the microplate Alamar Blue assay (MABA) method. Compared to the reference isoniazid and ethambutol, the synthesized compounds did not exhibit significant anti-TB activity, while the activities of the compounds were found in the range of 62.5–125 μg/mL against the Mycobacterium tuberculosis H37Rv strain (Poyraz et al., 2017).
Compound 33e (MIC: 1.95 μg/mL), which is one of the pyrrolidines synthesized for the preparation of N-benzoylthiourea derivatives used in the synthesis of thiazole-pyrrolidine derivatives and contains 4-chlorophenyl and indole groups in its structure, showed better anti-TB activity against Mycobacterium tuberculosis than the reference ethambutol (5 and 10 μg/mL). The anti-TB activity of compound 36 (MIC: 0.24 μg/mL), which was obtained by condensation of compound 33e with benzoyl isothiocyanate, was found to be eight times more potent than that of the starting compound 34e and better than that of the reference anti-TB drugs isoniazid and ethambutol. In addition, the anti-TB activity of compound 36, obtained by the addition of 4-bromomethoxy acetophenone to compound 34e, was found to be 0.48 μg/mL. Moreover, the antimicrobial activities of the compounds were tested against S. aureus, E. coli, A. baumannii, B. subtilis, and A. hydrophila strains. Compound 15c, which contains an indole heterocycle, together with a fused bicyclic pyrrolidine ring, was twice more active against A. baumannii than the reference antibacterial agent ampicillin (MIC: 125 μg/mL) with a MIC value of 62.5 μg/mL (Belveren et al., 2017).
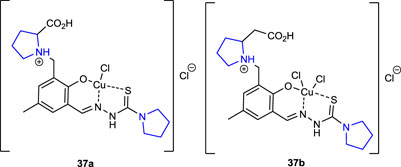
Dobrova et al. (2016) synthesized nickel(II), palladium(II), and copper(II) complexes of pyrrolidine thiosemicarbazone hybrids, evaluated their in vitro anticancer potential against A549 (lung cancer), CH1 (human ovarian carcinoma), and SW480 (colon cancer) cancer cell lines and noncancerous murine embryonal fibroblasts (NIH/3T3), and studied the cell death mechanism using the flow cytometry method. The outcomes were evaluated against a cisplatin control group. The thiosemicarbazone pyrrolidine–copper(II) complexes, with two pyrrolidine rings in their structure, were the most potent anticancer compounds against three cancer lines among the synthesized ligands and complexes. Copper complex 37a (IC50: 0.99 ± 0.09) was approximately three times more potent than cisplatin used as a control (IC50: 3.5 ± 0.3) against the SW480 cancer cell line, while copper complex 37b (IC50: 3.7 ± 0.1) showed approximate activity to cisplatin. The results of the research revealed that altering the coordinating groups of the ligands enhanced their antiproliferative activity.
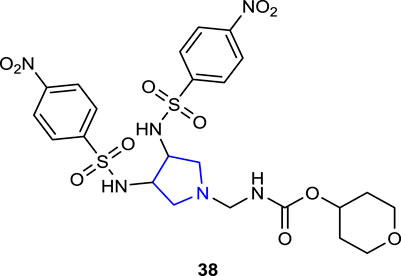
Kumar et al. (2016) synthesized sulfonylamino pyrrolidine derivatives and evaluated their in silico EGFR potential as well as their antibacterial and antifungal activities. Antibacterial activity studies were compared with those of reference cefaclor against S. aureus, E. coli, and P. aeruginosa bacteria. Compound 38, which contains two nitrophenyls together with the pyran ring as a substituent in its structure, showed the best activity against three bacteria [S. aureus (MIC: 3.11 μg/mL), E. coli (MIC: 6.58 μg/mL), and P. aeruginosa (MIC: 5.82 μg/mL)]. Antifungal activity studies were also performed against A. niger and C. albicans fungal strains using ketoconazole as the reference. Compound 38 also showed the highest activity against the respective fungal strains [A. niger (MIC: 1.78 μg/mL) and C. albicans (MIC: 3.18 μg/mL)]. Compared to the reference, in silico studies showed that the compounds could serve as EGFR inhibitors.
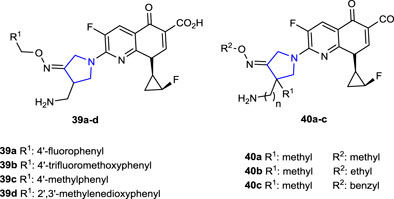
Huang et al. (2015) synthesized a series of 1-[(1R,2S)-2-fluorocyclopropyl]naphthyridone derivatives containing oxime-functionalized pyrrolidine and evaluated their antimicrobial activities. The antituberculosis (TB) activity of pyrrolidine derivatives was compared with that of the reference drugs isoniazid and rifampicin using the MABA method against the Mycobacterium tuberculosis (MTB) H37Rv ATCC 27294 (MTB-1) strain. Compounds 39a, 39c, and 39d, which contain 4-F, 4-CH3, and 2′,3′-methylenoxyl substituents, respectively, were found to be the most active compounds against the MTB-1 strain with a MIC value of <0.25 μg/mL. In addition, these most active compounds against the clinically isolated MDR-MTB 6133 (MTB-2) strain resistant to isoniazid and rifampicin showed much better anti-TB activity than the references (MIC: 0.043, 0.03, and 0.054 μg/mL, respectively). The antibacterial activity of the compounds was tested against a series of Gram-negative (E. coli, K. pneumoniae, and P. aeruginosa) and Gram-positive (S. aureus, MSSA: methicillin-sensitive S. aureus, MRSA: methicillin-resistant S. aureus, MSSE: methicillin-sensitive S. epidermidis, MRSE: methicillin-resistant S. epidermidis, S. pneumoniae, E. faecalis, and E. faecium) strains. Ciprofloxacin (CPFX), levofloxacin (LVFX), and moxifloxacin (MXFX) were used as references. Compounds 39b, 39c, and 40a–c showed better activity (MIC: <0.008–32 μg/mL) against Gram-positive bacteria than against Gram-negative bacteria than the references (MIC: 0.125–>128 μg/mL).
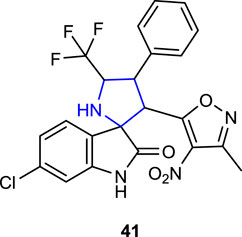
Liu et al. (2021) synthesized spiropyrrolidine oxindole derivatives carrying nitroisoxazole as novel GPX4/MDM2 inhibitors inhibiting breast cancer cell proliferation. Among the synthesized compounds, compound 6-Cl-substituted 41 (Ki: 0.24 ± 0.06 µM) exhibited the highest activity in suppressing MDM2-mediated p53 degradation as well as GPX4 levels in MCF-7 breast cancer cells.
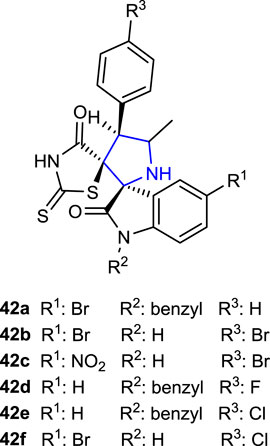
Toumi et al. (2021) designed rhodanine-substituted spirooxindole pyrrolidine compounds and tested their ability to inhibit the α-amylase enzyme, which is related to diabetes. While almost all compounds inhibit the α-amylase enzyme at a certain level, the compounds 42c (IC50: 1.59 ± 0.08 μg/mL), 42d (IC50: 1.67 ± 0.09 μg/mL), 42e (IC50: 1.63 ± 0.09 μg/mL), and 42f (IC50: 1.57 ± 0.10 μg/mL), bearing R1: NO2, H, H, and Br; R2: H, benzyl, benzyl, and H; and Ar: Br, F, Cl, and Cl substituents, respectively, showed approximately the same level of inhibition as the reference acarbose (IC50: 1.56 ± 0.09 μg/mL). Compound 42a (IC50: 1.49 ± 0.10 μg/mL), with a 4-bromo substituent on the isatin ring in its structure, was more active than the standard acarbose. In subsequent investigations of in vivo hypoglycemic activity, 42a–c and 42f were the most active compounds that decreased the blood glucose levels. The findings suggested that N-substituted groups attached to isatin and substituents on the phenyl ring may both affect enzyme function.
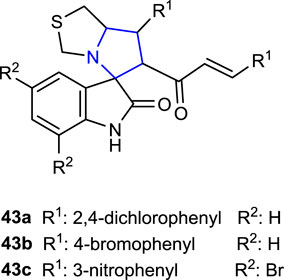
Islam et al. (2020a) synthesized two series of spiropyrrolidine-thiazolo-oxindole derivatives, namely, dibromo-substituted and non-substituted oxindole, and evaluated their anticancer activity against HepG2, MCF-7, and HCT-116 cell lines. The results were compared to those of cisplatin and reported. Thus, compounds 2,4-dichlorophenyl-substituted 43a (IC50: 0.85 ± 0.20 μg/mL) and 4-bromophenyl-substituted 43b (IC50: 0.80 ± 0.10 μg/mL) were discovered to be roughly 11 times more active against the HepG2 cell line than the reference (IC50: 9.00 ± 0.76 μg/mL) among the non-substituted oxindole compounds. In addition, compound 43a (IC50: 2.00 ± 0.60 μg/mL) was more active than the reference compound against the HCT-116 cell line, whereas compound 43b (IC50: 3.00 ± 0.50 μg/mL) displayed the same activity. One of the dibromo-substituted oxindole derivative compounds, 3-NO2-substituted 43c (IC50 HepG2: 5.00 ± 0.66, IC50 MCF-7: 4.00 ± 0.29, and IC50 HCT-116: 2.80 ± 0.20 μg/mL), showed higher activity against all three cell lines than the reference, as well as approximately twice better activity than cisplatin (IC50: 9.00 ± 0.29 μg/mL) against the MCF-7 cell line.
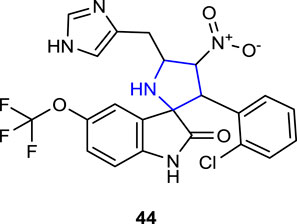
Bolous et al. (2019) designed and synthesized spirooxindole pyrrolidine-linked indole and imidazole heterocyclic hybrids and evaluated the advanced antifungal activity against clinically isolated fungal strains for compound 44, which was found to be the most active antifungal agent after broad-spectrum antifungal screening. In the tests, compound 44 containing OCF3 and 2-Cl was found to be the most active compound against the C. albicans strain with a MIC value of 4 μg/mL. Compound 44 was then tested against pathogenic strains of C. albicans, C. glabrata, C. tropicalis, C. parapsilosis, and Cryptococcus neoformans isolated from humans. These fluconazole-resistant organisms were inhibited by compound 44. In addition, compound 44 reduced the activity of the C. albicans strain, which gained resistance to antifungal agents by forming filaments and biofilms, by up to 25% at concentrations of 32 μg/mL and 64 μg/mL. The MTS assay was also used to determine the non-toxicity of compound 44 to the HCT116 mammalian cell line in vitro.
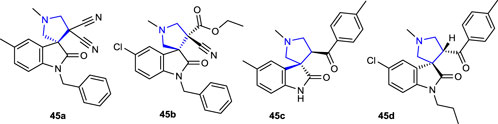
Huang et al. (2018) synthesized a series of spirooxindole pyrrolidine derivatives and evaluated their antibacterial activities against E. coli and S. aureus and inhibitory potency against the AChE enzyme. The most active compounds against E. coli bacteria were compounds 45b (R1: Cl and R2: CH2Ph) and 27c (R1: CH3, R2: H, and Ar: 4-CH3C6H4). The compounds did not show significant activity against the bacterial strains. Compounds 45a (R1: Cl and R2: CH2Ph; 94.66% ± 3.26%) and 45d (R1: Cl, R2: butyl, and Ar: 4-CH3C6H4; 64.95% ± 1.64%) showed the strongest inhibition against the AChE enzyme compared to the reference tacrine. The IC50 values of compounds 45a and 45d were found to be 69.07 ± 10.99 µM and 111.38 ± 10.11 µM, respectively.
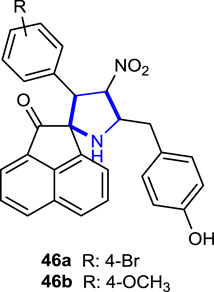
Kumar et al. (2018) synthesized a series of spiropyrrolidine analogs in a stereoselective manner and evaluated their anticancer effects against cancer and non-cancer cell lines. In anticancer activity studies, the mechanism of action of the compounds was investigated using non-cancer (PCS-130-010 and BRL-3A) and cancer (A549 and Jurkat cell types) cell lines and compared with the reference anticancer drug camptothecin (CPT) for 24- and 48-h periods. The compounds showed no significant loss of cell viability and were not toxic to non-cancer cells. Compound 46a with a 4-Br substitution was the most active, whereas compound 46b with a 4-OCH3 substituent was the least active. Moreover, cell death was noted to be an apoptotic pathway mediated by the increased activation of caspase-3 proteins, while halogen-substituted pyrrolidines were found to be more active than the others.
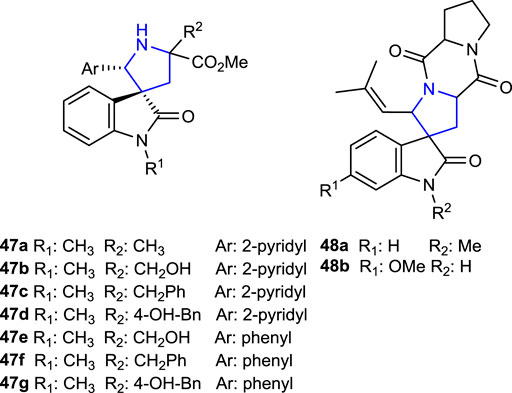
Millington et al. (2018) synthesized spirotryprostatin analogs 47a–g and 48a, b, known for their anticancer properties and containing a spirooxindole structure, by metal-catalyzed imine azomethine ylide cycloaddition reactions following by the Heck reaction.
Bharkavi et al. (2016) developed dispiro indeno pyrrolidine derivatives and tested their AChE inhibition, antituberculosis, and anticancer activity. Compound 49c (IC50: 1.07 μg/mL), which contains a 2,4-dichlorophenyl group, was found to be approximately 12 times more active than the reference cycloserine (IC50: 12.47 μg/mL) and 36 times more active than the reference pyrimethamine (IC50: 37.35 μg/mL) against Mycobacterium tuberculosis H37Rv. In addition, compound 49a (IC50: 55.79 ± 2.60 μg/mL), containing a 4-fluorophenyl substituent, exhibited the same activity as the standard doxorubicin (IC50: 55.91 ± 1.34 μg/mL) against CCRF-CM cell lines but did not show significant activity against HT29 and MCF7 cell lines. Moreover, compound 49b (IC50: 1.16 ± 0.1 μmol/L), containing 4-methoxyphenyl, was the most active compound against the AChE enzyme compared to the reference donepezil.

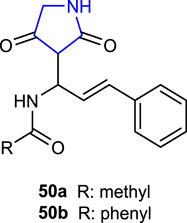
Al-Zharani et al. (2022) synthesized pyrrolidine-2,4-diones and evaluated mosquito larvicidal activities against Culex quinquefasciatus and compared with the reference permethrin. Compounds 50a (LD50 = 26.06 μg/mL), containing acetamide in its structure, and 50b (LD50: 26.89 μg/mL), containing a benzamide moiety, showed larvicidal activity comparable to the reference (LD50 = 26.14 μg/mL). The experimental results and the highest docking scores for compounds 50a and 50b against the 3OGN protein are consistent.
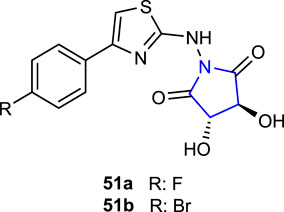
Kocabaş et al. (2021) synthesized a series of pyrrolidine-thiazole derivatives and evaluated their antibacterial activities and cytotoxicity. The antibacterial activities of the compounds were tested using the microdilution method against E. coli, S. typhimurium, B. cereus, and S. aureus strains, and their cytotoxicity was tested on L929 cells using the XTT method. Compound 51a (MIC: 21.70 ± 0.36 and 30.53 ± 0.42 μg/mL), containing the 4-fluorophenyl substituent attached to the thiazole ring, was compared with the reference gentamicin (MIC: 22.65 ± 0.21 and 22.17 ± 0.47 μg/mL), showing the best activity against B. cereus and S. aureus. In addition, compounds 51a, b, containing 4-bromophenyl and 4-fluorophenyl substituents, respectively, did not show toxicity at the concentrations performed.
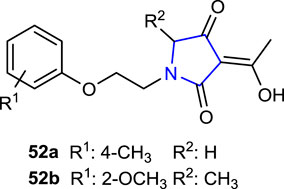
Chen et al. (2021) incorporated natural herbicide tetramic acids bearing pyrrolidine-2,4-dione with ether structures containing alkoxyalkyl and phenoxyethyl groups, which are widely known for their herbicide, fungicidal, and insecticide effects, into a single molecule and immediately evaluated their herbicidal activities using the Petri dish culture method in barnyard grass (Echinochloa crus-galli) and rape (Brassica campestris) model plants. Compounds 52a (84%), with a 4-CH3 substituent, and 52b (65.6%), with 2-OCH3, showed the highest inhibition rates.
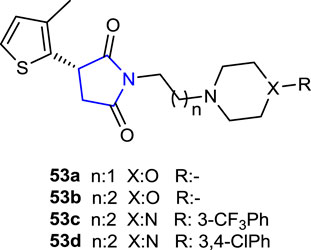
Góra et al. (2020) synthesized a series of 3-(3-methylthiophen-2-yl)-pyrrolidine-2,5-dione hybrids and evaluated their anticonvulsant and antinociceptive activities in vivo in animal models using MES, scPTZ, and psychomotor tests (6 Hz). The effects of compounds 53, which include 2-morpholinoethyl, 3-morpholinopropyl, 3-CF3Ph, and 3,4-dichlorophenyl substituents in their structures, on GABA transporters, sodium, and calcium channels were studied to determine the anticonvulsant action mechanisms. Compound 53b was the most active compound, with an ED50 of 62.14 mg/kg in the MES test and 75.59 mg/kg in the 6 Hz test compared to the reference valproic acid and ethosuximide. In addition, the hot plate and writhing tests evaluated the antinociceptive activities of these active compounds. The results showed that compound 53d demonstrated an analgesic effect comparable to that of aspirin at a dose of 30 mg/kg in the writhing test, whereas compounds 53c and 53d demonstrated central analgesic activity at the same dose in the hot plate test.
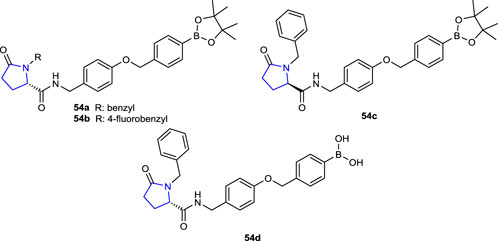
Gerokonstantis et al. (2020) synthesized 2-pyrrolidinone and pyrrolidine derivatives and evaluated the enzyme inhibition activities of autotaxin (ATX) isolated from melanoma cells, which are responsible for various pathological inflammations such as fibrosis, cancer, liver toxicity, and thrombosis due to hydrolysis of lysophosphatidylcholine (LPC). Among the compounds, compounds 54a (IC50: 0.05 µΜ), 54b (IC50: 0.12 µΜ), and 54c (IC50: 0.18 µΜ) (of which 54a, b, d had S configuration and 54c had R configurations), bearing benzyl, 4-fluorobenzyl, and benzyl, respectively, and 54d (IC50: 0.035 µΜ), which are boronic acid derivatives, could be considered the most potent ATX inhibitors. Compound 54d (IC50: 0.035 µΜ) with S configuration was the most active among these compounds.
Pyrrolidine alkaloids refer to five-membered nitrogen-containing heterocyclic ring systems and exhibit a wide variety of biological and pharmacological activities. Because of the versatile biological importance of pyrrolidine alkaloids, there are many reports and excellent reviews dealing with the source of these alkaloids, together with the chemistry and biological importance of pyrrolidine-type alkaloids 55–58 (Figure 3) (O'Hagan, 2000; Ibrahim SRM, 2015; Hosseinzadeh et al., 2018; Islam and Mubarak, 2020b; Jeelan Basha et al., 2022; Bhat et al., 2023b). Some of the pyrrolidine alkaloids known for their neurological and antioxidant effects (e.g., habbemines A and B and nicotine), together with a potential protective agent for newly prepared nicotine derivatives, were reported by Malczewska-Jaskola et al. (2016) against in vitro oxidative hemolysis and morphological injury of human erythrocytes.
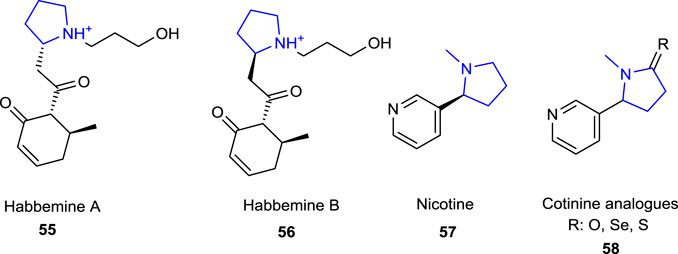
FIGURE 3. Naturally occurring pyrrolidine alkaloids.
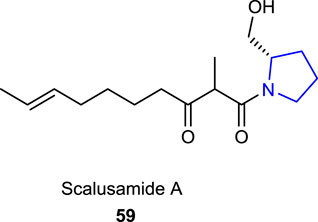
A marine-originated pyrrolidine alkaloid, scalusamide A 59, reported by Tsuda et al. (2005), which was obtained from the gastrointestinal tract of a marine fish, isolated from the cultured broth of the fungus Penicillium citrinum, and characterized by analytical techniques, was screened for its antifungal and antibacterial activities, which showed potential inhibition when tested against Penicillium brevicompactum, Cryptococcus neoformans, and Micrococcus luteus.
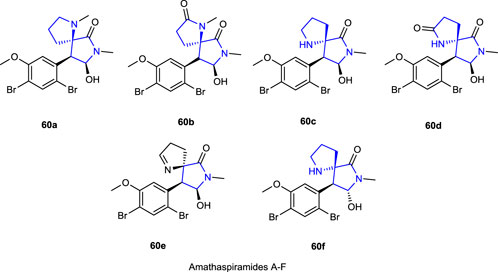
Some spiro-based structural skeleton pyrrolidine alkaloids and amathaspiramide analogs A-F 60 were isolated from marine bryozoan Amathia wilsoni by Morris and Prinsep (1999), and Shimokawa et al. (2016) studiedtheir antiproliferative activity against HCT116, Pc-3, MV4-11, and MiaPaCa-2 cell lines.
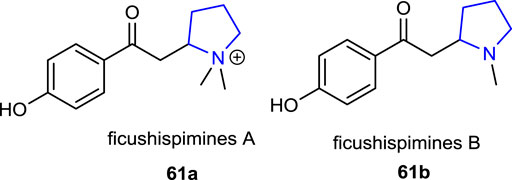
Shi et al. (2016) reported some biologically active new pyrrolidine alkaloids, ficushispimines A 61a and B 61b, which were isolated from the twigs of Ficus hispida, exhibiting glucosidase inhibitory effects.
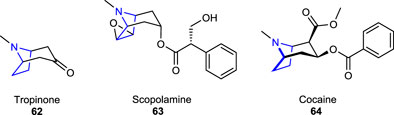
One of the pharmacologically and biologically important pyrrolidine alkaloids is the drug named anisomycin 6 (also known as flagecidin). It is an antibiotic isolated from various Streptomyces species (e.g., produced by bacteria Streptomyces griseus and Streptomyces hygrospinosus). This naturally occurring monohydroxypyrrolidine alkaloid acts to inhibit bacterial protein synthesis. It is used as an antiparasitic, antimicrobial, antineoplastic, and antifungal agent and as a DNA and protein synthesis inhibitor (blocks peptide bond formation). Anisomycin 6 has also been used for the treatment of trichomoniasis and amoebic dysentery and has shown in vitro potential antitumor activity against human tumor cell lines (Zheng et al., 2017).
Other biologically and pharmaceutically important tropane alkaloids, belonging to the pyrroline classes of plant alkaloids (subgroup of pyrrolidine), result from metabolic diversity and occur in phylogenetically distinct plant families that possess the tropane core structure [e.g., tropinone 62, scopolamine 63, and cocaine 64] (Kim et al., 2016). The anticholinergic drug scopolamine, a tropane alkaloid that could be obtained naturally or synthetically, was previously prescribed to treat motion sickness and postoperative nausea and vomiting (Apfel et al., 2010). Tropinone alkaloid, synthesized as a synthetic precursor of atropine, has the same tropane core structure as cocaine and atropine (Drager, 2006).
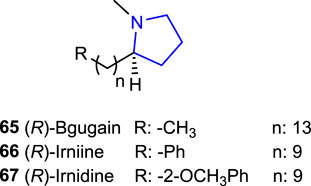
2-Alkylpyrrolidine derivatives [(R)-bgugaine 65, (R)-irnidine 66, and (R)-irnidine 67] are versatile pyrrolidine alkaloids isolated from the tubers of Arisarum vulgare, which revealed significant bioactivities such as apoptosis, cytolysis effects, and antimycotic, antimitotic, antibacterial, and antifungal activities (Melhaoui et al., 1992; Melhaoui et al., 1993; Melhaoui and Belouali, 1998; Rakba et al., 1999; Lamkadem et al., 2004; Majik et al., 2008), and cytotoxic effects and showed hepatotoxin activity in rat and human liver cell culture (Rakba et al., 1999). In addition, some of the (R)-enantiomers of these alkylpyrrolidine alkaloids have been reported to possess a binding affinity for DNA, together with induced significant DNA damage in a human hepatoblastoma (HepG2) cell line (Melhaoui, 1998; Rakba et al., 1999), and revealed electrophysiologic activity against MRC-5 fibroblasts at 10 µM (2.81 μg/mL) (Lamkadem et al., 2001).
Some naturally occurring pyrrolidine dicarboxylate derivatives, known as kainoids, exhibit both excitatory and excitotoxic activity and are used as pharmacological probes. Kainic acid/kainite 68, one of the kainoids which was first isolated from the seaweed Digenea simplex (Chekan et al., 2019), is a potent neuroexcitatory amino acid agonist that acts by activating receptors for glutamate as a neurotransmitter in the central nervous system (Carlson, 2013).
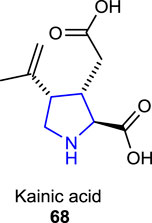
A naturally occurring neurotoxin, domoic acid 69, an analog of kainic acid-type pyrrolidine, which is produced by the alga called Pseudo-nitzschia australis, affects the brain and causes seizures and memory impairment and acts as a potent activator of the kainic receptors in the CNS (Clayton et al., 1999; Carcache et al., 2003). Some congeners of domoic acid (isodomoic acids) 70–73 were isolated from a red alga called Chondria armata, and their structures were deduced to be (E,E)- and (Z,E)-isomers of 2-carboxy-4-(5-carboxy-l-methyl-2-hexenylidene)-3-pyrrolidineacetic acid 74 after their structural determination (Zaman et al., 1997).
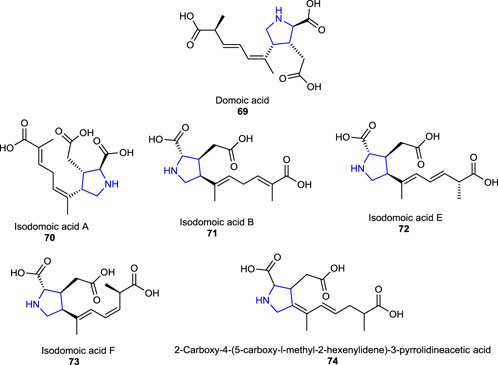
Some of the other pyrrolidine dicarboxylate derivatives, known as acromelic acids (A-C) 75–77, bearing a pyridone moiety, were isolated from a poisonous mushroom Clitocybe amoenolens/Clitocybe acromelalga and act as a neurotoxin, together with a role as an NMDA (N-methyl-D-aspartic acid) receptor agonist. Acromelic acid (C) was found to exhibit a lethal toxic effect on mice at a dose of 10 mg/kg (Fushiya et al., 1990; Carcache et al., 2003).
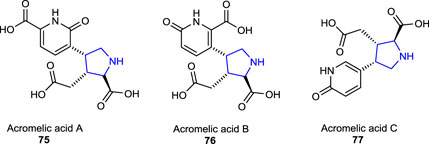
Recently, two tetracyclic functionalized pyrrolidine-type alkaloids, senecipyrrolidine 78 and emiline 79, were isolated from Jacobaea gigantea (Jacobaea/Senecio genera) (Asteraceae) by Mezache et al. (2019) and characterized using analytical techniques. The herbal plant J. gigantea is also known as an endemic herb that was found to grow in Algeria, Morocco, and Tunisia.
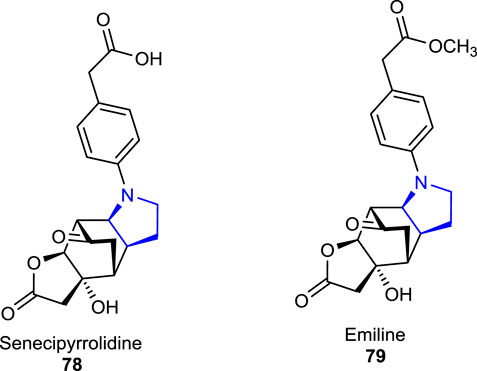
Three other naturally occurring pyrrolidine alkaloids, namely, acanthophoraine A 80, acanthophoraine B 81, and acanthophoraine C 82, were isolated from a red alga called Acanthophora spicifera, and their antibacterial activities were screened against the bacterial strains, including E. faecalis, S. aureus, P. aeruginosa, K. pneumoniae, A. baumannii, and E. coli, and no desirable effects were found on the tested bacteria (Lin et al., 2020; Guan et al., 2021).
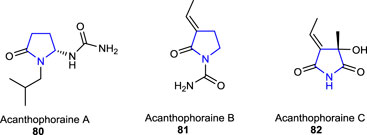
The hygrolines 83 and 84 and pseudohygrolines 85 and 86 are 2-substituted pyrrolidine alkaloids with two chiral centers constituting a 1,3-aminoalcohol unit, isolated from Solanaceae (S. tricolor), Schizanthus hookeri, Carallia brachiate, and genera Schizanthus and Erythroxyloncoca. These structurally simple 2-substituted naturally occurring pyrrolidine alkaloids are important pharmacophores due to their intriguing biological and pharmacological activity. Some of its derivatives have been found to show significant hallucinogenic characteristics and displayed antitrypanosomatid and antiplasmodial activities (Cretton et al., 2017; Christen et al., 2020; Cretton et al., 2021).
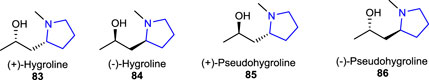
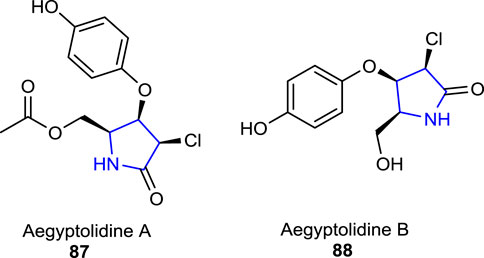
Ibrahim SRM (2015) reported potentially bioactive aegyptolidine A 87 and B 88 pyrrolidine alkaloids which were isolated from Aspergillus aegyptiacus (a species of the fungus in the genus Aspergillus). The studies have shown that these two alkaloids exhibited weak cytotoxic activity against the murine lymphoma cancer cell (L5178Y) line.
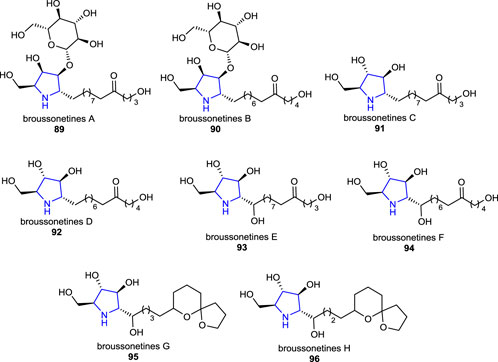
There are many structurally identified polyhydroxy pyrrolidine alkaloids that possess glycosidase inhibitory properties, which have been isolated from natural sources (Watson et al., 1997; Shibano et al., 1998; Elbein and Molyneux, 1999; Shibano et al., 1999; O'Hagan, 2000). Some of the well-known extensively studied representative examples of these alkaloids are broussonetines 89–96 (Broussonetia species), mainly isolated from the branches of Broussonetia kazinoki Sieb. (Moraceae). Shibano et al. reported the isolation, structural determination, and bioactivities of these series of pyrrolidine alkaloids called broussonetines A–H 89–96 and screened them for their inhibitory activities on some glycosidases (Shibano et al., 1997; Shibano et al., 1998).
This review focuses on the current progress in pyrrolidine derivatives, which show multiple pharmacological activities and functions in the treatment of several health disorders. Pyrrolidine-based therapeutic candidates are currently used in the therapy of some diseases. In view of the aforementioned information, combining pyrrolidine compounds, which are known to exhibit many biological activities, with different bioactive heterocyclic structures via molecular hybridization will contribute to the development of alternative molecules to drugs that are used in the treatment of diseases and have various limitations. The pyrrolidine skeleton continues to provide new formulas of synthetic and natural compounds with diverse biological activities. Many SAR studies have confirmed the crucial role of this heterocycle in the effective interaction with the natural target. It is noteworthy that, despite the overwhelming variety of substituents anchored to it, a selected number of them are ready to promote beneficial biological interaction. It is important not to discard minor effects in biomedical applications because the fine tuning of the atomic arrangement of the radicals bonded to the ring can raise a very important interaction. The chiral natural pool also provides new compounds, which can serve as starting points to design potent inhibitor agents and can also be useful for the inspiration for the search of many other drug surrogates. This field has wide scope, and extensive work is necessary to develop new drugs.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
We gratefully acknowledge financial support from Çukurova University (Project Nos: TSA-2022-15050, TSA-2021-13814 and FYL-2023-15939). The authors also thank the Spanish Ministerio de Ciencia, Innovación y Universidades (project RED 2018-102387-T), the Spanish Ministerio de Economía, Industria y Competitividad, Agencia Estatal de Investigación (AEI) and Fondo Europeo de Desarrollo Regional (FEDER, EU) (projects CTQ 2017-82935-P and PID 2019-107268 GB-I00), the Generalitat Valenciana (IDIFEDER/2021/013 and GVA-COVID19/2021/079), Medalchemy S. L. (Medalchemy-18T), and the University of Alicante (VIGROB-068 and UAUSTI21-05).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al-Zharani, M., Al-Eissa, M. S., Rudayni, H. A., SurendraKumar, R., Idhayadhulla, A., et al. (2022). Mosquito larvicidal activity of pyrrolidine-2, 4-dione derivatives: An investigation against Culex quinquefasciatus and molecular docking studies. Saudi J. Biol. Sci. 29 (4), 2389–2395. doi:10.1016/j.sjbs.2021.12.003
Apfel, C. C., Zhang, K., George, E., Shi, S., Jalota, L., Hornuss, C., et al. (2010). Transdermal scopolamine for the prevention of postoperative nausea and vomiting: A systematic review and meta-analysis. Clin. Ther. 32 (12), 1987–2002. doi:10.1016/j.clinthera.2010.11.014
Belabbes, A., Retamosa, M. G., Foubelo, F., Sirvent, A., Najera, C., Yus, M., et al. (2023). Pseudo-multicomponent 1,3-dipolar cycloaddition involving metal-free generation of unactivated azomethine ylides. Org. Biomol. Chem. 21 (9), 1927–1936. doi:10.1039/d3ob00023k
Belveren, S., Dondas, H. A., Ulger, M., Poyraz, S., Garcia-Minguens, E., Ferrandiz-Saperas, M., et al. (2017). Synthesis of highly functionalized 2-(pyrrolidin-1-yl)thiazole frameworks with interesting antibacterial and antimycobacterial activity. Tetrahedron 73 (48), 6718–6727. doi:10.1016/j.tet.2017.10.007
Belveren, S., Larranaga, O., Poyraz, S., Dondas, H. A., Ulger, M., Sahin, E., et al. (2019). From bioactive pyrrolidino[3,4-c] pyrrolidines to more bioactive pyrrolidino[3,4-b] pyrrolidines via ring-opening/ring-closing promoted by sodium methoxide. Synthesis-Stuttgart 51 (7), 1565–1577. doi:10.1055/s-0037-1611356
Benedetto Tiz, D., Bagnoli, L., Rosati, O., Marini, F., Santi, C., and Sancineto, L. (2022). FDA-approved small molecules in 2022: Clinical uses and their synthesis. Pharmaceutics 14 (11), 2538. doi:10.3390/pharmaceutics14112538
Bharkavi, C., Kumar, S. V., Ali, M. A., Osman, H., Muthusubramanian, S., and Perumal, S. (2016). A facile stereoselective synthesis of dispiro-indeno pyrrolidine/pyrrolothiazole–thiochroman hybrids and evaluation of their antimycobacterial, anticancer and AchE inhibitory activities. Bioorg. Med. Chem. Lett. 24 (22), 5873–5883. doi:10.1016/j.bmc.2016.09.044
Bhat, A. A., Singh, I., Tandon, N., and Tandon, R. (2023a). Structure activity relationship (SAR) and anticancer activity of pyrrolidine derivatives: Recent developments and future prospects (A review). Eur. J. Med. Chem. 246, 114954. doi:10.1016/j.ejmech.2022.114954
Bhat, A. A., Tandon, N., Singh, I., and Tandon, R. (2023b). Structure-activity relationship (SAR) and antibacterial activity of pyrrolidine based hybrids: A review. J. Mol. Struct. 1283, 135175. doi:10.1016/j.molstruc.2023.135175
Bhat, C., and Tilve, S. G. (2014). Recent advances in the synthesis of naturally occurring pyrrolidines, pyrrolizidines and indolizidine alkaloids using proline as a unique chiral synthon. RSC Adv. 4 (11), 5405–5452. doi:10.1039/C3RA44193H
Bolous, M., Arumugam, N., Almansour, A. I., Kumar, R. S., Maruoka, K., Antharam, V. C., et al. (2019). Broad-spectrum antifungal activity of spirooxindolo-pyrrolidine tethered indole/imidazole hybrid heterocycles against fungal pathogens. Bioorg. Med. Chem. Lett. 29 (16), 2059–2063. doi:10.1016/j.bmcl.2019.07.022
Carcache, L. M., Rodriguez, J., and Rein, K. S. (2003). The structural basis for kainoid selectivity at AMPA receptors revealed by low-mode docking calculations. Bioorg. Med. Chem. 11 (4), 551–559. doi:10.1016/s0968-0896(02)00448-0
Chekan, J. R., McKinnie, S. M. K., Moore, M. L., Poplawski, S. G., Michael, T. P., and Moore, B. S. (2019). Scalable biosynthesis of the seaweed neurochemical, kainic acid. Angew. Chem. Int. Ed. Engl. 58 (25), 8454–8457. doi:10.1002/anie.201902910
Chen, M., Geng, C. W., Han, L., Liu, Y., Yu, Y. K., Lu, A. M., et al. (2021). Design, synthesis, crystal structure, and herbicidal activity of novel pyrrolidine-2, 4-dione derivatives incorporating an alkyl ether pharmacophore with natural tetramic acids as lead compounds. New. J. Chem. 45 (12), 5621–5630. doi:10.1039/d1nj00119a
Christen, P., Cretton, S., Humam, M., Bieri, S., Muñoz, O., and Joseph-Nathan, P. (2020). Chemistry and biological activity of alkaloids from the genus Schizanthus. Phytochem. Rev. 19 (3), 615–641. doi:10.1007/s11101-018-9598-5
Clayton, E. C., Peng, Y. G., Means, L. W., and Ramsdell, J. S. (1999). Working memory deficits induced by single but not repeated exposures to domoic acid. Toxicon 37 (7), 1025–1039. doi:10.1016/s0041-0101(98)00230-x
Cretton, S., Genta-Jouve, G., Kaiser, M., Maser, P., Munoz, O., Burgi, T., et al. (2021). Hygroline derivatives from Schizanthus tricolor and their anti-trypanosomatid and antiplasmodial activities. Phytochemistry 192, 112957. doi:10.1016/j.phytochem.2021.112957
Cretton, S., Muñoz, O., Tapia, J., Marcourt, L., Maes, L., and Christen, P. (2017). Two new hygroline and tropane alkaloids isolated from Schizanthus hookeri and S. tricolor (Solanaceae). Nat. Prod. Commun. 12 (3), 1934578X1701200–358. doi:10.1177/1934578X1701200311
Dobrova, A., Platzer, S., Bacher, F., Milunovic, M. N., Dobrov, A., Spengler, G., et al. (2016). Structure-antiproliferative activity studies on l-proline- and homoproline-4-N-pyrrolidine-3-thiosemicarbazone hybrids and their nickel(ii), palladium(ii) and copper(ii) complexes. Dalton Trans. 45 (34), 13427–13439. doi:10.1039/c6dt02784a
Drager, B. (2006). Tropinone reductases, enzymes at the branch point of tropane alkaloid metabolism. Phytochemistry 67 (4), 327–337. doi:10.1016/j.phytochem.2005.12.001
Elbein, A. D., and Molyneux, R. J. (1999). Alkaloid glycosidase inhibitors. Comp. Nat. Prod. Chem., 129–160. doi:10.1016/B978-0-08-091283-7.00098-9
Frejat, F. O. A., Cao, Y., Zhai, H., Abdel-Aziz, S. A., Gomaa, H. A., Youssif, B. G., et al. (2022). Novel 1, 2, 4-oxadiazole/pyrrolidine hybrids as DNA gyrase and topoisomerase IV inhibitors with potential antibacterial activity. Arab. J. Chem. 15, 103538. doi:10.1016/j.arabjc.2021.103538
Fushiya, S., Sato, S., Kanazawa, T., Kusano, G., and Nozoe, S. (1990). Acromelic acid C. A new toxic constituent of clitocybe acromelalga: An efficient isolation of acromelic acids. Tetrahedron Lett. 31 (27), 3901–3904. doi:10.1016/S0040-4039(00)97501-4
Gerokonstantis, D. T., Nikolaou, A., Magkrioti, C., Afantitis, A., Aidinis, V., Kokotos, G., et al. (2020). Synthesis of novel 2-pyrrolidinone and pyrrolidine derivatives and study of their inhibitory activity against autotaxin enzyme. Bioorg. Med. Chem. Lett. 28 (2), 115216. doi:10.1016/j.bmc.2019.115216
Góra, M., Czopek, A., Rapacz, A., Dziubina, A., Głuch-Lutwin, M., Mordyl, B., et al. (2020). Synthesis, anticonvulsant and antinociceptive activity of new hybrid compounds: Derivatives of 3-(3-methylthiophen-2-yl)-pyrrolidine-2, 5-dione. Int. J. Mol. Sci. 21 (16), 5750. doi:10.3390/ijms21165750
Guan, Z. B., Liang, Y. Q., Lin, J. L., Liao, X. J., Xu, S. H., and Zhao, B. X. (2021). Two new pyrrolidine alkaloids from the red alga Acanthophora spicifera. Nat. Prod. Res. 35 (21), 3824–3829. doi:10.1080/14786419.2020.1741581
Guazzelli, L., D'Andrea, F., Sartini, S., Giorgelli, F., Confini, G., Quattrini, L., et al. (2019). Synthesis and investigation of polyhydroxylated pyrrolidine derivatives as novel chemotypes showing dual activity as glucosidase and aldose reductase inhibitors. Bioorg. Chem. 92, 103298. doi:10.1016/j.bioorg.2019.103298
Hosseinzadeh, Z., Ramazani, A., Hosseinzadeh, K., Razzaghi-Asl, N., and Gouranlou, F. (2018). An overview on chemistry and biological importance of pyrrolidinone. Curr. Org. Synth. 15 (2), 166–178. doi:10.2174/1570179414666170908165445
Huang, J., Liu, H., Liu, M., Zhang, R., Li, L., Wang, B., et al. (2015). Synthesis, antimycobacterial and antibacterial activity of l-[(1R,2S)-2-fluorocyclopropyl]naphthyridone derivatives containing an oxime-functionalized pyrrolidine moiety. Bioorg. Med. Chem. Lett. 25 (22), 5058–5063. doi:10.1016/j.bmcl.2015.10.027
Huang, Y., Min, W., Wu, Q. W., Sun, J., Shi, D. H., and Yan, C. G. (2018). Facile one-pot synthesis of spirooxindole-pyrrolidine derivatives and their antimicrobial and acetylcholinesterase inhibitory activities. New. J. Chem. 42 (19), 16211–16216. doi:10.1039/C8NJ03813A
Hussain, F., Khan, Z., Jan, M. S., Ahmad, S., Ahmad, A., Rashid, U., et al. (2019). Synthesis, in-vitro α-glucosidase inhibition, antioxidant, in-vivo antidiabetic and molecular docking studies of pyrrolidine-2, 5-dione and thiazolidine-2, 4-dione derivatives. Bioorg. Chem. 91, 103128. doi:10.1016/j.bioorg.2019.103128
Ibrahim Srm, M. G., and Mohamed, G. A. Moharram am, youssef dta (2015). aegyptolidines A and B: New pyrrolidine alkaloids from the fungus Aspergillus aegyptiacus. Phytochem. Lett. 12(12), 90–93. doi:10.1016/j.phytol.2015.03.001
Islam, M. S., Al-Majid, A. M., El-Senduny, F. F., Badria, F. A., Rahman, A. M., Barakat, A., et al. (2020a). Synthesis, anticancer activity, and molecular modeling of new halogenated spiro [pyrrolidine-thiazolo-oxindoles] derivatives. Appl. Sci. 10 (6), 2170. doi:10.3390/app10062170
Islam, M. T., and Mubarak, M. S. (2020b). Pyrrolidine alkaloids and their promises in pharmacotherapy. Adv. Tradit. Med. 20 (1), 13–22. doi:10.1007/s13596-019-00419-4
Jan, M. S., Ahmad, S., Hussain, F., Ahmad, A., Mahmood, F., Rashid, U., et al. (2020). Design, synthesis, in-vitro, in-vivo and in-silico studies of pyrrolidine-2, 5-dione derivatives as multitarget anti-inflammatory agents. Eur. J. Med. Chem. 186, 111863. doi:10.1016/j.ejmech.2019.111863
Jeelan Basha, N., Basavarajaiah, S. M., and Shyamsunder, K. (2022). Therapeutic potential of pyrrole and pyrrolidine analogs: An update. Mol. Divers. 26 (5), 2915–2937. doi:10.1007/s11030-022-10387-8
Kim, N., Estrada, O., Chavez, B., Stewart, C., and D'Auria, J. C. (2016). Tropane and granatane alkaloid biosynthesis: A systematic analysis. Molecules 21 (11), 1510. doi:10.3390/molecules21111510
Kocabaş, E., Sarıgüney, A., Erci, F., Çakır Koç, R., Kocabas, H., Torlak, E., et al. (2021). Synthesis, antibacterial and cytotoxic activities of new thiazole based pyrrolidine derivatives. Biointerface Res. Appl. Chem. 11 (4), 12178–12185. doi:10.33263/BRIAC114.1217812185
Kumar, B. S., Prasad, A. R. G., Reddy, P. R., and Ravindranath, L. K. (2016). Synthesis, antimicrobial and in silico EGFR inhibitory activity evaluation of sulfonylamino pyrrolidine derivatives. Pharm. Chem. J. 50 (7), 443–450. doi:10.1007/s11094-016-1467-1
Kumar, R. S., Almansour, A. I., Arumugam, N., Mohammad, F., Alshahrani, W. S., Kotresha, D., et al. (2018). Highly functionalized pyrrolidine analogues: Stereoselective synthesis and caspase-dependent apoptotic activity. RSC Adv. 8 (72), 41226–41236. doi:10.1039/c8ra07985d
Lamkadem, M., Aziz, M., Schwob, I., Rabier, J., Mimouni, J., Fontonge, R., et al. (2004). Cytotoxic effect and electrophysiological study on human MRC-5 fibroblasts of R-irniine, a natural alkylpyrrolidine alkaloid. Nat. Prod. Res. 18 (4), 311–318. doi:10.1080/14786410310001630500
Lamkadem, M., Melhaoui, A., Mimouni, J., and Fontonge, R. (2001). Cytotoxic effect and electrophysiologic activity of (R)-bgugaine, an alkylpyrrolidine alkaloid against MRC-5 fibroblasts. Toxicon 39 (4), 485–490. doi:10.1016/s0041-0101(00)00149-5
Li Petri, G., Raimondi, M. V., Spano, V., Holl, R., Barraja, P., and Montalbano, A. (2021). Pyrrolidine in drug discovery: A versatile scaffold for novel biologically active compounds. Top. Curr. Chem. 379 (5), 34. doi:10.1007/s41061-021-00347-5
Li, Z., Wang, X., Lin, Y., Wang, Y., Wu, S., Xia, K., et al. (2020). Design, synthesis, and evaluation of pyrrolidine based CXCR4 antagonists with in vivo anti-tumor metastatic activity. Eur. J. Med. Chem. 205, 112537. doi:10.1016/j.ejmech.2020.112537
Lin, J. L., Liang, Y. Q., Liao, X. J., Yang, J. T., Li, D. C., Huang, Y. L., et al. (2020). Acanthophoraine A, a new pyrrolidine alkaloid from the red alga Acanthophora spicifera. Nat. Prod. Res. 34 (14), 2065–2070. doi:10.1080/14786419.2019.1569008
Liu, S. J., Zhao, Q., Peng, C., Mao, Q., Wu, F., Zhang, F. H., et al. (2021). Design, synthesis, and biological evaluation of nitroisoxazole-containing spiro[pyrrolidin-oxindole] derivatives as novel glutathione peroxidase 4/mouse double minute 2 dual inhibitors that inhibit breast adenocarcinoma cell proliferation. Eur. J. Med. Chem. 217, 113359. doi:10.1016/j.ejmech.2021.113359
Łowicki, D., and Przybylski, P. (2022). Tandem construction of biological relevant aliphatic 5-membered N-heterocycles. Eur. J. Med. Chem. 235, 114303. doi:10.1016/j.ejmech.2022.114303
Lukowska-Chojnacka, E., Kowalkowska, A., Gizinska, M., Koronkiewicz, M., and Staniszewska, M. (2019). Synthesis of tetrazole derivatives bearing pyrrolidine scaffold and evaluation of their antifungal activity against Candida albicans. Eur. J. Med. Chem. 164, 106–120. doi:10.1016/j.ejmech.2018.12.044
Majik, M. S., Parameswaran, P. S., and Tilve, S. G. (2008). A concise synthesis of (R)-Bgugaine, a pyrrolidine alkaloid from Arisarum vulgare. J. Chem. Res. 2008 (3), 121–122. doi:10.3184/030823408x298481
Malczewska-Jaskola, K., Jasiewicz, B., and Mrowczynska, L. (2016). Nicotine alkaloids as antioxidant and potential protective agents against in vitro oxidative haemolysis. Chem. Biol. Interact. 243, 62–71. doi:10.1016/j.cbi.2015.11.030
Martinez-Bailen, M., Carmona, A. T., Cardona, F., Matassini, C., Goti, A., Kubo, M., et al. (2020). Synthesis of multimeric pyrrolidine iminosugar inhibitors of human β-glucocerebrosidase and α-galactosidase A: First example of a multivalent enzyme activity enhancer for Fabry disease. Eur. J. Med. Chem. 192, 112173. doi:10.1016/j.ejmech.2020.112173
Melhaoui, A. (1998). A new toxic alkylpyrrolidine alkaloid from Arisarum vulgare. Planta Med. 64 (5), 476–477. doi:10.1055/s-2006-957490
Melhaoui, A., and Belouali, H. (1998). DNA affinity of active alkaloids from Arisarum vulgare Targ. J. Ethnopharmacol. 62 (1), 67–71. doi:10.1016/s0378-8741(98)00052-x
Melhaoui, A., Jossang, A., and Bodo, B. (1992). Structure of irniine, a pyrrolidine alkaloid from Arisarum vulgare. J. Nat. Prod. 55 (7), 950–952. doi:10.1021/np50085a016
Melhaoui, A., Mallea, M., Jossang, A., and Bodo, B. (1993). Antibiotic and antifungal pyrrolidine alkaloids, from Arisarum vulgare. Nat. Prod. Lett. 2 (3), 237–242. doi:10.1080/10575639308043815
Meyers, M. J., Liu, J., Xu, J., Leng, F., Guan, J., Liu, Z., et al. (2019). 4-Aryl pyrrolidines as a novel class of orally efficacious antimalarial agents. Part 1: Evaluation of 4-aryl-N-benzylpyrrolidine-3-carboxamides. J. Med. Chem. 62 (7), 3503–3512. doi:10.1021/acs.jmedchem.8b01972
Mezache, N., Derbre, S., Laouer, H., Richomme, P., Seraphin, D., and Akkal, S. (2019). Senecipyrrolidine, an unusual pyrrolidine alkaloid isolated from Jacobaea gigantea (Desf) Pelser (Asteraceae). Nat. Prod. Res. 33 (15), 2182–2191. doi:10.1080/14786419.2018.1493584
Millington, E. L., Dondas, H. A., Fishwick, C. W. G., Kilner, C., and Grigg, R. (2018). Catalytic bimetalic [Pd(0)/Ag(I) Heck-1,3-dipolar cycloaddition cascade reactions accessing spiro-oxindoles. Concomitant in situ generation of azomethine ylides and dipolarophile. Tetrahedron 74 (27), 3564–3577. doi:10.1016/j.tet.2018.05.017
Moni, L., Banfi, L., Basso, A., Carcone, L., Rasparini, M., and Riva, R. (2015). Ugi and passerini reactions of biocatalytically derived chiral aldehydes: Application to the synthesis of bicyclic pyrrolidines and of antiviral agent telaprevir. J. Org. Chem. 80 (7), 3411–3428. doi:10.1021/jo502829j
Morris, B. D., and Prinsep, M. R. (1999). Amathaspiramides A-F, novel brominated alkaloids from the marine bryozoan amathia wilsoni. J. Nat. Prod. 62 (5), 688–693. doi:10.1021/np980410p
O'Hagan, D. (2000). Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids. Nat. Prod. Rep. 17 (5), 435–446. doi:10.1039/a707613d
Poyraz, S., Belveren, S., Ulger, M., Sahin, E., and Dondas, H. A. (2017). Synthesis, characterization, crystal structure, and antituberculosis activity of some novel polysubstituted aminocarbothiol/thiohydantoin-pyrrolidine derivatives. Monats. Chem. 148 (12), 2173–2182. doi:10.1007/s00706-017-2039-0
Poyraz, S., Canacankatan, N., Belveren, S., Yetkin, D., Kibar, K., Ulger, M., et al. (2018). Study of the anti(myco)bacterial and antitumor activities of prolinate and N-amidocarbothiolprolinate derivatives based on fused tetrahydropyrrolo[3,4-c]pyrrole-1,3(2H,3aH)-dione, bearing an indole ring. Monats. Chem. 149 (12), 2253–2263. doi:10.1007/s00706-018-2286-8
Poyraz, S., Döndaş, H. A., Sansano, J. M., Belveren, S., Yamali, C., Ülger, M., et al. (2023b). N-Benzoylthiourea-pyrrolidine carboxylic acid derivatives bearing an imidazole moiety: Synthesis, characterization, crystal structure, in vitro ChEs inhibition, and antituberculosis, antibacterial, antifungal studies. J. Mol. Struct. 1273 (134303), 134303. doi:10.1016/j.molstruc.2022.134303
Poyraz, S., Dondas, H. A., Yamali, C., Belveren, S., Demir, Y., Aydınoglu, S., et al. (2023a). Design, synthesis, biological evaluation and docking analysis of pyrrolidine benzenesulfonamides as carbonic anhydrase or acetylcholinesterase inhibitors and antimicrobial agents. J. Biomol. Struct. Dyn., 1–18. doi:10.1080/07391102.2023.2214224
Rakba, N., Melhaoui, A., Loyer, P., Guy Delcros, J., Morel, I., and Lescoat, G. (1999). Bgugaine, a pyrrolidine alkaloid from Arisarum vulgare, is a strong hepatotoxin in rat and human liver cell cultures. Toxicol. Lett. 104 (3), 239–248. doi:10.1016/s0378-4274(98)00375-0
Ruan, B., Zhang, Y., Tadesse, S., Preston, S., Taki, A. C., Jabbar, A., et al. (2020). Synthesis and structure-activity relationship study of pyrrolidine-oxadiazoles as anthelmintics against Haemonchus contortus. Eur. J. Med. Chem. 190, 112100. doi:10.1016/j.ejmech.2020.112100
Salve, M. T., and Jadhav, S. B. (2021). Synthesis, characterization, and evaluation of in vitro antidiabetic activity of novel pyrrolidine sulphonamide derivative. Int. J. Pharm. Investig. 11 (4), 374–378. doi:10.5530/ijpi.2021.4.67
Shi, Z. F., Lei, C., Yu, B. W., Wang, H. Y., and Hou, A. J. (2016). New alkaloids and α-glucosidase inhibitory flavonoids from Ficus hispida. Chem. Biodivers. 13 (4), 445–450. doi:10.1002/cbdv.201500142
Shibano, M., Kitagawa, S., Nakamura, S., Akazawa, N., and Kusano, G. (1997). Studies on the constituents of Broussonetia species. II. Six new pyrrolidine alkaloids, broussonetine A, B, E, F and broussonetinine A and B, as inhibitors of glycosidases from Broussonetia kazinoki Sieb. Chem. Pharm. Bull. 45 (4), 700–705. doi:10.1248/cpb.45.700
Shibano, M., Nakamura, S., Akazawa, N., and Kusano, G. (1998). Studies on the constituents of Broussonetia species. III. Two new pyrrolidine alkaloids, broussonetines G and H, as inhibitors of glycosidase, from Broussonetia kazinoki Sieb. Chem. Pharm. Bull. 46 (6), 1048–1050. doi:10.1248/cpb.46.1048
Shibano, M., Tsukamoto, D., Kusano, G., Shibano, M., Tsukamoto, D., and Kusano, G. (1999). A new pyrrolizidine alkaloid, Broussonetine N, as an inhibitor of glycosidase, from Broussonetia kazinoki Sieb. and absolute stereostructures of Broussonetines A and B. Chem. Pharm. Bull. 46(6), 907–908. doi:10.1248/cpb.47.907
Shimokawa, J., Chiyoda, K., Umihara, H., and Fukuyama, T. (2016). Antiproliferative activity of amathaspiramide alkaloids and analogs. Chem. Pharm. Bull. 64 (8), 1239–1241. doi:10.1248/cpb.c16-00256
Sirin, S., Duyar, H., Aslım, B., and Seferoğlu, Z. (2021). Synthesis and biological activity of pyrrolidine/piperidine substituted 3-amido-9-ethylcarbazole derivatives. J. Mol. Struct. 1242, 130687. doi:10.1016/j.molstruc.2021.130687
Sreekanth, K., and Jha, A. (2020). Microwave assisted synthesis and antimicrobial activity of novel pyrrolidine derivatives. Russ. J. Gen. Chem. 90 (1), 129–134. doi:10.1134/S107036322001020X
Toumi, A., Boudriga, S., Hamden, K., Sobeh, M., Cheurfa, M., Askri, M., et al. (2021). Synthesis, antidiabetic activity and molecular docking study of rhodanine-substitued spirooxindole pyrrolidine derivatives as novel α-amylase inhibitors. Bioorg. Chem. 106, 104507. doi:10.1016/j.bioorg.2020.104507
Tsuda, M., Sasaki, M., Mugishima, T., Komatsu, K., Sone, T., Tanaka, M., et al. (2005). Scalusamides A-C, new pyrrolidine alkaloids from the marine-derived fungus Penicillium citrinum. J. Nat. Prod. 68 (2), 273–276. doi:10.1021/np049661q
Vitaku, E., Smith, D. T., and Njardarson, J. T. (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57 (24), 10257–10274. doi:10.1021/jm501100b
Watson, A. A., Nash, R. J., Wormald, M. R., Harvey, D. J., Dealler, S., Lees, E., et al. (1997). Glycosidase-inhibiting pyrrolidine alkaloids from Hyacinthoides non-scripta. Phytochemistry 46 (2), 255–259. doi:10.1016/S0031-9422(97)00282-3
Zaman, L., Arakawa, O., Shimosu, A., Onoue, Y., Nishio, S., Shida, Y., et al. (1997). Two new isomers of domoic acid from a red alga, Chondria armata. Toxicon 35 (2), 205–212. doi:10.1016/s0041-0101(96)00123-7
Zhang, H., Wang, X., Mao, J., Huang, Y., Xu, W., Duan, Y., et al. (2018). Synthesis and biological evaluation of novel benzofuroxan-based pyrrolidine hydroxamates as matrix metalloproteinase inhibitors with nitric oxide releasing activity. Bioorg. Med. Chem. 26 (15), 4363–4374. doi:10.1016/j.bmc.2018.06.023
Zheng, X., Cheng, Q., Yao, F., Wang, X., Kong, L., Cao, B., et al. (2017). Biosynthesis of the pyrrolidine protein synthesis inhibitor anisomycin involves novel gene ensemble and cryptic biosynthetic steps. Proc. Natl. Acad. Sci. U. S. A. 114 (16), 4135–4140. doi:10.1073/pnas.1701361114
Zhou, P., Xiang, L., Zhao, D., Ren, J., Qiu, Y., and Li, Y. (2019). Synthesis, biological evaluation, and structure activity relationship (SAR) study of pyrrolidine amide derivatives as N-acylethanolamine acid amidase (NAAA) inhibitors. Medchemcomm 10 (2), 252–262. doi:10.1039/c8md00432c

Keywords: pyrrolidine, pyrrolidine 2,5-dione, natural products, cytotoxicity, pharmaceuticals
Citation: Poyraz S, Döndaş HA, Döndaş NY and Sansano JM (2023) Recent insights about pyrrolidine core skeletons in pharmacology. Front. Pharmacol. 14:1239658. doi: 10.3389/fphar.2023.1239658
Received: 13 June 2023; Accepted: 05 July 2023;
Published: 06 September 2023.
Edited by:
Heike Wulff, University of California, Davis, United StatesReviewed by:
Tao Shi, The Scripps Research Institute, United StatesCopyright © 2023 Poyraz, Döndaş, Döndaş and Sansano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: H. Ali Döndaş, YWRvbmRhc0BjdS5lZHUudHI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.