 Yingxue Yang1†
Yingxue Yang1† Yuanqian Qu
Yuanqian Qu Yang Zhou
Yang Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 10 July 2023
Sec. Experimental Pharmacology and Drug Discovery
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1208740
Cancer is one of the important factors threatening human health. Hence, it is essential to create novel potent drugs to treat it. Due to the strong correlation among histone deacetylase1 (HDAC1), speckle-type POZ protein (SPOP) and cancers, dual inhibition of HDAC1 and SPOP may be a promising strategy for cancer treatment. In this study, we successfully identified four potential dual-targeting HDAC1/SPOP candidate compounds with structure-based virtual screening. In vitro inhibition experiments confirmed that the four compounds had dual inhibitory effects on HDAC1 and SPOP. Among them, compound HS-2 had a stronger inhibitory effect on HDAC1 and SPOP than the positive controls. Further molecular dynamics simulations indicated that HS-2 could stably bind to HDAC1 and SPOP. In addition, MTT assay indicated that HS-2 inhibited the growth of tumor cells in the micromolar range. In vivo evaluation showed that HS-2 could obviously inhibit the growth of tumor in nude mice without obvious toxicity. These findings suggest that HS-2 is a novel and potent dual-targeting HDAC1/SPOP inhibitor for cancer treatment.
Cancer is a major public health problem and one of the greatest threats to human health all over the world (Siegel et al., 2022). In 2023, it is approximately estimated that 1,958,310 new cancer instances will be identified including 81,800 cases of kidney cancer and 106,970 cases of colon cancer (Siegel et al., 2023). Colon cancer has the fifth highest mortality and incidence rate in the world, accounting for one in every ten cancer cases (Han et al., 2022; Jia et al., 2022). The metastatic characteristics of both cancers are the leading cause of death in cancer patients, and patients with distant metastatic disease usually have poor survival rates (Pretzsch et al., 2019; Bray et al., 2021; Sung et al., 2021). Renal cell carcinoma (RCC) is one of the most prevalent types of malignant tumors that belong to urinary system cancer, almost representing 3% of all cancer cases (Padala et al., 2020; Han et al., 2021; Li et al., 2022). Renal cell carcinoma with clear cells (ccRCC) is the most prevalent form with a relatively poor prognosis, which accounts for more than 70% (Chen et al., 2020; Hoefflin et al., 2020; Buttner et al., 2022). Although cancer is highly resistant to chemotherapy agents, chemotherapy remains the standard of treatment for patients suffering from cancers (Che et al., 2021; Huang et al., 2022). Therefore, it seems urgent to find novel targeting agents to improve the therapeutic effect of cancers.
Histone deacetylases (HDACs) are a significant class of epigenetic enzymes that participates in cell regulation and expression of genes by taking away the acetyl groups from lysine residues of both histones and non-histones (Wang H. et al., 2020a; Lanzi et al., 2021; Zhang and Fu, 2021). To date, 18 human HDACs have been identified and classified into four groups based on their sequence homology with yeast histone deacetylase genes: class I (HDACs 1, 2, 3, and 8), class II (HDACs 4, 5, 6, 7, 9, and 10), and class IV (HDAC11) are zinc-dependent metalloproteins that hydrolyze amide bonds with water as a nucleophile, class III (Sirtuins) however, are NAD-dependent and do not respond to classical HDAC inhibitors (Liu S. S. et al., 2020b; He et al., 2020). Class I is highly expressed in tumor cells, especially HDAC1, HDAC2 and HDAC3 (Kato et al., 2009; Hoefflin et al., 2020; Que et al., 2021). HDACs cause cell cycle arrest in a p53 which acts as an oncogenic tumor suppressor protein independent manner by the transcriptional activation of cell cycle regulatory genes (Cha et al., 2009; Wang et al., 2023). Moreover, modification of p53 acetylation regulates the transcriptional activity and is associated with the control of apoptosis and autophagy, which are important for the treatment of malignant tumors (Mrakovcic et al., 2019). And HDAC inhibitors also inhibit angiogenesis and cell proliferation (Jin et al., 2021; Ou et al., 2022). Therefore, inhibition of HDACs is considered an attractive strategy for the treatment of cancers. Several HDAC inhibitors, such as Vorinostat, Panobinostat, and Romidepsin, have FDA approval for the treatment of lymphoma and myeloma (Chang et al., 2021; Lanzi et al., 2021). The clinical data on HDAC inhibitors as monotherapeutic treatments in solid tumors are poor, despite encouraging preclinical findings on these inhibitors (Pili et al., 2017; Mrakovcic et al., 2019). Therefore, we are considering the use of HDAC in combination administration for cancers.
Speckle-type POZ protein (SPOP) is a nuclear protein whose imbalance leads to abnormal regulation of cell proliferation, cell apoptosis and cell cycle (Wang Z. et al., 2020b; Clark and Burleson, 2020; Song et al., 2020). As an E3 ligase junction, SPOP can mediate E3 ubiquitin ligase recognition of substrate protein and catalyze protein ubiquitin by influencing the ubiquitin signaling pathway (Chen et al., 2016; Cho et al., 2016; Wettersten et al., 2017; Kaelin, 2022). At the same time, in the cytoplasm of ccRCC, SPOP is overexpressed, which boosts proliferation and eventually encourages tumorigenesis ultimately (Senft et al., 2018; Ju et al., 2019). In colon cancer cells, overexpression of SPOP significantly inhibits cell proliferation and migration by up-regulating E-cadherin and down-regulating Vimentin, MMP2 and MMP7 (Song et al., 2020). Most previous studies of SPOP have focused on its functions and molecular mechanisms, there have been few reports of SPOP inhibitors in cancer treatment to date (Dai et al., 2017; Zhang et al., 2017; Zhang et al., 2021). A small molecule SPOP inhibitor, SPOP-IN-6b has been reported to inhibit both the activity of SPOP and the carcinogenic signaling pathways and finally kill human ccRCC cells which depend on carcinogenic cytoplasmic SPOP (Guo et al., 2016). However, SPOP-IN-6b showed inhibitory activity in the micromolar range in both in vivo and in vitro, and its affinity and activity were low. Meanwhile, the clinical therapeutic efficacy remained to be explored (Guo et al., 2016). As a result, there is an urgent need to develop potent small molecule inhibitors of SPOP.
Although HDAC inhibitors have manageable side effects and are well tolerated by patients, they do not have enough activity for cancer therapy (Ho et al., 2020; Rausch et al., 2020). The combination of HDAC inhibitors with other anticancer agents is of great benefit in the treatment of cancers in a growing number of studies (Kiweler et al., 2020; Molina et al., 2020; Pili et al., 2020). They can enhance the inhibition of cell proliferation and cell growth, and also enhance the inhibition of angiogenesis in cancers (Rausch et al., 2020; Sharma et al., 2021). However, combination therapies may cause undesirable drug interactions and unnecessary drug toxicity (Zou et al., 2019; Ince and Eisen, 2021). To avoid the mentioned problem, we intend to combine HDAC1 with other targets to develop dual-targeting inhibitors. Even though SPOP serves as a regulatory hub in the treatment of cancers, few SPOP inhibitors are reported to date. Therefore, this study aims to develop novel potent HDAC1/SPOP dual-targeting inhibitors to avoid the limitations of single-target therapy and improve therapeutic efficacy.
Structure-based virtual screening is a useful and valuable silico technique and is generally recognized as an important method for finding new drug compounds (Zhou et al., 2021b; Sanachai et al., 2022). Compared with traditional screening methods, structure-based virtual screening improves the efficiency of screening large-scale compound databases and also provides more reasonable and accurate binding modes (Zhou et al., 2021a; Sirous et al., 2021). In previous studies, kinds of research on single-targeting inhibitors of HDAC or SPOP using structure-based virtual screening has been reported (Guo et al., 2016; Liu J. et al., 2020a; Sirous et al., 2020). Although virtual screening techniques have been utilized to identify SPOP or HDAC1 single-targeting inhibitors, there are currently no reports of dual-targeting HDAC1/SPOP inhibitors (Guo et al., 2016; Liu J. et al., 2020a). In this study, taking full advantage of the aforementioned virtual screening, we successfully identified the first dual-targeting HDAC1/SPOP inhibitor.
Throughout the study, we first preprocessed the ligands obtained from the Protein Data Bank were preprocessed. Secondly, PAINS filtering and drug-likeness screening were used to get the hit compounds, which were docked to HDAC1 and SPOP separately to screen potential dual-targeting compounds. Finally, four compounds with good docking scores were selected for the inhibitory activity assay, and the compounds with the best activity were selected for further biological evaluation to determine their anti-tumor activity.
All tumor cell lines including HCT-116, A498, 769-P, Caki-2, Ramos, OS-RC-2 and Ketr-3 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, United States). All cells were maintained in a humidified incubator with 5% CO2 and 95% air at 37 °C under standard conditions. Compounds were purchased from WuXi AppTec. HDAC1 and SPOP proteins were obtained from Abcam (Cambridge, MA, United States).
Docking-based virtual screening was performed using the molecular docking module in Molecular Operating Environment (MOE) Program. The X-ray 3D structures of HDAC1 (PDB ID: 5ICN) and SPOP (PDB ID: 3HQL) were obtained from the Protein Data Bank. In front of docking, the two proteins were prepared with the QuickPrep function in MOE program to optimize their protonation states and add hydrogen atoms. An in-house database contains 43,000 compounds. All these compounds are composed of 8,000 compounds from the commercially available SPECS database (http://www.specs.net) and 35,000 compounds reported in previous studies (Zhou et al., 2019). A total of 43,000 compounds from an in-house database were further converted into three-dimensional structures through the energy optimization algorithm of MOE and used for virtual screening (Zhou et al., 2019). The PAINS-Remover program was used to screen and remove Pan Assay Interference Compounds from the virtual screening compound database and remove these compounds in biological analysis to reduce the false positive rate of the compounds (Baell and Holloway, 2010). A total of 42,510 compounds were obtained after filtration. According to the modified drug-likeness properties, which means the molecular weight ≤600, the number of hydrogen bond donors ≤5, the number of hydrogen bond acceptors ≤10, the log P (log octanol/water partition coefficient) ≤5, SwissADME was used to filter 37,060 compounds for further virtual screening (Daina et al., 2014; 2017). After that, the docking validation was performed to demonstrate the similar congruence between the native and the docked pose. All compounds were connected to the HDAC1 active site by the Dock protocol of the MOE program and potential HDAC1-targeting compounds were screened by a reasonable Docking Scoring threshold, resulting in 117 compounds with binding free energy <−14.05 kcal/mol. Finally, the selected compounds were paired to the active site of SPOP, and then the optimal four compounds with potential dual-targeting HDAC1/SPOP were selected for further biological evaluation.
In vitro HDAC1 inhibition experiments were carried out as described previously (Li et al., 2014). In the 96-well plates, different concentrations of the tested compound (50 μL) were added to 10 μl of HDAC1 enzyme solution. After incubation at 37°C for 5 min, 40 μl of the fluorescent substrate Boc-Lys (acetyl)-AMC was then added to the mixture. The plates were incubated at 37°C for 0.5 h, and the developer (100 μl) containing trypsin and TSA was added to the mixture. Finally, the fluorescence intensity at wavelengths of 390 nm and 460 nm was measured 20 min later by a microplate reader (Perkin Elemer).
The enzymatic activity of SPOP was determined by a previously described fluorescence polarization (FP) assay (Guo et al., 2016). FITC-labeled peptide substrate puc_SBC1 (FITC-puc_SBC1, FITC-LACDEVTSTTSSSTA) was purchased (GL Biochem Ltd, Shanghai). Serial dilutions of competitors for SPOP23-337 were prepared from the 20 mM DMSO stocks. The diluted compound was added to the reaction mixture in 100 μl of HEPES (25 mM, pH 7.5) containing 10 μM SPOP and 50 nM FITC-puc_SBC1. After incubation for 1 h at 4°C, FP was measured on a microplate reader (Perkin Elemer) using the wavelengths of 480 nm for excitation and 535 nm for emission, respectively.
All experiments were performed in triplicate. Competition binding data were analyzed using GraphPad Prism 6.0 software (GraphPad Software Inc, San Diego, CA) and the inhibition constants (IC50) were calculated by nonlinear curve fitting.
The GROMACS (version 2021.5) program with periodic boundary conditions was used to simulate the MD of HS-2 in the AMBER99SB-ILDN force field to analyze the changes in system stability over time (Abraham et al., 2015; Pall et al., 2020). Firstly, single point charge (SPC) water molecules were used to solvate the compound in a cube box with 1.0 nm away from the compound, and Na+ and Cl− were used to replace the water molecules to neutralize the energy of the system. It then used the steepest descent algorithm and set 5,000 steps to minimize the energy system. A further NVT simulation of 100 ps was performed with a V-rescale thermostat to keep the system temperature at 300 K. The Parinello-Rahman regulator was then simulated with 100ps NPT to maintain the system pressure of 1 bar. Finally, the system was conducted to 50 ns MD simulation and the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) analysis were recorded. 10 ps time intervals were used to save the trajectory data.
According to the previously reported method (Namwan et al., 2022; Zhao et al., 2023), at a concentration of 5 × 104/ml, cell lines were planted in 96-well plates and cultivated for an entire night. The cells were incubated at 37 °C for 72 h while being exposed to various inhibitor concentrations. And then, MTT stock solution (5 mg/ml) was added to each well and incubated for an additional 4 h. Dimethyl sulfoxide (DMSO) was used to dissolve the insoluble crystals, and a microplate reader was used to detect absorbance at 570 nm. Survival ratio (%) was calculated using the following equation: survival ratio (%) = (Atreatment/Acontrol) × 100%. Data were analyzed using the analysis software GraphPad Prism 6.0 software (GraphPad Software Inc., San Diego, CA), and the half-maximal inhibitory concentration (IC50) values were calculated by the dose-response curve visualized using nonlinear regression (curve fit).
The tumor cells (106 cells/well) were seeded onto 6-well plates and allowed to culture for 24 h. The cells were incubated with various concentrations (0, 0.5 and 2 μM) of HS-2 at 37°C for 72 h. Tumor cells cells were washed and lysed with RIPA. Protein samples were detected by western blot as described previously (Zheng et al., 2021).
According to the previously reported in vivo inhibition method, we injected HCT-116 tumor cells (200 μl, 1 × 107 cells) into the right subcutaneous space of 6-week-old BALB/c nude mice (Experimental Animal Center of Yangzhou University (Yangzhou, China) (Zheng et al., 2021). All experimental protocols were approved by the Animal Ethics Committee of China Pharmaceutical University. When the tumors grew to 90–120 mm3, the mice were randomly divided into three groups (five mice per group) and intraperitoneally administered with vehicle (PBS buffer pH 7.4, volume capacity: 0.2 ml), compound HS-2 (volume capacity: 0.2 ml in PBS vehicle, 5 mg/kg), and compound HS-2 (volume capacity: 0.2 ml in PBS vehicle, 10 mg/kg) every 3 days for a total of six times. Tumor volume and weight were measured every 3 days. Tumor volume was calculated using the formula (c × c × d)/2 (c, the smallest diameter; d, the largest diameter).
In this study, potential dual-targeting HDAC1/SPOP inhibitors were identified from an in-house database, and the multi-step process of docking-based virtual screening was shown in Figure 1. A total of 43,000 compounds were processed for Energy Minimization in MOE, and the 2D structure of each compound was transformed into the 3D structure to prepare the 3D database. After that, the PAINS filter was applied to reduce the false positive rate of compounds and improve the efficiency of virtual screening. According to the modified drug-likeness properties as the basis for filtering to narrow down the filtering scope. Before docking, the validation of docking was performed in HDAC1 and SPOP respectively. The validation results were shown in Figures 2A, B, indicating the native and docking poses of both HDAC1 and SPOP are almost identical. Then, the Dock program was used to perform docking-based virtual screening of HDAC1 (PDB ID: 5ICN) and SPOP (PDB ID: 3HQL). The binding affinity between each compound and a target was determined by the size of the binding free energy. In general, a lower binding free energy indicated a stronger binding affinity. The peptide inhibitor of the HDAC1 crystal structure (PDB ID: 5ICN) was chosen as the positive control with a docking score of −14.05 kcal/mol. According to the scoring threshold of −14.05 kcal/mol, 117 compounds were obtained. Afterward, the 117 compounds below −14.05 kcal/mol were further docked into the active site of SPOP, and SPOP-IN-6b was selected as the positive control with the docking score of −8.40 kcal/mol. According to the scoring threshold of −8.40 kcal/mol, four potential dual-targeting candidate compounds simultaneously satisfying both scoring thresholds were obtained (Table 1). The structures of the compounds were shown in Figure 3. Finally, the drug-likeness parameters of 4 compounds were calculated (Table 2) and further biological research was carried out.
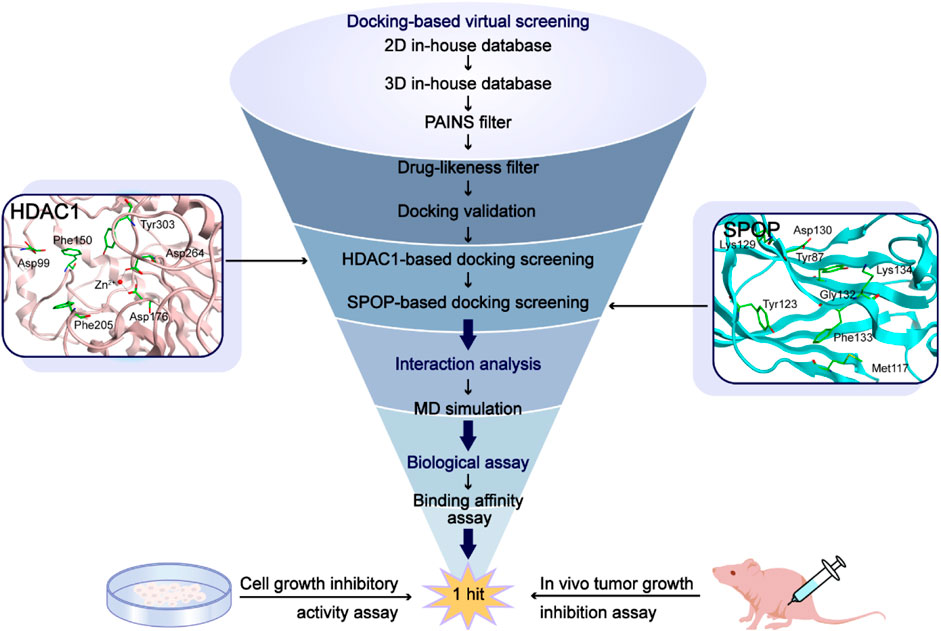
FIGURE 1. The workflow of the docking-based virtual screening process of dual-targeting HDAC1/SPOP inhibitors.
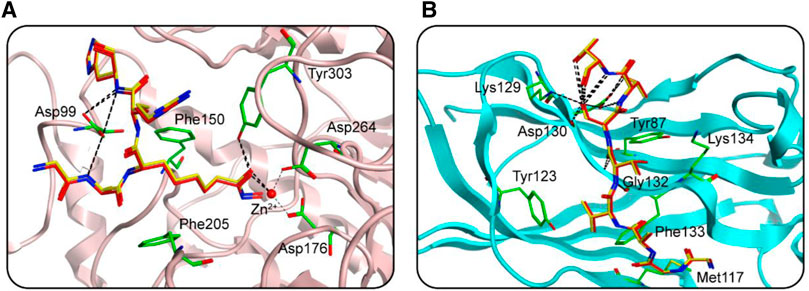
FIGURE 2. The docking validation before molecular docking. (A) The docking validation of HDAC1. (B) The docking validation of SPOP. (yellow for docking pose, red for native pose).
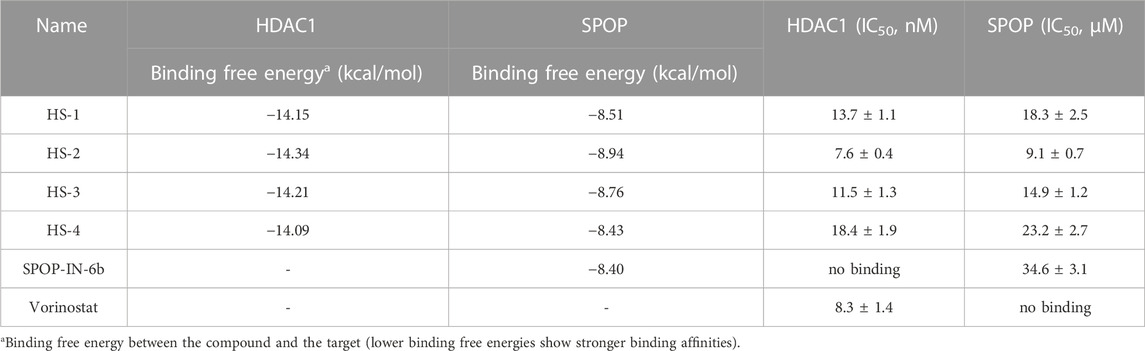
TABLE 1. The docking scores and biological data of four hit compounds.
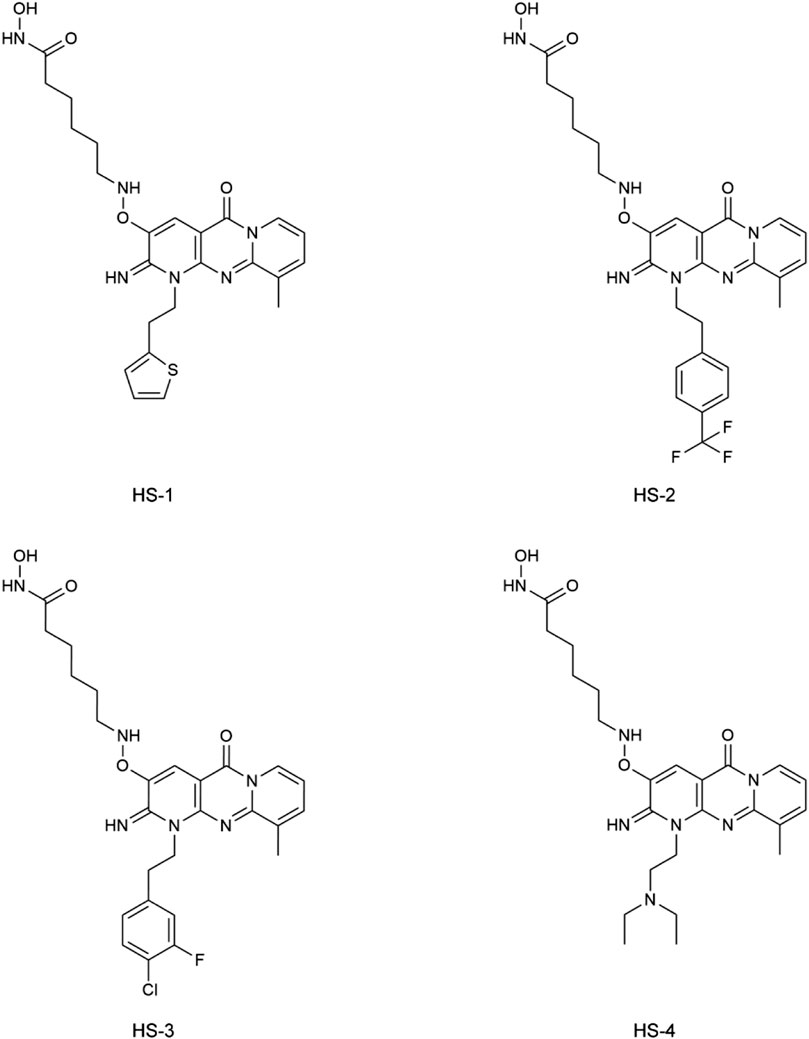
FIGURE 3. The chemical structures of four compounds.
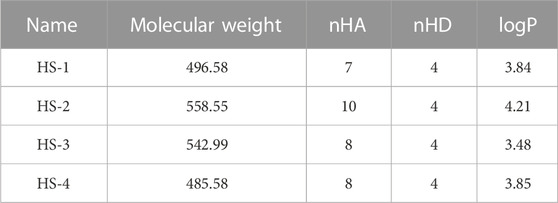
TABLE 2. The drug-like parameters of four compounds.
We studied the binding mode of HS 1–4 to the active sites of HDAC1 and SPOP. Figure 4 showed the docking and binding mode between HS 1–4 and HDAC1. In Figure 4, the compounds HS 1–4 were strongly coincident, and six amino acid residues (Asp99, Phe150, Asp176, Phe205, Asp264, and Tyr303) played an important role in the specific binding of each compound to HDAC1. As with the interaction between the native ligand and the zinc ion, the end-hydroxamic acid structure of each compound formed ionic bonds with the zinc ion, which also did the same with Asp176 and Asp264. Additionally, the compounds could establish hydrogen bonds with Asp99 and Tyr303, respectively. Ionic and hydrogen bonds could stabilize the binding of the compound to the active site and help to enhance the reactivity of the compound. It could be concluded that the screened compounds could be well combined with HDAC1. Figure 5 exhibited the docking and binding mode of HS 1–4 and SPOP. As shown in Figures 5A, C, E, G, HS 1–4 formed hydrogen bonds with eight amino acid residues, namely Tyr87, Met117, Tyr123, Lys129, Asp130, Gly132, Phe133 and Lys134, respectively. In addition, it could be seen from Figures 5B, D, F, H that the compound could be perfectly accommodated by the active pocket of SPOP. In summary, the docking results of HS 1–4 with HDAC1 and SPOP fully indicate that HS 1–4 interact with key amino acid residues of both HDAC1 and SPOP, thus demonstrating that HS 1–4 might be a potential dual-targeting inhibitor.
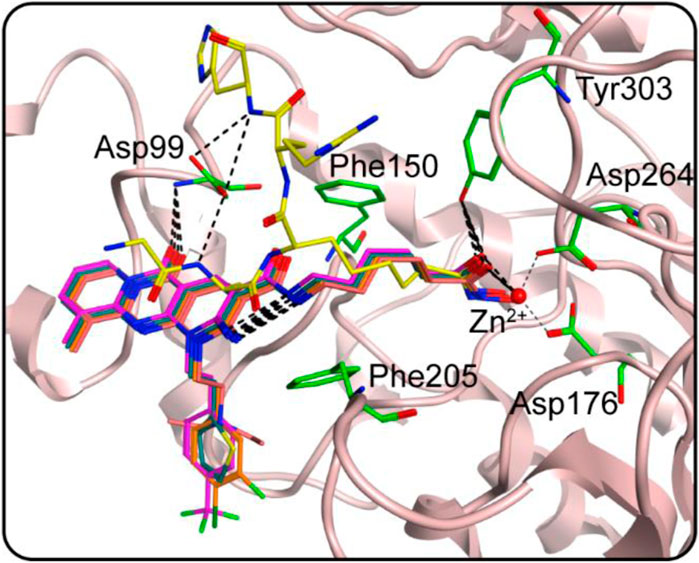
FIGURE 4. The docking poses of HS 1–4 at the HDAC1 active site (yellow for native ligand, dark cyan for HS-1, magenta for HS-2, orange for HS-3 and dark pink for HS-4). Hydrogen bonds are indicated by dashed black lines.

FIGURE 5. The docking poses of HS 1–4 at the SPOP active site were predicted. (A), (B) HS-1 and its corresponding surface map; (C, D) HS-2 and its corresponding surface map; (E, F) HS-3 and its corresponding surface map; (G, H) HS-4 and its corresponding surface map. Compounds are indicated by different colors (yellow for HS-1, purple for HS-2, orange for HS-3, and pink for HS-4), and SPOP is coded by cyan-blue. Hydrogen bonds are indicated by dashed black lines. The surface of SPOP is plotted by H-bond (purple), hydrophobic (green), and mildly polar (blue) regions.
To further assess the inhibitory effects of four selected compounds on the dual targets in further detail, in vitro inhibition experiments of four selected compounds were performed. SPOP-IN-6b and Vorinostat were selected as positive controls, respectively. As shown in Table 1, HS 1–4 showed inhibitory effects on both targets. The IC50 values of HDAC1 inhibition activity of HS 1–4 were 13.7, 7.6, 11.5, and 18.4 nM, and the IC50 values of SPOP inhibition activity of HS 1–4 were 18.3, 9.1, 14.9, and 23.2 μM, respectively. In the positive control, SPOP-IN-6b showed an inhibitory activity on SPOP without an inhibitory effect on HDAC1, while Vorinostat showed an inhibitory activity on HDAC1 without affecting SPOP activity. It was worth noting that among the four compounds, compound HS-2 had the strongest inhibitory activity against HDAC1 and SPOP. The inhibitory effect of HS-2 was stronger than that of the positive controls, and its IC50 value is about 1.1 times that of Vorinostat and 3.8 times that of SPOP-IN-6b. Therefore, the most promising inhibitor HS-2 was further used for molecular dynamics simulation.
HS-2-HDAC1 complex and HS-2-SPOP complex were simulated with 100 ns molecular dynamics to analyze the system binding stability with time. Figures 6A, B described the root mean square deviation (RMSD) of HS-2 in HDAC1 and SPOP complexes, respectively. RMSD is one of the most important factors to describe the stability of the MD simulation system. A lower RMSD value usually indicates better binding stability. The final equilibrium values of the HS-2-HDAC1 and HS-2-SPOP complexes were both below 0.4 nm, with mean RMSD values of roughly 0.20 and 0.26 nm, correspondingly. This demonstrates that HS-2-HDAC1 and HS-2-SPOP complexes are stable in molecular dynamics simulations and that HS-2 may bind to HDAC1 and SPOP concurrently and firmly. The RMSF of the amino acid residue C atom is then determined, and the stability of the system was analyzed by the flexibility of the amino acid residue. The RMSF value is typically associated with interactions between HDAC1/SPOP and HS-2, such as hydrogen bonding. In general, a low RMSF indicates minimal residue movement and consequently a stable system. As shown in Figures 6C, D, the high fluctuations of RMSF of the two complexes existed at the C-/N- ends which might be related to their few interactions. Furthermore, the key amino acid residues corresponding to HS-2-HDAC1 and HS-2-SPOP complexes showed limited fluctuations below 0.2 nm. The secondary structures of the complexes were represented in Figures 6E, F as Coil, B-Sheet, B-Bridge, Bend, Turn, A-Helix, 3-Helix, and 5-Helix in HS-2-HDAC1. These secondary structures were in a stable state of fluctuation within 50 ns. In conclusion, the results of the molecular dynamics simulation suggest that HS-2 may be firmly attached to the active pockets of HDAC1 and SPOP. Therefore, HS-2 could be a novel and effective dual-targeting inhibitor of HDAC1/SPOP.
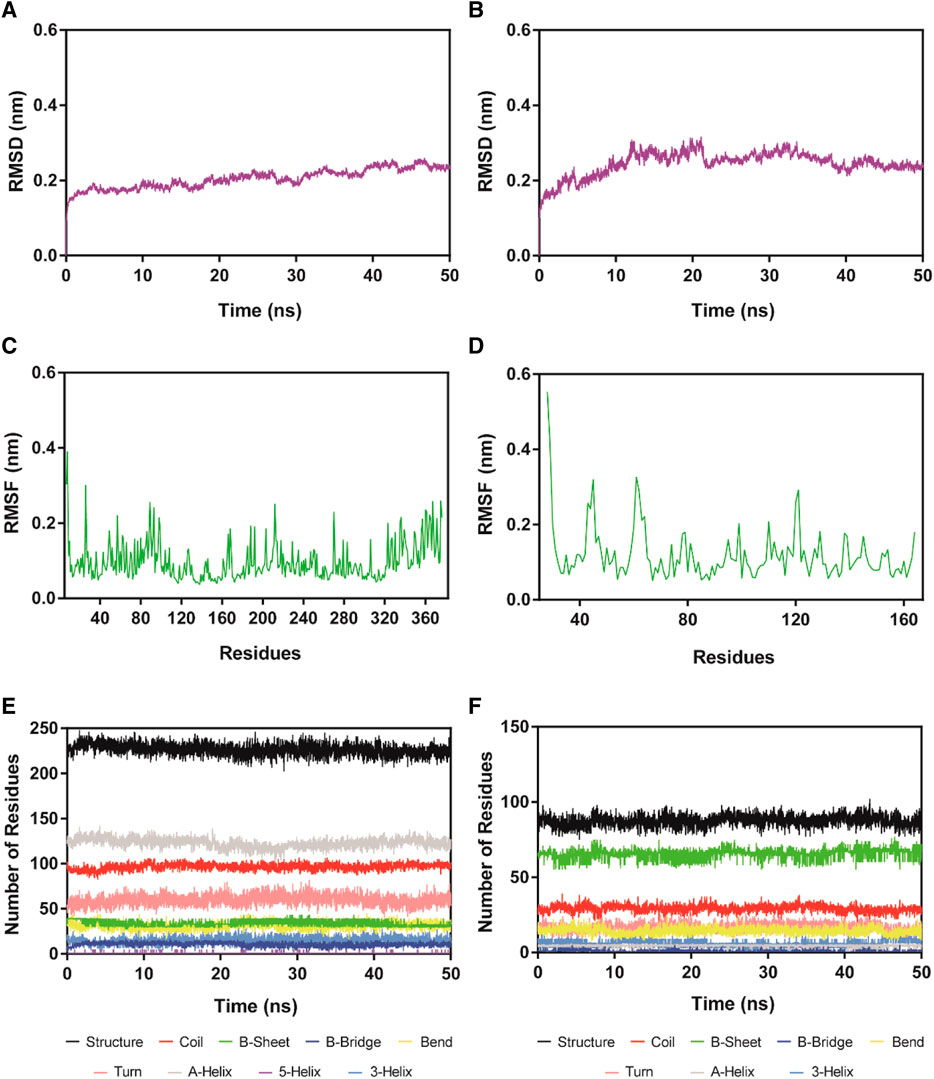
FIGURE 6. RMSD and RMSF of Cα atoms of HS-2-HDAC1 and HS-2-SPOP complexes and the secondary structures analysis of the complexes. (A, B) are the RMSD of HS-2 in complex with HDAC1 and SPOP, respectively. (C, D) are the RMSF of HS-2 in complexes with HDAC1 and SPOP, respectively. (E, F) are the secondary structures analysis of HS-2 in complex with HDAC1 and SPOP, respectively (black for Structure, red for Coil, green for B-Sheet, dark blue for B-Bridge, yellow for Bend, pink for Turn, grey for A-Helix, purple for 5-Helix and blue for 3-Helix).
To analyze the inhibitory effect of compound HS-2 on the growth of cancer-related cells furtherly, the IC50 value of HS-2 was determined by MTT assay. In general, a lower IC50 value indicated a stronger apoptosis-inducing ability of the drug. As shown in Table 3, HS-2 exhibited growth inhibitory activity on the Ketr-3 cell, A498 cell, 769-P cell, Caki-2 cell, Ramos cell, OS-RC-2 cell, and HCT-116 cell with IC50 values < 10 μM. Among them, HS-2 showed excellent anti-tumor activity against A498 cell, Caki-2 cell, and Ramos cell with IC50 values of 1.6, 1.8, and 1.9 μM, respectively. The results indicate the presence of the anti-tumor ability of HS-2. The SPOP and HDAC levels were further measured through western blot. We found that the SPOP and HDAC levels in HCT-116 and A498 cells was reduced upon the treatment of HS-2 (Supplementary Figures S1, S2).
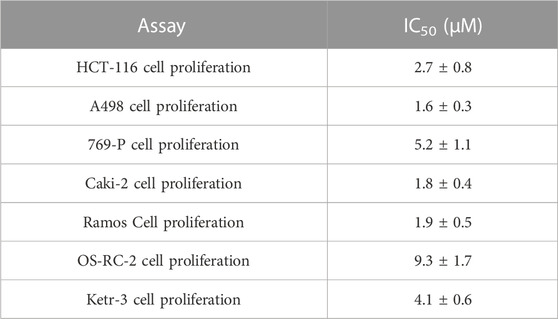
TABLE 3. Cell growth inhibition of HS-2.
To further demonstrate the anti-tumor ability of HS-2 in vivo, HCT-116 cells were transplanted into BALB/c nude mice to establish colon carcinoma models, and the model mice were divided into three groups: vehicle, compound HS-2 (5 mg/kg), and compound HS-2 (10 mg/kg). According to Figure 7A, mice given HS-2 at doses of 5 mg/kg and 10 mg/kg had significantly less tumor volume than the control group, indicating that HS-2 exerted a significant anti-tumor effect in vivo. In addition, mice treated with HS-2 at the dose of 10 mg/kg had a more pronounced trend of tumor volume reduction than those treated with the dose of 5 mg/kg, indicating that HS-2 inhibited tumor growth in a dose-dependent manner. As shown in Figure 7B, the nude mice in all groups were slowly gaining weight, with no significant differences. Therefore, the in vivo results show that compound HS-2 can effectively inhibit tumor growth with no significant toxicity.
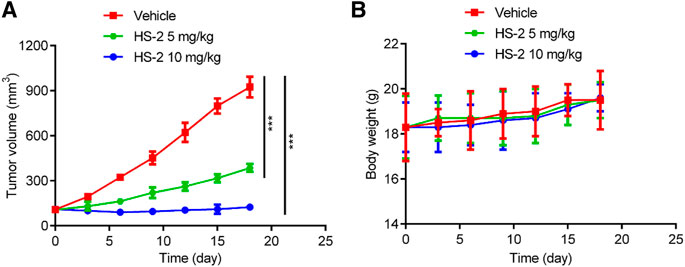
FIGURE 7. HS-2 shows strong antitumor activity to tumor cell-derived xenograft. (A) Changes in tumor volume. (B) Body weight of mice. The data are shown as mean ± SD, n = 5. ***p < 0.001 means a significant difference versus the vehicle group.
Cancer is not sensitive to radiotherapy and chemotherapy, and it has drug resistance and poor prognosis in most patients with advanced cancer (Srinivasan et al., 2015; Lai et al., 2021). HDAC1 and SPOP are closely related to the targeted therapy of cancers, and targeting HDAC1 and SPOP is effective for cancer treatment, which provides new therapeutic ideas and methods for the treatment of RCC. However, to date, there are no reports on dual-targeting inhibitors of HDAC1 and SPOP. In this study, a novel and potent HDAC1/SPOP dual-targeting inhibitor HS-2 was identified using a docking-based virtual screening strategy and molecular dynamics simulation. The interaction analysis showed that HS 1–4 could establish hydrogen bonds with Asp99 and Tyr303 and form ionic bonds with zinc ions to ensure the good docking activity of HS 1–4 with HDAC1. At the same time, the interaction of the compounds with the residues of Tyr87, Met117, Tyr123, Lys129, Asp130, Gly132, Phe133, and Lys134 ensures that it was fully accommodated in the SPOP active pocket. Further in vitro inhibition experiments showed that the four compounds exhibited inhibition of HDAC1 in the nanomolar range and SPOP in the micromolar range. Among them, compound HS-2 showed a stronger inhibitory effect on HDAC1 and SPOP than the positive controls. Therefore, HS-2 was used for molecular dynamics simulation. The results of molecular dynamics simulation showed that HS-2 could stably bind to both HDAC1 and SPOP. In the MTT assay, HS-2 showed growth inhibitory activity in the micromolar range against a range of cancer-related cells, which suggested the existence of potentially potent anti-tumor ability of compound HS-2. In vivo inhibition experiments showed that compound HS-2 inhibited tumor growth in a dose-dependent manner and had no obvious toxicity. Therefore, these results indicate that HS-2 may be a novel HDAC1 and SPOP dual-targeting inhibitor, which may be of value in the treatment of cancer.
The original contribution presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by the Animal Ethics Committee of China Pharmaceutical University.
YQ and YZ conceived and designed the experiments; YY, SC conducted experiments; QW and M-MN analyzed the data; and YY and SC wrote the manuscript. All authors contributed to the article and approved the submitted version.
This project is supported by the Youth Program in Science & Technology of Changzhou Health Committee (No. QN202026), and Qing Miao Talent Program of Changzhou Health Committee (No. CZQM2022010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1208740/full#supplementary-material
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25. doi:10.1016/j.softx.2015.06.001
Baell, J. B., and Holloway, G. A. (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53 (7), 2719–2740. doi:10.1021/jm901137j
Bray, F., Laversanne, M., Weiderpass, E., and Soerjomataram, I. (2021). The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 127 (16), 3029–3030. doi:10.1002/cncr.33587
Buttner, F. A., Winter, S., Stuhler, V., Rausch, S., Hennenlotter, J., Fussel, S., et al. (2022). A novel molecular signature identifies mixed subtypes in renal cell carcinoma with poor prognosis and independent response to immunotherapy. Genome Med. 14 (1), 105. doi:10.1186/s13073-022-01105-y
Cha, T. L., Chuang, M. J., Wu, S. T., Sun, G. H., Chang, S. Y., Yu, D. S., et al. (2009). Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin. Cancer Res. 15 (3), 840–850. doi:10.1158/1078-0432.CCR-08-1918
Chang, L., Ruiz, P., Ito, T., and Sellers, W. R. (2021). Targeting pan-essential genes in cancer: Challenges and opportunities. Cancer Cell 39 (4), 466–479. doi:10.1016/j.ccell.2020.12.008
Che, X., Su, W., Li, X., Liu, N., Wang, Q., and Wu, G. (2021). Angiogenesis pathway in kidney renal clear cell carcinoma and its prognostic value for cancer risk prediction. Front. Med. (Lausanne) 8, 731214. doi:10.3389/fmed.2021.731214
Chen, S. L., Huang, Q. S., Huang, Y. H., Yang, X., Yang, M. M., He, Y. F., et al. (2020). GYS1 induces glycogen accumulation and promotes tumor progression via the NF-κB pathway in Clear Cell Renal Carcinoma. Theranostics 10 (20), 9186–9199. doi:10.7150/thno.46825
Chen, W., Hill, H., Christie, A., Kim, M. S., Holloman, E., Pavia-Jimenez, A., et al. (2016). Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539 (7627), 112–117. doi:10.1038/nature19796
Cho, H., Du, X., Rizzi, J. P., Liberzon, E., Chakraborty, A. A., Gao, W., et al. (2016). On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 539 (7627), 107–111. doi:10.1038/nature19795
Clark, A., and Burleson, M. (2020). SPOP and cancer: A systematic review. Am. J. Cancer Res. 10 (3), 704–726.
Dai, X., Gan, W., Li, X., Wang, S., Zhang, W., Huang, L., et al. (2017). Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 23 (9), 1063–1071. doi:10.1038/nm.4378
Daina, A., Michielin, O., and Zoete, V. (2014). iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model 54 (12), 3284–3301. doi:10.1021/ci500467k
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717. doi:10.1038/srep42717
Guo, Z. Q., Zheng, T., Chen, B., Luo, C., Ouyang, S., Gong, S., et al. (2016). Small-molecule targeting of E3 ligase adaptor SPOP in kidney cancer. Cancer Cell 30 (3), 474–484. doi:10.1016/j.ccell.2016.08.003
Han, D., Yu, Z., Zhang, H., Liu, H., Wang, B., and Qian, D. (2021). Microenvironment-associated gene HSD11B1 may serve as a prognostic biomarker in clear cell renal cell carcinoma: A study based on TCGA, RT-qPCR, western blotting, and immunohistochemistry. Bioengineered 12 (2), 10891–10904. doi:10.1080/21655979.2021.1994908
Han, S., Wang, D., Huang, Y., Zeng, Z., Xu, P., Xiong, H., et al. (2022). A reciprocal feedback between colon cancer cells and Schwann cells promotes the proliferation and metastasis of colon cancer. J. Exp. Clin. Cancer Res. 41 (1), 348. doi:10.1186/s13046-022-02556-2
He, Y., Khan, S., Huo, Z., Lv, D., Zhang, X., Liu, X., et al. (2020). Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J. Hematol. Oncol. 13 (1), 103. doi:10.1186/s13045-020-00924-z
Ho, T. C. S., Chan, A. H. Y., and Ganesan, A. (2020). Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 63 (21), 12460–12484. doi:10.1021/acs.jmedchem.0c00830
Hoefflin, R., Harlander, S., Schafer, S., Metzger, P., Kuo, F., Schonenberger, D., et al. (2020). HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 11 (1), 4111. doi:10.1038/s41467-020-17873-3
Huang, J., Leung, D. K., Chan, E. O., Lok, V., Leung, S., Wong, I., et al. (2022). A global trend analysis of kidney cancer incidence and mortality and their associations with smoking, alcohol consumption, and metabolic syndrome. Eur. Urol. Focus 8 (1), 200–209. doi:10.1016/j.euf.2020.12.020
Ince, W., and Eisen, T. (2021). Combination therapies in clinical trials for renal cell carcinoma: How could they impact future treatments? Expert Opin. Investig. Drugs 30 (12), 1221–1229. doi:10.1080/13543784.2021.2014814
Jia, S. N., Han, Y. B., Yang, R., and Yang, Z. C. (2022). Chemokines in colon cancer progression. Semin. Cancer Biol. 86 (Pt 3), 400–407. doi:10.1016/j.semcancer.2022.02.007
Jin, N., George, T. L., Otterson, G. A., Verschraegen, C., Wen, H., Carbone, D., et al. (2021). Advances in epigenetic therapeutics with focus on solid tumors. Clin. Epigenetics 13 (1), 83. doi:10.1186/s13148-021-01069-7
Ju, L. G., Zhu, Y., Long, Q. Y., Li, X. J., Lin, X., Tang, S. B., et al. (2019). SPOP suppresses prostate cancer through regulation of CYCLIN E1 stability. Cell Death Differ. 26 (6), 1156–1168. doi:10.1038/s41418-018-0198-0
Kaelin, W. G. (2022). Von hippel-lindau disease: Insights into oxygen sensing, protein degradation, and cancer. J. Clin. Invest. 132 (18), e162480. doi:10.1172/JCI162480
Kato, T., Shimono, Y., Hasegawa, M., Jijiwa, M., Enomoto, A., Asai, N., et al. (2009). Characterization of the HDAC1 complex that regulates the sensitivity of cancer cells to oxidative stress. Cancer Res. 69 (8), 3597–3604. doi:10.1158/0008-5472.CAN-08-4368
Kiweler, N., Wunsch, D., Wirth, M., Mahendrarajah, N., Schneider, G., Stauber, R. H., et al. (2020). Histone deacetylase inhibitors dysregulate DNA repair proteins and antagonize metastasis-associated processes. J. Cancer Res. Clin. Oncol. 146 (2), 343–356. doi:10.1007/s00432-019-03118-4
Lai, Y., Tang, F., Huang, Y., He, C., Chen, C., Zhao, J., et al. (2021). The tumour microenvironment and metabolism in renal cell carcinoma targeted or immune therapy. J. Cell Physiol. 236 (3), 1616–1627. doi:10.1002/jcp.29969
Lanzi, C., Favini, E., Dal Bo, L., Tortoreto, M., Arrighetti, N., Zaffaroni, N., et al. (2021). Upregulation of ERK-EGR1-heparanase axis by HDAC inhibitors provides targets for rational therapeutic intervention in synovial sarcoma. J. Exp. Clin. Cancer Res. 40 (1), 381. doi:10.1186/s13046-021-02150-y
Li, M., Liu, G., Jin, X., Guo, H., Setrerrahmane, S., Xu, X., et al. (2022). Micropeptide MIAC inhibits the tumor progression by interacting with AQP2 and inhibiting EREG/EGFR signaling in renal cell carcinoma. Mol. Cancer 21 (1), 181. doi:10.1186/s12943-022-01654-1
Li, X., Inks, E. S., Li, X., Hou, J., Chou, C. J., Zhang, J., et al. (2014). Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J. Med. Chem. 57 (8), 3324–3341. doi:10.1021/jm401877m
Liu, J., Zhou, J., He, F., Gao, L., Wen, Y., Gao, L., et al. (2020a). Design, synthesis and biological evaluation of novel indazole-based derivatives as potent HDAC inhibitors via fragment-based virtual screening. Eur. J. Med. Chem. 192, 112189. doi:10.1016/j.ejmech.2020.112189
Liu, S. S., Wu, F., Jin, Y. M., Chang, W. Q., and Xu, T. M. (2020b). HDAC11: A rising star in epigenetics. Biomed. Pharmacother. 131, 110607. doi:10.1016/j.biopha.2020.110607
Molina, A. M., van der Mijn, J. C., Christos, P., Wright, J., Thomas, C., Dutcher, J. P., et al. (2020). NCI 6896: a phase I trial of vorinostat (SAHA) and isotretinoin (13-cis retinoic acid) in the treatment of patients with advanced renal cell carcinoma. Invest. New Drugs 38 (5), 1383–1389. doi:10.1007/s10637-019-00880-7
Mrakovcic, M., Kleinheinz, J., and Frohlich, L. F. (2019). p53 at the crossroads between different types of HDAC inhibitor-mediated cancer cell death. Int. J. Mol. Sci. 20 (10), 2415. doi:10.3390/ijms20102415
Namwan, N., Senawong, G., Phaosiri, C., Kumboonma, P., Somsakeesit, L. O., Samankul, A., et al. (2022). HDAC inhibitory and anti-cancer activities of curcumin and curcumin derivative CU17 against human lung cancer A549 cells. Molecules 27 (13), 4014. doi:10.3390/molecules27134014
Ou, Z., Lin, S., Qiu, J., Ding, W., Ren, P., Chen, D., et al. (2022). Single-nucleus RNA sequencing and spatial transcriptomics reveal the immunological microenvironment of cervical squamous cell carcinoma. Adv. Sci. (Weinh) 9 (29), e2203040. doi:10.1002/advs.202203040
Padala, S. A., Barsouk, A., Thandra, K. C., Saginala, K., Mohammed, A., Vakiti, A., et al. (2020). Epidemiology of renal cell carcinoma. World J. Oncol. 11 (3), 79–87. doi:10.14740/wjon1279
Pall, S., Zhmurov, A., Bauer, P., Abraham, M., Lundborg, M., Gray, A., et al. (2020). Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 153 (13), 134110. doi:10.1063/5.0018516
Pili, R., Adra, N., Damayanti, N., Logan, T. F., Narayan, V., Monk, P., et al. (2020). Immunomodulation by HDAC inhibition: Results from a phase I study with entinostat in combination with atezolizumab and bevacizumab in metastatic renal cell carcinoma patients. J. Clin. Oncol. 38 (15_Suppl. l), 5064. doi:10.1200/JCO.2020.38.15_suppl.5064
Pili, R., Liu, G., Chintala, S., Verheul, H., Rehman, S., Attwood, K., et al. (2017). Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: A multicentre, single-arm phase I/II clinical trial. Br. J. Cancer 116 (7), 874–883. doi:10.1038/bjc.2017.33
Pretzsch, E., Bösch, F., Neumann, J., Ganschow, P., Bazhin, A., Guba, M., et al. (2019). Mechanisms of metastasis in colorectal cancer and metastatic organotropism: Hematogenous versus peritoneal spread. J. Oncol. 2019, 7407190. doi:10.1155/2019/7407190
Que, Y., Zhang, X. L., Liu, Z. X., Zhao, J. J., Pan, Q. Z., Wen, X. Z., et al. (2021). Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J. Immunother. Cancer 9 (2), e001696. doi:10.1136/jitc-2020-001696
Rausch, M., Weiss, A., Zoetemelk, M., Piersma, S. R., Jimenez, C. R., van Beijnum, J. R., et al. (2020). Optimized combination of HDACI and TKI efficiently inhibits metabolic activity in renal cell carcinoma and overcomes sunitinib resistance. Cancers 12 (11), 3172. doi:10.3390/cancers12113172
Sanachai, K., Mahalapbutr, P., Hengphasatporn, K., Shigeta, Y., Seetaha, S., Tabtimmai, L., et al. (2022). Pharmacophore-based virtual screening and experimental validation of pyrazolone-derived inhibitors toward janus kinases. ACS Omega 7 (37), 33548–33559. doi:10.1021/acsomega.2c04535
Senft, D., Qi, J., and Ronai, Z. A. (2018). Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 18 (2), 69–88. doi:10.1038/nrc.2017.105
Sharma, R., Kadife, E., Myers, M., Kannourakis, G., Prithviraj, P., and Ahmed, N. (2021). Determinants of resistance to VEGF-TKI and immune checkpoint inhibitors in metastatic renal cell carcinoma. J. Exp. Clin. Cancer Res. 40 (1), 186. doi:10.1186/s13046-021-01961-3
Siegel, R. L., Miller, K. D., Fuchs, H. E., and Jemal, A. (2022). Cancer statistics, 2022. CA Cancer J. Clin. 72 (1), 7–33. doi:10.3322/caac.21708
Siegel, R. L., Miller, K. D., Wagle, N. S., and Jemal, A. (2023). Cancer statistics, 2023. CA Cancer J. Clin. 73 (1), 17–48. doi:10.3322/caac.21763
Sirous, H., Campiani, G., Brogi, S., Calderone, V., and Chemi, G. (2020). Computer-driven development of an in silico tool for finding selective histone deacetylase 1 inhibitors. Molecules 25 (8), 1952. doi:10.3390/molecules25081952
Sirous, H., Campiani, G., Calderone, V., and Brogi, S. (2021). Discovery of novel hit compounds as potential HDAC1 inhibitors: The case of ligand- and structure-based virtual screening. Comput. Biol. Med. 137, 104808. doi:10.1016/j.compbiomed.2021.104808
Song, Y., Xu, Y., Pan, C., Yan, L., Wang, Z. W., and Zhu, X. (2020). The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol. Cancer 19 (1), 2. doi:10.1186/s12943-019-1124-x
Srinivasan, R., Ricketts, C. J., Sourbier, C., and Linehan, W. M. (2015). New strategies in renal cell carcinoma: Targeting the genetic and metabolic basis of disease. Clin. Cancer Res. 21 (1), 10–17. doi:10.1158/1078-0432.CCR-13-2993
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Wang, H., Liu, Y. C., Zhu, C. Y., Yan, F., Wang, M. Z., Chen, X. S., et al. (2020a). Chidamide increases the sensitivity of refractory or relapsed acute myeloid leukemia cells to anthracyclines via regulation of the HDAC3 -AKT-P21-CDK2 signaling pathway. J. Exp. Clin. Cancer Res. 39 (1), 278. doi:10.1186/s13046-020-01792-8
Wang, J., Qu, C., Shao, X., Song, G., Sun, J., Shi, D., et al. (2023). Carrier-free nanoprodrug for p53-mutated tumor therapy via concurrent delivery of zinc-manganese dual ions and ROS. Bioact. Mater 20, 404–417. doi:10.1016/j.bioactmat.2022.06.005
Wang, Z., Song, Y., Ye, M., Dai, X., Zhu, X., and Wei, W. (2020b). The diverse roles of SPOP in prostate cancer and kidney cancer. Nat. Rev. Urol. 17 (6), 339–350. doi:10.1038/s41585-020-0314-z
Wettersten, H. I., Aboud, O. A., Lara, P. N., and Weiss, R. H. (2017). Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 13 (7), 410–419. doi:10.1038/nrneph.2017.59
Zhang, H., and Fu, L. (2021). The role of ALDH2 in tumorigenesis and tumor progression: Targeting ALDH2 as a potential cancer treatment. Acta Pharm. Sin. B 11 (6), 1400–1411. doi:10.1016/j.apsb.2021.02.008
Zhang, J., Gao, K., Xie, H., Wang, D., Zhang, P., Wei, T., et al. (2021). SPOP mutation induces DNA methylation via stabilizing GLP/G9a. Nat. Commun. 12 (1), 5716. doi:10.1038/s41467-021-25951-3
Zhang, P., Wang, D., Zhao, Y., Ren, S., Gao, K., Ye, Z., et al. (2017). Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 23 (9), 1055–1062. doi:10.1038/nm.4379
Zhao, M., Yang, K., Zhu, X., Gao, T., Yu, W., Liu, H., et al. (2023). Design, synthesis and biological evaluation of dual Topo II/HDAC inhibitors bearing pyrimido[5,4-b]indole and pyrazolo[3,4-d]pyrimidine motifs. Eur. J. Med. Chem. 252, 115303. doi:10.1016/j.ejmech.2023.115303
Zheng, L., Ren, R., Sun, X., Zou, Y., Shi, Y., Di, B., et al. (2021). Discovery of a dual tubulin and poly(ADP-ribose) polymerase-1 inhibitor by structure-based pharmacophore modeling, virtual screening, molecular docking, and biological evaluation. J. Med. Chem. 64 (21), 15702–15715. doi:10.1021/acs.jmedchem.1c00932
Zhou, Y., Chen, Y., Huang, X., Tan, Y., Hu, R., Li, C., et al. (2021a). A supramolecular nanomedicine based on bendamustine and MDM2-targeted D-peptide inhibitor for breast cancer therapy. Adv. Healthc. Mater 10 (21), e2100980. doi:10.1002/adhm.202100980
Zhou, Y., Chen, Y., Tan, Y., Hu, R., and Niu, M. M. (2021b). An NRP1/MDM2-targeted D-peptide supramolecular nanomedicine for high-efficacy and low-toxic liver cancer therapy. Adv. Healthc. Mater 10 (9), e2002197. doi:10.1002/adhm.202002197
Zhou, Y., Tang, S., Chen, T., and Niu, M. M. (2019). Structure-based pharmacophore modeling, virtual screening, molecular docking and biological evaluation for identification of potential poly (ADP-Ribose) polymerase-1 (PARP-1) inhibitors. Molecules 24 (23), 4258. doi:10.3390/molecules24234258
Keywords: cancer, histone deacetylase 1, speckle-type POZ protein, dualtargeting inhibitors, structure-based virtual screening
Citation: Yang Y, Chen S, Wang Q, Niu M-M, Qu Y and Zhou Y (2023) Identification of novel and potent dual-targeting HDAC1/SPOP inhibitors using structure-based virtual screening, molecular dynamics simulation and evaluation of in vitro and in vivo antitumor activity. Front. Pharmacol. 14:1208740. doi: 10.3389/fphar.2023.1208740
Received: 19 April 2023; Accepted: 29 June 2023;
Published: 10 July 2023.
Edited by:
Pierfausto Seneci, University of Milan, ItalyReviewed by:
Gulam M. Rather, The State University of New Jersey, United StatesCopyright © 2023 Yang, Chen, Wang, Niu, Qu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanqian Qu, eXVhbnFpYW4wNTE5QDE2My5jb20=; Yang Zhou, eWFuZ3pocGF0aG9sb2d5QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.