
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 31 May 2023
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1173542
 Shuying Xie1
Shuying Xie1 Shizhang Wei2Xiao Ma3Ruilin Wang4Tingting He4
Shizhang Wei2Xiao Ma3Ruilin Wang4Tingting He4 Zhao Zhang3
Zhao Zhang3 Ju Yang3
Ju Yang3 Jiawei Wang3Lei Chang1
Jiawei Wang3Lei Chang1 Manyi Jing5
Manyi Jing5 Haotian Li5
Haotian Li5 Xuelin Zhou6*
Xuelin Zhou6* Yanling Zhao1,5*
Yanling Zhao1,5*Hereditary cholestatic liver disease caused by a class of autosomal gene mutations results in jaundice, which involves the abnormality of the synthesis, secretion, and other disorders of bile acids metabolism. Due to the existence of a variety of gene mutations, the clinical manifestations of children are also diverse. There is no unified standard for diagnosis and single detection method, which seriously hinders the development of clinical treatment. Therefore, the mutated genes of hereditary intrahepatic cholestasis were systematically described in this review.
Abnormalities in the synthesis, secretion, metabolism, and excretion of bile acids can result in cholestasis, a condition also known as cholestatic hepatitis. Cholestasis can be categorized as intrahepatic or extrahepatic depending on its location, with hereditary and acquired forms of intrahepatic cholestasis. Hereditary cholestasis includes Progressive familial cholestasis (PFIC), benign recurrent intrahepatic cholestasis (BRIC) (van Mil et al., 2004), Alagille syndrome (McDaniell et al., 2006), Gilbert syndrome (van Dijk et al., 2015), Dubin-Johnson syndrome (Corpechot et al., 2020), and congenital biliary atresia (Bessho and Bezerra, 2011) in newborns. Acquired forms include intrahepatic cholestasis of pregnancy (ICP) (Williamson and Geenes, 2014), drug-induced cholestasis (Lucena et al., 2020), nonalcoholic fatty liver disease (NAFLD) (Targher et al., 2021), obstructive cholestasis (Strnad et al., 2017), TPN-induced cholestasis (Ghosh et al., 2021), primary biliary cirrhosis (PBC) (Floreani et al., 2015), and primary sclerosing cholangitis (PSC) (Yokoda and Carey, 2019). Extrahepatic cholestasis results from injury or obstruction of the extrahepatic bile duct caused by external injury or malignant cholangiocarcinoma (Gao et al., 2020a). Surgical reconstruction of the bile duct or removal of the obstruction can often relieve congenital biliary atresia. Given the complex pathogenesis of hereditary Intrahepatic Cholestasis (HIHC), this review focused on genetic mutations associated with this form of the condition. The categories and details of hereditary cholestasis are shown in Table 1.
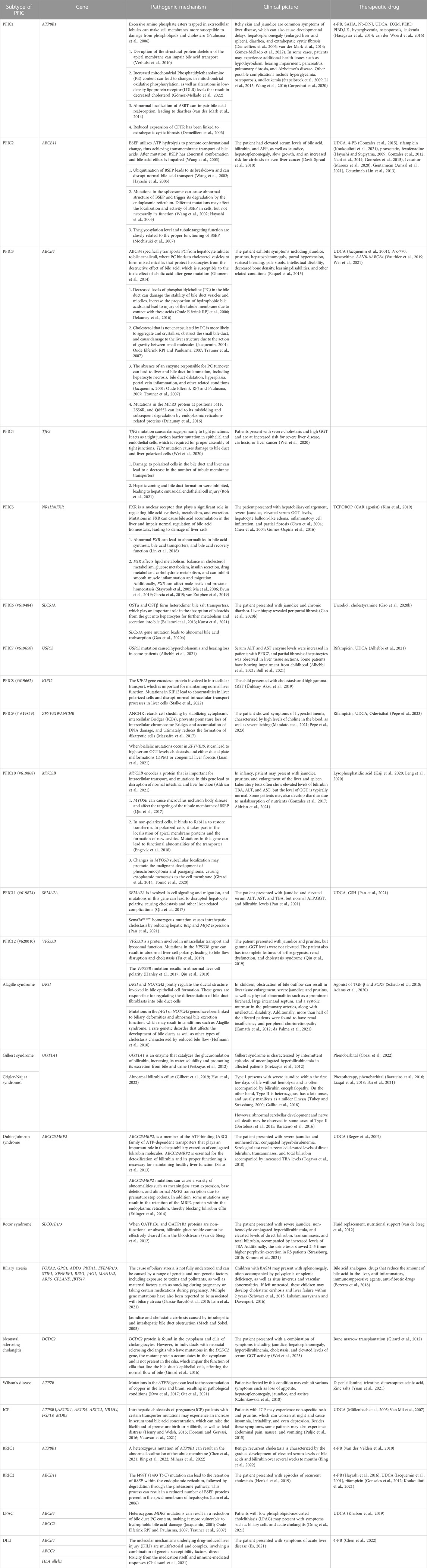
TABLE 1. Overviews of Hereditary cholestasis.
PFIC (progressive familial intrahepatic cholestasis) is a rare autosomal recessive disorder that affects liver function due to gene mutations. It typically presents as intrahepatic cholestasis in infancy and can lead to end-stage liver disease or failure by preschool age, with few patients surviving without liver transplantation (Yokoda and Carey, 2019).
Symptoms of PFIC usually appear within the first 1–2 months of life and include jaundice, pruritus, severe malabsorption, diarrhea, rickets, growth retardation, and liver and spleen enlargement. Fat-soluble vitamin absorption disorder can cause other symptoms, such as coagulation dysfunction (Amirneni et al., 2020). Notably, serum gamma-glutamyl transferase (GGT) levels are typically normal during cholestasis in PFIC1 and PFIC2, which is unusual for other cholestatic diseases (Poupon et al., 2000).
The most common types of PFIC (PFIC1/2/3) are caused by mutations in the ATP8B1, ABCB11, and ABCB4 genes. Other studies have identified mutations in tight junction proteins TJP2(tight junction protein 2) and NR1H4 as contributing to PFIC4/5, as well as mutations in MYO5B.
PFIC1, also known as Byler’s disease, is a rare disorder caused by mutations or deletions in the ATP8B1 gene. This gene encodes a protein called ATPase class I type 8B member 1 (ATP8B1), which is primarily found in the apical membrane of several types of cells including those in the intestine, stomach, bladder, hepatocytes, bile ducts, and intestines (Davit-Spraul et al., 2010). Under normal conditions, ATP8B1 helps maintain the asymmetry of the plasma membrane by moving excess lipids from the outer to the inner side of the cell. This process helps protect the cell from harmful hydrophobic bile salts. However, in the presence of ATP8B1 gene mutations, excess lipids may remain on the outer side of the cell membrane, making it more vulnerable to damage from phospholipids and cholesterol (Paulusma et al., 2006). ATP8B1 plays a role in forming polarized epithelial cells, which are necessary for the proper expression of the protein and formation of microvilli. In vitro studies using small interfering RNA (siRNA) have shown that inhibiting ATP8B1 expression can disrupt the structure of the membrane in polarized epithelial cells known as Caco2 cells. This disruption can lead to disorganization of the root tip and impair the function of microvilli (Verhulst et al., 2010). Studies on mice with ATP8B1 gene mutations have shown that their liver cells have reduced ability to transport hydrophobic bile acids, which can lead to cholestasis. These mice also had increased excretion of cholesterol and phosphatidylserine (PS) in the bile, indicating that the normal structure of the cell membrane is disrupted (Gómez-Mellado et al., 2022). This disruption can lead to a loss of asymmetry in the membrane lipids and affect the function of the canalicular membrane. The findings from the studies on mice with ATP8B1 gene mutations suggest that deficiencies in ATP8B1 can weaken the resistance of the canalicular membrane to hydrophobic bile salts. This can lead to abnormal bile salt excretion and cholestasis. Additionally, the abnormal structure of the cell membrane can also disrupt the skeleton of the apical excitatory protein, further impairing liver function (Paulusma et al., 2006). The abnormal structure of the cell membrane in ATP8B1-deficient liver cells can also impair the function of microvilli, which are important for proper bile acid transport (Verhulst et al., 2010). Additionally, the ability of ATP8B1 to transport bile acids and form micelles may be closely related to another protein called CDC50A (Paulusma et al., 2008; El Sherrif et al., 2013). ATP8B1 activity is regulated by its autoinhibitory state, which is controlled by the presence of other proteins such as CDC50A/B. When ATP8B1 forms a complex with these proteins, the autoinhibitory regions at the N and C termini are released, inducing ATP8B1 to enter a phosphorylated state known as E2P that is ready for substrate binding. Interestingly, the presence of bile acids can further promote the self-inhibitory release of ATP8B1. This suggests that the regulation of ATP8B1 activity is highly dependent on the concentration and nature of bile acids in the liver (Cheng et al., 2022).
Besides PFIC1, other disorders have been associated with ATP8B1 dysfunction. Studies have shown that ATP8B1 deficiency can increase the levels of phosphatidylethanolamine (PE) in liver mitochondria, which in turn stimulates mitochondrial oxidative phosphorylation and leads to increased levels of low-density lipoprotein receptor (LDLR). This may provide a possible explanation for the reduced plasma cholesterol levels observed in PFIC1 disease (Gómez-Mellado et al., 2022). Overall, these findings help explain how ATP8B1 deficiencies can lead to PFIC1 and highlight the importance of this protein for proper liver function (Paulusma et al., 2008; El Sherrif et al., 2013).
Mutations in either ATP8B1 or CDC50A in enterocytes can impair the localization of apical sodium-dependent bile acid transporter (ASBT) and affect bile acid reuptake. This impairment may be a cause of diarrhea in PFIC1 patients. ASBT is responsible for absorbing bile acids from the intestine back into the bloodstream, and its dysfunction can lead to malabsorption of bile acids, which can result in loose stools and diarrhea (van der Mark et al., 2014). Loss of ATP8B1 function and downregulation of cystic fibrosis transmembrane conductance regulator (CFTR) in biliary cells can lead to impaired bile acid excretion and may contribute to the development of extrahepatic cystic fibrosis in PFIC1 patients (Demeilliers et al., 2006). Researchers have explored the use of compounds such as 4-phenylbutyrate (4-PB), which target CFTR modulators, to alleviate intractable pruritus and improve the quality of life in these patients. However, caution should be taken when using such compounds as they may worsen the underlying cystic fibrosis condition (Hasegawa et al., 2014). Several compounds, including 4-PB, suberoylanilide hydroxamic acid (SAHA), and N-butyl deoxynojirimycin (NB-DNJ), have been found to improve the impaired plasma membrane transport function of ATP8B1. These compounds may hold promise as potential treatments for PFIC1 and related conditions. However, further research is needed to determine the safety and efficacy of these compounds in clinical settings. (van der Woerd et al., 2016). ATP8B1 mutations can lead to organ dysfunction outside of the liver, including hypothyroidism (Li et al., 2015), hearing impairment (Stapelbroek et al., 2009), pancreatitis, and pulmonary fibrosis (Wang et al., 2016). In addition, studies have suggested that ATP8B1 genetic variants may be related to resilience in Alzheimer’s disease (Corpechot et al., 2020), although further research is needed to confirm this association. These findings highlight the importance of early detection and proper management of ATP8B1-related conditions to prevent complications and improve patient outcomes.
Treatment options for patients with PFIC1 include ursodeoxycholic acid (UDCA), which can relieve pruritus and jaundice. Other drugs such as S-adenosyl-methionine have shown promise in treating related conditions like primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), and ICP. Glucocorticoids and other immunosuppressants may also be effective, but their use are limited due to potential side effects such as hyperglycemia, osteoporosis, and leukemia. Surgical procedures like partial external bile duct transfer (PEBD), partial internal bile duct shunt (PIBD), and ileal exclusion (I.E.,) have been used to reduce bile acid reabsorption and alleviate symptoms of cholestasis. PEBD has shown some therapeutic effect, with improvements in serum bile acid concentrations, liver function indexes, and slowed progression of liver fibrosis in most PFIC1/2 patients. PIBD transfers bile from the gallbladder to the colon, reducing bile acid reabsorption and lowering serum bile acid and bilirubin levels. I.E., bypasses the distal ileum to reduce bile acid reabsorption. Odevixibat, an oral agent that interrupts the enterohepatic circulation of bile acids, has shown effectiveness in reducing serum bile acid levels and pruritus, and improving quality of life in children with PFIC1/2. However, more research is needed to determine the long-term safety and efficacy of this drug (Thompson et al., 2022). Liver transplantation remains the most effective treatment for PFIC1, but a shortage of liver donors poses a significant challenge. While liver transplantation can provide a cure for PFIC1, it carries the risk of complications and requires lifelong immunosuppressive therapy. Therefore, alternative treatments such as medical therapy and surgical interventions are important options to consider before resorting to liver transplantation. Additionally, efforts to increase awareness about organ donation and improve organ allocation may help address the issue of donor shortages.
Studies have shown that the absence of ATP8B1 can result in incomplete polarization of human monocyte-derived macrophages (HMDMs) into M2c phenotype, which may aid in identifying PFIC1 patients without significant pathogenic mutations in ATP8B1. This suggests that ATP8B1 plays a role in promoting the differentiation of HMDMs into M2c macrophages. Therefore, analyzing the M2c phenotype of patient HMDMs may be helpful in the diagnosis of PFIC1 and BRIC1. Flow cytometry can be used to detect the M2c phenotype and determine whether there is an ATP8B1 gene mutation in PFIC1 patients. This approach may provide a non-invasive and reliable diagnostic tool for these rare liver diseases (Mizutani et al., 2021).
Both progressive familial intrahepatic cholestasis type 2 (PFIC2) and benign recurrent intrahepatic cholestasis (BRIC2) are caused by mutations in the ABCB11 gene, which encodes the BSEP protein. Compared to PFIC1, PFIC2 typically has an earlier onset and more rapid progression, often leading to end-stage liver disease or liver failure before adulthood. Patients with PFIC2 may have elevated plasma levels of bile acids, bilirubin, and alpha-fetoprotein (AFP), as well as symptoms such as jaundice, pruritus, liver and spleen enlargement, and slow growth. Liver biopsies of patients with PFIC2 often show varying degrees of inflammation, including dilation of bile ducts, loss of microvilli, structural abnormalities in mitochondria, accumulation of cell granules in the bile duct lumen, fibrosis, and eventually cirrhosis. Children with PFIC2 are also at increased risk of developing liver nodules, which can be dysplastic and may progress to cancer.
BSEP uses ATP hydrolysis to power conformational changes that enable transmembrane transport of substrates, which is necessary for BSEP to transport bile acids. The energy from ATP hydrolysis allows BSEP to change its shape and move substrates across the hepatocyte canalicular membrane and into bile for excretion (Wang et al., 2003). BSEP is expressed primarily in the liver and functions as the main bile salt export pump. Unlike other P-glycoproteins, which transport a variety of compounds, BSEP specifically catalyzes the transport of hydrophobic bile salts such as glycine- and taurine-conjugated bile acids across the canalicular membrane of hepatocytes. In addition to transporting bile acids, BSEP also plays a role in lipid secretion from hepatocytes into bile. This process is important for regulating bile acid homeostasis and preventing accumulation of toxic levels of bile acids in the liver. Impaired BSEP function can disrupt bile acid homeostasis and lead to cholestasis and liver damage, as seen in PFIC2 and BRIC. As the primary bile salt export pump in the liver, BSEP is an important target for developing therapies for cholestatic liver diseases (Hayashi et al., 2005; Hayashi et al., 2012).
The expression of BSEP mRNA and protein can vary widely among individuals and is tightly regulated by transcriptional mechanisms (Ho et al., 2010). Upon activation, farnesoid X receptor (FXR) forms a heterodimer with the retinoid X receptor alpha (RXRα) and binds to specific DNA sequences in the promoter region of the ABCB11 gene, known as the FXR response element (FXRE). This binding event leads to activation of BSEP transcription and increased expression levels of BSEP mRNA and protein. The degree of activation of BSEP transcription by FXR is positively correlated with the concentration of CDCA. In other words, higher concentrations of CDCA lead to greater activation of BSEP transcription and increased expression of BSEP (Plass et al., 2002; Baghdasaryan et al., 2014).
Bile acids not only serve as substrates for BSEP transport, but also play a role in regulating BSEP expression and localization. For example, the bile acid taurocholate can promote the targeting of BSEP to the canalicular membrane by activating the exchange protein directly activated by cAMP (EPAC)/liver kinase B1 (LKB1)/AMP-activated protein kinase (AMPK) signaling pathway (Homolya et al., 2014). In addition to bile acids, various signaling molecules and transcription factors can indirectly enhance BSEP transcription by activating FXR. For instance, liver receptor homolog 1 (LRH-1) and nuclear receptor coactivator 6 (NCOA6) are known to enhance BSEP expression through FXR activation (Ananthanarayanan et al., 2011). Regulation of BSEP localization and turnover also plays a role in regulating its function. BSEP is found to recycle between the plasma membrane and cytoplasm, with internalization and recycling being regulated by various mechanisms. When BSEP completes its function of transporting bile acids across the canalicular membrane, it can be internalized through clathrinid-mediated endocytosis and degraded by lysosomes. Alternatively, BSEP can be recycled back to the canalicular membrane in a Rab11-dependent manner when it next receives a transport signal. Understanding these mechanisms is important for developing therapies that can modulate BSEP function and improve outcomes in cholestatic liver diseases (Aida et al., 2014; Czuba et al., 2018). Some mutations can result in misfolded BSEP proteins that are recognized by the endoplasmic reticulum-associated degradation (ERAD) pathway. Misfolded BSEP proteins may arise due to various mechanisms, including spliceosome mutations, premature termination of transcription, or errors during protein folding. In the ERAD pathway, the misfolded BSEP protein is targeted for degradation via ubiquitination and subsequent proteasomal degradation in the cytoplasm. This leads to reduced levels of functional BSEP protein on the canalicular membrane and impaired bile flow, which can contribute to the development of PFIC2 (Wang et al., 2002; Hayashi et al., 2005). The expression of BSEP protein can be influenced by various factors, including post-translational modifications such as glycosylation. Basic studies have shown that the transport function of rat BSEP protein is dependent on its N-linked glycosylation, as inhibition of glycosylation can impair the targeting of BSEP to the canalicular membrane or lead to abnormal protein folding and degradation in the endoplasmic reticulum. In addition to glycosylation, mutations in the ABCB11 gene can also affect the targeting function of BSEP. For example, some mutations have been found to affect the interaction between BSEP and myelin and lymphocyte proteins, which are required for proper targeting of BSEP to the canalicular membrane (Mochizuki et al., 2007).
The protein expression level of BSEP can vary among patients with cholestatic liver diseases due to mutations in the ABCB11 gene. Different mutations may not necessarily affect the function of BSEP, but can alter its localization and activity within cells, resulting in distinct clinical phenotypes. For example, studies have shown that two mutations in the ABCB11 gene, p. A570T and p. R1050C, can lead to higher levels of BSEP expression in patients with BRIC2 compared to patients with PFIC2, but lower expression levels compared to patients with ICP (Lam et al., 2007). Ching-Wan Lam and others identified two novel mutations in exons 14 and 18 of the ABCB11 gene in patients with mild forms of PFIC2. The clinical presentation of these patients was relatively mild compared to classical PFIC2, suggesting a possible genotype-phenotype correlation in ABCB11 mutations (Lam et al., 2006). In addition, a study by Soroka et al. showed that the localization and turnover of BSEP on the canalicular membrane are regulated by various mechanisms, including vesicle release, anchoring, and internalization pathways. HCLS1-associated protein X-1 (HAX-1), epidermal growth factor receptor pathway substrate 15 (EPS15), adaptor protein-2 (AP-2), and intracellular calcium signaling via the IP3-receptor have been implicated in regulating BSEP trafficking to the canalicular membrane (Soroka and Boyer, 2014).
Some mutations can cause unstable increases in BSEP mRNA levels, protein misfolding and instability, loss of transport function, and impaired targeting to the canalicular membrane. To test possible mutated mRNA sites, exonic splicing enhancers (ESE) and exonic splicing silencers (ESdS) can be used. Various computational methods, such as ligand quantitative structure-activity relationship (QSAR), pharmacophore modeling, and nonlinear self-organizing mapping, have been established and validated to analyze high-throughput BSEP in vitro drug interaction datasets. While the underlying mechanisms are not yet fully understood, some studies suggest that cholesterol supplements may alleviate the progression of PFIC2. Further research is needed to elucidate the specific mechanisms by which cholesterol affects BSEP function and to determine whether this approach may be a viable therapeutic option for patients with cholestatic liver diseases (Engelmann et al., 2015). Various methods have been used to detect BSEP function and expression, including Xenopus oocytes, canalicular membrane vesicles (CMV), BSEP-expressing membrane vesicles, BSEP-expressing cell lines, and sandwich-cultured hepatocytes (SCH) (Cheng et al., 2016). The choice of detection method depends on the specific research question and available resources. Each method has advantages and limitations that must be considered when designing experiments to study BSEP and other hepatic transporters.
The treatment of progressive PFIC2 typically involves medications aimed at improving bile flow and reducing cholestasis. UDCA is considered the first-line therapy for PFIC2, as it can stimulate the transcription of BSEP and multidrug resistance-associated protein 2 (MRP2), stabilize liver cell membranes, inhibit hepatocyte apoptosis, and promote bile flow through biliary and hepatic changes. UDCA has demonstrated some therapeutic effect in early-stage PFIC2, but its efficacy is limited in advanced disease where there is severe depletion of BSEP on the canalicular membrane.4 -PB is another medication that has been used to treat severe forms of BSEP deficiency. It can partially correct the tubular localization of mutated BSEP, slow down the ubiquitination of BSEP, and relieve cholestasis in affected individuals. Clinical studies have shown that 4-PB can improve clinical symptoms in some patients with BSEP mutations. Other drugs that have shown promise in treating cholestatic liver diseases include pravastatin and fexofenadine (Hayashi and Sugiyama, 2009; Gonzales et al., 2012; Naoi et al., 2014; Gonzales et al., 2015), which have been reported to improve bile excretion and reduce serum bile acid levels in patients with BSEP deficiency. However, further studies are needed to determine the safety and efficacy of these medications in larger patient populations (Hirano et al., 2005; Matsushima et al., 2008). Ivacaftor, a drug used to treat cystic fibrosis, has shown promise as a potential therapy for BSEP deficiency. A study by Davit-Spraul et al. demonstrated that ivacaftor can improve BSEP function in patients with PFIC2 who carry the p. Gly308Asp missense mutation (Mareux et al., 2020). In addition, Rachida Amzal and others found that two other compounds, gentamicin and 4-PB, can increase BSEP expression on the canalicular membrane in vitro. The study used several cell lines, including HEK293, Can10, HepG2, and MDCK cells, to evaluate the effects of these compounds on BSEP localization and activity. Gentamicin, an antibiotic, was shown to enhance BSEP trafficking to the canalicular membrane by reducing protein degradation, while 4-PB was shown to increase BSEP expression through a different mechanism involving transcriptional activation (Amzal et al., 2021).
Liver transplantation is currently the only definitive treatment for PFIC2 with end-stage liver disease. However, PFIC2 can recur after transplantation, particularly in cases where the underlying cause of cholestasis is due to an autoimmune response against BSEP. This condition is known as autoimmune BSEP disease (AIBD), and it is associated with the presence of anti-BSEP antibodies in affected individuals. Detecting the presence of anti-BSEP antibodies can be a useful diagnostic tool for predicting the risk of PFIC2 recurrence after liver transplantation. Immunofluorescence (IF) or immunohistochemistry (IHC) staining of patient liver tissues is commonly used in clinical practice to identify the presence of these antibodies (Kubitz et al., 2015)Anti-BSEP antibody-associated cholestasis severity correlates with the level of anti-BSEP antibodies in the body. Reducing this antibody titer can improve cholestatic hepatitis within days by removing the antibody from plasma using immunosorbent therapy (Kubitz et al., 2016). Cetuximab has also been studied for its potential therapeutic effect on this condition by binding to plasma cells and BSEP antibodies. It is important to note that although reducing the antibody titer may help alleviate symptoms, it may not completely cure the disease. Therefore, ongoing monitoring and management are crucial for effective treatment (Lin et al., 2013).
Persistent cholestasis in children caused by ABCB4 (alias multidrug resistance protein 3 [MDR3]) mutations differs from that of PFIC1/2. The onset of PFIC3 is relatively late, often in adulthood, and presents with symptoms like jaundice, pruritus, hepatosplenomegaly, varicose bleeding caused by portal hypertension, acholic stools, intellectual disability, decreased bone density, and learning disabilities (Raquel et al., 2015). Treatment with UDCA is the first choice for PFIC3, but only 30% of patients respond to it (Jacquemin et al., 2001). Liver transplantation is often viewed as a last resort due to the challenges that come with rejection and postoperative liver degeneration, which can compromise the effectiveness of the procedure. However, other surgical treatments such as complete biliary metastasis, drug transfer of bile acid, and hepatocyte transplantation offer promising new options for treating cholestasis in affected patients (van der Woerd et al., 2017). ABCB4/MDR3 is mainly concentrated in the right lobe of the liver and plays a critical role in the transport of bile acids, phospholipids, and phosphatidylcholine (PC). It acts as a positive regulator of lipid secretion in bile by transporting PC specifically from the bilateral membranes of the tubules to the tubules of hepatocytes. Within the tubules of hepatocytes, PC combines with cholesterol vesicles to form mixed micelles that work synergistically with ATP8B1 to protect hepatocytes from the damaging effects of bile acids. Overexpression of MDR3 leads to increased fluorescence intensity of PC, but not other types of phospholipids, during transport from hepatocytes to the bile duct (Ghonem et al., 2014). In individuals with MDR3 mutations, the level of PC in the bile duct decreases, which disrupts the stability of bile duct vesicles and micelles. This disruption increases the proportion of hydrophobic bile acids and leads to damage caused by contact between these hydrophobic bile acids and the tubule membrane (Oude Elferink RPJ et al., 2006; Delaunay et al., 2016). Cholesterol without PC encapsulation has a greater tendency to aggregate and crystallize due to the gravitational forces between small molecules. This can lead to the obstruction of small bile ducts and damage to the liver structure. The loss of PC turnover enzyme can result in liver and bile duct inflammation, including hepatocyte necrosis, biliary duct expansion, hyperplasia, portal vein inflammation, and other related conditions (Jacquemin, 2001; Oude Elferink RPJ and Paulusma, 2007; Trauner et al., 2007).
The function of MDR3 is dependent on the specific site of mutation. In general, homozygous mutations lead to PFIC3, while heterozygous mutations result in low phospholipid-associated cholelithiasis (LPAC) (Poupon et al., 2010). Moreover, MDR3 mutations may also result in PBC, ICP, oral contraceptives-induced cholestasis and drug-induced liver injury (DILI) (Reichert and Lammert, 2018; Vries et al., 2020). Studies have revealed that mutations in specific sites, such as I541F, L556R, and Q855L, can cause misfolding of MDR3 protein, leading to its degradation by endoplasmic reticulum-related proteins. In contrast, mutations at other sites have little effect on MDR3 function (Delaunay et al., 2016). The I54IF mutation in the first nucleotide binding domain (NBD) of MDR3 leads to reduced expression of the protein in the tubule membrane, resulting in its retention in ER/Golgi compartments (Delaunay et al., 2009; Delaunay et al., 2016). In cases of the I541F mutation, MDR3 glycosylation is immature and the protein remains in the cytoplasm. However, at low temperatures, such as 27°C, MDR3 can be expressed on the plasma membrane, promoting its localization within the tubules and restoring normal function to the mutant protein (Delaunay et al., 2009). Some mutation sites in MDR3, such as G551D, S1251N, and G1349D, are similar to the gated mutation of CFTR or ABCC7. The use of CFTR enhancers like Ivacaftor (iVx-770) and Roscovitine (Vauthier et al., 2019) can promote normal processing of MDR3 and increase its expression in the tubule membrane (Delaunay et al., 2017). Recently, gene therapy has shown promising results in restoring phospholipid transport and re-expressing MDR3 protein by targeting liver hABCB4 mRNA in cells and PFIC3 mouse liver. In mice with PFIC3, liver inflammation, catheter reaction, and fibrosis were significantly reduced following this therapy (Wei et al., 2021). Adeno-associated virus (AAV) is a promising treatment option for PFIC3. After 12 weeks of injecting adeno-associated virus carrying Abcb4 into PFIC3 mice, all male mice showed a significant curative effect, while 50% of female mice showed improvement. After prolonged treatment, the symptoms of cholestasis in female mice also showed good remission (Weber et al., 2019). Recombinant adeno-associated virus vectors are considered a promising delivery system. The combination of recombinant adeno-associated virus (rAAV) with transposition-mediated somatic cell integration and single-dose injection into Abcb4−/− mice at an early age can prevent cholestatic cirrhosis and greatly reduce the incidence of tumors in Abcb4−/− mice (Siew et al., 2019). Estrogen and lecithin are also considered significant contributors to PFIC3. Estrogen disturbances can worsen PFIC3 symptoms, especially during pregnancy, leading to severe cholestasis and intractable pruritus. Albumin dialysis or molecular adsorbent recirculation systems (MARS) can alleviate hormone-induced cholestasis exacerbations (Lemoine et al., 2008). Dietary lecithin has been shown to reduce cholestasis symptoms in Abcb4−/− mice by decreasing serum levels of cholic acid and bilirubin, and reducing bile duct hyperplasia and activation of fibroblasts around the bile duct (Lamireau et al., 2007). Sem J Aronson et al. found that injecting AAV8-hABCB4 into Abcb4−/− mice stabilized the expression of ABCB4 protein in their liver, inhibiting the development of PFIC3. J M De Vree et al. discovered that transplantation of liver cells with normal expression of Mdr3 into Mdr2−/− mice could restore phospholipid secretion and improve liver pathological injury (De Vree et al., 2000; Aronson et al., 2019).
PFIC4 is a cholestatic disease caused by abnormal function of the TJP2 protein of Tight junction protein ZO-2. TJP2 plays a significant role in tight junctions and adhesion junctions, acting as a tight junction barrier in epithelial and endothelial cells and being necessary for the proper assembly of tight junctions. Mutations in TJP2 can also cause hyperkalemia and PFIC1. Patients with TJP2 mutations typically present with severe cholestasis and high GGT levels. Clinical studies have demonstrated that TJP2 mutations increase the risk of severe liver disease, cirrhosis, or liver cancer in adolescents and adults (Wei et al., 2020). ZO-1/2 deficient mice at 6 weeks of age exhibit a higher fatality rate due to damage to the bile duct and liver polarized cells, as well as a decreased number of transporters on the tubule membrane. Liver zonation and bile duct formation are also inhibited, which can lead to injury of hepatic sinusoidal endothelial cells (Itoh et al., 2021). In Tjp2 knockout mice, levels of Claudin domain-containing protein 1(CLDN1) were reduced, and luciferase isothiocyanate showed impaired tight junctions. Additionally, there were dilated tubules, reduced microvilli density, and decreased expression of the tubule membrane transporter BSEP (Xu et al., 2021). In vitro experiments have shown that inhibiting TJP2 expression using siRNA can inhibit the proliferation of L02 and HepG2 cells, induce cell apoptosis, and cause microfilament disturbance (Tang et al., 2021).
PFIC5 is a form of cholestasis that occurs in newborns and is caused by mutations in the nuclear receptor FXR/NR1H4. This condition can result in liver and gallbladder enlargement, severe jaundice, and elevated serum GGT levels (Chen et al., 2004). FXR is a bile acid receptor that binds to bile acids like CDCA, DCA, and DCA, indirectly inhibiting CYP7A1 and CYP8B1 while activating transcription factor MafG (MAFG). These factors all play a role in regulating bile acid synthesis, suppressing its production. Inhibition of Hepatic sodium/bile acid cotransporter (NTCP) reduces bile acid reabsorption, thereby lowering plasma levels of these compounds. FXR also directly recruits histone methyltransferase Histone-arginine methyltransferase CARM1 (CARM1) and activates BSEP to promote bile acid secretion. Additionally, inhibition of toll-like receptors can alleviate bile acid-induced cholestatic hepatitis (Lin et al., 2018). In basic studies, Fxr/Shp double-knockout mice have exhibited a cholestasis phenotype highly similar to human PFIC5. These mice showed severe cholestasis, an increased liver index, and an enlarged gallbladder. Liver pathology revealed increased duct reaction, balloon-like edema of hepatocytes, inflammatory cell infiltration, and partial fibrosis (Gomez-Ospina et al., 2016; Schady and Finegold, 2017). Extracellular vehicles (EVS) have emerged as a potential treatment for cholestatic hepatitis C. McDaniel K et al. found that miRNA Lethal 7, secreted by liver stem cell derived EVS (LSCEVS), can reduce catheter response and biliary fibrosis in mice with cholestasis. NR1H4 serves as a pivotal mediator of let-7 signal-related inflammation/fibrosis response (McDaniel et al., 2019). In addition to its role in regulating bile acid synthesis and cholestatic hepatitis, FXR has also been found to improve the autoimmune function of the central nervous system in an IL10-dependent manner (Hucke et al., 2016). However, Fxr/Shp knockout mice have exhibited severe cholestasis, an increased liver index, and gallbladder enlargement. Liver pathology analyses of these mice also revealed increased catheter response, hepatocyte balloon-like edema, inflammatory cell infiltration, and partial fibrosis (Kim et al., 2018). The expression of FXR can be influenced by various factors, including miRNA-192 (Krattinger et al., 2016) and miRNA-194 (Nie et al., 2017). In addition to its role in regulating bile acid synthesis and cholestatic hepatitis, FXR also plays a key role in lipid metabolism (van Zutphen et al., 2019), cholesterol metabolism balance (Byun et al., 2019), glucose metabolism (Ma et al., 2006), insulin secretion, drug metabolism, and carbohydrate metabolism (Stayrook et al., 2005). FXR has been found to inhibit smooth muscle inflammation and migration, as well as affect testis and prostate homeostasis (Garcia et al., 2019). Additionally, activation of FXR can lead to an extended lifespan through the activation of heterological metabolic pathways (Jiang et al., 2013). FXR also plays a crucial role in preventing parenteral nutrition-associated cholestasis (El Kasmi et al., 2022). However, abnormalities in FXR function can result in kidney damage. Basic studies have shown that Fxr−/− mice exhibit kidney damage, which may be related to cycNr1h4 (Lu et al., 2020). These studies also found that the activity of the ammonia detoxification enzyme in plasma was reduced, which limited the rate of ammonia decomposition (Massafra et al., 2017). Kang Ho Kim et al. investigated the effects of CAR agonists on cholestatic liver injury in a mouse model of PBC using Fxr/Shp double knockout (DKO) mice. They found that treatment with a CAR agonist reduced liver injury and inflammation, as well as decreased the expression of bile acid synthesis genes. The study suggests that CAR agonists may be a potential therapeutic strategy for treating PBC and other cholestatic liver diseases (Kim et al., 2019).
PFIC6 (Progressive Familial Intrahepatic Cholestasis type 6) is a rare genetic disorder that affects the liver and digestive system. The disorder is caused by mutations in the SLC51A gene which encodes the Organic solute transporter subunit alpha (OSTα) protein. OSTα does form a heterodimeric bile salt transporter with OSTβ, and they play an important role in the uptake of bile acids from the gut into hepatocytes for further metabolism and secretion into bile. It is true that the reduced bile acid reabsorption after SLC51A gene mutation leads to decreased bile flow and inhibition of fat digestion, which explains the decreased efficiency of bile acid reabsorption in PFIC6 patients (Ballatori et al., 2013; Kunst et al., 2021). This can result in increased hepatic bile acid synthesis and bile acid accumulation in the liver, leading to cholestatic liver injury and diarrhea (Gao et al., 2020b). However, it should be noted that the diarrhea seen in PFIC6 is mainly due to impaired bile acid transport and not directly related to the reduced efficiency of fat digestion. Additionally, the accumulation of bile acids in the liver can lead to liver damage and cirrhosis if left untreated.
PFIC7 (Progressive Familial Intrahepatic Cholestasis type 7) is associated with increased serum levels of the liver enzymes ALT and AST, and partial fibrosis of hepatocytes can be observed in liver tissue sections. Some patients may also develop hearing loss in childhood (Alhebbi et al., 2021; Bull et al., 2021). USP53 (ubiquitin specific peptidase 53) spliceosome variants can result in the premature termination of the protein, leading to the loss of its functional structure and abnormal metabolic and regulatory interactions. This can contribute to cholestasis in affected individuals (Gezdirici et al., 2023). Jing Zhang et al. did discover that USP53 mutations may play a role in some of the phenotypic features observed in cases of TJP2 defect (Zhang et al., 2020). TJP2 is involved in regulating cell junctions, which are important for maintaining the integrity of tissues such as the liver. Mutations in TJP2 can cause various liver diseases, including PFIC4. It has been suggested that USP53 may interact with TJP2 and regulate its function in the liver. Additionally, Olga Shatokhina et al. identified biallelic mutations in USP53 as one of the causes of cholestasis with low γ-GGT (gamma-glutamyltransferase) levels. Low γ-GGT levels are typically seen in patients with low bile flow and can be a sign of intrahepatic cholestasis (Shatokhina et al., 2021).
PFIC8 (Progressive Familial Intrahepatic Cholestasis type 8) is caused by mutations in the Kinesin-related protein 12(KIF12) gene. The researchers found that these mutations lead to disturbed hepatocyte polarity, which is a disruption in the alignment of cells within the liver tissue. This disruption can impair bile flow and lead to the accumulation of bile acids, causing cholestasis and other liver-related complications. The KIF12 gene does code for a protein involved in intracellular transport, which is important for maintaining normal liver function. Mutations in KIF12 can affect the proper functioning of this protein and disrupt the normal intracellular transport processes in hepatocytes (Ünlüsoy Aksu et al., 2019; Stalke et al., 2022).
Recent research by Angela Pepe et al. has identified a homozygous mutation in ANCHR (Abscission/NoCut checkpoint regulator, ZFYVE19) in a 6-year-old girl with chronic cholestasis. The patient exhibited hypercholinemia and severe itching, which were not consistent with classical pathogenic genes associated with PFIC. This newly discovered mutation has been classified as a novel non-syndromic phenotype of PFIC9 (OMIM #619849) (Pepe et al., 2023). ANCHR plays a key role in regulating neutron cell shedding during cell division. ANCHR delays cell shedding by stabilizing cytokinetic intercellular bridges (ICBs) to prevent premature extinction of intercellular chromosome bridges and accumulation of DNA damage, ultimately reducing the formation of dikaryotic cells (Massafra et al., 2017), (Bai et al., 2020). ANCHR interacts with Charged multivesicular body protein 4c (CHMP4C), which helps regulate the timing of abscission and prevents multinucleation in an Aurora-B-dependent manner. Additionally, ANCHR controls the late fall-off in DNA separation and midbody reaching through its interaction with BPS4 (Thoresen et al., 2014). Biallelic mutations in ZFYVE19 can lead to high serum GGT cholestasis and ductal plate malformations (DPM) or congenital liver fibrosis (Mandato et al., 2021). In vitro studies have shown that these mutations can cause centrality and axon abnormalities (Luan et al., 2021). Claudia Mandato et al. discovered that a homozygous senseless mutation in ZFYVE19 resulted in a truncated protein of 222 amino acids, leading to severe cholestasis and jaundice symptoms that manifested 2 months after birth. The patient also had elevated levels of serum bile acids and GGT (Mandato et al., 2021).
The researchers found that mutations in a gene called MYO5B were responsible for cholestasis and congenital diarrhea. MYO5B encodes a protein that is important for intracellular transport, and mutations in this gene lead to disruption of normal intestinal and liver function (Aldrian et al., 2021). In infancy, children with PFIC10 may present with jaundice, pruritus, and enlargement of the liver and spleen. Laboratory tests often show elevated levels of bilirubin, TBA, ALT, and AST, but the level of GGT is typically normal. Some patients may also develop diarrhea due to malabsorption of nutrients (Gonzales et al., 2017; Aldrian et al., 2021). MYO5B mutations have been associated with microvillus inclusion body disease (MVID), which affects the targeting of the tubule membrane by BSEP and leads to impaired bile acid excretion, ultimately causing cholestasis (Qiu et al., 2017). The formation of recycling endosomes can also be inhibited by MYO5B mutations, which interferes with protein localization between the Golgi apparatus and the cycling endosome (Tzaban et al., 2009; Overeem et al., 2020). This interference can lead to functional expression defects and ultimately result in cholestasis. MYO5B binds to Rabgtase, one of the largest families of proteins that regulate membrane transport, to help regulate specific membrane transport pathways. In non-polarized cells, it binds to Rab11a to recover transferrin, while in polarized cells, it plays a role in the localization of apical membrane proteins and the formation of a new lumen (Roland et al., 2011; Schneeberger et al., 2015). Patients with MYO5B mutation can exhibit impaired function of the apical circulatory corpuscle pathway in hepatocytes, leading to impaired targeting of BSEP to the tubule membrane (Engevik et al., 2018). This impairment can ultimately result in cholestasis. In addition, damage to the ileum caused by MYO5B mutations can lead to increased bile acid reabsorption (Girard et al., 2014). MYO5B is also implicated in the malignant development of some pheochromocytomas and paragangliomas, with changes in subcellular localization and metastasis from the cytoplasm to the cell membrane (Tomić et al., 2020). Additionally, lysophosphatidic acid has been shown to improve diarrhea symptoms in mice that are deficient in MYO5B (Kaji et al., 2020; Leng et al., 2020).
The researchers identified a homozygous R148W mutation in the Semaphorin 7A (SEMA7A) gene as a possible cause of PFIC in certain patients. SEMA7A is involved in cell signaling and migration, and mutations in this gene can lead to disrupted hepatocyte polarity, causing cholestasis and other liver-related complications. The findings highlight the importance of genetic testing for accurate diagnosis of cholestatic liver disease and the identification of potential treatment targets for these conditions. The study expands our understanding of the genetic basis of PFIC and provides new insights into the molecular mechanisms underlying bile flow disorders (Qiu et al., 2017).
PFIC12 (Progressive Familial Intrahepatic Cholestasis type 12) is caused by mutations in the VPS33B gene. VPS33B is a protein involved in intracellular transport and lysosomal function. Mutations in the VPS33B gene can result in abnormal liver cell polarity, leading to bile flow disruption and cholestasis. Patients with PFIC12 commonly present with additional symptoms beyond cholestasis. The joint curvature and renal dysfunction mentioned are characteristic features of another rare disorder called Arthrogryposis-Renal Dysfunction-Cholestasis (ARC) syndrome, which can be caused by mutations in the same VPS33B gene. It should be noted that not all patients with VPS33B mutations develop ARC syndrome, but those that do may present with these additional symptoms (Fu et al., 2019).
Alagille syndrome, also known as neonatal-associated jaundice, is a multi-system autosomal dominant genetic disease (Vandriel et al., 2023).
Obstruction of bile outflow in children can result in enlarged liver tissue, severe jaundice, pruritus, and several physical characteristics such as a prominent forehead, a large internasal septum, and a systolic murmur in the pulmonary arteries. Patients with this condition may also exhibit intellectual disability. More than half of the patients with this condition have been found to have renal insufficiency (Kamath et al., 2012), and peripheral chorioretinopathy can also occur (da Palma et al., 2021).
The majority of patients with this syndrome have mutations in the protein jagged-1 (JAG1) gene, while only 1% of patients have mutations in the neurogenic locus notch homolog protein 2 (NOTCH2) gene (McDaniell et al., 2006). JAG1 is a ligand of several Notch receptors and plays an essential role in proper Notch signaling. JAG1 is also involved in mammalian cardiovascular development, promoting the formation of blood vessels, aortic valves, and pulmonary valves (Andersson et al., 2018). JAG1 and NOTCH2 jointly regulate the ductal structure involved in bile epithelial cell formation. These genes are responsible for regulating the differentiation of bile duct fibroblasts into bile duct cells. Mutations in either JAG1 or NOTCH2 can lead to biliary deformity and abnormal bile excretion function (Hofmann et al., 2010). Researchers have identified specific targets that can either alleviate or aggravate the course of Alagille disease. For instance, transforming growth factor β85 signal and Transcription factor SOX-9 (SOX9) (Adams et al., 2020) can promote the formation of the biliary system in hepatocytes and potentially alleviate cholestatic liver disease. On the other hand, THBS2 can inhibit JAG1/NOTCH2 interaction, which may further exacerbate the progression of the disease (Tsai et al., 2016). Partial external bile duct drainage (PEBD) is currently an effective method for treating Alagille syndrome, as it has shown promising results in alleviating symptoms such as jaundice and pruritus. However, PEBD does not cure the underlying issues causing these symptoms and does not address the root cause of the disease. The problem of bile duct excretion in patients with Alagille syndrome has yet to be fully resolved (Olyaei et al., 2001). A Phase 2 clinical study found that maralixibat may have clinical significance for treating Alagille syndrome (Gonzales et al., 2021). Additionally, it has been suggested that enhancing JAGGED/NOTCH signaling through normal random variation, gene therapy, or NOTCH agonists may promote intrahepatic duct cell regeneration in patients with Alagille syndrome (Zhao et al., 2022).
Gilbert syndrome is a congenital autosomal recessive disorder characterized by mild unconjugated bilirubin metabolism disorder, which is caused by decreased UDP-glucuronosyltransferase 1A1 (UGT1A1) promoter activity (Gilbert et al., 2019; Hsu et al., 2022).
Patients with Gilbert syndrome typically have intermittent episodes of unconjugated hyperbilirubinemia, which means that there is excess bilirubin in the blood that has not been processed by the liver (Fretzayas et al., 2012). However, this is not due to hepatocellular disease (disease affecting the liver cells) or hemolysis (excessive breakdown of red blood cells), but rather a deficiency of the enzyme UDP-glucuronosyltransferase (UGT). This leads to impaired processing of bilirubin and its excretion into the bile, resulting in elevated levels of unconjugated bilirubin in the blood.
It is important to note that UGT1A1 homozygosity is only a predisposing factor for Gilbert syndrome, as its penetrance is only 50%, most likely due to the influence of modifier genes and the presence of specific haplotypes with other UGT1A1 genotypes.
UGT1A1 is an enzyme that catalyzes the glucuronidation of bilirubin, increasing its water solubility and promoting its excretion from bile and urine. UGT1A1 is primarily found in the liver, small intestine, and colon. Additionally, differential methylation of the −1 CpG site located within the upstream stimulatory factor response element that regulates UGT4A1 expression could explain a portion of the interindividual variability in UGT1A1 in hepatic glucuronidation. Thus, the diagnosis of GS is based on clinical symptoms and laboratory findings, rather than solely on genetic testing (Ehmer et al., 2012; Cooper et al., 2013; Yasar et al., 2013). Clinical studies in patients with Gilbert syndrome have shown that having slightly increased plasma bilirubin concentration or increased bilirubin bioavailability may be beneficial for alleviating metabolic syndrome, cardiovascular disease, and type 2 diabetes (Vitek et al., 2019). This beneficial effect may be due to the inhibition of platelet response and thrombosis by unbound plasma bilirubin. Mutations in ABCC2 have also been associated with other liver diseases, including LPAC, ICP, and BRIC (Kundur et al., 2015), however, the frequency of these mutations is similar to that in the general population (Corpechot et al., 2020).
Crigler-Najjar syndrome, also known as congenital non-obstructive non-hemolytic jaundice, is a genetic disease caused by autosomal mutations.
It is classified into two types: type I (recessive) and type II (dominant). Type I is homozygous and presents with severe jaundice within 1–4 days of birth without hemolysis. Due to the excellent lipid solubility of unbound bilirubin, it is easy for it to cross the blood-brain barrier, and high concentrations may cause bilirubin encephalopathy (Tukey and Strassburg, 2000). Type II Crigler-Najjar syndrome is a heterozygous form of the disorder that typically presents with mild disease and a late onset. Liver transplantation is an effective treatment for alleviating symptoms of CN1, but it is associated with potential complications, such as postoperative infection, immune rejection, and limited availability of donor organs (Gailite et al., 2018).
Recent basic studies have shown promise in treating CN1 using adeno-associated virus (AAV) injection of UGT1A1. While this method has demonstrated success in alleviating the progression of CN1 (Bortolussi et al., 2014; Villiger et al., 2018), there have been reports of brain damage and nucleoomycosis (Yueh et al., 2017) following AAV injection. These adverse effects may be related to the expression of P-glycoprotein, which is enriched in the blood-brain barrier and functions to pump bilirubin out of the brain. Pathological examination of tissues in patients with Crigler-Najjar syndrome has revealed abnormal development of the cerebellum, nerve cell death (Bortolussi et al., 2015), and myelin degeneration (Barateiro et al., 2016). These conditions have been positively associated with bilirubin encephalopathy. Recent research by Kevin A. Strauss et al. has shown that phototherapy can provide protective benefits for patients with CN1 if it is properly used (Strauss et al., 2020). Studies have found that both phototherapy and phenobarbital can effectively lower the levels of unconjugated bilirubin caused by UGT1A1 gene mutations. These treatments work by increasing the excretion and metabolism of bilirubin from the liver, reducing bilirubin accumulation in the blood and tissues. These findings emphasize the importance of early diagnosis and appropriate management of conditions such as Crigler-Najjar Syndrome Type II to prevent complications and improve patient outcomes (Barateiro et al., 2016; Liaqat et al., 2018; Bai et al., 2021; Cozzi et al., 2022).
Dubin-Johnson syndrome (DJs) is an autosomal recessive genetic disorder characterized by non-hemolytic, primarily conjugated hyperbilirubinemia and severe jaundice.
Serological tests typically reveal elevated levels of TBA, direct bilirubin, and glutamine transferase. Histomorphology observations of liver tissue in patients with DJs show dense pigmentation within liver cells. Immunohistochemical analyses of these tissues have revealed decreased levels of MRP2 and increased levels of BSEP (Togawa et al., 2018). Another study has shown that a mutation involving a hydrophobic amino acid, isoleucine, being replaced by phenylalanine results in a 75% reduction of MRP2 tubule membrane localization. This significantly reduces the efficiency of cholic acid efflux (Keitel et al., 2003).
Dubin-Johnson syndrome caused by ABCC2/MRP2 mutations can be due to various genetic abnormalities, including meaningless exon expression, base deletion, and abnormal transcription leading to premature stop codons. These mutations may lead to bilirubin efflux blockage due to retention of MRP2 protein in the endoplasmic reticulum (Erlinger et al., 2014). ABCC2/MRP2, is a member of the ATP-binding (ABC) family of ATP-dependent transporters that plays an important role in the hepatobiliary excretion of conjugated bilirubin molecules. ABCC2/MRP2 is essential for the detoxification of bilirubin and its proper functioning is necessary for maintaining healthy liver function (Saito et al., 2013). The function of ABCC2/MRP2’s nucleotide-binding domain can be affected by missense mutations, such as R768W and Q1382R. The R768W mutation results in abnormal translation and impaired sorting of MRP2 protein, while the Q1382R mutation affects substrate-induced ATP hydrolysis (Hashimoto et al., 2002). Additionally, the abnormal functioning of the PI3Kγ/AKT signaling pathway can lead to decreased utilization of MRP2 membrane surface, potentially impairing its ability to excrete bilirubin and other conjugated waste molecules (Beer et al., 2020). According to Rivka H Regev et al., UDCA was found to have a beneficial therapeutic effect on neonatal Dubin-Johnson syndrome (Regev et al., 2002).
Rotor Syndrome is an autosomal recessive genetic disease characterized by a deficiency in liver uptake and storage of bilirubin (Kimura et al., 2021).
Patients with Rotor Syndrome often present with a range of symptoms, including jaundice, hepatomegaly, and elevated levels of bilirubin and bile acids (Kimura et al., 2021).
Diagnosis of Rotor Syndrome typically involves laboratory tests such as bilirubin and bile acid measurements and imaging studies like MRI or CT scan (Kimura et al., 2021). Management of Rotor Syndrome may include supportive measures such as fluid replacement and nutritional support, as well as medications to reduce bilirubin and bile acid levels. The treatment approach may vary depending on the severity of symptoms and underlying conditions. Rotor Syndrome is thought to result from a deficiency in two transporter proteins, OATP1B1 and OATP1B3, that are responsible for the uptake of bilirubin glucuronide from the blood into the liver. In the absence of functional OATP1B1 and OATP1B3 proteins, bilirubin glucuronide cannot be efficiently cleared from the blood, resulting in hyperbilirubinemia, jaundice, and other symptoms associated with Rotor Syndrome (van de Steeg et al., 2012). Rotor syndrome presents as hyperbilirubinemia with clinical manifestations like those seen in Dubin-Johnson syndrome. However, in patients with rotor syndrome, liver histology was normal and there was no hyperpigmentation in the liver (Kumar et al., 2023). Urine tests in patients with Rotor syndrome have shown increased porphyrin excretion levels ranging from 2 to 5 times higher than the normal range (Strassburg, 2010). It should be noted that certain drugs, such as cyclosporine A, atorvastatin, and rifampicin, can potentially exacerbate symptoms in patients with Rotor Syndrome. These drugs may interfere with the processing and excretion of bilirubin and bile acids in the liver, leading to increased accumulation of these compounds in the blood and tissues. Therefore, caution should be exercised when prescribing medications to patients with Rotor Syndrome, and drug-induced liver injury should be monitored closely. It is important for patients with Rotor Syndrome to inform their healthcare providers about their condition and any medications they are taking, including over-the-counter drugs and herbal supplements (Morais and Machado, 2022).
Biliary atresia, also known as common bile duct dilatation, is a cystic malformation located near the site of common bile duct obstruction (Muise et al., 2006). The prevalence of congenital biliary atresia varies by geography and ethnicity There are significant regional differences in the incidence of BA, ranging from 1:15,000 to 19,000 in Europe and 1:5–10 000 in Japan and China. In addition, both intrahepatic and extrahepatic bile duct obstruction can lead to biliary atresia, but extrahepatic obstruction is typical (Mack and Sokol, 2005; Cavallo et al., 2022).
This condition can be classified into three types based on the location of the atresia. Type I biliary atresia involves no intrahepatic bile duct injury but varying degrees of loss or obstruction in the common bile duct. Choledochojejunostomy is a possible treatment option, and the prognosis for this type is generally good. Type II biliary atresia is associated with intrahepatic bile duct atresia but does not involve damage to the gallbladder or common bile duct. Type III biliary atresia involves hepatic portal atresia (Mieli-Vergani and Vergani, 2009). Biliary atresia can be classified into different types based on its etiology, including Biliary Atresia Splenic Malformation (BASM), cystic atresia, viral biliary atresia, and CMV-IgM+ve associated biliary atresia (Pang et al., 2021). Children with BASM may have malformations of the spleen (polysplenia or absence of spleen), heterotopic viscera (Schwarz et al., 2013), malformations of the heart, and abnormal blood vessels (Lakshminarayanan and Davenport, 2016). These abnormalities can contribute to the development of biliary atresia in affected children. Children with biliary atresia may benefit from Kasai portoenterostomy biliary drainage, which can reduce the need for liver transplantation in 80% of cases. The success of surgery is evaluated based on the level of bilirubin in plasma and the color of the stool (Mieli-Vergani and Vergani, 2009). However, if children still experience persistent cholestasis, cirrhosis combined with liver dysfunction, or portal hypertension after surgery, liver transplantation should be considered. Untreated children may develop cholestatic cirrhosis that progresses to liver failure within 2 years. The etiology of biliary atresia is not completely clear, but it can be caused by genetics, inflammation, viruses, poisons, and other factors (Asai et al., 2015). In addition, liver obstruction or fibrosis due to inflammation may occur in children with diseases such as rotavirus and reovirus (Szavay et al., 2002). Therefore, suppressing the inflammatory response may be a potential treatment option to slow the progression of biliary atresia (Superina et al., 2011).
In the vast majority of patients, currently referred to as isolated biliary atresia, there are still too few reported etiological or pathogenesis clues (Davenport et al., 2021). Isolated biliary atresia is a rare pediatric liver disease in which bile ducts in the liver become inflamed and blocked, leading to liver damage and eventually liver failure (Cui et al., 2013). While the exact cause of isolated biliary atresia is still unknown, research has suggested that it may be the result of both genetic and non-genetic factors (Garcia-Barceló et al., 2010; Lam et al., 2021). Microscopic examination of patients with biliary atresia showed that the cilia of bile duct cells were less in number and shorter in length, and the function of transporting bile acids was downregulated, leading to the accumulation of bile acids, which led to cytotoxicity and ciliary injury (Xia et al., 2006). Genes associated with cilia function such as MAN1A2, ARF6, CPLANE, and JBTS17 mutations can lead to bile duct dysplasia (Chu et al., 2012; So et al., 2020). Other studies have demonstrated that mutations in FOXA2, GPC1, ADD3, PKD1L1, EFEMP1/3, STIP1, XPNPEP1, REV1 and JAG1 genes can increase an individual’s genetic susceptibility to biliary atresia (Cui et al., 2013; Tsai et al., 2014; Tsai et al., 2015; Rajagopalan et al., 2020; Davenport et al., 2021). As for non-genetic mechanisms of pathogenesis, some studies have suggested that viral infections could play a role. Specifically, cytomegalovirus (CMV) has been found in the livers of infants with biliary atresia, leading some researchers to believe that it may trigger an inflammatory response that damages the bile ducts (Bezerra et al., 2014).
Other environmental factors that have been linked to isolated biliary atresia include exposure to toxins and pollutants, as well as maternal factors such as smoking during pregnancy or certain medications taken during pregnancy (Cavallo et al., 2022). In addition, abnormal expression of methylation and microRNA can lead to damage of biliary epithelial cells, which may be related to the pathogenesis of biliary atresia (He et al., 2022). Various treatment options, such as bile acid analogues, drugs that reduce the amount of bile acid in the liver, anti-inflammatory and immunosuppressive agents, and anti-fibrotic drugs, can provide relief from the symptoms of biliary atresia to a certain extent.
Neonatal sclerosing cholangitis (NSC) is a severe autosomal recessive liver disease that often results in decompensated biliary cirrhosis during childhood, requiring liver transplantation (Wei et al., 2023).
The patient presented with jaundice, poor growth, and pruritus (Girard et al., 2016).
DCDC2 gene mutations have been strongly associated with this condition (Wei et al., 2023). DCDC2 inhibits the classical WNT signaling pathway, regulates ciliary production and length, and is expressed in most organs, including bile duct epithelial cells. Under normal conditions, the DCDC2 protein is found in the cytoplasm and cilia of cholangiocytes. However, in individuals with neonatal sclerosing cholangitis who have mutations in the DCDC2 gene, the mutant protein accumulates in the cytoplasm and is not present in the cilia (Girard et al., 2016). The DCDC2 gene is responsible for producing a protein that plays an important role in the development and function of cells in the liver’s bile ducts. When the gene is mutated, this protein may not function properly, leading to inflammation and scarring in the bile ducts. These changes can cause symptoms such as jaundice, poor growth, and itching (Girard et al., 2016). Further research is needed to better understand the mechanisms behind this genetic association and develop effective treatments for this condition (Grammatikopoulos et al., 2016). Georg-Friedrich Vogel et al. found that a variation in the splicing site of the DCDC2 receptor (c.294–2A>G) can lead to intralobular cholestasis, large catheter sclerosis, and catheter plate deformity (Chen et al., 2018). The patient exhibited symptoms such as jaundice, hepatosplenomegaly, hyperbilirubinemia, cholestasis, and elevated levels of serum GGT activity. A liver biopsy revealed varying degrees of bile duct hyperplasia, portal inflammation, and/or fibrosis (Wei et al., 2023).
Wilson’s disease is an autosomal recessive genetic disorder that presents with loss of appetite, hepatosplenomegaly, jaundice, and ascites (Członkowska et al., 2018).
The abnormal copper metabolism in the body leads to deposition of ceruloplasmin in the liver, which can inhibit bile excretion and cause cholestasis. In severe cases, acute liver failure may occur. If the ceruloplasmin level is low, 24-h urine copper or blood copper tests should be performed. If the diagnosis is not confirmed, molecular genetic analysis of ATP7B can also be carried out to confirm the diagnosis (Kwo et al., 2017). In addition to ABCC2 mutations, other genetic mutations such as those in MMP9 can also contribute to the development of cholecystitis. In vitro studies have shown that Abcc12 mutation in zebrafish infants can lead to bile duct cell apoptosis. Knocking out MMP9 results in decreased interlobular bile duct numbers, increased serum AKP and TBIL levels, and aggravated bile duct cell apoptosis (Pham et al., 2021).
Treatment with copper chelators such as D-penicillamine, trientine, dimercaptosuccinic acid and Zinc salts has been shown to significantly improve liver injury and symptoms in patients with Wilson’s disease (Yuan et al., 2021).
ICP (Walker et al., 2020) is a unique complication that occurs during the second and third trimesters of pregnancy, with incidence rates ranging from 0.2% to 25% (Palmer et al., 2019).
Patients with ICP often experience non-specific rash and pruritus, which can be aggravated at night and lead to insomnia, irritability, and even depression. Abdominal pain, nausea, and vomiting may also occur in some cases. Pruritus usually resolves within 1–2 days after delivery, although in a few patients it can take up to a week. However, the recurrence rate of ICP in subsequent pregnancies is high, at about 60% (Puljic et al., 2015). Laboratory tests have shown that patients with ICP often have increased levels of serum alkaline phosphatase, bile acid, aspartate aminotransferase, and alanine aminotransferase. Due to malabsorption of lipid substances and lipid-soluble vitamins, patients with ICP may experience fatty stool, osteoporosis, hemolysis, and other related conditions. Elevated maternal bile acid levels in ICP can seriously affect the health of newborns and increase the risk of complications such as premature delivery, intrauterine asphyxia, meconium staining of amniotic fluid, and fetal bradycardia (Floreani and Gervasi, 2016). In addition to affecting maternal health, ICP can also have adverse effects on infant cardiac function. Infants born to mothers with untreated ICP may experience abnormal cardiac rhythms, and the severity of the condition is often proportional to maternal bile acid levels (Henry and Welsh, 2015; Vasavan et al., 2021). ICP has been reported to be strongly associated with hepatitis C virus (HCV) infection. HCV can impair the function of ABC protein transporters, leading to disruptions in bile acid transport and metabolism, which may contribute to the development of ICP in affected individuals (Reyes et al., 2000; Hinoshita et al., 2001; Iwata et al., 2011).
Several bile acid-related synthesis and transport proteins are involved in the development of ICP, including ATP8B1, ABCB11, ABCB4, ABCC2, NR1H4, FGF19, and MDR3. These proteins play important roles in regulating the metabolism and transport of bile acids in the liver, and mutations or functional abnormalities in these genes may contribute to the pathogenesis of ICP (Kawakita et al., 2015). In addition, 17α-estradiol can induce pregnancy-induced cholestasis by trans-inhibiting the tubulomembrane targeting activity of BSEP. The metabolites of 17α-estradiol are then exported from MRP2 to the tubule lumen. Although BSEP has high homology with MDR1 and MRP2, these two transporters cannot compensate for cholestasis caused by BSEP mutations because of their low affinity for human primary bile acids. MDR3 mutations are also closely associated with ICP, and approximately 16% of ICP cases are caused by mutations in this gene (Piechota and Jelski, 2020). FXR mutations have also been identified as potential contributors to the development of cholestasis, including ICP. In Caucasian patients with cholestasis, FXR mutations have been detected by sequencing. These mutations may be related to the FXR-BSEP pathway, which plays an important role in regulating bile acid homeostasis and transport in the liver. Further studies are needed to understand the precise mechanisms underlying the relationship between FXR mutations and ICP (Müllenbach et al., 2005; Van Mil et al., 2007). UDCA is the first-line treatment for ICP as it can reduce pruritus and jaundice in affected patients. However, its effectiveness varies among individuals. UDCA works by increasing the water solubility of bile acids, which reduces their harmful effects on hepatocytes and bile duct epithelial cells. It also protects the cell membrane of hepatocytes and decreases the likelihood of bile acids passing through the placenta to the fetus (Beuers et al., 2015). Although UDCA is often prescribed to reduce symptoms in patients with ICP, a study conducted by British scholars found that it may not effectively prevent adverse perinatal outcomes, as alanine aminotransferase and ICP were not strongly associated with an increased risk of preterm birth or stillbirth (Chappell et al., 2019). In cases where premature delivery is a concern, glucocorticoids may be recommended to reduce the risk of fetal respiratory insufficiency and intraventricular hemorrhage. These medications are typically administered before 34 weeks of gestation to optimize their effectiveness. An individual patient data (IPD) meta-analysis has found that the risk of stillbirth significantly increases when the serum bile acid concentration in patients with ICP reaches 100 μM/L or higher. This highlights the importance of timely diagnosis and treatment of ICP to prevent adverse outcomes for both the mother and the fetus (Ovadia et al., 2019). It is worth mentioning that Peter H Dixon et al. conducted a study to identify genetic mutations associated with ICP. They found that 11 genes were potentially linked to the development of the condition, including GCKR, ABCG5/8, ABCB11, SCARB2, ABCB1/4, CYP7A1, SERPNA1, ENPP7, TMEM147, SULT2A1, and HNF4A33. Further research is needed to better understand the role these genes play in the pathogenesis of ICP (Dixon et al., 2017; Liu et al., 2020).
Citrin deficiency is a genetic disorder caused by mutations in the SLC25A13 gene, which encodes an electrogenic aspartate/glutamate antiporter. The condition is inherited in an autosomal recessive pattern and can manifest in different stages. The first stage of Citrin deficiency is characterized by intrahepatic cholestasis in newborns. This represents an early-onset manifestation of the disease and is often associated with jaundice, hepatomegaly, and other related symptoms. Further stages of Citrin deficiency may develop later in life and can include neurological symptoms and other complications (Hayasaka, 2021).
DILI is an infrequent but significant adverse drug reaction that can lead to acute liver failure, making it a crucial cause of this condition (Chalasani et al., 2021; Fernandez-Checa et al., 2021).
In cases where DILI is caused by rifampin, treatment with 4-PB has been shown to have a protective effect. This protection is thought to arise from 4-PB inhibiting endoplasmic reticulum (ER) stress and preventing the ubiquitination of MRP2 in hepatocytes. By blocking these processes, 4-PB can help prevent the development of rifampin-induced hepatocyte injury and mitigate the risk of complications associated with DILI (Chen et al., 2022). Numerous genome-wide association studies (GWAS) focusing on DILI have identified various HLA alleles that exhibit a strong association with DILI, albeit being specific to certain drugs. For instance, HLA-B57:01 is linked to DILI caused by flucloxacillin reactions, while HLA-A02:01 and HLA-DRB115:01 are associated with amoxicillin-clavulanic acid (AC)-induced DILI. Additionally, HLA-B35:02 is correlated with minocycline-induced DILI, whereas HLA-A*33:01 is related to terbinafine-induced DILI. These HLA alleles may also potentially play a role in the development of DILI due to other drugs (Cirulli et al., 2019).
Elizabeth T Cirulli et al. conducted a study and found that multiple DILI and cholestasis may be associated with PTPN22 gene mutations. The PTPN22 gene encodes a protein involved in regulating immune cell function, which suggests that immune-mediated mechanisms may play a role in the pathogenesis of drug-induced liver injury and cholestasis.
LPAC is a rare disease that causes adult intrahepatic cholelithiasis, and it typically affects individuals under 40 years old. Clinical manifestations include biliary colic and acute cholangitis. More than half of LPAC patients have been found to have ABCB4 mutation, according to Catherine Dong et al. (2021). If left untreated, LPAC syndrome may progress to cholangiocarcinoma (Khabou et al., 2019).
BRIC is a rare genetic liver disorder that causes intermittent episodes of cholestasis, which is a condition where bile flow from the liver to the small intestine is blocked or impaired.
BRIC is caused by mutations in either the ATP8B1 gene (type 1) or the ABCB11 gene (type 2), both of which are involved in the transport of bile acids. The main difference between type 1 and type 2 BRIC is the underlying genetic mutation. Type 1 BRIC is caused by mutations in the ATP8B1 gene, which regulates bile salt export from the liver. Type 2 BRIC is caused by mutations in the ABCB11 gene, which encodes a protein called the BSEP that also plays a role in bile acid transport. Both types of BRIC can present with similar symptoms of intermittent attacks of cholestasis, including abdominal pain, jaundice, itching, and fatigue. However, there can be variations in the severity and frequency of these attacks depending on the specific gene mutation involved.
BRIC1 is associated with heterozygous mutations in ATP8B1, such as p. T888K (Bing et al., 2022), c.1429 + 2T>G (Mihara et al., 2022), c.1817T>C, and p. I606T (Chen et al., 2021). These mutations are thought to affect the tubular membrane localization of ATP8B1, leading to impaired bile acid transport and cholestasis. Unlike PFIC1, BRIC1/ICP is a milder form of the disease that can recur intermittently and is often responsive to medical therapy (Folmer et al., 2009). What’s worse, R Mullenbach et al. found that mutations at two heterozygous sites of ATP8B1 may lead to ICP, which shows the increasing phosphodiester signal and decreasing phosphomonoester/phosphodiester ratio (Müllenbach et al., 2005).
In contrast, BRIC is a milder form of the disease that typically presents with recurrent episodes of cholestasis and pruritus, but liver damage is generally mild. Diagnosis of both PFIC2 and BRIC typically involves genetic testing, and treatment options depend on the individual patient’s symptoms and disease severity (Henkel et al., 2019). Patients with BRIC can often be managed effectively with medical therapy, such as medications like UDCA and rifampicin, which Improve bile flow and reduce toxicity of bile acids (Koukoulioti et al., 2021). Other medications and interventions, such as bile acid sequestrants, may also be considered depending on the individual patient’s needs. Surgical interventions, such as liver transplantation or biliary diversion surgery, may be necessary for patients with PFIC2 who do not respond to medical therapy.
Children may develop liver fibrosis and even cirrhosis (Cui et al., 2013; Tsai et al., 2014; Tsai et al., 2015). The symptoms of HIHC usually appear during infancy or early childhood and may include jaundice, itching, pale stools, failure to thrive, and growth retardation (Liu et al., 2018; Schonfeld and Brown, 2019). Some forms of HIHC can also lead to progressive liver disease, cirrhosis, and liver failure. Treatment for HIHC generally involves supportive care, such as medication to manage, including UDCA, rifampin and 4-PB (van der Velden et al., 2010; Beuers et al., 2014). In severe cases, liver transplantation may be necessary. In some cases, 4-PB may be used to treat HIHC by increasing the expression of the affected transporter proteins on the canalicular membrane, improving bile flow, and reducing cholestasis-related complications (Naoi et al., 2014; van der Woerd et al., 2016). However, more research is needed to understand the efficacy of 4-PB treatment for HIHC. Mutations in transport proteins such as ATP8B1, BSEP, and MRP2 can result in abnormal positioning of functional proteins, consequently disrupting the normal transportation of bile acids. However, administering 4-PB can partially alleviate the symptoms caused by these proteins, enhance bile acid transport, and reduce the probability of complications relating to cholestasis (Hayashi and Sugiyama, 2007; van der Velden et al., 2010; Hasegawa et al., 2014; Gonzales et al., 2015; Amzal et al., 2021). Additionally, 4-PB is capable of regulating MRP2 ubiquitination by restraining ER stress. By enhancing the efflux of hepatotoxic drugs from the liver, this effect shields against DILI (Chen et al., 2022).
HIHC is a rare genetic disorder that affects the liver’s ability to secrete bile. It is caused by mutations in several different genes, including ATP8B1, ABCB11, ABCB4 and so on (Wang et al., 2003; Paulusma et al., 2006; Ghonem et al., 2014). These genes play important roles in the transport of bile acids and phospholipids from the hepatocytes into the bile canaliculi, and mutations in these genes can disrupt this process. NR1H4 mutation leads to abnormal bile acid synthesis rate (Hassan et al., 2022). SLC51A mutation results in reduced bile acid reabsorption efficiency (Ballatori et al., 2013; Kunst et al., 2021). Mutations in tight junction proteins (TJP2, USP53), KIF12, MYO5B, SEMA7A, and VPS33B lead to impaired hepatocyte polarization, affecting the number of tubular membrane transporters and reducing bile acid transport efficiency (Qiu et al., 2017; Fu et al., 2019; Ünlüsoy Aksu et al., 2019; Wei et al., 2020; Zhang et al., 2020; Aldrian et al., 2021; Stalke et al., 2022). JAG1 or NOTCH2 mutations are linked with biliary malformations and abnormal bile excretion (Hofmann et al., 2010). UGT1A1 and ABCC2, SLCO1B1/3 mutations result in abnormal bilirubin excretion and metabolism (Hashimoto et al., 2002; van de Steeg et al., 2012; Saito et al., 2013; Gilbert et al., 2019; Hsu et al., 2022). DCDC2 mutations are associated with cilia and cause restricted bile acid flow (Wei et al., 2023). ATP7B mutation leads to copper deposition, causing substantial liver damage that disrupts hepatocyte function (Kwo et al., 2017). Other genes including FOXA2, GPC1, ADD3, PKD1L, EFEMP1/3, STIP1 were found to be associated with biliary atresia. As shown in Figure 1, the molecular mechanisms behind hereditary intrahepatic cholestasis were summarized.
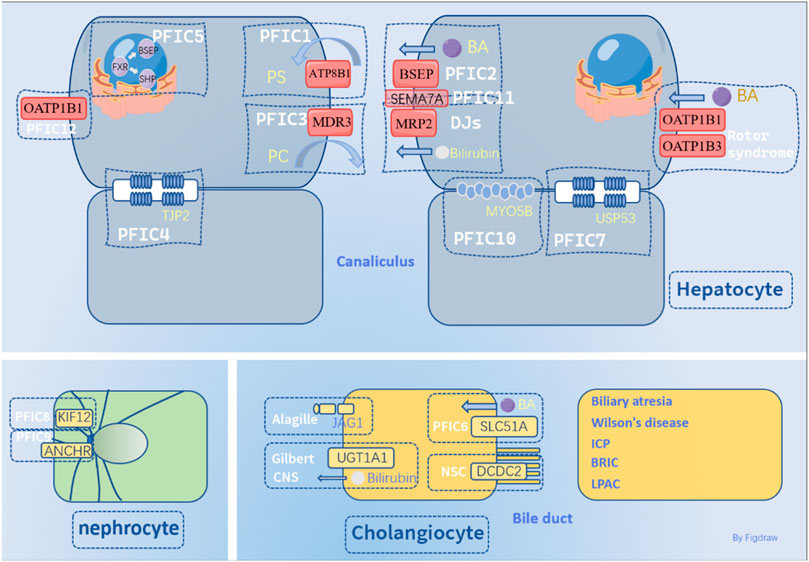
FIGURE 1. Molecular mechanism behind hereditary intrahepatic cholestasis. PFIC, progressive familial cholestasis; PS, Phosphatidylserine; PC, Phosphatidylcholine; BA, bile acid; DJs, Dubin-Johnson syndrome; ICP, Intrahepatic cholestasis of pregnancy; BRIC, Benign recurrent intrahepatic cholestasis; LPAC, Low phospholipid-associated cholelithiasis. This figure was technically supported by Figdraw.
SX conceived the study and drafted the manuscript. SW, XM, ZZ, JY, JW, LC, MJ, HL, participated in writing the manuscript. XZ and YZ made strict revisions to the manuscript. All authors contributed to the article and approved the submitted version.
This present research was supported by National Natural Science Foundation of China (82274187), the Major Program of the National Natural Science Foundation of China (82192915) and Major scientific research problems and key topics of medical technology problems of China Medical Education Association (2022KTZ016).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adams, J. M., Huppert, K. A., Castro, E. C., Lopez, M. F., Niknejad, N., Subramanian, S., et al. (2020). Sox9 is a modifier of the liver disease severity in a mouse model of Alagille syndrome. Hepatol. Balt. Md 71 (4), 1331–1349. doi:10.1002/hep.30912
Aida, K., Hayashi, H., Inamura, K., Mizuno, T., and Sugiyama, Y. (2014). Differential roles of ubiquitination in the degradation mechanism of cell surface-resident bile salt export pump and multidrug resistance-associated protein 2. Mol. Pharmacol. 85 (3), 482–491. doi:10.1124/mol.113.091090
Aldrian, D., Vogel, G. F., Frey, T. K., Ayyıldız Civan, H., Aksu, A. Ü., Avitzur, Y., et al. (2021). Congenital diarrhea and cholestatic liver disease: Phenotypic spectrum associated with MYO5B mutations. J. Clin. Med. 10 (3), 481. doi:10.3390/jcm10030481
Alhebbi, H., Peer-Zada, A. A., Al-Hussaini, A. A., Algubaisi, S., Albassami, A., AlMasri, N., et al. (2021). New paradigms of USP53 disease: Normal GGT cholestasis, BRIC, cholangiopathy, and responsiveness to rifampicin. J. Hum. Genet. 66 (2), 151–159. doi:10.1038/s10038-020-0811-1
Amirneni, S., Haep, N., Gad, M. A., Soto-Gutierrez, A., Squires, J. E., and Florentino, R. M. (2020). Molecular overview of progressive familial intrahepatic cholestasis. World J. Gastroenterol. 26 (47), 7470–7484. doi:10.3748/wjg.v26.i47.7470
Amzal, R., Thébaut, A., Lapalus, M., Almes, M., Grosse, B., Mareux, E., et al. (2021). Pharmacological premature termination codon readthrough of ABCB11 in bile salt export pump deficiency: An in vitro study. Hepatol. Balt. Md 73 (4), 1449–1463. doi:10.1002/hep.31476
Ananthanarayanan, M., Li, Y., Surapureddi, S., Balasubramaniyan, N., Ahn, J., Goldstein, J. A., et al. (2011). Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. Am. J. Physiol. Gastrointest. Liver Physiol. 300 (5), G771–G781. doi:10.1152/ajpgi.00499.2010
Andersson, E. R., Chivukula, I. V., Hankeova, S., Sjöqvist, M., Tsoi, Y. L., Ramsköld, D., et al. (2018). Mouse model of Alagille syndrome and mechanisms of Jagged1 missense mutations. Gastroenterology 154 (4), 1080–1095. doi:10.1053/j.gastro.2017.11.002
Aronson, S. J., Bakker, R. S., Shi, X., Duijst, S., Ten Bloemendaal, L., de Waart, D. R., et al. (2019). Liver-directed gene therapy results in long-term correction of progressive familial intrahepatic cholestasis type 3 in mice. J. Hepatol. 71 (1), 153–162. doi:10.1016/j.jhep.2019.03.021
Asai, A., Miethke, A., and Bezerra, J. A. (2015). Pathogenesis of biliary atresia: Defining biology to understand clinical phenotypes. Nat. Rev. Gastroenterol. Hepatol. 12 (6), 342–352. doi:10.1038/nrgastro.2015.74
Baghdasaryan, A., Chiba, P., and Trauner, M. (2014). Clinical application of transcriptional activators of bile salt transporters. Mol. Asp. Med. 37 (100), 57–76. doi:10.1016/j.mam.2013.12.001
Bai, J., Wioland, H., Advedissian, T., Cuvelier, F., Romet-Lemonne, G., and Echard, A. (2020). Actin reduction by MsrB2 is a key component of the cytokinetic abscission checkpoint and prevents tetraploidy. Proc. Natl. Acad. Sci. U. S. A. 117 (8), 4169–4179. doi:10.1073/pnas.1911629117
Bai, J., Li, L., Liu, H., Liu, S., Bai, L., Song, W., et al. (2021). UGT1A1-related bilirubin encephalopathy/kernicterus in adults. J. Clin. Transl. Hepatol. 9 (2), 180–186. doi:10.14218/JCTH.2020.00108
Ballatori, N., Christian, W. V., Wheeler, S. G., and Hammond, C. L. (2013). The heteromeric organic solute transporter, ostα-ostβ/SLC51: A transporter for steroid-derived molecules. Mol. Asp. Med. 34 (2-3), 683–692. doi:10.1016/j.mam.2012.11.005
Barateiro, A., Chen, S., Yueh, M. F., Fernandes, A., Domingues, H. S., Relvas, J., et al. (2016). Reduced myelination and increased glia reactivity resulting from severe neonatal hyperbilirubinemia. Mol. Pharmacol. 89 (1), 84–93. doi:10.1124/mol.115.098228
Beer, A. J., Hertz, D., Seemann, E., Beretta, M., Westermann, M., Bauer, R., et al. (2020). Reduced Mrp2 surface availability as PI3Kγ-mediated hepatocytic dysfunction reflecting a hallmark of cholestasis in sepsis. Sci. Rep. 10 (1), 13110. doi:10.1038/s41598-020-69901-3
Bessho, K., and Bezerra, J. A. (2011). Biliary atresia: Will blocking inflammation tame the disease? Annu. Rev. Med. 62, 171–185. doi:10.1146/annurev-med-042909-093734
Beuers, U., Kremer, A. E., Bolier, R., and Elferink, R. P. J. O. (2014). Pruritus in cholestasis: Facts and fiction. Hepatol. Balt. Md 60 (1), 399–407. doi:10.1002/hep.26909
Beuers, U., Trauner, M., Jansen, P., and Poupon, R. (2015). New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 62, S25–S37. doi:10.1016/j.jhep.2015.02.023
Bezerra, J. A., Spino, C., Magee, J. C., Shneider, B. L., Rosenthal, P., Wang, K. S., et al. (2014). Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: The START randomized clinical trial. JAMA 311 (17), 1750–1759. doi:10.1001/jama.2014.2623
Bezerra, J. A., Wells, R. G., Mack, C. L., Karpen, S. J., Hoofnagle, J. H., Doo, E., et al. (2018). Biliary atresia: Clinical and research challenges for the twenty-first century. Hepatol. Balt. Md 68 (3), 1163–1173. doi:10.1002/hep.29905
Bing, H., Li, Y. L., Li, D., Zhang, C., and Chang, B. (2022). Case report: A rare heterozygous ATP8B1 mutation in a BRIC1 patient: Haploinsufficiency? Front. Med. 9, 897108. doi:10.3389/fmed.2022.897108
Bortolussi, G., Zentillin, L., Vaníkova, J., Bockor, L., Bellarosa, C., Mancarella, A., et al. (2014). Life-long correction of hyperbilirubinemia with a neonatal liver-specific AAV-mediated gene transfer in a lethal mouse model of Crigler-Najjar Syndrome. Hum. Gene Ther. 25 (9), 844–855. doi:10.1089/hum.2013.233
Bortolussi, G., Codarin, E., Antoniali, G., Vascotto, C., Vodret, S., Arena, S., et al. (2015). Impairment of enzymatic antioxidant defenses is associated with bilirubin-induced neuronal cell death in the cerebellum of Ugt1 KO mice. Cell Death Dis. 6 (5), e1739. doi:10.1038/cddis.2015.113
Bull, L. N., Ellmers, R., Foskett, P., Strautnieks, S., Sambrotta, M., Czubkowski, P., et al. (2021). Cholestasis due to USP53 deficiency. J. Pediatr. Gastroenterol. Nutr. 72 (5), 667–673. doi:10.1097/MPG.0000000000002926
Byun, S., Jung, H., Chen, J., Kim, Y. C., Kim, D. H., Kong, B., et al. (2019). Phosphorylation of hepatic farnesoid X receptor by FGF19 signaling-activated Src maintains cholesterol levels and protects from atherosclerosis. J. Biol. Chem. 294 (22), 8732–8744. doi:10.1074/jbc.RA119.008360
Cavallo, L., Kovar, E. M., Aqul, A., McLoughlin, L., Mittal, N. K., Rodriguez-Baez, N., et al. (2022). The epidemiology of biliary atresia: Exploring the role of developmental factors on birth prevalence. J. Pediatr. 246, 89–94.e2. doi:10.1016/j.jpeds.2022.03.038
Chalasani, N. P., Maddur, H., Russo, M. W., Wong, R. J., and Reddy, K. R.Practice Parameters Committee of the American College of Gastroenterology (2021). ACG clinical guideline: Diagnosis and management of idiosyncratic drug-induced liver injury. Am. J. Gastroenterol. 116 (5), 878–898. doi:10.14309/ajg.0000000000001259
Chappell, L. C., Bell, J. L., Smith, A., Linsell, L., Juszczak, E., Dixon, P. H., et al. (2019). Ursodeoxycholic acid versus placebo in women with intrahepatic cholestasis of pregnancy (PITCHES): A randomised controlled trial. Lancet Lond Engl. 394 (10201), 849–860. doi:10.1016/S0140-6736(19)31270-X
Chen, F., Ananthanarayanan, M., Emre, S., Neimark, E., Bull, L. N., Knisely, A. S., et al. (2004). Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology 126 (3), 756–764. doi:10.1053/j.gastro.2003.12.013
Chen, H. L., Wu, S. H., Hsu, S. H., Liou, B. Y., Chen, H. L., and Chang, M. H. (2018). Jaundice revisited: Recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J. Biomed. Sci. 25 (1), 75. doi:10.1186/s12929-018-0475-8
Chen, H., Wu, D., Jiang, W., Lei, T., Lu, C., and Zhou, T. (2021). Case report: A novel homozygous variant identified in a Chinese patient with benign recurrent intrahepatic cholestasis-type 1. Front. Med. 8, 705489. doi:10.3389/fmed.2021.705489
Chen, J., Wu, H., Tang, X., and Chen, L. (2022). 4-Phenylbutyrate protects against rifampin-induced liver injury via regulating MRP2 ubiquitination through inhibiting endoplasmic reticulum stress. Bioengineered 13 (2), 2866–2877. doi:10.1080/21655979.2021.2024970
Cheng, Y., Woolf, T. F., Gan, J., and He, K. (2016). In vitro model systems to investigate bile salt export pump (BSEP) activity and drug interactions: A review. Chem. Biol. Interact. 255, 23–30. doi:10.1016/j.cbi.2015.11.029
Cheng, M. T., Chen, Y., Chen, Z. P., Liu, X., Zhang, Z., Chen, Y., Hou, W. T., and Zhou, C. Z. (2022). Structural insights into the activation of autoinhibited human lipid flippase ATP8B1 upon substrate binding. Proceedings of the National Academy of Sciences of the United States of America, 119 (14), e2118656119. doi:10.1073/pnas.2118656119
Chu, A. S., Russo, P. A., and Wells, R. G. (2012). Cholangiocyte cilia are abnormal in syndromic and non-syndromic biliary atresia. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 25 (5), 751–757. doi:10.1038/modpathol.2011.212
Cirulli, E. T., Nicoletti, P., Abramson, K., Andrade, R. J., Bjornsson, E. S., Chalasani, N., et al. (2019). A missense variant in PTPN22 is a risk factor for drug-induced liver injury. Gastroenterology 156 (6), 1707–1716.e2. doi:10.1053/j.gastro.2019.01.034
Cooper, D. N., Krawczak, M., Polychronakos, C., Tyler-Smith, C., and Kehrer-Sawatzki, H. (2013). Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 132 (10), 1077–1130. doi:10.1007/s00439-013-1331-2
Corpechot, C., Barbu, V., Chazouillères, O., Broué, P., Girard, M., Roquelaure, B., et al. (2020). Genetic contribution of ABCC2 to Dubin-Johnson syndrome and inherited cholestatic disorders. Liver Int. Off. J. Int. Assoc. Study Liver 40 (1), 163–174. doi:10.1111/liv.14260
Cozzi, L., Nuti, F., Degrassi, I., Civeriati, D., Paolella, G., and Nebbia, G. (2022). Gilbert or crigler-najjar syndrome? Neonatal severe unconjugated hyperbilirubinemia with P364L UGT1A1 homozygosity. Ital. J. Pediatr. 48 (1), 59. doi:10.1186/s13052-022-01251-4
Cui, S., Leyva-Vega, M., Tsai, E. A., EauClaire, S. F., Glessner, J. T., Hakonarson, H., et al. (2013). Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 144 (5), 1107–1115. doi:10.1053/j.gastro.2013.01.022
Członkowska, A., Litwin, T., Dusek, P., Ferenci, P., Lutsenko, S., Medici, V., et al. (2018). Wilson disease. Nat. Rev. Dis. Primer 4 (1), 21. doi:10.1038/s41572-018-0018-3
Czuba, L. C., Hillgren, K. M., and Swaan, P. W. (2018). Post-translational modifications of transporters. Pharmacol. Ther. 192, 88–99. doi:10.1016/j.pharmthera.2018.06.013
da Palma, M. M., Igelman, A. D., Ku, C., Burr, A., You, J. Y., Place, E. M., et al. (2021). Characterization of the spectrum of ophthalmic changes in patients with Alagille syndrome. Invest. Ophthalmol. Vis. Sci. 62 (7), 27. doi:10.1167/iovs.62.7.27
Davenport, M., Muntean, A., and Hadzic, N. (2021). Biliary atresia: Clinical phenotypes and aetiological heterogeneity. J. Clin. Med. 10 (23), 5675. doi:10.3390/jcm10235675
Davit-Spraul, A., Fabre, M., Branchereau, S., Baussan, C., Gonzales, E., Stieger, B., et al. (2010). ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): Phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatol. Balt. Md 51 (5), 1645–1655. doi:10.1002/hep.23539
De Vree, J. M., Ottenhoff, R., Bosma, P. J., Smith, A. J., Aten, J., and Oude Elferink, R. P. (2000). Correction of liver disease by hepatocyte transplantation in a mouse model of progressive familial intrahepatic cholestasis. Gastroenterology 119 (6), 1720–1730. doi:10.1053/gast.2000.20222
Delaunay, J. L., Durand-Schneider, A. M., Delautier, D., Rada, A., Gautherot, J., Jacquemin, E., et al. (2009). A missense mutation in ABCB4 gene involved in progressive familial intrahepatic cholestasis type 3 leads to a folding defect that can be rescued by low temperature. Hepatol. Balt. Md 49 (4), 1218–1227. doi:10.1002/hep.22775
Delaunay, J. L., Durand-Schneider, A. M., Dossier, C., Falguières, T., Gautherot, J., Davit-Spraul, A., et al. (2016). A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatol. Balt. Md 63 (5), 1620–1631. doi:10.1002/hep.28300
Delaunay, J. L., Bruneau, A., Hoffmann, B., Durand-Schneider, A. M., Barbu, V., Jacquemin, E., et al. (2017). Functional defect of variants in the adenosine triphosphate-binding sites of ABCB4 and their rescue by the cystic fibrosis transmembrane conductance regulator potentiator, ivacaftor (VX-770). Hepatol. Balt. Md 65 (2), 560–570. doi:10.1002/hep.28929
Demeilliers, C., Jacquemin, E., Barbu, V., Mergey, M., Paye, F., Fouassier, L., et al. (2006). Altered hepatobiliary gene expressions in PFIC1: ATP8B1 gene defect is associated with CFTR downregulation. Hepatol. Balt. Md 43 (5), 1125–1134. doi:10.1002/hep.21160
Dixon, P. H., Sambrotta, M., Chambers, J., Taylor-Harris, P., Syngelaki, A., Nicolaides, K., et al. (2017). An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci. Rep. 7 (1), 11823. doi:10.1038/s41598-017-11626-x
Dong, C., Condat, B., Picon-Coste, M., Chrétien, Y., Potier, P., Noblinski, B., et al. (2021). Low-phospholipid-associated cholelithiasis syndrome: Prevalence, clinical features, and comorbidities. JHEP Rep. Innov. Hepatol. 3 (2), 100201. doi:10.1016/j.jhepr.2020.100201
Ehmer, U., Kalthoff, S., Fakundiny, B., Pabst, B., Freiberg, N., Naumann, R., et al. (2012). Gilbert syndrome redefined: A complex genetic haplotype influences the regulation of glucuronidation. Hepatol. Balt. Md 55 (6), 1912–1921. doi:10.1002/hep.25561
El Kasmi, K. C., Ghosh, S., Anderson, A. L., Devereaux, M. W., Balasubramaniyan, N., D'Alessandro, A., et al. (2022). Pharmacologic activation of hepatic farnesoid X receptor prevents parenteral nutrition-associated cholestasis in mice. Hepatol. Balt. Md 75 (2), 252–265. doi:10.1002/hep.32101
El Sherrif, Y., Potts, J. R., Howard, M. R., Barnardo, A., Cairns, S., Knisely, A. S., et al. (2013). Hepatotoxicity from anabolic androgenic steroids marketed as dietary supplements: Contribution from ATP8B1/ABCB11 mutations? Liver Int. Off. J. Int. Assoc. Study Liver 33 (8), 1266–1270. doi:10.1111/liv.12216
Engelmann, G., Wenning, D., Herebian, D., Sander, O., Dröge, C., Kluge, S., et al. (2015). Two case reports of successful treatment of cholestasis with steroids in patients with PFIC-2. Pediatrics 135 (5), e1326–e1332. doi:10.1542/peds.2014-2376
Engevik, A. C., Kaji, I., Engevik, M. A., Meyer, A. R., Weis, V. G., Goldstein, A., et al. (2018). Loss of MYO5B leads to reductions in Na+ absorption with maintenance of CFTR-dependent Cl- secretion in enterocytes. Gastroenterology 155 (6), 1883–1897. doi:10.1053/j.gastro.2018.08.025
Erlinger, S., Arias, I. M., and Dhumeaux, D. (2014). Inherited disorders of bilirubin transport and conjugation: New insights into molecular mechanisms and consequences. Gastroenterology 146 (7), 1625–1638. doi:10.1053/j.gastro.2014.03.047
Es, B. (2021). Clinical management of patients with drug-induced liver injury (DILI). United Eur. Gastroenterol. J. 9 (7), 781–786. doi:10.1002/ueg2.12113
Fernandez-Checa, J. C., Bagnaninchi, P., Ye, H., Sancho-Bru, P., Falcon-Perez, J. M., Royo, F., et al. (2021). Advanced preclinical models for evaluation of drug-induced liver injury - consensus statement by the European Drug-Induced Liver Injury Network [PRO-EURO-DILI-NET]. J. Hepatol. 75 (4), 935–959. doi:10.1016/j.jhep.2021.06.021
Floreani, A., and Gervasi, M. T. (2016). New insights on intrahepatic cholestasis of pregnancy. Clin. Liver Dis. 20 (1), 177–189. doi:10.1016/j.cld.2015.08.010
Floreani, A., Franceschet, I., Perini, L., Cazzagon, N., Gershwin, M. E., and Bowlus, C. L. (2015). New therapies for primary biliary cirrhosis. Clin. Rev. Allergy Immunol. 48 (2-3), 263–272. doi:10.1007/s12016-014-8456-5
Folmer, D. E., van der Mark, V. A., Ho-Mok, K. S., and Paulusma, C. C.Oude Elferink RPJ (2009). Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatol. Balt. Md 50 (5), 1597–1605. doi:10.1002/hep.23158
Fretzayas, A., Moustaki, M., Liapi, O., and Karpathios, T. (2012). Gilbert syndrome. Eur. J. Pediatr. 171 (1), 11–15. doi:10.1007/s00431-011-1641-0
Fu, K., Wang, C., Gao, Y., Fan, S., Zhang, H., Sun, J., et al. (2019). Metabolomics and lipidomics reveal the effect of hepatic Vps33b deficiency on bile acids and lipids metabolism. Front. Pharmacol. 10, 276. doi:10.3389/fphar.2019.00276
Gailite, L., Rots, D., Pukite, I., Cernevska, G., and Kreile, M. (2018). Case report: Multiple UGT1A1 gene variants in a patient with crigler-najjar syndrome. BMC Pediatr. 18 (1), 317. doi:10.1186/s12887-018-1285-6
Gao, Y., Zhang, H., Zhou, N., Xu, P., Wang, J., Gao, Y., Jin, X., Liang, X., Lv, J., Zhang, Y., Tang, K., Ma, J., Zhang, H., Xie, J., Yao, F., Tong, W., Liu, Y., Wang, X., and Huang, B. (2020). Methotrexate-loaded tumour-cell-derived microvesicles can relieve biliary obstruction in patients with extrahepatic cholangiocarcinoma. Nature biomedical engineering, 4 (7), 743–753. doi:10.1038/s41551-020-0583-0
Gao, E., Cheema, H., Waheed, N., Mushtaq, I., Erden, N., Nelson-Williams, C., et al. (2020). Organic solute transporter alpha deficiency: A disorder with cholestasis, liver fibrosis, and congenital diarrhea. Hepatol. Balt. Md 71 (5), 1879–1882. doi:10.1002/hep.31087
Garcia, M., Thirouard, L., Monrose, M., Holota, H., De Haze, A., Caira, F., et al. (2019). Farnesoid X receptor alpha (FXRα) is a critical actor of the development and pathologies of the male reproductive system. Cell Mol. Life Sci. CMLS 76 (24), 4849–4859. doi:10.1007/s00018-019-03247-6
Garcia-Barceló, M. M., Yeung, M. Y., Miao, X. P., Tang, C. S. M., Cheng, G., Chen, G., et al. (2010). Genome-wide association study identifies a susceptibility locus for biliary atresia on 10q24.2. Hum. Mol. Genet. 19 (14), 2917–2925. doi:10.1093/hmg/ddq196
Gezdirici, A., Doğan, M., Özgüven, B. Y., and Akbulut, E.Kalaycik Şengül Ö (2023). Biallelic novel USP53 splicing variant disrupting the gene function that causes cholestasis phenotype and review of the literature. Mol. Syndromol. 13 (6), 471–484. doi:10.1159/000523937
Ghonem, N. S., Ananthanarayanan, M., Soroka, C. J., and Boyer, J. L. (2014). Peroxisome proliferator-activated receptor α activates human multidrug resistance transporter 3/ATP-binding cassette protein subfamily B4 transcription and increases rat biliary phosphatidylcholine secretion. Hepatol. Balt. Md 59 (3), 1030–1042. doi:10.1002/hep.26894
Ghosh, S., Devereaux, M. W., Anderson, A. L., Gehrke, S., Reisz, J. A., D'Alessandro, A., et al. (2021). NF-κB regulation of LRH-1 and ABCG5/8 potentiates phytosterol role in the pathogenesis of parenteral nutrition-associated cholestasis. Hepatol. Balt. Md 74 (6), 3284–3300. doi:10.1002/hep.32071
Gilbert, M. A., Bauer, R. C., Rajagopalan, R., Grochowski, C. M., Chao, G., McEldrew, D., et al. (2019). Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Hum. Mutat. 40 (12), 2197–2220. doi:10.1002/humu.23879
Girard, M., Franchi-Abella, S., Lacaille, F., and Debray, D. (2012). Specificities of sclerosing cholangitis in childhood. Clin. Res. Hepatol. Gastroenterol. 36 (6), 530–535. doi:10.1016/j.clinre.2012.04.003
Girard, M., Lacaille, F., Verkarre, V., Mategot, R., Feldmann, G., Grodet, A., et al. (2014). MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatol. Balt. Md 60 (1), 301–310. doi:10.1002/hep.26974
Girard, M., Bizet, A. A., Lachaux, A., Gonzales, E., Filhol, E., Collardeau-Frachon, S., et al. (2016). DCDC2 mutations cause neonatal sclerosing cholangitis. Hum. Mutat. 37 (10), 1025–1029. doi:10.1002/humu.23031
Gómez-Mellado, V. E., Chang, J. C., Ho-Mok, K. S., Bernardino Morcillo, C., Kersten, R. H. J., Oude Elferink, R. P. J., et al. (2022). ATP8B1 deficiency results in elevated mitochondrial phosphatidylethanolamine levels and increased mitochondrial oxidative phosphorylation in human hepatoma cells. Int. J. Mol. Sci. 23 (20), 12344. doi:10.3390/ijms232012344
Gomez-Ospina, N., Potter, C. J., Xiao, R., Manickam, K., Kim, M. S., Kim, K. H., et al. (2016). Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat. Commun. 7, 10713. doi:10.1038/ncomms10713
Gonzales, E., Grosse, B., Cassio, D., Davit-Spraul, A., Fabre, M., and Jacquemin, E. (2012). Successful mutation-specific chaperone therapy with 4-phenylbutyrate in a child with progressive familial intrahepatic cholestasis type 2. J. Hepatol. 57 (3), 695–698. doi:10.1016/j.jhep.2012.04.017
Gonzales, E., Grosse, B., Schuller, B., Davit-Spraul, A., Conti, F., Guettier, C., et al. (2015). Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatol. Balt. Md 62 (2), 558–566. doi:10.1002/hep.27767
Gonzales, E., Taylor, S. A., Davit-Spraul, A., Thébaut, A., Thomassin, N., Guettier, C., et al. (2017). MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease. Hepatol. Balt. Md 65 (1), 164–173. doi:10.1002/hep.28779
Gonzales, E., Hardikar, W., Stormon, M., Baker, A., Hierro, L., Gliwicz, D., et al. (2021). Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): A randomised phase 2 study. Lancet Lond Engl. 398 (10311), 1581–1592. doi:10.1016/S0140-6736(21)01256-3
Grammatikopoulos, T., Sambrotta, M., Strautnieks, S., Foskett, P., Knisely, A. S., Wagner, B., et al. (2016). Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J. Hepatol. 65 (6), 1179–1187. doi:10.1016/j.jhep.2016.07.017
Groen, A., Kunne, C., Jongsma, G., van den Oever, K., Mok, K. S., Petruzzelli, M., et al. (2008). Abcg5/8 independent biliary cholesterol excretion in Atp8b1-deficient mice. Gastroenterology 134 (7), 2091–2100. doi:10.1053/j.gastro.2008.02.097
Hanley, J., Dhar, D. K., Mazzacuva, F., Fiadeiro, R., Burden, J. J., Lyne, A. M., et al. (2017). Vps33b is crucial for structural and functional hepatocyte polarity. J. Hepatol. 66 (5), 1001–1011. doi:10.1016/j.jhep.2017.01.001
Hasegawa, Y., Hayashi, H., Naoi, S., Kondou, H., Bessho, K., Igarashi, K., et al. (2014). Intractable itch relieved by 4-phenylbutyrate therapy in patients with progressive familial intrahepatic cholestasis type 1. Orphanet J. Rare Dis. 9, 89. doi:10.1186/1750-1172-9-89
Hashimoto, K., Uchiumi, T., Konno, T., Ebihara, T., Nakamura, T., Wada, M., et al. (2002). Trafficking and functional defects by mutations of the ATP-binding domains in MRP2 in patients with Dubin-Johnson syndrome. Hepatol. Balt. Md 36 (5), 1236–1245. doi:10.1053/jhep.2002.36368
Hassan, H. M., Onabote, O., Isovic, M., Passos, D. T., Dick, F. A., and Torchia, J. (2022). Regulation of chromatin accessibility by the farnesoid X receptor is essential for circadian and bile acid homeostasis in vivo. Cancers 14 (24), 6191. doi:10.3390/cancers14246191
Hayasaka, K. (2021). Metabolic basis and treatment of citrin deficiency. J. Inherit. Metab. Dis. 44 (1), 110–117. doi:10.1002/jimd.12294
Hayashi, H., and Sugiyama, Y. (2007). 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatol. Balt. Md 45 (6), 1506–1516. doi:10.1002/hep.21630
Hayashi, H., and Sugiyama, Y. (2009). Short-chain ubiquitination is associated with the degradation rate of a cell-surface-resident bile salt export pump (BSEP/ABCB11). Mol. Pharmacol. 75 (1), 143–150. doi:10.1124/mol.108.049288
Hayashi, H., Takada, T., Suzuki, H., Akita, H., and Sugiyama, Y. (2005). Two common PFIC2 mutations are associated with the impaired membrane trafficking of BSEP/ABCB11. Hepatol. Balt. Md 41 (4), 916–924. doi:10.1002/hep.20627
Hayashi, H., Inamura, K., Aida, K., Naoi, S., Horikawa, R., Nagasaka, H., et al. (2012). AP2 adaptor complex mediates bile salt export pump internalization and modulates its hepatocanalicular expression and transport function. Hepatol. Balt. Md 55 (6), 1889–1900. doi:10.1002/hep.25591
Hayashi, H., Naoi, S., Hirose, Y., Matsuzaka, Y., Tanikawa, K., Igarashi, K., et al. (2016). Successful treatment with 4-phenylbutyrate in a patient with benign recurrent intrahepatic cholestasis type 2 refractory to biliary drainage and bilirubin absorption. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 46 (2), 192–200. doi:10.1111/hepr.12561
He, L., Chung, P. H. Y., Lui, V. C. H., Tang, C. S. M., and Tam, P. K. H. (2022). Current understanding in the clinical characteristics and molecular mechanisms in different subtypes of biliary atresia. Int. J. Mol. Sci. 23 (9), 4841. doi:10.3390/ijms23094841
Henkel, S. A., Squires, J. H., Ayers, M., Ganoza, A., Mckiernan, P., and Squires, J. E. (2019). Expanding etiology of progressive familial intrahepatic cholestasis. World J. Hepatol. 11 (5), 450–463. doi:10.4254/wjh.v11.i5.450
Henry, A., and Welsh, A. W. (2015). Monitoring intrahepatic cholestasis of pregnancy using the fetal myocardial performance index: A cohort study. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 46 (5), 571–578. doi:10.1002/uog.14769
Hinoshita, E., Taguchi, K., Inokuchi, A., Uchiumi, T., Kinukawa, N., Shimada, M., et al. (2001). Decreased expression of an ATP-binding cassette transporter, MRP2, in human livers with hepatitis C virus infection. J. Hepatol. 35 (6), 765–773. doi:10.1016/s0168-8278(01)00216-1
Hirano, M., Maeda, K., Hayashi, H., Kusuhara, H., and Sugiyama, Y. (2005). Bile salt export pump (BSEP/ABCB11) can transport a nonbile acid substrate, pravastatin. J. Pharmacol. Exp. Ther. 314 (2), 876–882. doi:10.1124/jpet.105.084830
Ho, R. H., Leake, B. F., Kilkenny, D. M., Meyer Zu Schwabedissen, H. E., Glaeser, H., Kroetz, D. L., et al. (2010). Polymorphic variants in the human bile salt export pump (BSEP; ABCB11): Functional characterization and interindividual variability. Pharmacogenet Genomics 20 (1), 45–57. doi:10.1097/FPC.0b013e3283349eb0
Hofmann, J. J., Zovein, A. C., Koh, H., Radtke, F., Weinmaster, G., and Iruela-Arispe, M. L. (2010). Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: Insights into Alagille syndrome. Dev. Camb Engl. 137 (23), 4061–4072. doi:10.1242/dev.052118
Homolya, L., Fu, D., Sengupta, P., Jarnik, M., Gillet, J. P., Vitale-Cross, L., et al. (2014). LKB1/AMPK and PKA control ABCB11 trafficking and polarization in hepatocytes. PloS One 9 (3), e91921. doi:10.1371/journal.pone.0091921
Hsu, P. W. C., Liao, P. C., Kao, Y. H., Lin, X. Y., Chien, R. N., Yeh, C. T., et al. (2022). The mutation hotspots at UGT1A locus may Be associated with gilbert’s syndrome affecting the Taiwanese population. Int. J. Mol. Sci. 23 (20), 12709. doi:10.3390/ijms232012709
Hucke, S., Herold, M., Liebmann, M., Freise, N., Lindner, M., Fleck, A. K., et al. (2016). The farnesoid-X-receptor in myeloid cells controls CNS autoimmunity in an IL-10-dependent fashion. Acta Neuropathol. Berl. 132 (3), 413–431. doi:10.1007/s00401-016-1593-6
Itoh, M., Terada, M., and Sugimoto, H. (2021). The zonula occludens protein family regulates the hepatic barrier system in the murine liver. Biochim. Biophys. Acta Mol. Basis Dis. 1867 (1), 165994. doi:10.1016/j.bbadis.2020.165994
Iwata, R., Baur, K., Stieger, B., Mertens, J. C., Daly, A. K., Frei, P., et al. (2011). A common polymorphism in the ABCB11 gene is associated with advanced fibrosis in hepatitis C but not in non-alcoholic fatty liver disease. Clin. Sci. Lond Engl. 1979 120 (7), 287–296. doi:10.1042/CS20100246
Jacquemin, E., De Vree, J. M., Cresteil, D., Sokal, E. M., Sturm, E., Dumont, M., et al. (2001). The wide spectrum of multidrug resistance 3 deficiency: From neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 120 (6), 1448–1458. doi:10.1053/gast.2001.23984
Jacquemin, E. (2001). Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: One gene for three diseases. Semin. Liver Dis. 21 (4), 551–562. doi:10.1055/s-2001-19033
Jiang, Y., Jin, J., Iakova, P., Hernandez, J. C., Jawanmardi, N., Sullivan, E., et al. (2013). Farnesoid X receptor directly regulates xenobiotic detoxification genes in the long-lived Little mice. Mech. Ageing Dev. 134 (9), 407–415. doi:10.1016/j.mad.2013.08.003
Kaji, I., Roland, J. T., Watanabe, M., Engevik, A. C., Goldstein, A. E., Hodges, C. A., et al. (2020). Lysophosphatidic acid increases maturation of brush borders and SGLT1 activity in MYO5B-deficient mice, a model of microvillus inclusion disease. Gastroenterology 159 (4), 1390–1405. doi:10.1053/j.gastro.2020.06.008
Kamath, B. M., Podkameni, G., Hutchinson, A. L., Leonard, L. D., Gerfen, J., Krantz, I. D., et al. (2012). Renal anomalies in Alagille syndrome: A disease-defining feature. Am. J. Med. Genet. A 158A (1), 85–89. doi:10.1002/ajmg.a.34369
Kawakita, T., Parikh, L. I., Ramsey, P. S., Huang, C. C., Zeymo, A., Fernandez, M., et al. (2015). Predictors of adverse neonatal outcomes in intrahepatic cholestasis of pregnancy. Am. J. Obstet. Gynecol. 213 (4), 570.e1–e8. doi:10.1016/j.ajog.2015.06.021
Keitel, V., Nies, A. T., Brom, M., Hummel-Eisenbeiss, J., Spring, H., and Keppler, D. (2003). A common Dubin-Johnson syndrome mutation impairs protein maturation and transport activity of MRP2 (ABCC2). Am. J. Physiol. Gastrointest. Liver Physiol. 284 (1), G165–G174. doi:10.1152/ajpgi.00362.2002
Khabou, B., Trigui, A., Boudawara, T. S., Keskes, L., Kamoun, H., Barbu, V., et al. (2019). A homozygous ABCB4 mutation causing an LPAC syndrome evolves into cholangiocarcinoma. Clin. Chim. Acta Int. J. Clin. Chem. 495, 598–605. doi:10.1016/j.cca.2019.06.007
Kim, K. H., Choi, J. M., Li, F., Arizpe, A., Wooton-Kee, C. R., Anakk, S., et al. (2018). Xenobiotic nuclear receptor signaling determines molecular pathogenesis of progressive familial intrahepatic cholestasis. Endocrinology 159 (6), 2435–2446. doi:10.1210/en.2018-00110
Kim, K. H., Choi, J. M., Li, F., Dong, B., Wooton-Kee, C. R., Arizpe, A., et al. (2019). Constitutive androstane receptor differentially regulates bile acid homeostasis in mouse models of intrahepatic cholestasis. Hepatol. Commun. 3 (1), 147–159. doi:10.1002/hep4.1274
Kimura, A., Kagawa, T., Takei, H., Maruo, Y., Sakugawa, H., Sasaki, T., et al. (2021). Rotor syndrome: Glucuronidated bile acidemia from defective reuptake by hepatocytes. Hepatol. Commun. 5 (4), 629–633. doi:10.1002/hep4.1660
Koukoulioti, E., Ziagaki, A., Weber, S. N., Lammert, F., and Berg, T. (2021). Long-term colestyramine treatment prevents cholestatic attacks in refractory benign recurrent intrahepatic cholestasis type 1 disease. Hepatology 74 (1), 522–524. doi:10.1002/hep.31671
Krattinger, R., Boström, A., Schiöth, H. B., Thasler, W. E., Mwinyi, J., and Kullak-Ublick, G. A. (2016). microRNA-192 suppresses the expression of the farnesoid X receptor. Am. J. Physiol. Gastrointest. Liver Physiol. 310 (11), G1044–G1051. doi:10.1152/ajpgi.00297.2015
Kubitz, R., Dröge, C., Kluge, S., Stross, C., Walter, N., Keitel, V., et al. (2015). Autoimmune BSEP disease: Disease recurrence after liver transplantation for progressive familial intrahepatic cholestasis. Clin. Rev. Allergy Immunol. 48 (2-3), 273–284. doi:10.1007/s12016-014-8457-4
Kubitz, R., Dröge, C., Kluge, S., Stindt, J., Stross, C., Häussinger, D., et al. (2016). High affinity anti-BSEP antibodies after liver transplantation for PFIC-2 - successful treatment with immunoadsorption and B-cell depletion. Pediatr. Transpl. 20 (7), 987–993. doi:10.1111/petr.12751
Kumar, A., Hashmi, M. F., and Mehta, D. (2023). “Rotor syndrome,” in StatPearls (Florida: StatPearls Publishing).
Kundur, A. R., Singh, I., and Bulmer, A. C. (2015). Bilirubin, platelet activation and heart disease: A missing link to cardiovascular protection in gilbert’s syndrome? Atherosclerosis 239 (1), 73–84. doi:10.1016/j.atherosclerosis.2014.12.042
Kunst, R. F., Verkade, H. J., and van de Graaf, S. F. J.Oude Elferink RPJ (2021). Targeting the four pillars of enterohepatic bile salt cycling; lessons from genetics and Pharmacology. Hepatol. Balt. Md 73 (6), 2577–2585. doi:10.1002/hep.31651
Kwo, P. Y., Cohen, S. M., and Lim, J. K. (2017). ACG clinical guideline: Evaluation of abnormal liver chemistries. Am. J. Gastroenterol. 112 (1), 18–35. doi:10.1038/ajg.2016.517
Lakshminarayanan, B., and Davenport, M. (2016). Biliary atresia: A comprehensive review. J. Autoimmun. 73, 1–9. doi:10.1016/j.jaut.2016.06.005
Lam, C. W., Cheung, K. M., Tsui, M. S., Yan, M. S. C., Lee, C. Y., and Tong, S. F. (2006). A patient with novel ABCB11 gene mutations with phenotypic transition between BRIC2 and PFIC2. J. Hepatol. 44 (1), 240–242. doi:10.1016/j.jhep.2005.09.013
Lam, P., Pearson, C. L., Soroka, C. J., Xu, S., Mennone, A., and Boyer, J. L. (2007). Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am. J. Physiol. Cell Physiol. 293 (5), C1709–C1716. doi:10.1152/ajpcell.00327.2007
Lam, W. Y., Tang, C. S. M., So, M. T., Yue, H., Hsu, J. S., Chung, P. H. Y., et al. (2021). Identification of a wide spectrum of ciliary gene mutations in nonsyndromic biliary atresia patients implicates ciliary dysfunction as a novel disease mechanism. EBioMedicine 71, 103530. doi:10.1016/j.ebiom.2021.103530
Lamireau, T., Bouchard, G., Yousef, I. M., Clouzeau-Girard, H., Rosenbaum, J., Desmoulière, A., et al. (2007). Dietary lecithin protects against cholestatic liver disease in cholic acid-fed Abcb4-deficient mice. Pediatr. Res. 61 (2), 185–190. doi:10.1203/pdr.0b013e31802d7780
Lemoine, M., Revaux, A., Francoz, C., Ducarme, G., Brechignac, S., Jacquemin, E., et al. (2008). Albumin liver dialysis as pregnancy-saving procedure in cholestatic liver disease and intractable pruritus. World J. Gastroenterol. 14 (42), 6572–6574. doi:10.3748/wjg.14.6572
Leng, C., Rings, E. H. H. M., de Wildt, S. N., and van Ijzendoorn, S. C. D. (2020). Pharmacological and parenteral nutrition-based interventions in microvillus inclusion disease. J. Clin. Med. 10 (1), 22. doi:10.3390/jcm10010022
Li, L., Deheragoda, M., Lu, Y., Gong, J., and Wang, J. (2015). Hypothyroidism associated with ATP8B1 deficiency. J. Pediatr. 167 (6), 1334–1339. doi:10.1016/j.jpeds.2015.08.037
Liaqat, A., Shahid, A., Attiq, H., Ameer, A., and Imran, M. (2018). Crigler-najjar syndrome type II diagnosed in a patient with jaundice since birth. J. Coll. Physicians Surg--Pak JCPSP 28 (10), 806–808.
Lin, H. C., Alvarez, L., Laroche, G., Melin-Aldana, H., Pfeifer, K., Schwarz, K., et al. (2013). Rituximab as therapy for the recurrence of bile salt export pump deficiency after liver transplantation. Liver Transpl. Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transpl. Soc. 19 (12), 1403–1410. doi:10.1002/lt.23754
Lin, Y. C., Wang, F. S., Yang, Y. L., Chuang, Y. T., and Huang, Y. H. (2018). MicroRNA-29a mitigation of toll-like receptor 2 and 4 signaling and alleviation of obstructive jaundice-induced fibrosis in mice. Biochem. Biophys. Res. Commun. 496 (3), 880–886. doi:10.1016/j.bbrc.2018.01.132
Liu, T., Wang, R. X., Han, J., Hao, C. Z., Qiu, Y. L., Yan, Y. Y., et al. (2018). Comprehensive bile acid profiling in hereditary intrahepatic cholestasis: Genetic and clinical correlations. Liver Int. Off. J. Int. Assoc. Study Liver 38 (9), 1676–1685. doi:10.1111/liv.13714
Liu, X., Lai, H., Zeng, X., Xin, S., Nie, L., Liang, Z., et al. (2020). Whole-exome sequencing reveals ANO8 as a genetic risk factor for intrahepatic cholestasis of pregnancy. BMC Pregnancy Childbirth 20 (1), 544. doi:10.1186/s12884-020-03240-z
Lu, C., Chen, B., Chen, C., Li, H., Wang, D., Tan, Y., et al. (2020). CircNr1h4 regulates the pathological process of renal injury in salt-sensitive hypertensive mice by targeting miR-155-5p. J. Cell Mol. Med. 24 (2), 1700–1712. doi:10.1111/jcmm.14863
Luan, W., Hao, C. Z., Li, J. Q., Wei, Q., Gong, J. Y., Qiu, Y. L., et al. (2021). Biallelic loss-of-function ZFYVE19 mutations are associated with congenital hepatic fibrosis, sclerosing cholangiopathy and high-GGT cholestasis. J. Med. Genet. 58 (8), 514–525. doi:10.1136/jmedgenet-2019-106706
Lucena, M. I., Sanabria, J., García-Cortes, M., Stephens, C., and Andrade, R. J. (2020). Drug-induced liver injury in older people. Lancet Gastroenterol. Hepatol. 5 (9), 862–874. doi:10.1016/S2468-1253(20)30006-6
Ma, K., Saha, P. K., Chan, L., and Moore, D. D. (2006). Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Invest. 116 (4), 1102–1109. doi:10.1172/JCI25604
Mack, C. L., and Sokol, R. J. (2005). Unraveling the pathogenesis and etiology of biliary atresia. Pediatr. Res. 57, 87R–94R. doi:10.1203/01.PDR.0000159569.57354.47
Mandato, C., Siano, M. A., Nazzaro, L., Gelzo, M., Francalanci, P., Rizzo, F., et al. (2021). A ZFYVE19 gene mutation associated with neonatal cholestasis and cilia dysfunction: Case report with a novel pathogenic variant. Orphanet J. Rare Dis. 16 (1), 179. doi:10.1186/s13023-021-01775-8
Mareux, E., Lapalus, M., Amzal, R., Almes, M., Aït-Slimane, T., Delaunay, J. L., et al. (2020). Functional rescue of an ABCB11 mutant by ivacaftor: A new targeted pharmacotherapy approach in bile salt export pump deficiency. Liver Int. Off. J. Int. Assoc. Study Liver 40 (8), 1917–1925. doi:10.1111/liv.14518
Massafra, V., Milona, A., Vos, H. R., Ramos, R. J. J., Gerrits, J., Willemsen, E. C. L., et al. (2017). Farnesoid X receptor activation promotes hepatic amino acid catabolism and ammonium clearance in mice. Gastroenterology 152 (6), 1462–1476. doi:10.1053/j.gastro.2017.01.014
Matsushima, S., Maeda, K., Hayashi, H., Debori, Y., Schinkel, A. H., Schuetz, J. D., et al. (2008). Involvement of multiple efflux transporters in hepatic disposition of fexofenadine. Mol. Pharmacol. 73 (5), 1474–1483. doi:10.1124/mol.107.041459
McDaniel, K., Wu, N., Zhou, T., Huang, L., Sato, K., Venter, J., et al. (2019). Amelioration of ductular reaction by stem cell derived extracellular vesicles in MDR2 knockout mice via lethal-7 microRNA. Hepatol. Balt. Md 69 (6), 2562–2578. doi:10.1002/hep.30542
McDaniell, R., Warthen, D. M., Sanchez-Lara, P. A., Pai, A., Krantz, I. D., Piccoli, D. A., et al. (2006). NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 79 (1), 169–173. doi:10.1086/505332
Mieli-Vergani, G., and Vergani, D. (2009). Biliary atresia. Semin. Immunopathol. 31 (3), 371–381. doi:10.1007/s00281-009-0171-6
Suzuki, H., Arinaga-Hino, T., Sano, T., Mihara, Y., Kusano, H., Mizuochi, T., et al. (2022). Case report: A rare case of benign recurrent intrahepatic cholestasis-type 1 with a novel heterozygous pathogenic variant of ATP8B1. Front. Med. 9, 891659. doi:10.3389/fmed.2022.891659
Mizutani, A., Sabu, Y., Naoi, S., Ito, S., Nakano, S., Minowa, K., et al. (2021). Assessment of adenosine triphosphatase phospholipid transporting 8B1 (ATP8B1) function in patients with cholestasis with ATP8B1 deficiency by using peripheral blood monocyte-derived macrophages. Hepatol. Commun. 5 (1), 52–62. doi:10.1002/hep4.1605
Mochizuki, K., Kagawa, T., Numari, A., Harris, M. J., Itoh, J., Watanabe, N., et al. (2007). Two N-linked glycans are required to maintain the transport activity of the bile salt export pump (ABCB11) in MDCK II cells. Am. J. Physiol. Gastrointest. Liver Physiol. 292 (3), G818–G828. doi:10.1152/ajpgi.00415.2006
Morais, M. B., and Machado, M. V. (2022). Benign inheritable disorders of bilirubin metabolism manifested by conjugated hyperbilirubinemia-A narrative review. United Eur. Gastroenterol. J. 10 (7), 745–753. doi:10.1002/ueg2.12279
Muise, A. M., Turner, D., Wine, E., Kim, P., Marcon, M., and Ling, S. C. (2006). Biliary atresia with choledochal cyst: Implications for classification. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 4 (11), 1411–1414. doi:10.1016/j.cgh.2006.07.005
Müllenbach, R., Bennett, A., Tetlow, N., Patel, N., Hamilton, G., Cheng, F., et al. (2005). ATP8B1 mutations in British cases with intrahepatic cholestasis of pregnancy. Gut 54 (6), 829–834. doi:10.1136/gut.2004.058115
Naoi, S., Hayashi, H., Inoue, T., Tanikawa, K., Igarashi, K., Nagasaka, H., et al. (2014). Improved liver function and relieved pruritus after 4-phenylbutyrate therapy in a patient with progressive familial intrahepatic cholestasis type 2. J. Pediatr. 164 (5), 1219–1227. doi:10.1016/j.jpeds.2013.12.032
Nie, H., Song, C., Wang, D., Cui, S., Ren, T., Cao, Z., et al. (2017). MicroRNA-194 inhibition improves dietary-induced non-alcoholic fatty liver disease in mice through targeting on FXR. Biochim. Biophys. Acta Mol. Basis Dis. 1863 (12), 3087–3094. doi:10.1016/j.bbadis.2017.09.020
Olyaei, A. J., de Mattos, A. M., and Bennett, W. M. (2001). Nephrotoxicity of immunosuppressive drugs: New insight and preventive strategies. Curr. Opin. Crit. Care 7 (6), 384–389. doi:10.1097/00075198-200112000-00003
Ott, P., Ala, A., Askari, F. K., Czlonkowska, A., Hilgers, R. D., Poujois, A., et al. (2021). Designing clinical trials in wilson’s disease. Hepatol. Balt. Md 74 (6), 3460–3471. doi:10.1002/hep.32074
Oude Elferink RPJ Paulusma, C. C. (2007). Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). Pflugers Arch. 453 (5), 601–610. doi:10.1007/s00424-006-0062-9
Oude Elferink RPJ Paulusma, C. C., and Groen, A. K. (2006). Hepatocanalicular transport defects: Pathophysiologic mechanisms of rare diseases. Gastroenterology 130 (3), 908–925. doi:10.1053/j.gastro.2005.08.052
Ovadia, C., Seed, P. T., Sklavounos, A., Geenes, V., Di Ilio, C., Chambers, J., et al. (2019). Association of adverse perinatal outcomes of intrahepatic cholestasis of pregnancy with biochemical markers: Results of aggregate and individual patient data meta-analyses. Lancet Lond Engl. 393 (10174), 899–909. doi:10.1016/S0140-6736(18)31877-4
Overeem, A. W., Li, Q., Qiu, Y. L., Cartón-García, F., Leng, C., Klappe, K., et al. (2020). A molecular mechanism underlying genotype-specific intrahepatic cholestasis resulting from MYO5B mutations. Hepatol. Balt. Md 72 (1), 213–229. doi:10.1002/hep.31002
Palmer, K. R., Xiaohua, L., and Mol, B. W. (2019). Management of intrahepatic cholestasis in pregnancy. Lancet Lond Engl. 393 (10174), 853–854. doi:10.1016/S0140-6736(18)32323-7
Pan, Q., Luo, G., Qu, J., Chen, S., Zhang, X., Zhao, N., et al. (2021). A homozygous R148W mutation in Semaphorin 7A causes progressive familial intrahepatic cholestasis. EMBO Mol. Med. 13 (11), e14563. doi:10.15252/emmm.202114563
Pang, X., Cao, J., Chen, S., Gao, Z., Liu, G., Chong, Y., et al. (2021). Unsupervised clustering reveals distinct subtypes of biliary atresia based on immune cell types and gene expression. Front. Immunol. 12, 720841. doi:10.3389/fimmu.2021.720841
Paulusma, C. C., Groen, A., Kunne, C., Ho-Mok, K. S., Spijkerboer, A. L., Rudi de Waart, D., et al. (2006). Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatol. Balt. Md 44 (1), 195–204. doi:10.1002/hep.21212
Paulusma, C. C., Folmer, D. E., Ho-Mok, K. S., de Waart, D. R., Hilarius, P. M., Verhoeven, A. J., et al. (2008). ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatol. Balt. Md 47 (1), 268–278. doi:10.1002/hep.21950
Pepe, A., Colucci, A., Carucci, M., Nazzaro, L., Bucci, C., Ranucci, G., et al. (2023). Case Report: Add-on treatment with odevixibat in a new subtype of progressive familial intrahepatic cholestasis broadens the therapeutic horizon of genetic cholestasis. Front. Pediatr. 11, 1061535. doi:10.3389/fped.2023.1061535
Pham, D. H., Kudira, R., Xu, L., Valencia, C. A., Ellis, J. L., Shi, T., et al. (2021). Deleterious variants in ABCC12 are detected in idiopathic chronic cholestasis and cause intrahepatic bile duct loss in model organisms. Gastroenterology 161 (1), 287–300.e16. doi:10.1053/j.gastro.2021.03.026
Piechota, J., and Jelski, W. (2020). Intrahepatic cholestasis in pregnancy: Review of the literature. J. Clin. Med. 9 (5), 1361. doi:10.3390/jcm9051361
Plass, J. R. M., Mol, O., Heegsma, J., Geuken, M., Faber, K. N., Jansen, P. L. M., et al. (2002). Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatol. Balt. Md 35 (3), 589–596. doi:10.1053/jhep.2002.31724
Poupon, R., Chazouillères, O., and Poupon, R. E. (2000). Chronic cholestatic diseases. J. Hepatol. 32, 129–140. doi:10.1016/s0168-8278(00)80421-3
Poupon, R., Barbu, V., Chamouard, P., Wendum, D., Rosmorduc, O., and Housset, C. (2010). Combined features of low phospholipid-associated cholelithiasis and progressive familial intrahepatic cholestasis 3. Liver Int. Off. J. Int. Assoc. Study Liver 30 (2), 327–331. doi:10.1111/j.1478-3231.2009.02148.x
Puljic, A., Kim, E., Page, J., Esakoff, T., Shaffer, B., LaCoursiere, D. Y., et al. (2015). The risk of infant and fetal death by each additional week of expectant management in intrahepatic cholestasis of pregnancy by gestational age. Am. J. Obstet. Gynecol. 212 (5), 667.e1–e5. doi:10.1016/j.ajog.2015.02.012
Qiu, Y. L., Gong, J. Y., Feng, J. Y., Wang, R. X., Han, J., Liu, T., et al. (2017). Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ-glutamyltransferase cholestasis. Hepatol. Balt. Md 65 (5), 1655–1669. doi:10.1002/hep.29020
Qiu, Y. L., Liu, T., Abuduxikuer, K., Hao, C. Z., Gong, J. Y., Zhang, M. H., et al. (2019). Novel missense mutation in VPS33B is associated with isolated low gamma-glutamyltransferase cholestasis: Attenuated, incomplete phenotype of arthrogryposis, renal dysfunction, and cholestasis syndrome. Hum. Mutat. 40 (12), 2247–2257. doi:10.1002/humu.23770
Rajagopalan, R., Tsai, E. A., Grochowski, C. M., Kelly, S. M., Loomes, K. M., Spinner, N. B., et al. (2020). Exome sequencing in individuals with isolated biliary atresia. Sci. Rep. 10 (1), 2709. doi:10.1038/s41598-020-59379-4
Raquel, G.-G., Andueza, S., Hierro, L., Martínez-Fernández, P., D'Agostino, D., Jara, P., et al. (2015). Functional analysis of ABCB4 mutations relates clinical outcomes of progressive familial intrahepatic cholestasis type 3 to the degree of MDR3 floppase activity. Gut 64 (1), 147–155. doi:10.1136/gutjnl-2014-306896
Regev, R. H., Stolar, O., Raz, A., and Dolfin, T. (2002). Treatment of severe cholestasis in neonatal Dubin-Johnson syndrome with ursodeoxycholic acid. J. Perinat. Med. 30 (2), 185–187. doi:10.1515/JPM.2002.025
Reichert, M. C., and Lammert, F. (2018). ABCB4 gene aberrations in human liver disease: An evolving spectrum. Semin. Liver Dis. 38 (4), 299–307. doi:10.1055/s-0038-1667299
Reyes, H., Báez, M. E., González, M. C., Hernández, I., Palma, J., Ribalta, J., et al. (2000). Selenium, zinc and copper plasma levels in intrahepatic cholestasis of pregnancy, in normal pregnancies and in healthy individuals, in Chile. J. Hepatol. 32 (4), 542–549. doi:10.1016/s0168-8278(00)80214-7
Roland, J. T., Bryant, D. M., Datta, A., Itzen, A., Mostov, K. E., and Goldenring, J. R. (2011). Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc. Natl. Acad. Sci. U. S. A. 108 (7), 2789–2794. doi:10.1073/pnas.1010754108
Saito, S., Obata, A., Kashiwagi, Y., Abe, K., and Murase, K. (2013). Dynamic contrast-enhanced MRI of the liver in Mrp2-deficient rats using the hepatobiliary contrast agent Gd-EOB-DTPA. Invest. Radiol. 48 (7), 548–553. doi:10.1097/RLI.0b013e3182856a06
Schady, D. A., and Finegold, M. J. (2017). Contemporary evaluation of the pediatric liver biopsy. Gastroenterol. Clin. North Am. 46 (2), 233–252. doi:10.1016/j.gtc.2017.01.013
Schaub, J. R., Huppert, K. A., Kurial, S. N. T., Hsu, B. Y., Cast, A. E., Donnelly, B., et al. (2018). De novo formation of the biliary system by TGFβ-mediated hepatocyte transdifferentiation. Nature 557 (7704), 247–251. doi:10.1038/s41586-018-0075-5
Schneeberger, K., Vogel, G. F., Teunissen, H., van Ommen, D. D., Begthel, H., El Bouazzaoui, L., et al. (2015). An inducible mouse model for microvillus inclusion disease reveals a role for myosin Vb in apical and basolateral trafficking. Proc. Natl. Acad. Sci. U. S. A. 112 (40), 12408–12413. doi:10.1073/pnas.1516672112
Schonfeld, E. A., and Brown, R. S. (2019). Genetic causes of liver disease: When to suspect a genetic etiology, initial lab testing, and the basics of management. Med. Clin. North Am. 103 (6), 991–1003. doi:10.1016/j.mcna.2019.07.003
Schwarz, K. B., Haber, B. H., Rosenthal, P., Mack, C. L., Moore, J., Bove, K., et al. (2013). Extrahepatic anomalies in infants with biliary atresia: Results of a large prospective north American multicenter study. Hepatol. Balt. Md 58 (5), 1724–1731. doi:10.1002/hep.26512
Shatokhina, O., Semenova, N., Demina, N., Dadali, E., Polyakov, A., and Ryzhkova, O. (2021). A two-year clinical description of a patient with a rare type of low-GGT cholestasis caused by a novel variant of USP53. Genes 12 (10), 1618. doi:10.3390/genes12101618
Siew, S. M., Cunningham, S. C., Zhu, E., Tay, S. S., Venuti, E., Bolitho, C., et al. (2019). Prevention of cholestatic liver disease and reduced tumorigenicity in a murine model of PFIC type 3 using hybrid AAV-piggyBac gene therapy. Hepatol. Balt. Md 70 (6), 2047–2061. doi:10.1002/hep.30773
So, J., Ningappa, M., Glessner, J., Min, J., Ashokkumar, C., Ranganathan, S., et al. (2020). Biliary-atresia-associated mannosidase-1-alpha-2 gene regulates biliary and ciliary morphogenesis and laterality. Front. Physiol. 11, 538701. doi:10.3389/fphys.2020.538701
Soroka, C. J., and Boyer, J. L. (2014). Biosynthesis and trafficking of the bile salt export pump, BSEP: Therapeutic implications of BSEP mutations. Mol. Asp. Med. 37, 3–14. doi:10.1016/j.mam.2013.05.001
Stalke, A., Sgodda, M., Cantz, T., Skawran, B., Lainka, E., Hartleben, B., et al. (2022). KIF12 variants and disturbed hepatocyte polarity in children with a phenotypic spectrum of cholestatic liver disease. J. Pediatr. 240, 284–291.e9. doi:10.1016/j.jpeds.2021.09.019
Stapelbroek, J. M., Peters, T. A., van Beurden, D. H. A., Curfs, J. H. A. J., Joosten, A., Beynon, A. J., et al. (2009). ATP8B1 is essential for maintaining normal hearing. Proc. Natl. Acad. Sci. U. S. A. 106 (24), 9709–9714. doi:10.1073/pnas.0807919106
Stayrook, K. R., Bramlett, K. S., Savkur, R. S., Ficorilli, J., Cook, T., Christe, M. E., et al. (2005). Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology 146 (3), 984–991. doi:10.1210/en.2004-0965
Strassburg, C. P. (2010). Hyperbilirubinemia syndromes (Gilbert-Meulengracht, crigler-najjar, dubin-johnson, and rotor syndrome). Best. Pract. Res. Clin. Gastroenterol. 24 (5), 555–571. doi:10.1016/j.bpg.2010.07.007
Strauss, K. A., Ahlfors, C. E., Soltys, K., Mazareigos, G. V., Young, M., Bowser, L. E., et al. (2020). Crigler-najjar syndrome type 1: Pathophysiology, natural history, and therapeutic frontier. Hepatol. Balt. Md 71 (6), 1923–1939. doi:10.1002/hep.30959
Strnad, P., Tacke, F., Koch, A., and Trautwein, C. (2017). Liver - guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 14 (1), 55–66. doi:10.1038/nrgastro.2016.168
Superina, R., Magee, J. C., Brandt, M. L., Healey, P. J., Tiao, G., Ryckman, F., et al. (2011). The anatomic pattern of biliary atresia identified at time of Kasai hepatoportoenterostomy and early postoperative clearance of jaundice are significant predictors of transplant-free survival. Ann. Surg. 254 (4), 577–585. doi:10.1097/SLA.0b013e3182300950
Szavay, P. O., Leonhardt, J., Czech-Schmidt, G., and Petersen, C. (2002). The role of reovirus type 3 infection in an established murine model for biliary atresia. Eur. J. Pediatr. Surg. Off. J. Austrian Assoc. Pediatr. Surg. Al Z Kinderchir 12 (4), 248–250. doi:10.1055/s-2002-34477
Tang, J., Tan, M., Deng, Y., Tang, H., Shi, H., Li, M., Ma, W., Li, J., Dai, H., Li, J., Zhou, S., Li, X., Wei, F., Ma, X., and Luo, L. (2021). Two Novel Pathogenic Variants of TJP2 Gene and the Underlying Molecular Mechanisms in Progressive Familial Intrahepatic Cholestasis Type 4 Patients. Frontiers in cell and developmental biology, 9, 661599. doi:10.3389/fcell.2021.661599
Targher, G., Tilg, H., and Byrne, C. D. (2021). Non-alcoholic fatty liver disease: A multisystem disease requiring a multidisciplinary and holistic approach. Lancet Gastroenterol. Hepatol. 6 (7), 578–588. doi:10.1016/S2468-1253(21)00020-0
Thompson, R. J., Arnell, H., Artan, R., Baumann, U., Calvo, P. L., Czubkowski, P., et al. (2022). Odevixibat treatment in progressive familial intrahepatic cholestasis: A randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol. Hepatol. 7 (9), 830–842. doi:10.1016/S2468-1253(22)00093-0
Thoresen, S. B., Campsteijn, C., Vietri, M., Schink, K. O., Liestøl, K., Andersen, J. S., et al. (2014). ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat. Cell Biol. 16 (6), 550–560. doi:10.1038/ncb2959
Togawa, T., Mizuochi, T., Sugiura, T., Kusano, H., Tanikawa, K., Sasaki, T., et al. (2018). Clinical, pathologic, and genetic features of neonatal dubin-johnson syndrome: A multicenter study in Japan. J. Pediatr. 196, 161–167. doi:10.1016/j.jpeds.2017.12.058
Tomić, T. T., Olausson, J., Rehammar, A., Deland, L., Muth, A., Ejeskär, K., et al. (2020). MYO5B mutations in pheochromocytoma/paraganglioma promote cancer progression. PLoS Genet. 16 (6), e1008803. doi:10.1371/journal.pgen.1008803
Trauner, M., Fickert, P., and Wagner, M. (2007). MDR3 (ABCB4) defects: A paradigm for the genetics of adult cholestatic syndromes. Semin. Liver Dis. 27 (1), 77–98. doi:10.1055/s-2006-960172
Tsai, E. A., Grochowski, C. M., Loomes, K. M., Bessho, K., Hakonarson, H., Bezerra, J. A., et al. (2014). Replication of a GWAS signal in a Caucasian population implicates ADD3 in susceptibility to biliary atresia. Hum. Genet. 133 (2), 235–243. doi:10.1007/s00439-013-1368-2
Tsai, E. A., Grochowski, C. M., Falsey, A. M., Rajagopalan, R., Wendel, D., Devoto, M., et al. (2015). Heterozygous deletion of FOXA2 segregates with disease in a family with heterotaxy, panhypopituitarism, and biliary atresia. Hum. Mutat. 36 (6), 631–637. doi:10.1002/humu.22786
Tsai, E. A., Gilbert, M. A., Grochowski, C. M., Underkoffler, L. A., Meng, H., Zhang, X., et al. (2016). THBS2 is a candidate modifier of liver disease severity in Alagille syndrome. Cell Mol. Gastroenterol. Hepatol. 2 (5), 663–675. doi:10.1016/j.jcmgh.2016.05.013
Tukey, R. H., and Strassburg, C. P. (2000). Human UDP-glucuronosyltransferases: Metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 40, 581–616. doi:10.1146/annurev.pharmtox.40.1.581
Tzaban, S., Massol, R. H., Yen, E., Hamman, W., Frank, S. R., Lapierre, L. A., et al. (2009). The recycling and transcytotic pathways for IgG transport by FcRn are distinct and display an inherent polarity. J. Cell Biol. 185 (4), 673–684. doi:10.1083/jcb.200809122
Ünlüsoy Aksu, A., Das, S. K., Nelson-Williams, C., Jain, D., Özbay Hoşnut, F., Evirgen Şahin, G., et al. (2019). Recessive mutations in KIF12 cause high gamma-glutamyltransferase cholestasis. Hepatol. Commun. 3 (4), 471–477. doi:10.1002/hep4.1320
van de Steeg, E., Stránecký, V., Hartmannová, H., Nosková, L., Hřebíček, M., Wagenaar, E., et al. (2012). Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J. Clin. Invest. 122 (2), 519–528. doi:10.1172/JCI59526
van der Mark, V. A., de Waart, D. R., Ho-Mok, K. S., Tabbers, M. M., Voogt, H. W., Oude Elferink, R. P. J., et al. (2014). The lipid flippase heterodimer ATP8B1-CDC50A is essential for surface expression of the apical sodium-dependent bile acid transporter (SLC10A2/ASBT) in intestinal Caco-2 cells. Biochim. Biophys. Acta 1842, 2378–2386. doi:10.1016/j.bbadis.2014.09.003
van der Velden, L. M., Stapelbroek, J. M., Krieger, E., van den Berghe, P. V. E., Berger, R., Verhulst, P. M., et al. (2010). Folding defects in P-type ATP 8B1 associated with hereditary cholestasis are ameliorated by 4-phenylbutyrate. Hepatol. Balt. Md 51 (1), 286–296. doi:10.1002/hep.23268
van der Woerd, W. L., Wichers, C. G. K., Vestergaard, A. L., Andersen, J. P., Paulusma, C. C., Houwen, R. H. J., et al. (2016). Rescue of defective ATP8B1 trafficking by CFTR correctors as a therapeutic strategy for familial intrahepatic cholestasis. J. Hepatol. 64 (6), 1339–1347. doi:10.1016/j.jhep.2016.02.001
van der Woerd, W. L., Houwen, R. H., and van de Graaf, S. F. (2017). Current and future therapies for inherited cholestatic liver diseases. World J. Gastroenterol. 23 (5), 763–775. doi:10.3748/wjg.v23.i5.763
van Dijk, R., Beuers, U., and Bosma, P. J. (2015). Gene replacement therapy for genetic hepatocellular jaundice. Clin. Rev. Allergy Immunol. 48 (2-3), 243–253. doi:10.1007/s12016-014-8454-7
van Mil, S. W. C., van der Woerd, W. L., van der Brugge, G., Sturm, E., Jansen, P. L. M., Bull, L. N., et al. (2004). Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology 127 (2), 379–384. doi:10.1053/j.gastro.2004.04.065
Van Mil, S. W. C., Milona, A., Dixon, P. H., Mullenbach, R., Geenes, V. L., Chambers, J., et al. (2007). Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology 133 (2), 507–516. doi:10.1053/j.gastro.2007.05.015
van Zutphen, T., Stroeve, J. H. M., Yang, J., Bloks, V. W., Jurdzinski, A., Roelofsen, H., et al. (2019). FXR overexpression alters adipose tissue architecture in mice and limits its storage capacity leading to metabolic derangements. J. Lipid Res. 60 (9), 1547–1561. doi:10.1194/jlr.M094508
Vandriel, S. M., Li, L. T., She, H., Wang, J. S., Gilbert, M. A., Jankowska, I., et al. (2023). Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatol. Balt. Md 77, 512–529. Published online August 29. doi:10.1002/hep.32761
Vasavan, T., Deepak, S., Jayawardane, I. A., Lucchini, M., Martin, C., Geenes, V., et al. (2021). Fetal cardiac dysfunction in intrahepatic cholestasis of pregnancy is associated with elevated serum bile acid concentrations. J. Hepatol. 74 (5), 1087–1096. doi:10.1016/j.jhep.2020.11.038
Vauthier, V., Ben Saad, A., Elie, J., Oumata, N., Durand-Schneider, A. M., Bruneau, A., et al. (2019). Structural analogues of roscovitine rescue the intracellular traffic and the function of ER-retained ABCB4 variants in cell models. Sci. Rep. 9 (1), 6653. doi:10.1038/s41598-019-43111-y
Verhulst, P. M., van der Velden, L. M., Oorschot, V., van Faassen, E. E., Klumperman, J., Houwen, R. H. J., et al. (2010). A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatol. Balt. Md 51 (6), 2049–2060. doi:10.1002/hep.23586
Villiger, L., Grisch-Chan, H. M., Lindsay, H., Ringnalda, F., Pogliano, C. B., Allegri, G., et al. (2018). Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 24 (10), 1519–1525. doi:10.1038/s41591-018-0209-1
Vitek, L., Bellarosa, C., and Tiribelli, C. (2019). Induction of mild hyperbilirubinemia: Hype or real therapeutic opportunity? Clin. Pharmacol. Ther. 106 (3), 568–575. doi:10.1002/cpt.1341
Vries, E. E., Mazzetti, M., Takkenberg, B., Mostafavi, N., Bikker, H., Marzioni, M., et al. (2020). Carriers of ABCB4 gene variants show a mild clinical course, but impaired quality of life and limited risk for cholangiocarcinoma. Liver Int. Off. J. Int. Assoc. Study Liver 40 (12), 3042–3050. doi:10.1111/liv.14662
Walker, K. F., Chappell, L. C., Hague, W. M., Middleton, P., and Thornton, J. G. (2020). Pharmacological interventions for treating intrahepatic cholestasis of pregnancy. Cochrane Database Syst. Rev. 7 (7), CD000493. doi:10.1002/14651858.CD000493.pub3
Wang, L., Soroka, C. J., and Boyer, J. L. (2002). The role of bile salt export pump mutations in progressive familial intrahepatic cholestasis type II. J. Clin. Invest. 110 (7), 965–972. doi:10.1172/JCI15968
Wang, R., Lam, P., Liu, L., Forrest, D., Yousef, I. M., Mignault, D., et al. (2003). Severe cholestasis induced by cholic acid feeding in knockout mice of sister of P-glycoprotein. Hepatol. Balt. Md 38 (6), 1489–1499. doi:10.1016/j.hep.2003.09.037
Wang, N. L., Li, L. T., Wu, B. B., Gong, J. Y., Abuduxikuer, K., Li, G., and Wang, J. S. (2016). The Features of GGT in Patients with ATP8B1 or ABCB11 Deficiency Improve the Diagnostic Efficiency. PloS one, 11 (4), e0153114. doi:10.1371/journal.pone.0153114
Weber, N. D., Odriozola, L., Martínez-García, J., Ferrer, V., Douar, A., Bénichou, B., et al. (2019). Gene therapy for progressive familial intrahepatic cholestasis type 3 in a clinically relevant mouse model. Nat. Commun. 10 (1), 5694. doi:10.1038/s41467-019-13614-3
Wei, C. S., Becher, N., Friis, J. B., Ott, P., Vogel, I., and Grønbæk, H. (2020). New tight junction protein 2 variant causing progressive familial intrahepatic cholestasis type 4 in adults: A case report. World J. Gastroenterol. 26 (5), 550–561. doi:10.3748/wjg.v26.i5.550
Wei, G., Cao, J., Huang, P., An, P., Badlani, D., Vaid, K. A., et al. (2021). Synthetic human ABCB4 mRNA therapy rescues severe liver disease phenotype in a BALB/c.Abcb4-/- mouse model of PFIC3. J. Hepatol. 74 (6), 1416–1428. doi:10.1016/j.jhep.2020.12.010
Wei, X., Fang, Y., Wang, J. S., Wang, Y. Z., Zhang, Y., Abuduxikuer, K., and Chen, L. (2023). Neonatal sclerosing cholangitis with novel mutations in DCDC2 (doublecortin domain-containing protein 2) in Chinese children. Frontiers in pediatrics, 11, 1094895. doi:10.3389/fped.2023.1094895
Williamson, C., and Geenes, V. (2014). Intrahepatic cholestasis of pregnancy. Obstet. Gynecol. 124 (1), 120–133. doi:10.1097/AOG.0000000000000346
Xia, X., Francis, H., Glaser, S., Alpini, G., and LeSage, G. (2006). Bile acid interactions with cholangiocytes. World J. Gastroenterol. 12 (22), 3553–3563. doi:10.3748/wjg.v12.i22.3553
Xu, J., Kausalya, P. J., Van Hul, N., Caldez, M. J., Xu, S., Ong, A. G. M., et al. (2021). Protective functions of ZO-2/tjp2 expressed in hepatocytes and cholangiocytes against liver injury and cholestasis. Gastroenterology 160 (6), 2103–2118. doi:10.1053/j.gastro.2021.01.027
Yasar, U., Greenblatt, D. J., Guillemette, C., and Court, M. H. (2013). Evidence for regulation of UDP-glucuronosyltransferase (UGT) 1A1 protein expression and activity via DNA methylation in healthy human livers. J. Pharm. Pharmacol. 65 (6), 874–883. doi:10.1111/jphp.12053
Yokoda, R. T., and Carey, E. J. (2019). Primary biliary cholangitis and primary sclerosing cholangitis. Am. J. Gastroenterol. 114 (10), 1593–1605. doi:10.14309/ajg.0000000000000268
Yuan, X. Z., Yang, R. M., and Wang, X. P. (2021). Management perspective of wilson’s disease: Early diagnosis and individualized therapy. Curr. Neuropharmacol. 19 (4), 465–485. doi:10.2174/1570159X18666200429233517
Yueh, M. F., Chen, S., Nguyen, N., and Tukey, R. H. (2017). Developmental, genetic, dietary, and xenobiotic influences on neonatal hyperbilirubinemia. Mol. Pharmacol. 91 (5), 545–553. doi:10.1124/mol.116.107524
Zhang, J., Yang, Y., Gong, J. Y., Li, L. T., Li, J. Q., Zhang, M. H., Lu, Y., Xie, X. B., Hong, Y. R., Yu, Z., Knisely, A. S., and Wang, J. S. (2020). Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: Clinical, histological and ultrastructural characterization. Liver international : official journal of the International Association for the Study of the Liver, 40 (5), 1142–1150. doi:10.1111/liv.14422
Zhao, C., Lancman, J. J., Yang, Y., Gates, K. P., Cao, D., Barske, L., et al. (2022). Intrahepatic cholangiocyte regeneration from an Fgf-dependent extrahepatic progenitor niche in a zebrafish model of Alagille Syndrome. Hepatol. Balt. Md 75 (3), 567–583. doi:10.1002/hep.32173

Keywords: heredity, intrahepatic cholestasis, genetic mutation, molecular function, therapy
Citation: Xie S, Wei S, Ma X, Wang R, He T, Zhang Z, Yang J, Wang J, Chang L, Jing M, Li H, Zhou X and Zhao Y (2023) Genetic alterations and molecular mechanisms underlying hereditary intrahepatic cholestasis. Front. Pharmacol. 14:1173542. doi: 10.3389/fphar.2023.1173542
Received: 03 March 2023; Accepted: 16 May 2023;
Published: 31 May 2023.
Edited by:
Pedro Miguel Rodrigues, Biodonostia Health Research Institute (IIS Biodonostia), SpainReviewed by:
Oscar Briz, University of Salamanca, SpainCopyright © 2023 Xie, Wei, Ma, Wang, He, Zhang, Yang, Wang, Chang, Jing, Li, Zhou and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuelin Zhou, cGV0ZXJ4bHpob3VAZ21haWwuY29t, emhvdXh1ZWxpbkBjY211LmVkdS5jbg==; Yanling Zhao, emhhb3lsMjg1NUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.