 Yang Li
Yang Li- 1State Key Laboratory of Biotherapy and Cancer Center, West China School of Basic Medical Sciences and Forensic Medicine, West China Hospital, and Collaborative Innovation Center for Biotherapy, Sichuan University, Chengdu, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children of Ministry of Education, Department of Reproductive Medicine, West China Second University Hospital of Sichuan University, Chengdu, China
Cancer treatment is hampered by resistance to conventional therapeutic strategies, including chemotherapy, immunotherapy, and targeted therapy. Redox homeostasis manipulation is one of the most effective innovative treatment techniques for overcoming drug resistance. Reactive oxygen species (ROS), previously considered intracellular byproducts of aerobic metabolism, are now known to regulate multiple signaling pathways as second messengers. Cancer cells cope with elevated amounts of ROS during therapy by upregulating the antioxidant system, enabling tumor therapeutic resistance via a variety of mechanisms. In this review, we aim to shed light on redox modification and signaling pathways that may contribute to therapeutic resistance. We summarized the molecular mechanisms by which redox signaling-regulated drug resistance, including altered drug efflux, action targets and metabolism, enhanced DNA damage repair, maintained stemness, and reshaped tumor microenvironment. A comprehensive understanding of these interrelationships should improve treatment efficacy from a fundamental and clinical research point of view.
1 Introduction
Although the risk of death from cancer has decreased continuously by 32% over the past 3 decades, cancer treatment remains a significant global public health issue that faces multiple challenges (Siegel et al., 2022). In the early stage of tumor development, most patients will respond completely or partially to chemotherapy, whereas in the late stage, the therapeutic effect is still unsatisfactory, mainly due to drug resistance (Miller et al., 2019). Drug resistance is generally divided into inherent resistance and acquired resistance. Specifically, intrinsic resistance is mainly caused by existing factors (e.g., gene mutations), while acquired resistance is caused by changes in tumor cells during treatment, including drug target expression, post-translational modification, and activation of bypass signaling (Holohan et al., 2013; Li et al., 2020). Recent studies have shown that the stress response induced during tumor treatment, especially oxidative stress, is one of the vital reasons for the acquired drug resistance of tumor cells (Cui et al., 2018; Qin et al., 2022).
Traditional and new treatments, including surgery, immunotherapy, radiotherapy, chemotherapy, and targeted therapy, are the main methods of cancer treatment at present (Ward et al., 2021). Among several novel therapeutic approaches to overcome drug resistance, manipulating redox homeostasis is efficient in various tumors (Cui et al., 2018; Hayes et al., 2020). The regulation of redox homeostasis is essential in maintaining cell survival and normal function, which is accomplished through a balance of Reactive oxygen species (ROS) production and elimination (Lennicke and Cochemé, 2021). ROS in cells are mainly derived from the mitochondrial electron transport chain, endoplasmic reticulum oxidase, and NADPH oxidase (NOXs) (Block and Gorin, 2012; Ashton et al., 2018; Jakubczyk et al., 2020). The elimination of ROS is accomplished by antioxidants, antioxidant enzymes, antioxidant transcription factors NRF2, and high abundance redox proteins (Harris and DeNicola, 2020). NRF2 is a transcription factor involved in cell homeostasis and plays a crucial role in regulating redox homeostasis, drug metabolism, energy metabolism, proteasome degradation, and other physiological processes (Huang et al., 2015). Meanwhile, NRF2 targets genes associated with cell defense, including antioxidant response elements (AREs) (Lu et al., 2016). However, in oxidative stress cells, oxidation of the KEAP1-Cul3-RbX1 complex destroys specific key cysteine residues in Keap1, thereby interfering with the ubiquitin degradation process of NRF2, resulting in enhanced transcriptional activation of genes, including GSH, heme oxygenase-1 (HO-1), NADP(H), and quinone oxidoreductase 1 (NQO1) (Itoh et al., 1997), and the enhancement of chemotherapy resistance (Wang et al., 2008). ROS can act as a double-edged sword: In normal cells, the activation of ROS signaling can reduce the possibility of the formation of cancerous lesions; but its abnormal activation in precancerous cells with high levels of ROS may benefit cancer cell survival (Hayes and McMahon, 2006; Lau et al., 2008; Wang et al., 2008; Sporn and Liby, 2012; Gañán-Gómez et al., 2013). When the production and elimination of intracellular ROS are in a state of dynamic equilibrium, the cells may survive and become redox homeostatic. Previously considered intracellular by products of aerobic metabolism, reactive oxygen species (ROS) have now been seen as second messengers regulating multiple signaling pathways (Forman, 2016). The disulfide bond, a chemical bond formed by sulfhydryl groups connecting two different cysteine residues in the peptide chain, can stabilize the spatial configuration of various receptors, enzymes, and other proteins (Hohl et al., 2012). It has been found that redox homeostasis of cells has a vital influence on the formation of reversible disulfide bonds and plays a crucial role in spatial structure and functional maintenance (Liu et al., 2016). Changes in redox modifications and redox-mediated pathways together mediate oxidative stress-induced tumor drug resistance.
Under normal physiological conditions, the body is in a state of redox homeostasis, that is, the balance between oxidants and antioxidants. Compared to normal cells, cancer cells react with higher ROS levels during treatment through upregulating their antioxidant system. This promotes tumor therapeutic resistance via various mechanisms, such as altered drug efflux, metabolism and targets, improved DNA damage repair, preserved stemness, and modified tumor microenvironment. A comprehensive understanding of their relationships should improve treatment efficacy from a fundamental and clinical research point of view. This review aims to summarize the molecular mechanisms of redox-regulated cancer drug resistance and explore potential clinical strategies to overcome drug resistance.
2 Redox signaling regulates drug efflux
Sufficient intracellular drug concentration is required for drug toxicity that can effectively kill tumor cells, and plasma membrane transporters regulate drug concentration by regulating drug influx and efflux. Drug influx is characterized by drug concentration on both sides of the cell membrane, the aid of drug transporters, and the requirement of energy (Joyce et al., 2015). Drug efflux has been widely studied in numerous species as a crucial intracellular detoxification mechanism (Mandal et al., 2017).
Several cell membrane transporters are crucial to chemotherapeutic efflux and influx and drug resistance. As defined by the main amino acid sequence, they all have widespread substrate molecular specificity, promoting the elimination of major cancer chemotherapeutic agents such as antimetabolites, taxanes, and topoisomerase inhibitors (Wong et al., 2014). Multidrug resistance protein 1 (MDR1), MDR-associated protein 1 (MRP1), and breast cancer resistance protein (BCRP) are three of them that have been widely examined in relation to cancer drug resistance to chemotherapy (Sharom, 2008; Fletcher et al., 2010). ATP-binding cassette (ABC) transporters serve on efflux pumps that actively transport drugs out of cells against concentration gradients, thus promoting MDR by increasing the efflux of anticancer drugs so that the intracellular drug concentration is less than the level of effective drug therapy (Yardley, 2013; Li et al., 2016). ROS modulate ABC transporters in a two-pronged way: moderate oxidative stress may be necessary for the induction of ABC transporters in tumor cells, whereas excessive oxidative stress may inhibit the transporter system (Yuan et al., 2022).
2.1 Redox changes the conformation of drug efflux transporters
Specifically, ABC transporters contain two cytoplasmic nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs), which can be merged into multidomain peptides in a great diversity of ways. Conversion between the two main conformations of NBD dimers: ATP binding induces each NBD to form a closed dimer, and ATP hydrolysis restores the dimer to an open configuration. Changes in the two configurations generate driving forces that facilitate drug transport, reducing intracellular drug levels to sublethal (Higgins, 2007).
The transport activity of MDR1 is related to the REDOX state of cysteine residues Cys431 and Cys1074 in NBD1 and NBD2, and the structures of the two binding sites are similar. MRP1 also has a topological structure, in addition to a transmembrane domain MSD0, which acts as a gate valve in the process of drug transport across the membrane. Cysteine residues in MSD0 establish disulfide bonds to dimerize MRP1, and their structural mutations may prevent dimer formation and interfere with drug transport. For instance, cysteine residues in MSD0 can be affected by drug induction (e.g., destruction of MRP1 by the reducing agent dithiothreitol (DTT)) (Maiti, 2012). Furthermore, ROS can be produced after treatment with some anticancer drugs to promote the upregulation of the antioxidant system in cancer cells, such as enhancing the expression of GSH. The cysteine residue of glutathione is its active group, which can bind to a multitude of drugs and toxins and promote the efflux of drugs through MRP1, thus having an integrated detoxification effect (Krause et al., 2007).
Human BCRP, belonging to ABCG2, is a semi-molecular transporter responsible for the delivery of multiple drugs, including mitoxantrone and SN-38. BCRP plays a key role in chemotherapeutic resistance as a homodimer drug transporter. Notably, there are three important amino acid residues in BCRP, Cys592, Cys603, and Cys608, located on the outer surface of the plasma membrane (Henriksen et al., 2005). ABCG2 cysteine residues Cys592 and Cys608 formed intramolecular disulfide bonds, regulating substrate specificity, whereas Cys603 and Cys608 created intermolecular disulfide bonds, determining plasma membrane localization (Wakabayashi et al., 2006). Intermolecular and intramolecular disulfide bridges are critical to the structural and functional integrity of ABCG2. According to research, when cysteine residues are muted owing to external factors, the spatial structure of the BCRP protein changes, affecting its activity and function to overcome multidrug resistance and improve drug sensitivity (Kage et al., 2005). ROS regulation of ABC transporters is a new method to overcome drug resistance in cells. These studies suggest that ROS inactivate transporters by affecting disulfide bonds in the transporter protein dimer, highlighting the potential of ROS to alter transporters to reduce drug efflux and thereby promote drug efficacy.
2.2 Redox altered transporter gene expression
Studies have shown that redox regulation of drug efflux transporters can be carried out through multiple pathways, and several transcriptional pathways that regulate gene expression induced by environmental factors have been identified (McMahon et al., 2003). ROS mediated by endoplasmic reticulum stress have been shown to upregulate MDR1 expression in hepatoma cells (Ledoux et al., 2003). In Caco-2 human colon cancer cells, MDR1 expression is increased under high-concentration treatment with H2O2 (Terada et al., 2014). In detail, the development of oxidative stress leads to the oxidation of the SH group, which upregulates NRF2. NRF2 mediates an increase in the content and activity of p-GP transporters, thereby limiting the increase in membrane permeability (Shchulkin et al., 2021). In response to NRF2 activation, fork-head box O (FOXO) can be activated through disulfide interactions with transporters. According to the study by Myatt and Lam. (2007), the inactivation of the transcription factor FOXO may lead to cell carcinogenesis, so the activation of FOXO may contribute to the expression of drug efflux transporters (Gomes et al., 2013). After chemotherapeutic agents are used, intracellular redox equilibrium is disrupted and ROS accumulate, promoting the activation of FOXOs and inducing the expression of transporter proteins. For example, paclitaxel (PTX) has been reported to induce drug resistance in ovarian malignant tumor cells via Trx1 and the transcription factor FOXO (Wang et al., 2015). Furthermore, oxidative stress can promote the transcription of multiple transporter protein genes, including MDR, MRPs, and BCRP, by promoting the translocation of apurinic-apyrimidinic endonuclease I (APE-1) or redox factor I (Ref-1). In addition, oxidative stress network activation affects disulfide bonds between PKA or PKC to increase the activity of protease, which in turn affects its oxidative phosphorylation substrate MDR1, thereby reducing cellular drug sensitivity (Blobe et al., 1993; Brennan et al., 2006; Kang and Yang, 2020). Moreover, there is increasing evidence that epigenetic regulation can influence drug resistance by altering the configuration of drug efflux pumps. For example, methylation of the MDR1 promoter region silences the transcription of its genes (Lertratanangkoon et al., 1997; Baker et al., 2005). Overall, oxidative stress affects drug efflux via redox modification and redox signaling; hence, an in-depth understanding of the molecular mechanism can aid in resolving the clinical drug resistance issue.
3 Redox homeostasis in tumor cell survival and death
3.1 DNA repair
DNA can be damaged by various endogenous and environmental agents, leading to its physical or chemical changes (Chatterjee and Walker, 2017). Although oxidative stress can damage cellular structures, DNA damage and genomic instability in the initial stage can drive the accumulation of carcinogenic changes that contribute to the development of cancer (Basu and Nohmi, 2018). Studies have found that ROS, as DNA damage agents during tumorigenesis, effectively increases cell mutation rate, thereby promoting carcinogenic transformation (Jackson and Loeb, 2001). ROS can generate DNA lesions in many ways, including base modification, nucleotide removal, DNA structural disruption, and DNA-protein crosslinking (Barnes et al., 2019). For example, ROS induce DNA fragmentation in sperm nuclear/mitochondrial genomes and reduce transcription levels (Bisht and Dada, 2017). Guanine is more easily oxidized than any other DNA base, and 8-hydroxy-2′-deoxyguanosine is the most usual form of oxidized guanine (Salehi et al., 2018). In addition, ROS can indirectly cause extra cyclic DNA damage by inducing lipid peroxidation (Yu et al., 2016). Oxidative stress also leads to mitochondrial DNA base lesions and degradation, an essential factor in mitochondrial gene mutations (Shokolenko et al., 2009).
The cell response to DNA damage is a complex process involving a variety of signaling networks and proteins that are activated or inactivated differentially in specific cancer types (Salehi et al., 2018). For a series of DNA damage events, from single-strand breaks (SSBs) to base alkylation events, tumor cells will initiate the corresponding DNA damage response (DDR) to resist this effect, thus maintaining tumor survival (Curtin, 2012; Lord and Ashworth, 2012). In normal cells, spontaneous DNA damage and repair are roughly balanced, and most DNA damage is repaired correctly. In tumor cells, however, this balance may change due to inadequate repair ability (Srinivas et al., 2019). Experiments have shown that ROS inhibit the activation of the DNA repair enzyme OGG1 (an 8-oxoG DNA glycosylase) by oxidizing critical cysteine residues such as Cys253 and Cys255 (Bravard et al., 2006). In addition, ROS can delay the recognition of damaged areas by affecting a range of sensor kinases (ATM and ATR) and downstream sensor kinases (CHK1 and CHK2) (Chang et al., 2015). H2AX has been shown to cluster around damage sites under the regulation of CTCF in the case of DNA damage, thus initiating DNA repair. Continuous oxidative stress accelerates the degradation of H2AX interceded by the E3 ubiquitin ligase RNF168, which enhances DNA damage in cancer cells (Gruosso et al., 2016). As ROS can inhibit the DDR of tumors to a certain extent, treatment with increased ROS may promote the death of cancer cells.
3.2 Apoptosis
Apoptosis is an evolutionarily conserved process that is essential for development and homeostasis. Furthermore, apoptosis is regulated by two main pathways, the death receptor pathway and the mitochondrial pathway, with ROS strongly implicated in the signal transduction of these pathways (Circu and Aw, 2010). Small amounts of ROS facilitate cancer cell development by activating several key factors in the cell cycle. A large amount of ROS generate oxidative stress that triggers programmed cell death, or apoptosis (Johar et al., 2015). In the case of tumors, cells lose their ability to undergo normal apoptotic induction, leading to uncontrolled proliferation (Mohammad et al., 2015). Studies have shown that VB1 increases the level of ROS in anti-BRAFi melanoma cells, leading to DNA cytotoxicity, G2/M cell cycle stagnate, and apoptosis (Liu et al., 2018). Bcl-2 family proteins are key factors regulating apoptosis, and their expression and transport to mitochondria are regulated by ROS (Li et al., 2004). After treating HepG2 cells with zinc oxide nanoparticles, reactive oxygen species changed mitochondrial membrane potential, degraded and downregulated Bcl-2, resulting in cell apoptosis (Sharma et al., 2012). In addition, doxorubicin promotes ROS accumulation in keratinocytes, inactivating ERK1/2 by oxidative modification of cysteine sulfonic acid, leading to the downregulation of Bcl-2 and induction of apoptosis (Luanpitpong et al., 2012). After sodium nitroprusside treatment, S-nitrosylation modification at Cys183 decreases the phosphorylation of ERK1/2, thus promoting apoptosis (Jin et al., 2018).
In various malignancies, accumulated ROS may activate p53 through the JNK signaling pathway, thereby promoting apoptosis (Shi et al., 2014). The cytosolic protein c-FLIP (cellular FLICE-inhibitory protein) is a death receptor-mediated apoptosis inhibitor that is upregulated in various cancers, thereby promoting the anti-apoptotic ability of tumor cells. ROS can affect the structural stability of the protein and promote its degradation (Wilkie-Grantham et al., 2013; Ming et al., 2023). In conclusion, increasing the ROS content in tumors to a certain extent through drugs may promote tumor cell apoptosis, thus reducing tumor drug resistance.
3.3 Necroptosis
Programmed cell necrosis is a mode of cell death mediated by death receptors and is closely regulated by intracellular signaling factors. This process involves the self-destruction of cells activated to prevent cell apoptosis from being blocked (Dhuriya and Sharma, 2018). At the core of necroptosis signaling is the activation of protein kinase receptor-interacting protein 3 (RIP3) by an upstream protein with the RIP homotypic interaction motif (RHIM) domain. This phosphorylates and activates mixed lineage kinase domain-like (MLKL), leading to cell membrane destruction and necrosis (Dondelinger et al., 2016). Ligands such as TNF and FasL can induce necroptosis, causing cell swelling and rupture. This releases damage-related molecular patterns (DAMPs), which are detected by the innate immune system as danger signals and trigger inflammatory responses (Orozco and Oberst, 2017).
Necroptosis has been associated with the inhibition of tumor development in some cancer types. For example, dysregulation of necroptosis has been linked to the development of acute myeloid leukemia and ovarian cancer. However, for esophageal, pancreatic, and colon cancer, necroptosis promotes tumor progression and metastasis. The underlying mechanism remains elusive (Wang et al., 2017). Studies have suggested that necroptosis is closely related to ROS and that they are positively correlated, which plays an important role in pathophysiological conditions such as tumors (Hsu et al., 2020). One study showed that ROS-mediated modification of residues of cysteine 257, 268, and 586 can lead to the formation of an intramolecular disulfide bond on RIP1, which promotes the autophosphorylation of RIP1 at serine 161 thereby effectively recruiting RIP3. These results suggests that ROS serves as an inducer of necroptosis (Zhang Y. et al., 2017). It was found that under hydrogen peroxide treatment, RIP3 could migrate to the endoplasmic reticulum and induce calcium ion imbalance, suggesting that oxidative stress could initiate programmed necrosis of cardiomyocytes through upregulation of RIP3 (Zhang T. et al., 2016). In addition, removal of ROS with butylated hydroxyanisole significantly reduced TNF-mediated necrosis in a mouse fibrosarcoma L929 cell line (Vanlangenakker et al., 2011). Although the molecular mechanism between necrosis and ROS has been established, effective therapies to inhibit cancer progression by simultaneously regulating necrosis and ROS need further investigation.
3.4 Autophagy
Autophagy is an intracellular catabolic process. Under nutrient deficiency or stress stimulation, the cell degrades misfolded proteins and damaged organelles, thereby providing the cell with energy (Klionsky et al., 2021). For tumors, the role of autophagy is two-sided and complex. In general, autophagy mainly inhibits tumors in the early stage of tumor formation, while it specifically protects and helps tumor cells survive (Rebecca and Amaravadi, 2016). Studies have shown that REDOX signal transduction and modification are closely related to autophagy. For example, ROS can regulate mTOR activity through various pathways, such as the LKB1/AMPK, HMGB1, and PI3K/Akt pathways, thus inducing autophagy (Gibson, 2013). ROS can also activate autophagy by phosphorylating JNK in human breast cancer cells (Sun et al., 2017). In addition, numerous autophagy-related proteins, including LC3-II, P62, and ATG protein, were activated in H9c2 cells treated with H2O2 (Ha et al., 2012). In laser therapy of cancer, accumulated ROS can increase beclin-1 expression, thereby promoting tumor autophagy and increasing its therapeutic resistance (Shu et al., 2016). However, several studies have shown that oxidative modifications of autophagy regulatory factors play an inhibitory role in autophagy. Oxidation of the autophagic proteins ATG4B on cysteine Cys292 and Cys361 inhibited the activity of tubulin 1A/1B LC3, leading to reduced autophagic flux (Zheng et al., 2020). In addition, oxidative modification of Cys263 and Cys572 inhibits LC3 lipidation (Frudd et al., 2018).
In addition to the two aspects of the influence of oxidative stress on autophagy, autophagy may mediate a negative feedback loop to prevent oxidative stress. Elevated ROS induces Ca2+ release by activating MCOLN1, leading to nuclear translocation of TFEB, which induces autophagy. Enhanced autophagy promotes the elimination of excess ROS (Zhang X. et al., 2016). If autophagy is insufficient to remove excess ROS, it may cause the death of tumor cells. Providing new targets may help accelerate the process of drug-resistant tumor therapy, and the specific role of oxidative stress on autophagy needs to be verified by further studies.
3.5 Cell survival
Moderate ROS promote cell survival and proliferation by means of multiple pathways, such as NF-κB, MAPK, and PI3K/AKT, which play a key role in tumor drug resistance (Figure 1). Activation of PI3K can lead to cell growth, cell proliferation, cell survival, etc. Modification of this pathway is closely related to the pathogenesis of most cancers (Noorolyai et al., 2019). The tumor suppressor gene PTEN is a negative regulator of the PI3K/AKT signaling pathway. Studies have found that oxidative stress can induce the inactivation of PTEN (oxidation on Cys124 and Cys71), thereby activating the PI3K and AKT pathways to promote cell survival (Leslie et al., 2003; Kim et al., 2018).
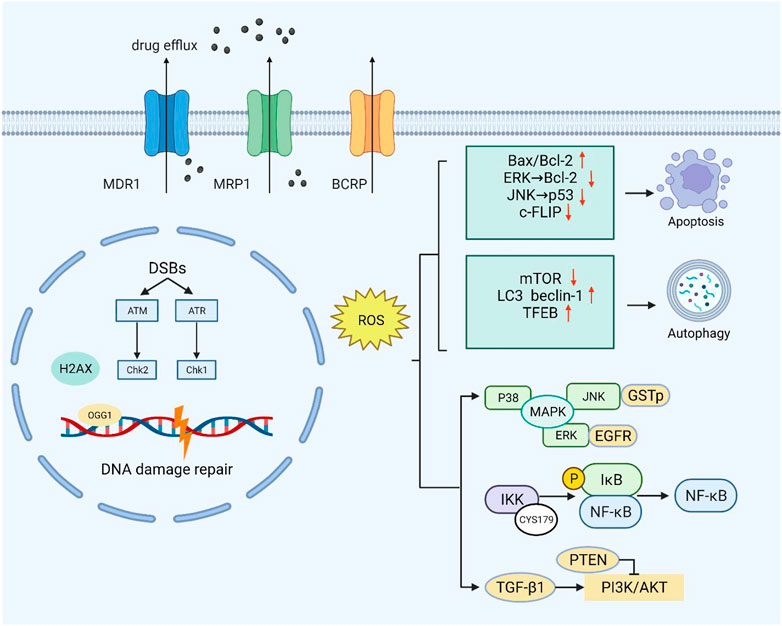
FIGURE 1. Moderate ROS levels can limit the anticancer activity of drugs by increasing drug efflux. Excessive ROS can cause endogenous DNA damage and inhibit the DNA damage repair response. At high ROS levels, death receptor-mediated and mitochondrial pathways are triggered to induce apoptosis. ROS can also inhibit the negative regulators of autophagy (mTOR) and increase the formation of autophagosomes by upregulating autophagy-related genes. At low levels, ROS act as second messengers by mediating multiple signaling pathways (PI3K/AKT, MAPK, and NF-κB) that contribute to cancer cell survival and proliferation.
The ROS-dependent activation of TGF-β1 was specific and restricted to the oxidation of Met253 on latency-associated peptide β (LAP-β) (Richter et al., 2015). Studies have shown that ROS upregulates and activates the PI3K/Akt signaling pathway through TGF-β1, leading to malignant changes in kidney cells (Lu et al., 2019). Intriguingly, PI3K/Akt also upregulates glutathione synthesis through NRF2 to resist oxidative stress in breast tumors (Lien et al., 2016).
NF-κB plays an irreplaceable role in cell survival and proliferation, and NF-κB activation can actuate drug resistance in a variety of tumors during treatment (Drain et al., 2021). Oxidative stress has the potential to regulate NF-κB activity, but the mechanism is not simple. Studies have shown that ROS can both promote and inhibit NF-κB activity, and in most cases, NF-κB activity is elevated in tumor cells (Nakajima and Kitamura, 2013). Berbamine-mediated excess ROS negatively regulates the NF-κB pathway by means of activation of NF-κB inhibitor (IκB) and oxidation of cysteine in the binding region of NF-κB to DNA, thereby inhibiting the development of bladder cancer (Han et al., 2021). In addition, ROS-mediated oxidation of Cys179 can inhibit IKK activity, which is essential for NF-κB activation (Reynaert et al., 2006).
The mitogen-activated protein kinase (MAPK) signaling pathway has four subfamilies: ERK, P38, JNK, and ERK5, which mediate reactions from plentiful stimuli (Latimer and Veal, 2016). Oxidative modification of Cys119 and Cys162 of p38 affected the dimerization of its key upstream activator mitogen-activated protein kinase 3 (MKK3) (Bassi et al., 2017). ROS have been shown to activate EGF receptors, which can stimulate the subsequent activation of the ERK signaling pathway (León-Buitimea et al., 2012). Recently, studies have shown that ginsenoside Rh4 can activate autophagy and apoptosis in colorectal tumor cells by activating the ROS/JNK/p53 pathway (Wu Q.et al., 2018). In addition, ROS can separate JNK from glutathione S-transferase pi (GSTp), which can inhibit the activation of JNK, thereby facilitating the JNK signaling pathway (Castro-Caldas et al., 2012).
In summary, the redox-mediated cell survival signaling pathway significantly supplements drug resistance mechanisms. Further research on ROS targets will help speed up the development process of studying the molecular mechanism of ROS-induced tumor resistance and formulate therapeutic strategies.
4 Stress homeostasis and drug resistance
4.1 Redox homeostasis
Tumor cells can enter a reversible drug-tolerant persister (DTP) state to avoid chemotherapy and targeted drug attack by a mechanism different from tumor resistance through genetic mutations. In the case of oxidative stress caused by drug stimulation, DTP cells rely heavily on regulating redox homeostasis to avoid cell death, which is regarded as a major cause of tumor drug resistance (Mikubo et al., 2021). On the one hand, tumor cells with acquired drug resistance can increase oxidative stress and maintain their active proliferation state. On the other hand, tumor cells can also activate the antioxidant defense system to avoid oxidative stress-mediated cell death (Chini et al., 2021). It was found that oxidative stress can promote NRF2-mediated activation of the antioxidant genes. Keap1 also inhibits the phosphorylation of IκBα by mediating IKKβ degradation, thus antagonizing oxidative stress induced by the NF-κB pathway. Oxidative stress also promotes FOXO entry into the nucleus and activates the expression of downstream antioxidant genes (Rojo de la Vega et al., 2018; Hayes et al., 2020). Glutathione (GSH), a crucial reductive force in cells, can significantly reduce oxidative damage and lead to tumor drug resistance (Bansal and Simon, 2018). In detail, cystine transporter xCT can promote the transport of cysteine into the cell and provide the raw material for GSH synthesis. Recent research suggested that oxidative stress can enhance the expression and activity of xCT through transcription and post-translational modification, thus affecting the synthesis of GSH (Daher et al., 2019). In addition, the CD44 variant promotes intracellular cystine transport and GSH synthesis by interacting with cystine transporter xCT, thus maintaining very low ROS levels in tumor stem cells (Ishimoto et al., 2011). In addition, some high-abundance redox proteins can buffer oxidative stress through non-classical antioxidant pathways mediated by self-oxidation modifications. For example, the active cysteine sites (Cys115 and Cys161) of CypA undergo oxidative modification to form intramolecular disulfide bonds, which contribute to the elimination of intracellular ROS. Further studies have found that PRDX2 can bind to CypA through disulfide bonds and promote the reduction of oxidized CypA, thus maintaining the reduction ability of colorectal cancer cells (Peng et al., 2021). Drug-resistant cells express the sterol lipid transporter NPC1L1 (Niemann-Pick C1-like 1) to compensate for the inhibition of GSH synthesis caused by the downregulation of xCT. Meanwhile, NPC1L1 can resist oxidative stress induced by tumor therapy to a certain extent by promoting the uptake of vitamin E and cholesterol (Zhang et al., 2022). ANXA2 can also enhance tumor cell survival by buffering excessive oxidative stress through oxidative modification and inducing autophagy (Wang et al., 2018). The above studies indicate that DTP cells can resist oxidative stress induced by tumor therapy by regulating intracellular redox-related pathways, which provides ideas for targeting antioxidant systems to overcome tumor drug resistance.
4.2 Metabolic reprogramming
In somatic cells, glucose is metabolized by the tricarboxylic acid cycle and then phosphorylated to provide energy. When the oxygen supply is insufficient, a large amount of pyruvate is produced by anaerobic glycolysis and converted to lactic acid to maintain the energy supply. Even when oxygen is abundant, tumor cells preferentially conduct glycolysis to meet the demand for energy and nutrients for rapid proliferation, a phenomenon known as the Warburg effect (Liberti and Locasale, 2016). This metabolic change is widely regarded as a hallmark of cancer (Potter et al., 2016). A growing body of research suggests a link between aerobic glycolysis and tumor growth and chemotherapy resistance (Sattler et al., 2010). In addition, this effect may not be due to defective mitochondrial respiration but to the upregulation of glycolytic enzymes and glucose transporters (Wangpaichitr et al., 2021). While intracellular ROS production and the maintenance of redox homeostasis largely depend on cellular metabolism, ROS can also affect tumor energy metabolism by regulating key metabolic enzymes (Quijano et al., 2016). Many glycolytic enzymes contain highly conserved cysteines that are susceptible to redox modification in response to ROS (Lennicke and Cochemé, 2021). Hexokinase (HK) is the first rate-limiting step in catalyzing glucose metabolism, promoting the immediate use of ATP (Heneberg, 2019). Using 2-DG as an HK2 inhibitor to sensitize tumor cells can improve the efficacy of the cytotoxic drugs doxorubicin and paclitaxel (Maschek et al., 2004). It has been found that dehydroascorbic acid (DHA) can covalently bind to the active cysteine of hexokinase 1 (HK1) and irreversibly lose its enzyme activity (Fiorani et al., 2000). Pyruvate kinase M2 (PKM2) is a glycolytic enzyme with less pyruvate kinase activity that can prevent the flow of glycolytic metabolites into the TCA cycle, thus maintaining the survival of tumor cells (Christofk et al., 2008). Silencing PKM2 can increase docetaxel accumulation in A549 lung cancer cells and enhance its antitumor effect (Shi et al., 2010). In lung cancer cells, hydrogen peroxide caused a stepwise increase in ROS levels and further inhibited PKM2 activity by oxidizing cysteine Cys358 (Anastasiou et al., 2011). Dihydroxyacetone phosphate (DHAP) isomerizes under the catalytic action of triose phosphate isomerase (TPI) to form glyceraldehyde-3-phosphate (G3P) (Pekel and Ari, 2020). It has been found that ROS may cause oxidation of TPI by affecting intramolecular disulfide bonds, and oxidative TPI is subsequently degraded, thus turning glycolysis into the pentose phosphate pathway (PPP) (Dumont et al., 2016). Elevated levels of GAPDH are considered a key factor in the maintenance of tumor glycolytic phenotypes. Under oxidative stress, Cys152 of GAPDH can be oxidized to form intermolecular disulfide bonds, leading to aggregation of GAPDH and cell death (Reisz et al., 2016). In addition, redox modification of cysteine inhibits the activity of GAPDH and shifts metabolic flux from glycolysis to the PPP pathway (Ralser et al., 2007). Since cancer cells generally express more ROS than normal cells, changes in glycolysis to the pentose-phosphate pathway enhance the reductive ability of tumor cells to some extent (Kuehne et al., 2015). In this case, ROS act as signaling molecules that regulate tumor metabolism in response to changes in the cellular environment. This also suggests that the combination of ROS regulators and metabolic recombination of tumor cells can provide a new idea for tumor therapy. In conclusion, ROS-mediated redox modification can regulate the function of metabolic enzymes, thereby regulating tumor metabolic reprogramming and playing a significant role in tumorigenesis and development (Figure 2).
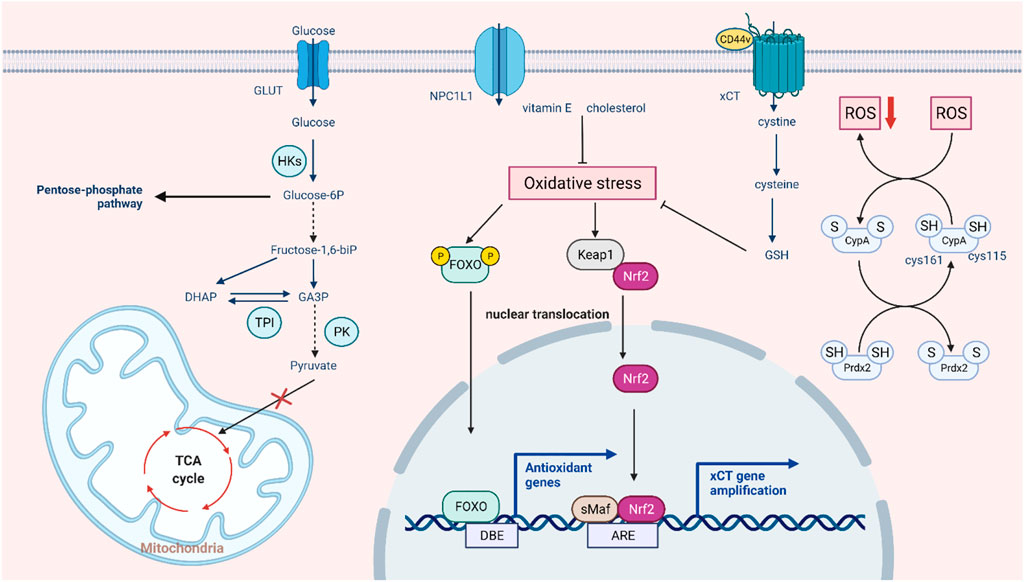
FIGURE 2. Intrinsic pathways involved in ROS-mediated drug resistance. In the aerobic state, tumor cells still preferentially choose glycolysis and conversion to lactic acid, and ROS regulate the three key enzymes in this process. In response to excessive oxidative stress, the glycolytic pathway partly switches to the pentose phosphate pathway, and the tumor also activates the antioxidant system to prevent cell death by allowing the translocation and activation of NRF2 and FOXO and enhancing XCT transport function to promote GSH synthesis. In addition, some non-classical antioxidant pathways, such as autooxidation modification of CypA and compensatory transport of NPC1L1, can also buffer oxidative stress.
5 EMT and cell stemness regulated by redox signaling
Epithelial-to-mesenchymal transition (EMT) plays a pivotal part in inducing tumor metastasis and promoting drug resistance (Chang, 2016; Ming et al., 2021). During EMT, epithelial cells lose their intercellular adhesion and cell polarity characteristics, as well as acquire motility, migration potential, and invasiveness (Brabletz et al., 2018). Cancer stem cells (CSCs) take an essential effect on tumor survival, proliferation, and metastasis. Specifically, CSCs can maintain cancer cell activity utilizing continuous self-updating and limitless proliferating, which are closely related to tumor drug resistance and recurrence (Phi et al., 2018). It was found that the signaling pathways activated in EMT are significantly similar to those that drive CSCs, such as the Wnt, Hedgehog, and Notch signaling pathways, which suggests that EMT is closely related to stem cell status in normal and tumor epithelial tissues (Du and Shim, 2016). In ionizing radiation, ROS activate a variety of EMT transcription factors, including ZEB1, Snail, STAT3, and HIF-1, thus promoting the metastasis and invasion of tumor cells (Lee et al., 2017). In breast cancer cells, poly ADP-ribose polymerase 3 (PARP3) plays an important role in inducing EMT and CSC phenotypes by regulating the TG2-snail-E-cadherin axis in response to ROS (Karicheva et al., 2016). It has been found that nicotine-induced elevated ROS levels alter EMT by regulating the AKT pathway in human renal epithelial cells to a certain extent and form stem cell-like spheroids (Chang and Singh, 2019). Paradoxically, after tumor stem cells are formed, intracellular R OS levels are low, which helps maintain the characteristics of CSCs (Qian et al., 2018). In addition, the stemness of CSCs was successfully reduced by increasing intracellular reactive oxygen species, and the efficiency of cisplatin therapy was improved (Chang et al., 2014). EMT-affected cells move from the primary tumor site to the surrounding stromal environment. The next is endovascular osmosis, which involves cancer cells entering the circulation and then being transported to distant organs. This process is closely linked to circulating tumor cells (CTCs) (Shibue and Weinberg, 2017). CTCs refer to tumor cells that are shed from solid tumor lesions and enter the bloodstream during tumor formation and development, reflecting tumor development and metastasis (Paoletti and Hayes, 2016). In blood circulation, the environmental factor that CTCs need to deal with is fluid shear stress (FSS), which can cause ROS generation, thus motivating CTCs to increase proliferation and behave more stem-like (Brown et al., 2021). The ERK pathway activates FSS-induced breast cancer cell survival, migration, and stemness alteration (Ma et al., 2017). In addition, ROS generated by PMN-MDSCs can promote the survival of CTCs by inducing the activation of the Notch signaling pathway (Sprouse et al., 2019). Different levels of ROS have different effects on the generation and maintenance of tumor stemness and the process of metastasis, which requires further studies to regulate ROS concentration to overcome drug-resistant tumors accurately (Figure 3).
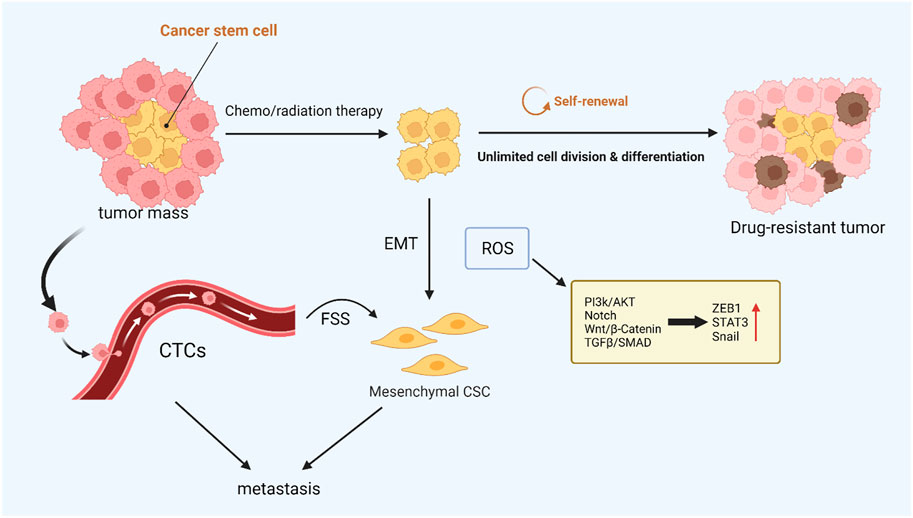
FIGURE 3. ROS-mediated cancer cell stemness. CSCs with low intracellular ROS levels are resistant to chemo- or radiotherapy, and CTCs are closely related to tumor metastasis. These resistant CSCs may contribute to local recurrence or metastasis via EMT. ROS can drive both EMT and stem cell phenotypes, and the activated signaling pathways are similar.
6 Tumor microenvironment and redox homeostasis
The tumor microenvironment comprises tumor cells, stromal cells, immune cells, secretory products (such as cytokines and chemokines) of corresponding cells, and non-cellular components in the extracellular matrix (ECM), which are closely related to tumor growth and metastasis. Hypoxia is one of the most prominent features of the tumor microenvironment and is associated with many tumor characteristics, including promoting cell proliferation, angiogenesis, metabolic reprogramming, and resistance to radiotherapy and chemotherapy (Jing et al., 2019; Bao and Wong, 2021). Reduced oxygen availability reduces the flow of electrons through the mitochondrial complex, allowing electrons to leak out of the transport chain and leading to the overproduction of ROS (Guzy et al., 2005). Angiogenesis in the tumor microenvironment is essential for cancer cell expansion and metastasis. Some evidence suggests that reactive oxygen species enhance angiogenesis. Under hypoxia, tumor cells express various angiogenic factors, such as vascular endothelial growth factor (VEGF), by activating HIF proteins (Xia et al., 2007). It has been found that inhibiting the expression of angiogenic markers and other oncogenic factors by reducing ROS can effectively suppress angiogenesis and dramatically inhibit tumor growth in HNSCC cells (Zheng et al., 2014). ROS can also induce tumor cells to secrete matrix metalloproteinases such as MMP-1, which promote vascular structure formation in the tumor microenvironment (Wartenberg et al., 2003).
Normal levels of ROS are important to T-cell activation and subsequent effects. For example, ROS regulate T cell activity by controlling IL-2 and IL-4 expression (Kaminski et al., 2010). Reducing T-cell-intrinsic ROS limits SENP7 cytosolic translocation and inhibits CD8+ T-cell metabolism and functional activity in human colorectal cancer cells (Wu et al., 2022). However, intolerable levels of ROS induced by mitochondrial dysfunction promote T-cell depletion, and lowering tumor hypoxia can limit T-cell exhaustion (Scharping et al., 2021). The redox status of T cells can also influence receptors on the surface of T cell membranes and thus affect signal transduction and distort immune responses (Kesarwani et al., 2013). Furthermore, immunosuppressive cells such as MDSCs increase ROS production in the tumor microenvironment, negatively affecting immune responses by stopping identification between the receptor TCR and MHC (Wei et al., 2015). T cells tend to become dysfunctional due to the immunosuppressive microenvironment. Hence, the treatment for regulating ROS or targeting its signaling may improve cancer immunotherapy. As a representative of stromal cells in the tumor microenvironment, cancer-associated fibroblasts (CAFs) are critical for increasing ROS levels in tumors (Chan et al., 2017).
On the other hand, ROS can reprogram the metabolic capacity of surrounding normal fibroblasts, turning them into the metabolic CAF phenotype in the TME (Zhang Z. et al., 2020). Oxidative ATM-mediated glycolysis enhancement in CAFs takes part in the metastasis of breast cancer cells (Sun et al., 2019). Another important feature of the tumor microenvironment is inflammation, which is closely related to the development of kinds of cancers, including gastric and nasopharyngeal cancer, and the efficacy of anticancer treatments (Zhao et al., 2021). Excessive ROS can promote the expression of proinflammatory cytokines by activating the transcription factors NF-KB and AP-1, which are sensitive to the redox state and may take part in activating the NLRP3 inflammasome (Hussain et al., 2016; Sho and Xu, 2019). Studies have shown that aloin alleviates LPS-induced inflammatory responses by inhibiting the ROS-mediated activation of the JAK1-STAT1/3 signaling pathway (Ma et al., 2018). In addition, ROS also form a regenerative feedback loop through autocrine TNF-α-mediated inflammatory cytokine/chemokine expression, which contributes to tumor progression (Blaser et al., 2016). In general, oxidative stress interacts with inflammation in the tumor microenvironment, and the specific mechanism still needs further investigation (Figure 4).
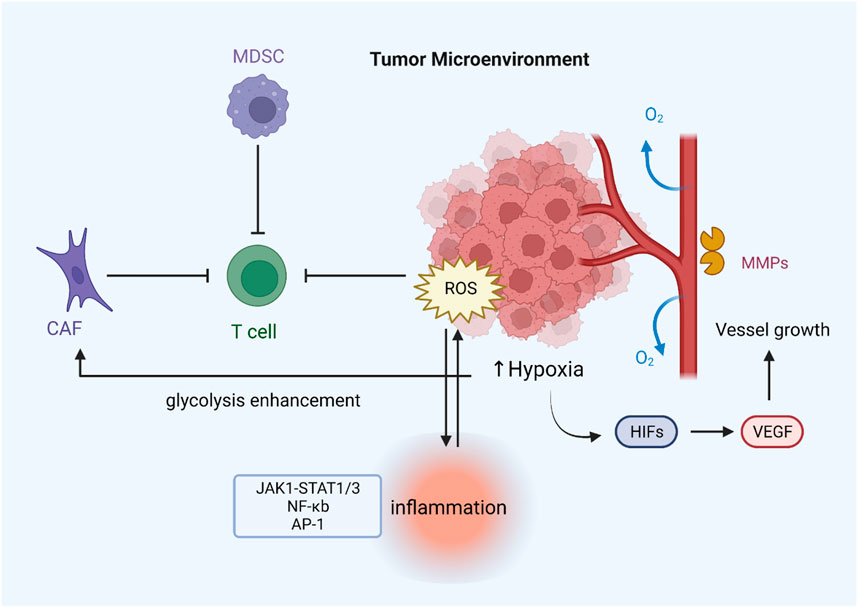
FIGURE 4. ROS regulate drug resistance by modulating the tumor microenvironment. Hypoxia can lead to ROS overproduction, which induces the expression of various angiogenic factors (VEGF) through the activation of HIF proteins, thereby promoting tumor angiogenesis. Increased release of MMP-1 contributes to the formation of vascular structures. MDSCs and CAFs can inhibit T-cell function by generating excessive ROS, thus leading to immune evasion of tumors. Increased ROS levels can also activate inflammation-related pathways and stimulate inflammasome formation.
7 Clinical application of redox regulators
The increase of ROS in tumor cells is of great significance for drug therapy (Forman and Zhang, 2021; Ming et al., 2022). Moderate levels of ROS can promote tumor signal transduction, cell proliferation, gene susceptibility to mutation, and other factors contributing to cancer progression. However, raising intracellular ROS levels to toxicity thresholds can overcome cellular antioxidant defenses and kill cancer cells, thus providing opportunities for the development of drug-resistant cancer therapies. Conventional chemotherapy selectively kills cancer cells by inducing excessive oxidative stress (Table 1). Anthracycline drugs such as doxorubicin, daunorubicin, and adriamycin have been reported to have anticancer activity in solid and hematological cancers because they block DNA synthesis and induce ROS production (Young et al., 1981). In addition, studies have shown that combining chemotherapy with some natural products can boost anticancer effects (Table 2). For example, the synergistic impact of anethole with adriamycin can promote ROS-mediated apoptosis in triple-negative breast cancer cells (Arumugam et al., 2021). The combination of doxorubicin and oxymatrine exerted superior synergistic effects on colorectal cancer cells in vitro and in vivo than either DOX or OMT alone (Pan et al., 2021). Platinum-based drugs, such as cisplatin and carboplatin, are known to maintain very high ROS levels, inducing DNA damage and cancer cell death by promoting oxidative stress, especially the overproduction of mitochondrial ROS and the reduction of intracellular antioxidants such as glutathione in some human malignancies (Mikuła-Pietrasik et al., 2019). However, oxidative stress caused by cisplatin can cause common side effects in normal cells, such as ototoxicity, which is caused by the excess production of ROS in cochlear cells (Sheth et al., 2017). Moreover, paclitaxel, which inhibits cell mitosis in tumor therapy, has been found to promote ROS generation by enhancing the activity of NADPH oxidase (NOX) (Alexandre et al., 2007). ROS-based treatments vary widely depending on tumor type, location, and stage of cancer development. Therapies that are effective for one type of tumor may not be effective for another type of tumor, and inappropriate application of ROS modulation therapy may not only be ineffective, but may even promote malignant progression. Only more refined regulation is expected to overcome the controversial side effects of pro-oxidative antitumor therapy (Wang et al., 2021).
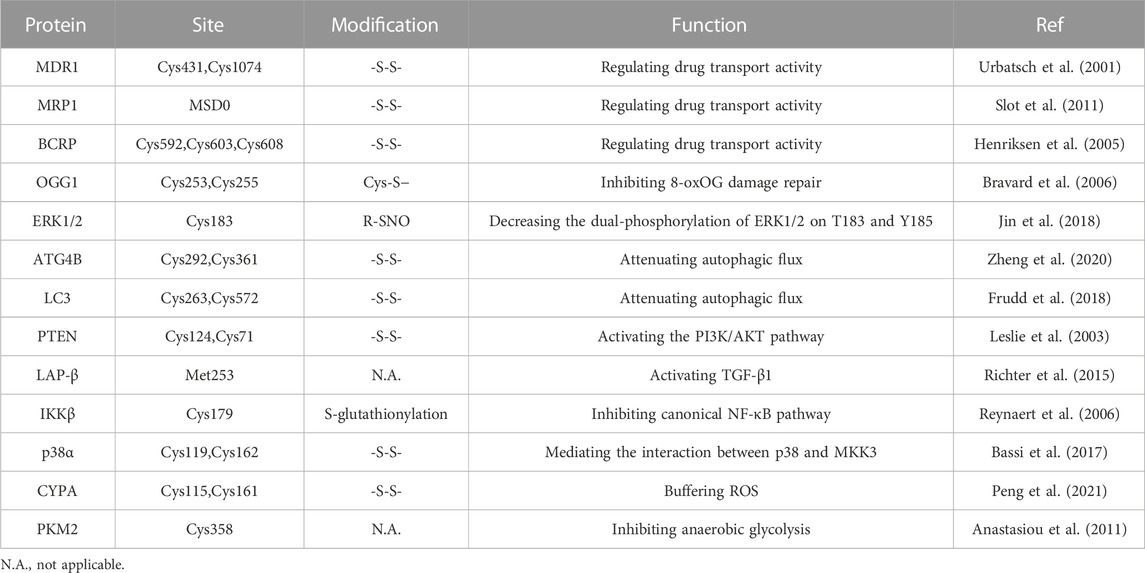
TABLE 1. Summary of redox sensors correlated with drug resistance.
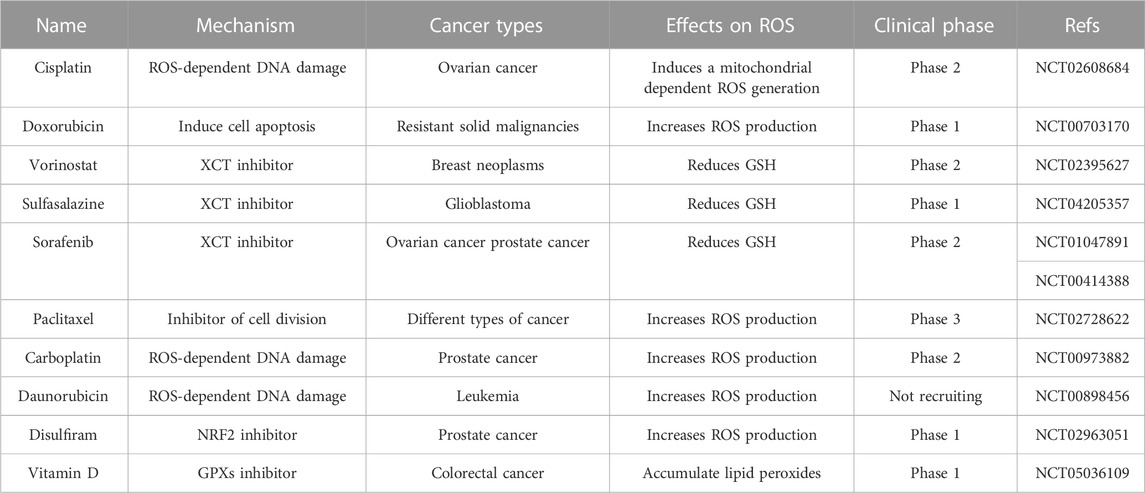
TABLE 2. Summary of redox-regulated drugs under clinical evaluation.
Ferroptosis is a regulatory manner of non-apoptotic cell death promoted by increased lipid ROS, which is closely related to cysteine metabolism and oxidative stress (Dixon et al., 2012). In detail, inhibition of the xCT system on the cell membrane reduces intracellular glutathione synthesis precursors, thereby indirectly inhibiting glutathione peroxidase 4 (GPX4), ultimately leading to the accumulation of fatal lipid peroxides and ferroptosis (Imai et al., 2017). The lethal metabolic imbalance caused by glutathione depletion or inactivation of GPX4 is characteristic of ferroptosis (Friedmann Angeli et al., 2014). There is increasing evidence that ROS-induced ferroptosis helps to inhibit tumor growth and increase sensitivity to chemotherapy. NRF2 activation contributes to the resistance of head and neck cancer (HNC) cells to sorafenib, while inhibition of the NRF2-ARE pathway can induce ferroptosis and reverse chemotherapeutic drug resistance in HNC cells (Roh et al., 2017). The researchers found that cisplatin-resistant ovarian cancer cells pretreated with erastin (a ferroptosis inducer) showed increased sensitivity to cisplatin, demonstrating the good synergistic effect of the two drugs (Sato et al., 2018). In addition, inducing ferroptosis by erastin was found to reverse cisplatin resistance in HNC cells and significantly enhance the anticancer activity of the first-line chemotherapeutic agents cytarabine and doxorubicin in HL60 cells (Yu et al., 2015; Roh et al., 2016). Therefore, ferroptosis may play a key role in tumorigenesis, and drugs or molecules that induce ferroptosis can be used as adjuvant chemotherapy to treat drug-resistant tumors in clinical practice. In conclusion, a more profound perception of the role of oxidative stress and ferroptosis in tumors will create new opportunities for diagnosis and therapeutic interventions in drug-resistant tumors.
8 Conclusion and perspectives
The main obstacle to the efficient treatment of human malignant tumors is the emergence of multidrug resistance during chemotherapy. In this review, we summarized the most recent studies on ROS and its effect on cancer drug resistance, including drug transport and metabolism, DNA damage repair, downstream adaptive responses, stemness maintenance, and tumor microenvironment (Table 3).
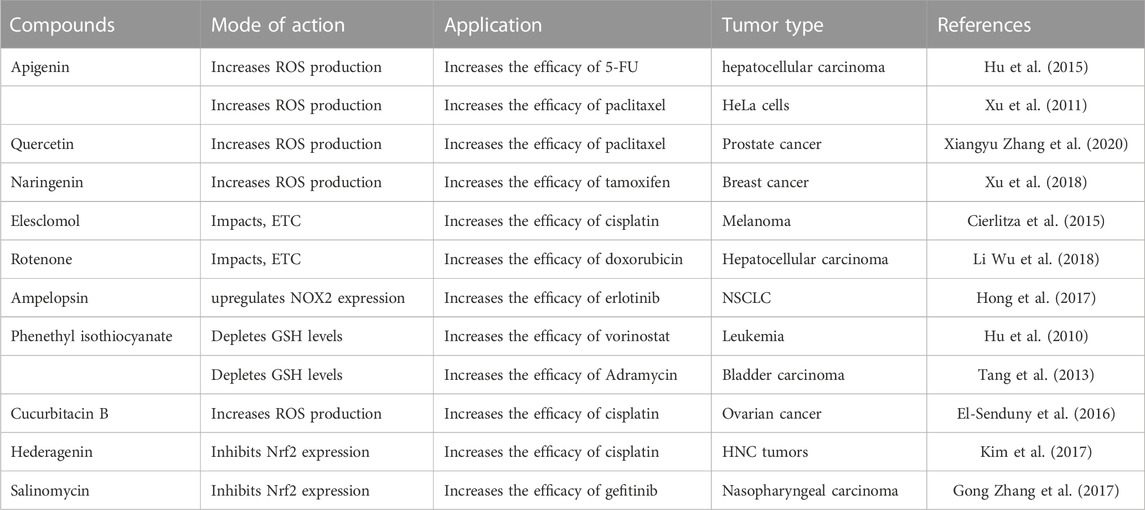
TABLE 3. Summary of compounds that enhance the efficacy of classical chemotherapeutic drugs.
Moderate oxidative stress may induce antioxidant genes overexpression and ABC transporter synthesis in cells. However, severe oxidative stress may disrupt the transporter system and aid in overcoming drug resistance. Redox modification alters the conformation of drug efflux transporters by affecting intermolecular disulfide bonds, lowering drug efflux, and enhancing therapeutic effectiveness potential. ROS-induced DNA damage can cause early tumor generation, yet its DDR-inhibiting effect may help overcome tumor drug resistance. Furthermore, moderate ROS activate pro-survival signaling pathways (PI3K/AKT, MAPK, and NF-κB) as second messengers. The mutual regulation of ROS and autophagy also promotes tumor survival. However, under oxidative stress, excessive ROS may accelerate tumor cell apoptosis. ROS can also influence tumor energy metabolism by regulating critical metabolic enzymes, skewing the tumor toward the pentose phosphate pathway to maintain its reducibility. The early EMT driving process requires a high concentration of ROS. Still, only a tiny amount of ROS is needed to maintain tumor stem cell features, indicating that more sophisticated redox regulation therapy is required.
In the tumor microenvironment, hypoxia-induced ROS can promote vascular reconstruction and immune escape. Currently, given the complexity of the tumor microenvironment, the specific mechanism of tumor drug resistance is still relatively unknown. Meanwhile, as an important feature of the tumor microenvironment, inflammation is closely related to oxidative stress, which can also promote tumor drug resistance. In general, a considerable body of evidence suggests that tumor drug resistance is associated with frequent variations in the redox state, implying that redox signaling can be used to explain the molecular mechanism of tumor drug resistance and provide therapeutic methods.
Author contributions
HZ designed this manuscript. YL and XZ drafted the manuscript. ZW and BL revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81601258) and the Chengdu Science and Technology Program (2021-YF05-01612-SN).
Acknowledgments
The authors acknowledge the BioRender (www.biorender.com), as figures in this review were created with the BioRender platform.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alexandre, J., Hu, Y., Lu, W., Pelicano, H., and Huang, P. (2007). Novel action of paclitaxel against cancer cells: Bystander effect mediated by reactive oxygen species. Cancer Res. 67 (8), 3512–3517. doi:10.1158/0008-5472.CAN-06-3914
Anastasiou, D., Poulogiannis, G., Asara, J. M., Boxer, M. B., Jiang, J. K., Shen, M., et al. (2011). Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Sci. (New York, NY) 334 (6060), 1278–1283. doi:10.1126/science.1211485
Arumugam, P., Sampathkumar, B., Perumalsamy, H., Balusamy, S. R., Ramesh, V., and Sundaravadevel, S. (2021). Synergistic effect of anethole and doxorubicin alleviates cell proliferation, cell cycle arrest, and ER stress and promotes ROS-mediated apoptosis in triple-negative breast cancer cells. J. Biochem. Mol. Toxicol. 35 (12), e22928. doi:10.1002/jbt.22928
Ashton, T. M., McKenna, W. G., Kunz-Schughart, L. A., and Higgins, G. S. (2018). Oxidative phosphorylation as an emerging target in cancer therapy. Clin. cancer Res. 24 (11), 2482–2490. doi:10.1158/1078-0432.CCR-17-3070
Baker, E. K., Johnstone, R. W., Zalcberg, J. R., and El-Osta, A. (2005). Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene 24 (54), 8061–8075. doi:10.1038/sj.onc.1208955
Bansal, A., and Simon, M. C. (2018). Glutathione metabolism in cancer progression and treatment resistance. J. Cell. Biol. 217 (7), 2291–2298. doi:10.1083/jcb.201804161
Bao, M. H., and Wong, C. C. (2021). Hypoxia, metabolic reprogramming, and drug resistance in liver cancer. Cells 10 (7), 1715. doi:10.3390/cells10071715
Barnes, R. P., Fouquerel, E., and Opresko, P. L. (2019). The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 177, 37–45. doi:10.1016/j.mad.2018.03.013
Bassi, R., Burgoyne, J. R., DeNicola, G. F., Rudyk, O., DeSantis, V., Charles, R. L., et al. (2017). Redox-dependent dimerization of p38α mitogen-activated protein kinase with mitogen-activated protein kinase kinase 3. J. Biol. Chem. 292 (39), 16161–16173. doi:10.1074/jbc.M117.785410
Basu, A. K., and Nohmi, T. (2018). Chemically-induced DNA damage, mutagenesis, and cancer. Int. J. Mol. Sci. 19 (4), 1767. doi:10.3390/ijms19061767
Bisht, S., and Dada, R. (2017). Oxidative stress: Major executioner in disease pathology, role in sperm DNA damage and preventive strategies. Front. Biosci. Sch. Ed. 9 (3), 420–447. doi:10.2741/s495
Blaser, H., Dostert, C., Mak, T. W., and Brenner, D. (2016). TNF and ROS crosstalk in inflammation. Trends Cell. Biol. 26 (4), 249–261. doi:10.1016/j.tcb.2015.12.002
Blobe, G. C., Sachs, C. W., Khan, W. A., Fabbro, D., Stabel, S., Wetsel, W. C., et al. (1993). Selective regulation of expression of protein kinase C (PKC) isoenzymes in multidrug-resistant MCF-7 cells. Functional significance of enhanced expression of PKC alpha. J. Biol. Chem. 268 (1), 658–664. doi:10.1016/s0021-9258(18)54202-2
Block, K., and Gorin, Y. (2012). Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 12 (9), 627–637. doi:10.1038/nrc3339
Brabletz, T., Kalluri, R., Nieto, M. A., and Weinberg, R. A. (2018). EMT in cancer. Nat. Rev. Cancer 18 (2), 128–134. doi:10.1038/nrc.2017.118
Bravard, A., Vacher, M., Gouget, B., Coutant, A., de Boisferon, F. H., Marsin, S., et al. (2006). Redox regulation of human OGG1 activity in response to cellular oxidative stress. Mol. Cell. Biol. 26 (20), 7430–7436. doi:10.1128/MCB.00624-06
Brennan, J. P., Bardswell, S. C., Burgoyne, J. R., Fuller, W., Schröder, E., Wait, R., et al. (2006). Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 281 (31), 21827–21836. doi:10.1074/jbc.M603952200
Brown, S. R., Bates, J. C., Avera, A. D., and Kim, Y. (2021). Relationship between stemness, reactive oxygen species, and epithelial-to-mesenchymal transition in model circulating tumor cells. Cells Tissues Organs 211, 282–293. doi:10.1159/000516574
Castro-Caldas, M., Carvalho, A. N., Rodrigues, E., Henderson, C., Wolf, C. R., and Gama, M. J. (2012). Glutathione S-transferase pi mediates MPTP-induced c-Jun N-terminal kinase activation in the nigrostriatal pathway. Mol. Neurobiol. 45 (3), 466–477. doi:10.1007/s12035-012-8266-9
Chan, J. S., Tan, M. J., Sng, M. K., Teo, Z., Phua, T., Choo, C. C., et al. (2017). Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell. death Dis. 8 (1), e2562. doi:10.1038/cddis.2016.492
Chang, C. W., Chen, Y. S., Chou, S. H., Han, C. L., Chen, Y. J., Yang, C. C., et al. (2014). Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 74 (21), 6291–6305. doi:10.1158/0008-5472.CAN-14-0626
Chang, J. C. (2016). Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Med. Baltim. 95, S20–S25. doi:10.1097/MD.0000000000004766
Chang, M. C., Lin, L. D., Wu, M. T., Chan, C. P., Chang, H. H., Lee, M. S., et al. (2015). Effects of camphorquinone on cytotoxicity, cell cycle regulation and prostaglandin E2 production of dental pulp cells: Role of ROS, ATM/Chk2, MEK/ERK and hemeoxygenase-1. PLoS One 10 (12), e0143663. doi:10.1371/journal.pone.0143663
Chang, Y. W., and Singh, K. P. (2019). Nicotine-induced oxidative stress contributes to EMT and stemness during neoplastic transformation through epigenetic modifications in human kidney epithelial cells. Toxicol. Appl. Pharmacol. 374, 65–76. doi:10.1016/j.taap.2019.04.023
Chatterjee, N., and Walker, G. C. (2017). Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen 58 (5), 235–263. doi:10.1002/em.22087
Chini, C. C. S., Zeidler, J. D., Kashyap, S., Warner, G., and Chini, E. N. (2021). Evolving concepts in NAD(+) metabolism. Cell. Metab. 33 (6), 1076–1087. doi:10.1016/j.cmet.2021.04.003
Christofk, H. R., Vander Heiden, M. G., Harris, M. H., Ramanathan, A., Gerszten, R. E., Wei, R., et al. (2008). The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452 (7184), 230–233. doi:10.1038/nature06734
Cierlitza, M., Chauvistré, H., Bogeski, I., Zhang, X., Hauschild, A., Herlyn, M., et al. (2015). Mitochondrial oxidative stress as a novel therapeutic target to overcome intrinsic drug resistance in melanoma cell subpopulations. Exp. Dermatol 24 (2), 155–157. doi:10.1111/exd.12613
Circu, M. L., and Aw, T. Y. (2010). Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 48 (6), 749–762. doi:10.1016/j.freeradbiomed.2009.12.022
Cui, Q., Wang, J. Q., Assaraf, Y. G., Ren, L., Gupta, P., Wei, L., et al. (2018). Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat 41, 1–25. doi:10.1016/j.drup.2018.11.001
Curtin, N. J. (2012). DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 12 (12), 801–817. doi:10.1038/nrc3399
Daher, B., Parks, S. K., Durivault, J., Cormerais, Y., Baidarjad, H., Tambutte, E., et al. (2019). Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. 79 (15), 3877–3890. doi:10.1158/0008-5472.CAN-18-3855
Dhuriya, Y. K., and Sharma, D. (2018). Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflammation 15 (1), 199. doi:10.1186/s12974-018-1235-0
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dondelinger, Y., Hulpiau, P., Saeys, Y., Bertrand, M. J. M., and Vandenabeele, P. (2016). An evolutionary perspective on the necroptotic pathway. Trends Cell. Biol. 26 (10), 721–732. doi:10.1016/j.tcb.2016.06.004
Drain, A. P., Zahir, N., Northey, J. J., Zhang, H., Huang, P. J., Maller, O., et al. (2021). Matrix compliance permits NF-κB activation to drive therapy resistance in breast cancer. J. Exp. Med. 218 (5), e20191360. doi:10.1084/jem.20191360
Du, B., and Shim, J. S. (2016). Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 21 (7), 965. doi:10.3390/molecules21070965
Dumont, S., Bykova, N. V., Pelletier, G., Dorion, S., and Rivoal, J. (2016). Cytosolic triosephosphate isomerase from Arabidopsis thaliana is reversibly modified by glutathione on cysteines 127 and 218. Front. plant Sci. 7, 1942. doi:10.3389/fpls.2016.01942
El-Senduny, F. F., Badria, F. A., El-Waseef, A. M., Chauhan, S. C., and Halaweish, F. (2016). Approach for chemosensitization of cisplatin-resistant ovarian cancer by cucurbitacin B. Tumour Biol. 37 (1), 685–698. doi:10.1007/s13277-015-3773-8
Fiorani, M., De Sanctis, R., Scarlatti, F., Vallorani, L., De Bellis, R., Serafini, G., et al. (2000). Dehydroascorbic acid irreversibly inhibits hexokinase activity. Mol. Cell. Biochem. 209 (1-2), 145–153. doi:10.1023/a:1007168032289
Fletcher, J. I., Haber, M., Henderson, M. J., and Norris, M. D. (2010). ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 10 (2), 147–156. doi:10.1038/nrc2789
Forman, H. J. (2016). Redox signaling: An evolution from free radicals to aging. Free Radic. Biol. Med. 97, 398–407. doi:10.1016/j.freeradbiomed.2016.07.003
Forman, H. J., and Zhang, H. (2021). Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20, 689–709. doi:10.1038/s41573-021-00233-1
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell. Biol. 16 (12), 1180–1191. doi:10.1038/ncb3064
Frudd, K., Burgoyne, T., and Burgoyne, J. R. (2018). Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 9 (1), 95. doi:10.1038/s41467-017-02352-z
Gañán-Gómez, I., Wei, Y., Yang, H., Boyano-Adánez, M. C., and García-Manero, G. (2013). Oncogenic functions of the transcription factor Nrf2. Free Radic. Biol. Med. 65, 750–764. doi:10.1016/j.freeradbiomed.2013.06.041
Gibson, S. B. (2013). Investigating the role of reactive oxygen species in regulating autophagy. Methods Enzymol. 528, 217–235. doi:10.1016/B978-0-12-405881-1.00013-6
Gomes, A. R., Zhao, F., and Lam, E. W. (2013). Role and regulation of the forkhead transcription factors FOXO3a and FOXM1 in carcinogenesis and drug resistance. Chin. J. Cancer 32 (7), 365–370. doi:10.5732/cjc.012.10277
Gruosso, T., Mieulet, V., Cardon, M., Bourachot, B., Kieffer, Y., Devun, F., et al. (2016). Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol. Med. 8 (5), 527–549. doi:10.15252/emmm.201505891
Guzy, R. D., Hoyos, B., Robin, E., Chen, H., Liu, L., Mansfield, K. D., et al. (2005). Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell. metab. 1 (6), 401–408. doi:10.1016/j.cmet.2005.05.001
Ha, J. H., Noh, H. S., Shin, I. W., Hahm, J. R., and Kim, D. R. (2012). Mitigation of H2O2-induced autophagic cell death by propofol in H9c2 cardiomyocytes. Cell. Biol. Toxicol. 28 (1), 19–29. doi:10.1007/s10565-011-9202-x
Han, C., Wang, Z., Chen, S., Li, L., Xu, Y., Kang, W., et al. (2021). Berbamine suppresses the progression of bladder cancer by modulating the ROS/NF-κB Axis. Oxid. Med. Cell. Longev. 2021, 8851763. doi:10.1155/2021/8851763
Harris, I. S., and DeNicola, G. M. (2020). The complex interplay between antioxidants and ROS in cancer. Trends Cell. Biol. 30 (6), 440–451. doi:10.1016/j.tcb.2020.03.002
Hayes, J. D., Dinkova-Kostova, A. T., and Tew, K. D. (2020). Oxidative stress in cancer. Cancer Cell. 38 (2), 167–197. doi:10.1016/j.ccell.2020.06.001
Hayes, J. D., and McMahon, M. (2006). The double-edged sword of Nrf2: Subversion of redox homeostasis during the evolution of cancer. Mol. Cell. 21 (6), 732–734. doi:10.1016/j.molcel.2006.03.004
Heneberg, P. (2019). Redox regulation of hexokinases. Antioxidants redox Signal. 30 (3), 415–442. doi:10.1089/ars.2017.7255
Henriksen, U., Ju, F., Litman, T., and Gether, U. (2005). Identification of intra- and intermolecular disulfide bridges in the multidrug resistance transporter ABCG2. J. Biol. Chem. 280 (44), 36926–36934. doi:10.1074/jbc.M502937200
Higgins, C. F. (2007). Multiple molecular mechanisms for multidrug resistance transporters. Nature 446 (7137), 749–757. doi:10.1038/nature05630
Hohl, M., Briand, C., Grütter, M. G., and Seeger, M. A. (2012). Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 19 (4), 395–402. doi:10.1038/nsmb.2267
Holohan, C., Van Schaeybroeck, S., Longley, D. B., and Johnston, P. G. (2013). Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 13 (10), 714–726. doi:10.1038/nrc3599
Hong, S. W., Park, N. S., Noh, M. H., Shim, J. A., Ahn, B. N., Kim, Y. S., et al. (2017). Combination treatment with erlotinib and ampelopsin overcomes erlotinib resistance in NSCLC cells via the Nox2-ROS-Bim pathway. Lung Cancer 106, 115–124. doi:10.1016/j.lungcan.2017.02.009
Hsu, S. K., Chang, W. T., Lin, I. L., Chen, Y. F., Padalwar, N. B., Cheng, K. C., et al. (2020). The role of necroptosis in ROS-mediated cancer therapies and its promising applications. Cancers (Basel) 12 (8), 2185. doi:10.3390/cancers12082185
Hu, X. Y., Liang, J. Y., Guo, X. J., Liu, L., and Guo, Y. B. (2015). 5-Fluorouracil combined with apigenin enhances anticancer activity through mitochondrial membrane potential (ΔΨm)-mediated apoptosis in hepatocellular carcinoma. Clin. Exp. Pharmacol. Physiol. 42 (2), 146–153. doi:10.1111/1440-1681.12333
Hu, Y., Lu, W., Chen, G., Zhang, H., Jia, Y., Wei, Y., et al. (2010). Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood 116 (15), 2732–2741. doi:10.1182/blood-2009-11-256354
Huang, Y., Li, W., Su, Z. Y., and Kong, A. N. (2015). The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 26 (12), 1401–1413. doi:10.1016/j.jnutbio.2015.08.001
Hussain, T., Tan, B., Yin, Y., Blachier, F., Tossou, M. C., and Rahu, N. (2016). Oxidative stress and inflammation: What polyphenols can do for us? Oxid. Med. Cell. Longev. 2016, 7432797. doi:10.1155/2016/7432797
Imai, H., Matsuoka, M., Kumagai, T., Sakamoto, T., and Koumura, T. (2017). Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr. Top. Microbiol. Immunol. 403, 143–170. doi:10.1007/82_2016_508
Ishimoto, T., Nagano, O., Yae, T., Tamada, M., Motohara, T., Oshima, H., et al. (2011). CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 19 (3), 387–400. doi:10.1016/j.ccr.2011.01.038
Itoh, K., Chiba, T., Takahashi, S., Ishii, T., Igarashi, K., Katoh, Y., et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. biophysical Res. Commun. 236 (2), 313–322. doi:10.1006/bbrc.1997.6943
Jackson, A. L., and Loeb, L. A. (2001). The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. 477 (1-2), 7–21. doi:10.1016/s0027-5107(01)00091-4
Jakubczyk, K., Dec, K., Kałduńska, J., Kawczuga, D., Kochman, J., and Janda, K. (2020). Reactive oxygen species - sources, functions, oxidative damage. Pol. Merkur Lek. 48 (284), 124–127.
Jin, L., Cao, Y., Zhang, T., Wang, P., Ji, D., Liu, X., et al. (2018). Effects of ERK1/2 S-nitrosylation on ERK1/2 phosphorylation and cell survival in glioma cells. Int. J. Mol. Med. 41 (3), 1339–1348. doi:10.3892/ijmm.2017.3334
Jing, X., Yang, F., Shao, C., Wei, K., Xie, M., Shen, H., et al. (2019). Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 18 (1), 157. doi:10.1186/s12943-019-1089-9
Johar, R., Sharma, R., Kaur, A., and Mukherjee, T. K. (2015). Role of reactive oxygen species in estrogen dependant breast cancer complication. Anticancer Agents Med. Chem. 16 (2), 190–199. doi:10.2174/1871520615666150518092315
Joyce, H., McCann, A., Clynes, M., and Larkin, A. (2015). Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin. Drug Metab. Toxicol. 11 (5), 795–809. doi:10.1517/17425255.2015.1028356
Kage, K., Fujita, T., and Sugimoto, Y. (2005). Role of Cys-603 in dimer/oligomer formation of the breast cancer resistance protein BCRP/ABCG2. Cancer Sci. 96 (12), 866–872. doi:10.1111/j.1349-7006.2005.00126.x
Kaminski, M. M., Sauer, S. W., Klemke, C. D., Süss, D., Okun, J. G., Krammer, P. H., et al. (2010). Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: Mechanism of ciprofloxacin-mediated immunosuppression. J. Immunol. 184 (9), 4827–4841. doi:10.4049/jimmunol.0901662
Kang, Q., and Yang, C. (2020). Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 37, 101799. doi:10.1016/j.redox.2020.101799
Karicheva, O., Rodriguez-Vargas, J. M., Wadier, N., Martin-Hernandez, K., Vauchelles, R., Magroun, N., et al. (2016). PARP3 controls TGFβ and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget 7 (39), 64109–64123. doi:10.18632/oncotarget.11627
Kesarwani, P., Murali, A. K., Al-Khami, A. A., and Mehrotra, S. (2013). Redox regulation of T-cell function: From molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal 18 (12), 1497–1534. doi:10.1089/ars.2011.4073
Kim, E. H., Baek, S., Shin, D., Lee, J., and Roh, J. L. (2017). Hederagenin induces apoptosis in cisplatin-resistant head and neck cancer cells by inhibiting the nrf2-ARE antioxidant pathway. Oxid. Med. Cell. Longev. 2017, 5498908. doi:10.1155/2017/5498908
Kim, J. H., Choi, T. G., Park, S., Yun, H. R., Nguyen, N. N. Y., Jo, Y. H., et al. (2018). Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell. Death Differ. 25 (11), 1921–1937. doi:10.1038/s41418-018-0165-9
Klionsky, D. J., Petroni, G., Amaravadi, R. K., Baehrecke, E. H., Ballabio, A., Boya, P., et al. (2021). Autophagy in major human diseases. EMBO J. 40 (19), e108863. doi:10.15252/embj.2021108863
Krause, M. S., Oliveira, L. P., Silveira, E. M., Vianna, D. R., Rossato, J. S., Almeida, B. S., et al. (2007). MRP1/GS-X pump ATPase expression: Is this the explanation for the cytoprotection of the heart against oxidative stress-induced redox imbalance in comparison to skeletal muscle cells? Cell. Biochem. Funct. 25 (1), 23–32. doi:10.1002/cbf.1343
Kuehne, A., Emmert, H., Soehle, J., Winnefeld, M., Fischer, F., Wenck, H., et al. (2015). Acute activation of oxidative pentose phosphate pathway as first-line response to oxidative stress in human skin cells. Mol. Cell. 59 (3), 359–371. doi:10.1016/j.molcel.2015.06.017
Latimer, H. R., and Veal, E. A. (2016). Peroxiredoxins in regulation of MAPK signalling pathways; sensors and barriers to signal transduction. Mol. Cells 39 (1), 40–45. doi:10.14348/molcells.2016.2327
Lau, A., Villeneuve, N. F., Sun, Z., Wong, P. K., and Zhang, D. D. (2008). Dual roles of Nrf2 in cancer. Pharmacol. Res. 58 (5-6), 262–270. doi:10.1016/j.phrs.2008.09.003
Ledoux, S., Yang, R., Friedlander, G., and Laouari, D. (2003). Glucose depletion enhances P-glycoprotein expression in hepatoma cells: Role of endoplasmic reticulum stress response. Cancer Res. 63 (21), 7284–7290.
Lee, S. Y., Jeong, E. K., Ju, M. K., Jeon, H. M., Kim, M. Y., Kim, C. H., et al. (2017). Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 16 (1), 10. doi:10.1186/s12943-016-0577-4
Lennicke, C., and Cochemé, H. M. (2021). Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell. 81 (18), 3691–3707. doi:10.1016/j.molcel.2021.08.018
León-Buitimea, A., Rodríguez-Fragoso, L., Lauer, F. T., Bowles, H., Thompson, T. A., and Burchiel, S. W. (2012). Ethanol-induced oxidative stress is associated with EGF receptor phosphorylation in MCF-10A cells overexpressing CYP2E1. Toxicol. Lett. 209 (2), 161–165. doi:10.1016/j.toxlet.2011.12.009
Lertratanangkoon, K., Wu, C. J., Savaraj, N., and Thomas, M. L. (1997). Alterations of DNA methylation by glutathione depletion. Cancer Lett. 120 (2), 149–156. doi:10.1016/s0304-3835(97)00300-5
Leslie, N. R., Bennett, D., Lindsay, Y. E., Stewart, H., Gray, A., and Downes, C. P. (2003). Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 22 (20), 5501–5510. doi:10.1093/emboj/cdg513
Li, B., Jiang, J., Assaraf, Y. G., Xiao, H., Chen, Z. S., and Huang, C. (2020). Surmounting cancer drug resistance: New insights from the perspective of N(6)-methyladenosine RNA modification. Drug Resist Updat 53, 100720. doi:10.1016/j.drup.2020.100720
Li, D., Ueta, E., Kimura, T., Yamamoto, T., and Osaki, T. (2004). Reactive oxygen species (ROS) control the expression of Bcl-2 family proteins by regulating their phosphorylation and ubiquitination. Cancer Sci. 95 (8), 644–650. doi:10.1111/j.1349-7006.2004.tb03323.x
Li, W., Zhang, H., Assaraf, Y. G., Zhao, K., Xu, X., Xie, J., et al. (2016). Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat 27, 14–29. doi:10.1016/j.drup.2016.05.001
Liberti, M. V., and Locasale, J. W. (2016). The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 41 (3), 211–218. doi:10.1016/j.tibs.2015.12.001
Lien, E. C., Lyssiotis, C. A., Juvekar, A., Hu, H., Asara, J. M., Cantley, L. C., et al. (2016). Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell. Biol. 18 (5), 572–578. doi:10.1038/ncb3341
Liu, N., Wang, K. S., Qi, M., Zhou, Y. J., Zeng, G. Y., Tao, J., et al. (2018). Vitexin compound 1, a novel extraction from a Chinese herb, suppresses melanoma cell growth through DNA damage by increasing ROS levels. J. Exp. Clin. Cancer Res. 37 (1), 269. doi:10.1186/s13046-018-0897-x
Liu, Y., Li, Q., Zhou, L., Xie, N., Nice, E. C., Zhang, H., et al. (2016). Cancer drug resistance: Redox resetting renders a way. Oncotarget 7 (27), 42740–42761. doi:10.18632/oncotarget.8600
Lord, C. J., and Ashworth, A. (2012). The DNA damage response and cancer therapy. Nature 481 (7381), 287–294. doi:10.1038/nature10760
Lu, M. C., Ji, J. A., Jiang, Z. Y., and You, Q. D. (2016). The keap1-nrf2-ARE pathway as a potential preventive and therapeutic target: An update. Med. Res. Rev. 36 (5), 924–963. doi:10.1002/med.21396
Lu, Q., Wang, W. W., Zhang, M. Z., Ma, Z. X., Qiu, X. R., Shen, M., et al. (2019). ROS induces epithelial-mesenchymal transition via the TGF-β1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Exp. Ther. Med. 17 (1), 835–846. doi:10.3892/etm.2018.7014
Luanpitpong, S., Chanvorachote, P., Nimmannit, U., Leonard, S. S., Stehlik, C., Wang, L., et al. (2012). Mitochondrial superoxide mediates doxorubicin-induced keratinocyte apoptosis through oxidative modification of ERK and Bcl-2 ubiquitination. Biochem. Pharmacol. 83 (12), 1643–1654. doi:10.1016/j.bcp.2012.03.010
Ma, S., Fu, A., Chiew, G. G., and Luo, K. Q. (2017). Hemodynamic shear stress stimulates migration and extravasation of tumor cells by elevating cellular oxidative level. Cancer Lett. 388, 239–248. doi:10.1016/j.canlet.2016.12.001
Ma, Y., Tang, T., Sheng, L., Wang, Z., Tao, H., Zhang, Q., et al. (2018). Aloin suppresses lipopolysaccharide-induced inflammation by inhibiting JAK1-STAT1/3 activation and ROS production in RAW264.7 cells. Int. J. Mol. Med. 42 (4), 1925–1934. doi:10.3892/ijmm.2018.3796
Maiti, A. K. (2012). Genetic determinants of oxidative stress-mediated sensitization of drug-resistant cancer cells. Int. J. Cancer 130 (1), 1–9. doi:10.1002/ijc.26306
Mandal, A., Agrahari, V., Khurana, V., Pal, D., and Mitra, A. K. (2017). Transporter effects on cell permeability in drug delivery. Expert Opin. Drug Deliv. 14 (3), 385–401. doi:10.1080/17425247.2016.1214565
Maschek, G., Savaraj, N., Priebe, W., Braunschweiger, P., Hamilton, K., Tidmarsh, G. F., et al. (2004). 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 64 (1), 31–34. doi:10.1158/0008-5472.can-03-3294
McMahon, M., Itoh, K., Yamamoto, M., and Hayes, J. D. (2003). Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 278 (24), 21592–21600. doi:10.1074/jbc.M300931200
Mikubo, M., Inoue, Y., Liu, G., and Tsao, M. S. (2021). Mechanism of drug tolerant persister cancer cells: The landscape and clinical implication for therapy. J. Thorac. Oncol. 16 (11), 1798–1809. doi:10.1016/j.jtho.2021.07.017
Mikuła-Pietrasik, J., Witucka, A., Pakuła, M., Uruski, P., Begier-Krasińska, B., Niklas, A., et al. (2019). Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. life Sci. CMLS 76 (4), 681–697. doi:10.1007/s00018-018-2954-1
Miller, K. D., Nogueira, L., Mariotto, A. B., Rowland, J. H., Yabroff, K. R., Alfano, C. M., et al. (2019). Cancer treatment and survivorship statistics. CA Cancer J. Clin. 69 (5), 363–385. doi:10.3322/caac.21565
Ming, H., Li, B., Jiang, J., Qin, S., Nice, E. C., He, W., et al. (2023). Protein degradation: Expanding the toolbox to restrain cancer drug resistance. J. Hematol. Oncol. 16 (1), 6. doi:10.1186/s13045-023-01398-5
Ming, H., Li, B., Tian, H., Zhou, L., Jiang, J., Zhang, T., et al. (2022). A minimalist and robust chemo-photothermal nanoplatform capable of augmenting autophagy-modulated immune response against breast cancer. Mater Today Bio 15, 100289. doi:10.1016/j.mtbio.2022.100289
Ming, H., Li, B., Zhou, L., Goel, A., and Huang, C. (2021). Long non-coding RNAs and cancer metastasis: Molecular basis and therapeutic implications. Biochim. Biophys. Acta Rev. Cancer 1875 (2), 188519. doi:10.1016/j.bbcan.2021.188519
Mohammad, R. M., Muqbil, I., Lowe, L., Yedjou, C., Hsu, H. Y., Lin, L. T., et al. (2015). Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 35, S78–s103. doi:10.1016/j.semcancer.2015.03.001
Myatt, S. S., and Lam, E. W. (2007). The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 7 (11), 847–859. doi:10.1038/nrc2223
Nakajima, S., and Kitamura, M. (2013). Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 65, 162–174. doi:10.1016/j.freeradbiomed.2013.06.020
Noorolyai, S., Shajari, N., Baghbani, E., Sadreddini, S., and Baradaran, B. (2019). The relation between PI3K/AKT signalling pathway and cancer. Gene 698, 120–128. doi:10.1016/j.gene.2019.02.076
Orozco, S., and Oberst, A. (2017). RIPK3 in cell death and inflammation: The good, the bad, and the ugly. Immunol. Rev. 277 (1), 102–112. doi:10.1111/imr.12536
Pan, D., Zhang, W., Zhang, N., Xu, Y., Chen, Y., Peng, J., et al. (2021). Oxymatrine synergistically enhances doxorubicin anticancer effects in colorectal cancer. Front. Pharmacol. 12, 673432. doi:10.3389/fphar.2021.673432
Paoletti, C., and Hayes, D. F. (2016). Circulating tumor cells. Adv. Exp. Med. Biol. 882, 235–258. doi:10.1007/978-3-319-22909-6_10
Pekel, G., and Ari, F. (2020). Therapeutic targeting of cancer metabolism with triosephosphate isomerase. Chem. Biodivers. 17 (5), e2000012. doi:10.1002/cbdv.202000012
Peng, L., Jiang, J., Chen, H. N., Zhou, L., Huang, Z., Qin, S., et al. (2021). Redox-sensitive cyclophilin A elicits chemoresistance through realigning cellular oxidative status in colorectal cancer. Cell. Rep. 37 (9), 110069. doi:10.1016/j.celrep.2021.110069
Phi, L. T. H., Sari, I. N., Yang, Y. G., Lee, S. H., Jun, N., Kim, K. S., et al. (2018). Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 5416923. doi:10.1155/2018/5416923
Potter, M., Newport, E., and Morten, K. J. (2016). The Warburg effect: 80 years on. Biochem. Soc. Trans. 44 (5), 1499–1505. doi:10.1042/BST20160094
Qian, X., Nie, X., Yao, W., Klinghammer, K., Sudhoff, H., Kaufmann, A. M., et al. (2018). Reactive oxygen species in cancer stem cells of head and neck squamous cancer. Semin. Cancer Biol. 53, 248–257. doi:10.1016/j.semcancer.2018.06.001
Qin, S., Li, B., Ming, H., Nice, E. C., Zou, B., and Huang, C. (2022). Harnessing redox signaling to overcome therapeutic-resistant cancer dormancy. Biochim. Biophys. Acta Rev. Cancer 1877 (4), 188749. doi:10.1016/j.bbcan.2022.188749
Quijano, C., Trujillo, M., Castro, L., and Trostchansky, A. (2016). Interplay between oxidant species and energy metabolism. Redox Biol. 8, 28–42. doi:10.1016/j.redox.2015.11.010
Ralser, M., Wamelink, M. M., Kowald, A., Gerisch, B., Heeren, G., Struys, E. A., et al. (2007). Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 6 (4), 10. doi:10.1186/jbiol61
Rebecca, V. W., and Amaravadi, R. K. (2016). Emerging strategies to effectively target autophagy in cancer. Oncogene 35 (1), 1–11. doi:10.1038/onc.2015.99
Reisz, J. A., Wither, M. J., Dzieciatkowska, M., Nemkov, T., Issaian, A., Yoshida, T., et al. (2016). Oxidative modifications of glyceraldehyde 3-phosphate dehydrogenase regulate metabolic reprogramming of stored red blood cells. Blood 128 (12), e32–e42. doi:10.1182/blood-2016-05-714816
Reynaert, N. L., van der Vliet, A., Guala, A. S., McGovern, T., Hristova, M., Pantano, C., et al. (2006). Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. U. S. A. 103 (35), 13086–13091. doi:10.1073/pnas.0603290103
Richter, K., Konzack, A., Pihlajaniemi, T., Heljasvaara, R., and Kietzmann, T. (2015). Redox-fibrosis: Impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox Biol. 6, 344–352. doi:10.1016/j.redox.2015.08.015
Roh, J. L., Kim, E. H., Jang, H., and Shin, D. (2017). Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 11, 254–262. doi:10.1016/j.redox.2016.12.010
Roh, J. L., Kim, E. H., Jang, H. J., Park, J. Y., and Shin, D. (2016). Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 381 (1), 96–103. doi:10.1016/j.canlet.2016.07.035
Rojo de la Vega, M., Chapman, E., and Zhang, D. D. (2018). NRF2 and the hallmarks of cancer. Cancer Cell. 34 (1), 21–43. doi:10.1016/j.ccell.2018.03.022
Salehi, F., Behboudi, H., Kavoosi, G., and Ardestani, S. K. (2018). Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 8 (1), 13902. doi:10.1038/s41598-018-32308-2
Sato, M., Kusumi, R., Hamashima, S., Kobayashi, S., Sasaki, S., Komiyama, Y., et al. (2018). The ferroptosis inducer erastin irreversibly inhibits system x(c)- and synergizes with cisplatin to increase cisplatin's cytotoxicity in cancer cells. Sci. Rep. 8 (1), 968. doi:10.1038/s41598-018-19213-4
Sattler, U. G., Hirschhaeuser, F., and Mueller-Klieser, W. F. (2010). Manipulation of glycolysis in malignant tumors: Fantasy or therapy? Curr. Med. Chem. 17 (2), 96–108. doi:10.2174/092986710790112657
Scharping, N. E., Rivadeneira, D. B., Menk, A. V., Vignali, P. D. A., Ford, B. R., Rittenhouse, N. L., et al. (2021). Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22 (2), 205–215. doi:10.1038/s41590-020-00834-9
Sharma, V., Anderson, D., and Dhawan, A. (2012). Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2). Apoptosis 17 (8), 852–870. doi:10.1007/s10495-012-0705-6
Sharom, F. J. (2008). ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 9 (1), 105–127. doi:10.2217/14622416.9.1.105
Shchulkin, A. V., Abalenikhina, Y. V., Erokhina, P. D., Chernykh, I. V., and Yakusheva, E. N. (2021). The role of P-glycoprotein in decreasing cell membranes permeability during oxidative stress. Biochem. Biokhimiia 86 (2), 197–206. doi:10.1134/S0006297921020085
Sheth, S., Mukherjea, D., Rybak, L. P., and Ramkumar, V. (2017). Mechanisms of cisplatin-induced ototoxicity and otoprotection. Front. Cell. Neurosci. 11, 338. doi:10.3389/fncel.2017.00338
Shi, H. S., Li, D., Zhang, J., Wang, Y. S., Yang, L., Zhang, H. L., et al. (2010). Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci. 101 (6), 1447–1453. doi:10.1111/j.1349-7006.2010.01562.x
Shi, Y., Nikulenkov, F., Zawacka-Pankau, J., Li, H., Gabdoulline, R., Xu, J., et al. (2014). ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell. Death Differ. 21 (4), 612–623. doi:10.1038/cdd.2013.186
Shibue, T., and Weinberg, R. A. (2017). EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 14 (10), 611–629. doi:10.1038/nrclinonc.2017.44
Sho, T., and Xu, J. (2019). Role and mechanism of ROS scavengers in alleviating NLRP3-mediated inflammation. Biotechnol. Appl. Biochem. 66 (1), 4–13. doi:10.1002/bab.1700
Shokolenko, I., Venediktova, N., Bochkareva, A., Wilson, G. L., and Alexeyev, M. F. (2009). Oxidative stress induces degradation of mitochondrial DNA. Nucleic acids Res. 37 (8), 2539–2548. doi:10.1093/nar/gkp100
Shu, C. W., Chang, H. T., Wu, C. S., Chen, C. H., Wu, S., Chang, H. W., et al. (2016). RelA-mediated BECN1 expression is required for reactive oxygen species-induced autophagy in oral cancer cells exposed to low-power laser irradiation. PLoS One 11 (9), e0160586. doi:10.1371/journal.pone.0160586
Siegel, R. L., Miller, K. D., Fuchs, H. E., and Jemal, A. (2022). Cancer statistics, 2022. CA Cancer J. Clin. 72 (1), 7–33. doi:10.3322/caac.21708
Slot, A. J., Molinski, S. V., and Cole, S. P. (2011). Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 50 (1), 179–207. doi:10.1042/bse0500179
Sporn, M. B., and Liby, K. T. (2012). NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 12 (8), 564–571. doi:10.1038/nrc3278
Sprouse, M. L., Welte, T., Boral, D., Liu, H. N., Yin, W., Vishnoi, M., et al. (2019). PMN-MDSCs enhance CTC metastatic properties through reciprocal interactions via ROS/Notch/Nodal signaling. Int. J. Mol. Sci. 20 (8), 1916. doi:10.3390/ijms20081916
Srinivas, U. S., Tan, B. W. Q., Vellayappan, B. A., and Jeyasekharan, A. D. (2019). ROS and the DNA damage response in cancer. Redox Biol. 25, 101084. doi:10.1016/j.redox.2018.101084
Sun, K., Tang, S., Hou, Y., Xi, L., Chen, Y., Yin, J., et al. (2019). Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine 41, 370–383. doi:10.1016/j.ebiom.2019.02.025
Sun, Z. L., Dong, J. L., and Wu, J. (2017). Juglanin induces apoptosis and autophagy in human breast cancer progression via ROS/JNK promotion. Biomed. Pharmacother. 85, 303–312. doi:10.1016/j.biopha.2016.11.030
Tang, K., Lin, Y., and Li, L. M. (2013). The role of phenethyl isothiocyanate on bladder cancer ADM resistance reversal and its molecular mechanism. Anat. Rec. Hob. 296 (6), 899–906. doi:10.1002/ar.22677
Terada, Y., Ogura, J., Tsujimoto, T., Kuwayama, K., Koizumi, T., Sasaki, S., et al. (2014). Intestinal P-glycoprotein expression is multimodally regulated by intestinal ischemia-reperfusion. J. Pharm. Pharm. Sci. 17 (2), 266–276.
Urbatsch, I. L., Gimi, K., Wilke-Mounts, S., Lerner-Marmarosh, N., Rousseau, M. E., Gros, P., et al. (2001). Cysteines 431 and 1074 are responsible for inhibitory disulfide cross-linking between the two nucleotide-binding sites in human P-glycoprotein. J. Biol. Chem. 276 (29), 26980–26987. doi:10.1074/jbc.M010829200
Vanlangenakker, N., Vanden Berghe, T., Bogaert, P., Laukens, B., Zobel, K., Deshayes, K., et al. (2011). cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell. Death Differ. 18 (4), 656–665. doi:10.1038/cdd.2010.138
Wakabayashi, K., Nakagawa, H., Adachi, T., Kii, I., Kobatake, E., Kudo, A., et al. (2006). Identification of cysteine residues critically involved in homodimer formation and protein expression of human ATP-binding cassette transporter ABCG2: A new approach using the flp recombinase system. J. Exp. Ther. Oncol. 5 (3), 205–222.
Wang, J., Yang, H., Li, W., Xu, H., Yang, X., and Gan, L. (2015). Thioredoxin 1 upregulates FOXO1 transcriptional activity in drug resistance in ovarian cancer cells. Biochim. Biophys. Acta 1852 (3), 395–405. doi:10.1016/j.bbadis.2014.12.002
Wang, K., Zhang, T., Lei, Y., Li, X., Jiang, J., Lan, J., et al. (2018). Identification of ANXA2 (annexin A2) as a specific bleomycin target to induce pulmonary fibrosis by impeding TFEB-mediated autophagic flux. Autophagy 14 (2), 269–282. doi:10.1080/15548627.2017.1409405
Wang, T., Jin, Y., Yang, W., Zhang, L., Jin, X., Liu, X., et al. (2017). Necroptosis in cancer: An angel or a demon? Tumour Biol. 39 (6), 1010428317711539. doi:10.1177/1010428317711539
Wang, X. J., Sun, Z., Villeneuve, N. F., Zhang, S., Zhao, F., Li, Y., et al. (2008). Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29 (6), 1235–1243. doi:10.1093/carcin/bgn095
Wang, Y., Qi, H., Liu, Y., Duan, C., Liu, X., Xia, T., et al. (2021). The double-edged roles of ROS in cancer prevention and therapy. Theranostics 11 (10), 4839–4857. doi:10.7150/thno.56747
Wangpaichitr, M., Theodoropoulos, G., Nguyen, D. J. M., Wu, C., Spector, S. A., Feun, L. G., et al. (2021). Cisplatin resistance and redox-metabolic vulnerability: A second alteration. Int. J. Mol. Sci. 22 (14), 7379. doi:10.3390/ijms22147379
Ward, R. A., Fawell, S., Floc'h, N., Flemington, V., McKerrecher, D., and Smith, P. D. (2021). Challenges and opportunities in cancer drug resistance. Chem. Rev. 121 (6), 3297–3351. doi:10.1021/acs.chemrev.0c00383
Wartenberg, M., Budde, P., De Mareés, M., Grünheck, F., Tsang, S. Y., Huang, Y., et al. (2003). Inhibition of tumor-induced angiogenesis and matrix-metalloproteinase expression in confrontation cultures of embryoid bodies and tumor spheroids by plant ingredients used in traditional Chinese medicine. Lab. Invest. 83 (1), 87–98. doi:10.1097/01.lab.0000049348.51663.2f
Wei, J., Zhang, M., and Zhou, J. (2015). Myeloid-derived suppressor cells in major depression patients suppress T-cell responses through the production of reactive oxygen species. Psychiatry Res. 228 (3), 695–701. doi:10.1016/j.psychres.2015.06.002
Wilkie-Grantham, R. P., Matsuzawa, S., and Reed, J. C. (2013). Novel phosphorylation and ubiquitination sites regulate reactive oxygen species-dependent degradation of anti-apoptotic c-FLIP protein. J. Biol. Chem. 288 (18), 12777–12790. doi:10.1074/jbc.M112.431320
Wong, K., Ma, J., Rothnie, A., Biggin, P. C., and Kerr, I. D. (2014). Towards understanding promiscuity in multidrug efflux pumps. Trends Biochem. Sci. 39 (1), 8–16. doi:10.1016/j.tibs.2013.11.002
Wu, L., Zhao, J., Cao, K., Liu, X., Cai, H., Wang, J., et al. (2018). Oxidative phosphorylation activation is an important characteristic of DOX resistance in hepatocellular carcinoma cells. Cell. Commun. Signal 16 (1), 6. doi:10.1186/s12964-018-0217-2
Wu, Q., Deng, J., Fan, D., Duan, Z., Zhu, C., Fu, R., et al. (2018). Ginsenoside Rh4 induces apoptosis and autophagic cell death through activation of the ROS/JNK/p53 pathway in colorectal cancer cells. Biochem. Pharmacol. 148, 64–74. doi:10.1016/j.bcp.2017.12.004
Wu, Z., Huang, H., Han, Q., Hu, Z., Teng, X. L., Ding, R., et al. (2022). SENP7 senses oxidative stress to sustain metabolic fitness and antitumor functions of CD8+ T cells. J. Clin. investigation 132 (7), e155224. doi:10.1172/JCI155224
Xia, C., Meng, Q., Liu, L. Z., Rojanasakul, Y., Wang, X. R., and Jiang, B. H. (2007). Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 67 (22), 10823–10830. doi:10.1158/0008-5472.CAN-07-0783
Xu, Y., Xin, Y., Diao, Y., Lu, C., Fu, J., Luo, L., et al. (2011). Synergistic effects of apigenin and paclitaxel on apoptosis of cancer cells. PLoS One 6 (12), e29169. doi:10.1371/journal.pone.0029169
Xu, Z., Huang, B., Liu, J., Wu, X., Luo, N., Wang, X., et al. (2018). Combinatorial anti-proliferative effects of tamoxifen and naringenin: The role of four estrogen receptor subtypes. Toxicology 410, 231–246. doi:10.1016/j.tox.2018.08.013
Yardley, D. A. (2013). Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int. J. Breast Cancer 2013, 137414. doi:10.1155/2013/137414
Young, R. C., Ozols, R. F., and Myers, C. E. (1981). The anthracycline antineoplastic drugs. N. Engl. J. Med. 305 (3), 139–153. doi:10.1056/NEJM198107163050305
Yu, Y., Cui, Y., Niedernhofer, L. J., and Wang, Y. (2016). Occurrence, biological consequences, and human health relevance of oxidative stress-induced DNA damage. Chem. Res. Toxicol. 29 (12), 2008–2039. doi:10.1021/acs.chemrestox.6b00265
Yu, Y., Xie, Y., Cao, L., Yang, L., Yang, M., Lotze, M. T., et al. (2015). The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2 (4), e1054549. doi:10.1080/23723556.2015.1054549
Yuan, T., Hu, J., Zhu, X., Yin, H., and Yin, J. (2022). Oxidative stress-mediated up-regulation of ABC transporters in lung cancer cells. J. Biochem. Mol. Toxicol. 36, e23095. doi:10.1002/jbt.23095
Zhang, Y., Su, S. S., Zhao, S., Yang, Z., Zhong, C. Q., Chen, X., et al. (2017). RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329. doi:10.1038/ncomms14329
Zhang, G., Wang, W., Yao, C., Ren, J., Zhang, S., and Han, M. (2017). Salinomycin overcomes radioresistance in nasopharyngeal carcinoma cells by inhibiting Nrf2 level and promoting ROS generation. Biomed. Pharmacother. 91, 147–154. doi:10.1016/j.biopha.2017.04.095
Zhang, T., Zhang, Y., Cui, M., Jin, L., Wang, Y., Lv, F., et al. (2016). CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 22 (2), 175–182. doi:10.1038/nm.4017
Zhang, X., Huang, J., Yu, C., Xiang, L., Li, L., Shi, D., et al. (2020). Quercetin enhanced paclitaxel therapeutic effects towards PC-3 prostate cancer through ER stress induction and ROS production. OncoTargets Ther. 13, 513–523. doi:10.2147/OTT.S228453
Zhang, X., Yu, L., and Xu, H. (2016). Lysosome calcium in ROS regulation of autophagy. Autophagy 12 (10), 1954–1955. doi:10.1080/15548627.2016.1212787
Zhang, Z., Gao, Z., Rajthala, S., Sapkota, D., Dongre, H., Parajuli, H., et al. (2020). Metabolic reprogramming of normal oral fibroblasts correlated with increased glycolytic metabolism of oral squamous cell carcinoma and precedes their activation into carcinoma associated fibroblasts. Cell. Mol. Life Sci. 77 (6), 1115–1133. doi:10.1007/s00018-019-03209-y
Zhang, Z., Qin, S., Chen, Y., Zhou, L., Yang, M., Tang, Y., et al. (2022). Inhibition of NPC1L1 disrupts adaptive responses of drug-tolerant persister cells to chemotherapy. EMBO Mol. Med. 14 (2), e14903. doi:10.15252/emmm.202114903
Zhao, H., Wu, L., Yan, G., Chen, Y., Zhou, M., Wu, Y., et al. (2021). Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target Ther. 6 (1), 263. doi:10.1038/s41392-021-00658-5
Zheng, J., Nagda, D. A., Lajud, S. A., Kumar, S., Mouchli, A., Bezpalko, O., et al. (2014). Biliverdin's regulation of reactive oxygen species signalling leads to potent inhibition of proliferative and angiogenic pathways in head and neck cancer. Br. J. Cancer 110 (8), 2116–2122. doi:10.1038/bjc.2014.98
Keywords: redox homeostasis, drug resistance, cancer therapy, reactive oxygen species, redox signaling
Citation: Li Y, Zhang X, Wang Z, Li B and Zhu H (2023) Modulation of redox homeostasis: A strategy to overcome cancer drug resistance. Front. Pharmacol. 14:1156538. doi: 10.3389/fphar.2023.1156538
Received: 01 February 2023; Accepted: 13 March 2023;
Published: 22 March 2023.
Edited by:
Claudia Corso, Bioredox, BrazilReviewed by:
Andrew C. B. Cato, Karlsruhe Institute of Technology (KIT), GermanySamson Mathews Samuel, Weill Cornell Medicine, Qatar
Copyright © 2023 Li, Zhang, Wang, Li and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huili Zhu, hlzhu78@139.com
†These authors have contributed equally to this work