 Noa Rosenberg1,2
Noa Rosenberg1,2 Sibren van den Berg1,2
Sibren van den Berg1,2 Nina N. Stolwijk1,2
Nina N. Stolwijk1,2 Bart A. W. Jacobs1,3,4Hendrika C. Post1,5
Bart A. W. Jacobs1,3,4Hendrika C. Post1,5 Anna M. G. Pasmooij6,7Saco J. de Visser1,8
Anna M. G. Pasmooij6,7Saco J. de Visser1,8 Carla E. M. Hollak1,2*
Carla E. M. Hollak1,2*- 1Medicine for Society, Platform at Amsterdam UMC—University of Amsterdam, Amsterdam, Netherlands
- 2Expertise Center for Inborn Errors of Metabolism, Department of Endocrinology and Metabolism, Amsterdam UMC, Amsterdam Gastroenterology Endocrinology Metabolism (AGEM) Research Institute, MetabERN, University of Amsterdam, Amsterdam, Netherlands
- 3Department of Pharmacy, Amsterdam UMC—University of Amsterdam, Amsterdam, Netherlands
- 4Department of Pharmacy and Pharmacology, The Netherlands Cancer Institute—Antoni van Leeuwenhoek, Amsterdam, Netherlands
- 5Department of Oncology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 6Dutch Medicines Evaluation Board, Utrecht, Netherlands
- 7Center for Blistering Diseases, European Reference Network-Skin Reference Center (ERN-Skin), University Medical Center Groningen, University of Groningen, Groningen, Netherlands
- 8Centre for Future Affordable & Sustainable Therapy Development (FAST), The Hague, Netherlands
Background: Novel or repurposed medicines for rare diseases often emerge from fundamental research or empirical findings in academia. However, researchers may be insufficiently aware of the possibilities and requirements to bring novel medicinal treatment options to the patient. This paper aims to provide an easily applicable, comprehensive roadmap designed for academic researchers to make medicines for rare diseases available for patients by addressing the relevant regulatory frameworks, including marketing authorization and alternative routes.
Methods: Key points of the regulatory chapters “Placing on the Market” and “Scope” of Directive 2001/83/EC relating to medicinal products for human use were summarized. Provisions in EU directives regarding blood products, radiopharmaceuticals, and herbal and homeopathic medicinal products were excluded. Cross-referencing to other provisions was included. European case-law was retrieved from the InfoCuria database to exemplify the implications of alternative routes.
Results: Medicines may only be placed on the market with a valid marketing authorization. To obtain such authorization in Europe, a “Common Technical Document” comprising reports on quality and non-clinical and clinical studies must be submitted to a “competent authority”, a national medicine agency or the European Medicines Agency. Timely interaction of academic researchers with regulators via scientific advice may lead to better regulatory alignment and subsequently a higher chance for approval of academic inventions. Furthermore, reimbursement by national payers could be essential to ensure patient access. Apart from the marketing authorization route, we identified multiple alternative routes to provide (early) access. These include off-label use, named-patient basis, compassionate use, pharmacy compounding, and hospital exemption for Advanced Therapy Medicinal Products.
Discussion: Aligning academic (non-)clinical studies on rare diseases with regulatory and reimbursement requirements may facilitate fast and affordable access. Several alternative routes exist to provide (early) pharmaceutical care at a national level, but case-law demonstrates that alternative routes should be interpreted strictly and for exceptional situations only. Academics should be aware of these routes and their requirements to improve access to medicines for rare diseases.
1 Introduction
In the Europe, access to medicines can be particularly challenging for patients with rare diseases, (Deticek et al., 2018). Since 2000, the European Commission (EC) has implemented policy to implement incentives for the development of medicines targeting these diseases via so-called orphan designations (European Medicines Agency, 2022a). One of these incentives is the 10-year market exclusivity upon authorization as an orphan medicinal product. To qualify for an orphan designation, the following criteria must be met (European Medicines Agency, 2022a):
1. Prevalence in the European Union (EU) is < 1:2,000
2. The disease is life-threatening or seriously debilitating
3. There are no satisfactory methods or—in case there is a satisfactory method—the product must be of significant benefit
Up until 2021, more than 200 orphan medicinal products, have entered the market in the EU (European Medicines Agency, 2020a). However, with 5,000 to 10,000 rare diseases (Haendel et al., 2020), many patients are left without pharmaceutical care.
New or repurposed medicines often emerge from fundamental research or empirical findings in academia (Oprea et al., 2011; van den Berg et al., 2021), but frequently do not reach the patient. This could be explained by misalignment with regulatory requirements (van den Berg et al., 2021), negative reimbursement decisions (Deticek et al., 2018), or lack of commercial interest. Academics could become more engaged along the drug development chain in an effort to improve this. For instance, academia-driven public-private partnerships could be explored that include social principles such as cost-saving and socially responsible pricing. Such collaborations might improve academic inventions reaching the market (Organisation for Economic Co-operation and Development, 2008; Padhy and Gupta, 2011; Nederlandse Federatie van Universitair Medische Centra, 2019; Pushpakom et al., 2019) or ameliorate complex and lengthy reimbursement negotiations that may result from initial prices exceeding national reimbursement thresholds, often in combination with the inevitable paucity of effectiveness data (Vella Bonanno et al., 2017; Deticek et al., 2018).
In striving for improvement of patient access to scientific inventions, academics should be acquainted with the drug development process and the associated regulatory aspects. Currently, knowledge of regulatory routes to patient access is frequently insufficient (Kallio et al., 2022) and technology transfer offices that support academics in expediting drug development generally focus on patenting and out-licensing (Van Norman and Eisenkot, 2017a; Van Norman and Eisenkot, 2017b). Therefore, improving awareness and education on ways to bring medicinal inventions to the patient and the corresponding regulatory framework is important (STARS, 2022).
The regulation of medicinal products started approximately a century ago and implementation has largely been driven by tragedies such as sulfanilamide (Can Med Assoc J., 1937; Western Journal of Medicine, 1938), thalidomide (Kim and Scialli, 2011), etc., understandably adding regulations thereby creating one of the most regulated industries focusing roughly on four parameters:
• Quality: Are the product characteristics consistent over time and consistent with the product for which safety and efficacy was demonstrated?
• Safety: When used as intended, are the potential side effects and/or risks acceptable?
• Efficacy: How well does the medicinal product achieve its intended clinical effect?
• Pharmacovigilance: What are the adverse effects when the authorized medicine is used in regular healthcare practice?
The regulations are traditionally focused on the industry applying for marketing authorization, commonly referred to as FDA- or EC-approval. However, interactions between academics and regulators are expanding and of particular value in rare diseases (Ruperto, 2005; European Medicines Agency, 2017). At present, the pharmaceutical legislation is under revision by the EC, which might create new opportunities for acadamics (European Commission, 2021). As academics frequently find novel interventions for these diseases with typically large unmet needs, they are increasingly participating in public private partnerships with the aim of obtaining market approval. Additionally, regulators can benefit from concentrated expertise in academic institutes for advice on regulatory decision-making.
So far, regulatory education for academics has been limited. The EMA has several resources to foster development from early phases in academic laboratories (European Medicines Agency, 2020b). Besides, multiple tools have emerged to support academic drug developers, such as the IRDiRC Orphan Drug Development Guidebook (Hechtelt Jonker et al., 2020), the Market Approval Navigator (Paul Janssen Futurelab Leiden, 2022a), and the educational Horizon 2020 project “Strengthening Training of Academia in Regulatory Science (STARS)” (Starokozhko et al., 2021). Several publications describe opportunities for academic researchers (Davies et al., 2017; Begley et al., 2021), including regulatory procedures, timelines, and fees to obtain an orphan designation and marketing authorization (Davies et al., 2017). These tools can assist academics in obtaining a marketing authorization, but do not encompass the overall helicopter view towards patient access.
This paper aims to provide an easily applicable, comprehensive roadmap designed for academic researchers to make medicines for rare diseases available for patients by addressing the relevant regulatory framework including marketing authorization and alternative medicine-to-patient routes.
2 Methods
The regulatory framework is composed of routes to make medicines for rare diseases available for patients through 1) marketing authorization route and 2) alternative medicine-to-patient routes.
2.1 Marketing authorization
Key points are summarized of the regulatory chapter “Placing on the Market” of the Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use (Directive). This Directive is the core legislative act of medicines in the EU and is publicly accessible via the EudraLex collection (European Commission, 2015). Provisions relating to medicinal products were selected, excluding those regarding blood products, radiopharmaceuticals, and herbal and homeopathic medicinal products. Cross-referencing to other provisions was included as well.
2.2 Alternative routes
Key points are summarized of the chapter “Scope” of the Directive 2001/83/EC. Provisions were included or excluded on the same criteria as described for the marketing authorization route. To better understand the interpretation of the alternative routes within the regulatory framework, we chose to describe purposively sampled case-law (judicial decisions) that exemplify the practical implications of the Directive. The InfoCuria database was used to retrieve judgments of the European Court of Justice (ECJ) in English that refer to the included provisions (InfoCuria Case Law, 2022).
The application of the legal framework and case-law for the academic healthcare professional are summarized as “implications.” To further illuminate complex regulatory matters we provided several illustrative examples of the described routes.
3 Results and implications
In this chapter, we outline the steps toward patient access following an academic invention through the marketing authorization route (Figure 1). Next, we will explain alternative medicine-to-patient routes with the aim to reaching access to both academic inventions, as well as to medicinal products developed by commercial parties (third parties) (Figure 2).
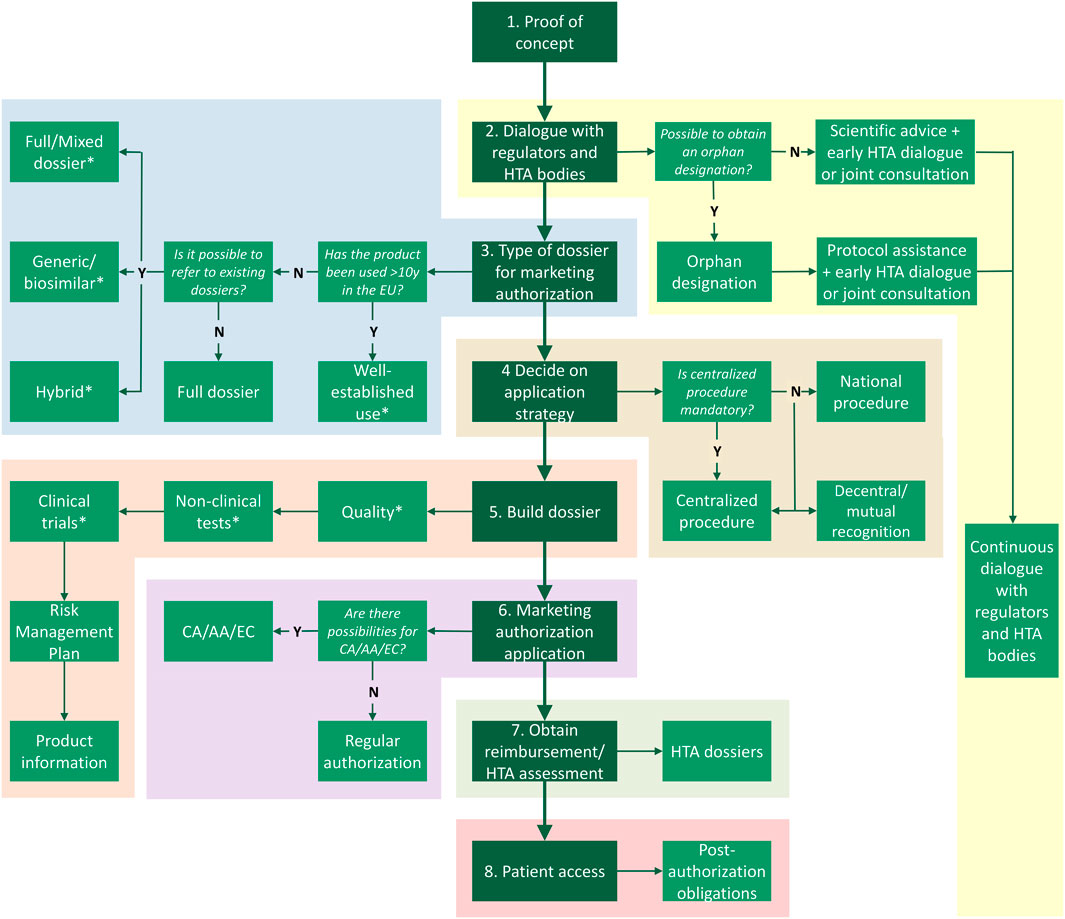
FIGURE 1. Route to patient access via marketing authorization. Following proof of concept there are multiple steps, including (1) dialogue with stakeholders, deciding what (2) type of dossier and (3) application procedure are suitable, (4) building the Common Technical Dossier, (5) submitting the marketing authorization application, (6) applying for the Health Technology Assessment (HTA) at a national HTA body, and (7) complying to post-authorization obligations such as pharmacovigilance. *Exemptions for certain (elements of) modules of the Common Technical Dossier may be applicable for authorized medicines. Y, yes; N, no; HTA, health technology assessment; CA, conditional approval; AA, accelerated assessment; EC, exceptional circumstances.
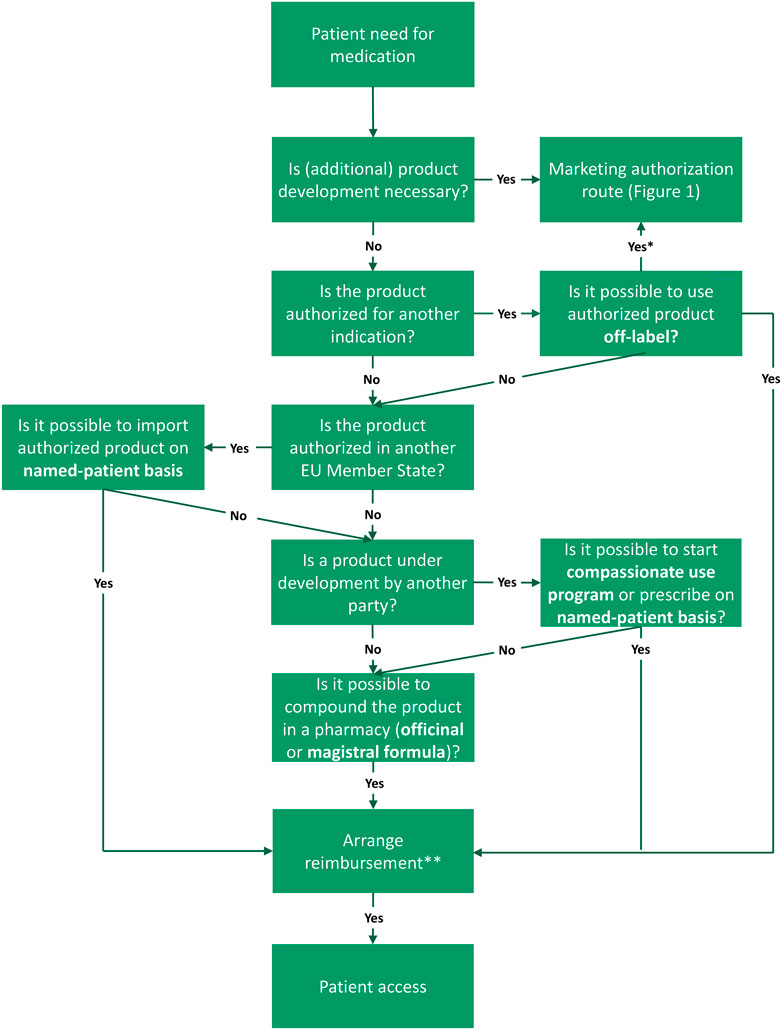
FIGURE 2. Routes to access following the need for a medicine in the European Union (EU), including alternative routes. There are multiple (decision) steps from unmet need to patient access (green boxes). Names of the routes are indicated in bold font. First step is that, in case of a patient need for medication, the academic should examine if product development is necessary, for example, because the product consists of a new active substance or a new formulation. When product development is needed, one should proceed with the marketing authorization route (see Figure 1). For products already authorized for another indication, off-label prescription is a viable option. However, the marketing authorization route can still be exploited to bring this indication on-label. If the product is authorized in another EU Member State, import on a named-patient basis could be suitable. In case the product is still under development by another party, it might be relevant to explore compassionate use programs or ways to prescribe an investigational product on named-patient basis. If these option are unattainable, pharmacy compounding via the magistral or officinal formula can offer a solution. In all cases, national reimbursement should be arranged in order to provide patient access. *Ideally, the marketing authorization route is executed to bring off-label uses on-label. **Reimbursement may following automatically, dependent on national legislation. Frequently, products facilitated via compassionate use programs are free of charge.
3.1 Marketing authorization
The most important rules regarding medicines in the EU are stipulated in Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use (from here footnotes refer to legal provisions). This Directive asserts that medicinal products may only be placed on the market with a valid marketing authorization1. According to the Directive, a medicinal product is defined as a substance—or a combination of substances—presented as having properties for treating or preventing a disease2 and applies when “prepared industrially”3. Steps to obtain marketing authorization and subsequent access to the authorized medicine will be explained in the following paragraphs.
3.1.1 Dossier
The first step towards marketing authorization is composing a dossier called “Common Technical Document” (CTD) which must be submitted to the European Medicines Agency (EMA) or a “competent authority”4–6, a national organization that is responsible for the authorization of medicines. This CTD consists of five modules, namely 1) administrative information, 2) summaries and overviews, 3) quality, 4) non-clinical study reports, and 5) clinical study reports7. The publicly available “CTD explained” animation clarifies the modules in the CTD and the compulsory steps to fill in this document (Paul Janssen Futurelab Leiden, 2022b).
1. Administrative information: The administrative information includes the orphan designation status, risk management plan, labeling, package leaflet, and required information about experts involved in the critical points related to quality, animal, and human investigations8.
2. The summaries and overviews: The summaries and overviews contain information on the quality of the product, and non-clinical and clinical aspects9.
3. The quality module: The quality module consists of all manufacturing details of the active substance and the finished product10. The difference between the active substance and the finished product is that the active substance(s) is the ingredient with pharmacological, immunological, or metabolic activity11 and the finished product is the dosage form, including all excipients and packaging material, which are required for marketing (e.g., a paper box with blisters that contain capsules)12.
4. The non-clinical study reports: The non-clinical study reports include pharmacological, pharmacokinetic and various toxicology studies13,14.
5. The clinical study reports: The clinical study reports contain pharmacokinetics, pharmacodynamics, and efficacy and safety studies14. In general, clinical trials should be performed as blinded randomized controlled clinical trials15. However, “exceptional circumstances” may be applicable for products that have an orphan designation and for which it is deemed impossible to provide complete (non-)clinical information16. This may be the case due to the rarity of a disease.
3.1.1.1 Implications for academia
Results of academic (non-)clinical studies might not only be relevant for scientific publications, but could also be part of a Common Technical Dossier. When expediting this, studies should be performed according to Good Manufacturing Practice (GMP), Good Laboratory Practice (GLP) and/or Good Clinical Practice (GCP) (European Medicines Agency, 2022b; European Medicines Agency, 2023a; European Medicines Agency, 2023b). By adhering to these mandatory international minimum quality standards for manufacturing, non-clinical and clinical aspects, academics can demonstrate that the product they investigate is consistent and reliable. To ensure that studies comply to further regulatory requirements, academics should consider relevant long-term (non-)clinical endpoints and gain scientific advice or protocol assistance from regulatory authorities at an early stage. Implementation of regulatory advice may increase the chance of obtaining marketing approval (Hofer et al., 2015), avoid unnecessary additional studies, and expand incentives for further development, investments, and potential partnerships. Cost reductions on advice for academia may be applicable at a national level (STARS, 2022) and is free for products with an orphan designation at the EMA (European Medicines Agency, 2020c). Thus, obtaining an orphan designation could be a strategic first step for academics researching rare diseases (Davies, 2017). To do so, they have to submit an application at the EMA, generally containing scientific documentation on the medical condition, prevalence, other methods for diagnosis, prevention or treatment of the condition, and a description of the stage of development (European Medicines Agency, 2022c).
3.1.2 Types of dossiers
The regulatory framework defines several legal bases for constructing the Common Technical Dossier. These legal bases describe various types of dossiers, of which some could be applicable to already authorized products and may exempt (a part of) studies17,18:
• Full dossier (Article 8(3)): Novel active substances require compiling a full dossier that consists of all above-mentioned modules.
• Full-mixed (Article 8(3)) or hybrid dossier (Article 10(3)): For known, repurposed, medicines used in a new indication or another application (e.g., method of administration), it is possible to constitute a full-mixed or hybrid dossier. In these cases, the dossier is a combination of studies and references to the original dossier19.
• Type II variation application: Current marketing authorization holders can submit a variation application to add a novel indication to the label. The application could be based on (non-)clinical studies, as well as bibliographic references20 (Commission Regulation (EC) No 1234/2008, 2008).
• Well-established use (Article 10a): Well-established use medicines are medicines that have been used in the EU for at least 10 years and for which efficacy and safety have been well established. This could be the case when a medicine has been used off-label. For this dossier, (non-)clinical tests and trials must be replaced by scientific literature21. In addition, for products with an orphan designation, systematic and documented use on a named-patient basis (see paragraph “Named-patient basis”) may be used in the dossier16.
• Generic medicines (Article 10(1)): Generic medicines are products with the same active substance and are bioequivalent to the original medicinal product. By proving this via a bioequivalency study, (non-)clinical studies can be replaced by references to the original dossier, but only after 8 years of data exclusivity on the original dossier have expired17. As long as this period has not expired, the applicant may only refer to the original dossier with the informed consent (Article 10c) of the current marketing authorization holder22,23.
• Biosimilars (Article 10(4)): Biosimilars are biological products that are highly similar to an approved biological medicine. Similar to generics, references to (non-)clinical studies24 may be made to the original dossier after the data exclusivity period. Contrary to generics, however, additional (non-)clinical studies are frequently necessary to prove the similarity in quality of the product25.
3.1.2.1 Implications for academia
Academic researchers should be aware of the existence of various types of dossiers and necessary studies. As academics are frequently involved in repurposing (Austin and Gadhia, 2017; Begley et al., 2021; Roessler et al., 2021), they may have a particular interest in full (Box 1), full-mixed and well-established use dossiers (Box 2). Academics could obtain assistance from a competent authority and investigate what type of data is necessary to complement references to an original dossier. The prevention of replicates of studies and ownership of additional data may be a cost-saving strategy and thus contribute to affordable medicines. Besides (non-)clinical study related elements, academics are up to date on scientific literature and commonly perform systematic reviews. These endeavors may not only be scientifically relevant, but also for drafting a dossier for medicines used off-label based on well-established use.
BOX 1 An illustrative example of academic endeavors in obtaining a full dossier.
BOX 2 An illustrative example of academic endeavors in obtaining a well-established use dossier.
3.1.3 Routes for application of the dossier
Once the dossier has been drafted, an application must be submitted5. There are four types of procedures to apply for marketing authorization, namely the centralized, national, decentralized, and mutual recognition procedures26.
• Centralized procedure: The centralized procedure refers to an application at the EMA which will lead to marketing authorization in all EU Member States27. This is mandatory for several types of medicines, including products with an orphan designation, medicines for oncological indications, biologicals, and cell- and gene therapy products28–31.
• National procedure: For the national procedure, the applicant will submit the dossier to a competent authority which will lead to marketing authorization in that Member State only.
• Decentralized and mutual recognition procedures: These procedures allow marketing authorization applications in multiple Member States concurrently or consecutively29.
3.1.3.1 Implications for academia
As the centralized route is mandatory for products with an orphan designation, the EMA may be the most relevant authority30 for academics that perform research on rare diseases. National authorities may provide additional support for academics and can be an initial point of contact (STARS, 2022).
3.1.4 During the assessment of the dossier: Accelerated access
When a marketing authorization application has been made, the concerned competent authority (or authorities) will assess the dossier on substantiated quality and the benefit-risk balance. There may be several routes on the European and national level to provide accelerated access31,32, in case of unmet medical need, for instance, accelerated assessment and conditional approval.
• Accelerated assessment: The accelerated assessment speeds up the reviewing process from 210 to 150 days. A request can only be submitted for products with a major public interest.
• Conditional approval: The conditional approval grants authorization based on less comprehensive clinical data than normally required. After additional obligations have been met, the conditional approval can be switched into a “regular” marketing authorization33–38.
• Exceptional circumstances: Exceptional circumstances can apply when it is deemed impossible to obtain comprehensive data on efficacy and safety due to rarity of the disease or ethical considerations34.
3.1.4.1 Implications for academia
When researchers are involved in the development of a medicine relating to a rare disease with an unmet need, they should be aware of the possibilities to initiate the previously mentioned options and apply for scientific advice as early as possible (particularly regarding exceptional circumstances) or other schemes that offer support during development, such as PRIME. This is a scheme that provides early and proactive support to optimize robust data generation and prepare for accelerated assessment (European Medicines Agency, 2022d; European Medicines Agency, 2022e).
3.1.5 From authorization to national reimbursement
Once an application is assessed and a marketing authorization is granted, the product may be placed on the market in the respective Member States35,36 and the marketing authorization holder should continue monitoring the safety of the medicine according to Good Pharmacovigilance Practice (GVP) (European Medicines Agency, 2022f) . Although the centralized procedure grants authorization in all EU Member States, there is no obligation to actually market adult medicines in every Member State. Furthermore, the individual Member States are responsible for national healthcare reimbursement schemes and may include or exclude the medicine from these schemes37. In general, national public organizations called Health Technology Assessment (HTA) bodies will scrutinize the evidence for effectiveness and cost-effectiveness as well as appropriate use, before recommending or deciding on reimbursement (Vreman et al., 2020; Schoot et al., 2022). To facilitate (fast) patient access, several initiatives for collaboration have been set up between regulators and HTA bodies, such as the parallel joint scientific consultation between the EMA and the European Network of Health Technology Assessment (EUnetHTA) (European Medicines Agency, 2022g).
3.1.5.1 Implications for academia
Early dialogue with HTA bodies and payers may be essential for academics, the industry38,39, and patients to expedite routes to reimbursement. Investigator-initiated research on efficacy, safety and effectiveness could be aligned with requirements from competent authorities and HTA bodies to obtain both marketing authorization and reimbursement. To harmonize this, academics could retrieve joint scientific advice. Obligations might be imposed regarding post-marketing data generation on safety, pregnancy and/or effectiveness. These data could be gathered via independent international registries (Jonker et al., 2017). Moreover, academics could be involved in the reimbursement assessment procedure of commercial products of third parties40,41. They can embrace a larger role by acting as clinical experts in committees involved in reimbursement recommendations or by proactively discussing appropriate use of the new product in clinical setting, but should be wary of potential conflicts of interest.
3.1.6 Data ownership and protection
Apart from obtaining marketing authorization, acquiring ownership of data and intellectual property is an important step in product development. The industry typically strives to obtain (licenses to) patents, data exclusivity, and/or market protection (KNAW, 2021), because this allows a company time to generate sufficient sales to recoup its investment by restricting other companies from entering the market. These tools could also be advantageous for academia in valorization and finding partners for non-academic aspects, such as manufacturing and distribution. Several options are included in the regulatory framework:
• Patents: Patents can be granted to novel technical inventions with an industrial application42. When medicines are protected by patents17,20,43,44, the patent holder earns the right to prevent others from commercially exploiting the invention for at least 20 years45. However, necessary clinical trials with the intention of applying for marketing authorization are exempt from this protection44. The publicly available patent portfolio calculator provides a fairly accurate estimate of all costs related to the filing of a patent application, the granting procedures and patent maintenance in a large number of countries around the world (Paul Janssen Futurelab Leiden, 2022c). Once received, the patent can be assigned or licensed to another party (European Patent Office, 2023). Frequently this is in return for royalties: a percentage of the revenues generated by the company exploiting the patented invention (European Patent Office, 2022).
• Data exclusivity: For novel authorized products data exclusivity of 8 years applies, which restricts references to an approved dossier (by generic or full-mixed dossiers)17.
• Market protection: market protection applies for up to 10 years, with the possibility to obtain a 1-year extension if a new indication is added to the marketing authorization within the first 8 years17. This means that a generic medicinal product may refer to the originator dossier after 8 years (at the end of data exclusivity), but may be placed on the market only after the marketing protection period has passed.
• Market exclusivity: For orphan medicinal products, a market exclusivity of up to 10 years will be active for particular authorized indication(s), regardless of patent protection. This means that it is, in principle, prohibited to market a similar medicinal product for the same indication(s)46–47.
3.1.6.1 Implications for academia
Academics may be insufficiently aware of the importance of the abovementioned protective tools and incentives. When aiming to bring treatments to patients, academics could consider ownership and protection of their data and intellectual property before publishing with the help of their technology transfer offices that are typically established to support academic researchers in the patenting process. When an institute has protected an invention, its (commercial) value increases. This could, in turn, lead to a lower risk and larger assurance of return on investments and gives the academic a stronger position for potential collaboration with industrial parties, for instance to set requirements on eventual patient access (KNAW, 2021). The same is true when academic institutes are prepared to co-invest in the development of a new medicinal product, i.e. by participating in clinical trials at a reduced cost rate, or to maintain disease registries that can be used to collect natural history47 data, safety and efficacy outcomes, and post-authorization evidence (Hollak et al., 2020). These independent registries are more cost-effective and have led to more completeness than industry-funded registries (Sirrs et al., 2021).
3.2 Alternative routes
Although a marketing authorization is mandatory to place a medicinal product on the market40, there are several provisions and exemptions in Directive 2001/83/EC that leave room for alternative medicine-to-patient routes. These alternative medicine-to-patient routes may provide both access to unauthorized products and/or uses following academic inventions, as well as access to commercial products from third parties (Figure 2). The following alternative routes will be discussed: off-label use, named-patient basis, compassionate use, magistral and officinal formulas, and the hospital exemption. The referenced case-law illustrates the interpretation of Directive 2001/83/EC (Supplementary Material S1).
3.2.1 Off-label
Off-label use is the use of an authorized medicinal product that is not in accordance with the Summary of Product Characteristics, such as different indication, dosage, duration of use or patient group (Nivel et al., 2017). As EU legislation does not cover off-label use, it is not prohibited to use a medicine for unauthorized indications (F. Hoffmann-La Roche Ltd and others v Autoritá Garante della Concorrenza e del Mercato, 2018), national legislation may apply and regulate off-label prescription (Nivel et al., 2017; Caminada et al., 2021).
3.2.1.1 Implications for academia
Off-label use may be a route to provide patients with a commercially available product after positive results from academic repurposing studies or initial empirical clinical findings (Box 3). Clinical practice shows that off-label use is common—for instance for pediatric uses—but varies in terms of underlying scientific substantiation (Nivel et al., 2017). National legislation may be in place, stipulating prerequisites for off-label prescription (Nivel et al., 2017; Caminada et al., 2021). In France, physicians are allowed to prescribe off-label medicines although they must justify their choice and inform the patient (Nivel et al., 2017). In Italy, there are several specific requirements, such as unmet need, support of completed phase II study and patient consent (Nivel et al., 2017). Legislation may also be in place for reimbursement of off-label use. In Netherlands, for example, the effectiveness of the product is a prominent element for reimbursement decisions (Nivel et al., 2017). As there is a large heterogeneity of off-label legislation between Member States (Caminada et al., 2021), national requirements should be scrutinized before academic repurposed inventions can be lawfully prescribed to patients. The possible shortcoming of evidence and unharmonized legislation may stress the need for bringing new off-label indications on-label, for instance by aligning investigator-initiated repurposing studies with regulatory and HTA requirements.
BOX 3 An illustrative example of off-label use.
3.2.2 Named-patient basis
For individual patients with special needs, a medicinal product can be provided on a “named-patient basis”. This means that Member States may allow unapproved medicines that are prescribed by a doctor based on therapeutic considerations to fulfill pharmaceutical needs48, when there are no authorized equivalents available (European Commission v Republic of Poland, 2012; Abcur AB v Apoteket Farmaci AB and Apoteket AB, 2015). This exception can be used to facilitate access in various ways. First, this route could be used for access to medicinal products that are still under development following academic inventions. Second, commercial investigational products could be supplied on a named-patient basis, for example, when a marketing authorization application in currently under review. Third, the named-patient basis can permit import (European Commission v Republic of Poland, 2012; F. Hoffmann-La Roche Ltd and others v Autoritá Garante della Concorrenza e del Mercato, 2018a) of medicines that are commercially available in other countries but do not have a marketing authorization in the country where the patient is treated. However, named-patient basis must remain exceptional and cannot be used to avoid using authorized products or omit obtaining marketing authorization for financial reasons (Supplementary Material S1) (European Commission v Republic of Poland, 2012; F. Hoffmann-La Roche Ltd and others v Autoritá Garante della Concorrenza e del Mercato, 2018a). Last, data gathered through the systematic and documented use of named-patient basis may be part of a well-established use dossier for orphan medicines16.
3.2.2.1 Implications for academia
Due to the individual character of the named-patient procedure, this can enable access to investigational products or products49 only available in another country for the treatment of patients with rare diseases or very small patient subsets in exceptional situations (Box 4). For this procedure, EU Member States impose various obligations, such as approval of prescriptions and physician’s statements, import permits, and pharmacovigilance (Kreeftmeijer-Vegter et al., 2013). If this is the intention to use data obtained on named-patient basis for a marketing authorization16, protocol assistance should be obtained from regulators on data collection to compose a positive benefit/risk balance. More guidance from regulators might be essential to improve use of aggregated named patient data for regulatory purposes. This could help academics bring medicines to patients and avoid unnecessary trials.
BOX 4 An illustrative example of named-patient import.
3.2.3 Compassionate use
Besides prescription of investigational products on named-patient basis, compassionate use is a way to provide access to an unauthorized medicine for a cohort of patients with a life-threatening or seriously debilitating disease who cannot be treated with an authorized therapy. To facilitate such program, the sponsor has to initiate this procedure with a competent authority when clinical trials are ongoing or when a marketing authorization application has been made49.
3.2.3.1 Implications for academia
When engaged in pre-marketing studies, physicians can stimulate manufacturers to apply for compassionate use programs to supply the product during or after the trials until the authorized medicine becomes readily available (Box 5). Steps to comply with these obligations should be taken in advance to make sure patients have continuous access. Furthermore, data collected via these programs could be used for regulatory filings and HTA assessments (Polak et al., 2020; Polak et al., 2022).
BOX 5 Illustrative example of compassionate use.
3.2.4 Magistral and officinal formulation
An alternative route to access is via pharmacy compounding. This is the preparation of medicinal product in a pharmacy (Abcur AB v Apoteket Farmaci AB and Apoteket AB, 2015). There are two types of pharmacy compounding that are exempt in Directive 2001/83/EC, namely the magistral formula and the officinal formula50,51:
• Magistral formula: the magistral formula is a medicine that is prepared according to a medical prescription for an individual patient50. This means magistral formulas may only be compounded for a specific patient after a physical examination of that patient and issuing the prescription (Abcur AB v Apoteket Farmaci AB and Apoteket AB, 2015).
• Officinal formula: the officinal formula is a medicine that is prepared in accordance with the pharmacopeia (the quality reference work of medicines) and is intended to be supplied to the patients served by the preparing pharmacy in question51. Case-law clarified that officinal formulas may only be delivered directly to patients of the pharmacy that prepared them and not to another pharmacy’s patients (Abcur AB v Apoteket Farmaci AB and Apoteket AB, 2015).
Pharmacy compounding via the magistral or officinal formula thus provides local access routes to unauthorized products or applications. This can be applicable to academic pharmaceutical inventions or to products that are similar to medicinal products authorized by third parties (Abcur AB v Apoteket Farmaci AB and Apoteket AB, 2015), for instance to overcome a temporary shortage.
3.2.4.1 Implications for academia
Often pharmacy preparations are compounded for individual patients with therapeutical needs, for example, a suitable formulation or dosage for a pediatric patient. Because of its individual character, the magistral formula forms an interesting local route for rare diseases with very few patients (Box 6). As the officinal formulas lack the individual element, they can be compounded before a prescription has been issued on (small) stock (Box 7) (Bruins, 2019). This makes the officinal formula more applicable to emergency settings or larger groups of patients. To be able to compound it is understandable that the active substance needs to be available and formulation should not violate patents, though many EU Member States exempt pharmacy preparations for individual cases from infringement (Visser, 2010).
BOX 6 An Illustrative example of a magistral formulation.
BOX 7 An illustrative example of an officinal formulation.
3.2.5 Hospital exemption for ATMPs
For advanced therapy medicinal products (ATMPs), which are gene therapy medicinal products, somatic cell medicinal products, or tissue-engineered products52, there is the possibility of a “hospital exemption.” In the case of an individual prescription (as discussed above), custom-made ATMPs may be exempt from marketing authorization53. These medicines are prepared on a non-routine basis according to specific quality standards and these medicines should be used in a hospital under the exclusive professional responsibility of a medical practitioner, within the same Member State as where they are manufactured53. The manufacturing needs to be authorized by national competent authorities and/or inspectorates (Coppens et al., 2020).
3.2.5.1 Implications
For academics, the hospital exemption provides a controlled local route for tailor-made ATMPs, such as personalized gene therapy for patients with very rare genetic disorders (Box 8). Besides, for seriously debilitating and life-threatening diseases, this may provide a route to commercially developed products from third parties that are not authorized yet. However, with varying national requirements on quality, efficacy and safety and subsequent concerns on potential public health impact (Coppens et al., 2020; Hills et al., 2020), the hospital exemption route should only be pursued with the utmost care.
BOX 8 An illustrative example of a hospital exemption.
3.2.6 Alternative routes and reimbursement
There are multiple alternative medicine-to-patient routes in the European regulatory framework, to which varying national rules apply, particularly regarding reimbursement (Rigter et al., 2021). Hence, it is imperative that the relevant national rules should be reviewed and, if applicable, corresponding organizations should be engaged from the onset to ensure patient access, e.g. through joint scientific advice. The differences between reimbursements of alternative medicine-to-patient routes can be exemplified by the Dutch system. Medicines used off-label are usually reimbursed automatically, (Zorginstituut Nederland, 2022) apart from medicines to which specific prescription restrictions apply. (Zorginstituut Nederland, 2022; Ministerie van Volksgezondheid Welzijn en Sport, 2022b). For other routes, several conditions must be met, i.e., that there are no authorized adequate alternative medicines available and that use of the product is rational pharmacology (Zorginstituut Nederland, 2022). Nonetheless, imported medicines for rare diseases that affect less than < 1:150,000 inhabitants are exempt from these conditions and will generally be compensated for.
4 Actionable recommendations
Based on the regulatory framework and relevant implications discussed, the following actionable recommendations can be set forth for the marketing authorization route and alternative medicine-patient routes.
Marketing authorization:
• Academics researching medicinal inventions for rare diseases should align research and clinical practice with regulatory requirements with the aim of obtaining marketing authorization and subsequent reimbursement to ensure long-term patient access. This potentiality can be bolstered when cooperating with technology transfer offices, industry, and patients.
• Academics should actively connect with regulators and HTA bodies to successfully align their research, for example, by applying for an orphan designation and/or scientific advice. This will also improve academia’s value in potential partnerships.
• Academics should not shy away from a more prominent position in drug development and become aware that this could support social terms regarding eventual access in collaborations.
• Universities/academic medical centers should establish regulatory offices or integrate knowledge on regulatory advice within existing Technology Transfer Offices and Centers of Entrepreneurship that help researchers navigate through the labyrinthine regulatory landscape.
Alternative routes:
• Clinicians should contact their pharmacists and relevant stakeholders such as healthcare insurance companies to discuss alternative routes and arrange coverage if necessary for local patient access.
• Academics could examine whether efficacy, safety and/or effectiveness data collected via alternative routes can serve the purpose of marketing approval and/or reimbursement.
5 Discussion
This regulatory roadmap is the first published guidance for academics to facilitate access to medicines for rare diseases. This framework indicates that there are several routes to patient access, both by developing an authorized medicinal product as well as via alternative medicine-to-patient routes, such as off-label or named-patient use. An authorized product offers the highest assurance of quality, efficacy, and safety and is the preferred option. To obtain marketing authorization for an academic invention, product development is necessary and a comprehensive dossier needs to be compiled and submitted to a competent authority (Davies et al., 2017). While this process requires extensive regulatory knowledge, often present in industry, academics should be aware that they can educate themselves and consequently expand their position. For instance, they can inquire scientific advice or protocol assistance, either central through the EMA or at a national level (STARS, 2022). Aligning investigator-initiated (non-)clinical studies with requirements for marketing authorizations and reimbursement could be crucial to determine end-points, improve chance of marketing approval and prevent access delays (Hofer et al., 2015; Ofori-Asenso et al., 2020). Besides streamlining research with formal requirements, academics could approach drug development trends proactively by considering establishing disease registries, patenting inventions, and data sharing (Van Norman and Eisenkot, 2017a; Van Norman and Eisenkot, 2017b; Sirrs et al., 2021; Karpen et al., 2021; Jansen-van der Weide et al., 2018; Jones et al., 2011; Schoenmakers et al., 2022). To support these effort, academic institutes could improve “in-house” regulatory knowledge to guide researchers through the regulatory roadmap.
In general, clinicians have alternative routes close-at-hand and can reach patient access through these alternatives typically together with their pharmacist. Case-law demonstrated, however, that these exceptional routes should be interpreted with caution and should not be executed because of financial interests. Furthermore, when proceeding with an alternative route, reassuring quality, safety and efficacy for patients should still be priority, similar to the essence of the regular framework. For example, scrutinizing strategies on how to utilize the resultant data for marketing authorization or reimbursement might be an activity more eminently dedicated to academics. The subsequent expanded position of academia in the drug development process could empower a more prominent stand in collaboration with regulators, HTA-bodies, and industry. This might even inforce an improved negotiation position on socially responsible terms when, for example, affordable pricing of an invention is what is deemed important from the academic perspective.
The current European legislation is built on the pharmaceutical industry that traditionally aimed for developing medicines for large patient populations and relatively recently consolidated incentives to develop medicines for rare diseases. Nowadays, we face a new “innovation wave” comprising of ATMPs and more personalized medicines (Konar et al., 2022). Medicinal products for rare diseases are a frontrunner in this paradigm shift from larger general indications to tailored personalized approaches, but are often authorized with less evidence on effectiveness evoking complicated reimbursement negotiations and impeded patient access (Malinowski et al., 2018). A modernization to adapt to the latest practice should also apply to the regulatory framework from authorization to reimbursement including bridging the gap between efficacy (authorization) and effectiveness in daily practice (reimbursement). The EU pharmaceutical legislation is currently under revision (European Commission, 2021)—of which the Clinical Trials Regulation entered into force beginning of 2022 (European Commision, 2014; European Medicines Agency, 2022h)—and will hopefully harmonize these components. Including possibilities to integrate alternative routes and marketing authorization routes in the EU legislation might help aligning academic endeavors and clinical practice with marketing authorization. The arena of (ultra-)rare diseases is an evident first step to do so, due to the (extremely) small patient populations, concentrated clinical expertise, and control of expenditures. Further research on the coalescence of routes, together with regulators, patients, and industry, is essential to sculpture future patient-centered legislation.
Author contributions
NR and CH contributed to the conception and design of the manuscript. NR drafted the manuscript. All authors contributed to interpretation of the data, manuscript revision, read and approved the submitted version.
Funding
This review is performed as part of a larger project “platform Medicijn voor de Maatschappij (platform Medicine for Society).” This platform is financially supported by a grant from “de Nationale Postcode Loterij” a National Lottery that distributes funds raised by this lottery for good causes primarily concerning health and welfare in Netherlands.
Acknowledgments
We would like to express our gratitude towards Joris Heus PhD (Innovation Exchange Amsterdam (IXA) Office AMC, Amsterdam UMC—University of Amsterdam, Amsterdam, Netherlands) and Arnold Versteeg LLM PhD for their help with interpreting legal and regulatory aspects.
Conflict of interest
CH is involved in pre-marketing research with Sanofi, Protalix, and Idorsia, outside the submitted work. NR, SB, BJ, NS, HP, SV, and CH are members of platform “Medicijn voor de Maatschappij”. This is an academic initiative that aims to support sustainable access to medicines for rare diseases. SV teaches Clinical Development at Paul Janssen Futurelab Leiden for academic and industry professionals.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed are the personal views of the authors and may not be understood nor quoted as being made on behalf of, or reflecting the position of, the Dutch Medicines Evaluation Board, Centre for Future Affordable & Sustainable Therapy development or Amsterdam UMC.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1142351/full#supplementary-material
Footnotes
1Directive 2001/83/EC art 6 para 1
2Directive 2001/83/EC art 1 para 2(a)
3Directive 2001/83/EC art 2 para 1
4Directive 2001/83/EC art 6 para 1
5Directive 2001/83/EC art 8 para 1
6Directive 2001/83/EC annex I para 1
7Directive 2001/83/EC annex I para 2
8Directive 2001/83/EC annex I part I-1
9Directive 2001/83/EC annex I part I-2
10Directive 2001/83/EC annex I part I-3
11Directive 2001/83/EC art 3a
12Directive 2001/83/EC Annex I part I art 3.2.2.1
13Directive 2001/83/EC annex I part I-4
14Directive 2001/83/EC annex I part I-5
15Directive 2001/83/EC annex I part I-5.2.5.1
16Directive 2001/83/EC Annex I Part III art 5
17Directive 2001/83/EC art 10 para 1
18Directive 2001/83/EC art 10 para 2(b)
19Directive 2001/83/EC arts 10 para 4
20Directive 2001/83/EC art 10a
21Directive 2001/83/EC art 10c
22Directive 2001/83/EC art 8(3)
23Directive 2001/83/EC art 10 para 3
24Directive 2001/83/EC art 10
25Directive 2001/83/EC annex I part II-4
26Directive 2001/83/EC art 28
27Directive 2001/83/EC annex I para 3
28Regulation (EC) No 726/2004 annex I para 1
29Regulation (EC) No 726/2004 annex I para 1a
30Regulation (EC) No 726/2004 annex I para 3
31Regulation (EC) No 726/2004 annex I para 4
32Directive 2001/83/EC art 26 para 1
33Regulation (EC) No 726/2004 art 14 para 9
34Regulation (EC) No 726/2004 art 14-a
35Commission Regulation (EC) No 507/2006 art 4
36Commission Regulation (EC) No 507/2006 art 5
37Commission Regulation (EC) No 507/2006 art 6
38Commission Regulation (EC) No 507/2006 art 7
39Regulation (EC) No 726/2004 art 14(8)
40Directive 2001/83/EC art 6
41Regulation (EC) No 726/2004 art 3 para 1
42Directive 2001/83/EC art 4 para 3
43The European Patent Convention art 52
44Directive 2001/83/EC art 10 para 6
45Regulation (EC) No 141/2000 art 8 para 1
46The European Patent Convention art 63 para 1
47Regulation (EC) No 141/2000 art 8 para 2
48Directive 2001/83/EC art 5 para 1
49Regulation (EC) No 726/2004 art 83
50Directive 2001/83/EC art 3 para 1
51Directive 2001/83/EC art 3 para 2
52Regulation (EC) No 1394/2007 art 2 para 1(a)
53Directive 2001/83/EC art 3 para 7
References
Abcur AB v Apoteket Farmaci AB and Apoteket AB (2015). Available from: https://curia.europa.eu/juris/document/document.jsf?text=&docid=165910&pageIndex=0&doclang=EN&mode=lst&dir=&occ=first&part=1&cid=7563958 (Accessed March 17, 2022).
Can Med Assoc J (1937). The elixir sulfanilamide-massengill. Canadian Medical Association Journal 37 (6). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC536239/pdf/canmedaj00186-0066a.pdf (Accessed 17, 2022).
Austin, B. A., and Gadhia, A. D. (2017). New therapeutic uses for existing drugs. Advances in Experimental Medicine and Biology 1031, 233–247. doi:10.1007/978-3-319-67144-4_14
Begley, C. G., Ashton, M., Baell, J., Bettess, M., Brown, M. P., Carter, B., et al. (2021). Drug repurposing: Misconceptions, challenges, and opportunities for academic researchers. Science Translational Medicine 13 (612), eabd5524. doi:10.1126/scitranslmed.abd5524
Bruins, B. J. (2019). “Brief van de Minister voor Medische zorg aan de Voorzitter van de Tweede kamer der Staten-generaal,” in Tweede kamer der Staten generaal (The Netherlands): Den Haag).
Caminada, R., Polano, M., Pasmooij, A. M. G., and Stoyanova-Beninska, V. (2021). Off-label prescription of medicines: What do we know about the legislation in EU member states? Rare Disease and Orphan Drugs Journal 1 (5). doi:10.20517/rdodj.2021.04
Coppens, D. G., Gardarsdottir, H., Bruin, M. L., Meij, P., Gm Leufkens, H., and Hoekman, J. (2020). Regulating advanced therapy medicinal products through the hospital exemption: An analysis of regulatory approaches in nine EU countries. Regenerative Medicine 15 (8), 2015–2028. doi:10.2217/rme-2020-0008
Davies, E. H., Fulton, E., Brook, D., and Hughes, D. A. (2017). Affordable orphan drugs: A role for not-for-profit organizations. British Journal of Clinical Pharmacology 83 (7), 1595–1601. doi:10.1111/bcp.13240
Deticek, A., Locatelli, I., and Kos, M. (2018). Patient access to medicines for rare diseases in European countries. Value Health 21 (5), 553–560. doi:10.1016/j.jval.2018.01.007
European Commission v Republic of Poland (2012). Available from: https://curia.europa.eu/juris/document/document.jsf?text=&docid=121168&pageIndex=0&doclang=en&mode=lst&dir=&occ=first&part=1&cid=7564923 (Accessed March 17, 2022).
European Commission, (2014). Clinical trials - regulation EU No 536/2014. Brussels. Available from: https://health.ec.europa.eu/medicinal-products/clinical-trials/clinical-trials-regulation-eu-no-5362014_en (Accessed December 8, 2022).
European Commission, (2015). EudraLex - volume 1 - pharmaceutical legislation for medicinal products for human use. Available from: [Accessed] https://ec.europa.eu/health/medicinal-products/eudralex/eudralex-volume-1_en.
European Medicines Agency, (2017). Patient registries initiative: Lessons learned from the recent workshops. Available from https://www.ema.europa.eu/en/documents/presentation/presentation-patient-registries-initiative-lessons-learned-recent-workshops-x-kurz-ema_en.pdf (Accessed December 7, 2022).
European Medicines Agency, (2020a). Orphan medicines figures 2000-2020. Amsterdam, Netherlands: European Medicines Agency. Available from: https://www.ema.europa.eu/en/documents/other/orphan-medicines-figures-2000-2020_en.pdf (Accessed May 18, 2021).
European Medicines Agency, (2020b). Academia. Available from https://www.ema.europa.eu/en/partners-networks/academia (Accessed July 1, 2022).
European Medicines Agency, (2020c). Press release: Academia developing medicines for rare diseases to receive free EMA scientific advice. Available from https://www.ema.europa.eu/en/news/academia-developing-medicines-rare-diseases-receive-free-ema-scientific-advice (Accessed December 7, 2022).
European Commission, (2021). Revision of the EU general pharmaceuticals legislation. Brussels. Available from https://ec.europa.eu/info/law/better-regulation/have-your-say/initiatives/12963-Revision-of-the-EU-general-pharmaceuticals-legislation/public-consultation_en (Accessed December 8, 2022).
European Medicines Agency, (2022a). Orphan designation: Overview. Available from: https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview (Accessed July 1, 2022).
European Medicines Agency, (2022b). Good Clinical Practice. Available from https://www.ema.europa.eu/en/human-regulatory/research-development/compliance/good-clinical-practice (Accessed February 1, 2023).
European Medicines Agency, (2022c). Procedural advice for orphan medicinal product designation: Guidance for sponsors. Available from https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/procedural-advice-orphan-medicinal-product-designation-guidance-sponsors_en.pdf (Accessed January 2, 2023).
European Medicines Agency, (2022d). Prime: Priority medicines. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines (Accessed October 24, 2022).
European Medicines Agency, (2022e). Prime: Analysis of the first 5 years’ experience Amsterdam. Available from https://www.ema.europa.eu/en/documents/report/prime-analysis-first-5-years-experience_en.pdf (Accessed December 7, 2022).
European Medicines Agency (2022f). Good Pharmacovigilance Practices, Available from https://www.ema.europa.eu/en/human-regulatory/post-authorisation/pharmacovigilance/good-pharmacovigilance-practices (Accessed February 1, 2023).
European Medicines Agency, (2022g). Parallel joint scientific consultation with regulators and health technology assessment bodies. Available from https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance/parallel-joint-scientific-consultation-regulators-health-technology-assessment-bodies (Accessed July 1, 2022).
European Medicines Agency, (2022h). Clinical trials regulation. Available from https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials/clinical-trials-regulation (Accessed December 8, 2022).
European Medicines Agency, (2023a). Good laboratory practice compliance. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/compliance/good-laboratory-practice-compliance (Accessed February 1, 2023).
European Medicines Agency, (2023b). Good Manufacturing Practice. Available from https://www.ema.europa.eu/en/human-regulatory/research-development/compliance/good-manufacturing-practice (Accessed February 1, 2023).
Directive 2001/83/EC of the European parlement and of the council of 6 november 2001 on the Community code relating to medicinal products for human use (2001). Official Journal of the European Union 311 (28), 67–11. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02001L0083-20210526 (Accessed March 17, 2022).
Regulation (EC) No 1394/2007 of the European parliament and of the council of 13 november 2007 on advanced therapy medicinal products and amending directive 2001/83/EC and regulation (EC) No 726/2004 (2007). Official Journal of the European Union 311. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02001L0083-20210526 (Accessed March 17, 2022)
European Patent Office, (2023). “Dealing with companies: Royalties,” in Inventors' handbook (München, Germany: European Patent Office).
European Patent Office, (2022). Part E, chapter XIV. Available from: https://www.epo.org/law-practice/legal-texts/html/guidelines/e/e_xiv_6_1.htm (Accessed December 7, 2022).
Novartis Farma SpA v Agenzia Italiana del Farmaco (AIFA) and Others (2018b). Available from: https://curia.europa.eu/juris/document/document.jsf?text=&docid=207947&pageIndex=0&doclang=EN&mode=lst&dir=&occ=first&part=1&cid=7565773 (Accessed March 17, 2022).
Haendel, M., Vasilevsky, N., Unni, D., Bologa, C., Harris, N., Rehm, H., et al. (2020). How many rare diseases are there? Nature Reviews Drug Discovery 19 (2), 77–78. doi:10.1038/d41573-019-00180-y
Hecht-Pharma GmbH v Hohenzollern Apotheke (2016). Winfried Ertelt. Available from: https://curia.europa.eu/juris/liste.jsf?num=C-276/15&language=EN (Accessed March 17, 2022).
Hechtelt Jonker, A., Hivert, V., Gabaldo, M., Batista, L., O'Connor, D., Aartsma-Rus, A., et al. (2020). Boosting delivery of rare disease therapies: The IRDiRC orphan drug development Guidebook. Nature Reviews Drug Discovery 19 (8), 495–496. doi:10.1038/d41573-020-00060-w
Hills, A., Awigena-Cook, J., Genenz, K., Ostertag, M., Butler, S., Eggimann, A. V., et al. (2020). An assessment of the hospital exemption landscape across European member states: Regulatory frameworks, use and impact. Cytotherapy 22 (12), 772–779.e1. doi:10.1016/j.jcyt.2020.08.011
Hofer, M. P., Jakobsson, C., Zafiropoulos, N., Vamvakas, S., Vetter, T., Regnstrom, J., et al. (2015). Regulatory watch: Impact of scientific advice from the European medicines agency. Nature Reviews Drug Discovery 14 (5), 302–303. doi:10.1038/nrd4621
F. Hoffmann-La Roche Ltd and others v Autoritá Garante della Concorrenza e del Mercato (2018a). Available from: https://curia.europa.eu/juris/document/document.jsf;jsessionid=8706003B703C57D13955C7AB24387852?text=&docid=198644&pageIndex=0&doclang=EN&mode=lst&dir=&occ=first&part=1&cid=168257 (Accessed March 3, 2022).
Hollak, C. E. M., Sirrs, S., van den Berg, S., van der Wel, V., Langeveld, M., Dekker, H., et al. (2020). Registries for orphan drugs: Generating evidence or marketing tools? Orphanet Journal of Rare Diseases 15 (1). doi:10.1186/s13023-020-01519-0
InfoCuria Case Law, (2022). Search Form. Available from: https://curia.europa.eu/juris/recherche.jsf?&cid=700453 (Accessed March 17, 2022).
Jansen-van der Weide, M. C., Gaasterland, C. M. W., Roes, K. C. B., Pontes, C., Vives, R., Sancho, A., et al. (2018). Rare disease registries: Potential applications towards impact on development of new drug treatments. Orphanet Journal of Rare Diseases 13 (1). doi:10.1186/s13023-018-0836-0
Jones, S., James, E., and Prasad, S. (2011). Disease registries and outcomes research in children: Focus on lysosomal storage disorders. Paediatric Drugs 13 (1), 33–47. doi:10.2165/11586860-000000000-00000
Jonker, C. J., van den Berg, H. M., Kwa, M. S. G., Hoes, A. W., and Mol, P. G. M. (2017). Registries supporting new drug applications. Pharmacoepidemiology and Drug Safety 26 (12), 1451–1457. doi:10.1002/pds.4332
Kallio, M. J., Starokozhko, V., Agricola, E., Burggraf, M., Hess, A., Ballensiefen, W., et al. (2022). Translating academic drug discovery into clinical development: A survey of the awareness of regulatory support and requirements among stakeholders in Europe. Clinical Pharmacology & Therapeutics 113, 349–359. doi:10.1002/cpt.2789
Karpen, S. R., White, J. K., Mullin, A. P., O'Doherty, I., Hudson, L. D., Romero, K., et al. (2021). Effective data sharing as a conduit for advancing medical product development. Therapeutic Innovation & Regulatory Science 55 (3), 591–600. doi:10.1007/s43441-020-00255-8
Kim, J. H., and Scialli, A. R. (2011). Thalidomide: The tragedy of birth defects and the effective treatment of disease. Toxicological Sciences 122 (1), 1–6. doi:10.1093/toxsci/kfr088
KNAW (2021). Efficiency gains through innovation in medicines development: How can science contribute? Amsterdam. Available from https://storage.knaw.nl/2022-06/20211005-Advies-Eficiency-through-innovation-web.pdf (Accessed January 3, 2023).
Konar, N. M., Karaismailoglu, S., and Karaismailoglu, E. (2022). Status and trends of personalized medicine research from 2000 to 2020: A bibliometric analysis. Current Medical Research and Opinion 38 (5), 837–846. doi:10.1080/03007995.2022.2052515
Kreeftmeijer-Vegter, A. R., van Veldhuizen, C. K., and de Vries, P. J. (2013). Roll out of intraveneous artesunate under named patient programmes in The Netherlands, Belgium and France. Orphanet Journal of Rare Diseases 8. doi:10.1186/1750-1172-8-150
Malinowski, K. P., Kawalec, P., Trabka, W., Sowada, C., and Pilc, A. (2018). Reimbursement of orphan drugs in Europe in relation to the type of authorization by the European medicines agency and the decision making based on health technology assessment. Frontiers in Pharmacology 9. doi:10.3389/fphar.2018.01263
Ministerie van Volksgezondheid Welzijn en Sport, (2022a). Besluit zorgverzekering. Den haag. Available from https://wetten.overheid.nl/BWBR0018492/2022-01-01/#Hoofdstuk2_Paragraaf1_Artikel2.8 (Accessed July 1, 2022).
Ministerie van Volksgezondheid Welzijn en Sport, (2022b). Regeling zorgverzekering. Den haag. Available from https://wetten.overheid.nl/BWBR0018715/2022-07-01 (Accessed July 1, 2022).
Nederlandse Federatie van Universitair Medische Centra, (2019). Tien principes voor maatschappelijk verantwoord licentiëren. Den haag. Available from https://www.nfu.nl/sites/default/files/2020-10/Tien_Principes_MVL.pdf (Accessed July 1, 2022).
Nivel, RIVM, and EPHA, (2017). Study on off-label use of medicinal products in the European Union. Brussels: European Commission. Available from https://www.nivel.nl/sites/default/files/bestanden/Report_OFF_LABEL_Nivel-RIVM-EPHA.pdf (Accessed January 3, 2023).
Ofori-Asenso, R., Hallgreen, C. E., and De Bruin, M. L. (2020). Improving interactions between health technology assessment bodies and regulatory agencies: A systematic review and cross-sectional survey on processes, progress, outcomes, and challenges. Frontiers in Medicine (Lausanne) 7, 582634. doi:10.3389/fmed.2020.582634
Oprea, T. I., Bauman, J. E., Bologa, C. G., Buranda, T., Chigaev, A., Edwards, B. S., et al. (2011). Drug repurposing from an academic perspective. Drug Discovery Today: Therapeutic Strategies 8 (3-4), 61–69. doi:10.1016/j.ddstr.2011.10.002
Organisation for Economic Co-operation and Development, (2008). “In pursuit of risk sharing and value for money,” in Public-private partnerships (Paris: OECD Publishing).
Padhy, B. M., and Gupta, Y. K. (2011). Drug repositioning: Re-investigating existing drugs for new therapeutic indications. Journal of Postgraduate Medicine 57 (2), 153–160. doi:10.4103/0022-3859.81870
Paul Janssen Futurelab Leiden (2022a). Market approval navigator. Leiden. Available from: https://marketapprovalnavigator.pauljanssenfuturelab.eu/login/ (Accessed July 1, 2022).
Paul Janssen Futurelab Leiden (2022b). CTD explained. Leiden. Available from https://www.pauljanssenfuturelab.eu/toolbox/ctd-explained/ (Accessed December 7, 2022).
Paul Janssen Futurelab Leiden (2022c). Patent portfolio calculator. Leiden. Available from: https://www.pauljanssenfuturelab.eu/toolbox/patent-portfolio-calculator/ (Accessed December 8, 2022).
Polak, T. B., Cucchi, D. G. J., van Rosmalen, J., and Uyl-de Groot, C. A. (2022). Real-world data from expanded access programmes in health technology assessments: A review of NICE technology appraisals. BMJ Open 12 (1), e052186. doi:10.1136/bmjopen-2021-052186
Polak, T. B., van Rosmalen, J., and Uyl-de Groot, C. A. (2020). Expanded Access as a source of real-world data: An overview of FDA and EMA approvals. British Journal of Clinical Pharmacology 86 (9), 1819–1826. doi:10.1111/bcp.14284
Pushpakom, S., Iorio, F., Eyers, P. A., Escott, K. J., Hopper, S., Wells, A., et al. (2019). Drug repurposing: Progress, challenges and recommendations. Nature Reviews Drug Discovery 18 (1), 41–58. doi:10.1038/nrd.2018.168
Rigter, T., Klein, D., Weinreich, S. S., and Cornel, M. C. (2021). Moving somatic gene editing to the clinic: Routes to market access and reimbursement in Europe. European Journal of Human Genetics 29, 1477–1484. doi:10.1038/s41431-021-00877-y
Rijksinstituut voor ziekte- en invaliditeitsverzekering, (2022a). Terugbetaling van magistrale bereidingen. Brussels. Available from: https://www.inami.fgov.be/nl/themas/kost-terugbetaling/door-ziekenfonds/geneesmiddel-gezondheidsproduct/terugbetalen/magistrale/Paginas/default.aspx#Contacten (Accessed December 8, 2022).
Rijksinstituut voor ziekte- en invaliditeitsverzekering, (2022b). Vergoedingsvoorwaarden voor magistrale bereidingen die enkel vergoedbaar zijn na machtiging door de adviserend arts. Brussels. Available from https://www.inami.fgov.be/SiteCollectionDocuments/lijst_magistrale_bereidingen_hoofdstukIV.pdf (Accessed December 8, 2022).
Roessler, H. I., Knoers, N., van Haelst, M. M., and van Haaften, G. (2021). Drug repurposing for rare diseases. Trends in Pharmacological Sciences 42 (4), 255–267. doi:10.1016/j.tips.2021.01.003
Ruperto, N. (2005). Prioritising drug development for children with rhumatologic diseases: The Paediatric RheumatologyInterNational Trials Organization (PRINTO) perspective. Available from https://www.ema.europa.eu/en/documents/presentation/presentation-session-27-examples-interactions-printo-nicola-ruperto_en.pdf (Accessed December 7, 2022).
Schoenmakers, D. H., Beerepoot, S., van den Berg, S., Adang, L., Bley, A., Boelens, J. J., et al. (2022). Modified Delphi procedure-based expert consensus on endpoints for an international disease registry for Metachromatic Leukodystrophy: The European Metachromatic Leukodystrophy initiative (MLDi). Orphanet Journal of Rare Diseases 17 (1). doi:10.1186/s13023-022-02189-w
Schoot, R. A., Otth, M. A., Frederix, G. W. J., Leufkens, H. G. M., and Vassal, G. (2022). Market access to new anticancer medicines for children and adolescents with cancer in Europe. European Journal of Cancer 165, 146–153. doi:10.1016/j.ejca.2022.01.034
Sirrs, S. M., Arthus, M. F., Bichet, D. G., Rockman-Greenberg, C., LeMoine, K., Morel, C. F., et al. (2021). Independent registries are cost-effective tools to provide mandatory postauthorization surveillance for orphan medicinal products. Value Health 24 (2), 268–273. doi:10.1016/j.jval.2020.10.006
Starokozhko, V., Kallio, M., Kumlin Howell, A., Makinen Salmi, A., Andrew-Nielsen, G., Goldammer, M., et al. (2021). Strengthening regulatory science in academia: STARS, an EU initiative to bridge the translational gap. Drug Discovery Today 26 (2), 283–288. doi:10.1016/j.drudis.2020.10.017
STARS (2022). STARS common strategy. Available from https://www.csa-stars.eu/files/STARS_Common_Strategy.pdf (Accessed December 7, 2022).
Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products(2008). OJL 334. Available from: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2008:334:0007:0024:en:PDF (Accessed July 1, 2022).
van den Berg, S., de Visser, S., Leufkens, H. G. M., and Hollak, C. E. M. (2021). Drug repurposing for rare diseases: A role for academia. Frontiers in Pharmacology 12, 746987. doi:10.3389/fphar.2021.746987
Van Norman, G. A., and Eisenkot, R. (2017a). Technology transfer: From the research bench to commercialization: Part 1: Intellectual property rights-basics of patents and copyrights. JACC: Basic to Translational Science 2 (1), 85–97. doi:10.1016/j.jacbts.2017.01.003
Van Norman, G. A., and Eisenkot, R. (2017b). Technology transfer: From the research bench to commercialization: Part 2: The commercialization process. JACC: Basic to Translational Science 2 (2), 197–208. doi:10.1016/j.jacbts.2017.03.004
Vella Bonanno, P., Ermisch, M., Godman, B., Martin, A. P., Van Den Bergh, J., Bezmelnitsyna, L., et al. (2017). Adaptive pathways: Possible next steps for payers in preparation for their potential implementation. Frontiers in Pharmacology 8. doi:10.3389/fphar.2017.00497
Visser, C. (2010). “Chapter 5: Patent exceptions and limitations in the health context,” in Exclusions from patentability and exceptions and limitations to patentees’ rights (Geneva, Switzerland: WIPO).
Vreman, R. A., Mantel-Teeuwisse, A. K., Hovels, A. M., Leufkens, H. G. M., and Goettsch, W. G. (2020). Differences in health technology assessment recommendations among European jurisdictions: The role of practice variations. Value Health 23 (1), 10–16. doi:10.1016/j.jval.2019.07.017
Western Journal of Medicine (1938). Elixir sulfanilamide-massengill: Report of the United States secretary of agriculture. Western Journal of Medicine 48 (1), 68–70. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1705805/pdf/calwestmed00370-0077.pdf (Accessed 7 1, 2022).
Zorginstituut Nederland, (2022). Niet-geregistreerde indicaties. Diemen. Available from https://www.farmacotherapeutischkompas.nl/algemeen/niet-geregistreerde-indicaties (Accessed December 8, 2022).
Keywords: regulatory framework, patient access, marketing authorization, alternative drug-to-market routes, rare diseases, orphan drugs
Citation: Rosenberg N, van den Berg S, Stolwijk NN, Jacobs BAW, Post HC, Pasmooij AMG, de Visser SJ and Hollak CEM (2023) Access to medicines for rare diseases: A European regulatory roadmap for academia. Front. Pharmacol. 14:1142351. doi: 10.3389/fphar.2023.1142351
Received: 11 January 2023; Accepted: 08 February 2023;
Published: 28 February 2023.
Edited by:
Sandor Kerpel-Fronius, Semmelweis University, HungaryReviewed by:
Segundo Mariz, European Medicines Agency, NetherlandsSam Salek, University of Hertfordshire, United Kingdom
Copyright © 2023 Rosenberg, van den Berg, Stolwijk, Jacobs, Post, Pasmooij, de Visser and Hollak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carla E. M. Hollak, Yy5lLmhvbGxha0BhbXN0ZXJkYW11bWMubmw=