 Beatrice Noce
Beatrice Noce Elisabetta Di Bello
Elisabetta Di Bello Rossella Fioravanti1*
Rossella Fioravanti1* Antonello Mai
Antonello Mai
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 02 February 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1120911
This article is part of the Research Topic New Insights into the Histone Lysine Specific Demethylase (LSD1) Inhibitors for Cancer Treatment View all 4 articles
Histone lysine-specific demethylase 1 (LSD1/KDM1A) was first identified in 2004 as an epigenetic enzyme able to demethylate specific lysine residues of histone H3, namely H3K4me1/2 and H3K9me1/2, using FAD as the cofactor. It is ubiquitously overexpressed in many types of cancers (breast, gastric, prostate, hepatocellular, and esophageal cancer, acute myeloid leukemia, and others) leading to block of differentiation and increase of proliferation, migration and invasiveness at cellular level. LSD1 inhibitors can be grouped in covalent and non-covalent agents. Each group includes some hybrid compounds, able to inhibit LSD1 in addition to other target(s) at the same time (dual or multitargeting compounds). To date, 9 LSD1 inhibitors have entered clinical trials, for hematological and/or solid cancers. Seven of them (tranylcypromine, iadademstat (ORY-1001), bomedemstat (IMG-7289), GSK-2879552, INCB059872, JBI-802, and Phenelzine) covalently bind the FAD cofactor, and two are non-covalent LSD1 inhibitors [pulrodemstat (CC-90011) and seclidemstat (SP-2577)]. Another TCP-based LSD1/MAO-B dual inhibitor, vafidemstat (ORY-2001), is in clinical trial for Alzheimer’s diseases and personality disorders. The present review summarizes the structure and functions of LSD1, its pathological implications in cancer and non-cancer diseases, and the identification of LSD1 covalent and non-covalent inhibitors with different chemical scaffolds, including those involved in clinical trials, highlighting their potential as potent and selective anticancer agents.
Lysine specific demethylase 1 (LSD1, KDM1A) was discovered in 2004 by the Shi’s group (Shi et al., 2004). Up to date, lysine demethylases (KDMs) according to their sequence homology and mechanism of action have been classified into two main classes: LSDs and the Jumonji-containing (JmjC) KDMs. The first enzymes belong to a superfamily of flavin adenine-dinucleotide (FAD)-dependent amine oxidases and comprise two isoforms, LSD1, the founding member, and LSD2, latterly discovered in 2009 (Ciccone et al., 2009). On the other hand, the JmjC KDMs are Fe (II)/2-oxoglutarate (2-OG)-dependent enzymes catalyzing the demethylation of mono-, di-, and trimethyl lysines, are structurally connected with nucleic acid oxygenases, and contain more than 20 members.
LSD1 specifically removes the methyl groups from mono- and dimethyl lysine 4 or lysine 9 of histone 3 (H3K4me1/2 and H3K9me1/2), behaving as either a repressor or activator of gene expression, respectively (Rotili and Mai, 2011). Mechanistically, single-electron oxidation of the mono- or dimethylated amine at the ε-position of lysine gives an iminium cation with simultaneous reduction of FAD to FADH2. After FADH2 reoxidation to FAD by O2, H2O2 is generated. The iminium cation is unstable and after easy hydrolysis furnish the amine with one methyl less and formaldehyde as by-product (Dai et al., 2020) (Figure 1). The key feature that allows this mechanism to happen is the lone pair of the N-methylated lysine. Hence, the trimethylated species H3K4me3 is not a substrate for LSD1, since the long pair is not available.
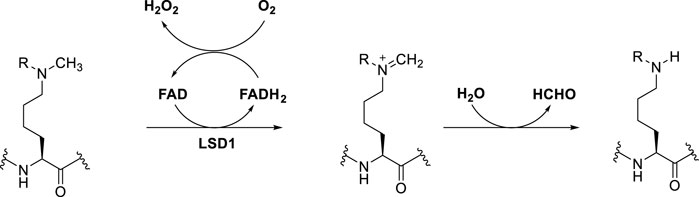
FIGURE 1. General catalytic mechanism of histone demethylation by LSD1.
LSD1 polypeptide chain can be divided into several functional regions: the N-flexible region; the SWIRM domain (small α-helical domain); the C-terminal amine oxidase domain (AOL), divided into two portions by the Tower domain (Chen et al., 2006). The N-flexible region has no predicted structural elements and is not necessary for catalysis, but it is essential for LSD1 nuclear localization. The SWIRM domain of LSD1 is useful as docking site to interact with other proteins and provides to retain LSD1 protein stability. It is characterized by six helical bundles and two-stranded β-sheets. These β-sheets help to form some interactions between SWIRM and AOL domains. The AOL domain - the catalytic part of LSD1 - comprises two lobes: in the first of them the oxidation reaction starts, through binding with the SWIRM domain containing the FAD-binding site, while the second one shows the site for substrate recognition. Both lobes establish a cavity in which the demethylation activity occurs into the catalytic center. The Tower domain, a typical antiparallel coiled coil, protrudes from the AOL domain and is formed by two α-helices. The Tower domain can act as an adaptor to recruit other proteins. Indeed, it is mandatory to allow the binding of the RE1-silencing transcription factor (CoREST), which is involved onto the formation of a heterodimeric complex with LSD1, increasing the enzyme activity (Figure 2) (Chen et al., 2006). This CoREST complex is important for its ability to demethylate the nucleosome complex of histones and DNA, because LSD1 alone can only demethylate the specific residue in peptides or bulk histones (Kim et al., 2020).
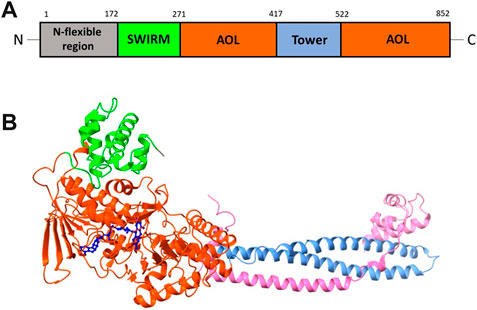
FIGURE 2. (A) Structural organization of LSD1 with the N-flexible region (grey) and the domains SWIRM (green), AOL (two parts, orange), and Tower (light blue). (B) X-ray structure of the LSD1/CoREST complex, with the domains SWIRM (green), AOL (orange) and Tower (light blue). The CoREST portion is depicted in pink. The FAD cofactor (blue) is located inside of the AOL domain (PDB ID: 3ABT).
LSD1, when associated with CoREST, histone deacetylases (HDACs), nucleosome remodeling and deacetylation complexes, and C-terminal binding proteins, demethylates H3K4me1/2 residues and works as transcriptional co-repressor (Lee et al., 2005) (Shi et al., 2005) (Wang et al., 2009) (Shi et al., 2003). In the presence of H3K9me1/2 residues (transcription repressor markers), LSD1 acts as a transcription co-activator by demethylation, associated to androgen and estrogen receptors (Metzger et al., 2005).
In most human cancer cells, LSD1 plays a pivotal role in reversible cellular processes such as epithelial-mesenchymal transition (EMT) (Nieto, 2009; Ambrosio et al., 2017b), contributing to cancer invasion and progression and inducing metastatic characteristics (Choi et al., 2015). Beyond histones, the discovery of a large amount of non-histone proteins have been identified as substrates of LSD1. The tumor suppressor p53 was the first non-histone LSD1 substrate identified (Huang et al., 2007). Afterwards, it has been shown that LSD1 regulates angiogenesis, cell cycle arrest, chromatin remodeling and proliferation of cancer cells by demethylation of HIF-1α (Lee et al., 2017), E2F1 (Kontaki and Talianidis, 2010), DNMT1 (Wang et al., 2009), and STAT3 (Yang et al., 2010). Most recently, AGO2 has been reported to be demethylated by LSD1, promoting protein stability and accumulating dsRNA expression to regulate tumor T cell response (Sheng et al., 2018). Thus, LSD1 is epigenetically involved in the control of many cellular processes, such as autophagy, stemness, differentiation, senescence, cell proliferation and motility, organogenesis, (Lan et al., 2008; Zheng et al., 2015; Chen et al., 2016; Hirano and Namihira, 2016; Ambrosio et al., 2017b), haematopoiesis, and neuronal and embryonic development (Whyte et al., 2012; Karakaidos et al., 2019).
Pathologically, LSD1 is involved in metastasis and tumorigenesis and has been found dysregulated and/or overexpressed in various cancer types. LSD1 was found significantly overexpressed in many solid tumours, including prostate (Etani et al., 2019), breast (Zhou et al., 2021), small cell lung cancer (Jin et al., 2019), bladder cancer (Hayami et al., 2011), medulloblastoma (Lee et al., 2019), neuroblastoma (Ambrosio et al., 2017a), glioma (Bailey et al., 2020b), and sarcomas (Bennani-Baiti et al., 2012), as well as hematological malignancies, such as acute myeloid leukemia (AML) (Zhang et al., 2021).
For instance, in prostate cancer (PCa) LSD1 inhibition can hamper the EMT process induced by androgens, leading to a delay in the transformation of PCa into castrate-resistant prostate cancer (Wang et al., 2015). In breast cancer, LSD1 and the KDM6A UTX, a Jumonji C demethylase, are co-expressed and co-localize with estrogen receptors (ERs). Dual LSD1/UTX inhibitors exerted anticancer activity in in vitro, ex vivo and in vivo breast cancer models. The same compounds gave downregulation of ERα, at both transcriptional and non-transcriptional levels, through modulation of miR-181a-5p expression (Benedetti et al., 2019; Benedetti et al., 2021). The reduction of activity of LSD1, obtained by either pharmacological or genetic approach, could prevent resistance to antitumor agents in breast as well as other cancer models (Lu et al., 2018; Sobczak et al., 2022). In blood cancers, various studies demonstrated the contribution of LSD1 to the onset and progression of AML (Lokken and Zeleznik-Le, 2012). In particular, LSD1 is able to increase the leukemic stem cells (LSCs)’ clonogenic activity and start their transcriptional actions, together with a reduction of myeloid differentiation. Indeed, LSD1 inhibition increases the levels of myeloid-lineage markers, including CD11b and CD86 (Fang et al., 2017). Both in vitro and in vivo studies reported the effects of LSD1 inhibitors in terms of anti-leukemic activity, reduction of LSCs growth, induction of cell differentiation and increased survival in mouse models of AML (Chen et al., 2012).
LSD1 overexpression has been also linked to resistance to chemotherapy, immunotherapy, and radiotherapy (Dagogo-Jack and Shaw, 2018). In varicella zoster virus (VZV) and herpes simplex virus (HSV), LSD1 depletion or inhibition led to repressive chromatin and viral cycle inhibition (Gu and Roizman, 2009) (Zhou et al., 2010). In sensory neurons, LSD1 inhibitors can block the HSV reactivation from latency, highlighting LSD1 as a crucial player in viral infection and reactivation (Liang et al., 2009). Finally, LSD1 has been reported to be involved in metabolic diseases (Hino et al., 2012) and in central nervous system disorders, such as Alzheimer’s disease and depression (Rusconi et al., 2016).
Resistance to immunotherapy is a complex and difficult mechanism and involves two types of cells, cancer cells and immune cells (Sharma et al., 2017). Overcoming resistance to immunotherapy means improving the ability of immune cells to recognize cancer cells, killing them (Schoenfeld and Hellmann, 2020). Targeting LSD1 could cause this effect, in particular reactivating critical immune checkpoint regulators and modulating T cells in cancer (Sheng et al., 2018) (Qin et al., 2019). Moreover, the inhibition of LSD1 suppressed stem cell-like properties and sensitized head and neck squamous cell carcinoma to PD-1 blockade (Han et al., 2021). For this reason, the combination of LSD1 inhibitors and anti-PD-1 agents could have a synergistic effect respect to the single-target inhibitor.
LSD1, in addition to the previously citated implications, has an important role in the viral transcription. In 2001, Sakane et al. (2011) showed that the LSD1/CoREST complex is linked to the HIV promoter and triggers, through K51 demethylation, the activity of Trans-Activator of Transcription (TAT), a regulatory protein essential for HIV replication. The involvement of LSD1 in viral infections of Herpes Virus was discovered in 2010. The α-herpesviruses, including HSV and VZV, after infection use cellular transcriptional coactivator host cell factor-1 to recruit LSD1 on the immediate early promoter. LSD1 inhibition leads to the accumulation of the chromatin in its condensed state blocking the viral cycle.
In addition to their inhibition of the LSD1 catalytic activity, some LSD1 inhibitors exert scaffolding functions leading, for instance, to the removal of the NK cells lytic capacity, through a potent reduction of oxidative phosphorylation (Bailey et al., 2020a), or to the disruption of the DRED complex, with increases of γ-globin and cellular HbF contents in vitro and in vivo, useful for innovative treatment of sickle cell disease (Holshouser et al., 2020).
LSD1 is overexpressed in many cancers, among which AML is one of the most represented (Schenk et al., 2012). LSD1 inhibitors gave highly variable results in AML cells, their effect can be improved by co-treatment with other drugs, including retinoic acid (RA) (McGrath et al., 2016).
It was demonstrated that the treatment with LSD1 inhibitors of cells of Acute Promyelocytic Leukemia (APL), a subtype of AML, does not induce growth arrest but increase sensitivity of the same cells to physiological concentrations of RA (Binda et al., 2010). In particular, in a panel of 21 AML cell lines (representing all subtypes), it was seen that the combination of RA with the LSD1 inhibitor MC2580 (4, see below) impacts on the viability of both LSD1 inhibitors-sensitive and -resistant AML cell lines. The RA treatment, joined to LSD1 inhibition, gives a differentiation not always dependent on changes in methylation levels of H3K4. The non-enzymatic role of LSD1 leads to a block of cell differentiation, but it can be removed by combination of LSD1 inhibitors with RA (Ravasio et al., 2020).
The combination between another LSD1 inhibitor (DDP38003) and RA was tested in vivo. All the control mice died within 3 weeks (median survival: 21 days). Treatments with RA alone or LSD1 inhibitor alone prolonged mice survival (median survival: 49 (RA) or 37 (LSD1i) days). The combination of RA and the LSD1 inhibitor strongly increased the therapeutic effect of the single agents alone (median survival, 70 days) (Ravasio et al., 2020).
Since the discovery of LSD1 in 2004, many LSD1 inhibitors have been described in literature, acting through covalent or non-covalent mechanism, alone or in combination with other therapeutic agents, to fight cancer and non-cancer diseases (Hojfeldt et al., 2013; Yang et al., 2018). Some of them have undergone clinical trials for cancer treatments.
Trans 2-phenylcyclopropylamine (tranylcypromine, TCP), known with the brand name of Parnate®, was first approved by the US Food and Drug Administration (FDA) in 1961 for patients with major depressive disorders. It displays high inhibitory activity against MAOs (Ki values = 19 (MAO-A) and 16 (MAO-B) μM) (Binda et al., 2010). Since the catalytic domain of LSD1 is similar at structural level with that of MAOs, and the molecular mechanism of catalysis is common between LSD1 and MAOs, several MAO inhibitors including TCP, pargyline, and phenelzine, were tested against LSD1. Phenelzine is a non-selective, irreversible MAO inhibitor in clinical use as antidepressant and anxiolytic agent. It has been reported to inhibit also LSD1 (Culhane et al., 2010), and entered clinical trials in combination with Abraxane, a nano formulation of Paclitaxel bound to albumin, for the treatment of metastatic or advanced breast cancer (Table 1). Early experimental findings highlighted TCP as the most valuable fragment despite its moderate LSD1 inhibitory activity (Ki = 271 µM) (Binda et al., 2010). Since TCP is an already approved drug, its repurposing as anticancer agent is a valuable strategy for drug discovery (Song et al., 2022). It has been postulated that TCP inactivates LSD1 through a single-electron transfer (SET) mechanism (Figure 3A). Depending on the cyclopropyl C-C bond involved in the homolysis, four different TCP-FAD adducts can be obtained through two distinct pathways. One initial step may consist in the opening of the cyclopropyl C1-C2 bond with the transfer of a single electron from the primary amine nitrogen of TCP to FAD, allowing the formation of a radical cation, followed by the formation of a stabilized benzyl radical. Such radical interacts with the C(4a) of the FAD cofactor leading to an iminium ion yielding the 3-phenylpropionaldehyde (A) after hydrolysis. Hence, this aldehyde intramolecularly can react with the N(5) of FAD to afford a hemiaminal intermediate (B), which gives the final unsaturated cyclic adduct (C) after dehydration (Figure 3A, pathway a). The second pathway involves the opening of the cyclopropyl C1-C3 bond, with the formation of a radical carbon and a concerted bond formation with the FAD C(4a) position leading to the production of a 2-phenylpropyl iminium ion, subsequently hydrolyzed to the corresponding aldehyde (D) (Figure 3A, pathway b). Despite the second route is energetically unfavorable, the adduct (D) is the major product detected and isolated during the MAO-B covalent inhibition by TCP (Schmidt and McCafferty, 2007; Yang et al., 2007). In 2010, we solved the crystal structures of (−)-TCP, (+)-TCP, (−)-4-Br-TCP, (+)-4-Br-cis 2-phenylcyclopropylamine and the two more potent LSD1 inhibitors 4 and 5 (see below) in complex with LSD1/CoREST (Binda et al., 2010), and we observed the formation of adducts in which the bond with the FAD factor occurred at the N(5) instead of the C(4) position, as proposed by Mimasu et al., in 2008 (Mimasu et al., 2008). The mechanism postulated for the formation of the N(5) adducts involves the attach of the C1-C2 bond of TCP on the N(5) position of FAD, with an initial formation of a stable benzylic cation. After, a 1,3 hydride shift occurs followed by hydrolysis of the iminium cation giving the adduct (E) in which FAD is acylated at N(5) (Figure 3B).
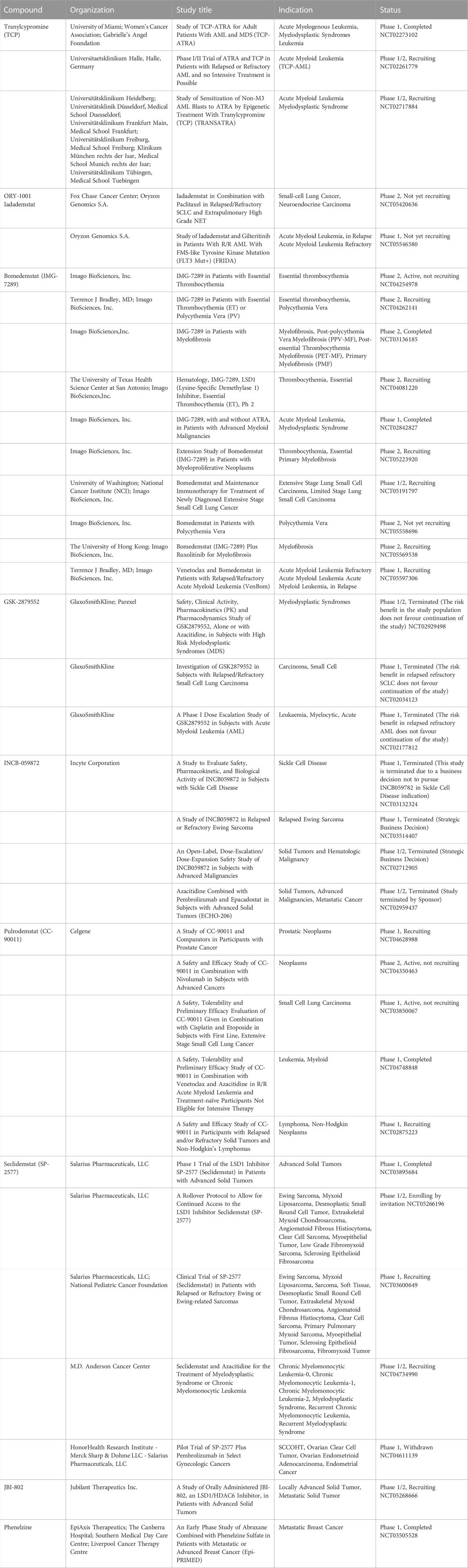
TABLE 1. Overview of LSD1 inhibitors currently in clinical investigations for cancer treatment.
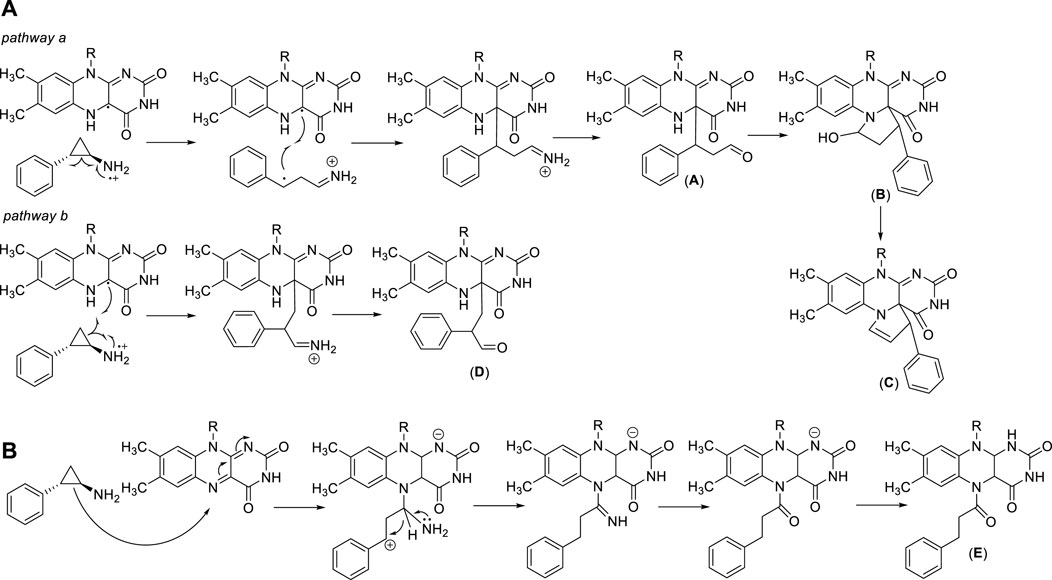
FIGURE 3. TCP-FAD adducts: proposed structures and mechanisms of formation with links at FAD C(4a) (A) or N(5) (B) position.
The covalent TCP-FAD adduct in LSD1 is in a hydrophobic pocket surrounded by Val333, His564, Thr335, Tyr761, Ala809, and Thr810 residues. The benzene ring of the adduct forms weak Van Der Waals bonds with the methyl groups of Thr810 and Thr335, but does not have other interactions with the previously named hydrophobic residues (Yang et al., 2007). For this reason, extensive structure-activity relationship (SAR) studies led to the discovery of numerous TCP-based irreversible LSD1 inhibitors with increased potency and selectivity, able to form strong covalent adducts with the flavin cofactor (Lee et al., 2006; Schmidt and McCafferty, 2007; Zheng et al., 2016).
We will summarize the results obtained with such TCP-based LSD1 inhibitors, and discuss their potential use as therapeutic drugs, with a focus on multi-targeting inhibitors and on compounds in clinical trials. We grouped these compounds into two clusters: i) derivatives substituted on the TCP phenyl ring, and ii) derivatives substituted on the TCP primary amino group.
From 2009, the TCP benzene ring has been substituted at ortho, meta, and/or para positions to produce potent LSD1 inhibitors. In 2010, Mimasu et al. described some derivatives with (cyclo)alkyl/arylalkyloxy residues at the ortho and eventually para positions, and halogen atoms at the meta position(s), of the TCP phenyl ring (Mimasu et al., 2010). The best result was obtained with S2101 (1) (Figure 4), an ortho-benzyloxy-meta/meta-difluoro substituted derivative exhibiting stronger anti-LSD1 potency than TCP (Ki = 0.61 μM). In 2009 and 2010, Ueda et al. (2009) and Binda et al. (2010) reported the same strategy to obtain potent LSD1 inhibitors: the insertion at para (Ueda, Binda) or meta (Ueda) positions of the TCP phenyl ring of hindered and branched amino acid-based substituents, linked through an ether (Ueda, NCL1 (2), NCL2 (3), Figure 4) or amide (Binda, MC2580 (4), Figure 4) function. NCL compounds revealed to be potent LSD1 inhibitors selective over MAOs (IC50 values: NCL1 = 2.5 (LSD1), 230 (MAO-A), 500 (MAO-B) µM; NCL2 = 1.9 (LSD1), 290 (MAO-A), >1,000 (MAO-B) µM), due to the presence of large groups attached to the TPC phenyl ring which cannot be accommodated by the MAOs active sites. The same compounds showed interesting anticancer properties, with a range of reduction of 50% cell growth from 6 to 67 μM in a panel of cancer cell lines (HeLa cervical cancer, HCT-116 colon cancer, PC-3 prostate cancer, KYSE-150 esophageal squamous cell carcinoma, and SH-SY5Y neuroblastoma). In Binda et al., the para position of the TCP ring was substituted with a simple aroyl amine or a N-benzyloxycarbonylamino acyl amine. MC2584 (5) and MC2580 (4) were identified as first lead compounds, with a Ki of 1.1 and 1.3 μM, respectively, and selectivity over MAO-B for MC2580. In APL cells, MC2580 displayed antiproliferative activity in synergy with RA (Binda et al., 2010). These two prototypes were after developed to furnish more potent, cell permeable and selective inhibitors (Fioravanti et al., 2020). Among them, MC3340 (6) (Figure 4) exhibited IC50LSD1 = 90 nM with selectivity over MAOs, and submicromolar potency against APL NB4 (IC50 = 0.6 μM) and AML MV4-11 (IC50 = 0.4 μM) cells (Fioravanti et al., 2020), and the 1S, 2R pure enantiomer 7 (Vianello et al., 2016) (Figure 4) showed IC50LSD1 = 84 nM, high selectivity over MAO-B, and huge anticlonogenic activity in mouse APL blasts and THP-1 cells. This last compound, when orally administered in vivo in a mouse APL model, appreciably increased survival of the treated mice (35% and 62% at the doses of 11.25 and 22.50 mg/kg, respectively) without apparent toxicity (Vianello et al., 2016). The replacement of the TCP benzene ring with pyrrole and indole moieties afforded MC3288 (8) and MC3382 (9) (Figure 4), which showed significant LSD1 inhibitory activity (IC50 = 0.032 µM and IC50 = 0.040 µM, respectively) and antiproliferative effects at low micromolar level against AML and APL cells (Rodriguez et al., 2015).
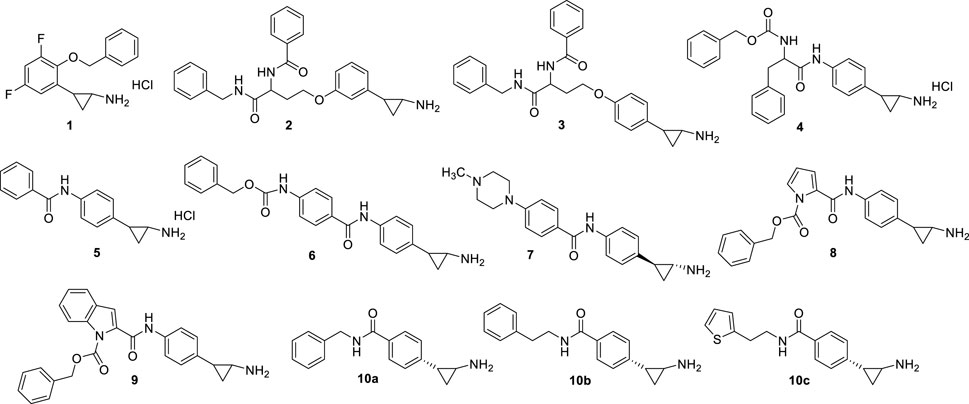
FIGURE 4. Covalent, TCP-based LSD1 inhibitors substituted on the phenyl ring.
In 2021, novel TCP analogues with a carboxamide at the para position of the benzene ring were developed by Borrello et al.. Some of them, containing a benzyl-, 2-phenethyl-, or 2-thienylethylamino carboxamide portion (10a-c) (Figure 4) were submicromolar inhibitors of the enzyme (10a: IC5010a = 0.3 µM; IC5010b = 0.4 µM; IC5010c = 0.6 µM) (Teresa Borrello et al., 2021).
Very often the epigenetic targets work in macromolecular and multicomponent complexes, formed by recruitment of different proteins and/or factors, able to activate or silence transcription. The idea to simultaneously inhibit different components of repressive complexes through a unique hybrid molecule could lead to a synergistic action for cancer treatment, by disruption of the complex and reactivation of transcription of tumor-suppressor genes. As in prostate cancer the two families of KDMs, LSD1 and JmjC enzymes (specifically, KDM4A/C), are co-expressed and colocalized with the androgen receptor, the design and synthesis of pan-demethylase inhibitors, able to hit at the same time both the families of demethylases, has a strong rationale for treatment of prostate cancer. Thus, the LSD1-inhibiting TCP portion was coupled with fragments able to inhibit the JmjC enzymes, such as the 4-carboxy-4′-carbomethoxy-2,2′-bipyridine and the 5-carboxy-8-hydroxyquinoline, to obtain multitargeting inhibitors (11 and 12) (Rotili et al., 2014). The two hybrid compounds induced arrest of proliferation and huge dose-dependent apoptosis in prostate cancer LNCaP and colon cancer HCT-116 cell lines, with slight or no apoptosis induction in mesenchymal progenitor cells, suggesting a cancer selectivity for these compounds. In the same cancer cell lines, 11 and 12 showed increased H3K4 and H3K9 methylation levels, confirming the inhibition of the two targets at the cellular level (Figure 5) (Rotili et al., 2014).
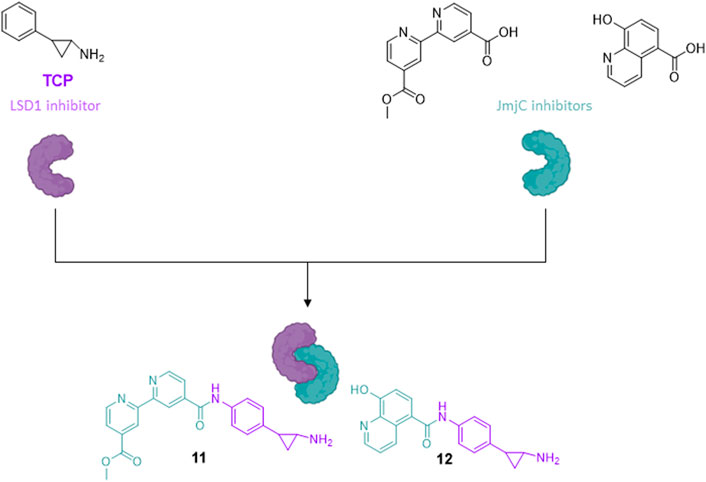
FIGURE 5. Hybrid compounds inhibitors of LSD1 and JmjC.
LSD1 physically interacts with the co-repressor CoREST and with HDAC1/2 to form a repressive CoREST complex (Pilotto et al., 2015; Zheng et al., 2015). Thus, the simultaneous blockage of LSD1 and HDAC1/2 represents a promising strategy to reactivate transcription against cancer, leading to combination studies and hybrid compounds development. The combination of TCP with the zinc-binding group of Entinostat, a well-known HDAC inhibitor belonging to the benzamide series and inhibiting specifically HDAC1-3, furnished hybrid molecules targeting the ternary CoREST complex and inhibiting both LSD1 and HDAC1 at submicromolar doses (Kalin et al., 2018).
In a panel of melanoma cells, the dual LSD1/HDAC compound Corin (Figure 6) (Kalin et al., 2018) exhibited higher inhibition of proliferation with respect to the relative single-target inhibitors (TCP for LSD1 and Entinostat for HDACs) and to their combination. Corin showed good pharmacokinetics and safety profile, and in melanoma SK-MEL-5 BALB/c mice it reduced by 61% tumor growth after 28 days without relevant toxicity. Moreover, Corin increased H3K9 acetylation and H3K4 dimethylation, induced some genes related to anticancer effects such as p21, CHOP, and MXD1, and reduced the level of the marker of proliferation Ki67 (Kalin et al., 2018). In diffuse intrinsic pontine glioma (DIPG) Corin was equally effective in vitro and in vivo, demonstrating the crucial effect of dual LSD1/HDAC inhibition also in this context (Anastas et al., 2019).
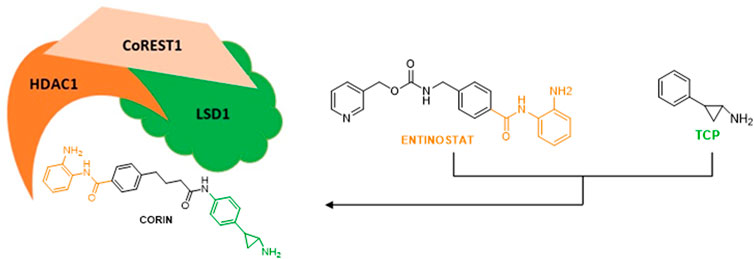
FIGURE 6. Schematic figure of CoREST1 complex and the inhibitor Corin.
Most of the LSD1 inhibitors currently undergoing clinical studies are TCP derivatives alkylated on the nitrogen atom. In 2013, some authors reported cyclic or linear peptides as LSD1 inhibitors (Tortorici et al., 2013; Kumarasinghe and Woster, 2014). Among them, Ogasawara et al. starting from the assumption that a peptide chain could be the vector of TCP gaining LSD1 inhibition, synthesized a series of peptide derivatives, including TCP-Lys-4 H3-21 showing potent LSD1 inhibition (IC50 = 0.16 μM) and >625 selectivity index over MAOs (Ogasawara et al., 2013). However, it was not very effective on cancer cells (GI50 values: 27 μM (HeLa) and >160 μM (SH-SY5Y)), probably due to poor cell permeability (Ogasawara et al., 2013). Afterwards, the same research group showed that the length of the peptide chain was very important for the inhibitory potency of compounds (Kakizawa et al., 2015). Thus, they reduced the peptide itself to a unique lysine residue, with acyl and amino groups at the α-amino and carboxy functions, and with the insertion of a phenylcyclopropyl moiety at the ε-amino group. The obtained NCD compounds exhibited submicromolar inhibitory activity against LSD1 (IC50 values: 0.30 (NCD18 (13)), 0.48 (NCD25 (14)) and 0.58 (NCD41 (15)) µM) (Figure 7), with preferences for either the 1R, 2S (NCD18 and NCD25) or 1S, 2R (NCD41) enantiomer, reflecting a different preferred binding mode in the LSD1 catalytic pocket. Among the three compounds, NCD41 displayed the highest antiproliferative activity against HeLa cervical cancer (GI50 = 4.1 μM) and neuroblastoma SH-SY5Y (GI50 = 2.4 μM) cells (Itoh et al., 2014).
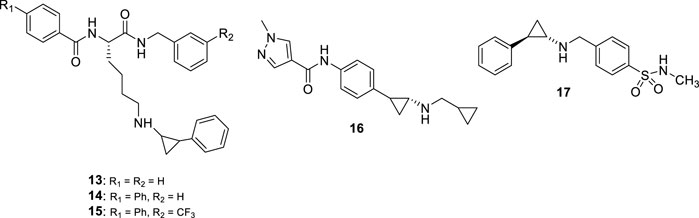
FIGURE 7. Covalent TCP-based LSD1 inhibitors substituted on the amino group.
In 2017, Takeda Pharmaceuticals disclosed a novel LSD1 inhibitor, T-3775440 (16) (Figure 7) showing potent LSD1 inhibition (IC50 = 20 nM) (Ishikawa et al., 2017a). In experimental studies, T-3775440 highly induced growth arrest in Acute Megakaryoblastic Leukaemia and AML cells through LSD1 inhibition. Moreover, the co-treatment of T-3775440 with the NEDD8-activating enzyme inhibitor pevonedistat displayed synergistic inhibition against AML cells (Ishikawa et al., 2017b).
In 2018, a number of N-alkylated TCP derivatives were synthesized, and some of them were found to be very potent (Schulz-Fincke et al., 2018). Among them, the N-methyl sulfonamide 17 (Figure 7) was the most potent LSD1 inhibitor (IC50 = 0.19 μM), with 90-fold selectivity over MAO-A and no inhibition towards MAO-B. Furthermore, such derivative was able to inhibit the formation of colonies of leukemic cells. Docking studies revealed that the large LSD1 binding pocket can admit the selective binding of lipophilic as well as polar groups, whereas in MAO-A the binding site is tighter and characterized by many lipophilic residues that do not allow the stacking with polar substituents (Schulz-Fincke et al., 2018). Other important TCP-based compounds substituted at the primary amino group entered clinical trials for the treatment of cancer diseases (see below).
The co-treatment of TCP and all-trans-retinoic acid (ATRA) is present in three clinical trials against AML and myelodysplastic syndromes (MDS) (Table 1). The combination therapy of TCP and ATRA displayed increased antileukemic effect compared with that of each drug alone (Wass et al., 2021).
ORY-1001 (Iadademstat, RG6016, R07051790) (Figure 8) is a potent LSD1 inhibitor (IC50 = 18 nM) developed by Oryzon Genomics, acting through irreversible binding to the LSD1 FAD cofactor. ORY-1001 is currently in clinical trials (Table 1) for the treatment of AML and solid tumors (Salamero et al., 2020). It is highly selective for LSD1 over MAOs and displayed unrivaled sub-nanomolar cellular activity in differentiation assays. In particular, it gave time and dose-dependent induction of the CD11b differentiation marker in mixed lineage leukemia (MLL)-AF9 cells. Interestingly, such effect preceded changes in H3K4me2 levels. Moreover, ORY-1001 decreased colony formation particularly on MLL-translocated cells, but also on other AML cells. In melanoma, the combination of ORY-1001 with an anti-PD1 antibody used for 22 days led to significant tumor growth inhibition, 54% higher than that with group treated with the anti-PD1 antibody alone (Maes et al., 2019).
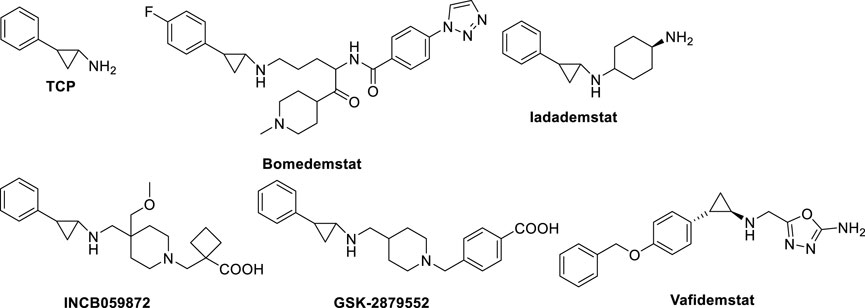
FIGURE 8. Irreversible LSD1 inhibitors in clinical trials for cancer treatment.
ORY-2001, or Vafidemstat (Figure 8), was reported by Oryzon Genomics and it is an orally bioavailable dual inhibitor of LSD1 and MAO-B (Cavalcanti et al., 2022). Due to this dual effect and its capability to cross the blood-brain barrier, ORY-2001 entered in 2019 the phase IIa clinical trial for the treatment of mild to moderate Alzheimer’s diseases (Mehndiratta and Liou, 2020), it being the only LSD1 inhibitor with an indication for treatment of neurogenerative diseases. In preclinical and clinical studies, ORY-2001 has been shown to be able to reestablish behavior impairments, decrease aggressiveness and social withdrawal and gain memory (EudraCT 2018-002140-88; EudraCT 2019-001436-54). In addition, ORY-2001 was effective in treatment of multiple sclerosis (EudraCT 2017-002838-23) and it represents the first epigenetic approach against this disease. Moreover, since in COVID-19 many inflammatory cytokines with immunomodulatory action are central to trigger the cytokine storm and the following high inflammation status, it was determined the tolerability and efficacy of ORY-2001 to prevent acute respiratory distress syndrome in patients with severe SARS-CoV-2 infection (EudraCT 2020-001618-39), due to the ability of ORY-2001 to reduce the levels of IL-6, IL-1β, and other relevant inflammatory cytokines. At the moment ORY-2001 is in Phase 2 clinical trials for mild to moderate Alzheimer’s Disease (NCT03867253) and for borderline personality disorder (NCT04932291).
Bomedemstat, also known as IMG-7289 (Figure 8) (Hiatt et al., 2022), is an oral small molecule irreversible LSD1 inhibitor (IC50 = 56.8 nM) developed by Imago BioSciences, in clinical trials for myeloid-related malignancies (Table 1). It is well tolerated in heterogeneous patients with advanced myelofibrosis (MF), improves symptomatology, reduces spleen sizes, and normalizes or improves blood cell counts in some patients (Dai et al., 2020). An ongoing, multi-center, open-label study involving IMG-7289 recently moved from a Phase I/IIa dose-range finding study to a Phase IIb study of administered orally once-daily in adult patients with intermediate-2 or high-risk MF resistant or intolerant to ruxolitinib, a selective JAK inhibitor. The high doses used for Phase II study (40–60 mg/day) are about 500-fold higher than those of ORY-1001, and surpass the 10 mg/day threshold typically used in the risk assessment protocol for idiosyncratic toxicity of covalent ligands, with increased risk for off-target effects (Lammert et al., 2008; Baillie, 2021).
GSK2879552 (Figure 8), an orally active N-alkylated derivate of TCP, was identified after the screening of 2.5 million compounds with an IC50 of 24 nM (Mohammad et al., 2015; Bauer et al., 2019), and the evaluation of its anticancer activity using more than 165 cell lines showed significant inhibition of AML and SCLC cell lines proliferation. In in vivo xenograft models, GSK2879552 was effective to inhibit the growth of NCI-H1417 SCLC cells without inducing thrombocytopenia and other hematologic toxicities (Mohammad et al., 2015). Two clinical phase 1 trials investigating the pharmacodynamics, pharmacokinetics, safety, and clinical activity of GSK2879552 in patients with relapsed/refractory SCLC (NCT02034123) and AML (NCT02177812) have been terminated. Besides, a phase I/II, open-label study evaluating the safety and clinical activity of GSK2879552 alone, or in combination with azacytidine, a well-known DNA methyltransferase inhibitor, in subjects with myelodysplasia, has also been terminated (NCT02929498). As shown on the website of clinicaltrials.gov, the risk/benefit ratio does not support further studies with this compound (Table 1). Additionally, GSK2879552 displayed synergy with ATRA in various subtypes of AML for cell differentiation, proliferation, and cytotoxicity (Smitheman et al., 2019).
INCB059872 (Figure 8), developed by Imago BioSciences, is another N-alkylated TCP-based LSD1 inhibitor currently under clinical trials to treat various cancers (Johnston et al., 2020). Currently, there are four clinical phase 1,2 studies for cancer therapy, for sickle cell disease (NCT03132324), relapsed Ewing sarcoma (NCT03514407), solid tumors and hematologic malignancy (NCT02712905), metastatic cancer (NCT02959437). However, all these studies have been terminated for strategic business decisions (Table 1). In human SCLC preclinical models, INCB059872 was effective in inducing growth arrest at nanomolar level (EC50 values = 47–377 nM).
Very recently, JBI-802 has been identified by Jubilant Therapeutics Inc. as a dual LSD1/HDAC6/8 inhibitor, featuring nanomolar inhibition of the above three targets (IC50 values: 50 nM (LSD1), 11 nM (HDAC6), and 98 nM (HDAC8)) and about 100-fold selectivity against other HDAC isoforms (Sivanandhan et al., 2020). In hematological cancers (such as AML, chronic lymphocytic leukemia, acute megakaryocytic leukemia, Z-138 lymphoblast cells, multiple myeloma cells) as well as in solid tumors (small cell lung cancer (SCLC) and sarcoma cells) JBI-802 displayed huge antiproliferative effect. Functional tests for target engagement studies performed both in vitro and in vivo showed significant dose-dependent increase in CD11b, CD86 and GFI1b (markers of LSD1 inhibition) and tubulin acetylation (marker of HDAC6 inhibition) levels (Sivanandhan et al., 2020). JBI-802 entered Phase 1/2 clinical trial for treatment of advanced and metastatic solid tumors (Table 1).
Additionally, Phenelzine (as written before) entered Phase 1 clinical trial for treatment of metastatic breast cancer in combination with nanoparticle albumin-bound Paclitaxel (Abraxane) (Table 1).
Compounds derived from TCP, showing irreversible LSD1 inhibition due to the formation of covalent bonds within the enzyme active site, are very effective but suffer from several side effects. Indeed, they can show significant affinities with several other targets beyond LSD1, including neurotransmitters, metabolizing enzymes, receptors, and transporters. Moreover, in addition to long-lasting on-target effects, they could in principle also induce prolonged off-target effects (Dai et al., 2020). Thus, researchers have turned to the design and synthesis of compounds that could establish a non-covalent bond with LSD1 and consequently should show a better safety profile. Many compounds have been designed, classified according to their chemical structure. Compound CBB1003 (18) (Figure 9) (IC50 = 10.5 µM) is part of a group of reversible LSD1 inhibitors containing guanidinium/amidinium groups. Such positively charged functions form strong hydrogen bonds with the negatively charged amino acid residues present in the active site of LSD1, and the hydrophobic substituents dock into the deep pocket close to the FAD cofactor. In addition, the nitro group of CBB1003 is likely to form a hydrogen bonding interaction with His564 (Wang et al., 2011). In further studies, CBB1003 displayed weak cell growth inhibition (IC50 = 250 μM) in colorectal cancer (CRC) through downregulation of leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5), a CRC stem cell marker involved in carcinogenesis (Hsu et al., 2015).
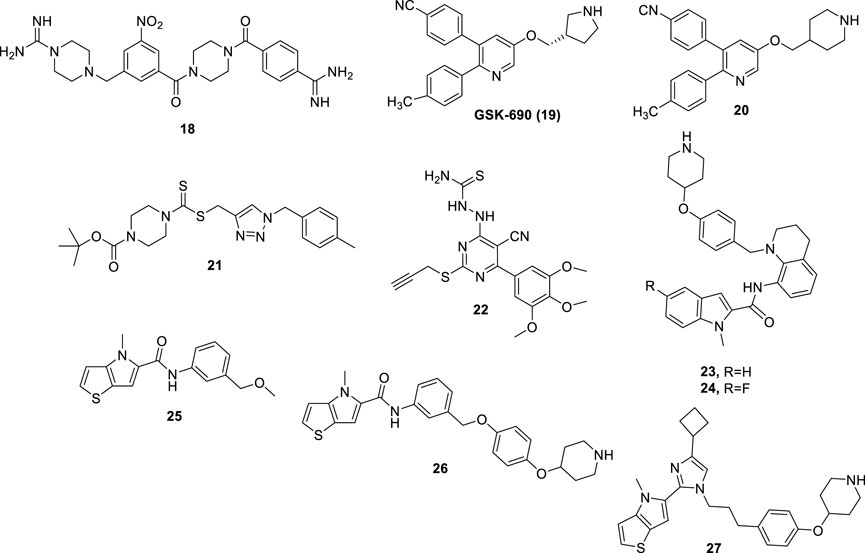
FIGURE 9. Reversible LSD1 inhibitors.
Continuing the search for derivatives that could show a potent and reversible inhibitory action against LSD1, numerous compounds with different chemical structures have been tested. GSK-690 (19) (Figure 9), a 2,3-diphenylpyridyn-5-ol derivative, was a potent LSD1 inhibitor (IC50 = 90 nM) in vitro and highly selective over MAO-A (IC50 >200 μM) (Hitchin et al., 2013). GSK-690, when tested in leukemia THP-1 and MV4-11 cells at 10 μM inhibited the relative clonogenic activities of almost 70% and 80%, respectively. Moreover, it demonstrated its LSD1 inhibition in cells by increasing the expression of CD86, a gene modulated by LSD1, in THP-1 cells (Mould et al., 2017).
Starting from the GSK-690 structure, numerous derivatives have been synthesized as novel LSD1 inhibitors (Wu et al., 2016). In 2015 the 3-(4-isocyanophenyl)-5-(piperidin-4-ylmethoxy)-2-(p-tolyl)pyridine 20 (Figure 9), in which a piperidine replaces the pyrrolidine ring, was shown to increase potency and selectivity over the prototype. Compound 20 displayed high antiproliferative effects in leukemia cell lines such as MV4−11 and Molm-13 (EC50 values = 0.36 and 3.4 μM, respectively) as well as in breast cancer cell lines such as MDA-MB-231 and MCF-7 (EC50 values = 5.6 and 3.6 μM, respectively). Against non-cancer WI-38 fibroblast cells, 20 exhibited less potency (EC50 = 26.6 μM), indicating appreciable cancer-selectivity. Finally, 20 gave accumulation of H3K4me2 in MV4-11 cells, as functional assay to demonstrate its LSD1 inhibition in a cellular context. Crystallographic studies (Niwa et al., 2018) performed on 20 complexed with LSD1/CoREST showed that the piperidine ring penetrated in the LSD1 active site and made interactions with the negatively charged residues Asn540 and Asp555. Moreover, the 4-cyanophenyl group was found deeply in the binding of the substrate pocket and made interactions with Lys661, while the 4-methylphenyl ring bound a hydrophobic cavity.
Several 1,2,3-triazolodithiocarbamates have been reported as novel LSD1 inhibitors (Zheng et al., 2013), with 21 (Figure 9) showing an IC50 of 2.1 μM in enzyme assay. In LSD1-overexpressing, low differentiated HGC-27 and MGC-803 human gastric cancer cell lines, 21 exhibited significant reduction of proliferation (IC50 values = 1.13 and 0.89 μM, respectively), without toxicity against GES-1 and SGC-7901 non-cancer gastric cell lines (IC50 values around 50 μM). In MGC-803 cells, 21 induced up to 44.7% apoptosis (1 μM) after 48 h, and inhibited migration and invasion at sub-toxic doses (0.25–0.02 μM). In MGC-803 xenograft mouse model, 21 at 20 mg/kg reduced the weight of tumor till 68.5%, with no changes in body weight.
Further new LSD1 inhibitors were developed in 2015: among them, the pyrimidine-thiosemicarbazide hybrid compound 22 (Figure 9) proved to be the most potent and selective (IC50LSD1 = 0.65 µM) (Ma et al., 2015). Furthermore, 22 was found to exert strong and selective cytotoxicity against MGC-803 and HGC-27 gastric cancer cells (IC50 = 4–8 μM). Compound 22 also showed inhibitory effects on cell invasion and migration, and tumor suppression and anti-metastatic effects in vivo without signs of evident toxicities.
Some indole derivatives (such as 23 and 24) were also described as reversible LSD1 inhibitors, potent at submicromolar/nanomolar levels. In blood as well as solid cancer cell lines, these compounds exhibited growth arrest at single-digit micromolar concentration (Wang et al., 2020) (Figure 9).
Some [1,2,3]triazolo [4,5-d]- and [1,2,3]triazolo [4,5-a]pyrimidines, variously substituted with arylthiol, arylamino, thiourea, thiosemicarbazide, and hydrazine moieties, displayed a non-covalent LSD1 inhibition at micromolar/submicromolar level, and inhibited proliferation of a panel of cancer cells at low micromolar doses (Li et al., 2017) (Li Z. et al., 2019) (Wang et al., 2017) (Wang et al., 2019) (Li et al., 2019c).
Starting from the thieno [3,2-b]pyrrole-5-carboxamide 25, selected through an HTS, an optimization process has been undertaken leading to 26, obtained by elongation of the methoxymethyl substituent at the meta position of the 25’s anilide portion to a 4-piperidinyloxy-4-phenoxymethyl chain. This change gave 18-fold increased potency against LSD1 (Sartori et al., 2017). Further structure-guided optimization furnished some thieno [3,2-b]pyrrole-5-carboxamides in which the long chain is moved from the meta to the ortho position of the anilide ring. Such derivatives showed single-digit nanomolar potency against LSD1 joined to high selectivity over MAOs, and single-digit micromolar (or lower) IC50 values against a panel of leukemia cells (MV4-11, THP-1, and NB4) (Vianello et al., 2017). The replacement of the carboxamide linkage with an imidazole (see compound 27 in Figure 9) led to picomolar LSD1 inhibition, together with nanomolar anti-clonogenic activity in THP-1 cells and in vivo good efficacy after oral administration in mouse leukemia models (Romussi et al., 2020).
The EGFR tyrosine kinase inhibitor and antineoplastic agent for the treatment of lung cancer (NSCLC) Osimertinib (28) (Figure 10) was shown to also inhibit LSD1 (IC50 = 3.98 μM). In the treatment of NSCLC, both LSD1 and EGFR are important drug targets. In NCI-H1975 NSCLC cells, Osimertinib reduced cell proliferation with IC50 = 3 μM and inhibited migration (Li et al., 2019b). In the same cell line, Osimertinib increased the methylation levels of H3K4me1/2 and abated the level of phospho-EGFR, showing its dual-targeting property in this cellular context. Docking studies revealed that Osimertinib occupies the LSD1 active pocket away from the FAD cofactor. The carbonyl group makes an hydrogen bond with Thr335, while the N,N-dimethyl group has a ionic interaction with Asp556 (Li et al., 2019b).
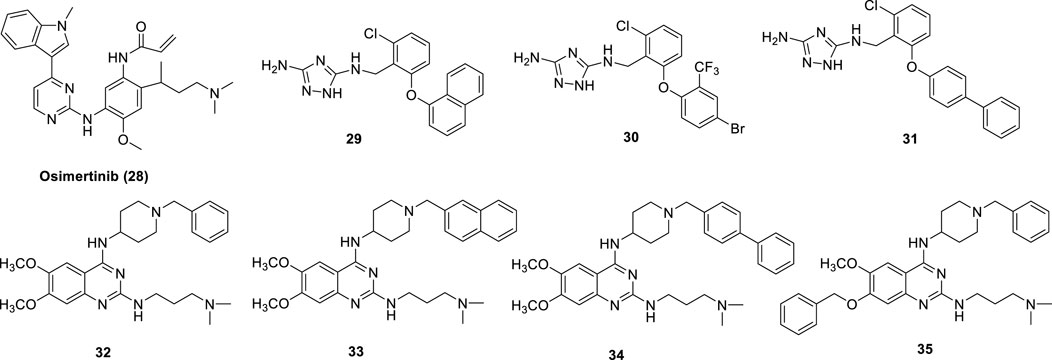
FIGURE 10. Reversible multi-targeting LSD1 inhibitors.
A new class of 3,5-diamino-1,2,4-triazoles have been reported as dual LSD1/spermine oxidase (SMOX) inhibitors (29-31) (Figure 10) (Holshouser et al., 2019). Really, these compounds showed up to 1000-fold difference between the inhibition values of the two targets, and sometimes they are more potent against MAOs than against LSD1.
In 2016, starting from a study on H3K9me1/2 methyltransferases inhibitors (Chang et al., 2010), our group identified MC3774 (32) (Figure 10) (Speranzini et al., 2016) as a compound able to inhibit LSD1 in a unprecedented way. Indeed, five copies of MC3774 stacked in a head-tail orientation at the entrance of the catalytic pocket of LSD1, not allowing the laying of the histone substrate (Speranzini et al., 2016). Furthermore, this complex extensively made interactions with negatively charged residues Glu559, Asp555, Asp557, Asp556, Asp553, and Glu387. MC3774 inhibited at the same time G9a and LSD1 (Ki values = 0.68 and 0.44 μM, respectively) and displayed micromolar/submicromolar anticancer activity against leukemia (THP-1 and MV4-11) and solid (breast MDA-MB-231 and rhabdomyosarcoma RD and RH30) cancer cells (Menna et al., 2022). Further chemical manipulation applied on the MC3774 structure furnished derivatives more potent against LSD1 (i.e., 33–35, Ki values = 0.16, 0.15, and 0.11, respectively) and less potent or totally inactive against G9a (Ki values = 2.9 (32), 1.2 (33), and 39.4 (34) μM) (Menna et al., 2022). Such derivatives revealed to be more potent than the parent compound against the above leukemia and solid cancer cells, thus suggesting a crucial role for LSD1 in the pathogenesis of these diseases (Figure 10).
Despite the huge work performed by researchers for discovery and identification of reversible LSD1 inhibitors (Dai et al., 2021), there are only few of them in clinical trials, i.e. Pulrodemstat (CC-90011) (Hollebecque et al., 2021) and Seclidemstat (SP-2577) (Soldi et al., 2020) (Figure 11).
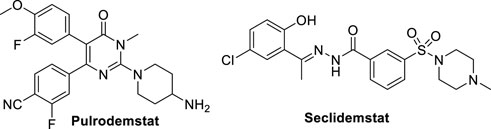
FIGURE 11. Reversible LSD1 inhibitors in clinical trials.
Pulrodemstat is the first orally active, reversible LSD1 inhibitor, developed by Celgene (Kanouni et al., 2020). Its structure, identified by a HTS, is similar to those of GSK-690 (19) and compound 20, in which the pyridine is replaced by a pyrimidinone ring, and the cyclic amine-methyloxy group by a 4-aminopiperidine. Pulrodemstat significantly inhibits LSD1 (IC50 = 0.30 nM) (Kanouni et al., 2020) and was effective in advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma, mainly in patients with neuroendocrine tumors. The pyrimidinone-based LSD1 inhibitor displayed growth arrest against cancer cells in vitro as well as in PDX models. Drug combinations of Pulrodemstat with Etoposide and Cisplatin on one side, and with Nivolumab on the other side, have been used against SCLC (NCT03850067) and advanced cancers (NCT04350463), respectively.
Seclidemstat, also known as SP-2577, is a potent reversible inhibitor of LSD1 (IC50 = 13 nM) with no action against MAOs (Sorna et al., 2013). Seclidemstat has received FDA fast track designation for lead drug candidate and has entered phase I clinical trial for the treatment of advanced solid tumors for patients with relapsed/refractory Ewing sarcoma. Reversible inhibitors could ameliorate some possible undesired effects that covalent inhibitors exert on erythropoiesis and establish a novel phenotype of LSD1 inhibition.
In Table 1 the various LSD1 inhibitors, both covalent and reversible, currently in clinical trials for cancer treatment are summarized. The most common causes of treatment failure in cancer patients are metastatic recurrence and therapy resistance.
In the last 10 years, LSD1 emerged as a potential therapeutic target particularly for the treatment of cancer. In AML and SCLC as well as in other cancer pathologies, elevated levels of LSD1 have been observed. Pharmacological inactivation of LSD1 with small molecule inhibitors showed suppression of cancer cell differentiation, proliferation, invasion, and migration. Numerous LSD1 inhibitors have been reported, with either irreversible or reversible mode of action. Most irreversible LSD1 inhibitors share the TCP structure as the pharmacophore group. They interact with the FAD cofactor to form a covalent bond, giving the irreversible LSD1 inhibition. To date, six TCP-based LSD1 irreversible inhibitors have entered clinical trials (TCP, GSK2879552, Bomedemstat (IMG-7289), Iadademstat (ORY-1001), INCB059872, and Vafidemstat (ORY-2001)). Although their potent and long-lasting effects, irreversible inhibitors have several side effects. For this reason, in the last years, many reversible LSD1 inhibitors have been studied and reported. Until now, only two of them have entered in clinical trials (Pulrodemstat (CC-90011) and Seclidemstat (SP-2577)).
Also, it is important and promising the molecular hybridization approach that combines different drug pharmacophoric moieties, in order to obtain novel dual compounds of two targets, or multitargeting compounds, able to improve potency and selectivity towards cancer cells, ameliorate pharmacokinetics and bioavailability, and possibly reduce adverse effects. Some promising dual-targeting LSD1 inhibitors have been summarized here, with better activity profiles respect to the corresponding single-target inhibitors and their combination.
Due to the important involvements of LSD1 in carcinogenesis, and to the numerous ways with which it interferes with various signaling pathways, targeting lysine demethylases and in particular LSD1 is becoming a promising treatment option for cancer patients.
BN and ED found the material; RF and AM checked and integrated the material; all the authors wrote the paper.
This work was supported by FISR 2019_00374 MeDyCa (AM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ambrosio, S., Sacca, C. D., Amente, S., Paladino, S., Lania, L., and Majello, B. (2017a). Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene 36, 6701–6711. doi:10.1038/onc.2017.267
Ambrosio, S., Sacca, C. D., and Majello, B. (2017b). Epigenetic regulation of epithelial to mesenchymal transition by the Lysine-specific demethylase LSD1/KDM1A. Biochim. Biophys. Acta Gene Regul. Mech. 1860, 905–910. doi:10.1016/j.bbagrm.2017.07.001
Anastas, J. N., Zee, B. M., Kalin, J. H., Kim, M., Guo, R., Alexandrescu, S., et al. (2019). Re-Programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell 36, 528–544. doi:10.1016/j.ccell.2019.09.005
Bailey, C. P., Figueroa, M., Gangadharan, A., Lee, D. A., and Chandra, J. (2020a). Scaffolding LSD1 inhibitors impair NK cell metabolism and cytotoxic function through depletion of glutathione. Front. Immunol. 11, 2196. doi:10.3389/fimmu.2020.02196
Bailey, C. P., Figueroa, M., Gangadharan, A., Yang, Y., Romero, M. M., Kennis, B. A., et al. (2020b). Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro Oncol. 22, 1302–1314. doi:10.1093/neuonc/noaa058
Baillie, T. A. (2021). Approaches to mitigate the risk of serious adverse reactions in covalent drug design. Expert Opin. Drug Discov. 16, 275–287. doi:10.1080/17460441.2021.1832079
Bauer, T. M., Besse, B., Martinez-Marti, A., Trigo, J. M., Moreno, V., Garrido, P., et al. (2019). Phase I, open-label, dose-escalation study of the safety, pharmacokinetics, pharmacodynamics, and efficacy of GSK2879552 in relapsed/refractory SCLC. J. Thorac. Oncol. 14, 1828–1838. doi:10.1016/j.jtho.2019.06.021
Benedetti, R., Dell'Aversana, C., De Marchi, T., Rotili, D., Liu, N. Q., Novakovic, B., et al. (2019). Inhibition of histone demethylases LSD1 and UTX regulates ERα signaling in breast cancer. Cancers (Basel) 11, 2027. doi:10.3390/cancers11122027
Benedetti, R., Papulino, C., Sgueglia, G., Chianese, U., De Marchi, T., Iovino, F., et al. (2021). Regulatory interplay between miR-181a-5p and estrogen receptor signaling cascade in breast cancer. Cancers (Basel) 13, 543. doi:10.3390/cancers13030543
Bennani-Baiti, I. M., Machado, I., Llombart-Bosch, A., and Kovar, H. (2012). Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing's sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum. Pathol. 43, 1300–1307. doi:10.1016/j.humpath.2011.10.010
Binda, C., Valente, S., Romanenghi, M., Pilotto, S., Cirilli, R., Karytinos, A., et al. (2010). Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 132, 6827–6833. doi:10.1021/ja101557k
Cavalcanti, F., Gonzalez-Rey, E., Delgado, M., Falo, C. P., Mestre, L., Guaza, C., et al. (2022). Efficacy of vafidemstat in experimental autoimmune encephalomyelitis highlights the kdm1a/RCOR1/HDAC epigenetic Axis in multiple sclerosis. Pharmaceutics 14, 1420. doi:10.3390/pharmaceutics14071420
Chang, Y., Ganesh, T., Horton, J. R., Spannhoff, A., Liu, J., Sun, A., et al. (2010). Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J. Mol. Biol. 400, 1–7. doi:10.1016/j.jmb.2010.04.048
Chen, Y., Jie, W., Yan, W., Zhou, K., and Xiao, Y. (2012). Lysine-specific histone demethylase 1 (LSD1): A potential molecular target for tumor therapy. Crit. Rev. Eukaryot. Gene Expr. 22, 53–59. doi:10.1615/critreveukargeneexpr.v22.i1.40
Chen, Y., Kim, J., Zhang, R., Yang, X., Zhang, Y., Fang, J., et al. (2016). Histone demethylase LSD1 promotes adipocyte differentiation through repressing wnt signaling. Cell Chem. Biol. 23, 1228–1240. doi:10.1016/j.chembiol.2016.08.010
Chen, Y., Yang, Y., Wang, F., Wan, K., Yamane, K., Zhang, Y., et al. (2006). Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proc. Natl. Acad. Sci. U. S. A. 103, 13956–13961. doi:10.1073/pnas.0606381103
Choi, H. J., Park, J. H., Park, M., Won, H. Y., Joo, H. S., Lee, C. H., et al. (2015). UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep. 16, 1288–1298. doi:10.15252/embr.201540244
Ciccone, D. N., Su, H., Hevi, S., Gay, F., Lei, H., Bajko, J., et al. (2009). KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461, 415–418. doi:10.1038/nature08315
Culhane, J. C., Wang, D., Yen, P. M., and Cole, P. A. (2010). Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J. Am. Chem. Soc. 132, 3164–3176. doi:10.1021/ja909996p
Dagogo-Jack, I., and Shaw, A. T. (2018). Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94. doi:10.1038/nrclinonc.2017.166
Dai, X. J., Liu, Y., Xiong, X. P., Xue, L. P., Zheng, Y. C., and Liu, H. M. (2020). Tranylcypromine based lysine-specific demethylase 1 inhibitor: Summary and perspective. J. Med. Chem. 63, 14197–14215. doi:10.1021/acs.jmedchem.0c00919
Dai, X. J., Liu, Y., Xue, L. P., Xiong, X. P., Zhou, Y., Zheng, Y. C., et al. (2021). Reversible lysine specific demethylase 1 (LSD1) inhibitors: A promising wrench to impair LSD1. J. Med. Chem. 64, 2466–2488. doi:10.1021/acs.jmedchem.0c02176
Etani, T., Naiki, T., Naiki-Ito, A., Suzuki, T., Iida, K., Nozaki, S., et al. (2019). NCL1, A highly selective lysine-specific demethylase 1 inhibitor, suppresses castration-resistant prostate cancer growth via regulation of apoptosis and autophagy. J. Clin. Med. 8, 442. doi:10.3390/jcm8040442
Fang, J., Ying, H., Mao, T., Fang, Y., Lu, Y., Wang, H., et al. (2017). Upregulation of CD11b and CD86 through LSD1 inhibition promotes myeloid differentiation and suppresses cell proliferation in human monocytic leukemia cells. Oncotarget 8, 85085–85101. doi:10.18632/oncotarget.18564
Fioravanti, R., Romanelli, A., Mautone, N., Di Bello, E., Rovere, A., Corinti, D., et al. (2020). Tranylcypromine-based LSD1 inhibitors: Structure-activity relationships, antiproliferative effects in leukemia, and gene target modulation. ChemMedChem 15, 643–658. doi:10.1002/cmdc.201900730
Gu, H., and Roizman, B. (2009). Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 83, 4376–4385. doi:10.1128/JVI.02515-08
Han, Y., Xu, S., Ye, W., Wang, Y., Zhang, X., Deng, J., et al. (2021). Targeting LSD1 suppresses stem cell-like properties and sensitizes head and neck squamous cell carcinoma to PD-1 blockade. Cell Death Dis. 12, 993. doi:10.1038/s41419-021-04297-0
Hayami, S., Kelly, J. D., Cho, H. S., Yoshimatsu, M., Unoki, M., Tsunoda, T., et al. (2011). Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 128, 574–586. doi:10.1002/ijc.25349
Hiatt, J. B., Sandborg, H., Garrison, S. M., Arnold, H. U., Liao, S. Y., Norton, J. P., et al. (2022). Inhibition of LSD1 with bomedemstat sensitizes small cell lung cancer to immune checkpoint blockade and T-cell killing. Clin. Cancer Res. 28, 4551–4564. doi:10.1158/1078-0432.CCR-22-1128
Hino, S., Sakamoto, A., Nagaoka, K., Anan, K., Wang, Y., Mimasu, S., et al. (2012). FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 3, 758. doi:10.1038/ncomms1755
Hirano, K., and Namihira, M. (2016). LSD1 mediates neuronal differentiation of human fetal neural stem cells by controlling the expression of a novel target gene, HEYL. Stem Cells 34, 1872–1882. doi:10.1002/stem.2362
Hitchin, J. R., Blagg, J., Burke, R., Burns, S., Cockerill, M. J., Fairweather, E. E., et al. (2013). Development and evaluation of selective, reversible LSD1 inhibitors derived from fragments. MedChemComm 4, 1513–1522. doi:10.1039/c3md00226h
Hojfeldt, J. W., Agger, K., and Helin, K. (2013). Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 12, 917–930. doi:10.1038/nrd4154
Hollebecque, A., Salvagni, S., Plummer, R., Isambert, N., Niccoli, P., Capdevila, J., et al. (2021). Phase I study of lysine-specific demethylase 1 inhibitor, CC-90011, in patients with advanced solid tumors and relapsed/refractory non-hodgkin lymphoma. Clin. Cancer Res. 27, 438–446. doi:10.1158/1078-0432.CCR-20-2380
Holshouser, S., Cafiero, R., Robinson, M., Kirkpatrick, J., Casero, R. A., Hyacinth, H. I., et al. (2020). Epigenetic reexpression of hemoglobin F using reversible LSD1 inhibitors: Potential therapies for sickle cell disease. ACS Omega 5, 14750–14758. doi:10.1021/acsomega.0c01585
Holshouser, S., Dunworth, M., Murray-Stewart, T., Peterson, Y. K., Burger, P., Kirkpatrick, J., et al. (2019). Dual inhibitors of LSD1 and spermine oxidase. Medchemcomm 10, 778–790. doi:10.1039/c8md00610e
Hsu, H. C., Liu, Y. S., Tseng, K. C., Yang, T. S., Yeh, C. Y., You, J. F., et al. (2015). CBB1003, a lysine-specific demethylase 1 inhibitor, suppresses colorectal cancer cells growth through down-regulation of leucine-rich repeat-containing G-protein-coupled receptor 5 expression. J. Cancer Res. Clin. Oncol. 141, 11–21. doi:10.1007/s00432-014-1782-4
Huang, J., Sengupta, R., Espejo, A. B., Lee, M. G., Dorsey, J. A., Richter, M., et al. (2007). p53 is regulated by the lysine demethylase LSD1. Nature 449, 105–108. doi:10.1038/nature06092
Ishikawa, Y., Gamo, K., Yabuki, M., Takagi, S., Toyoshima, K., Nakayama, K., et al. (2017a). A novel LSD1 inhibitor T-3775440 disrupts GFI1B-containing complex leading to transdifferentiation and impaired growth of AML cells. Mol. Cancer Ther. 16, 273–284. doi:10.1158/1535-7163.MCT-16-0471
Ishikawa, Y., Nakayama, K., Morimoto, M., Mizutani, A., Nakayama, A., Toyoshima, K., et al. (2017b). Synergistic anti-AML effects of the LSD1 inhibitor T-3775440 and the NEDD8-activating enzyme inhibitor pevonedistat via transdifferentiation and DNA rereplication. Oncogenesis 6, e377. doi:10.1038/oncsis.2017.76
Itoh, Y., Ogasawara, D., Ota, Y., Mizukami, T., and Suzuki, T. (2014). Synthesis, LSD1 inhibitory activity, and LSD1 binding model of optically pure lysine-PCPA conjugates. Comput. Struct. Biotechnol. J. 9, e201402002. doi:10.5936/csbj.201402002
Jin, Y., Ma, D., Gramyk, T., Guo, C., Fang, R., Ji, H., et al. (2019). Kdm1a promotes SCLC progression by transcriptionally silencing the tumor suppressor Rest. Biochem. Biophys. Res. Commun. 515, 214–221. doi:10.1016/j.bbrc.2019.05.118
Johnston, G., Ramsey, H. E., Liu, Q., Wang, J., Stengel, K. R., Sampathi, S., et al. (2020). Nascent transcript and single-cell RNA-seq analysis defines the mechanism of action of the LSD1 inhibitor INCB059872 in myeloid leukemia. Gene 752, 144758. doi:10.1016/j.gene.2020.144758
Kakizawa, T., Ota, Y., Itoh, Y., Tsumoto, H., and Suzuki, T. (2015). Histone H3 peptide based LSD1-selective inhibitors. Bioorg Med. Chem. Lett. 25, 1925–1928. doi:10.1016/j.bmcl.2015.03.030
Kalin, J. H., Wu, M., Gomez, A. V., Song, Y., Das, J., Hayward, D., et al. (2018). Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 9, 53. doi:10.1038/s41467-017-02242-4
Kanouni, T., Severin, C., Cho, R. W., Yuen, N. Y., Xu, J., Shi, L., et al. (2020). Discovery of CC-90011: A potent and selective reversible inhibitor of lysine specific demethylase 1 (LSD1). J. Med. Chem. 63, 14522–14529. doi:10.1021/acs.jmedchem.0c00978
Karakaidos, P., Verigos, J., and Magklara, A. (2019). LSD1/KDM1A, a gate-keeper of cancer stemness and a promising therapeutic target. Cancers (Basel) 11, 1821. doi:10.3390/cancers11121821
Kim, S. A., Zhu, J., Yennawar, N., Eek, P., and Tan, S. (2020). Crystal structure of the LSD1/CoREST histone demethylase bound to its nucleosome substrate. Mol. Cell 78, 903–914. doi:10.1016/j.molcel.2020.04.019
Kontaki, H., and Talianidis, I. (2010). Lysine methylation regulates E2F1-induced cell death. Mol. Cell 39, 152–160. doi:10.1016/j.molcel.2010.06.006
Kumarasinghe, I. R., and Woster, P. M. (2014). Synthesis and evaluation of novel cyclic Peptide inhibitors of lysine-specific demethylase 1. ACS Med. Chem. Lett. 5, 29–33. doi:10.1021/ml4002997
Lammert, C., Einarsson, S., Saha, C., Niklasson, A., Bjornsson, E., and Chalasani, N. (2008). Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: Search for signals. Hepatology 47, 2003–2009. doi:10.1002/hep.22272
Lan, F., Nottke, A. C., and Shi, Y. (2008). Mechanisms involved in the regulation of histone lysine demethylases. Curr. Opin. Cell Biol. 20, 316–325. doi:10.1016/j.ceb.2008.03.004
Lee, C., Rudneva, V. A., Erkek, S., Zapatka, M., Chau, L. Q., Tacheva-Grigorova, S. K., et al. (2019). Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nat. Commun. 10, 332. doi:10.1038/s41467-018-08269-5
Lee, J. Y., Park, J. H., Choi, H. J., Won, H. Y., Joo, H. S., Shin, D. H., et al. (2017). LSD1 demethylates HIF1α to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 36, 5512–5521. doi:10.1038/onc.2017.158
Lee, M. G., Wynder, C., Cooch, N., and Shiekhattar, R. (2005). An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437, 432–435. doi:10.1038/nature04021
Lee, M. G., Wynder, C., Schmidt, D. M., McCafferty, D. G., and Shiekhattar, R. (2006). Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 13, 563–567. doi:10.1016/j.chembiol.2006.05.004
Li, Z., Ding, L., Li, Z., Wang, Z., Suo, F., Shen, D., et al. (2019a). Development of the triazole-fused pyrimidine derivatives as highly potent and reversible inhibitors of histone lysine specific demethylase 1 (LSD1/KDM1A). Acta Pharm. Sin. B 9, 794–808. doi:10.1016/j.apsb.2019.01.001
Li, Z. H., Liu, X. Q., Geng, P. F., Suo, F. Z., Ma, J. L., Yu, B., et al. (2017). Discovery of [1,2,3]Triazolo[4,5-d]pyrimidine derivatives as novel LSD1 inhibitors. ACS Med. Chem. Lett. 8, 384–389. doi:10.1021/acsmedchemlett.6b00423
Li, Z. R., Suo, F. Z., Hu, B., Guo, Y. J., Fu, D. J., Yu, B., et al. (2019b). Identification of osimertinib (AZD9291) as a lysine specific demethylase 1 inhibitor. Bioorg Chem. 84, 164–169. doi:10.1016/j.bioorg.2018.11.018
Li, Z. R., Wang, S., Yang, L., Yuan, X. H., Suo, F. Z., Yu, B., et al. (2019c). Experience-based discovery (EBD) of aryl hydrazines as new scaffolds for the development of LSD1/KDM1A inhibitors. Eur. J. Med. Chem. 166, 432–444. doi:10.1016/j.ejmech.2019.01.075
Liang, Y., Vogel, J. L., Narayanan, A., Peng, H., and Kristie, T. M. (2009). Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 15, 1312–1317. doi:10.1038/nm.2051
Lokken, A. A., and Zeleznik-Le, N. J. (2012). Breaking the LSD1/kdm1a addiction: Therapeutic targeting of the epigenetic modifier in AML. Cancer Cell 21, 451–453. doi:10.1016/j.ccr.2012.03.027
Lu, Y., Liu, Y., Oeck, S., and Glazer, P. M. (2018). Hypoxia promotes resistance to EGFR inhibition in NSCLC cells via the histone demethylases, LSD1 and PLU-1. Mol. Cancer Res. 16, 1458–1469. doi:10.1158/1541-7786.MCR-17-0637
Ma, L. Y., Zheng, Y. C., Wang, S. Q., Wang, B., Wang, Z. R., Pang, L. P., et al. (2015). Design, synthesis, and structure-activity relationship of novel LSD1 inhibitors based on pyrimidine-thiourea hybrids as potent, orally active antitumor agents. J. Med. Chem. 58, 1705–1716. doi:10.1021/acs.jmedchem.5b00037
Maes, T., Mascaró, C., Sacilotto, N., Lufino, M. M., and Ciceri, F. (2019). Targeting KDM1A with iadademstat in combination with immunotherapy in an in vivo model of melanoma. J. Clin. Oncol. 37, e14248. doi:10.1200/jco.2019.37.15_suppl.e14248
McGrath, J. P., Williamson, K. E., Balasubramanian, S., Odate, S., Arora, S., Hatton, C., et al. (2016). Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 76, 1975–1988. doi:10.1158/0008-5472.CAN-15-2333
Mehndiratta, S., and Liou, J. P. (2020). Histone lysine specific demethylase 1 inhibitors. RSC Med. Chem. 11, 969–981. doi:10.1039/d0md00141d
Menna, M., Fiorentino, F., Marrocco, B., Lucidi, A., Tomassi, S., Cilli, D., et al. (2022). Novel non-covalent LSD1 inhibitors endowed with anticancer effects in leukemia and solid tumor cellular models. Eur. J. Med. Chem. 237, 114410. doi:10.1016/j.ejmech.2022.114410
Metzger, E., Wissmann, M., Yin, N., Muller, J. M., Schneider, R., Peters, A. H., et al. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439. doi:10.1038/nature04020
Mimasu, S., Sengoku, T., Fukuzawa, S., Umehara, T., and Yokoyama, S. (2008). Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 A. Biochem. Biophys. Res. Commun. 366, 15–22. doi:10.1016/j.bbrc.2007.11.066
Mimasu, S., Umezawa, N., Sato, S., Higuchi, T., Umehara, T., and Yokoyama, S. (2010). Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 49, 6494–6503. doi:10.1021/bi100299r
Mohammad, H. P., Smitheman, K. N., Kamat, C. D., Soong, D., Federowicz, K. E., Van Aller, G. S., et al. (2015). A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 28, 57–69. doi:10.1016/j.ccell.2015.06.002
Mould, D. P., Alli, C., Bremberg, U., Cartic, S., Jordan, A. M., Geitmann, M., et al. (2017). Development of (4-cyanophenyl)glycine derivatives as reversible inhibitors of lysine specific demethylase 1. J. Med. Chem. 60, 7984–7999. doi:10.1021/acs.jmedchem.7b00462
Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease: Old views and new perspectives. Int. J. Dev. Biol. 53, 1541–1547. doi:10.1387/ijdb.072410mn
Niwa, H., Sato, S., Hashimoto, T., Matsuno, K., and Umehara, T. (2018). Crystal structure of LSD1 in complex with 4-[5-(Piperidin-4-ylmethoxy)-2-(p-tolyl)pyridin-3-yl]benzonitrile. Molecules 23, 1538. doi:10.3390/molecules23071538
Ogasawara, D., Itoh, Y., Tsumoto, H., Kakizawa, T., Mino, K., Fukuhara, K., et al. (2013). Lysine-specific demethylase 1-selective inactivators: Protein-targeted drug delivery mechanism. Angew. Chem. Int. Ed. Engl. 52, 8620–8624. doi:10.1002/anie.201303999
Pilotto, S., Speranzini, V., Tortorici, M., Durand, D., Fish, A., Valente, S., et al. (2015). Interplay among nucleosomal DNA, histone tails, and corepressor CoREST underlies LSD1-mediated H3 demethylation. Proc. Natl. Acad. Sci. U. S. A. 112, 2752–2757. doi:10.1073/pnas.1419468112
Qin, Y., Vasilatos, S. N., Chen, L., Wu, H., Cao, Z., Fu, Y., et al. (2019). Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 38, 390–405. doi:10.1038/s41388-018-0451-5
Ravasio, R., Ceccacci, E., Nicosia, L., Hosseini, A., Rossi, P. L., Barozzi, I., et al. (2020). Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid-induced differentiation. Sci. Adv. 6, eaax2746. doi:10.1126/sciadv.aax2746
Rodriguez, V., Valente, S., Rovida, S., Rotili, D., Stazi, G., Lucidi, A., et al. (2015). Pyrrole- and indole-containing tranylcypromine derivatives as novel lysine-specific demethylase 1 inhibitors active on cancer cells. MedChemComm 6, 665–670. doi:10.1039/c4md00507d
Romussi, A., Cappa, A., Vianello, P., Brambillasca, S., Cera, M. R., Dal Zuffo, R., et al. (2020). Discovery of reversible inhibitors of KDM1A efficacious in acute myeloid leukemia models. ACS Med. Chem. Lett. 11, 754–759. doi:10.1021/acsmedchemlett.9b00604
Rotili, D., and Mai, A. (2011). Targeting histone demethylases: A new avenue for the fight against cancer. Genes Cancer 2, 663–679. doi:10.1177/1947601911417976
Rotili, D., Tomassi, S., Conte, M., Benedetti, R., Tortorici, M., Ciossani, G., et al. (2014). Pan-histone demethylase inhibitors simultaneously targeting Jumonji C and lysine-specific demethylases display high anticancer activities. J. Med. Chem. 57, 42–55. doi:10.1021/jm4012802
Rusconi, F., Grillo, B., Ponzoni, L., Bassani, S., Toffolo, E., Paganini, L., et al. (2016). LSD1 modulates stress-evoked transcription of immediate early genes and emotional behavior. Proc. Natl. Acad. Sci. U. S. A. 113, 3651–3656. doi:10.1073/pnas.1511974113
Sakane, N., Kwon, H. S., Pagans, S., Kaehlcke, K., Mizusawa, Y., Kamada, M., et al. (2011). Activation of HIV transcription by the viral Tat protein requires a demethylation step mediated by lysine-specific demethylase 1 (LSD1/KDM1). PLoS Pathog. 7, e1002184. doi:10.1371/journal.ppat.1002184
Salamero, O., Montesinos, P., Willekens, C., Perez-Simon, J. A., Pigneux, A., Recher, C., et al. (2020). First-in-Human phase I study of iadademstat (ORY-1001): A first-in-class lysine-specific histone demethylase 1A inhibitor, in relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 38, 4260–4273. doi:10.1200/JCO.19.03250
Sartori, L., Mercurio, C., Amigoni, F., Cappa, A., Faga, G., Fattori, R., et al. (2017). Thieno[3,2-b]pyrrole-5-carboxamides as new reversible inhibitors of histone lysine demethylase kdm1a/LSD1. Part 1: High-throughput screening and preliminary exploration. J. Med. Chem. 60, 1673–1692. doi:10.1021/acs.jmedchem.6b01018
Schenk, T., Chen, W. C., Gollner, S., Howell, L., Jin, L., Hebestreit, K., et al. (2012). Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611. doi:10.1038/nm.2661
Schmidt, D. M., and McCafferty, D. G. (2007). trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 46, 4408–4416. doi:10.1021/bi0618621
Schoenfeld, A. J., and Hellmann, M. D. (2020). Acquired resistance to immune checkpoint inhibitors. Cancer Cell 37, 443–455. doi:10.1016/j.ccell.2020.03.017
Schulz-Fincke, J., Hau, M., Barth, J., Robaa, D., Willmann, D., Kurner, A., et al. (2018). Structure-activity studies on N-Substituted tranylcypromine derivatives lead to selective inhibitors of lysine specific demethylase 1 (LSD1) and potent inducers of leukemic cell differentiation. Eur. J. Med. Chem. 144, 52–67. doi:10.1016/j.ejmech.2017.12.001
Sharma, P., Hu-Lieskovan, S., Wargo, J. A., and Ribas, A. (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. doi:10.1016/j.cell.2017.01.017
Sheng, W., LaFleur, M. W., Nguyen, T. H., Chen, S., Chakravarthy, A., Conway, J. R., et al. (2018). LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563. doi:10.1016/j.cell.2018.05.052
Shi, Y. J., Matson, C., Lan, F., Iwase, S., Baba, T., and Shi, Y. (2005). Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 19, 857–864. doi:10.1016/j.molcel.2005.08.027
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. doi:10.1016/j.cell.2004.12.012
Shi, Y., Sawada, J., Sui, G., Affar el, B., Whetstine, J. R., Lan, F., et al. (2003). Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738. doi:10.1038/nature01550
Sivanandhan, D., Rajagopal, S., Nair, S., Dhkar, R., Viswakarma, S., Siddiqui, A., et al. (2020). Abstract 1756: JBI-802, novel dual inhibitor of LSD1-HDAC6 for treatment of cancer. Cancer Res. 80, 1756. doi:10.1158/1538-7445.am2020-1756
Smitheman, K. N., Severson, T. M., Rajapurkar, S. R., McCabe, M. T., Karpinich, N., Foley, J., et al. (2019). Lysine specific demethylase 1 inactivation enhances differentiation and promotes cytotoxic response when combined with all-trans retinoic acid in acute myeloid leukemia across subtypes. Haematologica 104, 1156–1167. doi:10.3324/haematol.2018.199190
Sobczak, M., Strachowska, M., Gronkowska, K., and Robaszkiewicz, A. (2022). Activation of ABCC genes by Cisplatin depends on the CoREST occurrence at their promoters in A549 and MDA-MB-231 cell lines. Cancers (Basel) 14, 894. doi:10.3390/cancers14040894
Soldi, R., Ghosh Halder, T., Weston, A., Thode, T., Drenner, K., Lewis, R., et al. (2020). The novel reversible LSD1 inhibitor SP-2577 promotes anti-tumor immunity in SWItch/Sucrose-NonFermentable (SWI/SNF) complex mutated ovarian cancer. PLoS One 15, e0235705. doi:10.1371/journal.pone.0235705
Song, Y., Yang, X., and Yu, B. (2022). Repurposing antidepressants for anticancer drug discovery. Drug Discov. Today 27, 1924–1935. doi:10.1016/j.drudis.2021.10.019
Sorna, V., Theisen, E. R., Stephens, B., Warner, S. L., Bearss, D. J., Vankayalapati, H., et al. (2013). High-throughput virtual screening identifies novel N'-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 56, 9496–9508. doi:10.1021/jm400870h
Speranzini, V., Rotili, D., Ciossani, G., Pilotto, S., Marrocco, B., Forgione, M., et al. (2016). Polymyxins and quinazolines are LSD1/KDM1A inhibitors with unusual structural features. Sci. Adv. 2, e1601017. doi:10.1126/sciadv.1601017
Teresa Borrello, M., Benelkebir, H., Lee, A., Hin Tam, C., Shafat, M., Rushworth, S. A., et al. (2021). Synthesis of carboxamide-containing tranylcypromine analogues as LSD1 (KDM1A) inhibitors targeting acute myeloid leukemia. ChemMedChem 16, 1316–1324. doi:10.1002/cmdc.202000754
Tortorici, M., Borrello, M. T., Tardugno, M., Chiarelli, L. R., Pilotto, S., Ciossani, G., et al. (2013). Protein recognition by short peptide reversible inhibitors of the chromatin-modifying LSD1/CoREST lysine demethylase. ACS Chem. Biol. 8, 1677–1682. doi:10.1021/cb4001926
Ueda, R., Suzuki, T., Mino, K., Tsumoto, H., Nakagawa, H., Hasegawa, M., et al. (2009). Identification of cell-active lysine specific demethylase 1-selective inhibitors. J. Am. Chem. Soc. 131, 17536–17537. doi:10.1021/ja907055q
Vianello, P., Botrugno, O. A., Cappa, A., Dal Zuffo, R., Dessanti, P., Mai, A., et al. (2016). Discovery of a novel inhibitor of histone lysine-specific demethylase 1A (KDM1A/LSD1) as orally active antitumor agent. J. Med. Chem. 59, 1501–1517. doi:10.1021/acs.jmedchem.5b01209
Vianello, P., Sartori, L., Amigoni, F., Cappa, A., Faga, G., Fattori, R., et al. (2017). Thieno[3,2-b]pyrrole-5-carboxamides as new reversible inhibitors of histone lysine demethylase kdm1a/LSD1. Part 2: Structure-based drug design and structure-activity relationship. J. Med. Chem. 60, 1693–1715. doi:10.1021/acs.jmedchem.6b01019
Wang, J., Hevi, S., Kurash, J. K., Lei, H., Gay, F., Bajko, J., et al. (2009). The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 41, 125–129. doi:10.1038/ng.268
Wang, J., Lu, F., Ren, Q., Sun, H., Xu, Z., Lan, R., et al. (2011). Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 71, 7238–7249. doi:10.1158/0008-5472.CAN-11-0896
Wang, M., Liu, X., Guo, J., Weng, X., Jiang, G., Wang, Z., et al. (2015). Inhibition of LSD1 by Pargyline inhibited process of EMT and delayed progression of prostate cancer in vivo. Biochem. Biophys. Res. Commun. 467, 310–315. doi:10.1016/j.bbrc.2015.09.164
Wang, S., Li, Z. R., Suo, F. Z., Yuan, X. H., Yu, B., and Liu, H. M. (2019). Synthesis, structure-activity relationship studies and biological characterization of new [1,2,4]triazolo[1,5-a]pyrimidine-based LSD1/KDM1A inhibitors. Eur. J. Med. Chem. 167, 388–401. doi:10.1016/j.ejmech.2019.02.039
Wang, S., Zhao, L. J., Zheng, Y. C., Shen, D. D., Miao, E. F., Qiao, X. P., et al. (2017). Design, synthesis and biological evaluation of [1,2,4]triazolo[1,5-a]pyrimidines as potent lysine specific demethylase 1 (LSD1/KDM1A) inhibitors. Eur. J. Med. Chem. 125, 940–951. doi:10.1016/j.ejmech.2016.10.021
Wang, X., Zhang, C., Zhang, X., Yan, J., Wang, J., Jiang, Q., et al. (2020). Design, synthesis and biological evaluation of tetrahydroquinoline-based reversible LSD1 inhibitors. Eur. J. Med. Chem. 194, 112243. doi:10.1016/j.ejmech.2020.112243
Wass, M., Gollner, S., Besenbeck, B., Schlenk, R. F., Mundmann, P., Gothert, J. R., et al. (2021). A proof of concept phase I/II pilot trial of LSD1 inhibition by tranylcypromine combined with ATRA in refractory/relapsed AML patients not eligible for intensive therapy. Leukemia 35, 701–711. doi:10.1038/s41375-020-0892-z
Whyte, W. A., Bilodeau, S., Orlando, D. A., Hoke, H. A., Frampton, G. M., Foster, C. T., et al. (2012). Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225. doi:10.1038/nature10805
Wu, F., Zhou, C., Yao, Y., Wei, L., Feng, Z., Deng, L., et al. (2016). 3-(Piperidin-4-ylmethoxy)pyridine containing compounds are potent inhibitors of lysine specific demethylase 1. J. Med. Chem. 59, 253–263. doi:10.1021/acs.jmedchem.5b01361
Yang, G. J., Lei, P. M., Wong, S. Y., Ma, D. L., and Leung, C. H. (2018). Pharmacological inhibition of LSD1 for cancer treatment. Molecules 23, 3194. doi:10.3390/molecules23123194
Yang, J., Huang, J., Dasgupta, M., Sears, N., Miyagi, M., Wang, B., et al. (2010). Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. U. S. A. 107, 21499–21504. doi:10.1073/pnas.1016147107
Yang, M., Culhane, J. C., Szewczuk, L. M., Jalili, P., Ball, H. L., Machius, M., et al. (2007). Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 46, 8058–8065. doi:10.1021/bi700664y
Zhang, S., Liu, M., Yao, Y., Yu, B., and Liu, H. (2021). Targeting LSD1 for acute myeloid leukemia (AML) treatment. Pharmacol. Res. 164, 105335. doi:10.1016/j.phrs.2020.105335
Zheng, Y. C., Duan, Y. C., Ma, J. L., Xu, R. M., Zi, X., Lv, W. L., et al. (2013). Triazole-dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration. J. Med. Chem. 56, 8543–8560. doi:10.1021/jm401002r
Zheng, Y. C., Ma, J., Wang, Z., Li, J., Jiang, B., Zhou, W., et al. (2015). A systematic review of histone lysine-specific demethylase 1 and its inhibitors. Med. Res. Rev. 35, 1032–1071. doi:10.1002/med.21350
Zheng, Y. C., Yu, B., Jiang, G. Z., Feng, X. J., He, P. X., Chu, X. Y., et al. (2016). Irreversible LSD1 inhibitors: Application of tranylcypromine and its derivatives in cancer treatment. Curr. Top. Med. Chem. 16, 2179–2188. doi:10.2174/1568026616666160216154042
Zhou, G., Te, D., and Roizman, B. (2010). The CoREST/REST repressor is both necessary and inimical for expression of herpes simplex virus genes. mBio 2, e00313–e00310. doi:10.1128/mBio.00313-10
Keywords: histone demethylases, epigenetics, LSD1 inhibitors, dual-targeting compounds, cancer therapy
Citation: Noce B, Di Bello E, Fioravanti R and Mai A (2023) LSD1 inhibitors for cancer treatment: Focus on multi-target agents and compounds in clinical trials. Front. Pharmacol. 14:1120911. doi: 10.3389/fphar.2023.1120911
Received: 10 December 2022; Accepted: 20 January 2023;
Published: 02 February 2023.
Edited by:
Bin Yu, Zhengzhou University, ChinaReviewed by:
Patrick M. Woster, Medical University of South Carolina, United StatesCopyright © 2023 Noce, Di Bello, Fioravanti and Mai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rossella Fioravanti, cm9zc2VsbGEuZmlvcmF2YW50aUB1bmlyb21hMS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.