
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 02 February 2023
Sec. Pharmacogenetics and Pharmacogenomics
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1110460
This article is part of the Research Topic Insights in Pharmacogenetics and Pharmacogenomics: 2022 View all 4 articles
 Sofía Calleja1
Sofía Calleja1 Pablo Zubiaur1,2*Dolores Ochoa1
Pablo Zubiaur1,2*Dolores Ochoa1 Gonzalo Villapalos-García1
Gonzalo Villapalos-García1 Gina Mejia-Abril1
Gina Mejia-Abril1 Paula Soria-Chacartegui1
Paula Soria-Chacartegui1 Marcos Navares-Gómez1Alejandro de Miguel1Manuel Román1Samuel Martín-Vílchez1
Marcos Navares-Gómez1Alejandro de Miguel1Manuel Román1Samuel Martín-Vílchez1 Francisco Abad-Santos1,3*
Francisco Abad-Santos1,3*Venlafaxine pharmacokinetic variability and pharmacotherapy outcomes are well known to be related to CYP2D6 pharmacogenetic phenotype. In contrast, scarce pharmacogenetic information is available nowadays concerning desvenlafaxine, its active metabolite first marketed in 2012. The aim of this study was to evaluate the impact of 29 alleles in 12 candidate genes (e.g., CYP enzymes like CYP2D6, CYP3A4, or CYP2C19; ABC transporters like ABCB1; SLCO1B1; and UGT enzymes like UGT1A1) on desvenlafaxine pharmacokinetic variability and tolerability. Pharmacokinetic parameters and adverse drug reaction (ADR) incidence obtained from six bioequivalence clinical trials (n = 98) evaluating desvenlafaxine formulations (five with single dose administration and one with multiple-dose administration) were analyzed. No genetic polymorphism was related to pharmacokinetic variability or ADR incidence. Volunteers enrolled in the multiple-dose clinical trial also showed a higher incidence of ADRs, e.g., xerostomia or appetite disorders. Volunteers experiencing any ADR showed a significantly higher area under the time-concentration curve (AUC) than those not experiencing any ADR (5115.35 vs. 4279.04 ng*h/mL, respectively, p = 0.034). In conclusion, the strong dose-dependent relationship with the occurrence of ADRs confirms that the mechanism of action of desvenlafaxine is essentially dose-dependent.
Venlafaxine belongs to the group serotonin-noradrenaline re-uptake inhibitor (SNRI) antidepressant drugs. It was first marketed in 1995 and is widely used in the management of depression. Venlafaxine is metabolized by CYP2D6 resulting in its active metabolite desmethylvenlafaxine (i.e. desvenlafaxine), which was marketed in 2012 with the aim of reducing venlafaxine’s side effects (Food and Drug Administration, 2011). Desvenlafaxine daily dose ranges between 50 and 100 mg. All the pharmaceutical forms marketed are prolonged-release tablets. After oral administration, desvenlafaxine is highly absorbed, with a bioavailability of 80%. Maximum plasma concentrations (Cmax) are observed 7.5 h after oral administration (tmax) (Liebowitz and Tourian, 2010). The area under the time-concentration curve (AUC) in a 24 h dosing interval at steady state (AUCτ) with the 100 mg dose is 6747 ng*h/mL, and the Cmax is 376 ng/mL. The binding of desvenlafaxine to plasma proteins is low (30%) and the mean terminal half-life (t1/2), is approximately 11 h (Spanish Drug Agency, 2012). It is mostly metabolized by O-glucuronide conjugation by UDP-glucuronyltransferase (UGT) enzymes, including UGT1A1, UGT1A3, UGT2B4, UGT2B15, and UGT2B17. To a lesser extent, it undergoes oxidative metabolism by the cytochrome p450 (CYP) isoform CYP3A4 (Sproule et al., 2008; Preskorn et al., 2009; DrugBank, 2010). Venlafaxine structure contains a tertiary amine and a methoxy group, which can be N- and O-demethylated, respectively. The N-demethylation to N-desmethyl venlafaxine (NDV) is catalyzed by CYP3A4 and CYP2C19, while the O-demethylation to desvenlafaxine is catalyzed by CYP2D6 and CYP2C19 (Fogelman, 1999). When desvenlafaxine is administered instead of venlafaxine, the N-demethylation to N, O-didesmethyl venlafaxine is assumed to be mediated by means of CYP3A4 and CYP2C19 (pathway available at: https://www.pharmgkb.org/pathway/PA166014758).
CYP2D6 polymorphism conditions venlafaxine exposure and response. In fact, the Royal Dutch Pharmacist’s Association (KNMP) Pharmacogenomics Working Group (DPWG) includes the CYP2D6-venlafaxine drug-gene pair in their recommendations list. Briefly, an alternative drug that is not metabolized by CYP2D6 or a dose reduction should be considered for CYP2D6 intermediate metabolizers (IMs) and poor metabolizers (PMs) and a 150% dose increased should be considered for ultrarapid metabolizers (UMs) if necessary (Nichols et al., 2009; Nichols et al., 2013; Dutch Pharmacogenetics Working Group, 2022). In contrast, desvenlafaxine is apparently not affected by CYP2D6 phenotype and scarce pharmacogenetic information is available nowadays for this drug. One study evaluated the impact of ABCB1 rs1045642, ABCC1 rs212090, and UGT1A1 rs8175347 on desvenlafaxine dose requirements (Bousman et al., 2017). However, several additional pharmacogenetic biomarkers could be relevant concerning desvenlafaxine pharmacokinetics and/or dose requirements. Hence, the aim of this study was to evaluate the impact of 29 alleles in 12 candidate genes (i.e., ABCB1, ABCG2, CYP2B6, CYP2C19, CYP2C8, CYP2C9, CYP2D6, CYP3A4, CYP3A5, SLCO1B1, UGT1A1, and UGT2B15) on desvenlafaxine pharmacokinetic variability and tolerability.
The study population comprised 98 healthy volunteers enrolled in six different desvenlafaxine bioequivalence clinical trials performed at the Clinical Trial Unit of Hospital Universitario de La Princesa (UECHUP). All subjects included in the study signed two written informed consent forms, one for their participation in the clinical trial and another one for pharmacogenetic studies. They were free to withdraw from the study at any time. Study protocols were revised and approved by the Hospital’s Research Ethics Committee and by the Spanish Drugs Agency (AEMPS). Complying with Spanish and European legislation on research in humans, all of them were accomplished under the Good Clinical Practice guidelines and endorsing the Declaration of Helsinki. EudraCT numbers for clinical trials A to F were as follows: 2019-000628-17, 2019-002739-26, 2019-003272-39, 2019-004289-16, 2019-004882-41, and 2020-003002-31, respectively.
Inclusion criteria were as follows: age 18-55 years, being free from any psychiatric or organic conditions, having normal vital signs and electrocardiogram (ECG), normal medical records and physical examination and no clinically significant abnormalities in serology, hematology, coagulation, biochemistry, and urinalysis. Exclusion criteria comprised the following: presenting clinically significant biochemical alterations, having received pharmacological treatment in the last 15 days or any kind of medication in the 48 h prior to receiving the study drug, a body mass index (BMI) outside the 18.5–30 kg/m2 range, having donated blood in the previous month, history of sensitivity to any drug, suspected consumption of controlled substances, smokers, daily consumers of alcohol and/or acute alcohol poisoning in the last week, pregnant or breastfeeding women, having participated in a similar study within the previous 3 months and swallowing difficulty.
The current observational pharmacogenetic study was based on six independent phase I bioequivalence clinical trials (A, B, C, D, E, F). In three of them (A, E, F), two desvenlafaxine 100 mg prolonged release tablets formulations (one test formulation, T and one reference formulation, R) were administered once (single-dose); in two of them (B, D) two desvenlafaxine 50 mg prolonged release tablets formulations (T and R) were administered once (single-dose); in one of them (C), two desvenlafaxine 100 mg prolonged release tablets formulations (T and R) were administered several times (100 mg daily for 6 days), and the calculations to demonstrate bioequivalence were performed in steady-state. All of them were open-label, crossover, and randomized clinical trials, two sequence, two periods, with a wash-out period of at least 7 days (except for C clinical trial, without washout period between each period). The volunteers were hospitalized from 10 h before to 24 h after dosing in both periods. Formulations were administered by oral route under fasting conditions (A, B, C, E) or fed conditions (D, F) with 240 mL of water. Only the reference formulations (Pristiq® 50 or 100 mg prolonged release tablets, Pfizer, Spain) for each volunteer in each clinical trial were considered for this study.
Blood samples were collected in 3 mL EDTA-K2 tubes at several-points between predose and 72 h after dosing (A, B, D, E, F) or 24 h after dosing (C) (i.e., during an administration interval, τ). After the extraction (direct venipuncture or from an indwelling cannula), the tube was put into an ice-water bath. The tubes were centrifuged at 4°C during 10 min at 1900 G. Once centrifuged, 0.5 mL of plasma was aliquoted and stored in a freezer at −20°C ± 5°C, until its shipment to an external analytical laboratory. The analytical method involved a protein precipitation extraction procedure with 0.1% formic acid in methanol, and subsequent reversed phase high performance liquid chromatography coupled to a tandem mass spectrometry (HPLC-MS/MS), with a lower limit of quantification (LLOQ) of 1 ng/mL.
Pharmacokinetic parameters were estimated by non-compartmental methods using WinNonLin Professional Edition (version 8.1 for A, B, D, and E and version 8.3 for C and F, Scientific Consulting, Inc., Cary United States) pharmacokinetic program. Two pharmacokinetic parameters were obtained directly from raw data: tmax and Cmax (Cmax-ss in steady state for study C). The rest of the parameters are obtained by the trapezoidal rule or by non-compartmental methods. The AUC from zero to the last observed concentration time (t) (AUCt) and AUCƬ were calculated using the trapezoidal rule. The AUC from t time to infinity (AUCt→inf) was calculated dividing the concentration at t time (Ct) by the elimination constant (Ke), obtained by linear regression of the log-linear phase of the plasma level curve. The AUC from administration to infinity (AUC∞) was obtained adding AUCt to AUCt→inf. The t1/2 we calculated as -ln2/ke.
The safety assessment consisted of the evaluation of abnormalities in analytical values, blood (biochemistry, coagulation, haematology, serology) and urine parameters (urianalysis, pregnancy test, abuse drugs, and cotinine test), physical examination, vital signs (blood pressure and heart rate), serial ECG or any other clinically relevant event. Adverse events (AEs) spontaneously reported by volunteers or reported after open question were also collected. Causality was determined using the Spanish Pharmacovigilance System algorithm (Aguirre and García, 2016), according to five types of AE: definite, probable, possible, unlikely and unrelated. Only definite, probable, or possible AEs were considered as adverse drug reactions (ADRs) and included in the statistical analysis.
One EDTA-K2 tube used for pharmacokinetic profiling was reused for genotyping. After plasma separation, the cell concentrate was resuspended in NaCl 0.9% for the preservation of the blood. A Maxwell® RSC Instrument (Promega, United States) was used for the extraction of DNA. For the genotyping, a QuantStudio 12 k flex (ThermoFisher, United States) was used with two thermal blocks: the OpenArray for the genotyping of 24 out of 27 variants and the 96-well fast thermal-block for CNV determination and the genotyping of the remaining 3 (Zubiaur et al., 2020). Therefore, 27 variants in 12 genes were included in the present work, which allowed de inference of 29 alleles (see Table 1). Genes were selected based on their known involvement in desvenlafaxine pharmacokinetics (e.g., CYP2C19, ABCB1, or CYP3A4) or for their potential involvement with a more exploratory purpose (e.g., other relevant CYPs like CYP2D6 or CYP3A isoforms like CYP3A5, other transporters like ABCG2 and SLCO1B1 or other CYP2C enzymes like CYP2C19 or CYP2C9). Variants in potential candidate genes like UGT1A3 or UGT2B4 were ruled out as no variant is demonstrated to impact the enzyme’s function to date. A copy number variation (CNV) assay, which allows the identification of the gene deletion (*5) or gene duplications (xN), was performed for CYP2D6 as described previously (Belmonte et al., 2018). The following SNPs were manually genotyped with TaqMan assays following the manufacturer (ThermoFisher) recommendations: ABCG2 rs2231142, CYP3A4 rs67666821, and UGT2B15 rs1902023 (Table 1).
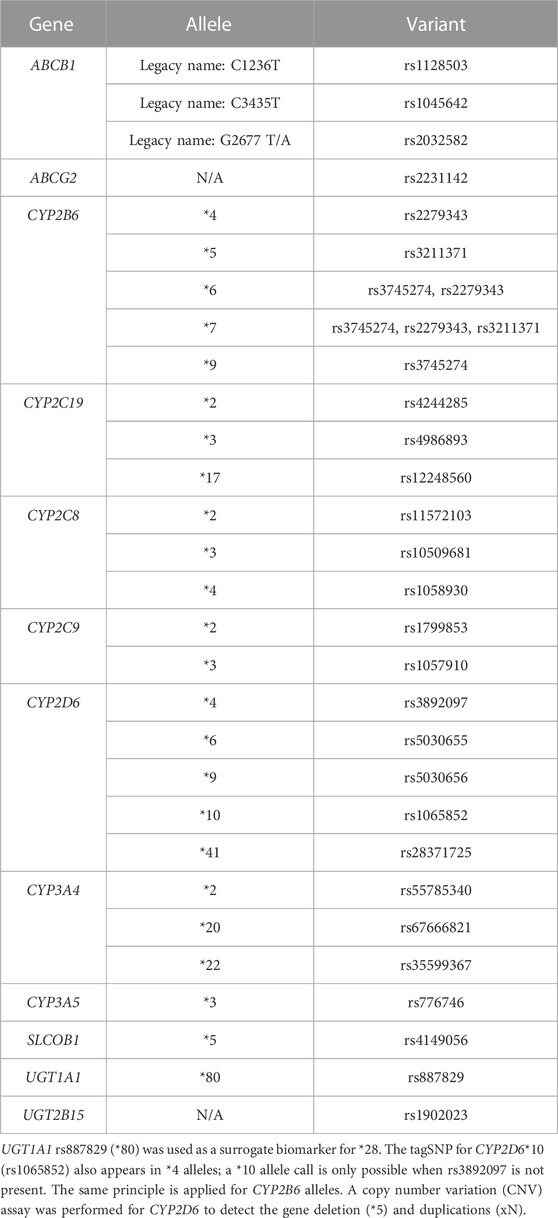
TABLE 1. Genes, alleles, and variants genotyped and analyzed.
Clinical Pharmacogenetics Implementation (CPIC) or DPWG pharmacogenetic guidelines were used to infer pharmacogenetic phenotypes when available: CYP2B6 (Desta et al., 2019), CYP2C19 (Hicks et al., 2015; Lee et al., 2022), CYP2C9 (Theken et al., 2020; Cooper-DeHoff et al., 2022), CYP2D6 (Hicks et al., 2015; Brown et al., 2019), CYP3A4 (Dutch Pharmacogenetics Working Group, 2021a), CYP3A5 (Birdwell et al., 2015), SLCOB1 (Cooper-DeHoff et al., 2022), UGT1A1 (Gammal et al., 2016; Dutch Pharmacogenetics Working Group, 2021b), ABCG2 (Cooper-DeHoff et al., 2022). ABCB1, CYP2C8, and UGT2B15 variants were individually analyzed as the pharmacogenetic phenotype has not been optimized to date.
The SPSS software was used for statistical analysis (version 26.0, SPSS Inc., Chicago, United States). Firstly, a univariate analysis was performed. For the comparison of means, a t-test (variables with two categories) or an ANOVA test followed by a Bonferroni post hoc (variables with three or more categories) was accomplished. For the comparison of the incidence of ADRs according to categorical variables, a Chi-squared or a Fisher exact test were used to infer statistical significance in contingence tables. Afterwards, each pharmacokinetic parameter or ADR were individually analyzed with a multivariate analysis by means of linear regression (pharmacokinetic parameters) or logistic regression (ADRs). Categorical variables with more than two categories, such as polymorphisms, were transformed to dummy variables. Moreover, any phenotype or genotype with p < 0.05 in the univariate analysis as well as race and sex (covariates) were included. Pharmacokinetic variables were logarithmically transformed for statistical inference to ensure a normal distribution. AUC and Cmax were divided by the dose/weight (DW) ratio to correct the impact of dose and weight on drug exposure. For the evaluation of ADRs, pharmacokinetic parameters without DW correction were included as independent variables in the logistic regression. The Bonferroni correction for multiple comparisons was applied.
A total of 98 healthy volunteers participated in the pharmacogenetic study. Mean age was 31.0 ± 8.9 years old, with evenly distributed proportions of males (n = 52) and females (n = 46) with 26 Caucasians and 72 healthy volunteers with Mixed race (including 71 Latin-Americans and one Black) (Table 2). Females exhibited lower height (p < 0.001) than males and lower weight (p < 0.001). In addition, the body mass index (BMI) of Caucasians was significantly lower than that of Mixed-race healthy volunteers (p = 0.010) (Table 2).
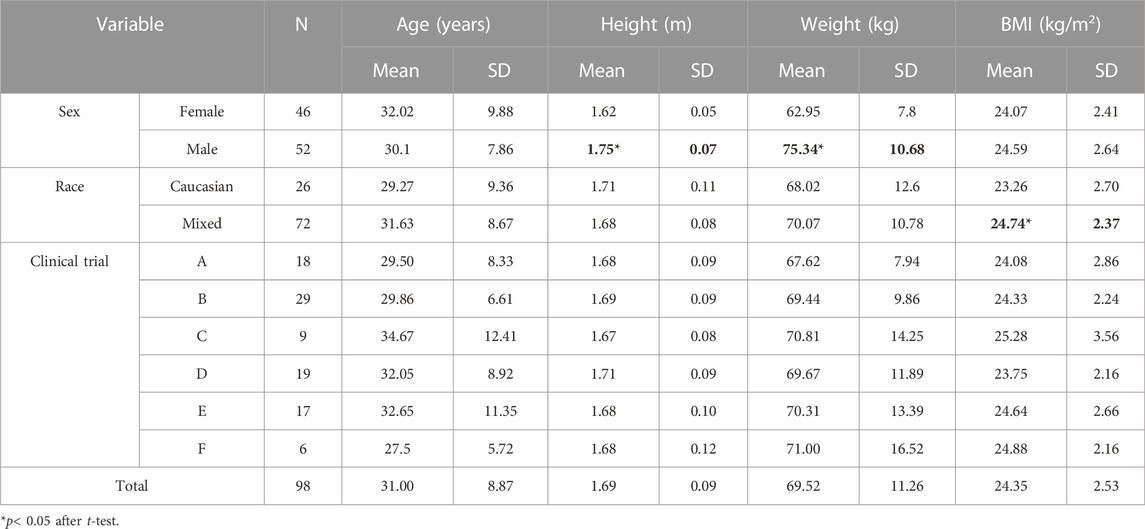
TABLE 2. Demographic characteristics of the study population according to sex, race, and clinical trial.
The total AUC∞ and Cmax were 4594.79 ± 1871.71 ng*h/mL and 187.19 ± 85.62 ng/mL respectively. Uncorrected mean (SD) AUC∞ and Cmax values are shown in Table 3 according to the clinical trial design.
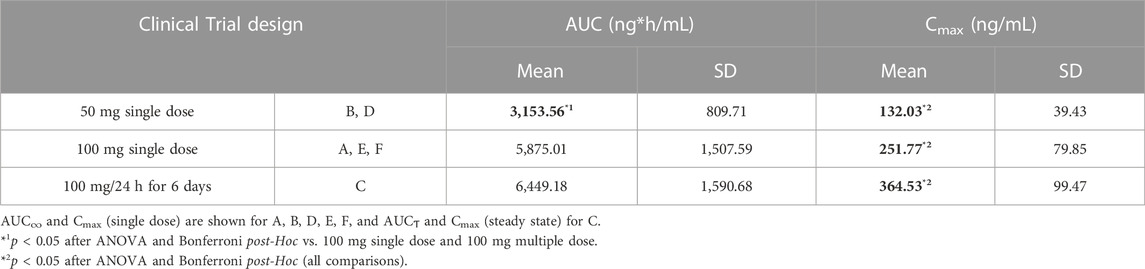
TABLE 3. Uncorrected AUC and Cmax values according to the study design.
Females showed higher Cmax/DW than males (p = 0.040). Caucasians showed a higher tmax than the Mixed-race healthy volunteers (p = 0.007) (Table 4). Several differences in pharmacokinetic parameters were observed according to the clinical trial design, with clinical trial C (the only one with multiple-dose drug administration) showing the highest Cmax/DW and t1/2 and lowest tmax. Finally, a higher Cmax/DW was observed in fed healthy volunteers compared to fasting (Table 4).
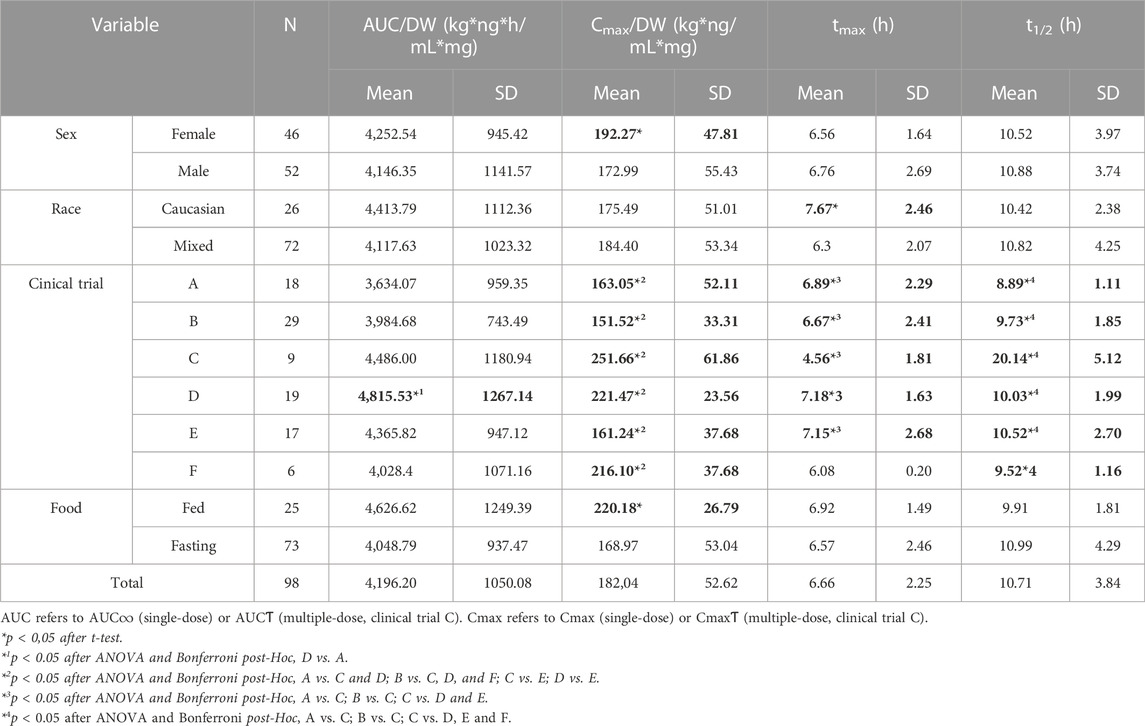
TABLE 4. Pharmacokinetic parameters according to sex, race, clinical trial, and food administration conditions.
CYP2C19 RMs showed a higher Cmax/DW compared to NMs (p = 0.021) and UGT1A1 PMs showed a higher tmax compared to IMs + NMs (p = 0.034) (Table 5). However, none of these associations remained significant after Bonferroni correction for multiple comparisons. No other association was observed, including CYP2D6. The mean and SD of pharmacokinetic parameters according to the remaining genotypes or phenotypes is shown in the Supplementary Table S1. Due to the scarcity of associations, a multivariate analysis was not possible.
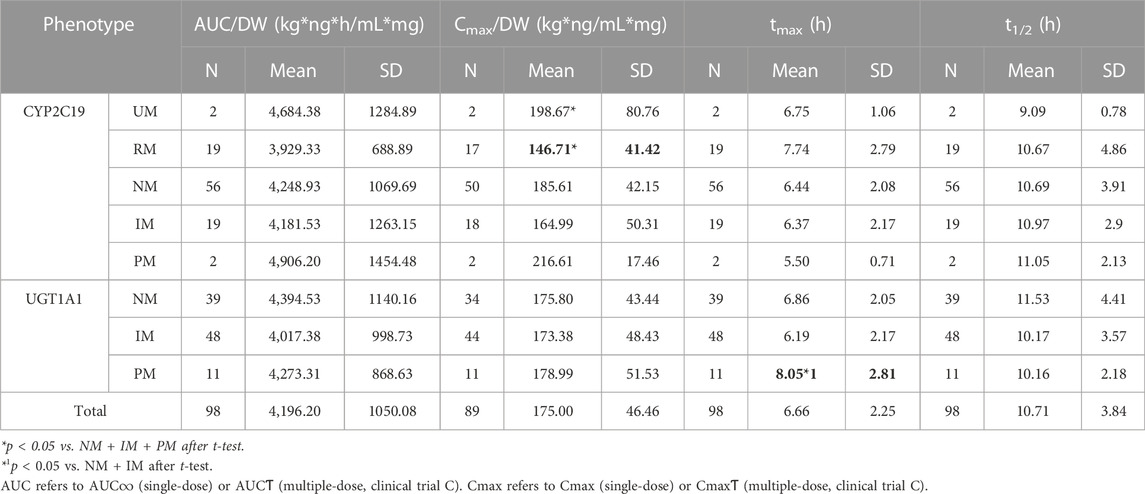
TABLE 5. Significant associations between CYP2C19 and UGT1A1 phenotypes and the pharmacokinetic parameters of desvenlafaxine.
Concerning drug safety, the incidence of any ADRs was significantly higher in the only clinical trial with multiple-dose administration (clinical trial C) (p = 0.022) (Table 6). The incidence of the following individual ADRs was consistently higher in clinical trial C: xerostomia and taste disorders, dizziness, somnolence, other sleeping disorders and decreased appetite (Table 6). Volunteers experiencing any ADR showed a significantly higher AUC∞ than those not experiencing any ADR (5115.35 ± 1931.97 h*ng/mL vs. 4279.04 ± 1776.43 h*ng/mL respectively, p = 0.034), but similar Cmax. Furthermore, the incidence of asthenia was 20% in individuals with ABCB1 rs203258 G/T genotype, 8.3% in those with A/A and 0% for those with G/G, G/A, T/T o A/T genotypes (p = 0.013). CYP2B6 RMs showed a higher incidence of asthenia (13% compared to 0% in NMs, IMs and PMs, p = 0.038). However, none of these associations remained significant after Bonferroni correction for multiple comparisons, therefore it was not possible to carry out the multivariate analysis.
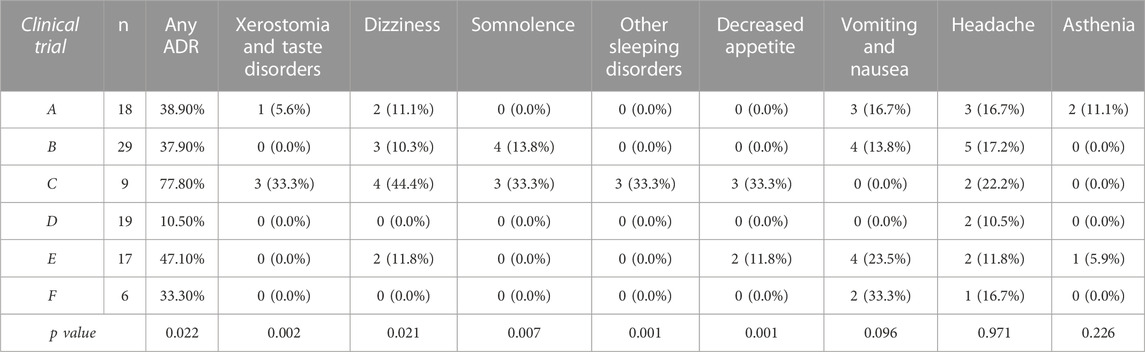
TABLE 6. Incidence of ADRs according to clinical trial design.
To date, numerous studies compared the efficacy and safety of antidepressants. In 2018, (Cipriani et al., 2018) published a comprehensive systematic review and meta-analysis of 21 antidepressant drugs and their use for the treatment of major depressive disorder. All antidepressants were found effective vs. placebo, with ORs ranging between 1.37 (for reboxetine) and 2.13 (for amitriptyline), with desvenlafaxine and venlafaxine showing intermediate effectiveness (OR = 1.49 and 1.78, respectively). In terms of dropout rate, both drugs showed a similar profile (ORs = 1.08 and 1.04 vs. placebo, respectively). Unfortunately, data comparing desvenlafaxine with other antidepressants is scarcely available, and placebo-controlled clinical trials are mainly available. With placebo as comparator, it seems that both drugs show a similar effectiveness profile, with desvenlafaxine showing a better safety profile (i.e., lower incidence of nausea) (Coleman et al., 2012). In a 8-week phase-III clinical trial, desvenlafaxine showed a 5% discontinuation rate compared to 6% for venlafaxine, and 13% for duloxetine (Tourian et al., 2009), which is consistent with desvenlafaxine having a slightly better safety profile. In summary, little evidence is available to justify the selection of other antidepressants in preference to desvenlafaxine. Otherwise, desvenlafaxine is one more drug in the therapeutic arsenal available to treat depression, and its selection over other antidepressant drugs depends on the clinician’s preference.
When comparing venlafaxine and desvenlafaxine, at equivalent doses, they could be considered similar therapeutic alternatives, as both compounds are potent serotonin and norepinephrine reuptake inhibitors. Venlafaxine, through CYP2D6, is converted to desvenlafaxine, hence, the pharmacological effects of desvenlafaxine can be expected when venlafaxine is administered; the only difference is that venlafaxine (the precursor) also inhibits dopamine reuptake and desvenlafaxine does not (Singh and Saadabadi, 2022). However, a clear drawback of venlafaxine therapy is the variability in response and safety associated with CYP2D6 phenotype. A priori, desvenlafaxine seems to have a better pharmacokinetic profile and a better dose-response and dose-safety relationship (Norman and Olver, 2021). In this work, not only did we confirm that the occurrence of ADRs caused by desvenlafaxine is associated with the time (multiple dose administration vs. single dose administration) and/or magnitude (AUC) of drug exposure, but we also clearly observed that the exposure to desvenlafaxine does not depend on the genetic polymorphism of candidate genes. The strong dose-dependent relationship with the occurrence of ADRs confirms that the mechanism of action of desvenlafaxine is essentially dose-dependent, compared to the biphasic dose-dependent mechanism of action of venlafaxine (Harvey et al., 2000). To our knowledge, this work is the first to analyze all potentially relevant pharmacogenes comprehensively and robustly with respect to the pharmacokinetics and safety of the drug. Although this might be somewhat obvious a priori, this does not hold for all drugs. For instance, voriconazole shows nonlinear pharmacokinetics, with the dose-response (or dose-safety) relationship exhibiting wide interpatient variability. This is typically explained by its limited capacity of elimination. Furthermore, CYP2C19 phenotype is a clear predictor of drug metabolism (Moriyama et al., 2017). Therefore, a reduction or increase in dose is not directly related to an increase in exposure or in the chances of achieving efficacy (Theuretzbacher et al., 2006; Zubiaur et al., 2021). Studies such as the present one are important to confirm this pharmacokinetic characteristic, which enables more predictable dosage adjustments of the drug.
Here, the healthy volunteers presented pharmacokinetic parameters consistent with previous literature. For instance, an AUC∞ of 5875.01 ng*h/mL was observed for the 100 mg formulation, compared to 6747 ng*h/mL and a Cmax of 251 ng/mL compared to 376 ng/mL (Spanish Drug Agency, 2012) (Spanish Drug Agency, 2012). Furthermore, the global tmax was 6.66 h compared to 7.5 h, and a t1/2 of 10.71 compared to 11 h (Spanish Drug Agency, 2012). Female healthy volunteers presented a higher Cmax/DW, which may be explained by sex-specific physiological differences in drug absorption (Soldin and Mattison, 2009). A clear dose-dependent relationship was observed in the variations of AUC and Cmax. Furthermore, several differences in pharmacokinetic variables were observed according to the clinical trial design. However, these differences were small, with the AUC/DW ranging from 86% to 115% with respect to mean AUC/DW, and the Cmax/DW ranging from 83% to 121%, except for the clinical trial C, which showed a 138% with respect to mean Cmax/DW. This difference can be explained by the fact that, in clinical trial C, several desvenlafaxine doses were administered and the residual concentration from previous drug administrations needs to be considered. In contrast, AUC∞ from single dose clinical trials and AUCƬ from the multiple-dose clinical trial were equivalent, as expected. The remaining differences related to the clinical trial design can be considered spurious and related to the low sample size for each clinical trial. Finally, fed healthy volunteers showed a higher Cmax/DW, an effect that is well described in desvenlafaxine’s drug label (Spanish Drug Agency, 2012).
As mentioned earlier, the incidence of ADRs was significantly higher in the only clinical trial with multiple-dose administration (clinical trial C). This may be explained by the higher observed Cmax/DW or the prolonged exposure to the drug. Furthermore, the AUC was identified as the best predictor of ADR development; the fact that drug exposure is the main predictor of drug tolerability is reassuring. Assuming linear pharmacokinetics, ADRs resulting from drug overexposure could be resolved with dose adjustments. This does not apply equally to venlafaxine, as certain patients (e.g., CYP2D6 UMs or PMs) will respond variably to similar adjustments. The association between CYP2B6 RMs and asthenia incidence can be considered spurious; should CYP2B6 metabolize desvenlafaxine, a lower rate of ADRs should be expected in individuals with a higher metabolic capacity. Alternatively, this would be explained if desvenlafaxine had an active metabolite responsible for this side effect. However, to our knowledge, this metabolite has not been described to date. Likewise, the association between ABCB1 rs203258 G/T and asthenia can only be explained as a spurious event because a heterozygous disadvantage is not possible for traits inherited in a codominant manner.
Several study limitations should be considered. The clinical trial study design did not allow to collect data on the long-term pharmacokinetics and tolerability or effectiveness. We were also unable to analyze the influence of Black race on desvenlafaxine pharmacokinetics because there was only one subject self-reported as Black. In addition, some Tier 1 variants in CYP2C9 and CYP2D6 were not included (Pratt et al., 2019; Pratt et al., 2021) thus the pharmacogenetic phenotype may not have been correctly inferred, albeit this possibility is very unlikely due to the low prevalence of these alleles (e.g., CYP2C9*8). In addition, among the UGT enzymes, only variants in UGT1A1 have been demonstrated to alter the enzyme’s function, while no variants with a functional impact are described for other important UGT candidates such as UGT1A3 or UGT2B4. Furthermore, these results may be interpreted with caution given the limited sample size, which implies that the statistical power is limited and may not be sufficient to detect pharmacogenetic effects, especially for rare polymorphisms. In addition, some associations are described that did not reach the significance level after Bonferroni correction for multiple comparisons, but these associations must be considered cautiously due to the exploratory nature of this work. In contrast, bioequivalence clinical trials offer a controlled setting for the evaluation of pharmacokinetic variability based on genetic polymorphism or demographics as confounding factors are avoided.
Desvenlafaxine pharmacokinetic parameters were unrelated to genetic polymorphism of the principal candidate pharmacogenes. In contrast, drug exposure in time and magnitude (AUC) were the principal predictors of ADR incidence, which confirms that the mechanism of action of desvenlafaxine is essentially dose-dependent. These two findings can be interpreted as therapeutic advantages of desvenlafaxine compared to venlafaxine.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Independent Ethics Board of Hopsital Universitario de La Princesa. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Independent Ethics Board of Hospital Universitario de La Princesa.
SC, PZ, and FA-S conceptualized the study; SC and PZ contributed to methodology; FA-S and PZ validated the study; SC and PZ were responsible for formal analysis; SC, PZ, PS-C, GV-G, MN-G and FA-S performed the observational pharmacogenetic study; FA-S provided resources; SC and PZ were responsible for data curation; SC and PZ prepared the original draft; all authors reviewed and edited the manuscript; SC and PZ visualized the study; PZ and FA-S supervised the study; PZ and FA-S were responsible for project administration; FA-S acquired funding. DO, GMA, AM, MR, SM-V, and FA-S participated as investigators of the clinical trials.
PS-C is financed by Universidad Autónoma de Madrid (FPI-UAM, 2021). GV-G is co-financed by Instituto de Salud Carlos III (ISCIII) and the European Social Fund (PFIS predoctoral grant, number FI20/00090). MN-G is financed by the ICI20/00131 grant, Acción Estratégica en Salud 2017–2020, ISCIII. PZ is financed by Universidad Autónoma de Madrid, Margarita Salas contract, grants for the requalification of the Spanish university system.
FA-S, DO , and MR have been consultants or investigators in clinical trials sponsored by the following pharmaceutical companies: Abbott, Alter, Aptatargets, Chemo, FAES, Farmalider, Ferrer, Galenicum, GlaxoSmithKline, Gilead, Italfarmaco, Janssen-Cilag, Kern, Normon, Novartis, Servier, Teva, and Zambon.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1110460/full#supplementary-material
Aguirre, C., and García, M. (2016). Evaluación de la causalidad en las comunicaciones de reacciones adversas a medicamentos. Algoritmo del Sistema Español de Farmacovigilancia. Med. Clínica 147, 461–464. doi:10.1016/j.medcli.2016.06.012
Belmonte, C., Ochoa, D., Román, M., Saiz-Rodríguez, M., Wojnicz, A., Gómez-Sánchez, C. I., et al. (2018). Influence of CYP2D6, CYP3A4, CYP3A5 and ABCB1 polymorphisms on pharmacokinetics and safety of aripiprazole in healthy volunteers. Basic Clin. Pharmacol. Toxicol. 122, 596–605. doi:10.1111/bcpt.12960
Birdwell, K., Decker, B., Barbarino, J., Peterson, J., Stein, C., Sadee, W., et al. (2015). Clinical pharmacogenetics implementation consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin. Pharmacol. Ther. 98, 19–24. doi:10.1002/cpt.113
Bousman, C. A., Müller, D. J., Ng, C. H., Byron, K., Berk, M., and Singh, A. B. (2017). Concordance between actual and pharmacogenetic predicted desvenlafaxine dose needed to achieve remission in major depressive disorder: A 10-week open-label study. Pharmacogenetics Genomics 27, 1–6. doi:10.1097/FPC.0000000000000253
Brown, J. T., Bishop, J. R., Sangkuhl, K., Nurmi, E. L., Mueller, D. J., Dinh, J. C., et al. (2019). Clinical pharmacogenetics implementation consortium guideline for cytochrome P450 (CYP) 2D6 genotype and atomoxetine therapy. Clin. Pharmacol. Ther. 106, 94–102. doi:10.1002/cpt.1409
Cipriani, A., Furukawa, T. A., Salanti, G., Chaimani, A., Atkinson, L. Z., Ogawa, Y., et al. (2018). Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 391, 1357–1366. doi:10.1016/S0140-6736(17)32802-7
Coleman, K. A., Xavier, V. Y., Palmer, T. L., Meaney, J. V., Radalj, L. M., and Canny, L. M. (2012). An indirect comparison of the efficacy and safety of desvenlafaxine and venlafaxine using placebo as the common comparator. CNS Spectr. 17, 131–141. doi:10.1017/S1092852912000648
Cooper-DeHoff, R. M., Niemi, M., Ramsey, L. B., Luzum, J. A., Tarkiainen, E. K., Straka, R. J., et al. (2022). The clinical pharmacogenetics implementation consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharma Ther. 111, 1007–1021. doi:10.1002/cpt.2557
Desta, Z., Gammal, R. S., Gong, L., Whirl-Carrillo, M., Gaur, A. H., Sukasem, C., et al. (2019). Clinical pharmacogenetics implementation consortium (CPIC) guideline for CYP2B6 and efavirenz-containing antiretroviral therapy. Clin. Pharmacol. Ther. 106, 726–733. doi:10.1002/cpt.1477
DrugBank (2010). Desvenlafaxine: Uses, interactions, mechanism of action. Available at: https://go.drugbank.com/drugs/DB06700.
Dutch Pharmacogenetics Working Group (DPWG) (2022). CYP2D6: Venlafaxine. 1538 to 1540. Available at: https://www.g-standaard.nl/risicoanalyse/B0001540.PDF.
Dutch Pharmacogenetics Working Group (DPWG) (2021a). CYP3A4: Quetiapine. 5991 to 5992. Available at: https://www.g-standaard.nl/risicoanalyse/B0005991.PDF.
Dutch Pharmacogenetics Working Group (DPWG) (2021b). UGT1A1: Irinotecan. 1691 to 1694. Available at: https://www.g-standaard.nl/risicoanalyse/B0001693.PDF.
Fogelman, S., Schmider, J., VenKataKrishnan, K., von Moltke, L. L., Harmatz, J. S., Shader, R. I., et al. (1999). O- and N-demethylation of venlafaxine in vitro by human liver microsomes and by microsomes from cDNA-transfected cells effect of metabolic inhibitors and SSRI antidepressants. Neuropsychopharmacology 20, 480–490. doi:10.1016/S0893-133X(98)00113-4
Food and Drug Administration (FDA) (2011). PRISTIQ® (desvenlafaxine) Extended-Release Tablets, oral - drug label. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021992s030lbl.pdf (Accessed May 14, 2022).
Gammal, R., Court, M., Haidar, C., Iwuchukwu, O., Gaur, A., Alvarellos, M., et al. (2016). Clinical pharmacogenetics implementation consortium (CPIC) guideline for UGT1A1 and atazanavir prescribing. Clin. Pharmacol. Ther. 99, 363–369. doi:10.1002/cpt.269
Harvey, A. T., Rudolph, R. L., and Preskorn, S. H. (2000). Evidence of the dual mechanisms of action of venlafaxine. Arch. Gen. Psychiatry 57, 503–509. doi:10.1001/archpsyc.57.5.503
Hicks, J., Bishop, J., Sangkuhl, K., Müller, D., Ji, Y., Leckband, S., et al. (2015). Clinical pharmacogenetics implementation consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin. Pharmacol. Ther. 98, 127–134. doi:10.1002/cpt.147
Lee, C. R., Luzum, J. A., Sangkuhl, K., Gammal, R. S., Sabatine, M. S., Stein, C. M., et al. (2022). Clinical pharmacogenetics implementation consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin Pharma Ther. 112, 959–967. doi:10.1002/cpt.2526
Liebowitz, M. R., and Tourian, K. A. (2010). Efficacy, safety, and tolerability of desvenlafaxine 50 mg/d for the treatment of major depressive disorder: A systematic review of clinical trials. Prim. Care Companion J. Clin. Psychiatry 12, PCC09r00845. doi:10.4088/PCC.09r00845blu
Moriyama, B., Obeng, A. O., Barbarino, J., Penzak, S., Henning, S., Scott, S., et al. (2017). Clinical pharmacogenetics implementation consortium (CPIC) guidelines for CYP2C19 and voriconazole therapy. Clin. Pharmacol. Ther. 102, 45–51. doi:10.1002/cpt.583
Nichols, A. I., Abell, M., Madelyn, A., Chen, Y., Behrle, J. A., Frick, G., et al. (2013). Effects of desvenlafaxine on the pharmacokinetics of desipramine in healthy adults. Int. Clin. Psychopharmacol. 28, 99–105. doi:10.1097/YIC.0b013e32835c1f49
Nichols, A. I., Fatato, P., Shenouda, M., Paul, J., Isler, J. A., Pedersen, R. D., et al. (2009). The effects of desvenlafaxine and paroxetine on the pharmacokinetics of the cytochrome P450 2D6 substrate desipramine in healthy adults. J. Clin. Pharmacol. 49, 219–228. doi:10.1177/0091270008326716
Norman, T. R., and Olver, J. S. (2021). Desvenlafaxine in the treatment of major depression: An updated overview. Expert Opin. Pharmacother. 22, 1087–1097. doi:10.1080/14656566.2021.1900826
Pratt, V. M., Cavallari, L. H., Del Tredici, A. L., Gaedigk, A., Hachad, H., Ji, Y., et al. (2021). Recommendations for clinical CYP2D6 genotyping allele selection: A joint consensus recommendation of the association for molecular pathology, college of American pathologists, Dutch pharmacogenetics working group of the royal Dutch pharmacists association, and the European society for Pharmacogenomics and personalized therapy. J. Mol. Diagn 23, 1047–1064. doi:10.1016/j.jmoldx.2021.05.013
Pratt, V. M., Cavallari, L. H., Del Tredici, A. L., Hachad, H., Ji, Y., Moyer, A. M., et al. (2019). Recommendations for clinical CYP2C9 genotyping allele selection: A joint recommendation of the association for molecular pathology and college of American pathologists. J. Mol. Diagn 21, 746–755. doi:10.1016/j.jmoldx.2019.04.003
Preskorn, S., Patroneva, A., Silman, H., Jiang, Q., Isler, J. A., Burczynski, M. E., et al. (2009). Comparison of the pharmacokinetics of venlafaxine extended release and desvenlafaxine in extensive and poor cytochrome P450 2D6 metabolizers. J. Clin. Psychopharmacol. 29, 39–43. doi:10.1097/JCP.0b013e318192e4c1
Singh, D., and Saadabadi, A. (2022). Venlafaxine,” in StatPearls (treasure island (FL): StatPearls publishing). Available at: http://www.ncbi.nlm.nih.gov/books/NBK535363/(Accessed November 9, 2022).
Soldin, O. P., and Mattison, D. R. (2009). Sex differences in pharmacokinetics and pharmacodynamics. Clin. Pharmacokinet. 48, 143–157. doi:10.2165/00003088-200948030-00001
Spanish Drug Agency (2012). PRISTIQ® (desvenlafaxine) Extended-Release Tablets, oral - drug label. Available at: https://cima.aemps.es/cima/pdfs/es/ft/75561/FT_75561.pdf (Accessed November 3, 2022).
Sproule, A. B., Hazra, M., and Pollock, G. B. (2008). Desvenlafaxine succinate for major depressive disorder. Drugs Today 44, 475–487. doi:10.1358/dot.2008.44.7.1227147
Theken, K. N., Lee, C. R., Gong, L., Caudle, K. E., Formea, C. M., Gaedigk, A., et al. (2020). Clinical pharmacogenetics implementation consortium guideline (CPIC) for CYP2C9 and nonsteroidal anti-inflammatory drugs. Clin. Pharmacol. Ther. 108, 191–200. doi:10.1002/cpt.1830
Theuretzbacher, U., Ihle, F., and Derendorf, H. (2006). Pharmacokinetic/pharmacodynamic profile of voriconazole. Clin. Pharmacokinet. 45, 649–663. doi:10.2165/00003088-200645070-00002
Tourian, K. A., Padmanabhan, S. K., Groark, J., Brisard, C., and Farrington, D. (2009). Desvenlafaxine 50 and 100 mg/d in the treatment of major depressive disorder: An 8-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial and a post hoc pooled analysis of three studies. Clin. Ther. 31, 1405–1423. doi:10.1016/j.clinthera.2009.07.006
Zubiaur, P., Kneller, L. A., Ochoa, D., Mejía, G., Saiz-Rodríguez, M., Borobia, A. M., et al. (2021). Evaluation of voriconazole CYP2C19 phenotype-guided dose adjustments by physiologically based pharmacokinetic modeling. Clin. Pharmacokinet. 60, 261–270. doi:10.1007/s40262-020-00941-8
Zubiaur, P., Saiz-Rodríguez, M., Ochoa, D., Navares-Gómez, M., Mejía, G., Román, M., et al. (2020). Effect of sex, use of pantoprazole and polymorphisms in SLC22A1, ABCB1, CES1, CYP3A5 and CYP2D6 on the pharmacokinetics and safety of dabigatran. Adv. Ther. 37, 3537–3550. doi:10.1007/s12325-020-01414-x
Keywords: desvenlafaxine, pharmacogenetics, major depressive disorder, precision medicine, pharmacokinetics, safety
Citation: Calleja S, Zubiaur P, Ochoa D, Villapalos-García G, Mejia-Abril G, Soria-Chacartegui P, Navares-Gómez M, de Miguel A, Román M, Martín-Vílchez S and Abad-Santos F (2023) Impact of polymorphisms in CYP and UGT enzymes and ABC and SLCO1B1 transporters on the pharmacokinetics and safety of desvenlafaxine. Front. Pharmacol. 14:1110460. doi: 10.3389/fphar.2023.1110460
Received: 28 November 2022; Accepted: 20 January 2023;
Published: 02 February 2023.
Edited by:
Elena García-Martín, University of Extremadura, SpainReviewed by:
Ingrid Fricke-Galindo, Instituto Nacional de Enfermedades Respiratorias-México (INER), MexicoCopyright © 2023 Calleja, Zubiaur, Ochoa, Villapalos-García, Mejia-Abril, Soria-Chacartegui, Navares-Gómez, de Miguel, Román, Martín-Vílchez and Abad-Santos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pablo Zubiaur, cGFibG8uenViaWF1ckB1YW0uZXM=; Francisco Abad-Santos, ZnJhbmNpc2NvLmFiYWRAdWFtLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.