 Eva Montané
Eva Montané Javier Santesmases
Javier Santesmases
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 30 January 2023
Sec. Pharmacoepidemiology
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1090707
Objectives: To describe the characteristics of safety alerts issued by the Spanish Medicines Agency (AEMPS) and the Spanish Pharmacovigilance System over a 7-year period and the regulatory actions they generated.
Methods: A retrospective analysis was carried out of drug safety alerts published on the AEMPS website from 1 January 2013 to 31 December 2019. Alerts that were not drug-related or were addressed to patients rather than healthcare professionals were excluded.
Results: During the study period, 126 safety alerts were issued, 12 of which were excluded because they were not related to drugs or were addressed to patients and 22 others were excluded as they were duplications of previous alerts. The remaining 92 alerts reported 147 adverse drug reactions (ADRs) involving 84 drugs. The most frequent source of information triggering a safety alert was spontaneous reporting (32.6%). Four alerts (4.3%) specifically addressed health issues related to children. ADRs were considered serious in 85.9% of the alerts. The most frequent ADRs were hepatitis (seven alerts) and congenital malformations (five alerts), and the most frequent drug classes were antineoplastic and immunomodulating agents (23%). Regarding the drugs involved, 22 (26.2%) were “under additional monitoring.” Regulatory actions induced changes in the Summary of Product Characteristics in 44.6% of alerts, and in eight cases (8.7%), the alert led to withdrawal from the market of medicines with an unfavorable benefit/risk ratio.
Conclusion: This study provides an overview of drug safety alerts issued by the Spanish Medicines Agency over a 7-year period and highlights the contribution of spontaneous reporting of ADRs and the need to assess safety throughout the lifecycle of medicines.
Drug safety is an important health concern that requires continuous evaluation throughout a drug’s lifecycle, not only during its development phase but also after it has entered the market. When a drug is approved, safety information is still limited. A major reason for this is the poor external validity of clinical trials, particularly for some conducted by the pharmaceutical industry (Rothwell, 2005). In these studies, patients exposed to the drug have been selected following strict inclusion and exclusion criteria, leading to significant differences between the population treated and the individuals likely to be seen in real clinical practice who tend to be older, have many comorbidities, and use polypharmacy (Martin et al., 2004). Some adverse drug reactions (ADRs) may only be detected after marketing authorization has been granted because of the mechanism of production, a long latency period, or very low incidence. Therefore, the safety of medicines should be continuously assessed even after their authorization and appearance on the market, through pharmacovigilance systems (European Medicines Agency, 2022a) in which governmental regulatory agencies typically play a key role. In the last 50 years, pharmacovigilance systems have been developed in many countries around the world (Uppsala Monitoring Centre, 2022). In the European Union (EU), pharmacovigilance is jointly managed by EU member states, the European Medicines Agency (EMA), and the European Commission (European Medicines Agency, 2022a).
When health professionals or patients suspect an ADR, a spontaneous report is sent to the local regulatory agency. In Spain, reports are first sent to the national pharmacovigilance system the Sistema Español de Farmacovigilancia de Medicamentos de uso Humano (SEFV-H). The SEFV-H consists of 17 regional pharmacovigilance centers and the Spanish Medicine Agency (Agencia Española de Medicamentos y Productos Sanitarios [AEMPS]). Then, reports are sent from the SEFV-H to EudraVigilance, the EMA’s pharmacovigilance network. Finally, spontaneous reports are included in Vigibase, the database of the World Health Organization Uppsala Monitoring Centre (Baldo et al., 2018).
To ensure that drugs continue to have an optimal benefit–risk balance after they are put on the market, they are typically subject to risk management plans involving post-authorization safety studies. However, spontaneous reports of ADRs are also essential in triggering alarm signals. Then, these signals need to be detected, validated, and confirmed following a management process (European Medicines Agency, 2022b). For this purpose, the EMA published new legislation concerning pharmacovigilance and risk management in 2010 and in the process created the Pharmacovigilance Risk Assessment Committee (PRAC), which is the EU-level committee responsible for assessing and monitoring the safety of medicinal products for human use across Europe. The PRAC came into existence at the end of 2012 and has since demonstrated its effectiveness in detecting and evaluating new drug safety signals (Potts et al., 2020). In Spain, the published law was enacted in 2013 that brought Spain in line with European legislation (Agencia estatal boletín oficial del estado, 2022) by promoting the creation of health data bases, conducting pharmacoepidemiological studies and evaluating ADR reports received by the SEFV-H.
In general, drug safety alerts are motivated by signals obtained from databases of pharmacovigilance systems and signals warning of ADRs detected in post-marketing studies and/or clinical trials. A signal “suggests a new potentially causal association or a new aspect of a known association between an intervention and an event or set of related events that is judged to be of sufficient likelihood to justify verificatory action” (European Medicines Agency, 2022b). The generated alerts provide information to health-care professionals or patients about the needed regulatory actions based on the signals such as new restrictions on the use of medicine in particular populations, the mandatory monitoring of patients using that medicine, or the withdrawal of the drug from the market (Farcaş et al., 2018).
In a recent study comparing drug safety alerts issued by national medicine regulators in Australia, Canada, the United Kingdom, and the United States from 2007 to 2016, major differences were found in the use of safety advisories by regulators, including their frequency, content, communication type, and focus (Perry et al., 2020). However, few previous studies have analyzed the characteristics of safety alerts issued by regulatory authorities, though data are available for a few other countries, including Portugal, the Netherlands, Brazil, and the Ivory Coast (Ferreira et al., 2020; Soares et al., 2015; Mol et al., 2010; N’Guessan-Irié et al., 2012). To our knowledge, no research has been published regarding alerts specifically issued by the Spanish Medicines Authority.
With the goal of filling the gap, this study attempted to describe the characteristics of safety warnings issued by the Spanish Medicines Agency and Pharmacovigilance System during a 7-year period after PRAC began to function and examine the regulatory actions taken as a result of each alert. Secondary goals included comparing the alerts issued by different institutions, assessing specific safety alerts related to drugs under ‘additional monitoring,’ and analyzing alerts according to the drug’s target population and medical area of focus.
This study is a retrospective analysis of drug safety alerts issued by the Spanish Medicines Agency and published on its website (Notas informativas and medicamentos de uso humano. Agencia Española de Medicamentos y Productos Sanitarios, 2022) during the 7-year period from 1 January 2013, after the launch of the PRAC to 31 December 2019, and before the COVID-19 pandemic started. This cut-off date was chosen to include homogenous safety alerts.
All of the safety alerts issued were selected and those involving medicines were included. Alerts that were not drug-related, warnings addressed to patients, and duplicated alerts were excluded. We considered an alert was duplicated when the ADR alert was similar and related to the same drug.
For each alert, the following descriptive variables were recorded.
- Year of publication
- Issuing agency: AEMPS or EMA
- Population targeted by the alert: children <18 years old, adults 18–65 years old, and elderly >65 years old
- Drugs involved: classification was based on the first level of the Anatomical Therapeutic Chemical (ATC) classification (WHO Collaborating Centre for Drug Statistics Methodology, 2018)
- Limitations on the prescription or dispensing of the drug: specifically whether it was limited to the hospital setting or not
- Prescribing specialists: the healthcare professionals most likely to prescribe the drug in question in the consensus view of the authors; for widely used drugs, prescribing professionals were assumed to be general practitioners
- Drugs under “additional monitoring:” this term is used by the EMA to denote medicines that are more intensively monitored than others, generally because there is less safety information available; they have an inverted black triangle [▼] displayed on their packaging and Summary of Product Characteristics (SmPC) (European Medicines Agency, 2022)
- Drug indication: A medical condition for which that medicine is prescribed
- Type of ADR: classified in accordance with the System Organ Classes (SOC) of the Medical Dictionary for Regulatory Activities (MedDRA) (MEDDRA, 2021)
- Seriousness of the ADR: a serious ADR was defined according to the International Conference on Harmonization (ICH) guideline E2D as one that is fatal, life-threatening, requires hospital admission or prolongation of hospital stay and causes persistent or significant disability/incapacity, congenital anomaly/congenital defect or medically important (European Medicines Agency, 2004)
- Sources of evidence: spontaneous reports, clinical trials, and/or observational studies
- Reiteration of the alert: whether it was a first-time alert or the reiteration of an alert issued before the study period
- Regulatory actions to be taken as noted in the alert such as new restrictions on use, risk minimization measures, changes in SmPC, or withdrawal from the market. Regulatory actions are based on the scientific decisions of an expert group responsible for assessing safety problems related to medicines such as the Committee for Medicinal Products for Human Use [CHMP]. When the group decides that the degree of risk is acceptable under the currently authorized conditions of use, the regulatory action is to provide information regarding the ADR and recommending risk minimization measures such as patient monitoring and follow-up. When the group decides that the risk is acceptable but only under certain conditions, the regulatory agency issues a restriction of use such as restricting the indications for which the medicine may be prescribed or the patient population to which it may be given. Any of these measures can lead to the modification and/or updating of the SmPC. When the group decides that the risk is unacceptable and the benefit/risk ratio is unfavorable, the drug is withdrawn from the market (Rodriguez Pascual, 2006).
The selection of alerts for inclusion in the study and the recording of study variables were conducted by the two researchers working independently (SJ and ME). Consensus was reached whenever discrepancies arose.
Descriptive statistics (counts and percentages) were used to characterize the variables assessed in this study. To compare the characteristics of alerts issued by AEMPS with those issued by the EMA (mainly by the PRAC), the Pearson chi-square or Fisher’s exact test for categorical variables was used. A bilateral p-value <0.05 was used to determine statistical significance.
All statistics were performed using the SPSS software package for Windows version 19.0 (SPSS Inc., Chicago, Ill).
An initial search of the EMA website yielded a total of 126 safety alerts. Thirty-four alerts were excluded (22 were duplicated alerts) (Figure 1). These duplicated alerts provide new safety data, corroborate drug risks reported by the PRAC when issued by the AEMPS, or include changes to the regulatory measures applied. Drugs involved in duplicated alerts were alemtuzumab, canagliflozin, cilostazol, codeine, cyproterone acetate, denosumab, diacerein, fentanyl, fingolimod, fusafungine, gliflozins, hydroxyethylstarch, idelalisib, methotrexate, mycophenolic acid, radium dichloride, strontium ranelate, tetrazepam, tofacitinib, ulipristal, valproic acid, and VPH vaccine. All duplicates (n = 22) were eliminated from the analyzed dataset, which resulted in a total of 92 remaining alerts involving 84 drugs.

FIGURE 1. Flow chart of included safety alerts.
Forty alerts (43.5%) were issued by the AEMPS, while the remainder were issued by the EMA (47 of which were specifically issued by the PRAC). Sources used to support safety alerts were spontaneous reporting of ADRs (30 alerts, 32.6%), clinical trials (16 alerts, 17.4%), observational studies (eight alerts, 8.7%), or multiple sources (21, 22.8%). A total of 17 alert (18.5%) sources were not specified.
A total of 147 ADRs were reported in the 92 safety alerts, ranging from one to four ADRs per alert; two or more ADRs were reported in 42 (45.6%) alerts. The most frequently reported ADRs were hepatitis (nine alerts), death (seven alerts), congenital malformation (six alerts), and drug inefficacy, arrhythmia, and arterial thrombosis (five alerts each). The remaining types of ARDs are detailed in Table1. The most frequent ADRs in terms of the MedDRA’s SOC classification were cardiac, vascular, and hepatobiliary disorders (19, 15, and 14 alerts, respectively) (Table 2).
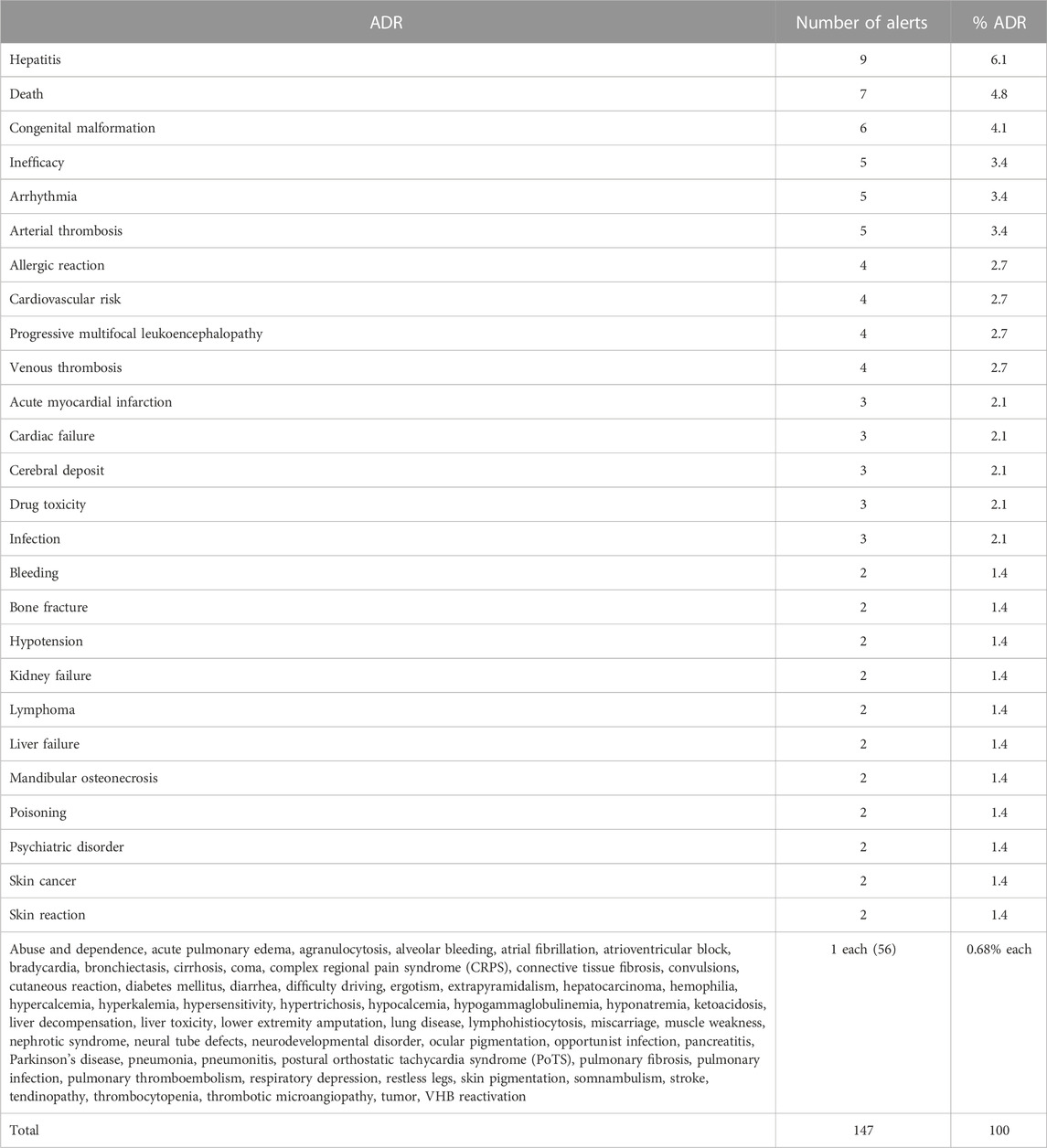
TABLE 1. Type of adverse drug reaction (ADR) related to the alert.
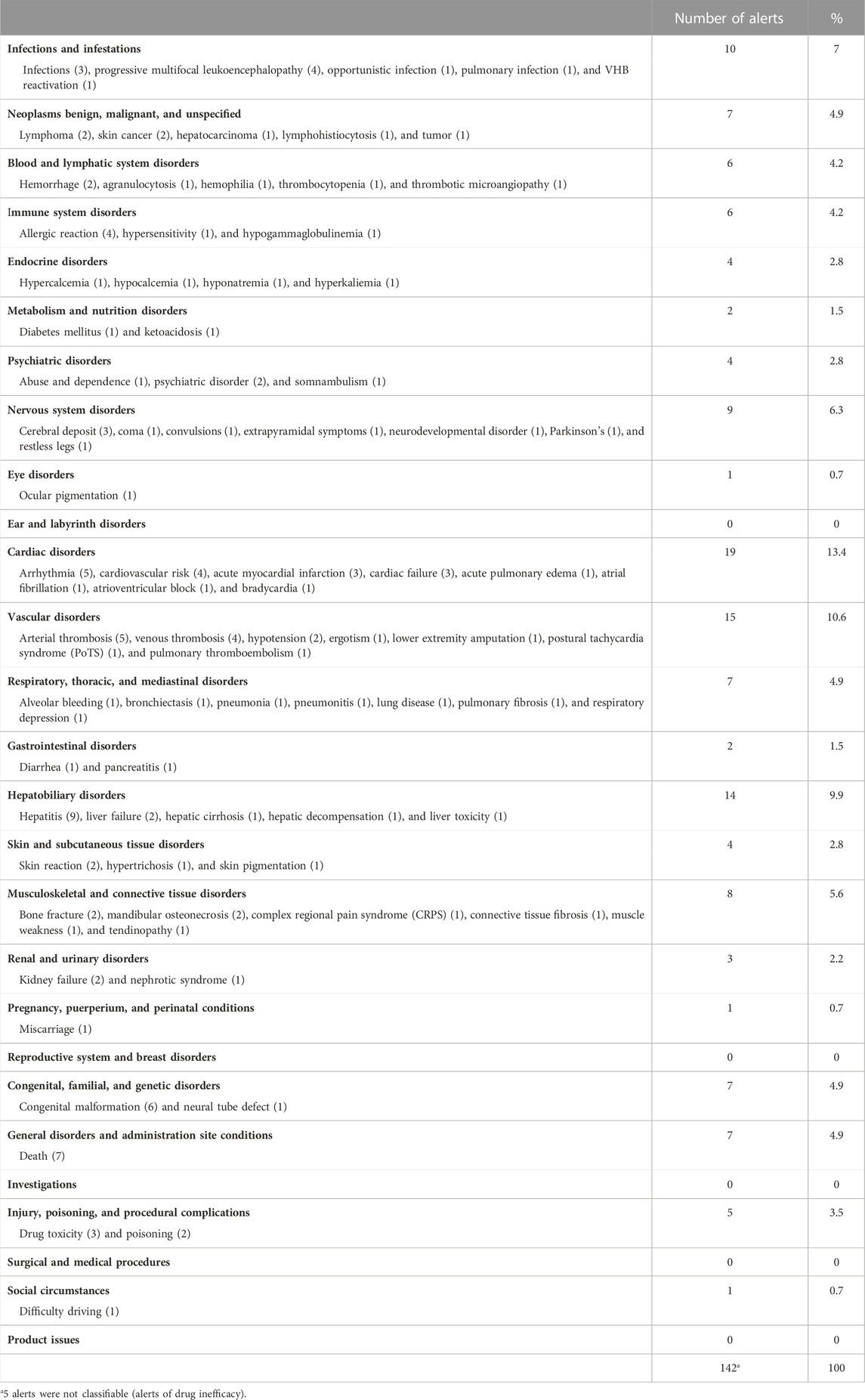
TABLE 2. ADRs classified according to MedDRA’s SOC terminology.
The most frequently alerted drugs (by ATC category) were L-Antineoplastic and immunomodulating agents (21 alerts, 23%), N-Nervous system (12, 13%), and J-Anti-infectives for systemic use (11, 12%) (Table 3). The most alerts (three) were issued for fingolimod (Table 4). The prescription of 37 drugs (45%) was restricted to the hospital setting.
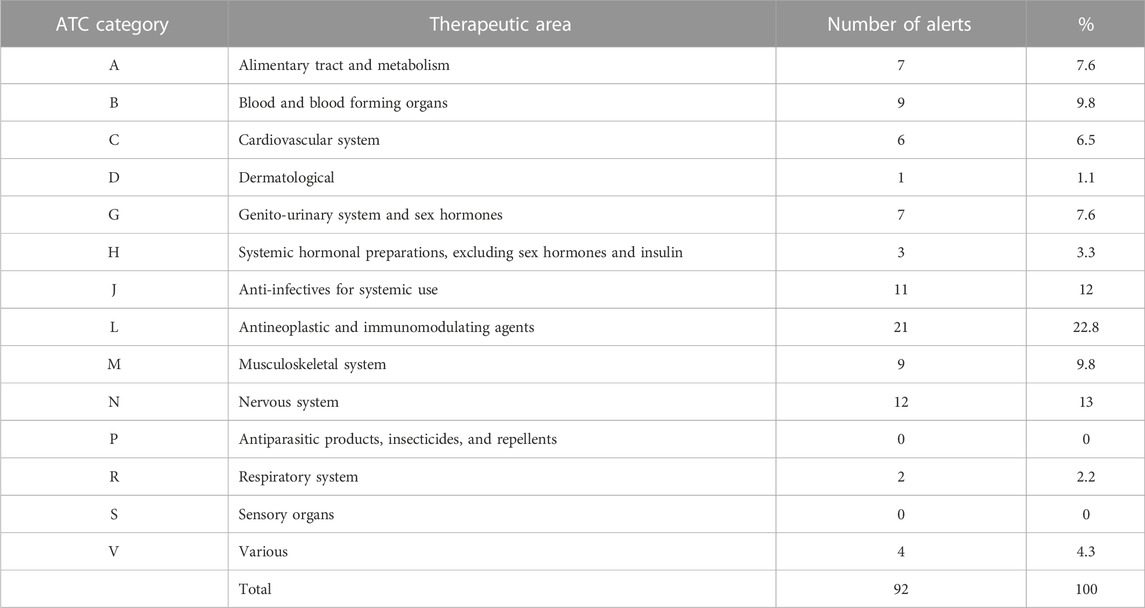
TABLE 3. Anatomical Therapeutic Chemical (ATC) drug classification system.
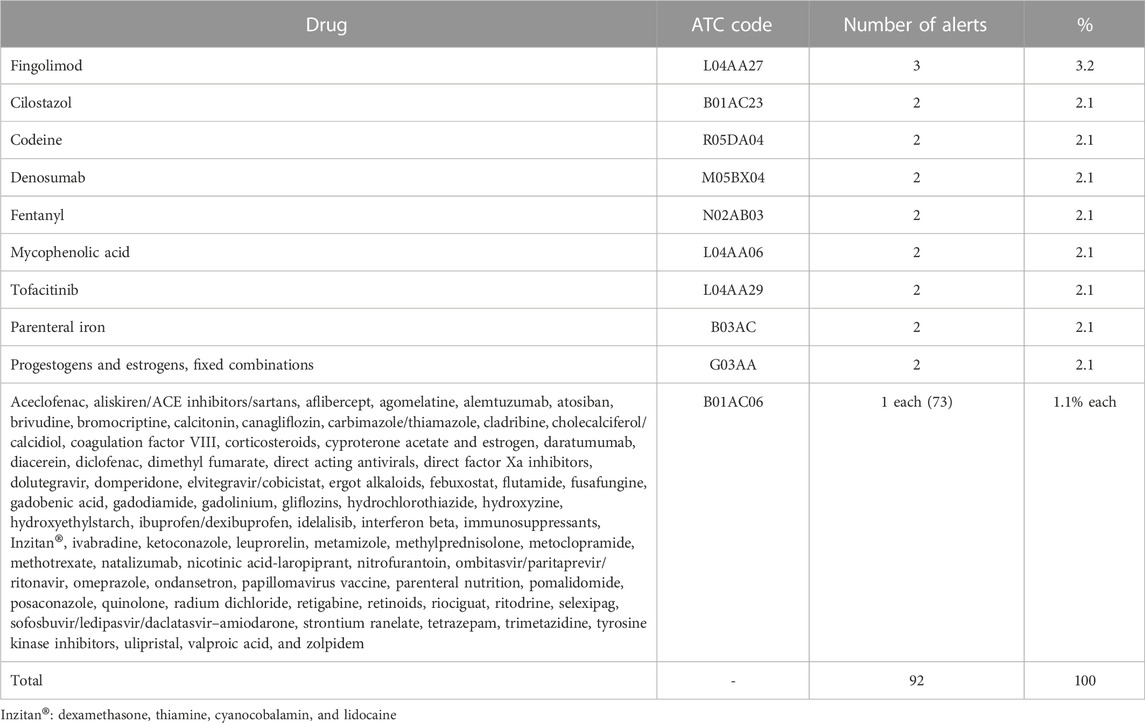
TABLE 4. Drugs involved in alerts.
With regard to drugs under ‘additional monitoring,’ a total of 26 alerts (28.3%) were related to 22 of these drugs (Table 5), and the most frequently involved drugs were fingolimod (three alerts) and tofacitinib (two alerts). Hepatitis or hepatobiliary disorders were the most frequent ADR alerts for this group of drugs (in connection with five drugs, 22.7%). The SmPC was modified for 12 of these drugs (54.5%), and one of them (strontium ranelate) was withdrawn from the market.
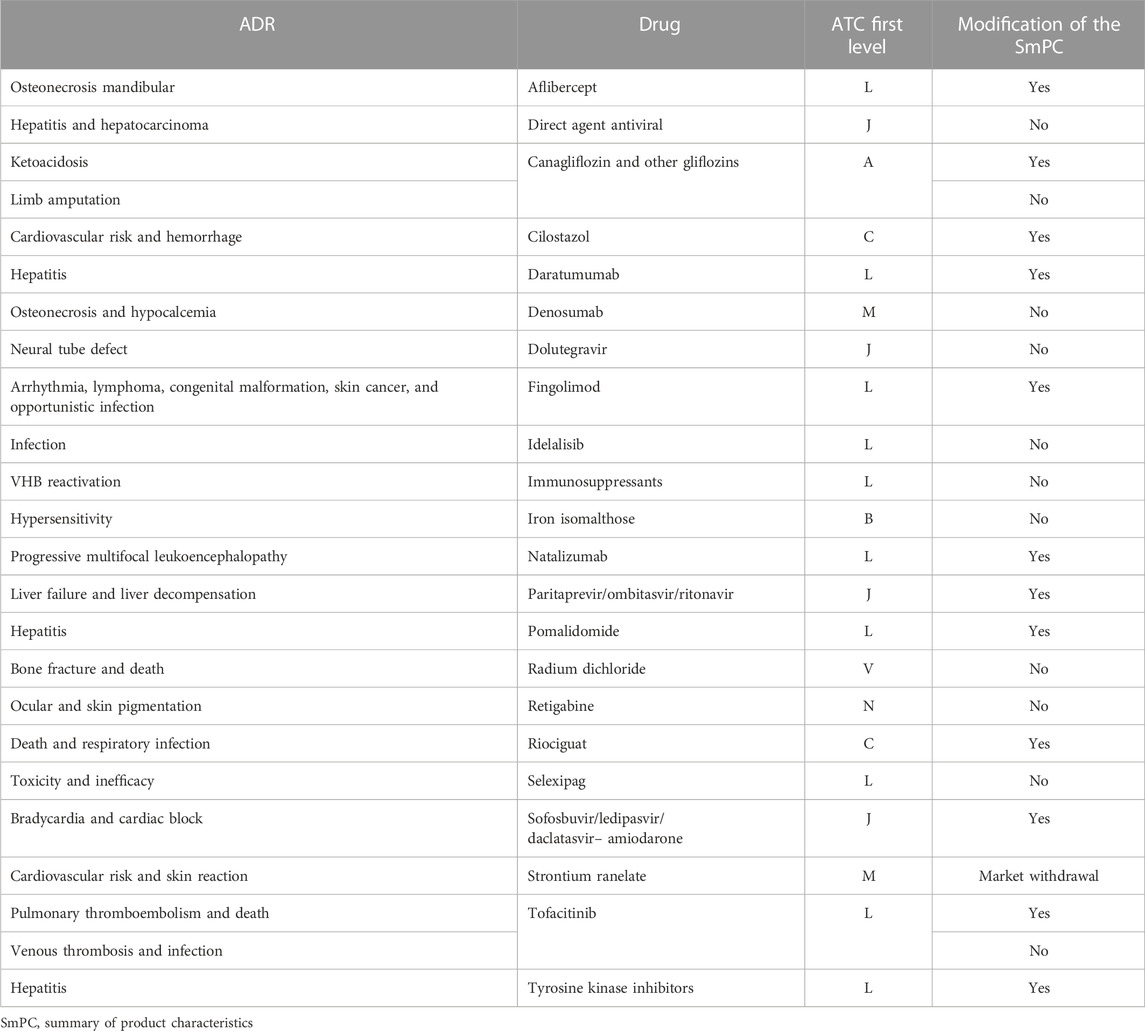
TABLE 5. Characteristics of alerts for drugs under ‘additional monitoring.’
All ADRs were considered serious, with the six exceptions being drug inefficacy, cerebral deposits of contrast, and hypertrichosis. Thirty-nine (42.4%) alerts were addressed to adult populations, 37 (40.2%) were addressed to the elderly population (whether exclusively or not), four (4.3%) alerts were specifically addressed to children or to children and adults, and eight (8.7%) alerts were targeted at populations of all ages. Alerts addressed to children are detailed in Table 6. Seven (7.6%) alerts were addressed to pregnant women and warned of consequences to the fetus (mainly congenital malformations). The features of these alerts are detailed in Table 7.
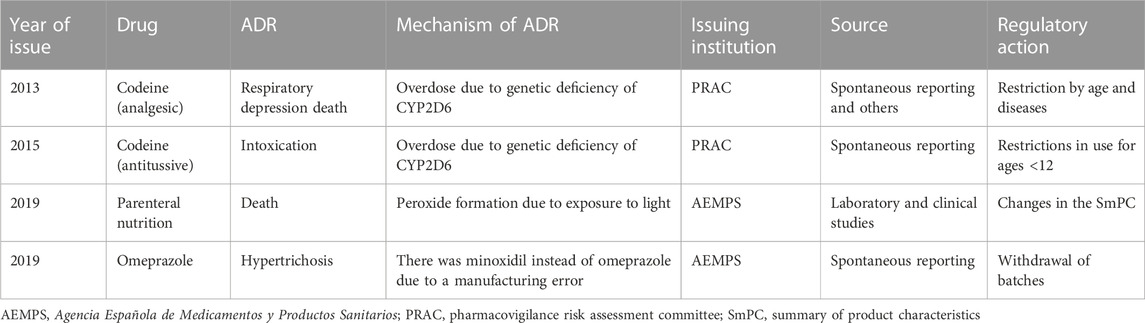
TABLE 6. Characteristics of alerts with drugs addressed to children.
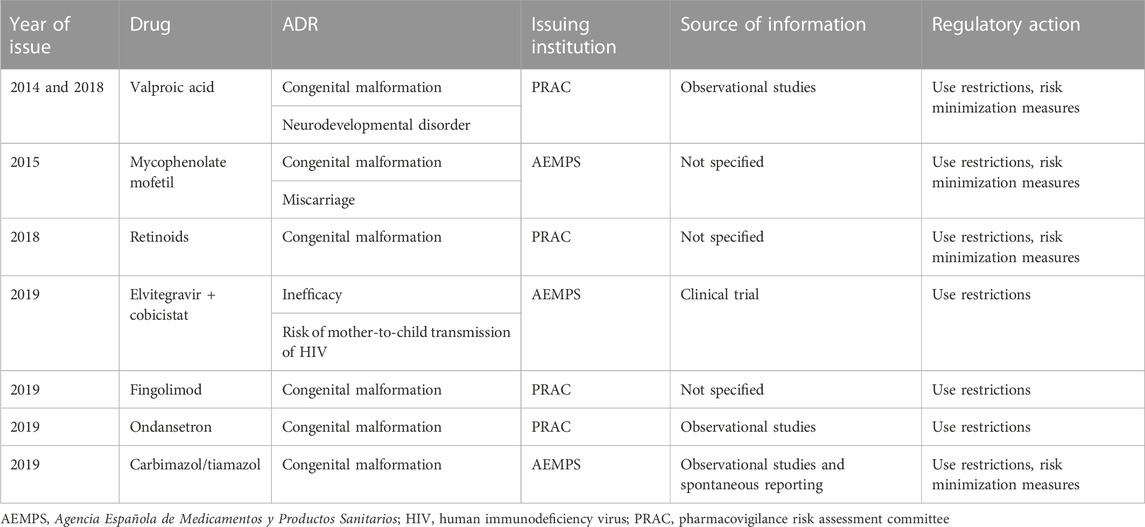
TABLE 7. Characteristics of alerts with drugs addressed to pregnant women.
The prescribing specialists were most often rheumatologists (10 alerts), neurologists, and hematologists (eight alerts each), although 11 alerts involved drugs to treat unspecific symptoms such as pain or fever, emesis, cough, rhinosinusitis, and urinary tract infection and therefore were likely of interest to general practitioners. The diseases most likely linked to safety alerts were any kind of pain (seven alerts), multiple sclerosis (six alerts), and osteoporosis (five alerts) (Table 8).
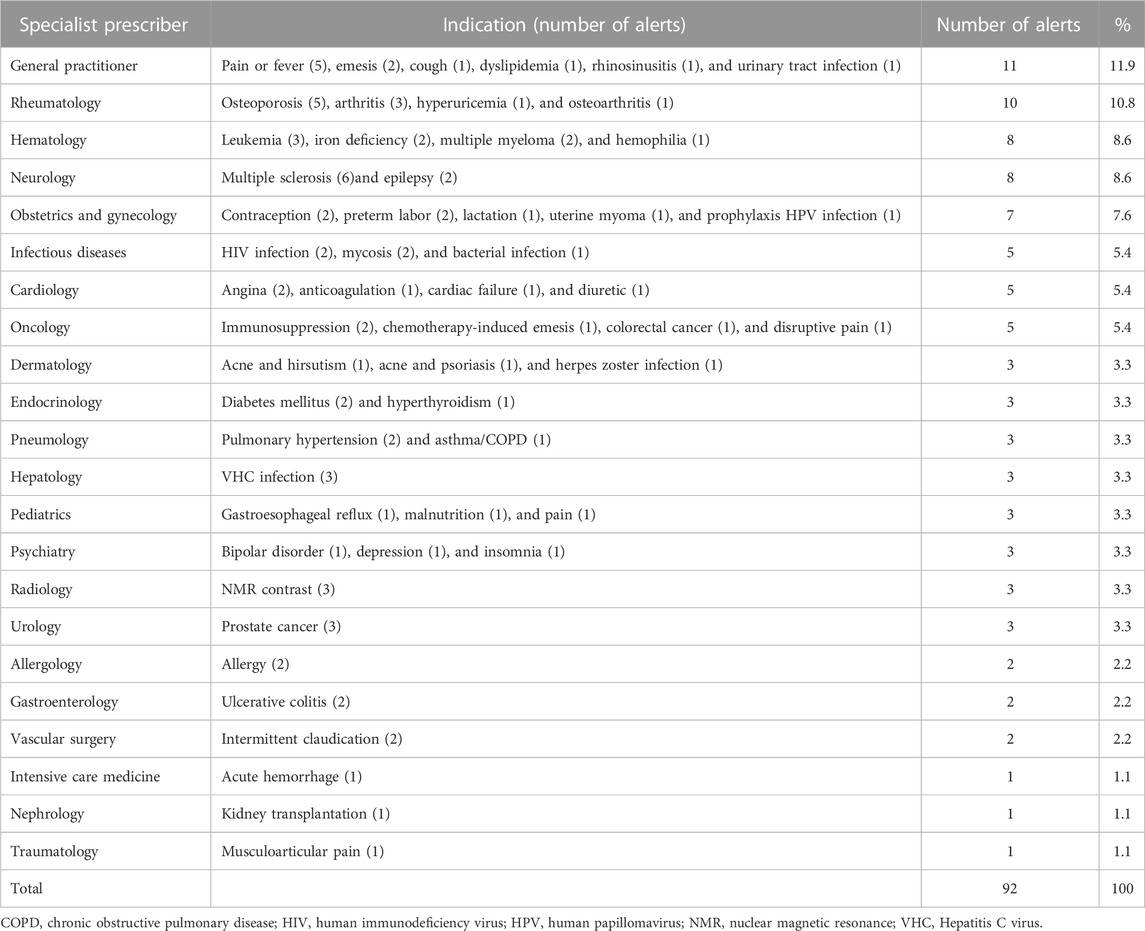
TABLE 8. Indication of drugs involved in alerts and potential specialist prescriber.
The most frequent regulatory actions derived from alerts were “restrictions in use” in 30 alerts (32.6%) and “reporting of ADRs and recommendation of patient monitoring and follow-up” in 14 alerts (15.2%). ‘Restrictions in use’ included restrictions related to the age of the population receiving the medicine, the specific disease being treated, and the duration of treatment and/or dosage. Combinations of these regulatory actions were present in 22 alerts (23.9%). The regulatory actions of 41 alerts (44.6%) involved changes in the SmPC, and eight (8.7%) required mandated withdrawal from the market of medicines with an unfavorable benefit/risk ratio. Only one of these withdrawn medicines was put on the market in the 5 years prior to the alert, while five had been on the Spanish market for more than 20 years (Table 9). Two batches of one drug were withdrawn from the market because they contained minoxidil instead of omeprazole due to an error in the manufacturing process.
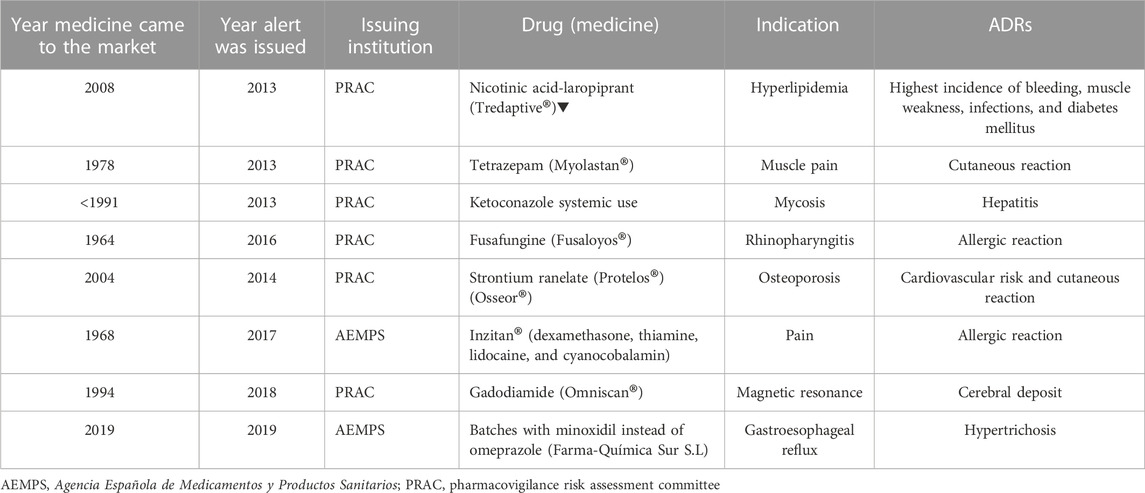
TABLE 9. Characteristics of medicines withdrawn from the market.
When safety alerts were compared by the issuing institution, some significant differences were found. Alerts issued by the AEMPS involved more drugs ‘under additional monitoring’ (42.5% vs. 17.3%; p < 0.008) and drugs restricted to a hospital setting (60% vs. 34.6%; p < 0.015), while alerts issued by the EMA were more often addressed to child populations (25% vs. 7.5%; p < 0.028).
The results of this study show that the Spanish Medicines Agency issued, on average, one or two drug safety alerts every month from 2013 to 2019, one out of five of these were alert duplications. The rate of alert issuance did not increase over time in this period, contrary to other studies conducted in Portugal and the Netherlands (Soares et al., 2015; Ferreira et al., 2020). Almost half of the alerts were issued by the national medicines agency, and some were subsequently issued by the EMA, indicating that the SEFV-H is a proactive local safety system and an effective national signal detection system that anticipates alerts issued by the PRAC (Farcas et al., 2020). Therefore, our findings are in line with those of other similar European studies. However, differences between European and other regulatory agencies (such as those in the US, Canada, or Australia) may lead to different risk assessments of a drug and hence to different regulatory actions (Farcaş et al., 2018; Bjerre et al., 2018; Bhasale et al., 2021).
In this study, we found that the main trigger leading to the generation of these alerts was spontaneous case reports, as indicated in other studies (Alves et al., 2013; Lester et al., 2013; Ishiguro et al., 2017). This underlines the importance and usefulness of spontaneous reporting and national pharmacovigilance systems in detecting issues of concern related to medicines and contributing to the generation of safety alerts during the marketing period (European Medicines Agency, 2022b). Other sources, such as clinical trials and/or post-marketing studies, were also necessary to identify or confirm safety issues in some alerts. More than one out of every four safety alerts concerned drugs under “additional monitoring,” that is, medicines authorized for use in the EU but are being monitored particularly closely by regulatory authorities either because the medicine has been recently approved or because there are limited data on its long-term use. These findings show that recently marketed drugs, with less safety information available, should be a priority for the reporting of suspected ADRs. Therefore, information about any possible ADRs must be collected as early as possible to further inform healthcare practitioners about the safety of these medicines.
Although most alerts were addressed the adult population, almost half were referred to drugs typically prescribed to elderly patients (whether exclusively or not), which is a population that frequently receives multiple drugs, has multiple morbidities, and is rarely included in clinical trials (Davies and O'Mahony, 2015). Pregnant women and children, who accounted for 12% of the analyzed alerts, are also “vulnerable” populations in terms of drug efficacy/effectiveness and safety. These alerts were specifically addressed to children involved with three drugs (codeine, parenteral nutrition, and minoxidil), which contrasts with another study where antidepressants were the only drug alerts related to children (Clavenna and Bonati, 2009). Spontaneous reporting in the pediatric population is crucial due to the limited number of clinical trials and the huge “off-label” use of drugs in situations where drug safety is not yet well-established (Conroy et al., 2000). Regarding alerts addressed to pregnant women, these mainly concerned the risk of congenital malformations, and the source of information was epidemiological studies (when specified). As pregnant women are traditionally excluded from clinical trials of therapeutics other than those related to diseases of childbirth, these data usually come from observational or pharmacoepidemiological studies (Irl and Hasford, 2020).
Rheumatologists, neurologists (mainly from multiple sclerosis units), and hematologists were most likely interested in the safety alerts. Moreover, almost half of the safety alerts involved drugs restricted to use in the hospital setting. Therefore, hospital pharmacovigilance programs must play an important role in disseminating safety alerts to their healthcare professionals. These results cannot be compared with those of other studies because studies with similar designs are lacking. Findings of this sort are also contingent on the study period assessed and are probably not informative about drug consumption or the number of drugs available for particular medical specialties. ADRs for which alerts are issued are unlikely to be identified in clinical trials because they are rare and unpredictable. In addition, the most frequent types of ADRs in this analysis were infections and neoplasms, which are type A effects related to the mechanism of action of antineoplastic agents, the most frequent drug class alerted, and consequently, these ADRs are frequent and predictable. In contrast to other studies, the drugs most frequently addressed in the alerts reported in this study were antidepressants, antidiabetics, anti-infectives, and anti-inflammatory drugs (Farcaş et al., 2018; Ferreira et al., 2020; Perry et al., 2020; Meddra, 2021). Hepatitis, death, and congenital malformations were the most frequent ADR alerts with the first two being the most frequently reported ADRs in other studies (Farcaş et al., 2018; Meddra, 2021). Nevertheless, information about birth defects is often lacking when a drug is marketed because pregnant women are excluded from clinical trials for ethical reasons. Both reported drugs and ADRs are in line with safety signals assessed by PRAC between 2014 and 2017 (Farcaş et al., 2018).
Regarding the regulatory actions resulting from these alerts, one-third led to a restriction in the use of the medicine, which is a finding that was similar to that reported in another study in which the most frequent restriction involved adding new contraindications for use (Perry et al., 2020). Some of the reported ADRs in this study were sufficiently serious and clinically important enough to require changes to their labeling or to be classified with an unfavorable benefit/risk ratio, leading to their withdrawal from the market. Changes or updates in the SmPC were frequent regulatory actions for almost half of the medicines in this and other studies (Lester et al., 2013; Ferreira et al., 2020; Perry et al., 2020). This was specifically true for nationally authorized drugs, which are more likely to be updated than EU-authorized products (van Hunsel et al., 2021). In contrast, drug withdrawal tended to be infrequent and occurred in approximately 5%–7% of alerted drugs, with several prior informational alerts preceding the suspension of the medicine (Soares et al., 2015; Ferreira et al., 2020). In our study, most of these drugs had been available in the Spanish market for decades. The causes of withdrawal of these drugs were heterogeneous, and for a few drugs the suspension of marketing authorization was due to multiple reasons. The most frequent reported causes for withdrawal from the market were liver, cardiac, and nervous system toxicity, and the median interval between the first report of an ADR pointing to withdrawal and actual withdrawal itself was approximately 1 year (Onakpoya et al., 2016a). However, there are discrepancies in the patterns of withdrawal of medicinal products from the market and withdrawals are inconsistent across countries (Onakpoya et al., 2016b). These findings underline the need for continuous monitoring of risks throughout the lifecycle of a medicine.
This study has several limitations. First, only the alerts issued in Spain were evaluated; however, evaluations and decisions taken by the EMA were involved in more than half of them. A second limitation was the short length of the study period of only 7 years; nonetheless, we consider this period sufficiently representative to characterize the issued alerts. The reason why we did not include alerts issued during 2020 and 2021 should be self-evident because the outbreak of the COVID-19 pandemic quickly changed the profile of alerts and their focus shifted to drugs used to treat the coronavirus and vaccines. Another limitation is that we did not include Direct Healthcare Professional Communications (formerly called ‘Dear Doctor Letter’), which are issued by pharmaceutical companies to inform healthcare providers of important drug-related safety issues and follow a different safety assessment process. Finally, we did not assess the impact of alerts on the prescription of the medicines concerned and trends in their consumption. Nevertheless, the impact of risk minimization has already been assessed in various other studies (The European Network of Centres for Pharmacoepidemiology and Pharmacovigilance).
Our study also has certain strengths. This is the first study to analyze drug alerts issued by the Spanish Medicines Agency. Additionally, it is the first study to devote special attention to alerts involving drugs under “additional monitoring” and also the first to break down the analysis according to the population targeted and the medical specialty involved.
Pharmacovigilance systems should ensure monitoring of all authorized medicines throughout their lifecycle in clinical use, and regulatory authorities should continuously review their benefit/risk ratio and issue an alert whenever a new signal or risk is detected. Over a 7-year period, almost one hundred alerts were issued by the Spanish’s Medicines Agency. Despite the limitations, spontaneous reporting was the most frequent source of information for the basis of alerts and reinforced the idea that such reporting is an essential tool for drug and patient safety. Drugs “under additional monitoring” were frequently implicated in alerts due to their unknown safety profile. The most frequent regulatory action resulting from an alert was changes in the SmPC, and one in ten alerts resulted in the withdrawal of the drug from the market. Our findings underline the important contribution of the spontaneous reporting of ADRs and the need for a safety assessment throughout the lifecycle of medicines, particularly when ‘vulnerable’ patient populations, such as children and pregnant women, are involved.
The raw data supporting the conclusions of this article will be made available by the authors without undue reservation.
EM: conceived the idea, contributed to the study design, conducted the literature search, reviewed the cases selected, contributed to data acquisition of included cases, analysis and interpretation of the data, and writing of the first draft of the manuscript. JS: contributed to the study design, reviewed the cases selected, contributed to data acquisition of included cases, analysis and interpretation of the data, and writing of the manuscript. All authors agree to be accountable for the content of this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agencia estatal boletín oficial del estado (2022). RD 577/2013 de farmacovigilancia de medicamentos de uso humano. Available at: https://www.boe.es/eli/es/rd/2013/07/26/577 (accessed Oct 23, 2022).
Alves, C., Macedo, A. F., and Marques, F. B. (2013). Sources of information used by regulatory agencies on the generation of drug safety alerts. Eur. J. Clin. Pharmacol. 69 (12), 2083–2094. doi:10.1007/s00228-013-1564-y
Baldo, P., Francescon, S., and Fornasier, G. (2018). Pharmacovigilance workflow in Europe and Italy and pharmacovigilance terminology. Int. J. Clin. Pharm. 40 (4), 748–753. doi:10.1007/s11096-018-0711-z
Bhasale, A. L., Sarpatwari, A., De Bruin, M. L., Lexchin, J., Lopert, R., Bahri, P., et al. (2021). Postmarket safety communication for protection of public health: A comparison of regulatory policy in Australia, Canada, the European union, and the United States. Clin. Pharmacol. Ther. 109 (6), 1424–1442. doi:10.1002/cpt.2010
Bjerre, L. M., Parlow, S., de Launay, D., Hogel, M., Black, C. D., Mattison, D. R., et al. (2018). Comparative, cross-sectional study of the format, content and timing of medication safety letters issued in Canada, the USA and the UK. BMJ Open 8 (10), e020150. doi:10.1136/bmjopen-2017-020150
Clavenna, A., and Bonati, M. (2009). Adverse drug reactions in childhood: a review of prospective studies and safety alerts. Arch. Dis. Child. 94 (9), 724–728. doi:10.1136/adc.2008.154377
Conroy, S., Choonara, I., ImPicciatore, P., Mohn, A., Arnell, H., Rane, A., et al. (2000). Survey of unlicensed and off label drug use in paediatric wards in European countries. European Network for Drug Investigation in Children. BMJ 320 (7227), 79–82. doi:10.1136/bmj.320.7227.79
Davies, E. A., and O'Mahony, M. S. (2015). Adverse drug reactions in special populations - the elderly. Br. J. Clin. Pharmacol. 80 (4), 796–807. doi:10.1111/bcp.12596
European Medicines Agency (2004). ICH topic E 2 D. Post approval safety data management. CPMP/ICH/3945/03. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-12.pdf (accessed Oct 23, 2022).
European Medicines Agency (2022). Medicines under additional monitoring. European Medicines Agency. Available at: https://www.ema.europa.eu/en/human-regulatory/post-authorisation/pharmacovigilance/medicines-under-additional-monitoring (accessed Oct 23, 2022).
European Medicines Agency (2022a). Pharmacovigilance: Overview. European Medicines Agency. Available at: https://www.ema.europa.eu/en/human-regulatory/overview/pharmacovigilance-overview (accessed Oct 23, 2022).
European Medicines Agency (2022b). Guideline on good pharmacovigilance practices (GVP) Module IX—signal management. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-ix-signal-management-rev-1_en.pdf (accessed Oct 23, 2022).
Farcaş, A., Mahalean, A., Bulik, N. B., Leucuta, D., and Mogoșan, C. (2018). New safety signals assessed by the pharmacovigilance risk assessment committee at EU level in 2014-2017. Expert Rev. Clin. Pharmacol. 11 (10), 1045–1051. doi:10.1080/17512433.2018.1526676
Farcas, A., Balcescu, T., Anghel, L., Bucsa, C., and Mogoșan, C. (2020). A description of medicines-related safety issues evaluated through a referral procedure at the EU level after 2012. Expert Opin. Drug Saf. 19 (6), 755–762. doi:10.1080/14740338.2020.1744561
Ferreira, V. C., Ferreira, G. C., and Baldoni, A. O. (2020). Thirteen years of medication alerts issued by the Brazilian Health Regulatory Agency (ANVISA): What is the profile? J. Eval. Clin. Pract. 26 (3), 957–961. doi:10.1111/jep.13233
Irl, C., and Hasford, J. (2020). Assessing the safety of drugs in pregnancy: the role of prospective cohort studies. Drug Saf. 22 (3), 169–177. doi:10.2165/00002018-200022030-00001
Ishiguro, C., Misu, T., Iwasa, E., and Izawa, T. (2017). Analysis of safety-related regulatory actions by Japan's pharmaceutical regulatory agency. Pharmacoepidemiol. Drug Saf. 26 (11), 1314–1320. doi:10.1002/pds.4252
Lester, J., Neyarapally, G. A., Lipowski, E., Graham, C. F., Hall, M., and Dal Pan, G. (2013). Evaluation of FDA safety-related drug label changes in 2010. Pharmacoepidemiol. Drug Saf. 22 (3), 302–305. doi:10.1002/pds.3395
Martin, K., Begaud, B., Latry, P., Miremont-Salame, G., Fourrier, A., and Moore, N. (2004). Differences between clinical trials and postmarketing use. Br. J. Clin. Pharmacol. 57 (1), 86–92. doi:10.1046/j.1365-2125.2003.01953.x
MEDDRA (2021). Introductory guide MedDRA version 24.0. Available at: https://www.meddra.org/how-to-use/support-documentation/english (accessed Oct 23, 2022).
Mol, P. G., Straus, S. M. J. M., Piening, S., de Vries, J. T. N., de Graeff, P. A., and Haaijer-Ruskamp, F. M. (2010). A decade of safety-related regulatory action in the Netherlands: a retrospective analysis of direct healthcare professional communications from 1999 to 2009. Drug Saf. 33 (6), 463–474. doi:10.2165/11532840-000000000-00000
N'Guessan-Irié, A. G., Yavo, J. C., Guillaume Amari, A. S., and Yapi, A. D. (2012). Summarizing of medicinal alerts in ivory Coast from 2001 till 2010. Therapie 67 (3), 251–256. doi:10.2515/therapie/2012029
Notas informativas, medicamentos de uso humano. Agencia Española de Medicamentos y Productos Sanitarios (2022). Available at: https://www.aemps.gob.es/acciones-informativas/notas-informativas-medicamentos-de-uso-humano/?cat=266&tag=seguridad-8 (accessed Oct 23, 2022).
Onakpoya, I. J., Heneghan, C. J., and Aronson, J. K. (2016a). Worldwide withdrawal of medicinal products because of adverse drug reactions: a systematic review and analysis. Crit. Rev. Toxicol. 46 (6), 477–489. doi:10.3109/10408444.2016.1149452
Onakpoya, I. J., Heneghan, C. J., and Aronson, J. K. (2016b). Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med. 1417 (1), 1056. doi:10.1186/s12916-016-0553-2
Perry, L. T., Bhasale, A., Fabbri, A., Lexchin, J., Puil, L., Joarder, M., et al. (2020). A descriptive analysis of medicines safety advisories issued by national medicines regulators in Australia, Canada, the United Kingdom and the United States - 2007 to 2016. Pharmacoepidemiol. Drug Saf. 29 (9), 1054–1063. doi:10.1002/pds.5072
Potts, J., Genov, G., Segec, A., Raine, J., Straus, S., and Arlett, P. (2020). Improving the safety of medicines in the European union: From signals to action. Clin. Pharmacol. Ther. 107 (3), 521–529. doi:10.1002/cpt.1678
Rodriguez Pascual, A. (2006). “Alertas y reacciones adversas de medicamentos,” in Curso de Actualización Pediatria (Madrid: Exlibris Ediciones), 69–76. En: AEPap ed.
Rothwell, P. M. (2005). External validity of randomised controlled trials: "to whom do the results of this trial apply? Lancet 365 (9453), 82–93. doi:10.1016/S0140-6736(04)17670-8
Soares, S., Roque, F., Teixeira Rodrigues, A., Figueiras, A., and Herdeiro, M. T. (2015). Safety alerts: An observational study in Portugal. Clin. Ther. 37 (9), 2122–2128. doi:10.1016/j.clinthera.2015.07.015
The European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP). “ENCePP guide on methodological standards in Pharmacoepidemiology,” in Chapter 15.4. Methods for pharmacovigilance impact research. Available at: https://www.encepp.eu/standards_and_guidances/methodologicalGuide15_4.shtml (Accessed Nov 7, 2022).
Uppsala Monitoring Centre (2022). The WHO programme for international drug monitoring. Available at: https://www.who-umc.org/global-pharmacovigilance/who-programme-for-international-drug-monitoring/(accessed Oct 23, 2022).
van Hunsel, F., de Jong, E., Gross-Martirosyan, L., and Hoekman, J. (2021). Signals from the Dutch national spontaneous reporting system: Characteristics and regulatory actions. Pharmacoepidemiol. Drug Saf. 30 (8), 1115–1122. doi:10.1002/pds.5246
WHO Collaborating Centre for Drug Statistics Methodology (2018). Guidelines for ATC classification and DDD assignment 2019. Oslo, Norway. Available at: http://www.whocc.no (accessed Oct 23, 2022).
Keywords: pharmacovigilance, safety-related regulatory actions, adverse drug reaction, spontaneous reporting systems, post-marketing surveillance
Citation: Montané E and Santesmases J (2023) Characteristics of drug safety alerts issued by the Spanish Medicines Agency. Front. Pharmacol. 14:1090707. doi: 10.3389/fphar.2023.1090707
Received: 05 November 2022; Accepted: 11 January 2023;
Published: 30 January 2023.
Edited by:
Fátima Roque, Instituto Politécnico da Guarda, PortugalReviewed by:
Xavier Kurz, EMA, NetherlandsCopyright © 2023 Montané and Santesmases. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eva Montané, ZW1vbnRhbmUuZ2VybWFuc3RyaWFzQGdlbmNhdC5jYXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.