 Lara J. Bou Malhab
Lara J. Bou Malhab Habiba Alsafar
Habiba Alsafar Saleh Ibrahim4
Saleh Ibrahim4 Mohamed Rahmani
Mohamed Rahmani
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 20 February 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1086946
Proteolysis targeting chimeras (PROTACs) are heterobifunctional small molecules that uses the proteasome ubiquitin system to target proteins of interest and promote their degradation with remarkable selectivity. Importantly, unlike conventional small molecule inhibitors, PROTACs have proven highly effective in targeting undruggable proteins and those bearing mutations. Because of these considerations, PROTACs have increasingly become an emerging technology for the development of novel targeted anticancer therapeutics. Interestingly, many PROTACs have demonstrated a great potency and specificity in degrading several oncogenic drivers. Many of these, following extensive preclinical evaluation, have reached advanced stages of clinical testing in various cancers including hematologic malignancies. In this review, we provide a comprehensive summary of the recent advances in the development of PROTACs as therapeutic strategies in diverse hematological malignancies. A particular attention has been given to clinically relevant PROTACs and those targeting oncogenic mutants that drive resistance to therapies. We also discus limitations, and various considerations to optimize the design for effective PROTACs.
Hematological malignancies are a highly heterogeneous group of blood cancers caused by abnormal differentiation of hematopoietic stem cells. Despite the remarkable advances in targeted therapy in hematological malignancies, chemotherapy is still the most common strategy. However, a major concern of chemotherapy is the side effects and long-term sequelae. Targeted therapies have been primarily employing either monoclonal antibodies or small molecules inhibitors. However, each of these approaches has advantages and disadvantages (Lee et al., 2018; Wilkes, 2018). Monoclonal antibodies are known for their high selectivity, high binding affinity, prolonged pharmacokinetic profile, and efficacy in blocking extracellular protein-protein interactions (Lu et al., 2020). Nevertheless, they are large units, which restrict them from crossing cell membrane and, consequently, their use is largely limited to cell surface targets. In addition, oral bioavailability of monoclonal antibodies is quite limited due to their poor ability to cross the intestinal epithelium and their susceptibility to the proteolytic degradation by digestive enzymes (Singh et al., 2008). On the other hand, small molecule inhibitors can be easily administered orally and are able to target intracellular proteins due to their cellular permeability. However, small molecules inhibitors recognize specific pockets or active sites within the protein targets which are lacking in the majority of human proteins particularly transcription factors, non-enzymatic proteins, and scaffold proteins (Toure and Crews, 2016; An and Fu, 2018).
While a number of new small molecule inhibitors and monoclonal antibodies have shown great activities in various hematological malignancies (Podhorecka et al., 2014; Hou et al., 2021; Sochacka-Cwikla et al., 2021), targeted protein degradation using Proteolysis Targeting Chimeras (PROTACs) has emerged as a promising approach in these as also in other types of malignancies (He et al., 2020a). This strategy exploits the ubiquitin-proteasome system (UPS) to target various proteins of interest for degradation. Notably, several PROTAC compounds have entered clinical evaluations in various tumors including hematological malignancies. While the BTK degrader NX-2127 has recently entered phase 1a/b clinical trial in patients with relapsed and refractory B-cell malignancies (NCT04830137), ARV-471 and ARV-110 have shown promising results in phase1/2 clinical trial in locally advanced or metastatic ER+/HER2− breast cancer (NCT04072952) or metastatic castration-resistant prostate cancer (NCT03888612) respectively. In this review, we discuss the recent advances, limitations, and future directions of PROTACs in hematological malignancies.
The ubiquitin-proteasome system (UPS) is the main proteolytic system in eukaryotes that controls proteins degradation and regulates different cellular processes such as stress responses, DNA repair, cell proliferation, apoptosis, etc. This has made the UPS a powerful and essential machine in maintaining protein quality control and homeostasis (Park et al., 2020a).
The first step of a protein degradation by UPS is its modification with ubiquitin tag, a signal for recognition and degradation by the proteasome 26S subunit (Park et al., 2020b). The ubiquitination process involves the covalent attachment of ubiquitin to lysine residues on the substrate protein via a three steps enzymatic cascade reaction involving the E1, E2, and E3 enzymes. First, ubiquitin is activated by the E1 ubiquitin-activating enzyme (E1) following a covalent linkage between the carboxyl-terminus of ubiquitin and a cysteine residue on the E1 enzyme forming a thioester bond (E1-ubq). Then ubiquitin is transferred to an E2 conjugating enzyme (E2-ubq), and lastly, E3 ligases transfer ubiquitin from the E2 to the substrate. There are three families of structurally and functionally distinct E3 ubiquitin ligases: 1) The Really Interesting New Gene (RING), which constitutes the largest family of E3 ligases. These E3 ligases are multi-subunit complexes that use specific Cullins as central molecular scaffolds to recruit the targeted substrates and bring them in a close proximity to the ubiquitin-charged E2 (E2-ubq) enzymes. In this case, the ubiquitin is directly transferred from E2-ubq to the substrate without the necessity to form a thioester bond with ubiquitin (Mani and Gelmann, 2005). 2) the Homologous to E6-AP Caboxy Terminus (HECT) family: HECT E3 ligases undergo a catalytic cysteine-dependent trans-thiolation reaction with E2-ubq forming an intermediate covalent E3-ubq bond prior to ubiquitin transfer to the substrate (Huibregtse et al., 1995; Scheffner et al., 1995). 3) The RING-Between-RING (RBR) family: These ligases have two canonical RING domains RING1 and RING2 linking an in-between RING (IBR) domain (Wenzel et al., 2011). They need at least four cycles of tagging the substrate to form a polyubiquitin chain allowing its recognition and degradation by the 26S proteasome system (Hershko et al., 1983; Voges et al., 1999; Ciechanover, 2005; Schulman and Harper, 2009).
Protein ubiquitination is a very dynamic and highly reversible process. It is often counteracted by deubiquitinating enzymes (DUBs), which remove the ubiquitin chain from the targeted substrate preventing its degradation (Komander et al., 2009).
Proteolysis targeting chimeras (PROTACs) is a strategy that induces the degradation of target proteins using the ubiquitin-proteasome system as illustrated in Figure 1. PROTACs are heterobifunctional small molecules containing three chemical elements: A ligand that binds the protein of interest, a second ligand that recruits an E3 ubiquitin ligase, and a linker that conjugates these two ligands (Gao et al., 2020). Contrary to small molecule inhibitors, PROTACs technology eliminates the targeted protein through degradation instead of its inhibition (Ciulli and Trainor, 2021). Once the complex (target substrate-PROTAC-E3 ligase) is formed, the E3 ligase employs an E2 ubiquitin-conjugating enzyme to transfer ubiquitin to the substrate. The poly-ubiquitinated substrate will be recognized and degraded by the proteasome system (Paiva and Crews, 2019).
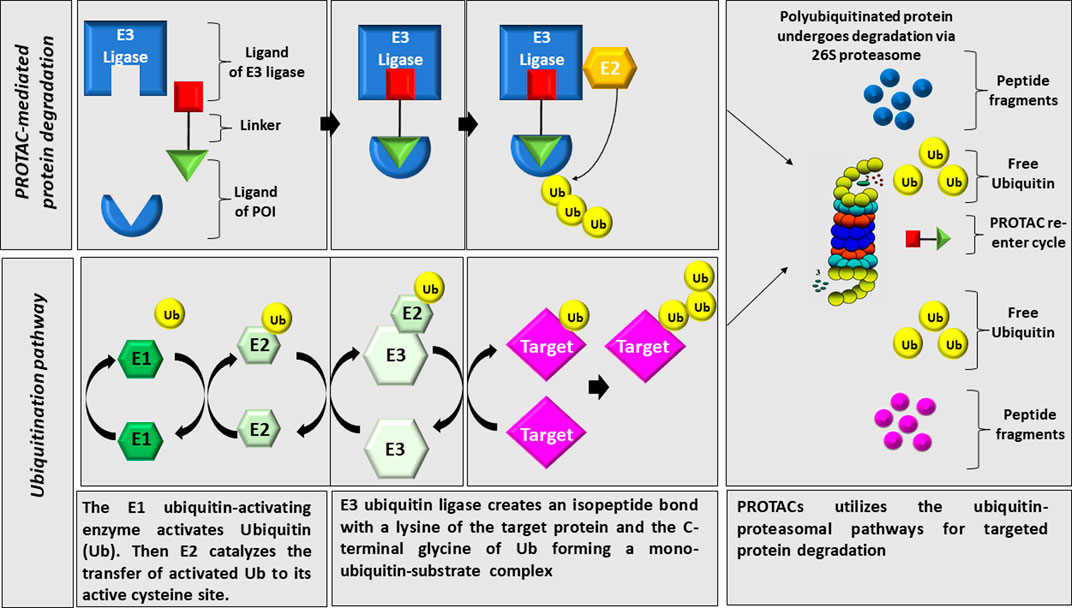
FIGURE 1. Illustration of PROTAC technology. PROTACs utilizes the ubiquitin-proteasomal pathways for targeted protein degradation through linking an E3 ligase to the protein of interest leading to its polyuiquitination and proteasomal degradation.
Despite the abundance of E3 ligases (more than 600) expressed in human cells, only few of them have been used in PROTAC technology to degrade target proteins (Fisher and Phillips, 2018). The field has tremendously evolved since the use of the first PROTAC by Sakamoto et al., in 2001 where a poorly permeable phospho-peptide moiety was employed to hijack Skp1-Cullin-F box complex (SCFß-TRCP) to degrade methionine aminopeptidase-2 (MetAp-2) (Sakamoto et al., 2001). In 2003, the same group have developed PROTACs that can target the estrogen receptor-alpha (ER-α) or androgen receptor (AR). These first developed PROTACs were peptide-based with a high molecular weight and very limited cell permeability (Sakamoto et al., 2003). From that point onwards, a number of substrate receptors of E3 ligases were discovered including inhibitors of apoptosis proteins (IAPs), Cereblon (CRBN), and Von Hippel-Lindau (VHL) among others. Many of these have been explored to generate permeable and biologically active PROTACs capable of degrading selected proteins in target cells. The CRBN and VHL have been the most successfully utilized E3 ligase substrate receptors in PROTAC system in hematological malignancies (Ito et al., 2010; Itoh et al., 2010; Buckley et al., 2012a).
CRBN has successfully been used in the development of PROTAC compounds targeting many proteins in different diseases including various cancers (Bricelj et al., 2021). It is a 442-amino acid protein that acts as a substrate receptor within the Cullin-4-RING E3 ubiquitin ligase (CRL4) complex (Ito et al., 2010). In addition to the molecular scaffold CUL4, this complex also includes the adaptor protein DDB1 (damages DNA-binding protein 1), and the ROC1 protein which recruits the ubiquitin-loaded E2 enzymes (Higa and Zhang, 2007; Jackson and Xiong, 2009; Ito and Handa, 2020) (Figure 2). It is important to note that major advances in this field have been catalyzed by the discoveries from the work of Benjamin Ebert’s group that the direct molecular target of thalidomide and other derived immunomodulatory drugs (IMiDs) such as lenalidomide and pomalidomide is CRBN E3 ligase (Kronke et al., 2014a; Kronke et al., 2014b; Kronke et al., 2015; Sievers et al., 2018). These studies revealed, for the first time, that binding of IMiDs to CRBN leads to increased recruitment of the zinc finger transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) to the E3 complex leading to their subsequent ubiquitination and proteasomal degradation (Kronke et al., 2014b; Chamberlain et al., 2014; Zhu et al., 2014; Kronke et al., 2015). This mechanism is believed to play a major role in the clinical activities of the immunomodulatory drugs (Kronke et al., 2014b; Chamberlain et al., 2014; Kronke et al., 2015).
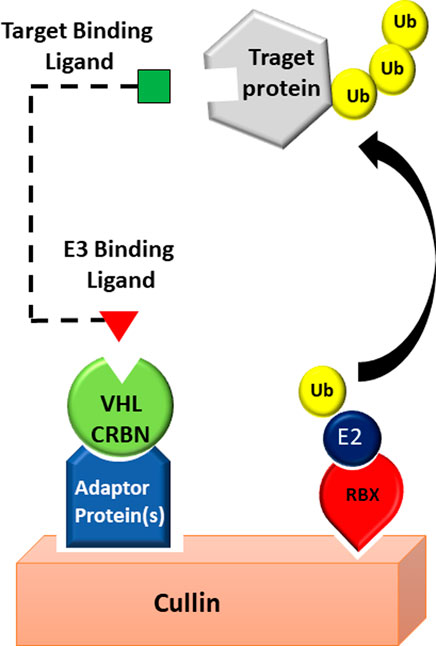
FIGURE 2. PROTAC technology exploits CRL complex. POI degradation via a CRBN or VHL based E3 ligase, where the POI is brought in close proximity to an E3 ligase.
Unlike CRBN which is a part of the Cullin-4-RING E3 ubiquitin ligase, VHL plays a central role in Cullin2 RING E3 ubiquitin ligase complex (CRL2VHL), a multiprotein complex containing the molecular scaffold CUL2, elongin B, elongin C, and Rbx-1, also known as ROC1 (Figure 2). VHL is the subunit that binds specifically to the target proteins (Czyzyk-Krzeska and Meller, 2004) and promote their proteasomal degradation. Among many of the substrates targeted by VHL E3 ligase, the hypoxia-inducible factor (HIF)-1α is the best characterized. VHL has been successfully utilized in PROTAC systems to target and degrade many proteins (Sun et al., 2019a). The initially developed VHL-based PROTACs utilized 5 to 7 amino acids long peptides derived from HIF-1α protein (Lee et al., 2007; Schneekloth et al., 2008) as molecular scaffold for the E3 ligase (VHL) instead of small-molecules and they are, therefore, referred to as “bioPROTACs”. The discovery of small-molecule mimetics of the HIF-1α peptide has led to a significant improvement in PROTACs design (Buckley et al., 2012a; Buckley et al., 2012b). One of the first PROTACs using small molecules as VHL-recruiting scaffold was designed to target the bromodomain proteins (BRDs) (Zengerle et al., 2015). In these studies, the bromodomain inhibitor JQ1 was used as BRD4-recruiting scaffold.
IAPs and mouse double minute 2 (MDM2) E3 ligases which are highly expressed in hematological malignancies, have also been of particular interest for the design of PROTACs in this disease and were extensively reviewed elsewhere (Xi et al., 2019; He et al., 2020a).
Over the last two decades, a rapidly growing number of PROTACs have been developed in various hematological malignancies (Table 1).
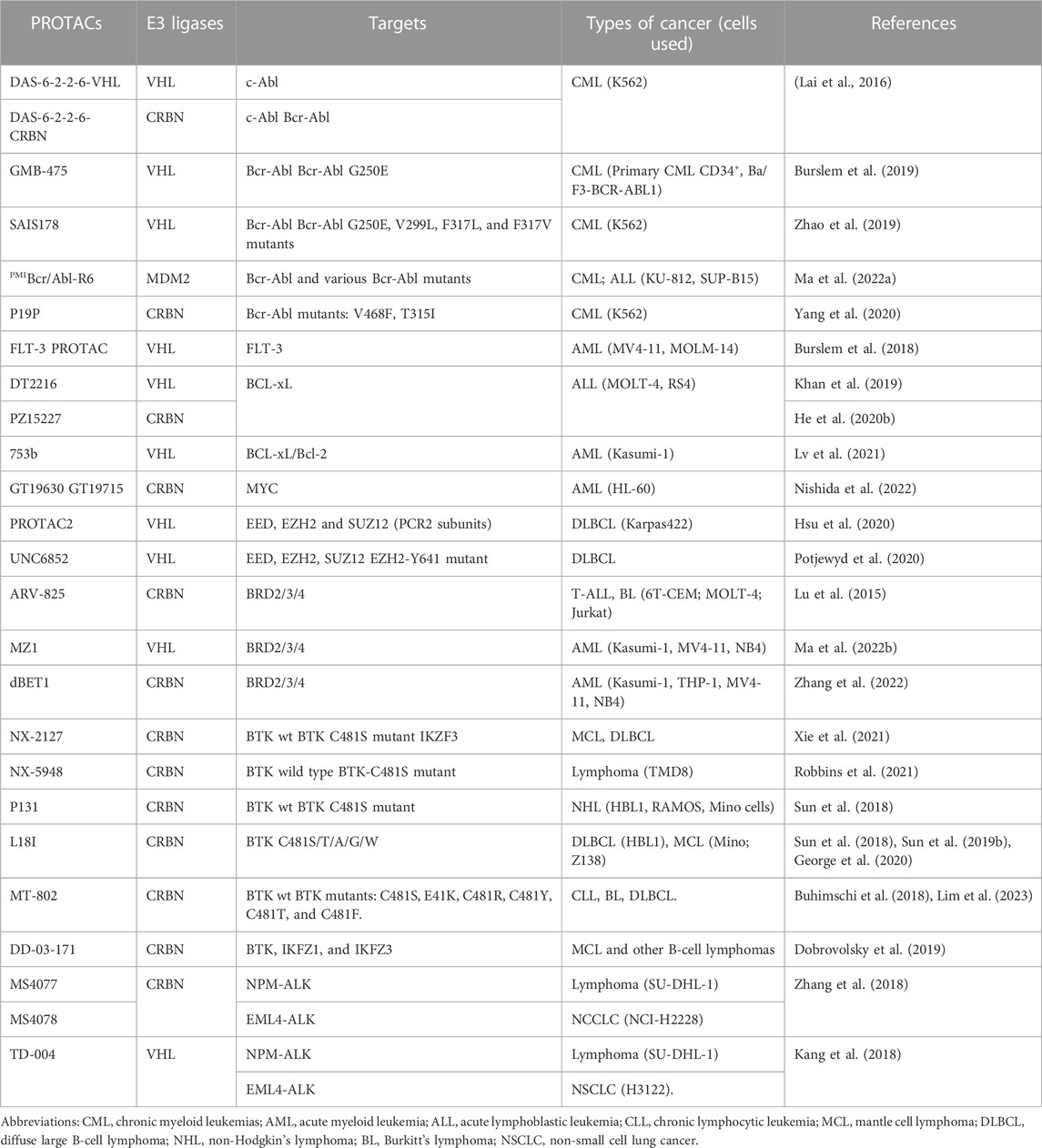
TABLE 1. Representative PROTACs in hematological malignancies.
While several BCR-ABL inhibitors (e.g., imatinib, dasatinib, bosutinib, nilotinib, asciminib) are highly effective in chronic myeloid leukemia (CML) (Wei et al., 2010; Keskin et al., 2016), many patients develop drug resistance due to mutations in the BCR-ABL gene (Hantschel et al., 2012). Efforts aiming at overcoming drug resistance in CML, have led to the development of diverse BCR-ABL PROTACs, some of which have shown great activity and selectivity towards various forms of BCR-ABL including mutants that confer resistance to the BCR-ABL inhibitors. It is important note that these studies have revealed that the capability of a PROTAC to induce the degradation of a given target does not only depend on its capacity to bind to the target, but it is determined by the cooperation of a collection of factors including the inhibitor warhead, the E3 ligase substrate receptor, the nature and the length of the linker, and the point of attachment of the linker on the PROTAC units.
In this regard, Crews group reported in 2016 the first BCR-ABL degraders (DAS-6-2-2-6) based on bosutinib or dasatinib capable of degrading c-ABL and BCR-ABL by using either CRBN or VHL E3 ligase substrate receptors (Lai et al., 2016). These studies revealed that treatment of the CML cell line K562 with 1 uM dasatinib-based DAS-VHL PROTAC led to the degradation of more than 65% of c-ABL protein, but it was ineffective against BCR-ABL. Noteworthy, DAS-VHL effectively engage its target Bcr-ABL as reflected by decreased Bcr-ABL downstream signaling, but did not lead to the degradation of this kinase. Interestingly, when CRBN was used instead of VHL, identical concentration of DAS-CRBN led to the degradation of both c-ABL (>85%) and BCR-ABL proteins (more than 85% and 60% respectively). Few years later, in 2019, new PROTAC called GMB-475 was developed using the GNF5, a small-molecule allosteric inhibitor which binds with high affinity to the myristoyl pocket of ABL kinase. Such compound has the ability to degrade both wild type BCR-ABL and BCR-ABL bearing certain mutations at nanomolar concentrations (Burslem et al., 2019). GMB-475 was particularly effective in degrading BCR-ABL bearing G250E mutation and exhibited a marked antiproliferative activity in cells with such mutation. In addition, GMB-475 exhibited a highly selectivity toxicity toward primary CML CD34+ cells, versus normal hematopoietic progenitor CD34+ cells.
Another BCR-ABL PROTAC, SIAIS178, in which dasatinib was linked to a VHL ligand was developed by Zhao and colleagues (Zhao et al., 2019). In contrast to DAS-VHL PROTAC developed earlier, SIAIS178 showed efficient degradation of BCR-ABL with a DC50 value of 8.5 nM and an antiproliferative effect with an IC50 of 24 nM in K562 cells. The differential activity of these dasatinib- and VHL-based PROTACs is probably due to the linker optimization achieved in SIAIS178. In vivo studies showed a significant tumor regression following exposure to SIAIS178 in a K562-derived xenograft tumor model. It is important to note that, in addition to its activity in wild type BCR-ABL, SIAIS178 also successfully recognized and degraded several clinically relevant resistance-conferring BCR-ABL mutations such as G250E, V299L, F317L, and F317V, but not T315I. Importantly, Yang et al., reported a series of PROTACs (e.g., P19P) capable of degrading dasatinib-resistant T315I and asciminib-resistant V468F mutations in BCR-ABL (Yang et al., 2020).
In a more recent study, Ma et al. have developed a compound referred to as PMIBcr/Abl-R6 that has the potential to degrade BCR-ABL regardless of its mutation status (Ma et al., 2022a). PMIBcr/Abl-R6 is a dual-targeting PROTAC with a particular design involving an MDM2/p53 inhibitor peptide sequence and Bcr/Abl tetramerization domain. PMIBcr/Abl-R6 interacts with Bcr/Abl oligomerization domain and binds the ubiquitin E3 ligase MDM2 with high affinity. Consequently, PMIBcr/Abl-R6 has the potential to degrade all forms of Bcr-Abl (p210, p190, p185) and Bcr/Abl mutants including the T315I, and to activate p53 (Ma et al., 2022a). In fact, PMIBcr/Abl-R6 showed a significant efficacy in various primary samples isolated from patients with CML and acute lymphoblastic leukemia (ALL), in association with Bcr/Abl degradation and p53 activation (Ma et al., 2022a). Noteworthy, one of these ALL patients bears Y253H, E255K/V, and T315I mutations.
FLT-3 gene mutations are the most common mutations in acute myeloid leukemia (AML) (Kiyoi and Naoe, 2002). Midostaurin and gilteritinib are two FDA approved FLT-3 inhibitors especially for patients with FLT-3 mutated AML (Antar et al., 2020). Unfortunately, due to the development of secondary resistance (McMahon et al., 2019), high doses of the inhibitor should be administered to achieve an efficient clinical response which creates off-target toxicities (Grunwald and Levis, 2013). A recent study by Burslem et al., has described a VHL-recruiting FLT-3 PROTAC employing the FLT-3 inhibitor quizartinib. Such compound exhibited high efficiency in degrading FLT-3 ITD in MV4-11 and MOLM-14 AML cells both in vitro and in in vivo xenograft model (Burslem et al., 2018).
Apoptosis is a well-characterized and highly regulated mechanism of cell death involving both mitochondrial and non-mitochondrial pathways (Hafezi and Rahmani, 2021). Any deregulation in this mechanism leads to several diseases including tumorigenesis (Plati et al., 2011). Mitochondrial apoptosis is regulated by protein-protein interactions among BCL-2 family members, which control mitochondrial outer membrane permeabilization (MOMP). BCL-2 family members are divided into two functionally and structurally distinct groups: Antiapoptotic proteins (BCL-xL, Mcl-1, Bcl-W, and BFL-1/A1) and pro-apoptotic proteins (BIK, BIM, BID, BAD, BMF, HRK, NOXA, PUMA, BAX and BAK) (Hafezi and Rahmani, 2021). Various BCL-XL or BCL-xL/BCL-2 inhibitors were developed (e.g., Navitoclax also referred to as ABT-263), however, most of them engendered on-target and dose-dependent platelet toxicities as a consequence of the essential role that BCL-xL plays in human platelets survival.
To overcome platelets toxicity of these inhibitors, a PROTAC approach based on VHL or CRBN and ABT-263 have led to the generation of a number of chemically and biologically active compounds among which DT2216 and PZ15227 were the most promising.
DT2216, a VHL-recruiting ABT-263-based PROTAC was recently developed by Khan and colleagues (Khan et al., 2019) and has demonstrated greater affinity to BCL-xL, and yet, showed much lower toxicity to platelets compared to the parent compound ABT-263. Importantly, the anti-tumor activity of DT2216 was considerably more potent than that of ABT-263 in AML cells both in vitro and in in vivo xenograft mouse model. In addition, DT2216 caused only mild reduction in platelet counts and no sign of reactive thrombocytopenia was observed in mice exposed to this agent (Khan et al., 2019). In a subsequent study, the same group has applied a series of modifications involving different types of linker and varying the attachment points on ABT-263 and E3 ligase ligands. These efforts have led to the development of a dual BCL-2/BCL-XL degrader 753b which exhibits considerable increase in potency compared to the BCL-XL targeting DT2216. These studies also provided evidence that the accessibility of lysines on a target protein is critical in determining the selectivity and potency of a PROTAC for such protein (Lv et al., 2021).
Interestingly, similar results were obtained using another ABT-263-based PROTAC PZ15227 which targets BCL-XL to CRBN E3 ligase for degradation (He et al., 2020b). In vitro studies using AML cell lines, revealed that in contrast to ABT-263, PZ15227 exhibits a significant selective toxicity toward malignant cells versus platelets. This was also recapitulated in in vivo xenograft mouse model. Importantly, PZ15227 resulted in only a moderate thrombocytopenia compared to a similar dose of ABT-263.
The low toxicity of PROTAC compounds to platelets would likely be explained, at least in part, by the low expression levels of the VHL and CRBN E3 ligases in these cells.
Myc is one of the most frequently dysregulated genes in human cancer including hematological malignancies. It is overexpressed through a variety of mechanisms particularly chromosomal rearrangements. Despite that Myc oncogenic potential has been clearly demonstrated for decades, targeting such transcriptional factor has been challenging. It is important to note that PROTAC approach has not been very successful in directly targeting Myc so far, and the most effective way to interfere with Myc activity is to inhibit its transcriptional regulators such as BET bromodomain protein 4 (BRD4). Importantly, some PROTAC compounds such as the CRBN-based GT19630 and GT19715 have shown potent preliminary activity in AML both in vitro and in vivo mouse model (Nishida et al., 2022). Specifically, GT19630 and GT19715 effectively degraded c-Myc protein in HL-60 cells with an IC50 1.4 nM and 1.8 nM respectively. In a xenograft mouse model with HL-60 cells, very low dose GT19630 (0.3mg/kg/bid) resulted in a marked c-Myc degradation and tumor growth inhibition. Of note, GT19715 showed greater activity in venetoclax resistant MV4-11 cells, which exhibit increased c-Myc level, compared to venetoclax-sensitive parental cells (Nishida et al., 2022).
The bromodomain and extraterminal (BET) domain protein family which includes BRD2, BRD3, BRD4 has been linked to the development of many tumors including hematological malignancies (Zuber et al., 2011; Alsarraj and Hunter, 2012; Segura et al., 2013; Asangani et al., 2014; Valent and Zuber, 2014; Liao et al., 2016; Wu et al., 2021). These considerations have prompted the search for small molecule inhibitors against BRD4 and some of these compounds have been leveraged to generate PROTACs against this oncogenic factor. Among these, ARV-825 PROTAC in which the BRD4 inhibitor OTX015 was linked to the E3 ligase CRBN binding inhibitor pomalidomide. AV-825 was highly effective in inducing BRD4 proteasomal degradation (Lu et al., 2015). In addition to BRD4, ARV-825 also induces the degradation of BRD3 and BRD2 in T-ALL cells. Importantly, ARV-825 suppressed T cell acute lymphoblastic leukemia (T-ALL) cell proliferation in vitro via cell cycle arrest and apoptosis. Such effect is more potent than those of BRD4 inhibitors such as JQ1, dBET1, and OTX015. ARV-825 was also very effective in reducing tumor growth in xenograft mouse model. Mechanistic studies have revealed that ARV-825 inhibited cell proliferation through BET and c-Myc depletion in vitro as well as in vivo (Wu et al., 2021).
A very recent study in AML reported a highly promising PROTAC compound MZ1, which targets and efficiently degrades BRD2, BRD3, and BRD4 proteins in various AML cell lines, and markedly suppress tumor growth in a xenograft mouse model (Ma et al., 2022b). MZ1 also downregulates cMyc which is positively regulated by BRD (Ma et al., 2022b). Another recently developed CRBN-based PROTAC dBET1 has also shown very potent activity in degrading BRD2, BRD3, and BRD4 proteins in association with a potent anti-tumor activity in various AML cell lines (Zhang et al., 2022).
BTK plays an essential role in B cell receptor (BCR) mediated B cells activation and proliferation (Mohamed et al., 2009). Ibrutinib, a BTK inhibitor, has been approved for the treatment of mantle cell lymphoma (MCL) and has been clinically evaluated as monotherapy or in combination in several malignancies including activated B cell-like (ABC) DLBCL (Pan et al., 2007). Unfortunately, MCL patients often develop drug resistance to ibrutinib due to C481S missense BTK mutation (Woyach et al., 2014). Recently two CRBN-based PROTAC BTK degraders NX-2127 and NX-5948 have entered clinical evaluations in a phase 1a/b study in patients with relapsed and refractory B-cell malignancies, whose disease progressed after at least 2 prior lines of therapy (NCT04830137 and NCT05131022 respectively).
In 2018, two PROTACs for ibrutinib-resistant BTK degradation were developed, P131 and L18I (Sun et al., 2018; Sun et al., 2019b). These studies, showed that these CRBN-based PROTACs were capable of degrading both the wild-type and ibrutinib-resistant C481S BTK protein at low concentrations. Interestingly unlike ibrutinib, BTK PROTACs showed a potent antiproliferative activities in cell bearing C481S BTK mutant (Sun et al., 2018). The antiproliferative activity of these compounds in DLBCL and MCL with wild-type BTK was also superior than that of ibrutinib (Sun et al., 2018). In addition, L18I, which is a second generation of BTK PROTAC, was not only highly effective against C481S BTK protein, but also potently degrades several other clinically relevant C481 mutations in B-cell tumors with a DC50 < 50 nM. More importantly, L18I induced rapid regression of C481S BTK HBL-1-derived xenograft tumors (George et al., 2020).
In another study, Crews’s group developed another highly effective BTK degrader, MT-802 (DC50: 14.9 nM) (Buhimschi et al., 2018; Lim et al., 2023). In contrast to ibrutinib, MT-802 was able to reduce the pool of active BTK in primary cells isolated from chronic lymphocytic leukemia (CLL) patients bearing C481S mutation (Buhimschi et al., 2018).
A number of other highly active BTK-specific degrader has been recently developed including DD-04-015, which degrade efficiently BTK only after 4 h exposure (Huang et al., 2018). Such compound was further optimized into DD-03-171 which has the ability to degrade the C481S BTK mutant and showed enhanced anti-proliferative effects on mantle cell lymphoma cells in vitro (5.1 nM) and significant efficacy towards patient-derived xenografts in vivo (Dobrovolsky et al., 2019).
Anaplastic lymphoma kinase (ALK) is a tyrosine kinase receptor found activated due to different genetic alterations (chromosomal translocations, substitution mutations and gene amplification) in many cancers including anaplastic large-cell lymphomas (ALCL) and diffuse large B-cell lymphomas (Webb et al., 2009). The most common ALK genetic alteration in lymphomas is NPM–ALK fusion protein that results from t(2;5) chromosomal rearrangement. Such aberration leads to ligand-independent constitutive activation of this tyrosine kinase. ALK is also frequently rearranged (EML4–ALK) in some solid tumors particularly non-small-cell lung carcinoma (NSCLC). Various ALK degraders were recently developed including MS4077, MS4078, TD-004. These PROTACs are capable of efficiently degrading NPM-ALK in lymphoma as well as EML4-ALK fusion proteins in NSCLC cells in association with a marked inhibition of cell growth (Kang et al., 2018; Zhang et al., 2018).
Polycomb repressive complex 2 (PRC2) has been widely linked to hematologic malignancies (Martin-Perez et al., 2010; Takamatsu-Ichihara and Kitabayashi, 2016; Iwama, 2017). PRC2 protein is composed of EZH2 (enhancer of zeste homolog 2), EED, SUZ12, RBAP46/48 and AEBP2 subunits (Herviou et al., 2016). The EZH2 is the catalytic subunit of PRC2. It catalyzes histone H3 methylation, a process in which both EED and SUZ12 subunits are required (Cao et al., 2002). Importantly, Loss-of function mutations in EZH2 and SUZ12 genes, which encode for central PCR2 components of PRC2 are frequently observed in patients with T-ALL (Ntziachristos et al., 2012; Simon et al., 2012; Zhang et al., 2012). Although effective EZH2 inhibitors exists, preclinical results show drug resistance due to the secondary mutations in both wild type and mutant EZH2 alleles (Gibaja et al., 2016). Two different PROTACs were generated using VHL ligand using different EED inhibitors and different linkers. PROTAC2 developed by Bloecher’s group in 2020, lead to a selective degradation of EED, EZH2 and SUZ12 reducing the proliferation of EZH-dependent tumor cells Karpas422 (Hsu et al., 2020). UNC6852, another EZH2 degrader that was developed by James group has shown similar results against the wild type EZH2, and interestingly also against the Y641N mutant EZH2 (Potjewyd et al., 2020).
Although cell permeability and target selectivity are major limitations of PROTAC system in therapeutics development, such approach has been highly effective in targeting and degrading many targets in various diseases including cancer. It is important to note that: 1) PROTACs are not equally effective in all tissues. For example, in a study by Zorba et al., a CRBN-based PROTAC compound (compound 10) effectively degrades BTK in mice spleen but not in the lungs despite similar compound delivery to both organs (Zorba et al., 2018). This could reflect tissue-specific differences in the expression of E3 ligases or other components of the ubiquitin system. 2) An important consideration when developing a PROTAC is the degree of expression of a particular E3 ubiquitin ligase, not only in the target cells but also in normal cells. It is conceivable that a high expression of an E3 ubiquitin ligase in aberrant cells makes it a good candidate to use in a PROTAC system. However, if normal cells also express high level of such factor the resulting PROTAC may lead to increased toxicity. 3) Another important consideration came from studies by Lv et al., using computational modelling which revealed that the PROTAC complex has highly selective ubiquitination activity toward lysine residues located on a defined position on the target protein (Lv et al., 2021). As both the selectivity and potency of a PROTAC is determined by the selectivity and potency of ubiquitination machinery, it is highly important to keep these considerations in mind when designing PROTACs.
Another important consideration when it comes to PROTAC design is the choice of the warhead which is critical for the potency and selectivity of PROTACs. This has been highlighted by the work from Lai and colleagues (Lai et al., 2016) in which they showed that dasatinib-based VHL-recruiting PROTACs degrades c-Abl but not Bcr-Abl, in contrast, dasatinib-based CRBN-recruiting PROTACs have the ability to target and degrade both c-Abl and Bcr-Abl. However, when the Bcr-Abl inhibitor bosutinib was used as a warhead instead of dasatinib in VHL-recruiting PROTACs, the activity of such PROTAC was lost against both c-Abl and Bcr-Abl (Lai et al., 2016).
Given the impressive preclinical activities, the wealth of PROTAC compounds diversity, and the numerous clinical trials testing them, such promising technology is poised to become a new therapeutic approach. However, to date most PROTACs are based on either CRBN or VHL. To enhance our ability to effectively and selectively degrade a wide range of protein targets, it would be critical to explore other E3 ubiquitin ligases and better understand the mechanisms of PROTAC off targets. While the PROTAC approach has particularly been focusing on cancer, such approach should also be explored in other diseases.
Overall, further improvement of the specificity and efficacy of these class of compounds will likely be the key for accelerating the development of this strategy in various types of cancer including hematological malignancies and potentially other diseases.
Conceptualization: MR and LJBM; writing original draft: LJBM and MR; critical revision and editing: MR, LJBM, SI, and HA. All authors have read and agreed to the published version of the manuscript.
This work was supported by Khalifa University award # FSU-2022-008.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alsarraj, J., and Hunter, K. W. (2012). Bromodomain-containing protein 4: A dynamic regulator of breast cancer metastasis through modulation of the extracellular matrix. Int. J. Breast Cancer 2012, 670632. doi:10.1155/2012/670632
An, S., and Fu, L. (2018). Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 36, 553–562. doi:10.1016/j.ebiom.2018.09.005
Antar, A. I., Otrock, Z. K., Jabbour, E., Mohty, M., and Bazarbachi, A. (2020). FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 34 (3), 682–696. doi:10.1038/s41375-019-0694-3
Asangani, I. A., Dommeti, V. L., Wang, X., Malik, R., Cieslik, M., Yang, R., et al. (2014). Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 510 (7504), 278–282. doi:10.1038/nature13229
Bricelj, A., Steinebach, C., Kuchta, R., Gutschow, M., and Sosic, I. (2021). E3 ligase ligands in successful PROTACs: An Overview of syntheses and linker attachment points. Front. Chem. 9, 707317. doi:10.3389/fchem.2021.707317
Buckley, D. L., Gustafson, J. L., Van Molle, I., Roth, A. G., Tae, H. S., Gareiss, P. C., et al. (2012). Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1α. Angew. Chem. Int. Ed. Engl. 51 (46), 11463–11467. doi:10.1002/anie.201206231
Buckley, D. L., Van Molle, I., Gareiss, P. C., Tae, H. S., Michel, J., Noblin, D. J., et al. (2012). Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc. 134 (10), 4465–4468. doi:10.1021/ja209924v
Buhimschi, A. D., Armstrong, H. A., Toure, M., Jaime-Figueroa, S., Chen, T. L., Lehman, A. M., et al. (2018). Targeting the C481S ibrutinib-resistance mutation in bruton's tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57 (26), 3564–3575. doi:10.1021/acs.biochem.8b00391
Burslem, G. M., Schultz, A. R., Bondeson, D. P., Eide, C. A., Savage Stevens, S. L., Druker, B. J., et al. (2019). Targeting BCR-ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation. Cancer Res. 79 (18), 4744–4753. doi:10.1158/0008-5472.CAN-19-1236
Burslem, G. M., Song, J., Chen, X., Hines, J., and Crews, C. M. (2018). Enhancing antiproliferative activity and selectivity of a FLT-3 inhibitor by proteolysis targeting chimera conversion. J. Am. Chem. Soc. 140 (48), 16428–16432. doi:10.1021/jacs.8b10320
Cao, R., Wang, L., Wang, H., Xia, L., Erdjument-Bromage, H., Tempst, P., et al. (2002). Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298 (5595), 1039–1043. doi:10.1126/science.1076997
Chamberlain, P. P., Lopez-Girona, A., Miller, K., Carmel, G., Pagarigan, B., and Chie-Leon, B. (2014). Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 21 (9), 803–809. doi:10.1038/nsmb.2874
Ciechanover, A. (2005). Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 6 (1), 79–87. doi:10.1038/nrm1552
Ciulli, A., and Trainor, N. (2021). A beginner’s guide to PROTACs and targeted protein degradation. Biochem. 43 (5), 74–79. doi:10.1042/bio_2021_148
Czyzyk-Krzeska, M. F., and Meller, J. (2004). von Hippel-lindau tumor suppressor: Not only HIF's executioner. Trends Mol. Med. 10 (4), 146–149. doi:10.1016/j.molmed.2004.02.004
Dobrovolsky, D., Wang, E. S., Morrow, S., Leahy, C., Faust, T., Nowak, R. P., et al. (2019). Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 133 (9), 952–961. doi:10.1182/blood-2018-07-862953
Fisher, S. L., and Phillips, A. J. (2018). Targeted protein degradation and the enzymology of degraders. Curr. Opin. Chem. Biol. 44, 47–55. doi:10.1016/j.cbpa.2018.05.004
Gao, H., Sun, X., and Rao, Y. (2020). PROTAC technology: Opportunities and challenges. ACS Med. Chem. Lett. 11 (3), 237–240. doi:10.1021/acsmedchemlett.9b00597
George, B., Chowdhury, S. M., Hart, A., Sircar, A., Singh, S. K., Nath, U. K., et al. (2020). Ibrutinib resistance mechanisms and treatment strategies for B-cell lymphomas. Cancers (Basel) 12 (5), 1328. doi:10.3390/cancers12051328
Gibaja, V., Shen, F., Harari, J., Korn, J., Ruddy, D., Saenz-Vash, V., et al. (2016). Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 35 (5), 558–566. doi:10.1038/onc.2015.114
Grunwald, M. R., and Levis, M. J. (2013). FLT3 inhibitors for acute myeloid leukemia: A review of their efficacy and mechanisms of resistance. Int. J. Hematol. 97 (6), 683–694. doi:10.1007/s12185-013-1334-8
Hafezi, S., and Rahmani, M. (2021). Targeting BCL-2 in cancer: Advances, challenges, and perspectives. Cancers (Basel) 13 (6), 1292. doi:10.3390/cancers13061292
Hantschel, O., Warsch, W., Eckelhart, E., Kaupe, I., Grebien, F., Wagner, K. U., et al. (2012). BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat. Chem. Biol. 8 (3), 285–293. doi:10.1038/nchembio.775
He, Y., Khan, S., Huo, Z., Lv, D., Zhang, X., Liu, X., et al. (2020). Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J. Hematol. Oncol. 13 (1), 103. doi:10.1186/s13045-020-00924-z
He, Y., Zhang, X., Chang, J., Kim, H. N., Zhang, P., Wang, Y., et al. (2020). Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 11 (1), 1996. doi:10.1038/s41467-020-15838-0
Hershko, A., Heller, H., Elias, S., and Ciechanover, A. (1983). Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258 (13), 8206–8214. doi:10.1016/s0021-9258(20)82050-x
Herviou, L., Cavalli, G., Cartron, G., Klein, B., and Moreaux, J. (2016). EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 7 (3), 2284–2296. doi:10.18632/oncotarget.6198
Higa, L. A., and Zhang, H. (2007). Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2, 5. doi:10.1186/1747-1028-2-5
Hou, J. Z., Ye, J. C., Pu, J. J., Liu, H., Ding, W., Zheng, H., et al. (2021). Novel agents and regimens for hematological malignancies: Recent updates from 2020 ASH annual meeting. J. Hematol. Oncol. 14 (1), 66. doi:10.1186/s13045-021-01077-3
Hsu, J. H., Rasmusson, T., Robinson, J., Pachl, F., Read, J., Kawatkar, S., et al. (2020). EED-targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 complex. Cell Chem. Biol. 27 (1), 41–46.e17. doi:10.1016/j.chembiol.2019.11.004
Huang, H. T., Dobrovolsky, D., Paulk, J., Yang, G., Weisberg, E. L., Doctor, Z. M., et al. (2018). A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 25 (1), 88–99 e6. doi:10.1016/j.chembiol.2017.10.005
Huibregtse, J. M., Scheffner, M., Beaudenon, S., and Howley, P. M. (1995). A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U. S. A. 92 (7), 2563–2567. doi:10.1073/pnas.92.7.2563
Ito, T., Ando, H., Suzuki, T., Ogura, T., Hotta, K., Imamura, Y., et al. (2010). Identification of a primary target of thalidomide teratogenicity. Science 327 (5971), 1345–1350. doi:10.1126/science.1177319
Ito, T., and Handa, H. (2020). Molecular mechanisms of thalidomide and its derivatives. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 96 (6), 189–203. doi:10.2183/pjab.96.016
Itoh, Y., Ishikawa, M., Naito, M., and Hashimoto, Y. (2010). Protein knockdown using methyl bestatin-ligand hybrid molecules: Design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 132 (16), 5820–5826. doi:10.1021/ja100691p
Iwama, A. (2017). Polycomb repressive complexes in hematological malignancies. Blood 130 (1), 23–29. doi:10.1182/blood-2017-02-739490
Jackson, S., and Xiong, Y. (2009). CRL4s: The CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 34 (11), 562–570. doi:10.1016/j.tibs.2009.07.002
Kang, C. H., Lee, D. H., Lee, C. O., Du Ha, J., Park, C. H., and Hwang, J. Y. (2018). Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun. 505 (2), 542–547. doi:10.1016/j.bbrc.2018.09.169
Keskin, D., Sadri, S., and Eskazan, A. E. (2016). Dasatinib for the treatment of chronic myeloid leukemia: Patient selection and special considerations. Drug Des. Devel Ther. 10, 3355–3361. doi:10.2147/DDDT.S85050
Khan, S., Zhang, X., Lv, D., Zhang, Q., He, Y., Zhang, P., et al. (2019). A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 25 (12), 1938–1947. doi:10.1038/s41591-019-0668-z
Kiyoi, H., and Naoe, T. (2002). FLT3 in human hematologic malignancies. Leuk. Lymphoma 43 (8), 1541–1547. doi:10.1080/1042819021000002866
Komander, D., Clague, M. J., and Urbe, S. (2009). Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10 (8), 550–563. doi:10.1038/nrm2731
Kronke, J., Fink, E. C., Hollenbach, P. W., MacBeth, K. J., Hurst, S. N., Udeshi, N. D., et al. (2015). Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523 (7559), 183–188. doi:10.1038/nature14610
Kronke, J., Hurst, S. N., and Ebert, B. L. (2014). Lenalidomide induces degradation of IKZF1 and IKZF3. Oncoimmunology 3 (7), e941742. doi:10.4161/21624011.2014.941742
Kronke, J., Udeshi, N. D., Narla, A., Grauman, P., Hurst, S. N., McConkey, M., et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343 (6168), 301–305. doi:10.1126/science.1244851
Lai, A. C., Toure, M., Hellerschmied, D., Salami, J., Jaime-Figueroa, S., Ko, E., et al. (2016). Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 55 (2), 807–810. doi:10.1002/anie.201507634
Lee, H., Puppala, D., Choi, E. Y., Swanson, H., and Kim, K. B. (2007). Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: A useful chemical genetic tool. Chembiochem 8 (17), 2058–2062. doi:10.1002/cbic.200700438
Lee, Y. T., Tan, Y. J., and Oon, C. E. (2018). Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 834, 188–196. doi:10.1016/j.ejphar.2018.07.034
Liao, Y. F., Wu, Y. B., Long, X., Zhu, S. Q., Jin, C., Xu, J. J., et al. (2016). High level of BRD4 promotes non-small cell lung cancer progression. Oncotarget 7 (8), 9491–9500. doi:10.18632/oncotarget.7068
Lim, Y. S., Yoo, S. M., Patil, V., Kim, H. W., Kim, H. H., Suh, B., et al. (2023). Orally bioavailable BTK PROTAC active against wild-type and C481 mutant BTKs in human lymphoma CDX mouse models. Blood Adv. 7 (1), 92–105. doi:10.1182/bloodadvances.2022008121
Lu, J., Qian, Y., Altieri, M., Dong, H., Wang, J., Raina, K., et al. (2015). Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22 (6), 755–763. doi:10.1016/j.chembiol.2015.05.009
Lu, R. M., Hwang, Y. C., Liu, I. J., Lee, C. C., Tsai, H. Z., Li, H. J., et al. (2020). Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 27 (1), 1. doi:10.1186/s12929-019-0592-z
Lv, D., Pal, P., Liu, X., Jia, Y., Thummuri, D., Zhang, P., et al. (2021). Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat. Commun. 12 (1), 6896. doi:10.1038/s41467-021-27210-x
Ma, B., Feng, H., Feng, C., Liu, Y., Zhang, H., Wang, J., et al. (2022). Kill two birds with one stone: A multifunctional dual-targeting protein drug to overcome imatinib resistance in philadelphia chromosome-positive leukemia. Adv. Sci. (Weinh). 9 (13), e2104850. doi:10.1002/advs.202104850
Ma, L., Wang, J., Zhang, Y., Fang, F., Ling, J., Chu, X., et al. (2022). BRD4 PROTAC degrader MZ1 exerts anticancer effects in acute myeloid leukemia by targeting c-Myc and ANP32B genes. Cancer Biol. Ther. 23 (1), 1–15. doi:10.1080/15384047.2022.2125748
Mani, A., and Gelmann, E. P. (2005). The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. 23 (21), 4776–4789. doi:10.1200/JCO.2005.05.081
Martin-Perez, D., Piris, M. A., and Sanchez-Beato, M. (2010). Polycomb proteins in hematologic malignancies. Blood 116 (25), 5465–5475. doi:10.1182/blood-2010-05-267096
McMahon, C. M., Ferng, T., Canaani, J., Wang, E. S., Morrissette, J. J. D., Eastburn, D. J., et al. (2019). Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 9 (8), 1050–1063. doi:10.1158/2159-8290.CD-18-1453
Mohamed, A. J., Yu, L., Backesjo, C. M., Vargas, L., Faryal, R., Aints, A., et al. (2009). Bruton's tyrosine kinase (btk): Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 228 (1), 58–73. doi:10.1111/j.1600-065X.2008.00741.x
Nishida, Y., Scruggs, D. A., Ayoub, E., Patsilevas, T., Ruvolo, V. R., Mak, P. Y., et al. (2022). AML-147 C-MYC targeting by degradation: Novel dual c-myc/GSPT1 degrader GT19715 exerts profound cell kill in vitro and in vivo in acute myeloid leukemia and lymphomas. Clin. Lymphoma Myeloma Leuk. 22, S218. doi:10.1016/s2152-2650(22)01230-7
Ntziachristos, P., Tsirigos, A., Van Vlierberghe, P., Nedjic, J., Trimarchi, T., Flaherty, M. S., et al. (2012). Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 18 (2), 298–301. doi:10.1038/nm.2651
Paiva, S. L., and Crews, C. M. (2019). Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 50, 111–119. doi:10.1016/j.cbpa.2019.02.022
Pan, Z., Scheerens, H., Li, S. J., Schultz, B. E., Sprengeler, P. A., Burrill, L. C., et al. (2007). Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem 2 (1), 58–61. doi:10.1002/cmdc.200600221
Park, J., Cho, J., and Song, E. J. (2020). Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 43 (11), 1144–1161. doi:10.1007/s12272-020-01281-8
Park, J., Cho, J., and Song, E. J. (2020). Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Archives Pharmacal Res. 43 (11), 1144–1161. doi:10.1007/s12272-020-01281-8
Plati, J., Bucur, O., and Khosravi-Far, R. (2011). Apoptotic cell signaling in cancer progression and therapy. Integr. Biol. (Camb). 3 (4), 279–296. doi:10.1039/c0ib00144a
Podhorecka, M., Markowicz, J., Szymczyk, A., and Pawlowski, J. (2014). Target therapy in hematological malignances: New monoclonal antibodies. Int. Sch. Res. Not. 2014, 701493. doi:10.1155/2014/701493
Potjewyd, F., Turner, A. W., Beri, J., Rectenwald, J. M., Norris-Drouin, J. L., Cholensky, S. H., et al. (2020). Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem. Biol. 27 (1), 47–56 e15. doi:10.1016/j.chembiol.2019.11.006
Robbins, D. W., Noviski, M., Rountree, R., Tan, M., Brathaban, N., Ingallinera, T., et al. (2021). Nx-5948, a selective degrader of BTK with activity in preclinical models of hematologic and brain malignancies. Blood 138, 2251. doi:10.1182/blood-2021-147473
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric molecules that target proteins to the skp1-cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 98 (15), 8554–8559. doi:10.1073/pnas.141230798
Sakamoto, K. M., Kim, K. B., Verma, R., Ransick, A., Stein, B., Crews, C. M., et al. (2003). Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell Proteomics 2 (12), 1350–1358. doi:10.1074/mcp.T300009-MCP200
Scheffner, M., Nuber, U., and Huibregtse, J. M. (1995). Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 373 (6509), 81–83. doi:10.1038/373081a0
Schneekloth, A. R., Pucheault, M., Tae, H. S., and Crews, C. M. (2008). Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med. Chem. Lett. 18 (22), 5904–5908. doi:10.1016/j.bmcl.2008.07.114
Schulman, B. A., and Harper, J. W. (2009). Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10 (5), 319–331. doi:10.1038/nrm2673
Segura, M. F., Fontanals-Cirera, B., Gaziel-Sovran, A., Guijarro, M. V., Hanniford, D., Zhang, G., et al. (2013). BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 73 (20), 6264–6276. doi:10.1158/0008-5472.CAN-13-0122-T
Sievers, Q. L., Petzold, G., Bunker, R. D., Renneville, A., Slabicki, M., Liddicoat, B. J., et al. (2018). Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362 (6414), eaat0572. doi:10.1126/science.aat0572
Simon, C., Chagraoui, J., Krosl, J., Gendron, P., Wilhelm, B., Lemieux, S., et al. (2012). A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 26 (7), 651–656. doi:10.1101/gad.186411.111
Singh, R., Singh, S., and Lillard, J. W. (2008). Past, present, and future technologies for oral delivery of therapeutic proteins. J. Pharm. Sci. 97 (7), 2497–2523. doi:10.1002/jps.21183
Sochacka-Cwikla, A., Maczynski, M., and Regiec, A. (2021). FDA-approved drugs for hematological malignancies-the last decade review. Cancers (Basel) 14 (1), 87. doi:10.3390/cancers14010087
Sun, X., Gao, H., Yang, Y., He, M., Wu, Y., Song, Y., et al. (2019). PROTACs: Great opportunities for academia and industry. Signal Transduct. Target Ther. 4, 64. doi:10.1038/s41392-019-0101-6
Sun, Y., Ding, N., Song, Y., Yang, Z., Liu, W., Zhu, J., et al. (2019). Degradation of Bruton's tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 33 (8), 2105–2110. doi:10.1038/s41375-019-0440-x
Sun, Y., Zhao, X., Ding, N., Gao, H., Wu, Y., Yang, Y., et al. (2018). PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 28 (7), 779–781. doi:10.1038/s41422-018-0055-1
Takamatsu-Ichihara, E., and Kitabayashi, I. (2016). The roles of Polycomb group proteins in hematopoietic stem cells and hematological malignancies. Int. J. Hematol. 103 (6), 634–642. doi:10.1007/s12185-016-2011-5
Toure, M., and Crews, C. M. (2016). Small-molecule PROTACS: New approaches to protein degradation. Angew. Chem. Int. Ed. Engl. 55 (6), 1966–1973. doi:10.1002/anie.201507978
Valent, P., and Zuber, J. (2014). BRD4: A BET(ter) target for the treatment of AML? Cell Cycle 13 (5), 689–690. doi:10.4161/cc.27859
Voges, D., Zwickl, P., and Baumeister, W. (1999). The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068. doi:10.1146/annurev.biochem.68.1.1015
Webb, T. R., Slavish, J., George, R. E., Look, A. T., Xue, L., Jiang, Q., et al. (2009). Anaplastic lymphoma kinase: Role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev. Anticancer Ther. 9 (3), 331–356. doi:10.1586/14737140.9.3.331
Wei, G., Rafiyath, S., and Liu, D. (2010). First-line treatment for chronic myeloid leukemia: Dasatinib, nilotinib, or imatinib. J. Hematol. Oncol. 3, 47. doi:10.1186/1756-8722-3-47
Wenzel, D. M., Lissounov, A., Brzovic, P. S., and Klevit, R. E. (2011). UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474 (7349), 105–108. doi:10.1038/nature09966
Wilkes, G. M. (2018). Targeted therapy: Attacking cancer with molecular and immunological targeted agents. Asia Pac J. Oncol. Nurs. 5 (2), 137–155. doi:10.4103/apjon.apjon_79_17
Woyach, J. A., Furman, R. R., Liu, T. M., Ozer, H. G., Zapatka, M., Ruppert, A. S., et al. (2014). Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 370 (24), 2286–2294. doi:10.1056/NEJMoa1400029
Wu, S., Jiang, Y., Hong, Y., Chu, X., Zhang, Z., Tao, Y., et al. (2021). BRD4 PROTAC degrader ARV-825 inhibits T-cell acute lymphoblastic leukemia by targeting 'Undruggable' Myc-pathway genes. Cancer Cell Int. 21 (1), 230. doi:10.1186/s12935-021-01908-w
Xi, M., Chen, Y., Yang, H., Xu, H., Du, K., Wu, C., et al. (2019). Small molecule PROTACs in targeted therapy: An emerging strategy to induce protein degradation. Eur. J. Med. Chem. 174, 159–180. doi:10.1016/j.ejmech.2019.04.036
Xie, H., Liu, J., Alem Glison, D. M., and Fleming, J. B. (2021). The clinical advances of proteolysis targeting chimeras in oncology. Explor Target Antitumor Ther. 2 (6), 511–521. doi:10.37349/etat.2021.00061
Yang, Y., Gao, H., Sun, X., Sun, Y., Qiu, Y., Weng, Q., et al. (2020). Global PROTAC toolbox for degrading BCR-ABL overcomes drug-resistant mutants and adverse effects. J. Med. Chem. 63 (15), 8567–8583. doi:10.1021/acs.jmedchem.0c00967
Zengerle, M., Chan, K. H., and Ciulli, A. (2015). Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10 (8), 1770–1777. doi:10.1021/acschembio.5b00216
Zhang, C., Han, X. R., Yang, X., Jiang, B., Liu, J., Xiong, Y., et al. (2018). Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). Eur. J. Med. Chem. 151, 304–314. doi:10.1016/j.ejmech.2018.03.071
Zhang, J., Ding, L., Holmfeldt, L., Wu, G., Heatley, S. L., Payne-Turner, D., et al. (2012). The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481 (7380), 157–163. doi:10.1038/nature10725
Zhang, K., Gao, L., Wang, J., Chu, X., Zhang, Z., Zhang, Y., et al. (2022). A novel BRD family PROTAC inhibitor dBET1 exerts great anti-cancer effects by targeting c-MYC in acute myeloid leukemia cells. Pathol. Oncol. Res. 28, 1610447. doi:10.3389/pore.2022.1610447
Zhao, Q., Ren, C., Liu, L., Chen, J., Shao, Y., Sun, N., et al. (2019). Discovery of SIAIS178 as an effective BCR-ABL degrader by recruiting von hippel-lindau (VHL) E3 ubiquitin ligase. J. Med. Chem. 62 (20), 9281–9298. doi:10.1021/acs.jmedchem.9b01264
Zhu, Y. X., Braggio, E., Shi, C. X., Kortuem, K. M., Bruins, L. A., Schmidt, J. E., et al. (2014). Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 124 (4), 536–545. doi:10.1182/blood-2014-02-557819
Zorba, A., Nguyen, C., Xu, Y., Starr, J., Borzilleri, K., Smith, J., et al. (2018). Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 115 (31), E7285–E7292. doi:10.1073/pnas.1803662115
Keywords: PROTACs, hematologic malignancies, resistance, VHL, CRBN
Citation: Bou Malhab LJ, Alsafar H, Ibrahim S and Rahmani M (2023) PROTACs: Walking through hematological malignancies. Front. Pharmacol. 14:1086946. doi: 10.3389/fphar.2023.1086946
Received: 01 November 2022; Accepted: 06 February 2023;
Published: 20 February 2023.
Edited by:
Andrei Leitao, Institute of Chemistry of São Carlos, University of São Paulo, BrazilReviewed by:
Ketankumar Patel, St. John’s University, United StatesCopyright © 2023 Bou Malhab, Alsafar, Ibrahim and Rahmani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamed Rahmani, bW9oYW1lZC5yYWhtYW5pQGt1LmFjLmFl; Lara J. Bou Malhab, SWJvbm1hbGhhYkBzaGFyamFoLmFjLmFl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.