 Chong Song1
Chong Song1 Xian Long
Xian Long
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 16 March 2023
Sec. Inflammation Pharmacology
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1081334
This article is part of the Research TopicAutophagy in Inflammation Related Diseases, Volume IIView all 7 articles
Non-alcoholic fatty liver disease (NAFLD) is common chronic metabolic liver disorder which is associated with fat accumulation in the liver. It causes a wide range of pathological effects such as insulin resistance, obesity, hypertension, diabetes, non-alcoholic steatohepatitis (NASH) and cirrhosis, cardiovascular diseases. The molecular mechanisms that cause the initiation and progression of NAFLD remain fully unclear. Inflammation is regarded as a significant mechanism which could result in cell death and tissue injury. Accumulation of leukocytes and hepatic inflammation are important contributors in NAFLD. Excessive inflammatory response can deteriorate the tissue injury in NAFLD. Thus, inhibition of inflammation improves NAFLD by reducing intrahepatic fat content, increasing β-oxidation of fatty acids, inducing hepato-protective autophagy, overexpressing peroxisome proliferator-activated receptor- γ (PPAR-γ), as well as attenuating hepatocyte apoptosis and increasing insulin sensitivity. Therefore, understanding the molecules and signaling pathways suggests us valuable information about NAFLD progression. This review aimed to evaluate the inflammation in NAFLD and the molecular mechanism on NAFLD.
Non-alcoholic fatty liver disease (NAFLD) is one of the most common chronic metabolic liver diseases recognized globally, with a prevalence of 25.2% (Ye et al., 2020). NAFLD includes a variety of benign reversible pathological changes including liver steatosis, and could also progress to more severe and irreversible pathological changes (Cobbina and Akhlaghi, 2017). 24% of patients can develop non-alcoholic steatohepatitis (NASH) and Cirrhosis; less than 15% of patients may eventually develop liver failure and hepatocellular carcinoma from cirrhosis (Schwabe et al., 2020). Non-alcoholic fatty liver disease (NAFLD) is a prominent cause of liver-related morbidity and mortality (Younossi et al., 2016; Albhaisi and Noureddin, 2021). Non-alcoholic steatohepatitis (NASH), the advanced form of NAFL, is illustrated by steatosis, inflammation, hepatocyte injury (e.g., ballooning), with or without fibrosis, which could progress to cirrhosis, fibrosis, liver-related complications and even hepatocellular carcinoma, which is seriously life-threatening (Huby and Gautier, 2022). Much progress has been made in understanding the pathogenesis of NASH and the complex and multifactorial (Hardy et al., 2016; Machado and Diehl, 2016) molecular pathways during development and progression. It has attracted more and more attention from scholars at home and abroad. It is currently believed that the mechanism of liver inflammation is an important driving force for the occurrence and development of NASH. This paper intended to provide an overview of the recent advances in this related field.
NASH is considered the progressive form of NAFLD and is characterized by liver steatosis, inflammation, hepatocellular injury and different degrees of fibrosis (Schuster et al., 2018). Accumulating studies demonstrated that a pivotal issue relates to the identification of those factors that trigger inflammation, thus fuelling the transition from NAFLD to NASH (Guo et al., 2022; An et al., 2023). These triggers of inflammtion in liver might be associated with their origins outside the liver as well as inside the organ, both of which accelerate to NASH development (Han et al., 2022). Although the marked progress in unravelling the mechanism that underlie NAFLD development and progression has made, yet the elucidation of its mechanism about the pathogenesis remains limited. Thus, the identification of aberrent factors that trigger inflammation, thereby preceding the transition from NAFLD to NASH. Several lines of studies have demonstrated that hepatic inflammatory response is regarded as an important driving force during the hepatic progression. Furthermore, extrahepatic factors including adipose tissue dysfunction and the gut could trigger the inflammation and exacerbate the tissue injury (Figure 1).
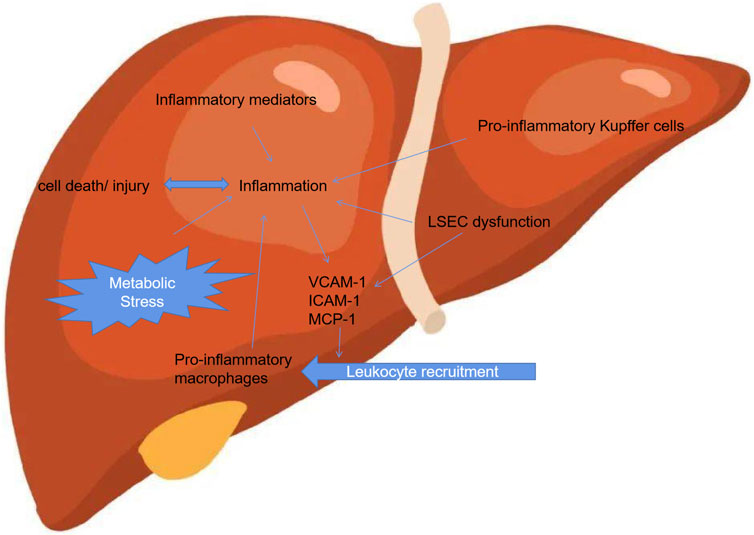
FIGURE 1. Inflammation associated with NAFLD.
A large number of studies have demonstrated that fructose could act as a pro-inflammatory factor, up-regulating liver inflammatory genes, reducing liver mitochondrial β-oxidation and ATP (Adenosine Triphosphate) levels, causing metabolic dysfunction and promoting the development of NASH (Lim et al., 2010; Rom et al., 2020). Increased intake of high fructose is associated with liver fibrosis, dyslipidemia, endoplasmic reticulum stress, hepatocyte ballooning, inflammation, and impaired insulin signaling (Jensen et al., 2018; Pouwels et al., 2022).
Increased intake of dietary saturated fatty acids induces de novo fat synthesis, promoting endoplasmic reticulum stress and cell apoptosis. Saturated fatty acids are lipotoxic to the liver and could cause NASH in mice alone or in combination with a high-fructose diet (Arrese et al., 2016; Lastuvkova et al., 2021). Free cholesterol in liver samples from NASH patients was higher than healthy controls. The results showed that the liver in a cholesterol diet-induced NASH mouse model exhibited mitochondrial dysfunction, increased reactive oxygen species (ROS) due to excessive lipid accumulation, and endoplasmic reticulum stress, and the inclusion of hepatocytes (Al-Rasadi et al., 2015). A series of chain reactions such as activation of the death pathway induce a strong inflammatory response (Arrese and Karpen, 2010; Asrih and Jornayvaz, 2013). Furthermore, accumulation of free cholesterol in Kupffer cells and hepatic stellate cells activates inflammation and promotes fibrosis. Conversely, removing cholesterol from a high-fat and high-cholesterol diet in mice reduced plasma VLDL levels and prevented liver inflammation and ballooning (Al-Rasadi et al., 2015). Linoleic acid, the most abundant n-6 polyunsaturated fatty acid in the Western diet, is the immediate precursor to oxidized linoleic acid metabolites, a group of bioactive lipids (Van Name et al., 2020). Lowering dietary linoleic acid levels can reduce the synthesis and/or accumulation of oxidized linoleic acid metabolites in humans. Oxidative metabolites from arachidonic acid and linoleic acid are associated with markers of steatosis, liver injury, and insulin sensitivity, and the relevant oxidative metabolites are more abundant in the plasma of NASH patients than NAFLD patients (Civelek and Podszun, 2022; Gao et al., 2022). These data suggest that oxidized linoleic acid metabolites play a crucial role in NASH.
NASH-related metabolic dysfunction is closely associated with insulin resistance and is a major driver of steatosis (Parthasarathy et al., 2020). Progressive adipose tissue dysfunction and insulin resistance may be key factors promoting the development of NASH. Visceral adipose tissue-released macrophages produced chemokines and cytokines involved in inflammation and fibrosis in human NASH. In a mouse model of NASH, mRNA levels of adipose tissue macrophage-related and inflammation-related genes were increased (Osorio-Conles et al., 2021). Adipose tissue dysfunction could lead to the accumulation of inflammatory cells in visceral adipose tissue, causing liver damage and the secretion of pro-inflammatory related mediators, including tumor necrosis factor (TNF), interleukin-1β (IL-1β) and IL-6; in turn, these inflammations promoted impaired insulin signaling (Daryabor et al., 2020). Adipose tissue insulin resistance was higher in NASH patients than in patients with simple steatosis. Furthermore, adipose tissue insulin resistance was associated with liver and muscle insulin resistance and liver fibrosis (Maximos et al., 2015; Rotman and Neuschwander-Tetri, 2017). The results also confirmed that obesity-associated inflammation induces insulin resistance (IR), which is central to NAFLD or hepatic steatosis. Prediction of HOMA values by SCGF-β levels, likely mediated by markers of inflammation, characterises a recent study, shedding some light on mechanisms inducing/worsening IR of male patients with obesity-related NAFLD (Tarantino et al., 2020).
As one of the largest endocrine organs in the body, adipose tissue could secrete inflammatory factors such as TNF and IL-6, as well as adipokines such as adiponectin and leptin, which further drove the progression of NASH. In the collected serum of NASH patients, it could be found that the content of leptin and adiponectin in vivo is negatively correlated (Zhao et al., 2018; Fan et al., 2020). In addition, adiponectin could inhibit hepatic steatosis, reduce insulin resistance, and has anti-inflammatory, anti-apoptotic and insulin-sensitizing functions. Furthermore, in addition to promoting inflammation, TNF may also increase insulin resistance and promote NASH progression. IL-6 promotes insulin resistance through multiple mechanisms, and NASH severity, fibrosis stage, and insulin resistance are associated. The study found that leptin in the early stage of NASH may prevent hepatic steatosis by inhibiting de novo synthesis of intrahepatic fat. However, as the disease progressed, leptin has pro-inflammatory and pro-fibrotic abilities (Jiménez-Cortegana et al., 2021). Therefore, leptin may have a bidirectional effects on NASH. However, this hypothesis has not been tested in humans.
The occurrence of NASH is related to intestinal flora imbalance and intestinal barrier dysfunction. The body regulates intestinal barrier permeability through the expression of CX3C-chemokine receptor 1 (CX3CR1). Studies have shown that deletion of CX3CR1 impairs intestinal barrier function and exacerbates the occurrence of NASH in a mouse model (Schneider et al., 2019; Ni et al., 2022). In addition, junctional adhesion molecule A expression levels were decreased and mucosal inflammation was increased in the colonic mucosa of NASH patients. In mice lacking junctional adhesion molecule A, increased intestinal permeability enables bacterial translocation to the liver and drives NASH. Therefore, disruption of intestinal barrier integrity may predispose patients to hepatic inflammation and contribute to the development of NASH (Zhang et al., 2020). Whether intestinal permeability is responsible for endotoxin exposure is unclear, but endotoxin induces an inflammatory response by activating inflammatory cells in the liver. Studies have found that NASH patients have higher blood endotoxin levels than healthy individuals (Sinakos et al., 2022). However, whether the permeability of the gut epithelium to gut microbial endotoxins is diet-induced has not been established. Furthermore, topical intestinal anti-inflammatory drugs fed mice on a high-fat diet reduced intestinal inflammation while improving intestinal permeability and hepatic steatosis (She et al., 2022). However, studies addressing the association between endotoxemia and the histological features of NASH remain controversial (Nagata et al., 2022; Wang et al., 2022). Therefore, more accurate biomarkers are needed to rigorously assess the contribution of endotoxemia to NASH. Gut microbes could be influenced by diet, which varies from person to person and over time. Therefore, more research is needed to reveal the association between the gut microbiome and NASH.
Bile acids are involved in the development of NAFLD and NASH (Jia et al., 2021). First, bile acids have important regulatory effects on lipid and carbohydrate metabolism, and if dysregulated, it could lead to glucose and lipid imbalances in the body and promote inflammation and fibrosis (Sheng et al., 2022). In addition, bile acids and gut microbiota were interrelated. On the one hand, changes in bile acid metabolism could affect the composition of the microbiota; on the other hand, gut dysbiosis affects bile acid composition and promoted the development of NAFLD and NASH. Defects in hepatic bile acid transport have been found in experimental models of NASH, and cholestasis may occur, leading to liver injuries and inflammation (van Golen et al., 2018). Several studies have found that NASH patients had higher levels of bile acids than NAFLD, and that altered bile acid metabolism was associated with the histological features of NASH (Abdelmalek, 2021). Therefore, alterations in bile acid metabolism may play a pro-inflammatory role in the pathogenesis of NASH, but the mechanism are needed to be investigated in further studies.
Oxidative stress is a common feature of chronic liver disease and plays a critical role in NASH progression. Oxidative stress markers correlate with neutrophil number and degree of liver injury. ROS disrupts the lysosomal membrane and releases proteases into the cytoplasm, triggering apoptosis and necrosis (Zhang et al., 2021). In liver, Kupffer cells are a major source of ROS, which is normally produced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. ATP activates Kupffer cells leading to ROS generation. Some lipid peroxidation products could activate hepatic stellate cells; in turn, hepatic stellate cells could produce ROS. During ER stress, mitochondrial ROS production increases, and Ca2+ leaked from the ER is taken up by mitochondria. Ca2+ uptake leads to the release of cytochrome C, which further eliminates the electron transport chain function and leads to increased ROS overproduction (Jangra et al., 2022).
Lipotoxicity is one of the major mechanisms of hepatocyte dysfunction and contributes to the progression of NASH (Horn et al., 2022). Previous studies have suggested that triglyceride accumulation in hepatocytes could cause NALFD, followed by oxidative stress and lipid peroxidation, promoting inflammation and fibrosis (Svegliati-Baroni et al., 2019). However, this argument has been challenged in animal models and clinical trials in recent years. There were data suggesting that triglyceride accumulation may actually be a protective mechanism against lipotoxicity. For example, inhibition of triglyceride synthesis in mice reduced hepatic steatosis but exacerbated liver injury and fibrosis (Newberry et al., 2017). Furthermore, triglyceride accumulation was not sufficient to cause inflammation in the liver, whereas other lipotoxic metabolites were a major contributor to disease progression. In general, animal fats are rich in saturated fatty acids palmitate and stearate, which are directly toxic to cells (Packard et al., 2020), while monounsaturated fatty acids could prevent saturated fatty acid-induced cytotoxicity (Urso and Zhou, 2021). Saturated fatty acids could induce hepatic endoplasmic reticulum stress and promote inflammasome activation, which could also activate amino-terminal kinase (JNK) and mitochondrial death pathways to induce hepatocyte apoptosis (Luci et al., 2020; Zhu et al., 2022). Increased hepatocyte free fatty acids could form lipotoxic intermediates in the liver, such as ceramides, diacylglycerols, lysophosphatidylcholines, oxidized fatty acids, and cholesterol metabolites, which act like ROS (Sowton et al., 2020). In addition to saturated fatty acids, excess cholesterol could also induce hepatotoxicity and be associated with NASH progression (Coué et al., 2019). The cellular damaging effects of cholesterol were primarily mediated by sensitizing cells to other cell death signals. Excessive free cholesterol accumulated in hepatocytes, Kupffer cells, hepatic stellate cells, disrupts membrane fluidity and caused glutathione loss in mitochondria, leading to oxidative stress, mitochondrial dysfunction and ATP depletion, and cellular apoptosis and necrosis. In addition, cholesterol crystals have been reported to be present in the lipid droplet membranes of hepatocytes in vivo and clinical NASH (Farrell et al., 2018; Horn et al., 2022). These crystals activated the inflammasome and acted in Kupffer cells and macrophages, leading to the production of IL-1β and TNF (Ioannou et al., 2017; Wree et al., 2018). Taken together, Kupffer cells and hepatic stellate cells could accumulate free cholesterol, thereby activating signaling of liver inflammation and fibrosis, leading to the development of NAFLD to NASH.
In the process of NAFLD progression to NASH, mitochondrial dysfunction is one of the important mechanisms. One of the main causes of mitochondrial deterioration in NASH is fatty acid oxidation and lipotoxicity (Carnagarin et al., 2021). The ever-increasing flux of fatty acids through mitochondria and the TCA cycle generated harmful ROS that attack the mitochondrial respiratory chain and mitochondrial DNA, and inhibit mitochondrial respiratory chain complex activity (Mooli and Ramakrishnan, 2022). Over time, mitochondria gradually became dysfunctional, triggering oxidative stress, ATP depletion, and disruption of mitochondrial integrity, all of which lead to hepatocyte death (Manne et al., 2018). Interestingly, both mitochondrial DNA and oxidized mitochondrial DNA released into the cytoplasm during programmed cell death cause inflammasome activation. These results provided a link between mitochondrial dysfunction, apoptosis, and inflammasome activation.
Free fatty acids derived from adipose tissue breakdown serve as natural nuclear receptor ligands and play a major role in the regulation of NASH-related lipid metabolism and inflammation (Yadati et al., 2021). Polyunsaturated fatty acids and their derivatives could activate peroxisome proliferator-activated receptor-α (PPARα), which could induce the expressions of genes involved in fatty acid oxidation and insulin sensitivity, and downregulate nuclear factor κB (NF-κB) level (Geethangili et al., 2021; López-Salazar et al., 2021). However, in a mouse model of NASH, a diet fed polyunsaturated fatty acids activates PPARα signaling and inhibits hepatic de novo fat synthesis, but does not prevent steatohepatitis (Ishida et al., 2021), which is likely to high levels of hepatic lipid peroxidation in this model abolished the protective effect of PPARα activation and resulted in lipotoxic hepatocyte injury and inflammatory cells recruitment. Similar to PPARα, PPARγ activation attenuates inflammation through multiple mechanisms, including decreased NF-κB activity and decreased TNF-α and IL-1β synthesis in monocytes and macrophages (Liu et al., 2022). As a master regulator of macrophage polarization, activation of PPARγ promotes the transition of macrophages from an M1-dominant phenotype to an M2 phenotype, thereby attenuating inflammation in experimental NASH (Luo et al., 2017; Valenzuela et al., 2017). Furthermore, PPARδ directly controls the inflammatory response of Kupffer cells in the liver. Other members of the nuclear receptor family, such as liver X receptors (LXRs), are also critical for the control of fatty acid synthesis and inflammation (Cai et al., 2017; Welch et al., 2022).
Taken together, two factors, intrahepatic and extrahepatic, could trigger the transition from isolated steatosis to NASH, and especially, determining the important role of hepatic inflammatory response during this process. Lipotoxicity causes cell apoptosis through peroxidative stress, endoplasmic reticulum stress, autophagy, etc., resulting in liver cell damage or death, and further leads to insulin resistance, which is the dominant factor in fat metabolism disorders. A vicious cycle of hepatic fat accumulation-liver injury-insulin resistance-lipid metabolism disorder. In addition, the imbalance of intestinal microbial homeostasis leads to the production of a large number of bacterial products, including short-chain fatty acids, lipopolysaccharides, deoxyribonucleic acid, polyamine metabolites, resulting in the accumulation of a large amount of fat in the liver, causing liver cell injury, inflammatory response, insulin resistance and liver fibrosis. On the contrary, the inflammatory response triggered by liver cell injury could chemotactic the infiltration of monocytes and lymphocytes, and participate in the formation and development of liver inflammation. Recently, although substantial progress has been made in revealing the mechanisms of NASH development, there are still no effective remedies available for alleviating or treating the disease. This requires further studies with integrated metabolomic, proteomic and epigenetic biology approaches to reveal the molecular features of NASH, searches for biomarkers of NASH, and identify more rational strategies.
CS and XL conceived, designed, or planned the ided. CS, XL, JH, and YH collected and read the literature. CS drafted the manuscript. CS and YH revised the manuscript. All authors read and approved the final manuscript.
This study was partly funded by National Natural Science Foundation of Hunan Province (2020JJ7090).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelmalek, M. F. (2021). Nonalcoholic fatty liver disease: Another leap forward. Nat. Rev. Gastroenterol. Hepatol. 18 (2), 85–86. doi:10.1038/s41575-020-00406-0
Al-Rasadi, K., Rizzo, M., Montalto, G., and Berg, G. (2015). Nonalcoholic fatty liver disease, cardiovascular risk, and carotid inflammation. Angiology 66 (7), 601–603. doi:10.1177/0003319714557353
Albhaisi, S., and Noureddin, M. (2021). Targeting inflammation in NASH. Front. Endocrinol. (Lausanne) 12, 767314. doi:10.3389/fendo.2021.767314
An, L., Wirth, U., Koch, D., Schirren, M., Drefs, M., Koliogiannis, D., et al. (2023). Metabolic role of autophagy in the pathogenesis and development of NAFLD. Metabolites 13 (1), 101. doi:10.3390/metabo13010101
Arrese, M., and Karpen, S. J. (2010). Nuclear receptors, inflammation, and liver disease: Insights for cholestatic and fatty liver diseases [J]. Clin. Pharmacol. Ther. 87 (4), 473–478. doi:10.1038/clpt.2010.2
Arrese, M., Cabrera, D., Kalergis, A. M., and Feldstein, A. E. (2016). Innate immunity and inflammation in NAFLD/NASH. Dig. Dis. Sci. 61 (5), 1294–1303. doi:10.1007/s10620-016-4049-x
Asrih, M., and Jornayvaz, F. R. (2013). Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J. Endocrinol. 218 (3), R25–R36. doi:10.1530/JOE-13-0201
Cai, C., Zhu, X., Li, P., Li, J., Gong, J., Shen, W., et al. (2017). NLRP3 deletion inhibits the non-alcoholic steatohepatitis development and inflammation in kupffer cells induced by palmitic acid. Inflammation 40 (6), 1875–1883. doi:10.1007/s10753-017-0628-z
Carnagarin, R., Tan, K., Adams, L., Matthews, V. B., and Kiuchi, M. G. (2021). Metabolic dysfunction-associated fatty liver disease (MAFLD)-A condition associated with heightened sympathetic activation. Int. J. Mol. Sci. 22 (8), 4241. doi:10.3390/ijms22084241
Civelek, M., and Podszun, M. C. (2022). Genetic factors associated with response to vitamin E treatment in NAFLD. Antioxidants (Basel) 11 (7), 1284. doi:10.3390/antiox11071284
Cobbina, E., and Akhlaghi, F. (2017). Non-alcoholic fatty liver disease (NAFLD) - pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 49 (2), 197–211. doi:10.1080/03602532.2017.1293683
Coué, M., Falewee, J., Aguesse, A., and Croyal, M. (2019). Spirulina liquid extract protects against fibrosis related to non-alcoholic steatohepatitis and increases ursodeoxycholic acid. Nutrients 11 (1), 194. doi:10.3390/nu11010194
Daryabor, G., Atashzar, M. R., Kabelitz, D., Meri, S., and Kalantar, K. (2020). The effects of type 2 diabetes mellitus on organ metabolism and the immune system. Front. Immunol. 11, 1582. doi:10.3389/fimmu.2020.01582
Fan, H., Diao, H., Lu, Y., Xie, J., and Cheng, X. (2020). The relation between serum adipose differentiation-related protein and non-alcoholic fatty liver disease in type 2 diabetes mellitus. Ther. Adv. Endocrinol. Metab. 11, 2042018820969025. doi:10.1177/2042018820969025
Farrell, G. C., Haczeyni, F., and Chitturi, S. (2018). Pathogenesis of NASH: How metabolic complications of overnutrition favour lipotoxicity and pro-inflammatory fatty liver disease. Adv. Exp. Med. Biol. 1061, 19–44. doi:10.1007/978-981-10-8684-7_3
Gao, Y., Jiang, D., Wang, C., An, G., Zhu, L., and Cui, C. (2022). Comprehensive analysis of metabolic changes in male mice exposed to sodium valproate based on GC-MS analysis. Drug Des. Devel Ther. 16, 1915–1930. doi:10.2147/DDDT.S357530
Geethangili, M., Lin, C. W., Mersmann, H. J., and Ding, S. T. (2021). Methyl brevifolincarboxylate attenuates free fatty acid-induced lipid metabolism and inflammation in hepatocytes through AMPK/NF-κB signaling pathway. Int. J. Mol. Sci. 22 (18), 10062. doi:10.3390/ijms221810062
Guo, X., Yin, X., Liu, Z., and Wang, J. (2022). Non-alcoholic fatty liver disease (NAFLD) pathogenesis and natural products for prevention and treatment. Int. J. Mol. Sci. 23 (24), 15489. doi:10.3390/ijms232415489
Han, J., Lee, C., Hur, J., and Jung, Y. (2022). Current therapeutic options and potential of mesenchymal stem cell therapy for alcoholic liver disease. Cells 12 (1), 22. doi:10.3390/cells12010022
Hardy, T., Oakley, F., and Anstee, Q. M. (2016). Nonalcoholic fatty liver disease: Pathogenesis and disease spectrum. Annu. Rev. Pathol. 11, 451–496. doi:10.1146/annurev-pathol-012615-044224
Horn, C. L., Morales, A. L., Savard, C., Farrell, G. C., and Ioannou, G. N. (2022). Role of cholesterol-associated steatohepatitis in the development of NASH. Hepatol. Commun. 6 (1), 12–35. doi:10.1002/hep4.1801
Huby, T., and Gautier, E. L. (2022). Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 22 (7), 429–443. doi:10.1038/s41577-021-00639-3
Ioannou, G. N., Subramanian, S., Chait, A., Haigh, W. G., Yeh, M. M., Farrell, G. C., et al. (2017). Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J. Lipid Res. 58 (6), 1067–1079. doi:10.1194/jlr.M072454
Ishida, N., Yamada, H., and Hirose, M. (2021). Euphausia pacifica (north pacific krill): Review of chemical features and potential benefits of 8-HEPE against metabolic syndrome, dyslipidemia, NAFLD, and atherosclerosis. Nutrients 13 (11), 3765. doi:10.3390/nu13113765
Jangra, A., Kothari, A., Sarma, P., Medhi, B., Omar, B. J., and Kaushal, K. (2022). Recent advancements in antifibrotic therapies for regression of liver fibrosis. Cells 11 (9), 1500. doi:10.3390/cells11091500
Jensen, T., Abdelmalek, M. F., Sullivan, S., Nadeau, K. J., Green, M., Roncal, C., et al. (2018). Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 68 (5), 1063–1075. doi:10.1016/j.jhep.2018.01.019
Jia, W., Wei, M., Rajani, C., and Zheng, X. (2021). Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell. 12 (5), 411–425. doi:10.1007/s13238-020-00804-9
Jiménez-Cortegana, C., García-Galey, A., Tami, M., Del Pino, P., Carmona, I., López, S., et al. (2021). Role of leptin in non-alcoholic fatty liver disease. Biomedicines 9 (7), 762. doi:10.3390/biomedicines9070762
Lastuvkova, H., Faradonbeh, F. A., Schreiberova, J., Hroch, M., Mokry, J., Faistova, H., et al. (2021). Atorvastatin modulates bile acid homeostasis in mice with diet-induced nonalcoholic steatohepatitis. Int. J. Mol. Sci. 22 (12), 6468. doi:10.3390/ijms22126468
Lim, J. S., Mietus-Snyder, M., Valente, A., Schwarz, J. M., and Lustig, R. H. (2010). The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 7 (5), 251–264. doi:10.1038/nrgastro.2010.41
Liu, H., Zang, C., Shang, J., Zhang, Z., Wang, L., Yang, H., et al. (2022). Gardenia jasminoides J. Ellis extract GJ-4 attenuates hyperlipidemic vascular dementia in rats via regulating PPAR-γ-mediated microglial polarization. Food Nutr. Res. 66, 66. doi:10.29219/fnr.v66.8101
López-Salazar, V., Tapia, M. S., Tobón-Cornejo, S., Díaz, D., Alemán-Escondrillas, G., Granados-Portillo, O., et al. (2021). Consumption of soybean or olive oil at recommended concentrations increased the intestinal microbiota diversity and insulin sensitivity and prevented fatty liver compared to the effects of coconut oil. J. Nutr. Biochem. 94, 108751. doi:10.1016/j.jnutbio.2021.108751
Luci, C., Bourinet, M., Leclère, P. S., Anty, R., and Gual, P. (2020). Chronic inflammation in non-alcoholic steatohepatitis: Molecular mechanisms and therapeutic strategies. Front. Endocrinol. (Lausanne) 11, 597648. doi:10.3389/fendo.2020.597648
Luo, W., Xu, Q., Wang, Q., Wu, H., and Hua, J. (2017). Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 7, 44612. doi:10.1038/srep44612
Machado, M. V., and Diehl, A. M. (2016). Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 150 (8), 1769–1777. doi:10.1053/j.gastro.2016.02.066
Manne, V., Handa, P., and Kowdley, K. V. (2018). Pathophysiology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Clin. Liver Dis. 22 (1), 23–37. doi:10.1016/j.cld.2017.08.007
Maximos, M., Bril, F., Portillo Sanchez, P., Lomonaco, R., Orsak, B., Biernacki, D., et al. (2015). The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease [J]. Hepatology 61 (1), 153–160. doi:10.1002/hep.27395
Mooli, R. G. R., and Ramakrishnan, S. K. (2022). Emerging role of hepatic ketogenesis in fatty liver disease. Front. Physiol. 13, 946474. doi:10.3389/fphys.2022.946474
Nagata, N., Chen, G., Xu, L., and Ando, H. (2022). An update on the chemokine system in the development of NAFLD. Med. Kaunas. 58 (6), 761. doi:10.3390/medicina58060761
Newberry, E. P., Xie, Y., Kennedy, S. M., Graham, M. J., Crooke, R. M., Jiang, H., et al. (2017). Prevention of hepatic fibrosis with liver microsomal triglyceride transfer protein deletion in liver fatty acid binding protein null mice. Hepatology 65 (3), 836–852. doi:10.1002/hep.28941
Ni, Y., Zhuge, F., Ni, L., Nagata, N., Yamashita, T., Mukaida, N., et al. (2022). CX3CL1/CX3CR1 interaction protects against lipotoxicity-induced nonalcoholic steatohepatitis by regulating macrophage migration and M1/M2 status. Metabolism 136, 155272. doi:10.1016/j.metabol.2022.155272
Osorio-Conles, Ó., Vega-Beyhart, A., Ibarzabal, A., Balibrea, J. M., Graupera, I., Rimola, J., et al. (2021). A distinctive NAFLD signature in adipose tissue from women with severe obesity. Int. J. Mol. Sci. 22 (19), 10541. doi:10.3390/ijms221910541
Packard, C. J., Boren, J., and Taskinen, M. R. (2020). Causes and consequences of hypertriglyceridemia. Front. Endocrinol. (Lausanne) 11, 252. doi:10.3389/fendo.2020.00252
Parthasarathy, G., Revelo, X., and Malhi, H. (2020). Pathogenesis of nonalcoholic steatohepatitis: An overview. Hepatol. Commun. 4 (4), 478–492. doi:10.1002/hep4.1479
Pouwels, S., Sakran, N., Graham, Y., and Pintar, T. (2022). Non-alcoholic fatty liver disease (NAFLD): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr. Disord. 22 (1), 63. doi:10.1186/s12902-022-00980-1
Rom, O., Liu, Y., Liu, Z., Zhao, Y., Wu, J., Ghrayeb, A., et al. (2020). Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci. Transl. Med. 12, eaaz2841. doi:10.1126/scitranslmed.aaz2841
Rotman, Y., and Neuschwander-Tetri, B. A. (2017). Liver fat accumulation as a barometer of insulin responsiveness again points to adipose tissue as the culprit. Hepatology 65 (4), 1088–1090. doi:10.1002/hep.29094
Schneider, K. M., Mohs, A., Kilic, K., Candels, L. S., Elfers, C., Bennek, E., et al. (2019). Intestinal microbiota protects against MCD diet-induced steatohepatitis. Int. J. Mol. Sci. 20 (2), 308. doi:10.3390/ijms20020308
Schuster, S., Cabrera, D., Arrese, M., and Feldstein, A. E. (2018). Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 15 (6), 349–364. doi:10.1038/s41575-018-0009-6
Schwabe, R. F., Tabas, I., and Pajvani, U. B. (2020). Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158 (7), 1913–1928. doi:10.1053/j.gastro.2019.11.311
She, Y., Wang, K., Makarowski, A., Mangat, R., Tsai, S., Willing, B. P., et al. (2022). Effect of high-fat and low-fat dairy products on cardiometabolic risk factors and immune function in a low birthweight swine model of diet-induced insulin resistance. Front. Nutr. 9, 923120. doi:10.3389/fnut.2022.923120
Sheng, W., Ji, G., and Zhang, L. (2022). The effect of lithocholic acid on the gut-liver Axis. Front. Pharmacol. 13, 910493. doi:10.3389/fphar.2022.910493
Sinakos, E., Liava, C., and Loomba, R. (2022). Emerging advances in the pharmacologic treatment of nonalcoholic steatohepatitis and related cirrhosis. Ann. Gastroenterol. 35 (3), 213–225. doi:10.20524/aog.2022.0704
Sowton, A. P., Padmanabhan, N., Tunster, S. J., McNally, B. D., Murgia, A., Yusuf, A., et al. (2020). Mtrr hypomorphic mutation alters liver morphology, metabolism and fuel storage in mice. Mol. Genet. Metab. Rep. 23, 100580. doi:10.1016/j.ymgmr.2020.100580
Svegliati-Baroni, G., Pierantonelli, I., Torquato, P., Marinelli, R., Ferreri, C., Chatgilialoglu, C., et al. (2019). Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 144, 293–309. doi:10.1016/j.freeradbiomed.2019.05.029
Tarantino, G., Citro, V., Balsano, C., Capone, D., Vincenzo, C., and Clara, B. (2020). Could SCGF-beta levels Be associated with inflammation markers and insulin resistance in male patients suffering from obesity-related NAFLD? Diagn. (Basel) 10 (6), 395. doi:10.3390/diagnostics10060395
Urso, C. J., and Zhou, H. (2021). Palmitic acid lipotoxicity in microglia cells is ameliorated by unsaturated fatty acids. Int. J. Mol. Sci. 22 (16), 9093. doi:10.3390/ijms22169093
Valenzuela, R., Illesca, P., Echeverría, F., Espinosa, A., Rincón-Cervera, M. Á., and Ortiz, M. (2017). Molecular adaptations underlying the beneficial effects of hydroxytyrosol in the pathogenic alterations induced by a high-fat diet in mouse liver: PPAR-α and Nrf2 activation, and NF-κB down-regulation. Food Funct. 19;8(4):1526–1537. doi:10.1039/c7fo00090a
van Golen, R. F., Olthof, P. B., Lionarons, D. A., Reiniers, M. J., Alles, L. K., Uz, Z., et al. (2018). FXR agonist obeticholic acid induces liver growth but exacerbates biliary injury in rats with obstructive cholestasis. Sci. Rep. 8 (1), 16529. doi:10.1038/s41598-018-33070-1
Van Name, M. A., Savoye, M., Chick, J. M., Galuppo, B. T., Feldstein, A. E., Pierpont, B., et al. (2020). A low ω-6 to ω-3 PUFA ratio (n-6:n-3 PUFA) diet to treat fatty liver disease in obese youth. J. Nutr. 150 (9), 2314–2321. doi:10.1093/jn/nxaa183
Wang, L., Cao, Z. M., Zhang, L. L., Li, J. M., and Lv, W. L. (2022). The role of gut microbiota in some liver diseases: From an immunological perspective. Front. Immunol. 13, 923599. doi:10.3389/fimmu.2022.923599
Welch, R. D., Billon, C., Losby, M., Bedia-Diaz, G., Fang, Y., Avdagic, A., et al. (2022). Emerging role of nuclear receptors for the treatment of NAFLD and NASH. Metabolites 12 (3), 238. doi:10.3390/metabo12030238
Wree, A., McGeough, M. D., Inzaugarat, M. E., Eguchi, A., Schuster, S., Johnson, C. D., et al. (2018). NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 67 (2), 736–749. doi:10.1002/hep.29523
Yadati, T., Bitorina, A., Oligschlaeger, Y., Gijbels, M. J., and Mohren, R. (2021). Inhibition of extracellular cathepsin D reduces hepatic lipid accumulation and leads to mild changes in inflammationin NASH mice. Front. Immunol. 12, 675535. doi:10.3389/fimmu.2021.675535
Ye, Q., Zou, B., Yeo, Y. H., Li, J., Huang, D. Q., Wu, Y., et al. (2020). Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 5 (8), 739–752. doi:10.1016/S2468-1253(20)30077-7
Younossi, Z. M., Koenig, A. B., Abdelatif, D., Fazel, Y., Henry, L., and Wymer, M. (2016). Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64 (1), 73–84. doi:10.1002/hep.28431
Zhang, N., Tao, J., Gao, L., Bi, Y., Li, P., Wang, H., et al. (2020). Liraglutide attenuates nonalcoholic fatty liver disease by modulating gut microbiota in rats administered a high-fat diet. Biomed. Res. Int. 18, 2947549. doi:10.1155/2020/2947549
Zhang, V. X., Sze, K. M., Chan, L. K., Ho, D. W., Tsui, Y. M., Chiu, Y. T., et al. (2021). Antioxidant supplements promote tumor formation and growth and confer drug resistance in hepatocellular carcinoma by reducing intracellular ROS and induction of TMBIM1. Cell Biosci. 11 (1), 217. doi:10.1186/s13578-021-00731-0
Zhao, Y., Wang, F., Gao, L., Xu, L., Tong, R., Lin, N., et al. (2018). Ubiquitin-specific protease 4 is an endogenous negative regulator of metabolic dysfunctions in nonalcoholic fatty liver disease in mice. Hepatology 68 (3), 897–917. doi:10.1002/hep.29889
Keywords: non-alcoholic fatty liver diseases, non-alcoholic steatohepatitis, inflammation, peroxisome proliferator-activated receptor- γ (PPAR-γ), insulin resistance
Citation: Song C, Long X, He J and Huang Y (2023) Recent evaluation about inflammatory mechanisms in nonalcoholic fatty liver disease. Front. Pharmacol. 14:1081334. doi: 10.3389/fphar.2023.1081334
Received: 27 October 2022; Accepted: 28 February 2023;
Published: 16 March 2023.
Edited by:
Elena Talero, Sevilla University, SpainReviewed by:
Yuanli Chen, Hefei University of Technology, ChinaCopyright © 2023 Song, Long, He and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongpan Huang, eW9uZ3Bhbmh1YW5neXh5QDE2My5jb20=; Jianbin He, aGpiMDkxOWhoQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.