
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 10 November 2022
Sec. Inflammation Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.999851
This article is part of the Research TopicThe Potential Drug Regulation in Arthritic DisordersView all 11 articles
 Hao Tang1,2†
Hao Tang1,2† Kunpeng Qin1†
Kunpeng Qin1† Anquan Wang1
Anquan Wang1 Shuang Li1
Shuang Li1 Sheng Fang3
Sheng Fang3 Weilu Gao1
Weilu Gao1 Ming Lu1
Ming Lu1 Wei Huang4*
Wei Huang4* Hui Zhang1*
Hui Zhang1* Zongsheng Yin1*
Zongsheng Yin1*Osteoarthritis (OA) is a chronic degenerative joint disease characterized by articular cartilage destruction. The pathological mechanisms are complex; in particular, inflammation, autophagy, and apoptosis are often involved. 3,3-Diindolylmethane (DIM), a phytoconstituent extracted from cruciferous vegetables, has various effects such as anti-inflammatory, antioxidant and anti-apoptotic. However, the effects of DIM on osteoarthritic chondrocytes remain undetermined. In this study, we simulated a lipopolysaccharide (LPS)-induced osteoarthritis model in human primary chondrocytes. We found that LPS stimulation significantly inhibited autophagy, induced chondrocyte apoptosis and extracellular matrix (ECM) degradation, which could be ameliorated by DIM. DIM inhibited the expression of a disintegrin and metalloproteinase with thrombospondin motif 5 (ADAMTS-5), matrix metalloproteinase 13 (MMP13), cleaved caspase-3, Bax, and p62, and increased the expression level of collagen II, aggrecan, Bcl-2, light chain 3 Ⅱ (LC3 Ⅱ), and beclin-1. Mechanistic studies showed that DIM increased chondrocyte autophagy levels by inhibiting the activation of PI3K/AKT/mTOR pathway. In mice destabilization of the medial meniscus (DMM) model, immunohistochemical analysis showed that DIM inhibited the expression of p-PI3K and cleaved caspase-3, increased the expression of LC3 Ⅱ. Furthermore, DIM relieved joint cartilage degeneration. In conclusion, our findings demonstrate for the first time that DIM inhibits LPS-induced chondrocyte apoptosis and ECM degradation by regulating the PI3K/AKT/mTOR-autophagy axis and delays OA progression in vivo.
Osteoarthritis (OA) is a degenerative joint disease that affects older people worldwide, characterized by progressive destruction and inflammation of articular cartilage (Glyn-Jones et al., 2015). The incidence of OA is reported to be increasing year by year, and the condition is a major cause of disability in older adults. Contemporary treatment mainly improves symptoms, and there is no effective treatment to prevent the progression of OA (Maudens et al., 2018). Therefore, it is clinically important to develop more convenient and effective interventions to slow down progression of the disease (Goldring, 2006). Degenerative changes of articular cartilage are the most important pathological changes in OA, and articular cartilage consists of only chondrocytes and extracellular matrix (ECM). The main matrix components of the ECM are collagen II and aggrecan, and the main ECM catabolic enzymes are matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) (Cucchiarini et al., 2016). Under normal physiological conditions, chondrocytes maintain the balance of ECM synthesis and catabolism to ensure the structural and functional integrity of cartilage (Musumeci et al., 2011a).
Recently, an increasing number of studies have shown that OA has diverse causative factors and a complex pathogenesis and determined that the release of inflammatory mediators causes inhibition of cellular autophagy, resulting in increased apoptosis, which ultimately leads to an imbalance in ECM metabolism and accelerates OA development (Li et al., 2019). Previous study has reported that apoptosis is positively correlated with the severity of cartilage destruction and matrix depletion in human OA tissue specimens (Musumeci et al., 2015). Chondrocytes freshly isolated from human OA cartilage displayed morphological evidence of apoptosis, while those from normal donors did not have any apoptotic cell signatures. These findings suggest that OA chondrocytes exhibit distinct apoptotic propensities (Musumeci et al., 2011a; Musumeci et al., 2011b). In addition, when chondrocyte homeostasis is unbalanced, the release of inflammatory mediators and catabolic enzymes is accelerated, the destruction of ECM speeds up, and OA progression occurs (Zhao et al., 2020).
Autophagy is a mechanism and a dynamic cellular process whereby damaged cellular components (such as organelles and proteins) are wrapped by membrane structures to form autophagic vesicles, which then fuse with lysosomes, and whose contents are degraded and recycled to maintain normal cellular metabolism (He and Klionsky, 2009; Ravikumar et al., 2010; Polewska, 2012; Li et al., 2016). It has been reported that normal cartilage tissue expresses abundant LC3 II, Beclin1 and other important autophagy-related proteins, suggesting that autophagy may be involved in maintaining the normal physiological function and structural integrity of cartilage tissue (Sasaki et al., 2012; Zhang et al., 2015; Qin et al., 2017). In contrast, the expression of autophagy-related proteins is reduced in human OA chondrocytes (Loeser, 2011; Lian et al., 2018). Activation of autophagy increases the expression levels of both collagen II and aggrecan while reducing those of ADAMTS-5 and MMP-13 in chondrocytes and ameliorating arthritis progression (Qin et al., 2017; Huang et al., 2020; Lin et al., 2021). Furthermore, inhibition of autophagy in mice chondrocytes exacerbated arthritis progression (Bouderlique et al., 2016), while intra-articular injection of rapamycin to activate autophagy in a mice destabilization of the medial meniscus (DMM) model improved cartilage degeneration (Takayama et al., 2014).
3,3-diindolylmethane (DIM) is a natural product found in cruciferous edible plants, such as cauliflower, cabbage, and broccoli. Previous studies have reported that DIM has several preventive effects, especially displaying anti-tumor, anti-inflammatory, antioxidant and free radical scavenging roles (Luo et al., 2018; Ye et al., 2021). Recent studies have determined that DIM also protects against kidney, heart, and liver damage. It has been reported that DIM attenuated carbon tetrachloride–induced acute liver injury in mice by inhibiting inflammatory responses and apoptosis and by modulating oxidative stress (Munakarmi et al., 2020). DIM has been reported to attenuate oxidative stress–induced apoptosis in hippocampal neurons (Lee et al., 2019) and protect neuronal cells from inflammation and brain tissue ischemia (Rzemieniec et al., 2016; Lee et al., 2019). In addition, DIM attenuates lipopolysaccharide (LPS)-induced inflammatory responses and apoptosis in cardiomyopathy (Luo et al., 2018) and induces protective autophagy in prostate cancer (Draz et al., 2017). In a rat model of rheumatoid arthritis, DIM was observed to block osteoclast formation (Dong et al., 2010) and, through inhibition of the MAPK and AKT/mTOR pathways, inhibit synovial fibroblast proliferation, migration, invasion, and inflammatory factor release and attenuate experimental arthritis progression (Du et al., 2019).
Although previous studies have extensively investigated the function of DIM, the potential role of DIM in osteoarthritis and its effects on chondrocytes remain undetermined. In the present study, we observed the effects of DIM on LPS stimulation-induced chondrocyte autophagy, apoptosis, and ECM metabolism in vitro and explored the possible molecular mechanisms. The potential therapeutic function of DIM was also evaluated in a DMM mice model of OA.
DIM was purchased from MCE (HY-15758, Monmouth Junction, NJ, United States) (purity >99%). LPS is currently the main pro-inflammatory inducer (Mayeux, 1997), which is commonly used to induce arthritis models in chondrocytes (Yoshino et al., 2000; Lorenz et al., 2013; Ding et al., 2019; Li et al., 2020; Zeng et al., 2021). So, we used LPS (10 µg/ml, Sigma-Aldrich) to treat human primary chondrocytes to simulate OA models. Six groups were established. The control group contained chondrocytes without any treatment. The negative control group contained chondrocytes to which only DIM (40 µM) was added. The LPS group contained chondrocytes only treated with LPS (10 ug/ml). The LPS + DIM group contained the chondrocytes treated with different concentrations of DIM (10, 20, 40 µM) for 2 h, and then LPS was added in the medium to stimulate chondrocytes for 24 h (without replacing the medium). We used 40 µM of DIM for the detection of immunofluorescence, flow cytometry, monodansylcadaverine (MDC) staining, transmission electron microscopy (TEM) and glycosaminoglycan (GAG) experiments.
In vivo, mice were randomly divided into a sham-operated group, an OA group (DMM), and a DIM-treated OA group (DMM + DIM) (n = 10 in each group). Mice DMM model creation took place in the OA group, and the DIM-treated OA group received a 50 mg/kg/day dose of DIM (via daily intraperitoneal injection for 8 weeks). Mice in the Sham and DMM groups received equal amounts of saline. All animals were executed 8 weeks after surgery, and cartilage samples were collected for immunological and histological analysis.
The collection of cartilage tissue involving human OA was approved by the Medical Ethics Committee of the First Affiliated Hospital of Anhui Medical University (ethics no. PJ2022-04-55), and all participants signed an informed consent form. Articular cartilage was obtained from patients who had undergone total knee arthroplasty at the First Affiliated Hospital of Anhui Medical University. The specimens were rapidly transferred to the operating table and washed 3–5 times with phosphate-buffered saline (PBS) in a sterile environment, and the cartilage tissue was then cut into bone pieces of 1 mm3 in size and then digested with 0.25% trypsin (Beyotime, Shanghai, China) for 30 min, followed by 0.2% collagenase Ⅱ (Sigma-Aldrich) in a 37°C incubator overnight. The digested chondrocytes were then resuspended and inoculated in culture flasks containing Dulbecco’s modified Eagle medium/nutrient mixture F-12, 10% fetal bovine serum, and 1% penicillin/streptomycin antibiotics; when 80%–90% fusion was achieved, the cells were then digested using 0.25% trypsin solution, centrifuged, resuspended, and passaged. The complete medium was changed every 2 days, and only cells of the first or second generation were used for the experiments to avoid phenotypic changes.
Chondrocytes were inoculated in 24-well plates and cultured for 24 h, then washed three times with PBS solution for 5 min/time and subsequently fixed in 4% paraformaldehyde solution for 20 min. After being washed with PBS, they were treated with 1% toluidine blue solution (Solarbio, Beijing, China) for 1 h at room temperature, washed with PBS again, dried and placed on slides, and then sealed with neutral gum. All chondrocytes were observed under a microscope (Tissue FAXS Plus S; Tissue Gnostics, Vienna, Austria) and photographed.
The cytotoxicity of DIM on human chondrocytes was assayed with a cell counting kit 8 (CCK-8) (Beyotime). First, human chondrocytes were cultured in 96-well plates (5×103 cells/well) for 24 h with different concentrations of DIM (0, 1, 5, 10, 20, 40, or 80 μM) for 24 or 48 h. After each well washed with PBS, 100 μL new medium containing 10% CCK8 was added to each well, incubated at 37°C for 2 h. The absorbance was then detected at 450 nm using a microplate reader (Leica Microsystems, Wetzlar, Germany).
The treated cells were washed with enzyme-free water, and total cellular RNA was subsequently extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, United States), RNA purity was assessed according to a 260-/280-nm ratio using a NanoDrop One device (Thermo Fisher Scientific, Waltham, MA, United States), and RNA concentrations were measured using a reverse transcription kit (Takara Bio, Kusatsu, Japan) A 10-µL reaction system was set up to reverse-transcribe the extracted RNA to complementary DNA. Subsequently, qRT-PCR of the target genes was performed according to the SYBR® Premix Ex Taq™ II kit (Takara Bio), and data analysis was performed with the Light Cycler 96 software (Roche, Alameda, CA, United States). The target gene messenger RNA levels were normalized to GAPDH levels, and the data obtained were analyzed using the 2−ΔΔCT method. All experiments were repeated three times. The primer sequences were provided by Sangon (Shanghai, China) and are listed in Supplementary Table S1.
A western blot technique was used to detect the expressions of related proteins. The treated cells were collected, radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime) and phenylmethylsulfonyl fluoride (PMSF) (Beyotime) were added, and the mixture was placed on ice for 30 min to lyse. Then, the supernatant was centrifuged and subsequently protein concentration was measured by BCA method (Beyotime) and added to 5 × sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) protein loading buffer (Beyotime) and boiled for 10 min to obtain the total protein. Each well was loaded with 20ug protein, and then under the same GAPDH condition, the loading volume of each sample was calculated according to the required loading amount of protein and the sample protein concentration. The protein was subsequently separated by SDS-PAGE, then transferred to a polyvinylidene fluoride membrane (Millipore, Burlington, MA, United States) in constant flow. The membranes were subsequently closed with 5% skim milk for 2 h at room temperature. washed with tris-buffered saline and polysorbate 20 and incubated with the specific primary antibody overnight at 4°C, then washed and incubated with the corresponding specific secondary antibody for 2 h at room temperature, respectively. Finally, all band signals were detected using ECL ultrasensitive chemiluminescence reagents (Thermo Fisher Scientific, Waltham, MA, United States) and detected using the ImageJ version 1.53c software (U.S. National Institutes of Health, Bethesda, MD, United States) for quantitative analysis. The following antibodies were used in the western blot analysis: anti–collagen Ⅱ antibody (1:1000, 28459-1-AP, Proteintech Group, Rosemont, IL, United States), anti-aggrecan antibody (1:1000, 13880-1-AP, Proteintech Group), anti–ADAMTS-5 antibody (1:1000, Ab41037, Abcam, Cambridge, United Kingdom), anti–MMP-13 antibody (1:1000, 18165-1-AP, Proteintech Group), anti–LC3 A/B antibody (1:1000, 12741, CST, Danvers, MA, United States), anti-p62 antibody (1:10000, Ab109012, Abcam), anti–beclin-1 antibody (1:1000, Ab62557, Abcam), anti–cleaved caspase-3 antibody (1:1000, 5A1E, CST), anti-Bax antibody (1:5000, Ab32503, Abcam), anti–Bcl-2 antibody (1:1000, Ab32124, Abcam), anti–p-PI3K antibody (1:1000, ab151549, Abcam), anti–PI3K antibody (1:1000, 3358, CST), anti-AKT antibody (1:1000, 4691, CST), anti–phospho-AKT antibody (1:2000, 4060, CST), anti-mTOR antibody (1:5000, Ab32028, Abcam), anti–phospho-mTOR antibody (1:5000, Ab109268, Abcam), and anti-GAPDH antibody (1:10000, 60004-1-lg, Proteintech Group).
The treated cells were washed with PBS, then underwent 4% paraformaldehyde fixation for 20 min, were washed with PBS three more times, and underwent 0.3% Triton X-100 permeabilized for 15 min at room temperature. The cells were then closed with 10% bovine serum albumin (BSA) (goat serum blocking solution, Beyotime) for 1 h, then were washed with PBS and incubated overnight at 4°C in a wet box with the appropriate specific primary antibody, as follows: anti–collagen Ⅱ antibody (1:300, 28459-1-AP, Proteintech Group), anti–MMP-13 antibody (1: 300, 18165-1-AP, Proteintech Group), anti–LC3 A/B antibody (1:200, 12741, CST), anti–cleaved caspase-3 antibody (1:400, 5A1E, CST), or anti–p-PI3K antibody (1:300, Ab151549, Abcam). After three washes, the cells were incubated with fluorescein isothiocyanate (FITC) or rhodamine-labeled secondary antibody (1:100ZF-0311/ZF-0316, ZSGB-BIO, Beijing, China) for 1 h at room temperature and protected from light. Subsequently, 4′,6-diamidino-2-phenylindole (DAPI) staining solution (Beyotime) was added to label cell nuclei for 5 min, and finally anti-fluorescence quenching mounting solution was added dropwise. Images were observed and obtained under an automatic positive fluorescence microscope (DM6B; Leica, Wetzlar, Germany) and were quantified using ImageJ version 1.53c (U.S. National Institutes of Health, Bethesda).
MDC is one of the most used fluorescent probes for cellular autophagy detection. It can specifically label autophagosomes through ion capture and specific binding to membrane lipids (Biederbick et al., 1995; Niemann et al., 2000; Niemann et al., 2001; Vázquez and Colombo, 2009). During our study, treated chondrocyte crawls were fixed in 4% paraformaldehyde for 20 min, washed three times with PBS, and incubated with MDC staining solution (Beyotime) for 60 min at 37°C in an incubator protected from light. Then, we washed them three times with assay buffer. Green fluorescence was observed under an automatic positive fluorescence microscope (DM6B; Leica).
The treated cells were collected, then the samples were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (P885738, Macklin, Shanghai, China) at 4°C overnight. After washing with phosphate buffer, the samples were fixed in phosphate buffer with 1% OsO4 at 4°C for 2 h and rinsed thoroughly with ddH2O. The 2% aqueous uranyl acetate was used for en bloc staining for 2 h and then the samples were serially dehydrated with 50%, 70%, 90% and 100% alcohol and 100% acetone and embedded in epoxy resin for making the blocks of samples. Silver sections were cut with an ultramicrotome (EM UC7, Leica; thickness 70–90 nm), stained with lead citrate and uranyl acetate, and observed with an electron microscope (Talos L120C G2, Thermo Scientific, MA, United States).
Safranin O is a cationic dye that binds polyanions and binds to GAGs in chondrocytes to give them a red color, with the intensity of the red color being directly proportional to the GAG content. It is used to assess the content of GAGs in chondrocytes and can reflect the ability of chondrocytes to perform anabolic and catabolic activities. We washed the treated chondrocytes with PBS and fixed them with 4% paraformaldehyde for 20 min, then washed them with PBS and incubated them with safranin O staining solution (Solarbio, Beijing, China) for 30 min at room temperature. The cells were then washed with PBS and observed by microscopy (Tissue FAXS Plus S; Tissue Gnostics), and photographs were taken.
The treated chondrocytes were collected in flow tubes, washed twice with cold PBS, and resuspended with 400 μL of 1 × annexin V conjugate using the annexin V-FITC/propidium iodide double-stained apoptosis assay kit (BestBio, Shanghai, China), followed by the addition of 5 μL of annexin V-FITC staining solution and incubation for 15 min at 4°C under light-proof conditions. Finally, 5 μL of propidium iodide (PI) staining solution was added and incubated for 3 min at 4°C under light-proof conditions, followed by flow cytometry (BD Celesta, San Jose, CA, United States). Flow analysis was performed with FlowJo version 10.6.0 (FlowJo LLC, Ashland, OR, United States).
Thirty 10-week-old C57BL/6 male wild-type mice were obtained from the Animal Center of Anhui Medical University. All surgical interventions, treatments, and postoperative animal care procedures were administered in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were performed in strict accordance with the requirements of the Animal Ethics Committee of Anhui Medical University (ethics no. LISC20190738). All the mice in the experiment were housed in standard experimental cages with a 12-h light/dark cycle and were freely access to water and standard food. Mice OA models were created by DMM surgery. Anesthesia was performed by intraperitoneal injection of an appropriate amount of 2% pentobarbital, followed by exposure of the joint capsule medial to the patellar tendon of the right knee and dissection of the medial meniscus tibial ligament with microsurgical scissors. In the sham group, only the joint capsule was incised, and the medial meniscal tibial ligament was not treated.
The obtained knee joints were fixed in 4% paraformaldehyde for 24 h, then decalcified, paraffin-embedded, cut into 5-μm sections, dewaxed, and rehydrated. Antigen repair was performed using the E enzyme method (DIG-3008; MXB, Fuzhou China), and appropriate amounts of rabbit- or mouse-derived endogenous peroxidase blocker (PV-6001/PV-6002, ZSGB-BIO) were added and incubated at room temperature for 10 min. After rinsing with PBS, an appropriate amount of primary antibody was added dropwise and incubated at 37°C for 60 min, using the appropriate specific primary antibody, as follows: anti–LC3 A/B antibody (1:300, 12741, CST), anti–cleaved caspase-3 antibody (1:1000, 5A1E, CST), anti-p-PI3K antibody (1:100, Ab151549, Abcam), anti–collagen Ⅱ antibody (1:800, 28459-1-AP, Proteintech Group), anti-aggrecan antibody (1:200, 13880-1-AP, Proteintech Group), anti–ADAMTS-5 antibody (1: 200, DF13268, Affinity), anti–MMP-13 antibody (1:200, 18165-1-AP, Proteintech Group). This was followed by the addition of the corresponding enzyme-labeled goat anti-rabbit or mouse immunoglobulin G polymer (PV-6001/PV-6002, ZSGB-BIO) dropwise to the section, then incubation at room temperature for 20 min; the addition of DAB (ZLI-9018, ZSGB-BIO) color development solution. Finally, re-staining, dehydration, transparency, and sealing of the section. The staining results were observed and interpreted by a qualified pathologist under a light microscope.
The obtained mice knee sections were dewaxed, hydrated, and stained with hematoxylin and eosin (H and E) staining as well as safranin O/fast green staining (Servicebio, Wuhan, China) to assess cartilage destruction. The extent of cartilage degeneration in the stained sections was assessed using the Osteoarthritis Research Society International (OARSI) scoring system (Glasson et al., 2010). The extent of synovial tissue changes in the stained sections was assessed using the Krenn Synovitis Score Criteria (Krenn et al., 2002; Krenn et al., 2006).
Eight weeks after DMM, mice were placed on a digital X-ray machine (Labscope, Glenbrook Technologies lnc, Randolph, NJ, United States) to undergo X-ray frontal and lateral imaging of all knee joints to assess the joint space, cartilage surface sclerosis, and bone formation. X-ray machine settings were 25 kV and 0.1 mA.
All experiments were performed at least three times, and data are presented as mean ± standard deviation (SD) values. Statistical analysis was performed using the Prism version 9.0 software (GraphPad Software, San Diego, CA, United States). Statistically significant differences between the two groups were analyzed by t test, one-way ANOVA was used to compare multiple data groups. p < 0.05 was statistically significant.
First, we used toluidine blue staining and immunofluorescence staining to identify the isolated primary chondrocytes. Toluidine blue staining showed that the proteoglycans in the chondrocytes stained blue-purple, and the chondrocytes were spindle-shaped (Figure 1A). In addition, immunofluorescence staining showed greenish collagen II in the cytoplasm of chondrocytes and no positive staining in cell nuclei (Figure 1B). These two staining methods confirmed that the primary cells extracted from articular cartilage were chondrocytes.
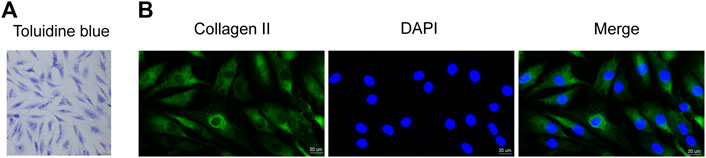
FIGURE 1. Identification of human primary chondrocytes and the effect of DIM on human OA chondrocyte viability (A) Proteoglycans in human primary chondrocytes were stained purple by toluidine blue (scale bar, 50 μm) (B) Collagen II immunofluorescence staining showed that the collagen II was stained green, while cell nuclei were stained blue by DAPI (scale bar, 20 μm).
The chemical structure of DIM is illustrated in Figure 2A. To clarify the cytotoxic effect of DIM on human chondrocytes, we assayed the cell viability by CCK-8 assay. Different concentrations of DIM (0, 1, 5, 10, 20, 40, and 80 μM) were incubated with chondrocytes for 24 and 48 h. After DIM treatment of chondrocytes for 24 and 48 h, there was no significant cytotoxic effect on primary chondrocytes at concentrations of 0–40 μM, but the high concentration of 80 μM reflected significant cytotoxicity and affected cell viability (Figures 2B,C). Therefore, the safe concentrations of 0, 10, 20, or 40 μM of DIM were used in subsequent related experiments.
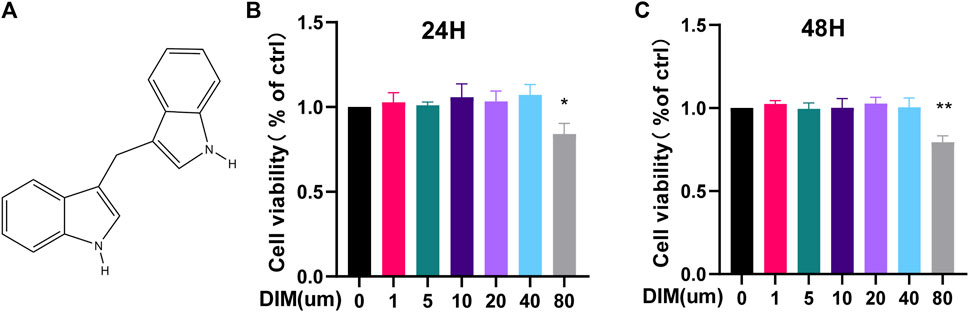
FIGURE 2. (A) Chemical structure of DIM (B,C) The cytotoxicity of DIM on chondrocytes was examined at various concentrations of DIM (0, 1,5, 10, 20, 40, and 80 μM) for 24 and 48 h by using the CCK-8 assay. The data are presented as mean ± SD values from 3 independent experiments. Using one-way ANOVA, * p <0.05, ** p <0.01 vs. the control.
We used western blot analysis (Figures 3A,B) and qRT-PCR (Figure 3C) to detect the expression of the following anabolic and catabolic indicators in cartilage: collagen II, aggrecan, ADAMTS-5, and MMP-13. Collagen II and aggrecan were significantly reduced in the LPS group, while ADAMTS-5 and MMP-13 were significantly increased. Pre-treatment with DIM reversed the downregulation of collagen II and aggrecan and the upregulation of ADAMTS-5 and MMP-13 induced by LPS stimulation in a dose-dependent relationship. We further examined the deposition of GAGs in human chondrocytes using safranin O staining, and GAG expression appeared significantly reduced in LPS-treated chondrocytes compared to controls, whereas DIM treatment ameliorated the LPS-induced loss of GAGs (Figure 3D, Supplementary Figure S2). In addition, we detected the expressions of collagen II and MMP-13 by immunofluorescence, and the results of fluorescence analysis were consistent with the western blot results (Figures 3E,F). Overall, the results showed that DIM ameliorated LPS-induced human primary chondrocytes anabolic and catabolic imbalance, which in turn reflected that DIM improved LPS-induced ECM degradation.
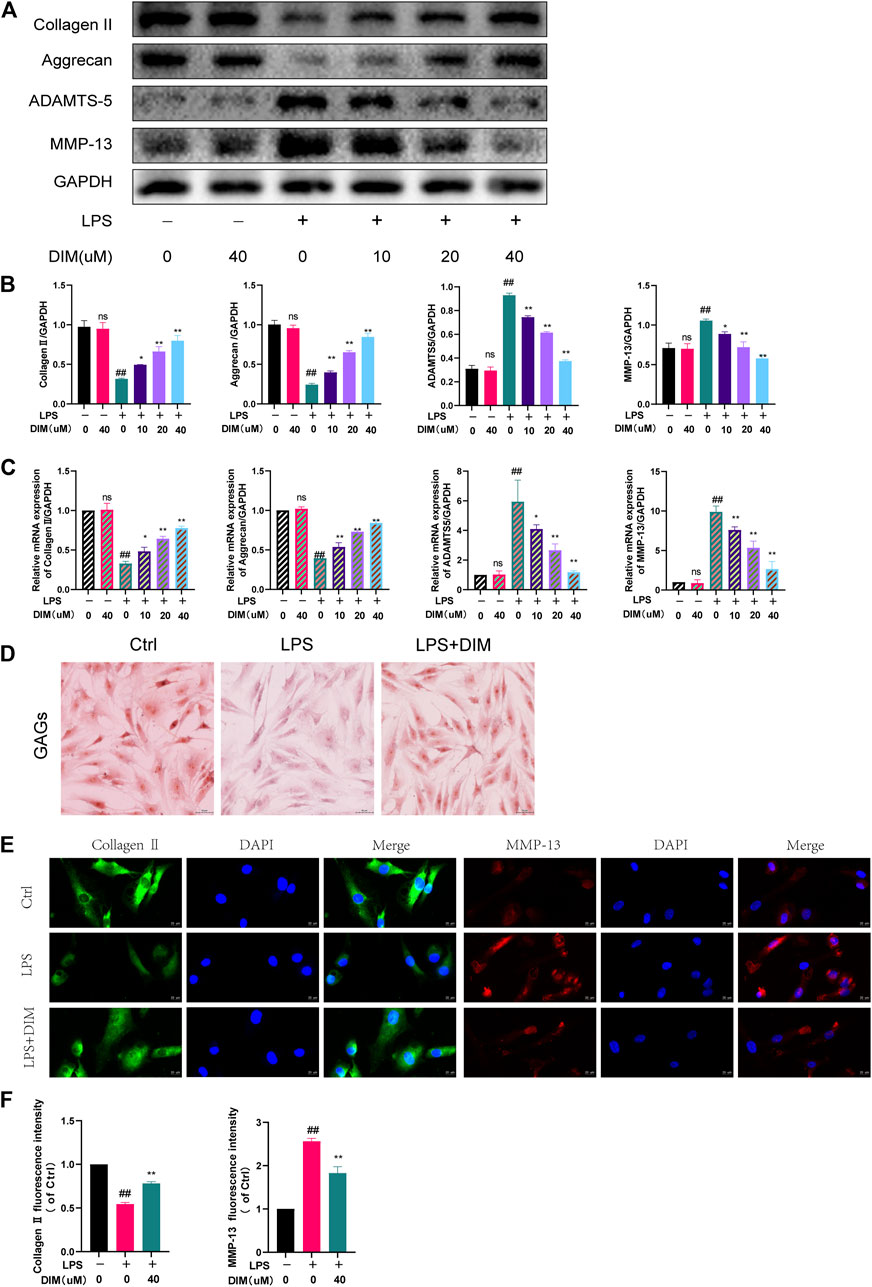
FIGURE 3. DIM alleviates LPS-induced ECM degradation in human chondrocytes (A,B) Effects of DIM on the protein expression levels of collagen II, aggrecan, ADAMTS-5, and MMP-13 in human chondrocytes treated as above were measured by western blotting and quantitation (C) Effects of DIM on the messenger RNA expression levels of collagen II, aggrecan, ADAMTS-5, and MMP-13 in human chondrocytes treated as above were measured by qRT-PCR (D) Safranin O staining of GAGs in human primary chondrocytes in each group (scale bar, 50 μm) (E) After treatment, immunofluorescence staining showed the changes in the fluorescence intensity of collagen II and MMP-13 in each group (scale bar, 20 µm) (F) The fluorescence intensity was measured with the ImageJ software (U.S. National Institutes of Health, Bethesda). The data are presented as mean ± SD values from three independent experiments. Using one-way ANOVA, NS, no statistical difference. # p <0.05 vs. the control; # # p <0.01 vs. the control; * p <0.05 vs. the LPS group; ** p < 0.01 vs. the LPS group.
We detected the expressions of Bax, Bcl-2, and cleaved caspase-3 by western blot analysis and further detected the expression of cleaved caspase-3 by immunofluorescence staining. Compared to the control group, LPS stimulation upregulated Bax and cleaved caspase-3 expression, while Bcl-2 expression was significantly down-regulated. In contrast, the expressions of Bax and cleaved caspase-3 were significantly down-regulated in the LPS group with the addition of DIM pre-treatment, while Bcl-2 expression was significantly increased (Figures 4A,B). Similarly, immunofluorescence staining for labeled cleaved caspase-3 results showed that DIM treatment decreased the intensity of cleaved caspase-3 (Figures 4C,D). In addition, apoptotic cells were examined by flow cytometry, and LPS caused an increase in chondrocyte apoptosis compared to the control group. However, the percentage of LPS-induced apoptosis was significantly reduced following pre-treatment with 40 μM of DIM (Figures 4E,F). These data suggest that DIM has an anti-apoptotic effect in human chondrocytes.
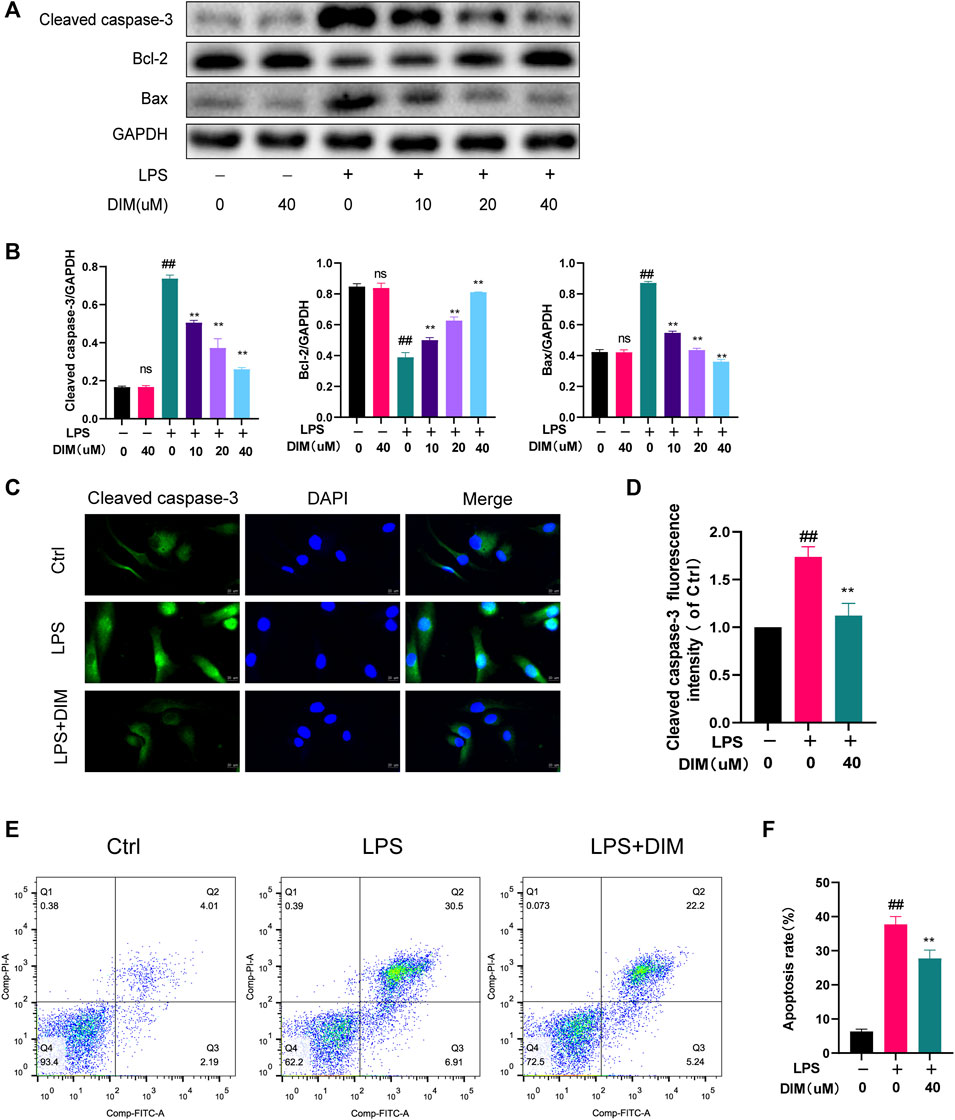
FIGURE 4. DIM attenuates LPS-induced apoptosis in human chondrocytes (A,B) Effects of DIM on the protein expression levels of cleaved caspase-3, Bcl-2, and Bax in human chondrocytes treated as above were measured by western blotting and quantitation (C) After treatment, immunofluorescence staining showed the changes in the fluorescence intensity of cleaved caspase-3 in each group (scale bar, 20 µm) (D) The fluorescence intensity was measured with the ImageJ software (U.S. National Institutes of Health, Bethesda) (E,F) After treatment, the results of flow cytometry detection of apoptotic cells in each group and quantitation. The data are presented as mean ± SD values from three independent experiments. Using one-way ANOVA, NS, no statistical difference. # p <0.05 vs. the control; # # p <0.01 vs. the control; * p <0.05 vs. the LPS group; ** p <0.01 vs. the LPS group.
Autophagy is a common mechanism for the removal of redundant or damaged organelles during development and aging. Moreover, autophagy plays a key role in regulating energy cycling and cellular homeostasis in chondrocytes (Mizushima and Levine, 2020). To investigate whether DIM activates autophagy in chondrocytes, we performed western blot analysis to assess changes in beclin-1, LC3 II, and p62 protein levels in human chondrocytes under different treatment conditions, which play a key role in the onset of autophagy (Glick et al., 2010; Boya et al., 2013). In LPS-treated primary human chondrocytes, a significant increase in LC3 II and beclin-1 levels and a significant decrease in p62 levels were observed in cells pre-treated with increased DIM (Figures 5A,B). Then, immunofluorescence staining results showed that DIM pre-treatment significantly increased the expression of LC3 II in LPS-stimulated chondrocytes (Figures 5C,D). In addition, MDC is an eosinophilic fluorescent dye that is specific for autophagosome formation. we assessed autophagic flux by MDC staining and transmission electron microscopy to observe the effect of DIM as well as that of LPS on the number of autophagosomes in chondrocytes; the intensity of MDC staining was diminished and the number of autophagosomes was reduced in LPS-treated chondrocytes compared to the blank control group. The intensity of MDC staining and the number of autophagosomes were significantly increased in primary human chondrocytes in the LPS + DIM group compared to the LPS group (Figures 5E,F). Based on these results, DIM could ameliorate the LPS-induced inhibition of autophagy and restore chondrocyte autophagy levels.
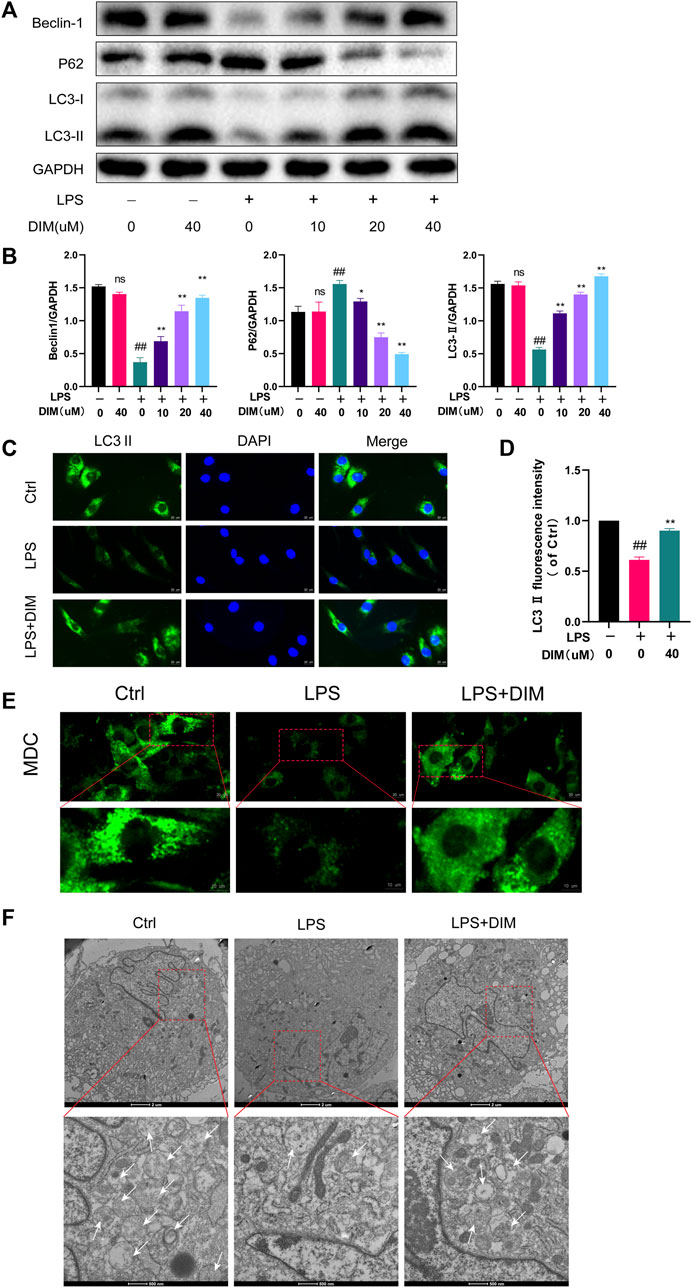
FIGURE 5. DIM relieves the LPS-induced inhibition of autophagy (A,B) Effects of DIM on the protein expression levels of beclin-1, LC3 II, and p62 in human chondrocytes treated as above were measured by western blotting and quantitation (C) After treatment, immunofluorescence staining showed the changes in the fluorescence intensity of LC3 II in each group (scale bar, 20 µm) (D) The fluorescence intensity was measured with the ImageJ software (U.S. National Institutes of Health, Bethesda) (E) After treatment, the effects of DIM and LPS on the number of chondrocytes autophagosomes were observed by MDC staining (punctate green staining represents autophagosomes; scale bar, 20 μm; partially enlarged image scale bar, 10 µm) (F) Microstructural detection of autophagosomes by transmission electron microscopy (White arrows indicate the formation of autophagosomes. Scale bar = 2 μm; partially enlarged image scale bar, 500 nm). The data are presented as mean ± SD values from three independent experiments. NS, no statistical difference; # p <0.05 vs. the control; # # p <0.01 vs. the control; * p <0.05 vs. the LPS group; ** p <0.01 vs. the LPS group.
It is well known that the PI3K/AKT/mTOR signaling pathway is a key regulatory pathway for autophagy, and activation of the PI3K/AKT/mTOR pathway in OA is associated with autophagy inhibition (Xue et al., 2017). To confirm whether the mechanism of enhanced autophagy by DIM is related to the PI3K/AKT/mTOR signaling pathway, p-PI3K, p-AKT, and p-mTOR were detected by western blot analysis. The results showed that LPS treatment activated the PI3K/AKT/mTOR pathway compared to the control group, and the expressions of p-PI3K, p-AKT, and p-mTOR were significantly increased; meanwhile, the addition of DIM pre-treatment to the LPS group dose-dependently down-regulated the expressions of p-PI3K, p-AKT, and p-mTOR but did not affect the expressions of total PI3K, total AKT, or total mTOR (Figure 6A). The western blotting quantification analysis were based on the ratio of phosphorylated proteins (p-PI3K, p-AKT and p-mTOR) to total proteins (PI3K, AKT and mTOR) (Figure 6B). Immunofluorescence staining showed that DIM pre-treatment significantly inhibited p-PI3K activation (Figures 6C,D). These data suggest that the DIM can Inhibit activation of the PI3K/AKT/mTOR pathway in human chondrocytes.
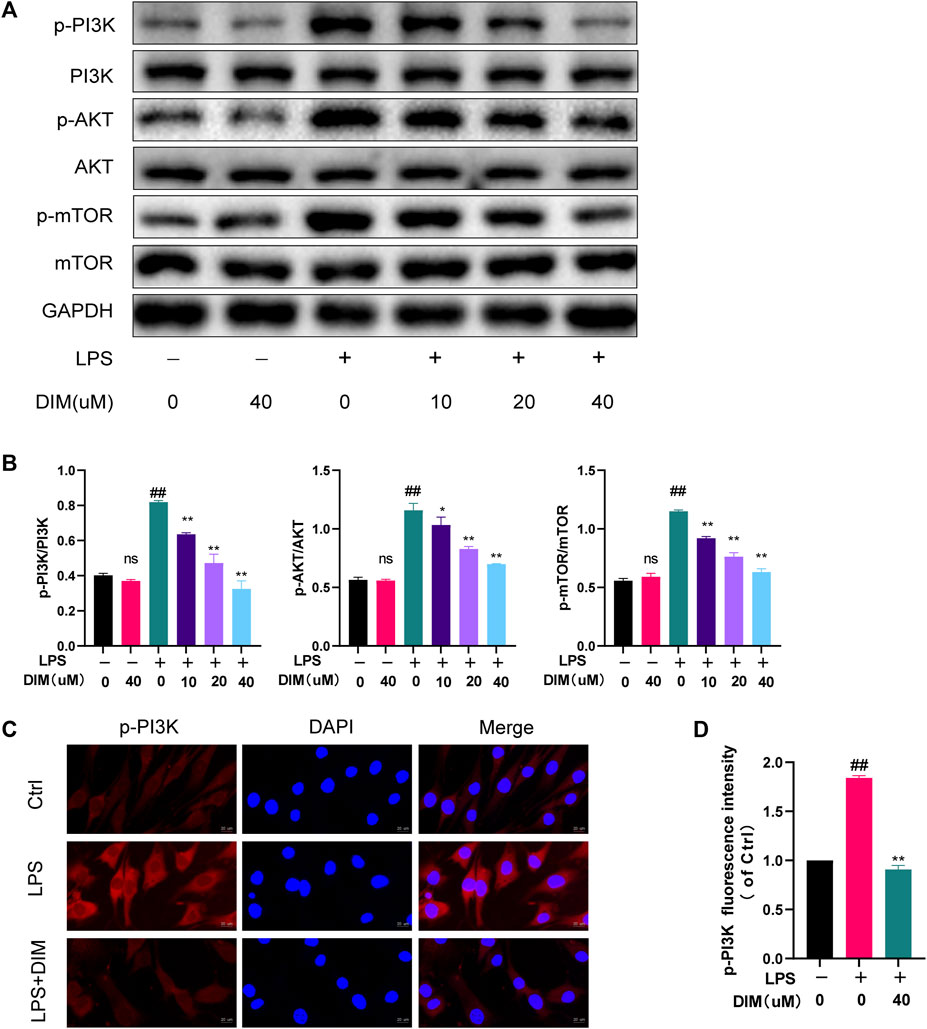
FIGURE 6. DIM inhibits the activation of the PI3K/AKT/mTOR signaling pathway (A,B) Effects of DIM on the protein expression levels of p-PI3K, total PI3K, p-AKT, total AKT, p-mTOR, and total mTOR in human chondrocytes treated as above were measured by western blotting and quantitation (C) After treatment, immunofluorescence staining showed the changes in the fluorescence intensity of p-PI3K in each group (scale bar, 20 µm) (D) The fluorescence intensity was measured with the ImageJ software (U.S. National Institutes of Health, Bethesda). The data are presented as mean ± SD values from three independent experiments. Using one-way ANOVA, NS, no statistical difference; # p <0.05 vs. the control; # # p <0.01 vs. the control; * p < 0.05 vs. the LPS group; ** p < 0.01 vs. the LPS group.
To determine whether DIM-enhanced autophagy was mediated by the PI3K/AKT/mTOR pathway, the pathway activator 740Y-P (HY-P0175, MCE, Monmouth Junction, NJ, United States) and inhibitor LY294002 (HY-10108, MCE, Monmouth Junction, NJ, United States) were used for functional reversion experiments (Figures 7A,B). DIM increased LC3 II and beclin-1 expression levels and decreased p62 levels in the LPS group, but the increase in autophagy triggered by DIM was eliminated with 740Y-P, while the increase in autophagy triggered by DIM was enchanced with LY294002. Similar results were obtained for immunofluorescence labeling of LC3 II. Compared to the LPS + DIM group, the green fluorescence intensity of LC3 II decreased after 740Y-P use and increased after LY294002 use (Figures 7C,D). In addition, we observed the number of autophagosomes by MDC staining and transmission electron microscopy in chondrocytes. Compared to the LPS + DIM group, The intensity of MDC staining and the number of autophagosomes were significantly decreased after 740Y-P used and increased after LY294002 used (Figures 7E,F). These data suggested that the regulatory effect of DIM on autophagy was mediated via the PI3K/AKT/mTOR pathway.
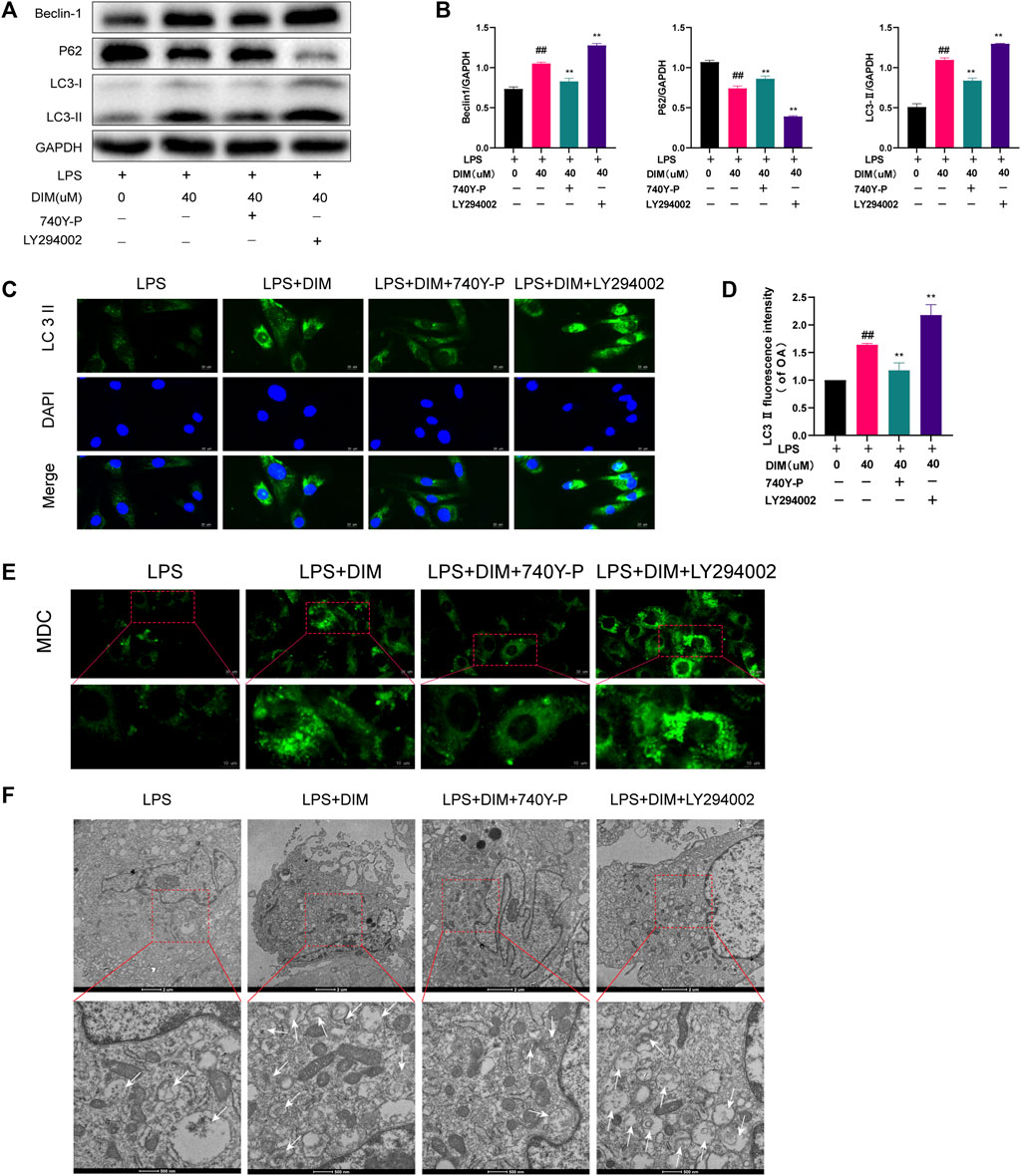
FIGURE 7. DIM enhances autophagy levels by modulating the PI3K/AKT/mTOR signaling pathway (A,B) Compared to the LPS + DIM group, the addition of PI3K activator (740Y-P) after treatment triggered a reduction in the level of autophagy, while the addition of PI3K inhibitor LY294002 increased the level of autophagy. Levels of autophagic marker proteins (beclin-1, p62, and LC3 II) were measured by western blotting and quantitation (C) The fluorescence intensity of LC3 II was detected by immunofluorescence staining (scale bar, 20 µm) (D) The fluorescence intensity was measured with the ImageJ software (U.S. National Institutes of Health, Bethesda) (E) The number of chondrocytes autophagosomes was observed by MDC staining (punctate green staining represents autophagosomes; scale bar, 20 μm; partially enlarged image scale bar, 10 µm) (F) Microstructural detection of autophagosomes by transmission electron microscopy (White arrows indicate the formation of autophagosomes. Scale bar = 2 µm; partially enlarged image scale bar, 500 nm). The data are presented as mean ± SD values from three independent experiments. Using one-way ANOVA, NS, no statistical difference; # p < 0.05 vs. the LPS group; # # p < 0.01 vs. the LPS group; * p < 0.05 vs. the LPS + DIM group; ** p < 0.01 vs. the LPS + DIM group.
We used chloroquine (CQ), an autophagy inhibitor (HY-17589A, MCE), and rapamycin (Rapa), an autophagy activator (HY-10219, MCE), to determine whether autophagy was involved in the inhibitive effects of DIM on apoptosis. Compared to the LPS + DIM group, CQ effectively abolished the effects of DIM on reducing cleaved caspase-3 and Bax expression levels as well as increasing Bcl-2 expression levels. Compared to the LPS + DIM group, Rapa further reduced cleaved caspase-3 and Bax expression levels as well as increased Bcl-2 expression levels (Figures 8A,B). Additionally, we also detected the expression level of cleaved caspase-3 by immunofluorescence staining (Figures 8C,D) and the apoptosis of chondrocytes by flow cytometry (Figures 8E,F). Compared to the LPS + DIM group, the fluorescence intensity of cleaved caspase-3 and the chondrocyte apoptosis were significantly increased after CQ treatment. In contrast, both the fluorescence intensity of cleaved caspase-3 and chondrocyte apoptosis were reduced after Rapa treatment. These data suggested that inhibition of chondrocyte apoptosis by DIM in LPS-treated chondrocytes is mediated by autophagy.
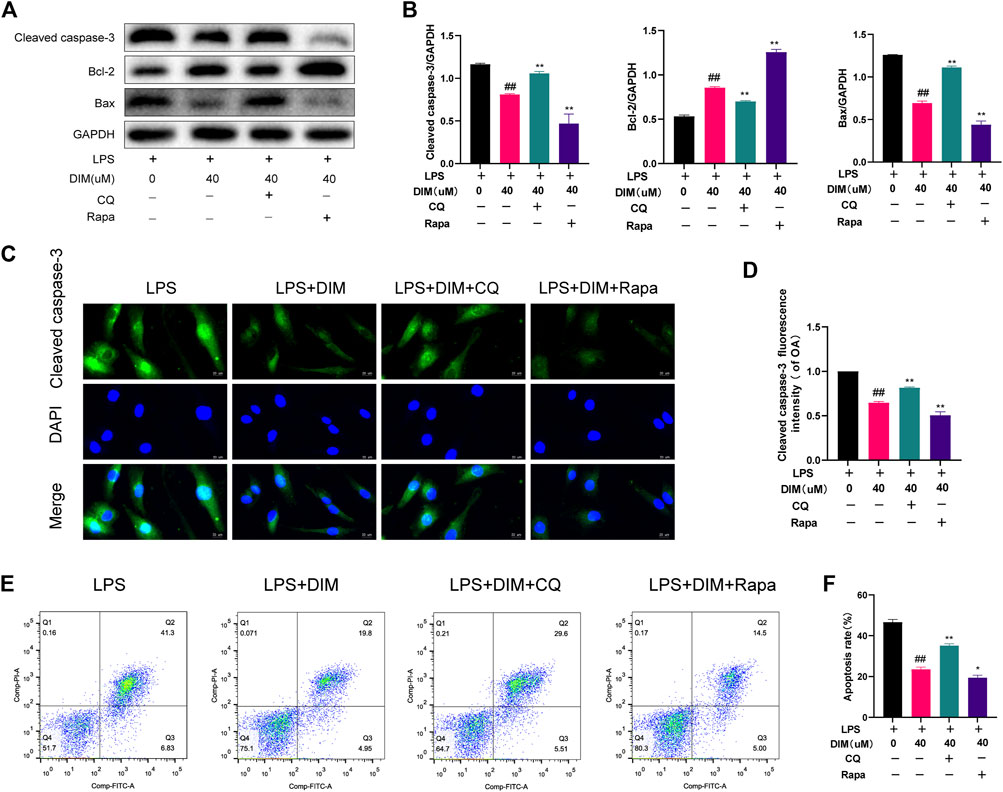
FIGURE 8. Activation of autophagy by DIM ameliorates LPS-induced chondrocyte apoptosis (A,B) Compared to the LPS + DIM group, the addition of an autophagy inhibitor (CQ) after treatment increased chondrocyte apoptosis, while the addition of an autophagy activator (Rapa) reduced chondrocyte apoptosis. Apoptosis marker proteins (cleaved caspase-3, Bcl-2, and Bax) were measured by western blotting and quantitation (C) The fluorescence intensity of cleaved caspase-3 was detected by immunofluorescence staining (scale bar, 20 µm) (D) the fluorescence intensity was measured with the ImageJ software (U.S. National Institutes of Health, Bethesda) (E,F) The apoptosis rate of chondrocytes was detected by flow cytometry and quantitation. The data are presented as mean ± SD values from three independent experiments. Using one-way ANOVA, NS, no statistical difference; # p <0.05 vs. the LPS group; # # p <0.01 vs. the LPS group; * p <0.05 vs. the LPS + DIM group; ** p <0.01 vs. the LPS + DIM group.
We evaluated the therapeutic effect of DIM using a mice DMM model. The morphological and histological changes of the model mice were observed by H and E staining, safranin O/fast green staining, and X-ray analysis. X-ray (Figure 9A) and Hematoxylin and eosin staining (Figure 9B) results revealed that, compared to the Sham group, the joint space of the mice in the DMM group was significantly narrowed; the cartilage surface was hardened, rough, and uneven; and osteophyte counts were increased. However, DIM treatment alleviated this pathological manifestation. In addition, safranin O/fast green staining showed that, compared to the Sham group, the DMM group had significant loss of proteoglycan in the articular cartilage and aggravated cartilage erosion; however, these changes were improved after DIM treatment (Figure 9C). The OARSI scores and Synovitis Scores were consistent with the above pathological results, indicating that the DMM mice model treated with DIM were significantly different from the OA group, and the OARSI scores and Synovitis Scores of the DMM + DIM group were significantly lower than those of the OA group (Figure 9D, Supplementary Figure S3). Immunohistochemical results showed that the average optical densities (AODs) of p-PI3K and cleaved caspase-3 in the DMM group were significantly higher than those in the sham group, while the AOD of LC3 II was lower than that in the sham group. Compared to the DMM group, the AOD of LC3 II was significantly increased after DIM treatment, while the AODs of p-PI3K and cleaved caspase-3 were significantly decreased (Figures 9E,F). The AODs of MMP-13 and ADAMTS-5 in the DMM group were significantly higher than those in the sham group. Compared to the DMM group, the AODs of MMP-13 and ADAMTS-5 were significantly decreased after DIM treatment (Supplementary Figure S4). In addition, immunohistochemical staining analysis of Collagen II and Aggrecan showed that the AODs of Collagen II and Aggrecan in articular cartilage were lower in the DMM group than those in the sham group. Compared to the DMM group, the AODs of Collagen II and Aggrecan were significantly increased after DIM treatment (Supplementary Figure S5), consistent with the in vitro results.
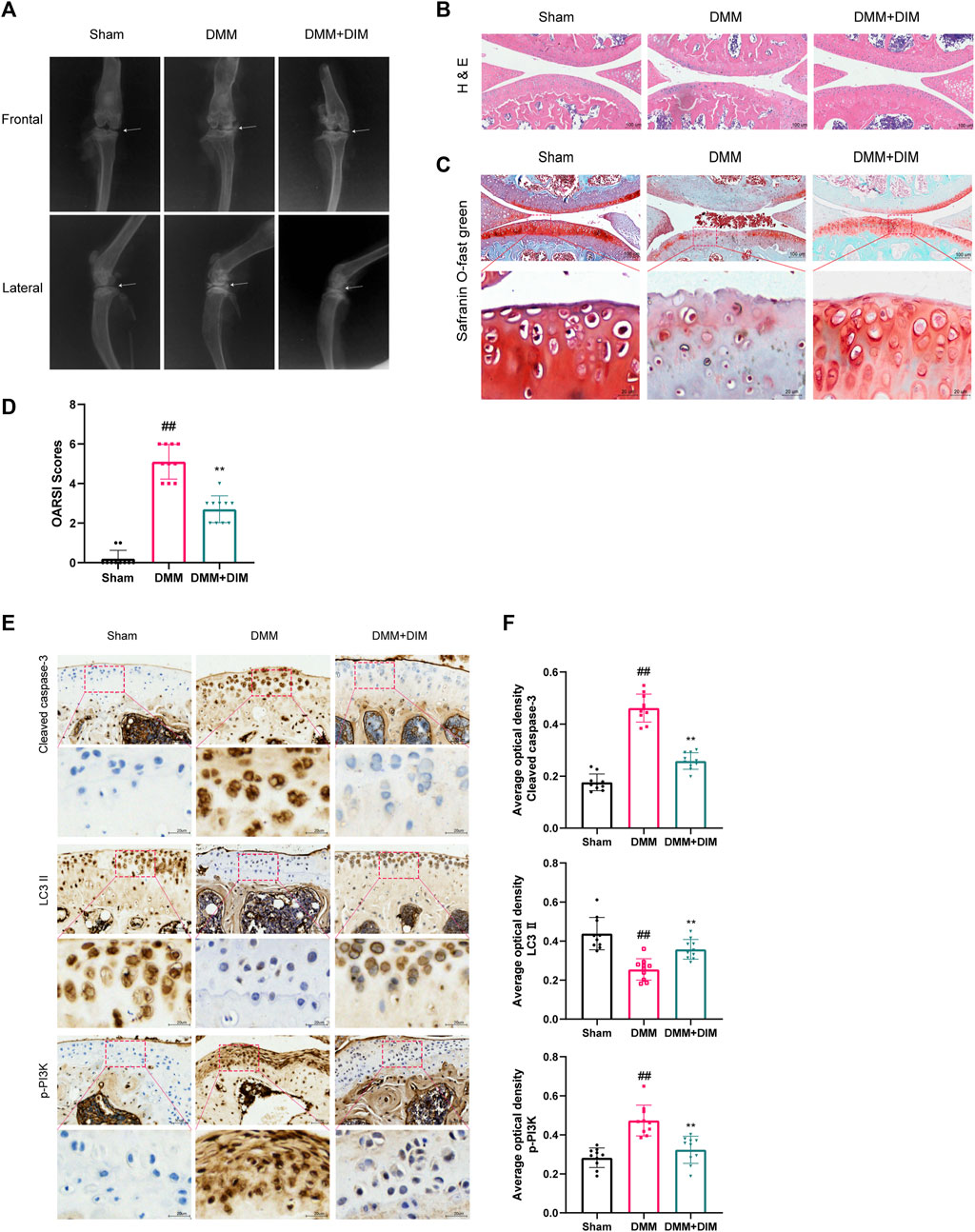
FIGURE 9. DIM alleviates OA development in the DMM mice model (A) Assessment of knee OA progression in a mice DMM model was conducted by digital X-ray imaging. Joint space narrowing was evident in the OA group (white arrows) (B) H and E staining of different experimental groups (scale bar, 100 µm) (C) Safranin O/fast green staining was used for the assessment of cartilage destruction (scale bar, 100 μm; partially enlarged image scale bar, 20 µm) (D) Mice articular cartilage OARSI scores (E,F) Immunohistochemical staining of cleaved caspase-3, LC3 II, and p-PI3K expressions in cartilage samples of different experimental groups (scale bar, 50 μm; partially enlarged image scale bar, 20 µm) and AODs were analyzed by the ImageJ software (U.S. National Institutes of Health, Bethesda). The data are presented as mean ± SD values from three independent experiments (n = 10 in each group). Using one-way ANOVA, NS, no statistical difference; # p < 0.05 vs. the Sham group; ## p < 0.01, vs. the Sham group, * p < 0.05 vs. the DMM group; ** p < 0.01 vs. the DMM group.
OA is the most common joint disease and the leading cause of disability worldwide. It can limit the patient’s daily activities, such as walking and running and other dependent behaviors, and severely affecting their quality of life, causing a significant social burden, with a high prevalence in the elderly population. Recent epidemiological findings show that the prevalence of OA is 9.6% in elder men and 18% in elder women (Woolf and Pfleger, 2003; Verlaan et al., 2018). The most affected site of OA is the knee joint (Fransen et al., 2011). OA is caused by a variety of factors and is an extremely complex pathogenic process that involves several different pathophysiological mechanisms, including increased inflammatory stimulation, an imbalance in chondrocyte metabolism, increased apoptosis, and degradation of the cartilage matrix (Li et al., 2019). Current treatments are limited to oral medications, which can relieve joint swelling and pain but cannot completely cure the development of OA, and joint replacement surgery is still required in the long term, so there is still a need to develop safer and more effective drugs to treat OA (Vaishya et al., 2016).
DIM is a natural Fat-soluble small molecule compound harvested from cruciferous vegetables which belongs to the class of indole glucosinolate (Maruthanila et al., 2014; Shi et al., 2017). Due to its lipophilic nature, DIM can directly cross the cell membrane into the cytoplasm to exert biological activity (Jellinck et al., 1993; Carpenter et al., 2016; Khan et al., 2019; Ong and Amstad, 2019; Martinotti et al., 2020). DIM can intracellularly regulate a variety of signaling pathways as well as signaling enzymes (Laiakis et al., 2020). For example, DIM can block PI3K/Akt/MTOR/NF-κB signaling (Ahmad et al., 2013), activate AMP-activated protein kinases (Chen et al., 2012), inhibit nuclear factor kappa B (Cho et al., 2008; Weng et al., 2012), inhibit cell cycle protein-dependent kinases (Kim et al., 2012), and reduce androgen receptor levels (Palomera-Sanchez et al., 2017). In recent years, an increasing number of studies have explored the chondroprotective effects of plant components (Sukhikh et al., 2021). DIM has been found to have various health-promoting benefits. However, it remains unclear whether DIM has any effect on OA chondrocytes, which is explored in depth in this paper. We investigated the role and mechanism of DIM in arthritis both in vivo and in vitro.
It has been reported that chondrocytes are the only cell type that constitutes articular cartilage and are responsible for the synthesis and secretion of cartilage ECM macromolecules, such as collagen II and aggrecan (Goldring and Marcu, 2009; Musumeci et al., 2015; Guilak et al., 2018). In addition, chondrocytes also can synthesize matrix degrading enzymes, such as MMP-13 and ADAMTS-5 (Vincenti and Brinckerhoff, 2002; Rowan et al., 2008; Takahata et al., 2019; Meng et al., 2020). Currently, the synthesis and degradation status of cartilage matrix is mainly reflected indirectly by detecting anabolic and catabolic marker proteins as well as proteoglycan moieties in chondrocytes (Chang et al., 2019; Huang et al., 2020; Lin et al., 2021). Under normal physiological conditions, chondrocytes maintain a balance between the synthesis and degradation of ECM components to ensure the structural and functional integrity of cartilage (Musumeci et al., 2011a). However, in the pathogenesis of OA, chondrocytes produce excess matrix-degrading enzymes to damage the extracellular matrix, particularly MMP-13 and ADAMTS-5 (Kapoor et al., 2011). MMP-13 is a subclass of collagenase with the function of cleaving ECM collagen II (Bramono et al., 2004), while ADAMTS-5 is a zinc protein hydrolase that destroys aggrecan (Gendron et al., 2007). The decrease in matrix component synthesis and the increase in chondrocyte matrix breakdown by catabolic proteins resulted in loss of cartilage matrix components and increased cartilage destruction. It has been reported that therapeutic substances targeting MMP-13 and ADAMTS-5 may be ideal agents for the treatment of OA (Burrage et al., 2006; Mead and Apte, 2018). In the present study, we found that DIM acts against LPS-induced chondrocyte matrix degradation with a protective effect, and DIM significantly inhibited the expression of catabolic indicators MMP-13 and ADAMTS-5 and increased the expression of anabolic indicators collagen II and aggrecan in cartilage ECM.
Increased apoptosis is another major cause of matrix degradation. Apoptosis is a form of programmed cell death that can be activated through several different pathways, including death receptor–mediated and mitochondria-dependent apoptosis (Musumeci et al., 2011a; Zamli and Sharif, 2011). Among the biomarkers of OA, inflammatory mediators play a clear role in chondrocyte apoptosis. The synthesis of pro-inflammatory cytokines, which contribute to increased apoptosis, is involved in the degeneration of the cartilage ECM (Wu et al., 2007; Musumeci et al., 2011b). Indeed, in human OA tissue specimens, dissolution and calcification of the descending ECM correlate with apoptosis, and the rate of apoptosis is positively correlated with the severity of OA (Musumeci et al., 2011a). Abnormal mechanical stress on normal cartilage can lead to chondrocyte apoptosis and result in cartilage degeneration and loss (Kim et al., 2002). In addition, the literature reports that DIM exerts neuroprotective effects through the production of brain-derived neurotrophic factor and antioxidant enzymes in oxidative stress–induced apoptosis in hippocampal neurons (Lee et al., 2019). We found that chondrocyte apoptosis was significantly increased after LPS stimulation, while pre-treatment with the addition of DIM reversed LPS-induced chondrocyte apoptosis and revealed an increase in anti-apoptotic protein levels (Bcl-2) and a decrease in pro-apoptotic protein levels (Bax and cleaved caspase-3). This indicates that DIM can also play an anti-apoptotic role in chondrocytes.
Our previous studies have demonstrated that ECM degradation can be improved by activating autophagy in OA chondrocytes (Huang et al., 2020; Wang et al., 2022). Next, we conducted an in-depth study of the mechanism by which DIM inhibits LPS-induced chondrocyte apoptosis in vitro. Previously, it was reported that, in human prostate cancer cells, DIM induced cytoprotective autophagy through the induction of AMPK activation by AEG-1 (Draz et al., 2017). DIM inhibited the proliferation of gastric cancer cells through the miR-30e/ATG5 pathway involved in autophagy control (Ye et al., 2016). Therefore, we hypothesized that the molecular mechanism by which DIM exerts its protective effect may be related to restoring the level of chondrocyte autophagy. Cellular autophagy is an important protective mechanism for cells to maintain homeostasis and survival in the internal environment (Mizushima, 2007; He and Klionsky, 2009; Ryter et al., 2013), and changes in inflammation, starvation, pathogen infection, and endoplasmic reticulum stress can cause alterations in autophagy. In addition, autophagy is known to regulate a variety of cellular processes, such as apoptosis, pathogen removal, antigen presentation, and inflammation and is associated with many human diseases (Levine and Kroemer, 2008; Doria et al., 2013). Autophagy has been widely reported to be associated with the development of OA, and autophagic flux has been suggested as a possible therapeutic target for OA (He and Cheng, 2018; Huang et al., 2020; Xu et al., 2021). In vitro, we found that autophagy in chondrocytes after LPS stimulation was significantly decreased and LC3 II and beclin-1 protein expression levels were decreased, p62 protein expression was increased, the number of autophagic vesicles was decreased, and autophagic flux was blocked, but these phenomena were significantly reversed after the application of DIM treatment. We employed CQ and Rapa, as a specific inhibitor and activators of autophagy, to determine whether DIM regulates apoptosis through autophagy. We found that DIM ameliorated LPS-induced apoptosis, but CQ reversed this phenomenon; while activation of autophagy by Rapa further inhibited apoptosis, acting synergistically with DIM. Our results indicate that DIM inhibits LPS-induced chondrocyte apoptosis by restoring the level of chondrocyte autophagy.
The literature reports that the PI3K/AKT/mTOR signaling pathway regulates a variety of cellular processes, including cellular autophagy, metabolism, inflammation, metabolism, angiogenesis, and the cell cycle (Malemud, 2015). It is also an important regulator of chondrocyte autophagic flux, and inhibition of PI3K/AKT/mTOR signaling pathway activation to promote increased autophagy is an effective strategy to improve OA symptoms (Xue et al., 2017; He and Cheng, 2018; Han et al., 2021). In our study, we found that the PI3K/AKT/mTOR signaling pathway was activated after LPS stimulation of chondrocytes compared to the normal group, and the expressions of p-PI3K, p-AKT, and p-mTOR were increased, while DIM treatment significantly inhibited LPS-induced activation of this pathway. Moreover, we employed the PI3K agonist 740Y-P and PI3K inhibitor LY294002 to determine whether DIM regulates autophagy through PI3K/AKT/mTOR signal pathway. We found that activation of the PI3K/AKT/mTOR pathway by 740Y-P reversed DIM-induced autophagy recovery, while LY294002 inhibited the activation of the PI3K/AKT/mTOR pathway and further enhanced autophagy in chondrocytes, synergizing with DIM. The results showed that DIM enhanced the autophagy level of chondrocytes by inhibiting the PI3K/AKT/mTOR signaling pathway.
In vivo, we used the DMM method to establish a C57BL/6 mice OA model, which was assessed by hematoxylin and eosin staining, X-ray, safranin O/fast green staining, and immunohistochemistry. We found that, compared to mice in the sham group, the model mice in the DMM group showed severe cartilage destruction in the knee joint with rough surfaces, massive proteoglycan loss, reduced autophagy protein levels, increased apoptotic protein levels, and activated pathway proteins, while DIM treatment improved these symptoms, reversed chondrocyte apoptosis and ECM degradation, and reduced the OARSI score in DMM mice. The results of in vivo and in vitro were generally consistent. Taken together, the present study showed that DIM, as an easily accessible botanical component, not only improved the degeneration of articular cartilage in mice, but also had a significant protective effect on inflammation-induced chondrocyte destruction. Therefore, it is of great value to further investigate the potential applications of DIM in the treatment of osteoarthritis.
We found that DIM can act as a protective agent for chondrocytes. More importantly, our results reveal the mechanism by which DIM exerts its chondroprotective effects (Figure 10). DIM can reduce inflammation-induced chondrocytes apoptosis and extracellular matrix degradation by activating PI3K/AKT/mTOR-mediated autophagy. This study provides new guiding directions for DIM as a promising drug for the treatment of OA.
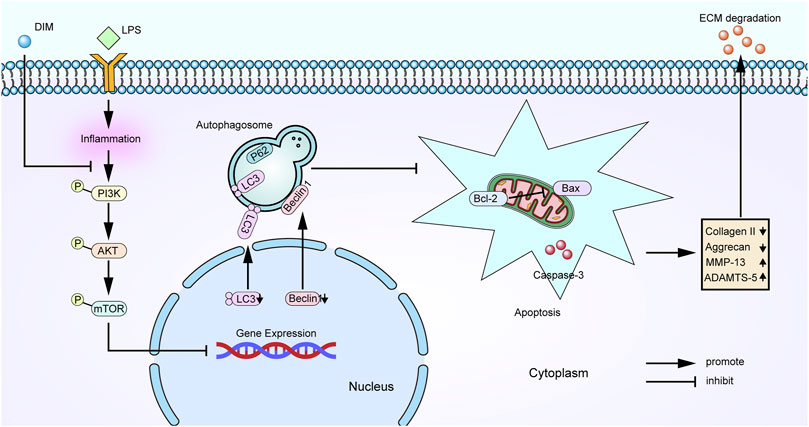
FIGURE 10. Schematic representation of the role of DIM in OA chondrocytes.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The study was conducted in accordance with the Declaration of Helsinki and approved by the Medical Ethics Committee of the First Affiliated Hospital of Anhui Medical University (protocol code PJ2022-04-55) for studies involving humans. The animal study protocol was approved by the Animal Ethics Committee of Anhui Medical University (protocol code LISC20190738).
HT and KQ: Conceptualization, Validation, Data curation and Writing–original draft. AW: Methodology. SL: Formal analysis. SF: Software. WG: Resources. ML: Investigation. WH, HZ, and ZY: Writing–review and editing, Funding acquisition, Supervision, Visualization.
This study was supported by the Provincial Natural Science Foundation of Anhui (2008085MH247) and Provincial University Natural Science Foundation of Anhui (KJ 2021A0299).
We thank WS, YK, RZ, JW, YR, and MW for their technical guidance during the experiments. We also thank the Center for Scientific Research of Anhui Medical University for providing valuable experimental facilities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor YC declared a shared parent affiliation with the author(s) at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.999851/full#supplementary-material
Ahmad, A., Biersack, B., Li, Y., Kong, D., Bao, B., Schobert, R., et al. (2013). Targeted regulation of PI3K/Akt/mTOR/NF-κB signaling by indole compounds and their derivatives: Mechanistic details and biological implications for cancer therapy. Anticancer. Agents Med. Chem. 13 (7), 1002–1013. doi:10.2174/18715206113139990078
Biederbick, A., Kern, H. F., and Elsässer, H. P. (1995). Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol. 66 (1), 3–14.
Bouderlique, T., Vuppalapati, K. K., Newton, P. T., Li, L., Barenius, B., and Chagin, A. S. (2016). Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann. Rheum. Dis. 75 (3), 627–631. doi:10.1136/annrheumdis-2015-207742
Boya, P., Reggiori, F., and Codogno, P. (2013). Emerging regulation and functions of autophagy. Nat. Cell Biol. 15 (7), 713–720. doi:10.1038/ncb2788
Bramono, D. S., Richmond, J. C., Weitzel, P. P., Kaplan, D. L., and Altman, G. H. (2004). Matrix metalloproteinases and their clinical applications in orthopaedics. Clin. Orthop. Relat. Res. 428, 272–285. doi:10.1097/01.blo.0000144166.66737.3a
Burrage, P. S., Mix, K. S., and Brinckerhoff, C. E. (2006). Matrix metalloproteinases: Role in arthritis. Front. Biosci. 11, 529–543. doi:10.2741/1817
Carpenter, T. S., Parkin, J., and Khalid, S. (2016). The free energy of small solute permeation through the Escherichia coli outer membrane has a distinctly asymmetric profile. J. Phys. Chem. Lett. 7 (17), 3446–3451. doi:10.1021/acs.jpclett.6b01399
Chang, S. H., Mori, D., Kobayashi, H., Mori, Y., Nakamoto, H., Okada, K., et al. (2019). Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-κB pathway. Nat. Commun. 10 (1), 1442. doi:10.1038/s41467-019-09491-5
Chen, D., Banerjee, S., Cui, Q. C., Kong, D., Sarkar, F. H., and Dou, Q. P. (2012). Activation of AMP-activated protein kinase by 3, 3'-Diindolylmethane (DIM) is associated with human prostate cancer cell death in vitro and in vivo. PLoS One 7 (10), e47186. doi:10.1371/journal.pone.0047186
Cho, H. J., Seon, M. R., Lee, Y. M., Kim, J., Kim, J. K., Kim, S. G., et al. (2008). 3, 3'-Diindolylmethane suppresses the inflammatory response to lipopolysaccharide in murine macrophages. J. Nutr. 138 (1), 17–23. doi:10.1093/jn/138.1.17
Cucchiarini, M., de Girolamo, L., Filardo, G., Oliveira, J. M., Orth, P., Pape, D., et al. (2016). Basic science of osteoarthritis. J. Exp. Orthop. 3 (1), 22. doi:10.1186/s40634-016-0060-6
Ding, Y., Wang, L., Zhao, Q., Wu, Z., and Kong, L. (2019). MicroRNA-93 inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting the TLR4/NF-κB signaling pathway. Int. J. Mol. Med. 43 (2), 779–790. doi:10.3892/ijmm.2018.4033
Dong, L., Xia, S., Gao, F., Zhang, D., Chen, J., and Zhang, J. (2010). 3, 3'-Diindolylmethane attenuates experimental arthritis and osteoclastogenesis. Biochem. Pharmacol. 79 (5), 715–721. doi:10.1016/j.bcp.2009.10.010
Doria, A., Gatto, M., and Punzi, L. (2013). Autophagy in human health and disease. N. Engl. J. Med. 368 (19), 1845–1846. doi:10.1056/NEJMc1303158
Draz, H., Goldberg, A. A., Titorenko, V. I., Tomlinson Guns, E. S., Safe, S. H., and Sanderson, J. T. (2017). Diindolylmethane and its halogenated derivatives induce protective autophagy in human prostate cancer cells via induction of the oncogenic protein AEG-1 and activation of AMP-activated protein kinase (AMPK). Cell. Signal. 40, 172–182. doi:10.1016/j.cellsig.2017.09.006
Du, H., Zhang, X., Zeng, Y., Huang, X., Chen, H., Wang, S., et al. (2019). A novel phytochemical, DIM, inhibits proliferation, migration, invasion and TNF-alpha induced inflammatory cytokine production of synovial fibroblasts from rheumatoid arthritis patients by targeting MAPK and AKT/mTOR signal pathway. Front. Immunol. 10, 1620. doi:10.3389/fimmu.2019.01620
Fransen, M., Bridgett, L., March, L., Hoy, D., Penserga, E., and Brooks, P. (2011). The epidemiology of osteoarthritis in Asia. Int. J. Rheum. Dis. 14 (2), 113–121. doi:10.1111/j.1756-185X.2011.01608.x
Gendron, C., Kashiwagi, M., Lim, N. H., Enghild, J. J., Thøgersen, I. B., Hughes, C., et al. (2007). Proteolytic activities of human ADAMTS-5: Comparative studies with ADAMTS-4. J. Biol. Chem. 282 (25), 18294–18306. doi:10.1074/jbc.M701523200
Glasson, S. S., Chambers, M. G., Van Den Berg, W. B., and Little, C. B. (2010). The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr. Cartil. 18 (3), S17–S23. doi:10.1016/j.joca.2010.05.025
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: Cellular and molecular mechanisms. J. Pathol. 221 (1), 3–12. doi:10.1002/path.2697
Glyn-Jones, S., Palmer, A. J. R., Agricola, R., Price, A. J., Vincent, T. L., Weinans, H., et al. (2015). Osteoarthritis. Lancet 386 (9991), 376–387. doi:10.1016/s0140-6736(14)60802-3
Goldring, M. B., and Marcu, K. B. (2009). Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 11 (3), 224. doi:10.1186/ar2592
Goldring, M. B. (2006). Update on the biology of the chondrocyte and new approaches to treating cartilage diseases. Best. Pract. Res. Clin. Rheumatol. 20 (5), 1003–1025. doi:10.1016/j.berh.2006.06.003
Guilak, F., Nims, R. J., Dicks, A., Wu, C. L., and Meulenbelt, I. (2018). Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol. 71-72, 40–50. doi:10.1016/j.matbio.2018.05.008
Han, G., Zhang, Y., and Li, H. (2021). The combination treatment of curcumin and probucol protects chondrocytes from TNF-alpha induced inflammation by enhancing autophagy and reducing apoptosis via the PI3K-Akt-mTOR pathway. Oxid. Med. Cell. Longev. 2021, 5558066. doi:10.1155/2021/5558066
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi:10.1146/annurev-genet-102808-114910
He, W., and Cheng, Y. (2018). Inhibition of miR-20 promotes proliferation and autophagy in articular chondrocytes by PI3K/AKT/mTOR signaling pathway. Biomed. Pharmacother. 97, 607–615. doi:10.1016/j.biopha.2017.10.152
Huang, W., Cheng, C., Shan, W. S., Ding, Z. F., Liu, F. E., Lu, W., et al. (2020). Knockdown of SGK1 alleviates the IL-1β-induced chondrocyte anabolic and catabolic imbalance by activating FoxO1-mediated autophagy in human chondrocytes. FEBS J. 287 (1), 94–107. doi:10.1111/febs.15009
Jellinck, P. H., Makin, H. L., Sepkovic, D. W., and Bradlow, H. L. (1993). Influence of indole carbinols and growth hormone on the metabolism of 4-androstenedione by rat liver microsomes. J. Steroid Biochem. Mol. Biol. 46 (6), 791–798. doi:10.1016/0960-0760(93)90320-v
Kapoor, M., Martel-Pelletier, J., Lajeunesse, D., Pelletier, J. P., and Fahmi, H. (2011). Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7 (1), 33–42. doi:10.1038/nrrheum.2010.196
Khan, A., Wang, C., Sun, X., Killpartrick, A., and Guo, M. (2019). Physicochemical and microstructural properties of polymerized whey protein encapsulated 3, 3'-diindolylmethane nanoparticles. Molecules 24 (4), E702. doi:10.3390/molecules24040702
Kim, H. T., Lo, M. Y., and Pillarisetty, R. (2002). Chondrocyte apoptosis following intraarticular fracture in humans. Osteoarthr. Cartil. 10 (9), 747–749. doi:10.1053/joca.2002.0828
Kim, S. J., Lee, J. S., and Kim, S. M. (2012). 3, 3'-Diindolylmethane suppresses growth of human esophageal squamous cancer cells by G1 cell cycle arrest. Oncol. Rep. 27 (5), 1669–1673. doi:10.3892/or.2012.1662
Krenn, V., Morawietz, L., Burmester, G. R., Kinne, R. W., Mueller-Ladner, U., Muller, B., et al. (2006). Synovitis score: Discrimination between chronic low-grade and high-grade synovitis. Histopathology 49 (4), 358–364. doi:10.1111/j.1365-2559.2006.02508.x
Krenn, V., Morawietz, L., Häupl, T., Neidel, J., Petersen, I., König, A., et al. (2002). Grading of chronic synovitis--a histopathological grading system for molecular and diagnostic pathology. Pathol. Res. Pract. 198 (5), 317–325. doi:10.1078/0344-0338-5710261
Laiakis, E. C., McCart, E. A., Deziel, A., Rittase, W. B., Bouten, R. M., Jha, J., et al. (2020). Effect of 3, 3'-diindolylmethane on pulmonary injury following thoracic irradiation in CBA mice. Health Phys. 119 (6), 746–757. doi:10.1097/hp.0000000000001257
Lee, B. D., Yoo, J. M., Baek, S. Y., Li, F. Y., Sok, D. E., and Kim, M. R. (2019). 3, 3'-diindolylmethane promotes BDNF and antioxidant enzyme formation via TrkB/akt pathway activation for neuroprotection against oxidative stress-induced apoptosis in hippocampal neuronal cells. Antioxidants (Basel) 9 (1), E3. doi:10.3390/antiox9010003
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132 (1), 27–42. doi:10.1016/j.cell.2007.12.018
Li, H., Xie, S., Li, H., Zhang, R., and Zhang, H. (2020). LncRNA MALAT1 mediates proliferation of LPS treated-articular chondrocytes by targeting the miR-146a-PI3K/Akt/mTOR axis. Life Sci. 254, 116801. doi:10.1016/j.lfs.2019.116801
Li, Y. S., Zhang, F. J., Zeng, C., Luo, W., Xiao, W. F., Gao, S. G., et al. (2016). Autophagy in osteoarthritis. Jt. Bone Spine 83 (2), 143–148. doi:10.1016/j.jbspin.2015.06.009
Li, Y., Wu, Y., Jiang, K., Han, W., Zhang, J., Xie, L., et al. (2019). Mangiferin prevents TBHP-induced apoptosis and ECM degradation in mouse osteoarthritic chondrocytes via restoring autophagy and ameliorates murine osteoarthritis. Oxid. Med. Cell. Longev. 2019, 8783197. doi:10.1155/2019/8783197
Lian, W. S., Ko, J. Y., Wu, R. W., Sun, Y. C., Chen, Y. S., Wu, S. L., et al. (2018). MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 9 (9), 919. doi:10.1038/s41419-018-0994-y
Lin, Z., Miao, J., Zhang, T., He, M., Wang, Z., Feng, X., et al. (2021). JUNB-FBXO21-ERK axis promotes cartilage degeneration in osteoarthritis by inhibiting autophagy. Aging Cell 20 (2), e13306. doi:10.1111/acel.13306
Loeser, R. F. (2011). Aging and osteoarthritis. Curr. Opin. Rheumatol. 23 (5), 492–496. doi:10.1097/BOR.0b013e3283494005
Lorenz, W., Buhrmann, C., Mobasheri, A., Lueders, C., and Shakibaei, M. (2013). Bacterial lipopolysaccharides form procollagen-endotoxin complexes that trigger cartilage inflammation and degeneration: Implications for the development of rheumatoid arthritis. Arthritis Res. Ther. 15 (5), R111. doi:10.1186/ar4291
Luo, Q., Yang, A., Cao, Q., and Guan, H. (2018). 3, 3'-Diindolylmethane protects cardiomyocytes from LPS-induced inflammatory response and apoptosis. BMC Pharmacol. Toxicol. 19 (1), 71. doi:10.1186/s40360-018-0262-x
Malemud, C. J. (2015). The PI3K/akt/PTEN/mTOR pathway: A fruitful target for inducing cell death in rheumatoid arthritis? Future Med. Chem. 7 (9), 1137–1147. doi:10.4155/fmc.15.55
Martinotti, C., Ruiz-Perez, L., Deplazes, E., and Mancera, R. L. (2020). Molecular dynamics simulation of small molecules interacting with biological membranes. Chemphyschem 21 (14), 1486–1514. doi:10.1002/cphc.202000219
Maruthanila, V. L., Poornima, J., and Mirunalini, S. (2014). Attenuation of carcinogenesis and the mechanism underlying by the influence of indole-3-carbinol and its metabolite 3, 3'-diindolylmethane: A therapeutic marvel. Adv. Pharmacol. Sci. 2014, 832161. doi:10.1155/2014/832161
Maudens, P., Jordan, O., and Allémann, E. (2018). Recent advances in intra-articular drug delivery systems for osteoarthritis therapy. Drug Discov. Today 23 (10), 1761–1775. doi:10.1016/j.drudis.2018.05.023
Mayeux, P. R. (1997). Pathobiology of lipopolysaccharide. J. Toxicol. Environ. Health 51 (5), 415–435. doi:10.1080/00984109708984034
Mead, T. J., and Apte, S. S. (2018). ADAMTS proteins in human disorders. Matrix Biol. 71-72, 225–239. doi:10.1016/j.matbio.2018.06.002
Meng, P., Zhang, F., Zhang, Y., Wei, H., Tan, S., Guo, X., et al. (2020). ADAMTS4 and ADAMTS5 may be considered as new molecular therapeutic targets for cartilage damages with Kashin-Beck Disease. Med. Hypotheses 135, 109440. doi:10.1016/j.mehy.2019.109440
Mizushima, N. (2007). Autophagy: Process and function. Genes Dev. 21, 2861–2873. doi:10.1101/gad.1599207
Mizushima, N., and Levine, B. (2020). Autophagy in human diseases. N. Engl. J. Med. 383 (16), 1564–1576. doi:10.1056/NEJMra2022774
Munakarmi, S., Chand, L., Shin, H. B., Jang, K. Y., and Jeong, Y. J. (2020). Indole-3-Carbinol derivative DIM mitigates carbon tetrachloride-induced acute liver injury in mice by inhibiting inflammatory response, apoptosis and regulating oxidative stress. Int. J. Mol. Sci. 21 (6), E2048. doi:10.3390/ijms21062048
Musumeci, G., Aiello, F. C., Szychlinska, M. A., Di Rosa, M., Castrogiovanni, P., and Mobasheri, A. (2015). Osteoarthritis in the XXIst century: Risk factors and behaviours that influence disease onset and progression. Int. J. Mol. Sci. 16 (3), 6093–6112. doi:10.3390/ijms16036093
Musumeci, G., Loreto, C., Carnazza, M. L., and Martinez, G. (2011a). Characterization of apoptosis in articular cartilage derived from the knee joints of patients with osteoarthritis. Knee Surg. Sports Traumatol. Arthrosc. 19 (2), 307–313. doi:10.1007/s00167-010-1215-0
Musumeci, G., Loreto, C., Carnazza, M. L., Strehin, I., and Elisseeff, J. (2011b). OA cartilage derived chondrocytes encapsulated in poly(ethylene glycol) diacrylate (PEGDA) for the evaluation of cartilage restoration and apoptosis in an in vitro model. Histol. Histopathol. 26 (10), 1265–1278. doi:10.14670/hh-26.1265
Niemann, A., Baltes, J., and Elsässer, H. P. (2001). Fluorescence properties and staining behavior of monodansylpentane, a structural homologue of the lysosomotropic agent monodansylcadaverine. J. Histochem. Cytochem. 49 (2), 177–185. doi:10.1177/002215540104900205
Niemann, A., Takatsuki, A., and Elsässer, H. P. (2000). The lysosomotropic agent monodansylcadaverine also acts as a solvent polarity probe. J. Histochem. Cytochem. 48 (2), 251–258. doi:10.1177/002215540004800210
Ong, I. L. H., and Amstad, E. (2019). Selectively permeable double emulsions. Small 15 (44), e1903054. doi:10.1002/smll.201903054
Palomera-Sanchez, Z., Watson, G. W., Wong, C. P., Beaver, L. M., Williams, D. E., Dashwood, R. H., et al. (2017). The phytochemical 3, 3'-diindolylmethane decreases expression of AR-controlled DNA damage repair genes through repressive chromatin modifications and is associated with DNA damage in prostate cancer cells. J. Nutr. Biochem. 47, 113–119. doi:10.1016/j.jnutbio.2017.05.005
Polewska, J. (2012). Autophagy--molecular mechanism, apoptosis and cancer. Postepy Hig. Med. Dosw. 66, 921–936. doi:10.5604/17322693.1021109
Qin, N., Wei, L., Li, W., Yang, W., Cai, L., Qian, Z., et al. (2017). Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J. Pharmacol. Sci. 134 (3), 166–174. doi:10.1016/j.jphs.2017.06.002
Ravikumar, B., Sarkar, S., Davies, J. E., Futter, M., Garcia-Arencibia, M., Green-Thompson, Z. W., et al. (2010). Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90 (4), 1383–1435. doi:10.1152/physrev.00030.2009
Rowan, A. D., Litherland, G. J., Hui, W., and Milner, J. M. (2008). Metalloproteases as potential therapeutic targets in arthritis treatment. Expert Opin. Ther. Targets 12 (1), 1–18. doi:10.1517/14728222.12.1.1
Ryter, S. W., CloonanSuzanne, M., Choi , , and Augustine, M. K. (2013). Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 36 (1), 7–16. doi:10.1007/s10059-013-0140-8
Rzemieniec, J., Litwa, E., Wnuk, A., Lason, W., Krzeptowski, W., and Kajta, M. (2016). Selective aryl hydrocarbon receptor modulator 3, 3'-diindolylmethane impairs AhR and ARNT signaling and protects mouse neuronal cells against hypoxia. Mol. Neurobiol. 53 (8), 5591–5606. doi:10.1007/s12035-015-9471-0
Sasaki, H., Takayama, K., Matsushita, T., Ishida, K., Kubo, S., Matsumoto, T., et al. (2012). Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 64 (6), 1920–1928. doi:10.1002/art.34323
Shi, H., Xu, X., Zhang, B., Xu, J., Pan, Z., Gong, A., et al. (2017). 3, 3'-Diindolylmethane stimulates exosomal Wnt11 autocrine signaling in human umbilical cord mesenchymal stem cells to enhance wound healing. Theranostics 7 (6), 1674–1688. doi:10.7150/thno.18082
Sukhikh, S., Noskova, S., Ivanova, S., Ulrikh, E., Izgaryshev, A., and Babich, O. (2021). Chondroprotection and molecular mechanism of action of phytonutraceuticals on osteoarthritis. Molecules 26 (8), 2391. doi:10.3390/molecules26082391
Takahata, Y., Nakamura, E., Hata, K., Wakabayashi, M., Murakami, T., Wakamori, K., et al. (2019). Sox4 is involved in osteoarthritic cartilage deterioration through induction of ADAMTS4 and ADAMTS5. Faseb J. 33 (1), 619–630. doi:10.1096/fj.201800259R
Takayama, K., Kawakami, Y., Kobayashi, M., Greco, N., Cummins, J. H., Matsushita, T., et al. (2014). Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res. Ther. 16 (6), 482. doi:10.1186/s13075-014-0482-4
Vaishya, R., Pariyo, G. B., Agarwal, A. K., and Vijay, V. (2016). Non-operative management of osteoarthritis of the knee joint. J. Clin. Orthop. Trauma 7 (3), 170–176. doi:10.1016/j.jcot.2016.05.005
Vázquez, C. L., and Colombo, M. I. (2009). Assays to assess autophagy induction and fusion of autophagic vacuoles with a degradative compartment, using monodansylcadaverine (MDC) and DQ-BSA. Methods Enzymol. 452, 85–95. doi:10.1016/s0076-6879(08)03606-9
Verlaan, L., Boekesteijn, R. J., Oomen, P. W., Liu, W. Y., Peters, M. J. M., Witlox, M. A., et al. (2018). Biomechanical alterations during sit-to-stand transfer are caused by a synergy between knee osteoarthritis and obesity. Biomed. Res. Int. 2018, 3519498. doi:10.1155/2018/3519498
Vincenti, M. P., and Brinckerhoff, C. E. (2002). Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: Integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 4 (3), 157–164. doi:10.1186/ar401
Wang, A., Fang, S., Zhong, L., Lu, M., Zhou, H., Huang, W., et al. (2022). Shikonin, a promising therapeutic drug for osteoarthritis that acts via autophagy activation. Int. Immunopharmacol. 106, 108563. doi:10.1016/j.intimp.2022.108563
Weng, J. R., Bai, L. Y., Chiu, C. F., Wang, Y. C., and Tsai, M. H. (2012). The dietary phytochemical 3, 3'-diindolylmethane induces G2/M arrest and apoptosis in oral squamous cell carcinoma by modulating Akt-NF-κB, MAPK, and p53 signaling. Chem. Biol. Interact. 195 (3), 224–230. doi:10.1016/j.cbi.2012.01.003
Woolf, A. D., and Pfleger, B. (2003). Burden of major musculoskeletal conditions. Bull. World Health Organ. 81 (9), 646–656. doi:10.1590/S0042-96862003000900007
Wu, G. J., Chen, T. G., Chang, H. C., Chiu, W. T., Chang, C. C., and Chen, R. M. (2007). Nitric oxide from both exogenous and endogenous sources activates mitochondria-dependent events and induces insults to human chondrocytes. J. Cell. Biochem. 101 (6), 1520–1531. doi:10.1002/jcb.21268
Xu, K., He, Y., Moqbel, S. A. A., Zhou, X., Wu, L., and Bao, J. (2021). SIRT3 ameliorates osteoarthritis via regulating chondrocyte autophagy and apoptosis through the PI3K/Akt/mTOR pathway. Int. J. Biol. Macromol. 175, 351–360. doi:10.1016/j.ijbiomac.2021.02.029
Xue, J. F., Shi, Z. M., Zou, J., and Li, X. L. (2017). Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed. Pharmacother. 89, 1252–1261. doi:10.1016/j.biopha.2017.01.130
Ye, Y., Fang, Y., Xu, W., Wang, Q., Zhou, J., and Lu, R. (2016). 3, 3'-Diindolylmethane induces anti-human gastric cancer cells by the miR-30e-ATG5 modulating autophagy. Biochem. Pharmacol. 115, 77–84. doi:10.1016/j.bcp.2016.06.018
Ye, Y., Li, X., Wang, Z., Ye, F., Xu, W., Lu, R., et al. (2021). 3, 3'-Diindolylmethane induces gastric cancer cells death via STIM1 mediated store-operated calcium entry. Int. J. Biol. Sci. 17 (5), 1217–1233. doi:10.7150/ijbs.56833
Yoshino, S., Sasatomi, E., and Ohsawa, M. (2000). Bacterial lipopolysaccharide acts as an adjuvant to induce autoimmune arthritis in mice. Immunology 99 (4), 607–614. doi:10.1046/j.1365-2567.2000.00015.x
Zamli, Z., and Sharif, M. (2011). Chondrocyte apoptosis: A cause or consequence of osteoarthritis? Int. J. Rheum. Dis. 14 (2), 159–166. doi:10.1111/j.1756-185X.2011.01618.x
Zeng, R., Lu, X., Lin, J., Ron, Z., Fang, J., Liu, Z., et al. (2021). FOXM1 activates JAK1/STAT3 pathway in human osteoarthritis cartilage cell inflammatory reaction. Exp. Biol. Med. 246 (6), 644–653. doi:10.1177/1535370220974933
Zhang, Y., Vasheghani, F., Li, Y. H., Blati, M., Simeone, K., Fahmi, H., et al. (2015). Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 74 (7), 1432–1440. doi:10.1136/annrheumdis-2013-204599
Keywords: 3, 3′-diindolylmethane, osteoarthritis, apoptosis, autophagy, chondrocyte
Citation: Tang H, Qin K, Wang A, Li S, Fang S, Gao W, Lu M, Huang W, Zhang H and Yin Z (2022) 3,3′-diindolylmethane inhibits LPS-induced human chondrocytes apoptosis and extracellular matrix degradation by activating PI3K-Akt-mTOR-mediated autophagy. Front. Pharmacol. 13:999851. doi: 10.3389/fphar.2022.999851
Received: 21 July 2022; Accepted: 10 October 2022;
Published: 10 November 2022.
Edited by:
Yan Chang, Anhui Medical University, ChinaReviewed by:
Anna Scanu, University of Padua, ItalyCopyright © 2022 Tang, Qin, Wang, Li, Fang, Gao, Lu, Huang, Zhang and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Huang, emdrZGh3ZWlAdXN0Yy5lZHUuY24=; Hui Zhang, emhhbmdodWlAYWhtdS5lZHUuY24=; Zongsheng Yin, eWluem9uZ3NoZW5nQHNpbmEuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.