 Cheng Wang
Cheng Wang Xianghuang Mei3
Xianghuang Mei3 Yuting Yang
Yuting Yang Zhenguo Zeng
Zhenguo Zeng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 28 November 2022
Sec. Inflammation Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.994625
This article is part of the Research Topic Phytochemicals Targeting Autophagy in Treatment of Bacterial Infection and Malignancies View all 5 articles
Acute lung injury (ALI) is a severe clinical disorder characterized by dysregulated inflammatory responses, leading to high rates of morbidity and mortality. Cinobufagin, a primary component isolated from cinobufotalin, exerts strong anticancer effects. However, there are few reports on its role in ALI, and it is unclear whether cinobufagin affects lipopolysaccharide (LPS)-induced ALI. Therefore, the present study aimed to investigate the effect of cinobufagin on LPS-induced ALI and to assess its potential mechanism of action. The results showed that cinobufagin alleviated lung histopathological changes and protected the permeability of lung tissues in LPS-induced ALI. In addition, cinobufagin effectively suppressed inflammatory responses through the induction of autophagy in LPS-induced ALI cells and in a mouse model. Moreover, cinobufagin enhanced autophagy through the p53/mTOR pathway in LPS-induced ALI. Herein, it was reported for the first time that cinobufagin inhibited the inflammatory response of LPS-induced ALI, which laid the foundation for further understanding and development of cinobufagin as a potential new drug for ALI.
Acute lung injury (ALI) is a severe clinical disorder with a high morbidity and mortality rate. Pathological characteristics include rapid alveolar injury, severe hypoxemia, and non-cardiogenic pulmonary edema, which may develop into acute respiratory distress syndrome (ARDS) (Thompson et al., 2017). Bacterial infections are the primary cause of ALI (Kumar, 2020). Lipopolysaccharide (LPS), the main component of the outer cell wall of Gram-negative bacteria, is a key factor in the development of ALI/ARDS (Chaudhuri et al., 2007). Exposure to LPS can lead to the infiltration of inflammatory cells and the generation of cytokines and chemokines, which subsequently results in the initiation and propagation of an inflammatory response in ALI/ARDS (Cheng et al., 2018). Therefore, LPS has been widely used as a model for experimental ALI (Chen et al., 2010). Current ALI treatment strategies include reducing bacterial count and ameliorating organ damage caused by excessive inflammation. Despite improvements in intensive care and mechanical ventilation regimens, the mortality rates as a consequence of pneumonia or sepsis associated with ALI remain high (Butt et al., 2016). Therefore, investigating new and effective means of treating ALI is crucial for improving the health and survival of patients.
Autophagy is a major catabolic process that transports proteins, cytoplasmic components, and organelles to lysosomes for degradation or recycling (Deretic, 2021). The main autophagy-related proteins are LC3-II, Beclin-1, and ATG5 (Dikic and Elazar, 2018) and the key signaling pathways regulating autophagy include the p53/mechanistic target of rapamycin complex 1 (mTORC1) and B-cell lymphoma 2 (Bcl-2) protein families (Maiuri et al., 2010; Houston, 2019). Autophagy is an early adaptive mechanism by which tissues clear organelles or proteins to maintain cell homeostasis. Excessive autophagy results in cell death (Barth et al., 2010). While autophagy plays a vital role in LPS-induced ALI, its specific mechanism remains unclear.
Cinobufotalin is a monomer from Bufo bufo gargarizans Cantor and Bufo melanostictus Schneider extracts used in traditional Chinese medicine (Zhang et al., 2020). Cinobufagin, a primary component isolated from cinobufotalin, has been shown to exert antitumor effects on a number of malignant tumors such as hepatocellular carcinoma, colorectal cancer, lung cancer, and osteosarcoma (Qi et al., 2011; Dai et al., 2018; Bai et al., 2021; Zhang et al., 2022). A previous study showed that cinobufagin attenuated bleomycin-induced pulmonary fibrosis in mice (Li et al., 2019). However, the effect of cinobufagin on LPS-induced ALI remains unclear. Therefore, the aim of the present study was to clarify whether cinobufagin exerts anti-inflammatory effects in LPS-induced ALI. We showed that cinobufagin could induce autophagy to inhibit the production of inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, in LPS-induced ALI, and the p53/mTOR pathway could be involved in cinobufagin-induced autophagy.
Cinobufagin was purchased from the X-Y Biotechnology Company (Shanghai, China). Dimethyl sulfoxide (DMSO) and LPS were purchased from Sigma-Aldrich (St. Louis, MO, United States). Cinobufagin was dissolved in dimethyl sulfoxide to a final concentration of 100 mmol/L. Pifithrin-u was purchased from Selleckchem (Houston, United States). Cell Counting Kit-8 (CCK-8) was purchased from Tongren Institute of Chemistry (Dalian, China); Annexin V-FITC was purchased from BD Biosciences; and ELISA test kits for IL-1B, IL-6, and TNF-a were purchased from Thermo Fisher Scientific (Waltham, MA, United States). TRIzol reagent was obtained from Invitrogen (Carlsbad, CA). Prime Script RT Master Mix and TB Green Premix Ex Taq were purchased from Takara Bio Inc. (Dalian, China). A BCA protein assay kit was purchased from Tiangen Biotech (Beijing, China). RIPA buffer was purchased from Beyotime (Shanghai, China). Primary antibodies against LC3B, Beclin-1, p53, phospho-p53, mTOR, phospho-mTOR, and HRP-linked anti-rabbit IgG were purchased from Cell Signaling Technology (Beverly, MA, United States). Antibody against GAPDH was purchased from Proteintech (Wuhan, China).
Animal experimental procedures were approved by the Animal Care and Use Committee of the First Hospital of Nanchang University. C57/BL mice were obtained from SJA Laboratory Animal Company (Hunan, China), housed in a specific pathogen-free (SPF) facility with a light/dark cycle of 12 h/12 h, and randomly assigned to four groups: control, LPS (5 mg/kg), LPS cinobufagin (7.5 mg/kg), and 3–MA (15 mg/kg) LPS + cinobufagin (n = 6 per group). An ALI model was established by intratracheal administration of LPS. 3-Methyladenine (3-MA) was administered 1 h before cinobufagin intervention, and cinobufagin was administered 1 h before LPS treatment. The control group was administered equal amounts of PBS simultaneously. Two days after LPS injection, mice were sacrificed for subsequent assays.
Human bronchial epithelial (HBE) cells were purchased from American Type Culture Collection (CRL-2741) and cultured in RPMI 1640 (Gibco, United States) containing 10% fetal bovine serum (FBS, Gibco, United States) in a 5% CO2 incubator at 37°C. Cells were digested with 0.25% trypsin and passaged according to the experimental requirements. Cells were pretreated with 3-MA (1 μM) or cinobufagin (2 μM) for 2 h. Cells were then incubated with LPS (100 μg/ml) for 24 h and harvested for further analysis.
Cell viability in the various treatment groups was assessed using a CCK8 assay. HBE cells in the logarithmic phase were inoculated into 96-well plates at a density of 8 × 103 cells/well overnight. Next, culture medium containing 10 ml of CCK-8 solution was added to each well. After incubation for 2 h, the absorbance (optical density value) of each well at a wavelength of 450 nm was measured using a microplate reader (Waltham, MA, United States).
HBE cells were seeded in six-well plates at a density of 10 × 104 cells/well. Following incubation with 3-MA (1 μM) or cinobufagin (2 μM) for 2 h, HBE cells were treated with LPS (100 μg/mg) in complete medium for 24 h. Cells were digested with trypsin and washed twice with pre-cooled PBS. Next, the cells were resuspended in 100 μL binding buffer, mixed with 5 μL annexin V-FITC and 5 μL PI, and incubated for 15 min in the dark. Following the addition of 400 μL binding buffer, apoptotic cells were detected by flow cytometry.
The protein levels of IL-1β, IL-6, and TNF-α in the bronchoalveolar lavage fluid (BALF) and cell supernatant were detected using an ELISA kit according to the manufacturer’s instructions.
Total RNA was extracted from the cells using TRIzol reagent. Reverse transcription was performed with total RNA (1,000 ng) using SYBR®Premix Ex Taq™ (TaKaRa, Shiga, Japan). qRT-PCR was performed using TB Green Premix Ex Taq (TaKaRa, Shiga, Japan) following the manufacturer’s instructions. Each sample was assayed in triplicate. The ΔΔCt method was used to quantify and compare mRNA levels to GAPDH. The primers used are listed in Table 1.

TABLE 1. Primers for quantitative real-time PCR.
Protein concentration was determined using a BCA protein assay kit. A total of 20 μg of protein was obtained from each group and separated using SDS-PAGE. After electrophoresis, the proteins were electrotransferred to nitrocellulose membranes and blocked with 5% skimmed milk at room temperature for 1 h. Different primary antibodies were added according to the experimental requirements and incubated overnight at 4°C. After washing with TBS-Tween-20 (TBS-T), the membranes were incubated with secondary antibody at room temperature for 1 h, and subsequently washed with TBS-T. The protein bands were visualized using SH-Cute 523 (Hangzhou, China).
Lung tissues were fixed in a 4% paraformaldehyde solution for more than 2 days. Thereafter, the tissues were dehydrated in water through alcohol and embedded in paraffin. A series of microsections were cut using a microtome and stained with hematoxylin and eosin (HE). The extent of lung injury was scored by two independent pathologists.
Following treatment of mice with LPS, cinobufagin, and 3-MA at different concentrations, fresh lung tissues were weighed to obtain the wet weight. The lung tissues were then dried for 48 h and weighed to determine their dry weight. Subsequently, the lung W/D ratio was calculated. Each experiment was repeated at least thrice.
Bronchoalveolar lavage fluid was collected by intratracheal injection of 1 ml of pre-cold PBS and centrifuged at 1,000 rpm for 10 min. After red blood cells were lysed, the remaining cells were washed, resuspended in PBS, counted, and the protein concentration in the supernatant was assessed using a BCA protein assay kit.
HBE cells were fixed with 4% paraformaldehyde. The cells were permeabilized with 0.3% Triton X-100, blocked with 1% bovine serum albumin (BSA), and incubated with antibodies against LC3B (1:500) overnight at 4°C. After washing with PBS thrice, the cells were incubated with secondary antibodies for 1 h. Next, the cell nuclei were stained with 4’6’-diamino-2-phenylindole (DAPI) and fluorescence images were captured using a confocal laser scanning microscope (Leica, Germany).
Unless otherwise stated, data are presented as the mean standard error of one representative of three independent experiments, each of which was conducted in triplicate. The Student’s t-test was performed to evaluate the differences between the two groups. Qualitative variables were compared using the chi-square (χ2) test, and p < 0.05 was considered to be statistically significant (*p < 0.05; ** p < 0.01; *** p < 0.001). Graphics were generated using the GraphPad Prism 8.1.1 software. All statistical analyses were performed using SPSS software (IBM SPSS Statistics 21.0).
Lung injury in mice was assessed by HE staining of lung tissue 48 h after intratracheal LPS injection. First, we separately explored the effects of different concentrations of cinobufagin (1.875, 3.75 mg/kg and 7.5 mg/kg) on mice and found that none of them caused significant inflammatory changes compared to the control group (Figures 1A,B). Therefore, 7.5 mg/kg was selected for subsequent experiments. In addition, as shown in Figures 1 C–D, the control group showed normal lung tissue structure. In the LPS-induced group, lung tissue was severely damaged with a destroyed alveolar structure, thickened pulmonary septum, and edematous and inflammatory cells infiltrated in the alveoli and interstitium. Pathological processes were ameliorated by cinobufagin (7.5 mg/kg) in mice, and these phenomena were reversed by 3-MA treatment.
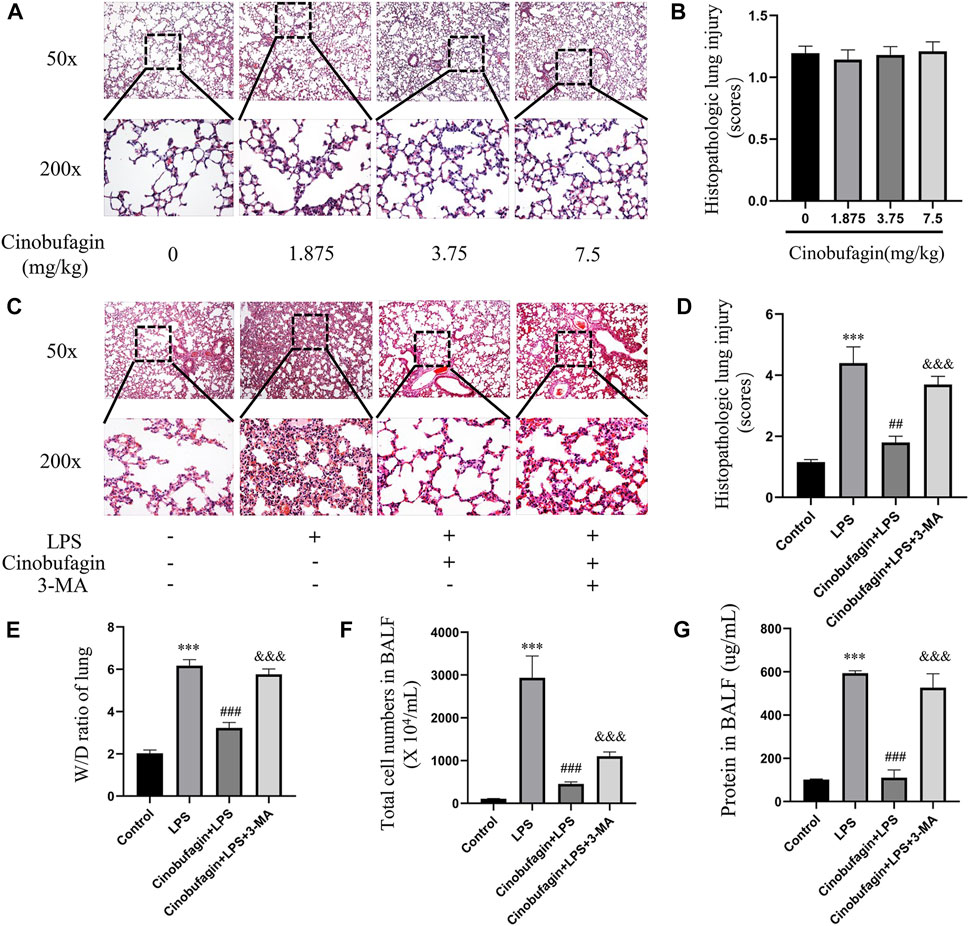
FIGURE 1. Effects of cinobufagin on LPS-induced histopathologic changes and edema in LPS-induced ALI in the lungs. (A) 24 h after different concentrations of cinobufagin injection, the lungs in each group were prepared for histological evaluation. Representative histological section of the lungs was stained by HE staining, magnification (50x, bar = 100 μm; 200x, bar = 50 μm). (B) The lung injury scores were determined. (C) 48 h after LPS injection, the lungs in each group were prepared for histological evaluation. (D) The lung injury scores were determined. (E) Lung tissues were weighed to calculate the W/D ratio. (F) The total cells detected by flow cytometry in BALF. (G) BCA assay was used to determine the total protein concentration in BALF. The values presented are mean ± SEM, ***p < 0.001 compared with control group. ##p < 0.01, ###p < 0.001 in comparison to LPS group. &&&p < 0.001 versus the cinobufagin + LPS group.
In ALI, when the epithelial or endothelial cell barrier is damaged, increased permeation of macromolecules and fluids into the interstitium is noted (Johnson and Matthay, 2010). The wet-to-dry weight ratio of the lungs, total cells, and BALF protein concentration were markers used to assess edema in ALI. Forty-8 h after LPS injection, the wet-to-dry weight ratio of the lung tissue, total cells, and BALF protein concentration increased. However, cinobufagin (7.5 mg/kg) blocked this increase, and the inhibitory effects of cinobufagin were reversed by 3-MA (Figures 1E–G). Taken together, the data shows that cinobufagin relieves the severity of increased lung permeability in LPS-induced ALI.
Inflammatory cytokines play key roles in LPS-induced ALI. We measured the levels of IL-1β, IL-6, and TNF-α in BALF to assess the severity of inflammatory responses in the lungs using ELISA. As shown in Figures 2A–C, the levels of IL-1β, IL-6, and TNF-α in BALF were significantly elevated in the LPS group compared to the control group. However, the cinobufagin-treated group in LPS-induced ALI showed significantly lower levels of IL-1β, IL-6, and TNF-α. Additionally, the inhibitory effects of cinobufagin were reversed by 3-MA. Thus, these results show that cinobufagin decreases LPS-induced lung inflammatory responses through the activation of autophagy in vivo.
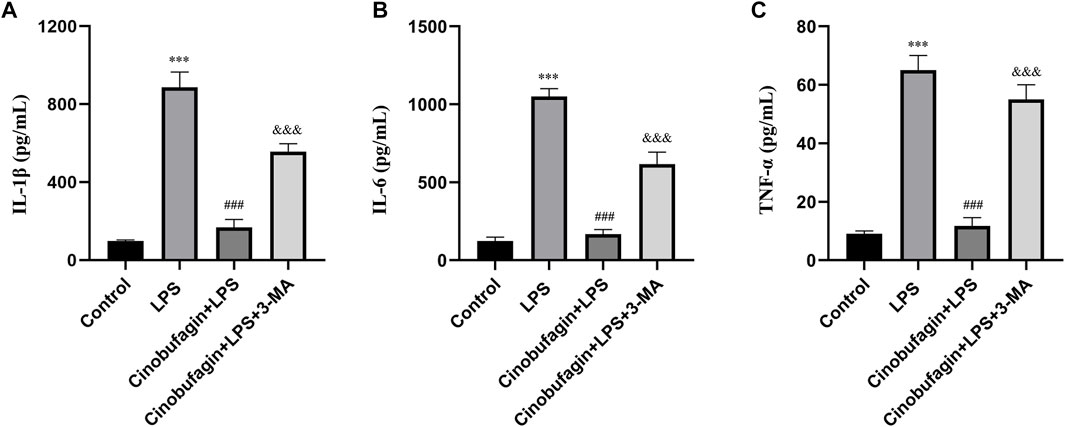
FIGURE 2. Cinobufagin inhibits lung inflammatory injury through autophagy activation in vivo. (A) IL-1β in BALF 48 h after LPS-induced ALI. (B) IL-6 in BALF 48 h after LPS-induced ALI. (C) TNF-α in BALF 48 h after LPS-induced ALI. The values presented are mean ± SEM, ***p < 0.001 compared with control group. ###p < 0.001 in comparison to LPS group. &&&p < 0.001 versus the cinobufagin + LPS group.
To elucidate the effect of cinobufagin on autophagy in LPS-induced lung injury, we first evaluated the protein levels of LC3-II/I and Beclin-1 using western blotting. As shown in Figures 3A–C, the expression of LC3-II/I and Beclin-1 was significantly decreased in the LPS group. Moreover, pretreatment with cinobufagin further enhanced the levels of LC3-II/I and Beclin-1 in an LPS-induced ALI mouse model. Importantly, autophagy activation induced by cinobufagin was reversed by 3-MA in vivo. Our results indicate that cinobufagin enhances autophagy in an LPS-induced ALI mouse model.
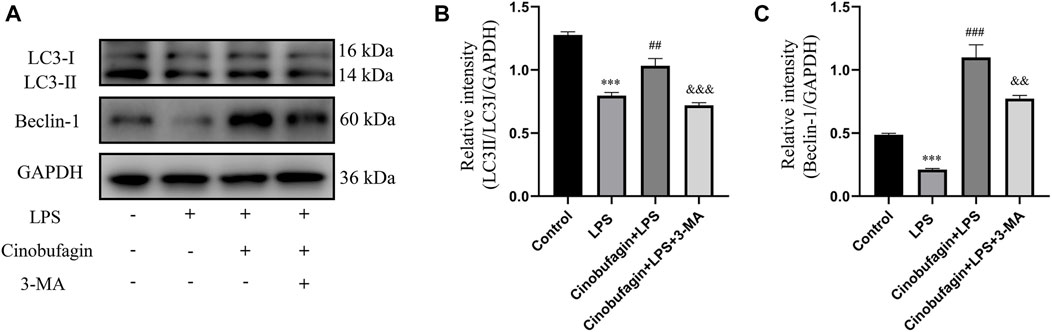
FIGURE 3. Cinobufagin induces autophagy in vivo. (A) Western blot detected the levels of LC3-II/I and Beclin-1 in lung tissues. (B–C) Quantitative analysis of LC3-II/I and Beclin-1 were shown in bar graphs, respectively. The values presented are mean ± SEM, ***p < 0.001 compared with control group. ##p < 0.01, ###p < 0.001 in comparison to LPS group. &&p < 0.01, &&&p < 0.001 versus the cinobufagin + LPS group.
To further identify the mechanisms of autophagy activated by cinobufagin in LPS-induced ALI in vivo, we investigated the p53/mTOR pathway. Cinobufagin treatment (7.5 mg/kg) resulted in increased levels of p-p53 and decreased levels of p-mTOR proteins. While these effects were reversed by 3-MA treatment, total p53 and mTOR levels did not change significantly in the LPS-induced ALI mouse model (Figures 4A–D). These results suggest that the p53/mTOR pathway governs cinobufagin-induced enhancement of autophagy in ALI in vivo.
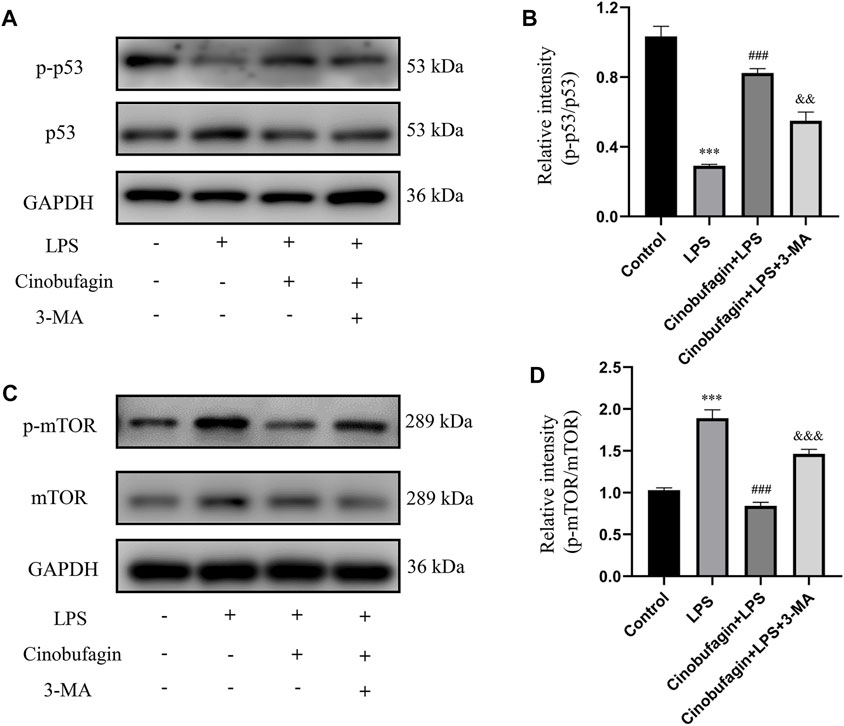
FIGURE 4. Cinobufagin induces autophagy through regulating the p53/mTOR pathway in vivo. (A–B) The levels of p-p53 and p53, and (C, D) p-mTOR and mTOR were evaluated by Western blot. The values presented are mean ± SEM, ***p < 0.001 compared with control group. ##p < 0.01, ###p < 0.001 in comparison to LPS group. &&p < 0.01, &&&p < 0.001 versus the cinobufagin + LPS group.
The epithelium is considered the primary defender of the airway and alveolus and forms a strong natural barrier against infectious and non-infectious stimuli in the pathogenesis and treatment of ALI (Crosby and Waters, 2010). Therefore, we used HBE cells for subsequent experiments. The effect of cinobufagin on the viability of HBE cells was assessed using the CCK8 assay. Cinobufagin demonstrated no significant cytotoxic effect on HBE cell viability (Figure 5A). To determine the working concentration of LPS, HBE cells were treated with varying concentrations of LPS (0, 50, 100, 200, and 400 μg/ml) for 24 h, and cell viability was assessed. LPS decreased the viability of HBE cells in a dose-dependent manner (Figure 5B). Based on these results, we used 100 μg/ml LPS for subsequent experiments. Next, we determined whether cinobufagin exerts a protective effect on HBE cells exposed to LPS. We pre-treated HBE cells with varying concentrations of cinobufagin (2 h) prior to LPS (100 μg/ml) exposure for 24 h, in which cinobufagin increased the decline in cell viability as a result of LPS challenge (Figure 5C). Thereafter, 3-MA, an autophagy inhibitor, was used to detect whether autophagy inhibition could decrease the viability of HBE cells. Results indicated that 3-MA (2 μM) reduced the viability of the cells (Figure 5D). Based on these results, we employed 2 μM cinobufagin and 1 μM 3-MA in subsequent experiments. To determine whether cinobufagin could increase the viability of HBE cells and whether 3-MA reversed the protective effect of cinobufagin, the cells were incubated with 100 μg/ml LPS, 2 μM cinobufagin, and/or 1 μM 3-MA. Cell viability was significantly increased with pre-treatment of cinobufagin and reversed by 3-MA (Figure 5E). To assess whether cinobufagin could increase the protective action against the cytotoxic effects of LPS and whether 3-MA reversed the protective effect, we pre-treated HBE cells with cinobufagin or 3-MA (2 h), followed by incubation with LPS (100 μg/ml) for 24 h. Apoptosis was assessed by PI and Annexin V-FITC double staining using flow cytometry. As shown in Figures 5 F–G, the rate of apoptosis was significantly increased by pretreatment with cinobufagin and reversed by 3-MA and cinobufagin treatment simultaneously in LPS-induced HBE cells.
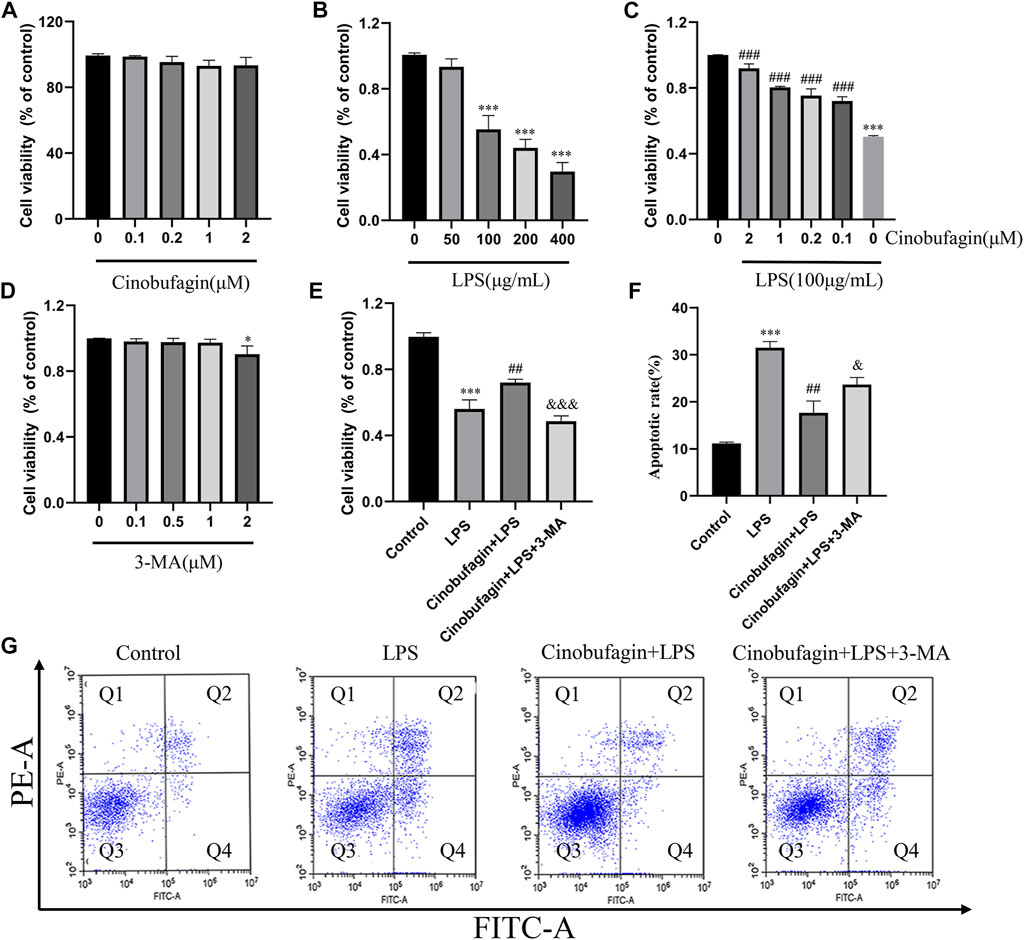
FIGURE 5. Cinobufagin promotes cell viability and protects LPS-induced apoptosis in HBE cells. (A–E) Viability of HBE cells was estimated using the CCK8 assay. (F) Quantitative analysis of cell apoptosis in HBE cells. (G) Apoptosis of HBE cells was stained with Annexin-VFITC and PI and analyzed by flow cytometry. The values presented are mean ± SEM, ***p < 0.001 compared with control group. ##p < 0.01, ###p < 0.001 in comparison to LPS group. &< 0.05, &&p < 0.01, &&&p < 0.001 versus the cinobufagin + LPS group.
The lung epithelium is essential for LPS-induced inflammatory responses (Gropper and Wiener-Kronish, 2008). We examined whether cinobufagin could affect the production of inflammatory cytokines in HBE cells using quantitative RT-PCR. As shown in Figures 6A–C, compared with the LPS group, cinobufagin signficantly suppressed the production of the proinflammatory cytokines, IL-1β, IL-6 and TNF-α; however, the inhibitory effects of cinobufagin were reversed by 3-MA in HBE cells treated with LPS. Similarly, cytokine secretion in HBE cells was detected by ELISA, which showed that the levels of IL-1β, IL-6, and TNF-α were also markedly attenuated by cinobufagin treatment. However, the inhibitory effects of cinobufagin were reversed by 3-MA (Figures 6D–F).
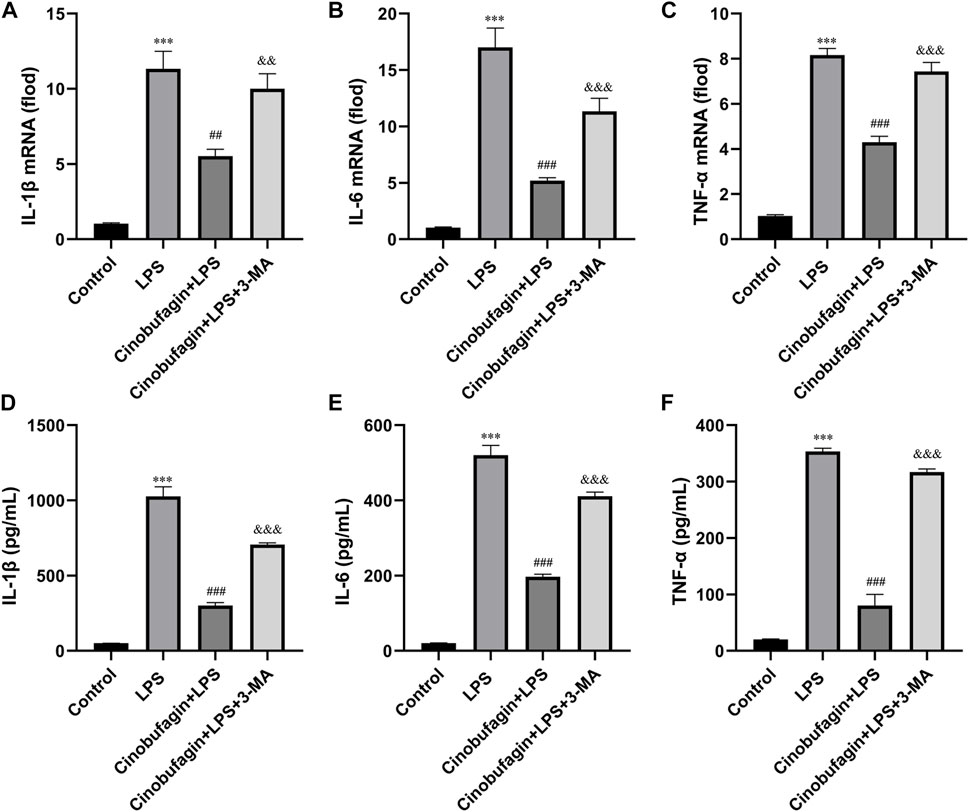
FIGURE 6. Cinobufagin decreased proinflammatory cytokine production in LPS-induced HBE cells. HBE cells were pre-treated with 2 μM cinobufagin or 1 μM 3-MA for 2 h before LPS (100 μg/ml) for 24 h, cells were collected to isolate total RNA, the mRNA expression level of (A) IL-1β, (B) IL-6, (C) TNF-α were measured by quantitative PCR. The secretion of (D) IL-1β, (E) IL-6, and (F) TNF-α in the supernatants was assayed by ELISA. The values presented are mean ± SEM, ***p < 0.001 compared with control group. ##p < 0.01, ###p < 0.001 in comparison to LPS group. &&p < 0.01, &&&p < 0.001 versus the cinobufagin + LPS group.
To investigate whether the protective effect of cinobufagin on HBE cells was associated with autophagy activation, we measured the levels of several key autophagy-related proteins by western blotting. Treatment with LPS significantly downregulated the ratio of LC3-II/LC3-I and the level of Beclin-1, suggesting the suppression of autophagy (Figures 7A–C). However, cinobufagin activated autophagy, and 3-MA reversed this effect in LPS-induced HBE cells. Subsequently, pre-treatment with cinobufagin obviously upregulated the fluorescence intensity of LC3 observed under CLSM (Figure 7D), indicating autophagy activation in the cells treated with LPS. In addition, the fluorescence intensity of LC3 significantly decreased in the 3-MA group. These results indicated that cinobufagin could induce autophagy in ALI in vitro.
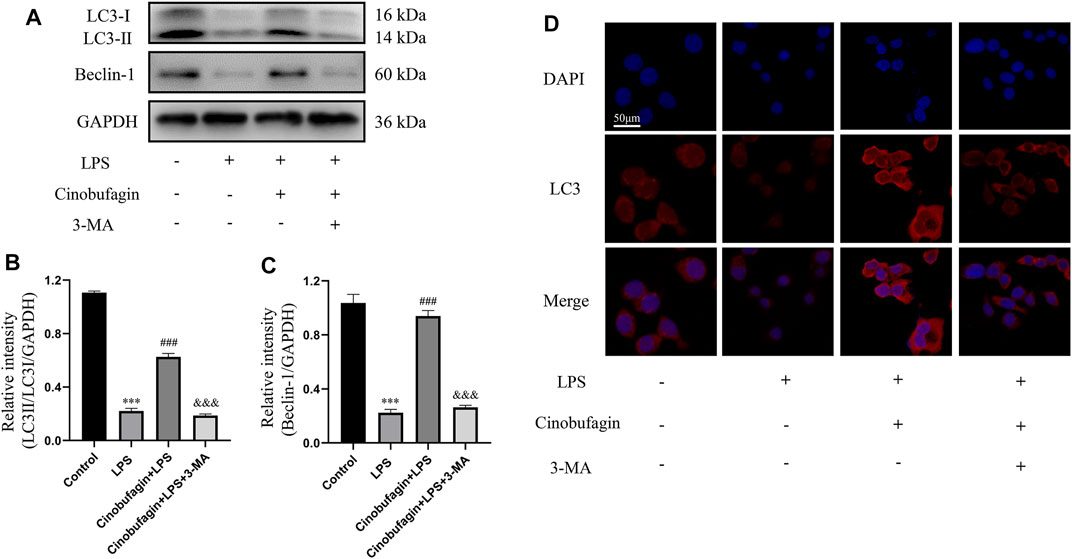
FIGURE 7. Cinobufagin induces autophagy in LPS-induced HBE cells. (A–C) The levels of LC3-II/I and Beclin-1 were detected by Western blot in HBE cells, and the bar graphs showed the quantitative analysis results of the corresponding protein in each group, respectively. (D) The representative images of the fluorescence of LC3 in HBE cells (magnification ×50). The values presented are mean ± SEM, ***p < 0.001 compared with control group. ##p < 0.01, ###p < 0.001 in comparison to LPS group. &&p < 0.01, &&&p < 0.001 versus the cinobufagin + LPS group.
The possible mechanisms of autophagy activation by cinobufagin in LPS-induced HBE cells were further investigated. We assessed the p53/mTOR pathway, a classic autophagy-related pathway. Notably, treatment with LPS significantly decreased the phosphorylation of p53 and increased the phosphorylation of mTOR compared to the control group. However, it was observed that cinobufagin activated the p53/mTOR pathway by significantly increasing the phosphorylation of p53 (p-p53) and decreasing the phosphorylation of mTOR (p-mTOR). These effects were reversed by 3-MA (Figures 8A–D). To verify the effect of the p53/mTOR signaling pathway in this study, the p53 inhibitor pifithrin-μ was used to evaluate the protective effect of cinobufagin against LPS-induced ALI. When combining pifithrin-μ with cinobufagin, the proteins p-p53, p53, p-mTOR, Beclin1, and LC3B-II/I were significantly restored (Figure 8E). These results suggest that cinobufagin induced autophagy, at least partly through the regulation of the p53/mTOR pathway in HBE cells treated with LPS.
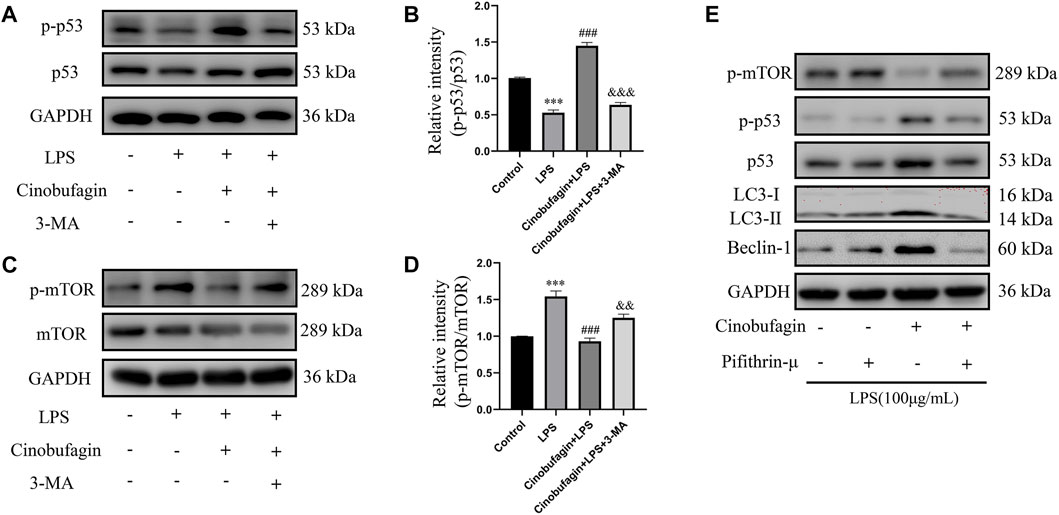
FIGURE 8. Cinobufagin enhances autophagy through the p53/mTOR pathway in vitro. Western blot detected the levels of p-p53, p53, p-mTOR, mTOR, LC3B, Beclin1 (A–E). The values presented are mean ± SEM, ***p < 0.001 compared with control group. ###p < 0.001 in comparison to LPS group. &&p < 0.01, &&&p < 0.001 versus the cinobufagin + LPS group.
Acute lung injury is caused by a variety of factors that cause alveolar epithelial cells and capillary endothelial cells to undergo apoptosis and sloughing, triggering lung tissue to develop diffuse edema and other pathological changes. These changes eventually manifest as acute hypoxic respiratory insufficiency and other symptoms (Rubenfeld et al., 2005). ALI is mainly characterized by progressive hypoxemia, respiratory distress, and heterogeneous exudative lesions on lung imaging. If left untreated, ALI can progress to ARDS and involve various tissues and organs, including the heart and kidney (Thompson et al., 2017). Currently, there are no definitive treatments for ALI in clinical practice, however, primary disease treatment and respiratory support is advocated.
With increasing awareness of the molecular mechanisms of disease occurrence and development, the discovery of active pharmaceutical ingredients from natural products for the treatment of various diseases has become a major goal for modern medical workers (Arulselvan et al., 2016; Evans et al., 2020; Atanasov et al., 2021). Cinobufagin, a natural product extracted from toad venom, is one of the major active ingredients in cinobufotalin injections which are used to treat intermediate and advanced tumors. However, the effects of cinobufagin on ALI remain unclear. In this study, we found that cinobufagin increased the viability and decreased the production of TNF-α, IL-1β, and IL-6 in LPS-induced HBE cells. Interestingly, cinobufagin reduced LPS-induced TNF-α to a greater degree than IL-6 and IL-8, which requires further exploration. Cinobufagin inhibited the secretion of inflammatory cytokines in an ALI mouse model. In addition, cinobufagin had little effect on the W/D ratio and BALF protein concentration when compared to the significant decrease in total number of cells. Based on these findings, we further studied the effect of cinobufagin on ALI and attempted to reveal its potential mechanism.
In recent years, autophagy has attracted considerable attention because of its effects on various physiological and pathological processes. Autophagy involves various autophagy-related proteins that engulf and kill pathogens to protect cells and inhibit the secretion of inflammasomes and inflammatory factors (Lo et al., 2013; Shibutani et al., 2015). According to a previous study, autophagy maintains the integrity of the endothelial barrier during LPS-induced lung injury (Zhang et al., 2018). Zhao reported that autophagy activation is involved in the pathophysiological process of sepsis, and alleviates excessive cytokine release and lung injury in sepsis (Zhao et al., 2019). Herein, we found that cinobufagin could activate autophagy in an LPS-induced ALI model. Moreover, cinobufagin significantly increased the protein ratio of LC3-II/LC3-I and the protein level of Beclin-1, as well as upregulated the fluorescence intensity of LC3B. During autophagy, LC3-I is converted to LC3-II, and the ratio of LC3-II/LC3-I is taken as a quantitative index of autophagic activity. Therefore, our study showed that cinobufagin activates autophagy in LPS-induced ALI. Importantly, the activation of autophagy by cinobufagin was reversed by 3-MA treatment. On the basis of these reports and our previous results, we hypothesized that cinobufagin regulates the expression of inflammatory cytokines by regulating autophagy in ALI. After inhibition of autophagy in vitro and in vivo, the cytokines IL-1β, IL-6, and TNF-α were inhibited. In conclusion, the anti-inflammatory effects of cinobufagin may be related to the induction of autophagy.
Many previous studies have found that the p53 signaling pathway plays a vital role in cell growth, apoptosis, autophagy, and many other cell functions (Chen, 2016; Guo et al., 2016). While p53 protein can activate autophagy, autophagy also inhibits p53 levels and function. This association is functionally intertwined and important for stress response, metabolism, and cancer (White, 2016; Yang et al., 2020). Previous studies have shown that mTOR is a major inhibitor of autophagy (Laplante and Sabatini, 2012). Similarly, it has been reported that activation of mTOR promotes LPS-induced ALI in lung epithelium (Hu et al., 2016). In terms of mechanism, there are multiple pathways involved in the regulation of mTOR, such as p53, MAPK, PI3K-Akt, and AMPK. According to the former study, chitooligosaccharides, could inhibit tumor progression and induce autophagy through the activation of the p53/mTOR pathway in osteosarcoma (Pan et al., 2021). In addition, Uzhytchak et al. found that iron oxide nanoparticle-induced autophagic flux was regulated by the interplay between the p53-mTOR axis in hepatic cells (Uzhytchak et al., 2020). Importantly, our results showed that cinobufagin increased p-p53 protein levels and reduced p-mTOR protein levels in LPS-induced ALI, while total p53 and mTOR showed no evident changes. Pifithrin-μ, a p53 inhibitor, was used to treat the HBE cells. When combining pifithrin-μ with cinobufagin, the proteins p-p53, p53, p-mTOR, Beclin1, and LC3B-II/I were significantly restored, indicating that cinobufagin induced autophagy at least party through the p53/mTOR pathway. Therefore, as shown in Figure 9, cinobufagin improved LPS-induced ALI by regulating the p53/mTOR pathway via autophagy activation.
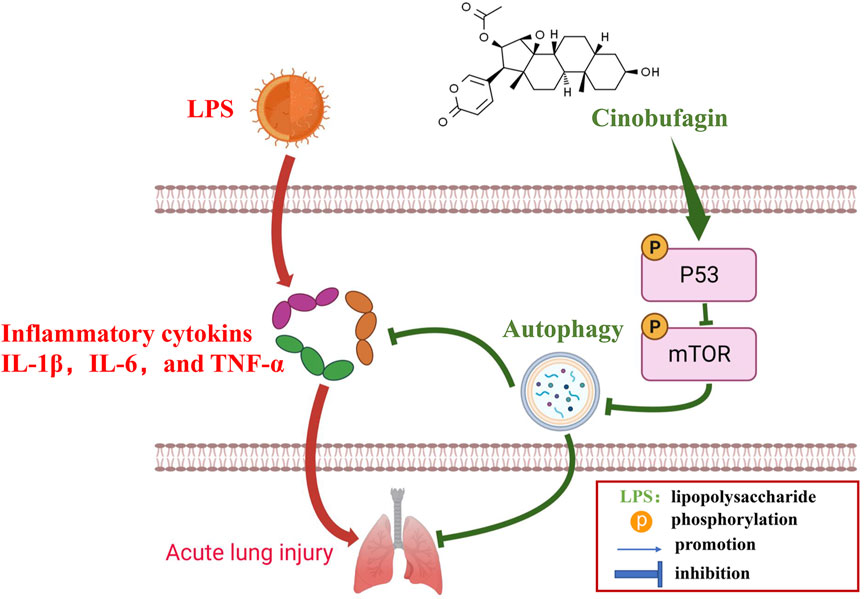
FIGURE 9. A diagram depicting the mechanism of Cinobufagin ameliorates LPS-induced ALI by activating autophagy through regulating of the p53/mTOR pathway.
In summary, we demonstrated that cinobufagin effectively suppressed inflammation and alleviated LPS-induced ALI in vivo and in vitro, at least to a large extent, through regulation of the p53/mTOR pathway via autophagy activation. Thus, cinobufagin may be an effective treatment for ALI in clinical practice.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by The Animal Care and Use Committee of the First Hospital of Nanchang University.
ZZ designed the research study. CW and XM performed the research. YW and YY analyzed the data. CW and XM wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
This work was supported by the National Natural Science Foundation of China (82060361, 81760351).
We would like to thank Editage (www.editage.cn) for English language editing. We are grateful for the support of Jiangxi Postgraduate Innovation Special Fund Project (YC2022-B057) for this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.994625/full#supplementary-material
Arulselvan, P., Fard, M. T., Tan, W. S., Gothai, S., Fakurazi, S., Norhaizan, M. E., et al. (2016). Role of antioxidants and natural products in inflammation. Oxid. Med. Cell. Longev. 2016, 5276130. doi:10.1155/2016/5276130
Atanasov, A. G., Zotchev, S. B., Dirsch, V. M., and Supuran, C. T. (2021). Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 20 (3), 200–216. doi:10.1038/s41573-020-00114-z
Bai, Y., Wang, X., Cai, M., Ma, C., Xiang, Y., Hu, W., et al. (2021). Cinobufagin suppresses colorectal cancer growth via STAT3 pathway inhibition. Am. J. Cancer Res. 11 (1), 200–214.
Barth, S., Glick, D., and Macleod, K. F. (2010). Autophagy: Assays and artifacts. J. Pathol. 221 (2), 117–124. doi:10.1002/path.2694
Butt, Y., Kurdowska, A., and Allen, T. C. (2016). Acute lung injury: A clinical and molecular review. Arch. Pathol. Lab. Med. 140 (4), 345–350. doi:10.5858/arpa.2015-0519-RA
Chaudhuri, N., Whyte, M. K., and Sabroe, I. (2007). Reducing the toll of inflammatory lung disease. Chest 131 (5), 1550–1556. doi:10.1378/chest.06-2869
Chen, H., Bai, C., and Wang, X. (2010). The value of the lipopolysaccharide-induced acute lung injury model in respiratory medicine. Expert Rev. Respir. Med. 4 (6), 773–783. doi:10.1586/ers.10.71
Chen, J. (2016). The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 6 (3), a026104. doi:10.1101/cshperspect.a026104
Cheng, N., Liang, Y., Du, X., and Ye, R. D. (2018). Serum amyloid A promotes LPS clearance and suppresses LPS-induced inflammation and tissue injury. EMBO Rep. 19 (10), e45517. doi:10.15252/embr.201745517
Crosby, L. M., and Waters, C. M. (2010). Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 298 (6), L715–L731. doi:10.1152/ajplung.00361.2009
Dai, G., Zheng, D., Guo, W., Yang, J., and Cheng, A. Y. (2018). Cinobufagin induces apoptosis in osteosarcoma cells via the mitochondria-mediated apoptotic pathway. Cell. Physiol. biochem. 46 (3), 1134–1147. doi:10.1159/000488842
Deretic, V. (2021). Autophagy in inflammation, infection, and immunometabolism. Immunity 54 (3), 437–453. doi:10.1016/j.immuni.2021.01.018
Dikic, I., and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell. Biol. 19 (6), 349–364. doi:10.1038/s41580-018-0003-4
Evans, L. W., Stratton, M. S., and Ferguson, B. S. (2020). Dietary natural products as epigenetic modifiers in aging-associated inflammation and disease. Nat. Prod. Rep. 37 (5), 653–676. doi:10.1039/c9np00057g
Gropper, M. A., and Wiener-Kronish, J. (2008). The epithelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 14 (1), 11–15. doi:10.1097/MCC.0b013e3282f417a0
Guo, Z., Kong, Q., Liu, C., Zhang, S., Zou, L., Yan, F., et al. (2016). DCAF1 controls T-cell function via p53-dependent and -independent mechanisms. Nat. Commun. 7, 10307. doi:10.1038/ncomms10307
Hu, Y., Lou, J., Mao, Y. Y., Lai, T. W., Liu, L. Y., Zhu, C., et al. (2016). Activation of MTOR in pulmonary epithelium promotes LPS-induced acute lung injury. Autophagy 12 (12), 2286–2299. doi:10.1080/15548627.2016.1230584
Johnson, E. R., and Matthay, M. A. (2010). Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 23 (4), 243–252. doi:10.1089/jamp.2009.0775
Kumar, V. (2020). Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front. Immunol. 11, 1722. doi:10.3389/fimmu.2020.01722
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell. 149 (2), 274–293. doi:10.1016/j.cell.2012.03.017
Li, X., Bi, Z., Liu, S., Gao, S., Cui, Y., Huang, K., et al. (2019). Antifibrotic mechanism of cinobufagin in bleomycin-induced pulmonary fibrosis in mice. Front. Pharmacol. 10, 1021. doi:10.3389/fphar.2019.01021
Lo, S., Yuan, S. S., Hsu, C., Cheng, Y. J., Chang, Y. F., Hsueh, H. W., et al. (2013). Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Ann. Surg. 257 (2), 352–363. doi:10.1097/SLA.0b013e318269d0e2
Maiuri, M. C., Galluzzi, L., Morselli, E., Kepp, O., Malik, S. A., and Kroemer, G. (2010). Autophagy regulation by p53. Curr. Opin. Cell. Biol. 22 (2), 181–185. doi:10.1016/j.ceb.2009.12.001
Pan, Z., Cheng, D. D., Wei, X. J., Li, S. J., Guo, H., and Yang, Q. C. (2021). Chitooligosaccharides inhibit tumor progression and induce autophagy through the activation of the p53/mTOR pathway in osteosarcoma. Carbohydr. Polym. 258, 117596. doi:10.1016/j.carbpol.2020.117596
Qi, F., Inagaki, Y., Gao, B., Cui, X., Xu, H., Kokudo, N., et al. (2011). Bufalin and cinobufagin induce apoptosis of human hepatocellular carcinoma cells via Fas- and mitochondria-mediated pathways. Cancer Sci. 102 (5), 951–958. doi:10.1111/j.1349-7006.2011.01900.x
Rubenfeld, G. D., Caldwell, E., Peabody, E., Weaver, J., Martin, D. P., Neff, M., et al. (2005). Incidence and outcomes of acute lung injury. N. Engl. J. Med. 353 (16), 1685–1693. doi:10.1056/NEJMoa050333
Shibutani, S. T., Saitoh, T., Nowag, H., Münz, C., and Yoshimori, T. (2015). Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 16 (10), 1014–1024. doi:10.1038/ni.3273
Thompson, B. T., Chambers, R. C., and Liu, K. D. (2017). Acute respiratory distress syndrome. N. Engl. J. Med. 377 (6), 562–572. doi:10.1056/NEJMra1608077
Uzhytchak, M., Smolková, B., Lunova, M., Jirsa, M., Frtús, A., Kubinová, Š., et al. (2020). Iron oxide nanoparticle-induced autophagic flux is regulated by interplay between p53-mTOR Axis and bcl-2 signaling in hepatic cells. Cells 9 (4), E1015. doi:10.3390/cells9041015
White, E. (2016). Autophagy and p53. Cold Spring Harb. Perspect. Med. 6 (4), a026120. doi:10.1101/cshperspect.a026120
Yang, Y., Karsli-Uzunbas, G., Poillet-Perez, L., Sawant, A., Hu, Z. S., Zhao, Y., et al. (2020). Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes. Dev. 34 (9-10), 688–700. doi:10.1101/gad.335570.119
Zhang, D., Zhou, J., Ye, L. C., Li, J., Wu, Z., Li, Y., et al. (2018). Autophagy maintains the integrity of endothelial barrier in LPS-induced lung injury. J. Cell. Physiol. 233 (1), 688–698. doi:10.1002/jcp.25928
Zhang, J., Hong, Y., Jiang, L., Yi, X., Chen, Y., Liu, L., et al. (2020). Global metabolomic and lipidomic analysis reveal the synergistic effect of bufalin in combination with cinobufagin against HepG2 cells. J. Proteome Res. 19 (2), 873–883. doi:10.1021/acs.jproteome.9b00681
Zhang, L., Liang, B., Xu, H., Gong, Y., Hu, W., Jin, Z., et al. (2022). Cinobufagin induces FOXO1-regulated apoptosis, proliferation, migration, and invasion by inhibiting G9a in non-small-cell lung cancer A549 cells. J. Ethnopharmacol. 291, 115095. doi:10.1016/j.jep.2022.115095
Keywords: acute lung injury, cinobufagin, lipopolysaccharide, autophagy, p53, mTOR
Citation: Wang C, Mei X, Wu Y, Yang Y and Zeng Z (2022) Cinobufagin alleviates lipopolysaccharide-induced acute lung injury by regulating autophagy through activation of the p53/mTOR pathway. Front. Pharmacol. 13:994625. doi: 10.3389/fphar.2022.994625
Received: 15 July 2022; Accepted: 15 November 2022;
Published: 28 November 2022.
Edited by:
Jian Zhang, Tianjin Medical University, ChinaReviewed by:
Lulong Bo, Naval Medical University, ChinaCopyright © 2022 Wang, Mei, Wu, Yang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenguo Zeng, emVuZ3poZW5ndW9AbmN1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.