
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 07 September 2022
Sec. Inflammation Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.992734
This article is part of the Research TopicFerroptosis as a Novel Therapeutic Target for Inflammation-Related DiseasesView all 8 articles
 Leyao Xiao1,2
Leyao Xiao1,2 Huanshao Huang1,2
Huanshao Huang1,2 Shuhao Fan1,2Biying Zheng1,2Jianguo Wu1,2
Shuhao Fan1,2Biying Zheng1,2Jianguo Wu1,2 Junai Zhang1,2
Junai Zhang1,2 Jiang Pi1,2*
Jiang Pi1,2* Jun-Fa Xu1,2*
Jun-Fa Xu1,2*To date, it has been confirmed that the occurrence and development of infectious diseases are tightly associated with regulatory cell death processes, such as apoptosis, autophagy, and necroptosis. Ferroptosis, as a newly discovered form of regulatory cell death characterized by iron-dependent lipid peroxidation, is not only closely associated with tumor progression, but is also found to be tightly related to the regulation of infectious diseases, such as Tuberculosis, Cryptococcal meningitis, Malaria and COVID-2019. The emerging critical roles of ferroptosis that has been found in infectious disease highlight ferroptosis as a potential therapeutic target in this field, which is therefore widely expected to be developed into new therapy strategy against infectious diseases. Here, we summarized the underlying mechanisms of ferroptosis and highlighted the intersections between host immunity and ferroptosis. Moreover, we illuminated the roles of ferroptosis in the occurrence and progression of different infectious diseases, which might provide some unique inspiration and thought-provoking perspectives for the future research of these infectious diseases, especially for the development of ferroptosis-based therapy strategy against infectious diseases.
Cell is the basic unit of the biological system, which not only provides the necessary energy and nutrition for physiological events, but also acts as the indispensable one for the host’s immunity against invasion. Various regulated cell death modalities are tightly associated with the pathological processes in many diseases. Strikingly, ferroptosis, a newly discovered cell death, accompanied by iron independence lipid peroxidation (LPO), has become an eye-catching topic nowadays (Dixon et al., 2012). The mechanism, morphology, and genomics of ferroptosis have been proved to be different from the well-known programmed cell death such as apoptosis, autophagy, and scorching death. Although lots of efforts have been made to depict the functions and mechanisms of ferroptosis, some critical mechanisms and related regulatory functions have not yet been fully explored, which requires more in-depth researches.
Over the past few years, shreds of evidences have shown that the close relationship between ferroptosis and cancer, neurodegenerative diseases, ischemia-reperfusion diseases, and kidney diseases (Stockwell et al., 2017; Stockwell et al., 2020; Wang et al., 2021b), however, there are relatively few studies describing the roles of ferroptosis in infectious diseases. Meanwhile, viruses, bacteria, and other pathogens in nature have coexisted with human beings for a long time, such as the world pandemic of COVID-19 (Kalra and Chawla, 2020), the growing bacterial drug resistance (Chen and Chen, 2021) and dangerous virus-related cancer (Ding et al., 2021) have brought endless suffering to people. Though many achievements and progresses have been made by human beings in the combat with infectious diseases, more works are still needed to be done, such as the updating of preventive measures and treatment strategies. Therefore, it is necessary to explore more in-depth understanding of the occurrence and development of infectious diseases. Potentially, ferroptosis may be a novel therapeutic target to develop more effective adjuvant therapies against infectious diseases.
It is conceivable, but not fully demonstrated, that ferroptosis triggered in infectious diseases acting a double-edged sword role that it may be caused by pathogens for survival or can be exploited as potential therapeutics. In this review, we briefly described the present understanding of ferroptosis induction and execution, and also highlighted the relationships between ferroptosis and infectious diseases so as to further understand the functions of ferroptosis in infectious diseases. Particularly, we hypothesized the therapeutic potential of ferroptosis in these diseases based on the current researches. We hope this review could enhance our understanding of ferroptosis in infectious diseases by exploring how ferroptosis contributes to host control of pathogens, how ferroptosis is triggered by some pathogens to promote disease development, and how ferroptosis can be controlled to defend against infectious diseases.
Lipid peroxidation is one of the most prominent features of ferroptosis, which is composed of a series of free radical chain reactions (Figure 1). Lipid peroxidation can be described generally as a process under which oxidants such as free radicals attack lipids containing carbon-carbon double bond(s), especially polyunsaturated fatty acids (PUFAs) (Minotti and Aust, 1989; Halliwell and Chirico, 1993; Cheng and Li, 2007). Free radicals, such as hydroxyl radicals (•OH) produced by the Fenton reaction (Fe2+ + H2O2 → Fe3+ + •OH + OH−), can be excessively produced due to an excess of ferrous iron (Cheng and Li, 2007). •OH is an essential substance for the initiation of lipid peroxidation and is capable of causing oxidative damage to cells. PUFAs are more susceptible to •OH compared with other intracellular lipids, the reaction between them generates LOOH, and ferrous iron catalyzes the cleavage of LOOH, again producing numerous ROS, such as LOO•, alkoxyl (LO•), or epoxy peroxyl radical (loo•), which further boost the oxidative stress in cell.(Halliwell and Chirico, 1993; Cheng and Li, 2007). As a result, this destructive lipid peroxidation disrupts the integrity and fluidity of the lipid bilayer of the cell membrane and ultimately leads to cell damage or death.
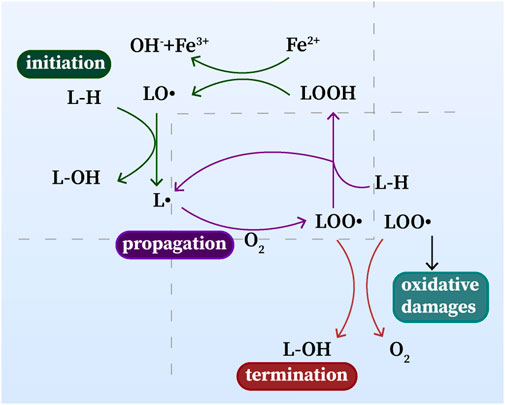
FIGURE 1. Lipid peroxidation: initiation, propagation and termination. L•: lipid radical; LO•: alkoxyl radical; L-OH, lipid alcohol; L–H, lipid; LOOH, lipid hydroperoxide; LOO•: epoxy peroxyl radical.
Glutathione peroxidase 4 (GPX4), a selenoprotein, one of the most well-known key factors in the regulation of ferroptosis, can reduce the level of intracellular lipid peroxide via lipids detoxification (Brigelius-Flohe and Maiorino, 2013; Yang et al., 2014). Glutathione (GSH) is used as a cofactor of GPX4 to assist GPX4, which converts toxic lipid hydroperoxides to non-toxic lipid alcohols. Thus, the reduction of GSH level will lead to the inhibition of GPX4, which suppresses the host capacity to repair peroxidized lipids and causes the occurrence of ferroptosis. Meantime, diverse ferroptosis inhibitors and inducers are applied to different kinds of research nowadays (Tables 1, 2) For example, as one of the representatives of class II ferroptosis-inducers (FINs) different from erastin, 1S, 3R-RSL 3(RSL3) can directly inhibit GPX4 without reducing the level of GSH to trigger ferroptosis. Knockdown of intracellular GPX4 can increase the level of intracellular lipid peroxides accompanied by ferroptosis, while iron chelator DFO and vitamin E prevent ferroptosis in GPX4 knockdown cells, reflecting the importance of GPX4 in protecting cells from excessive toxic lipid peroxides (Yang et al., 2014).
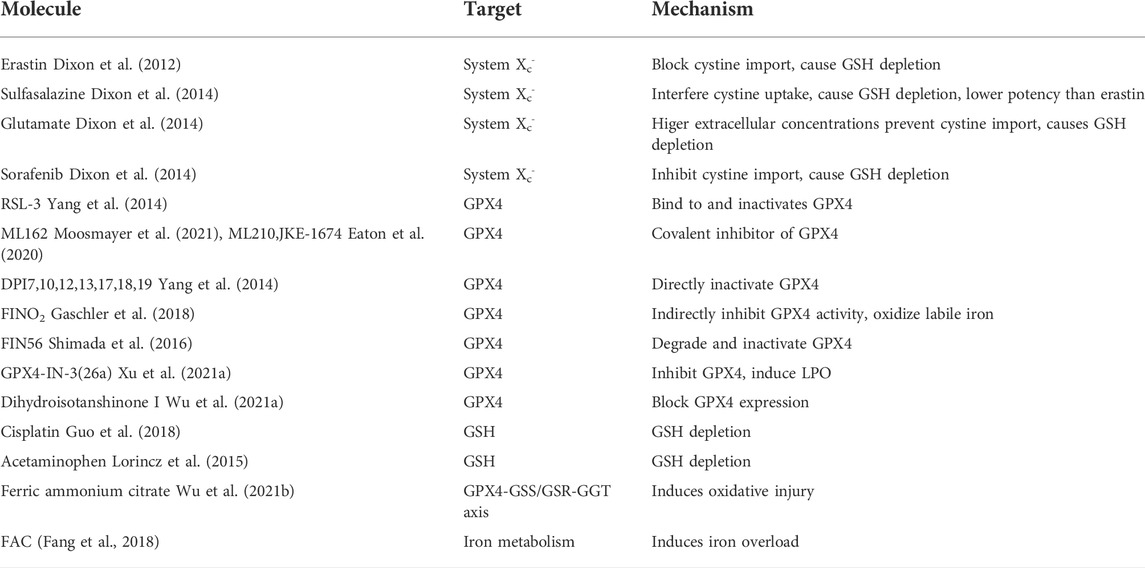
TABLE 1. Ferroptosis inducers.
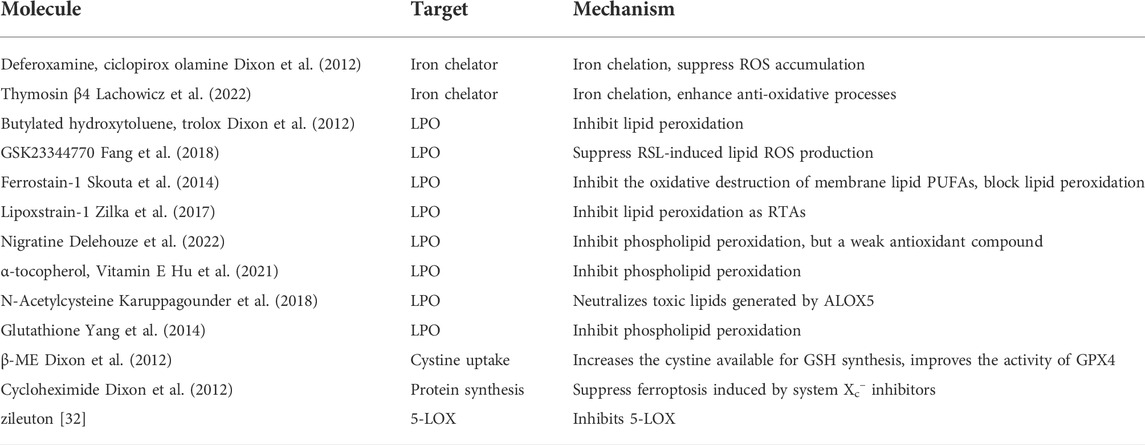
TABLE 2. Ferroptosis inhibitors.
Due to the important roles of selenium in GPX4, there is also a close relationship between selenium and ferroptosis (Ingold et al., 2018). Selenoprotein GPX4 is composed of selenocysteine, which is similar in structure to ordinary cysteine, except that the sulfur is replaced by selenium at the active site of cysteine. Via replacing the active site of GPX4 selenocysteine with sulfur to cysteine, Ingold et al. found that the oxidative stress in the cell was intensified after adding H2O2, and the active site was oxidized to sulfonic acid (SO2/3H), which inactivated GPX4 and eventually induced ferroptosis. This work strongly suggests that selenium plays an indispensable role in helping GPX4 resist ferroptosis by regulating lipid peroxidation.
Moreover, Zhang Y et al. discovered a new pathway regulating GPX4 independently with the level of intracellular GSH (Zhang et al., 2021). Mammalian rapamycin complex 1 (mTORC1) plays an important role in regulating protein synthesis, cell growth, lipid metabolism, autophagy, and other biological activities. In the absence of intracellular cystine, the activity of mTORC1 will be inhibited and the amount of mTORC1 localized to lysosomes will be decreased, thereby reducing the level of intracellular GPX4.
Meanwhile, GPX4 is not the only intracellular antioxidant molecule inhibiting ferroptosis. Ferroptosis inhibitor protein 1 (FSP1) is an in vitro NADPH-dependent coenzyme Q10 (CoQ) oxidoreductase localized on the cell membrane and is able to inhibit lipid peroxidation in a different way (Bersuker et al., 2019). The supplementation of exogenous FSP1 could promote the reduced CoQ to enhance the antioxidant function of capturing free radicals, thereby inhibiting LPO and restoring the resistance of FSP1 knockdown cells to ferroptosis (Bersuker et al., 2019). Similar to GPX4 and FSP1, the lipid detoxification phospholipase iPLA2β can inhibit p53-induced ferroptosis by resisting lipid peroxidation (Chen et al., 2021), and avoid ferroptosis by reducing its substrate 15-HpETE-PE, a ferroptosis-promoting factor (Sun et al., 2021).
Glutamine metabolism plays essential roles in cell biosynthesis, cell proliferation and cell death. The heterodimer of amino acid transporter solute carrier family 7 member 11 (SLC7A11) and amino acid transporter solute carrier family 3 member 2 (SLC3A2) is the main component of the sodium-dependent cystine/glutamate transporter (systemXc−) localized on cell membrane (Koppula et al., 2021), which exchanges cystine and glutamic acid at a ratio of 1:1. Cystine enters the cell and converts to cysteine for the synthesis of GSH, and SLC7A11 can regulate cystine uptake and participate in glutamine cycle metabolism (Zhang et al., 2021), thus promoting GPX4 protein synthesis. Consistently, Gao et al. illustrated that two specific amino acids, cystine and cysteine, were indispensable for the synthesis of intracellular GSH, and ROS was increased once in the absence of them (Gao et al., 2015). Meantime, glutamate-cysteine ligase catalytic subunit (GCLC), a catalytic enzyme involved in GSH synthesis pathway, can repress the sensitivity of cells to cystine and cysteine deficiency-induced ferroptosis (Kang et al., 2021). In addition, BECN1 (beclin 1) can directly bind to SLC7A11 and inhibit its function for the uptake of cystine, which leads to the lack of intracellular GSH and the accumulation of lipid peroxidation, triggering ferroptosis eventually (Song et al., 2018).
Iron is required for the execution of ferroptosis, thus, iron homeostasis is inseparable from the occurrence of ferroptosis. To date, a variety of related molecules have been found to regulate and maintain intracellular iron homeostasis (Halliwell and Chirico, 1993; Anderson and Vulpe, 2009). Transferrin receptor (TfR) is a prominent molecule for storing excess intracellular iron. The deficiency of TfR is capable of stimulating chronic iron accumulation and increasing the sensitivity of cells to ferroptosis. For example, the lack of TfR in hepatocytes is accompanied by ferroptosis, which aggravates the degree of cell damage (Yu et al., 2020).
Poly(C)-binding proteins 1 (PCBP1), an iron chaperone from one of the four homologous RNA-binding protein families in KH domain superfamily, can directly bind iron and combine with ferritin to complete the storage of intracellular iron (Shi et al., 2008). Evidence showed that the lipid peroxidation and product 4-Hydroxynonenol (4-HNE) were increased in PCBP1 knockout mouse liver cells and the level of GPX4 was also increased accordingly (Protchenko et al., 2021). Knockout of PCBP1 led to the failure of its function as an iron chaperone, and the increased free ferrous iron in the cell thus catalyzed the production of ROS through the Fenton reaction, resulting in the accumulation of lipid peroxides. This research also pointed out that PCBP1 could control the redox response in labile iron pool (LIP), thereby inhibiting iron-induced LPO or cell death under physiological conditions. Importantly, PCBP1 has the ability to repress ferritinophagy-mediated ferroptosis via inhibiting ferritinophagy through silencing BECN1 mRNA and binding with ALOX15 to attenuate the susceptibility of cells to ferroptosis (Lee et al., 2022).
In recent years, the relationship between autophagy and ferroptosis has engaged much attention, and the view that ferroptosis is an autophagy-dependent death is emerged. Many studies have reported that ferritinophagy can trigger ferroptosis (Hou et al., 2016; Zhou B. et al., 2020). Ferritin, an important protein for storing iron (Theil, 2004), can avoid cell damage caused by Fenton reaction via combining with excess ferrous iron (Minotti and Aust, 1989; Halliwell and Chirico, 1993; Cheng and Li, 2007). Notably, nuclear receptor coactivator (NCOA4), the selective carrier receptor of ferritinophagy, can drive ferritinophagy via releasing excess iron into the cell to induce autophagy degradation of ferritin (Mancias et al., 2014). Physiologically, the intracellular iron content can be compensated by ferritinophagy via the NCOA4 pathway. At the same time, down-regulation of NCOA4 prevents excessive ROS production to inhibit the occurrence of ferritinophagy and ferroptosis, while over-expression of NCOA4 causes the opposite performance. What’s more, the hypoxic environment can inhibit NCOA4-regulated ferroptosis and avoid cell death (Fuhrmann et al., 2020).
Meanwhile, a paper proposed that ATG5, an autophagy-related gene, regulated ferritinophagy and then induced ferroptosis (Hou et al., 2016). Eunhee Park et al. subsequently confirmed this view, they discovered that erastin could induce autophagy-related cell death, which further lead to iron-dependent ferroptosis by degradation of ferritin and induction of TfR1 (Park and Chung, 2019). Also, the inhibition of autophagy can downregulate intracellular iron content and weaken the degree of lipid peroxidation in ferroptosis, illuminating that ferritinophagy is a vital process involved in ferroptosis. Meanwhile, they considered the occurrence of autophagy as the consequence for the increase of intracellular ROS induced by erastin. Moreover, another group (Ma et al., 2017) indicated that the increase of iron-dependent ROS could also cause a similar phenomenon, suggesting that the inducement of autophagy in ferroptosis can be diversified. In addition, ferroptosis and autophagy can also occur at different time periods, implying that ferroptosis can also occur independently from autophagic cell death.
Acyl-CoA synthetase long-chain family member 4 (ACSL4) has been confirmed as a key ferroptosis gene, playing a crucial role in the synthesis of long-chain PUFA-CoA (Doll et al., 2017; Kagan et al., 2017). Pharmacological inhibition of ACSL4 in tumor cells has been proved to show inhibition effects on ferroptosis. Also, knocking out ASCL4 in cells reduced the level of PUFAs and eliminated the inhibitory effect of RSL3 on GPX4, thereby inhibiting ferroptosis (Doll et al., 2017), implicating that ASCL4 played a key role in RSL-3-induced ferroptosis. Notably, ASCL4 has the ability to enrich cell membrane lipids, thereby increasing the susceptibility in cell to ferroptosis. Moreover, research showed that protein kinase PKCβII, a lipid peroxide sensing molecule, promoted and amplified ferroptosis-related lipid peroxidation by phosphorylating ASCL4 and accelerated the occurrence of ferroptosis (Zhang H. L. et al., 2022). Meanwhile, Cytochrome P450 oxidoreductase (POR) can also catalyze lipid peroxidation of PUFAs, thereby promoting ferroptosis (Zou et al., 2020).
Lipoxygenase (LOX) catalyzes the production of lipid peroxides, as proof, Arachidonate 12-Lipoxygenase (ALOX12), an isoform of the mammalian lipoxygenase family, can inhibit p53-regulated ferroptosis via inhibiting lipid synthesis function by specifically binding to SLC7A11, which abolishes the function of p53 suppressing tumor growth through ferroptosis (Chu et al., 2019). Also, over-expression of 5-LOX, p12-LOX, and 15-LOX-1 could enhance cellular susceptibility to ferroptosis by catalyzing lipid peroxidation, but these LOXs are likely to boost the initial stage of ferroptosis by promoting the formation of lipid hydroperoxides (Shah et al., 2018).
What’s more, cellular energy stress might also be closely bound up with ferroptosis. For instance, activation of AMP-activated protein kinase (AMPK) caused by energy stress can inhibit the synthesis of certain anabolism such as PUFAs, which further suppresses ferroptosis (Lee et al., 2020). However, another study showed opposite results, which pointed out that AMPK helped BECN1 inhibit the transport of cystine by SCL7A11, and ultimately provoked ferroptosis (Song et al., 2018). These two opposite findings and the differences in the mechanisms involved may not be sufficient to clarify the precise role of energy stress in ferroptosis, but both indicated the critical roles of energy stress in ferroptosis.
Intriguingly, p53 was firstly found to suppress tumor growth via the induction of ferroptosis instead of the canonical way like apoptosis (Jiang et al., 2015). They illustrated that p53 inhibited cystine uptake by repressing the expression of SLC7A11, which sensitized cells to ferroptosis. Significantly, Jiang et al. not only proposed a new mechanism of tumor suppression but also disclosed the latent relationship between p53 and ferroptosis. More importantly, this non-canonical p53 activities provides a brand-new perspective for other researches in many diseases. Also, they further uncover that p53 acetylation is crucial for p53-Mediated ferroptosis and tumor suppression (Wang et al., 2016). However, it is still a mystery that how p53 orchestrates the ferroptotic responses while it executes its mission. Later, it was confirmed that SAT1 (spermidine/spermine N1 -acetyltransferase 1) gene induced by p53 could trigger lipid peroxidation and sensitize cells to ferroptosis (Ou et al., 2016). Moreover, ALOX15 induced by SAT1 is the executioner of the occurrence of ferroptosis. Meanwhile, p53-ALOX12 axis as we mentioned before and BRD7 (bromodomain-containing protein 7)-P53-SLC25A28 (solute carrier family 25 member 28) axis is also a part of the regulation mechanism of p53-induced ferroptosis (Chu et al., 2019; Zhang et al., 2020).
However, p53 can play a double-edged sword role in mediating ferroptosis. p53 stabilization suppress ferroptosis in response to systemXc− inhibition, which is different from previously identified function of p53 as a positive regulator of ferroptosis (Tarangelo et al., 2018). At the same time, TP53 surprisingly limits erastin-induced ferroptosis via blocking dipeptidyl-peptidase-4 (DPP4) activity (Xie et al., 2017). Increasing studies about p53-related ferroptosis have been published (Liu and Gu, 2022), gradually becoming an indispensable puzzle piece of the whole quest to explore the mechanisms of ferroptosis.
To date, mounting evidence has showed that the mechanisms and functions of ferroptosis (Figure 2) and its complicated and diverse relations with mutiple diseases. Intriguingly, these mechanisms may shed light on therapeutic development in different diseases in the future.
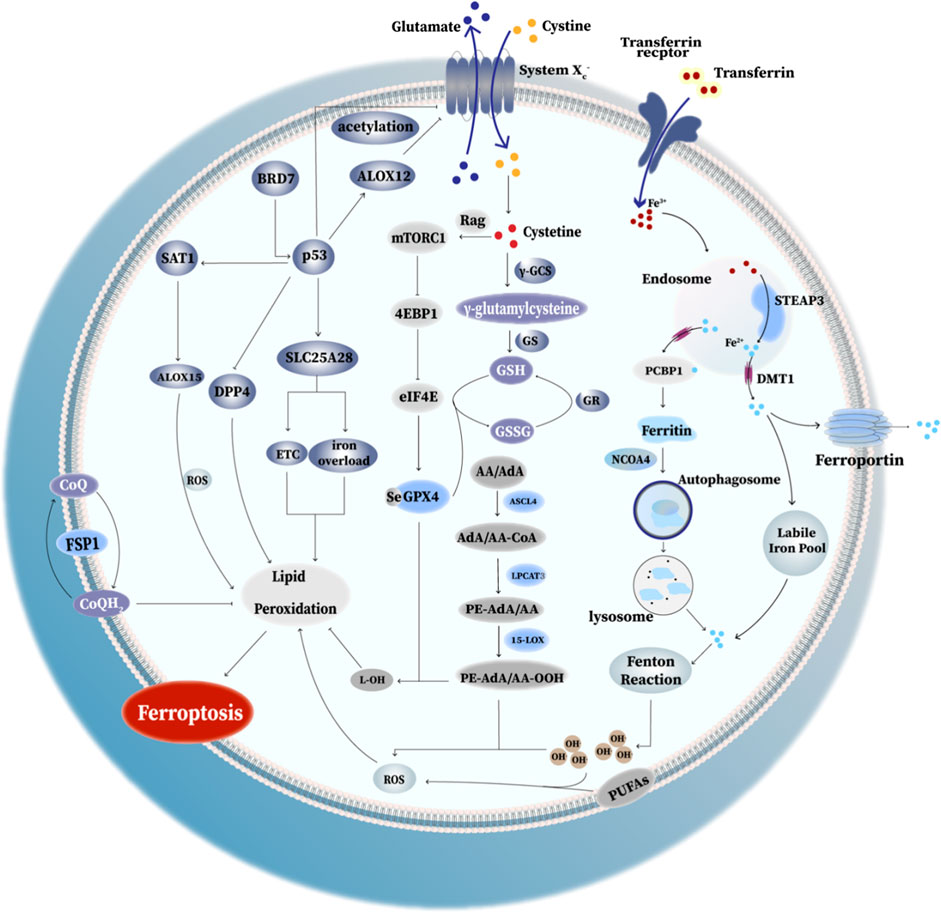
FIGURE 2. Mechanism of ferroptosis. I. Ferritinophagy-related ferroptosis: the degradation of ferritin via autophagy causes the iron dysregulation, which can lead to iron overload and trigger ferroptosis. II. GPX4 anti-ferroptosis way: GPX4 converts toxic lipid hydroperoxides to non-toxic lipid alcohols. SystemXc− exchanges cystine and glutamate in a ratio of 1:1. Cystine enters the cell and converts to cysteine for the synthesis of GSH, thus promoting GPX4 protein synthesis and enhancing its antioxidant function. III. Rag-mTORC1-4EBP signaling axis: mammalian target of rapamycin complex 1 (mTORC1) and promotes GPX4 protein synthesis at least partly through the Rag-mTORC1-4EBP signaling axis. (Rag(Ras-related GTPase): Rag GTPases play important roles in mTORC1 activation in response to amino-acid stimulation; eIF4E: eukaryotic initiate factor 4E; 4EBP(eIF4E binding protein): binding to eIF4E and thus decreased GPX4 level. IV. p53-mediated ferroptosis: (1) acetylation of p53 is crucial for p53-induced ferroptosis; (2) SAT1 activated by p53 induces ferroptosis via promoting ALOX15 expression;(3) the elevated BRD7 expression promote p53 mitochondrial translocation, leading to the interaction between mitochondrial p53 and SLC25A28, which could lead to the abnormal accumulation of redox-active iron and hyperfunction of electron transfer chain (ETC). (4) p53 promote ALOX12 binding with SLC7A11, eventually causing the inhibition of SystemXc− and triggering ferroptosis. (5) p53 inhibit erastin-induced ferroptosis via blocking the activity of DPP4. V. FSP1: FSP1 locates in the plasma membrane where it functions as an oxidoreductase that reduces coenzyme Q10 (CoQ). Reduced CoQ acts as a lipophilic radical-trapping antioxidant that halts the propagation of lipid peroxides, thus inhibiting ferroptosis. VI. PE-OOH as a ferroptotic death signal: ferroptosis involves a highly organized oxygenation center, wherein oxidation in endoplasmic-reticulum-associated compartments occurs on only one class of phospholipids (phosphatidyletha- nolamines (PEs)) and is specific toward two fatty acyls—arachidonoyl (AA) and adrenoyl (AdA). Moreover, several key enzymes like ASCL4, LPCAT3 and 15-LOX play a key role in proferroptotic system.
Innate immune cells, the first line of defense against infection, recognize pathogen-related molecular patterns (PAMPs) through pattern recognition receptors (PRRs) (i.e., Toll-like receptors (TLRs) and NOD-like receptors (NLRs)), thus initiating basic, simple, and rapid defensive responses to fight pathogen invasions (Thaiss et al., 2016). Damage-associated molecular patterns (DAMPs) (i.e., high mobility group protein B1 (HMGB1)) can be released from the host to initiate the corresponding inflammatory response against infections (Zindel and Kubes, 2020), such as regulating the inflammatory responses induced by ferroptosis (Wen et al., 2019). HMGB1 could increase through autophagy and bind to advanced glycosylation end-product specific receptor (AGER) to promote macrophage release tumor necrosis factor (TNF) in response to erastin, sorafenib, RSL3, and FIN56 (ferroptosis inducer 56) induced ferroptosis. In addition, knockdown of HMGB1 reduced erastin-induced ROS generation and suppressed TfR1 expression through the RAS-JNK/p38 pathway, thereby regulating ferroptosis (Ye et al., 2019).
TLRs are able to recognize PAMPs and DAMPs, and initiate corresponding immune responses, such as the immune clearance function of macrophages. Research believed that the efficiency of macrophages to clear apoptotic cells was better than that of ferroptotic cells (Kloditz and Fadeel, 2019). However, there is also research considered that SAPE-OOH (1-steaoryl-2-15-HpETE-sn-glycero-3-phosphatidyletha- nolamine), an oxidized phospholipid molecule on the membrane of ferroptotic cells, could be specifically recognized by TLR2, and thus provoked macrophages to phagocytose ferroptotic cells as immune clearance (Luo et al., 2021). Meanwhile, they found that macrophages in TLR2 knockout mice also phagocytosed ferroptotic cells well, suggesting that in addition to TLR2 recognition of SAPE-OOH, there were other pathways to maintain the phagocytic clearance of ferroptotic cells. Significantly, Gao’s team observed that ferroptosis inducers could induce ferroptosis in intracellular bacteria, thereby assisting macrophages to inhibit the growth of intracellular bacteria (Ma et al., 2022). In addition, they also dynamically monitored ferroptosis markers in macrophages after 12 and 24 h of bacteria infection and discovered that these markers increased during early infection, but dropped back to normal at the late stage. These results imply that ferroptosis has the potential to be served as a potential therapeutic target for intracellular bacteria at the early stage of infection.
What’s more, with the stimulation of pathogen infection, TLR recognition, and interferon signal regulation, macrophages usually differentiate into M1 macrophages equipped with pro-inflammatory effects, while M2 macrophages have the anti-inflammation and tissue repair effects (Murray, 2017). Also, a study showed that KRAS protein-encapsulated exosomes, secreted by pancreatic ductal adenocarcinoma mouse cancer cells that mediated by autophagy-dependent ferroptosis, could combine with AGER on the surface of macrophages, eventually promoting the differentiation of macrophages toward M2 (Dai et al., 2020). While macrophages perform functions such as endocytosis, the cells in aerobic environment yet can’t lead to ferroptosis in M1 macrophages, because there are a large number of inducible nitric oxide synthase (iNOS) in the activated M1 cells, which plays an antioxidant role similar to GPX4 (Kapralov et al., 2020). The high level of iNOS eventually makes M1 macrophages be resistant to ferroptosis, while conversely, M2 macrophages who lack iNOS is more sensitive to ferroptosis. At the same time, M1 macrophages can exert this protective function to protect neighboring cells from ferroptosis by regulating their resistance. Ferroptosis-related metabolism, such as glutathione metabolism, also has an impact on macrophage polarization. Adequate glutamine contributes to M2 differentiation, if glutamine is deficient, the number of M2 cells would decrease with the down-regulation of related genes, leading to the significant down-regulation of TCA (tricarboxylic acid) cycle transcriptional activity and autophagy disorders (Jha et al., 2015). Obviously, the relationship between ferroptosis, ferroptosis-related metabolism and macrophage polarization, and the ability to resist infection require more detailed researches.
One might wonder whether there are different correlations between macrophage and ferroptosis. Indeed, macrophages not only play a momentous role in anti-infection immunity but also gobble up aging or injured red blood cells and regulate iron homeostasis. Different polarization types of macrophages come with different functions in regulating iron metabolism (Cairo et al., 2011). It is widely known that iron contributes to the growth of invading bacteria. Under the stage of infection, M1 macrophages actively save iron in cells to limit the growth of extracellular bacteria by reducing the expression of ferroportin1 (FPN1), the only known iron exporter, and increasing extracellular iron uptake (Ganz, 2009). Consistently, iron deficiency can inhibit the growth of intracellular bacteria (Paradkar et al., 2008). For example, IFN-γ (Interferon-γ) limits the number of intracellular bacteria by reducing iron intake and increasing FPN1 expression (Nairz et al., 2008). In addition, contrary to the function of M1 macrophages, M2 macrophages tend to release iron to extracellular cells through FPN1 (Recalcati and Cairo, 2021).
Neutrophil is another important member of innate immune system, which counteracts pathogens via some mechanisms, such as phagocytosis and the production of NETs (neutrophil extracellular traps) (Castanheira and Kubes, 2019). Many published papers have told us the correlations between ferroptosis and neutrophil in cancer, but there are still limited information in infectious diseases. Proverbially, NETs are weapons used by neutrophils to fight against microbes (Brinkmann et al., 2004). However, in sepsis-associated acute lung injury (ALI), NETs contribute to the pathological progression through inducing ferroptosis in alveolar epithelial cells (Zhang H. et al., 2022).
A paper illustrating precise cell death pathways and signaling events orchestrate early inflammation after heart transplantation is thought-provoking (Li et al., 2019). They suggested that ferroptosis initiated neutrophil recruitment through TLR4/Trif signaling pathways, which reflected the possibilities of ferroptosis and relevant signaling events affecting innate immune function. Also, neutrophil-triggered ferroptosis occurred in tumor cell and showed positive feedback for tumor progression (Yee et al., 2020). These works collectively suggested that there might also be some critical roles for neutrophil-triggered ferroptosis against infections, which still need to be further confirmed.
Tremendous progress has been made to defend against pathogens invasion for a long time, a variety of vaccines have been developed to guard against virus infection or to reduce the symptoms of infection, but not all individuals can produce high titers of antibodies to successfully prevent disease after vaccination. Proverbially, T cell immunity is indispensable for defending against virus invasion. Matsushita et al. found that the immune function and virus clearance ability of GPX4-deficient T-cell mice were impaired after being infected with lymphocytic chorititis virus (Matsushita et al., 2015). Here, GPX4 deficiency caused abnormal lipid peroxidation, which led to T cell ferroptosis, thus weakening T cell immunological responses against virus infection. However, vitamin E, which has an antioxidant function, could restore the number of T cells in mouse and enhance their antiviral response. After that, Yao’s team reported that the occurrence of ferroptosis in follicular helper T(TFH) cells in mouse and human tonsils after ovalbumin injection, with higher levels of lipid ROS, MDA (malondialdehyde), and 4-HNE in TFH cell compared with non-TFH cells (Yao Y. et al., 2021). In addition, the same phenomenon was also found in human peripheral blood. Furthermore, combined with the result of Matsushita et al. (Matsushita et al., 2015), they listed some opinions: I. GPX4 selectively takes part in TFH cell immune responses, II. Enhanced TCR signal increases the sensitivity of TFH cell to ferroptosis, III. TFH cell immune response can be regulated by ferroptosis. Also, with the treatment of selenium, the GPX4 expression and the number of TFH cell were increased, and the efficiency of antibody responses in mice and teenagers after vaccination were elevated. In summary, these results reveal that “selenium-GPX4-ferroptosis” plays a crucial role in regulating TFH cell homeostasis, and can be targeted to enhance the immune response of the human body after vaccination, providing ideas and possibilities for preventing viral infection and enhancing the immune effects of the vaccine (Figure 3A). What’s more, antigen-specific memory CD4+ T cells can persist and confer rapid and efficient protection from microbial reinfection. mTORC2 (Mammalian rapamycin complex 1) is critical for long-term persistence of virus-specific memory CD4+ T cells, which ablation will induce aberrant mitochondrial ROS accumulation and ensue ferroptosis-causative lipid peroxidation (Wang et al., 2022).
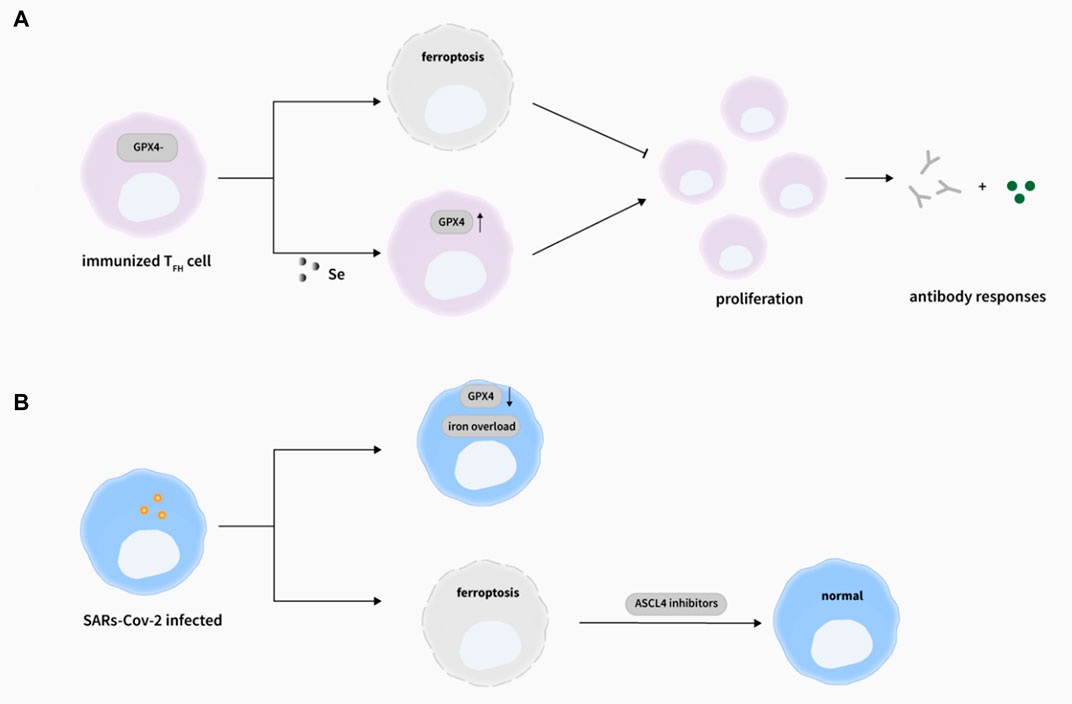
FIGURE 3. Ferroptosis and viral infections. (A) The deletion of GPX4 in T cells selectively abrogated TFH cells functions via ferroptosis in immunized mice. Importantly, selenium supplementation cloud enhance GPX4 expression in T cells, promoting TFH cell proliferation and boosting antibody responses in immunized mice. (Yao Y. et al., 2021). (B) During SARs-Cov-2 infection, the Gpx4 mRNA level was decreased (Wang et al., 2021a) and iron overload was observed (Zhou C. et al., 2020), and the induction of ferroptosis by SARS-Cov-2 could be rescued by ASCL4 inhibitors (Kung et al., 2022)
Meanwhile, the expression of p53 in U251 cells infected by Newcastle-disease-virus (NDV) was up-regulated, which led to the decrease of SLC7A11 and GPX4 protein levels, while knockdown of p53 reduced the ROS level (Kan et al., 2021). And NCOA4-related ferritinophagy was triggered by NDV, which could increase the level of intracellular ROS and ferrous iron, and then trigger ferroptosis, indicating that NDV could kill tumor cells in a ferroptotic way by inducing ferritinophagy or inhibiting SLC7A11.
What’s more, lipid metabolism is involved in viral replication by regulation of the formation, assembly, and release of replicative organelles. ASCL4, a key factor involved in lipid metabolism and ferroptosis, greatly promoted the replication of some enteroviruses and coronaviruses (Kung et al., 2022). In ASCL4 knockout cells, the viral titers of enteroviruses such as CV-A6, coronaviruses such as Cov-229E, influenza virus (IAV), and Zika virus were lower than those in the normal cells. In addition, enterovirus CV-A6 and coronaviruses such as CoV-229E, CoV-NL63, CoV-OC43, CoV-HKU1, and SARS-CoV-2 could induce ferroptosis in cells by regulating ASCL4 functions. Consistently, the inhibition of ASCL4 can repress virus replication effectively by inhibiting ferroptosis, which implicates the significance of ASCL4-induced ferroptosis in viral replication.
Previous studies have implied that ferroptosis might participate in the pathogenesis of COVID-19 (Figure 3B), as proof, iron overload was relatively associated with the COVID-19 infection (Zhou C. et al., 2020). Increased hepcidin may raise the risk of ferroptosis occurrence by iron accumulation in cellular, which certainly suggest the therapeutic potential of iron chelators. Ferroptosis inhibitors includes iron chelators, like the famous one DFO (deferoxamine), has been approved by the FDA for the treatment of iron overload (Meyer, 2006). Meanwhile, the induction of ferroptosis by SARS-Cov-2 could be rescued by ASCL4 inhibitors (Kung et al., 2022). Also, SARS-Cov-2 showed inhibitory effects on the expression of the selenoprotein GPX4 mRNA level in vivo(Wang et al., 2021a). Undoubtedly, targeting ferroptosis will pose some new perspectives for preventing and controlling viral infections. At the same time, the influence of SARS-CoV-2 and possible immunosuppressive drugs can cause the temporal inhibition of the humanbody immunological function, thus resulting in active TB caused by reactivation or infection of M. tuberculosis (Yang and Lu, 2020). Consequently, the co-infection of different pathogens requires more attentions in the future.
Previous studies have focused on the relationship between iron and Mycobacterium tuberculosis (Mtb), although excess iron could be extremely toxic to Mtb (Imlay et al., 1988; Byrd, 1997; Rodriguez, 2006), sufficient iron to some extent is beneficial for the growth and propagation of Mtb in the host (Gangaidzo et al., 2001; Schaible and Kaufmann, 2004). High concentrations of vitamin C could inhibit the growth of MDR (multiple drug resistance)-Mtb in the medium, leading to an increase of iron concentration in the bacteria with more ROS generated through the Fenton reaction, which in turn promoted lipid synthesis, caused DNA damage, changed the redox homeostasis, and ultimately inhibited the growth of MDR-Mtb (Vilcheze et al., 2013). However, Amaral et al. firstly discovered that Mtb-induced macrophage necrosis had many characteristics of ferroptosis, accompanied by iron overload, lipid peroxidation, and GPX4 downregulation in H37Rv-infected macrophages in both vivo and in vitro, which greatly increased the number of cell death (Amaral et al., 2019) (Figure 4A). And importantly, a reduction in granulomatous inflammation was observed in tissue sections with the treatment of ferrotstain-1 (Fer-1), and bacterial load in the lung of mice was also decreased, suggesting that inhibition of ferroptosis might contribute to the host cell resistance to Mtb to a certain extent. Meanwhile, BACH-1, a transcription factor, can disrupt iron homeostasis and redox by inhibiting GSH synthesis or the expression of LIP homeostasis-related genes, thereby promoting ferroptosis (Nishizawa et al., 2020). Combined with this, they observed that the number of intracellular bacteria and lung necrosis areas were reduced after the knockout of BACH-1 (Amaral et al., 2020). Consistently, the deficiency of BACH-1 in iron overload and Mtb-infected macrophages could resist the impairment of iron overload and ferroptosis (Aberman et al., 2021). Commonly, the above studies suggest that BACH-1-related ferroptosis may be a potential target for the effective treatment of tuberculosis. But interestingly, ferroptosis can also help macrophages kill intracellular bacteria such as s.aureas (Ma et al., 2022) (Figure 4B), which is contrary to the above studies to some degree, indicating that the mechanism of ferroptosis in macrophage resistance to intracellular bacteria still needs to be further explored.
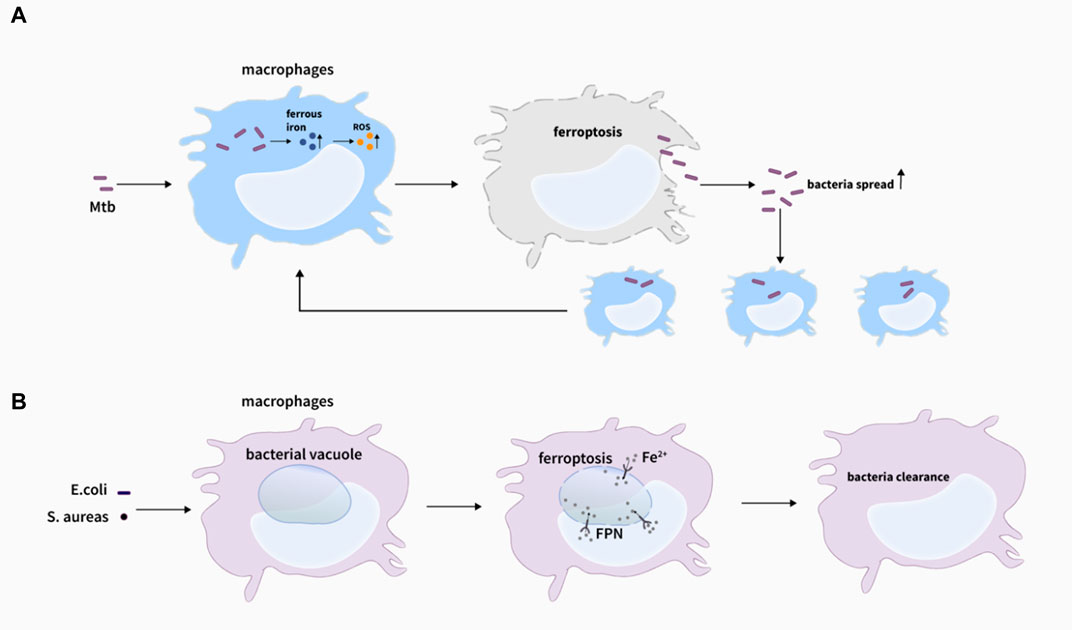
FIGURE 4. Ferroptosis and bacteria infections. (A) Lipid peroxidation induces plasma membrane destabilization, leading to ferroptosis-mediated cell death with M. tuberculosis infection, and ferroptosis drives macrophages necrosis and allows M. tuberculosis to thrive and spread, which promote the infection (Amaral et al., 2019). (B) Ferrous iron could be delivered to the intracellular bacterial vacuole via inward FPN transportation, eventually inducing ferroptosis-like death of bacteria, which assists killing bacteria of macrophages (Ma et al., 2022).
Commonly, 15-lipoxygenases (15LOXes) can induce ferroptosis by catalyzing lipid peroxidation (Kagan et al., 2017). In general, lipoxygenase can’t be produced in bacteria, but surprisingly, 15LOXes can be expressed in P. aeruginosa (Vance et al., 2004). Moreover, Dar et al. firstly proposed that 15-LOXes in P. aeruginosa could catalyze lipid peroxidation in human bronchial epithelial cells, thereby inducing ferroptosis and further spreading to surrounding cells and tissues (Dar et al., 2018). Oxidative stress caused by P. aeruginosa is one of the most important causes of cystic fibrosis in the human lung as P. aeruginosa can elevate ROS in cystic fibrosis airway epithelial cells. This could lead to lipid peroxidation and ultimately trigger ferroptosis, while Fer-1 and other ferroptosis inhibitors could improve the pathological condition of the diseased airway (Ousingsawat et al., 2021), implicating that the inhibition of ferroptosis was of great significance in the treatment of P. aeruginosa.
In addition, some studies have also revealed the important role of ferroptosis in the development of sepsis. For example, the inhibition of lipopolysaccharide-induced ferritinophagy-related ferroptosis could improve cardiac function and survival prognosis in mice with lipopolysaccharide-induced cardiac injury (Li et al., 2020). At the same time, itaconate, a metabolite produced during inflammatory macrophage activation, could inhibit ferroptosis by up-regulating Nrf2 (Nuclear Factor erythroid 2-Related Factor 2) levels, thereby alleviating the symptoms of acute lung injury (ALI) and the presence of macrophages in the lung tissue infiltration (He et al., 2022).
Cryptococcal meningitis (CM) is one of the most common clinical fungal infections, especially in AIDS patients with immunodeficiency. Notably, iron accumulation and lipid peroxidation occurred in the brains of CM patients (Xu X. et al., 2021), and ferritin levels in the cerebrospinal fluid were significantly elevated (Campbell et al., 1986). Consistently, iron overload exacerbates the condition of CM patients (Barluzzi et al., 2002). Meantime, after Cryptococcus infection of alveolar macrophages, significant intracellular lipid peroxidation occurred, and a large number of high-density lipid droplets were observed under electron microscopy (Gross et al., 2000). In addition, Cryptococcus infection of activated macrophages in the cerebrospinal fluid of AIDS patients could promote the release of pro-inflammatory cytokines and chemokines, which further promoted the expression of DMT1 and FPN1, and indirectly increased the extracellular iron uptake level of cells, resulting in an intracellular iron surplus (Urrutia et al., 2013; Jarvis et al., 2015). To date, no studies have directly pointed out the role of iron excess and lipid peroxidation in cryptococcus infection. However, considering that excess iron can induce lipid peroxidation, it can be assumed that there may be potential connections among iron accumulation, ROS accumulation, ferroptosis, and cryptococcus infection, which requires further explorations.
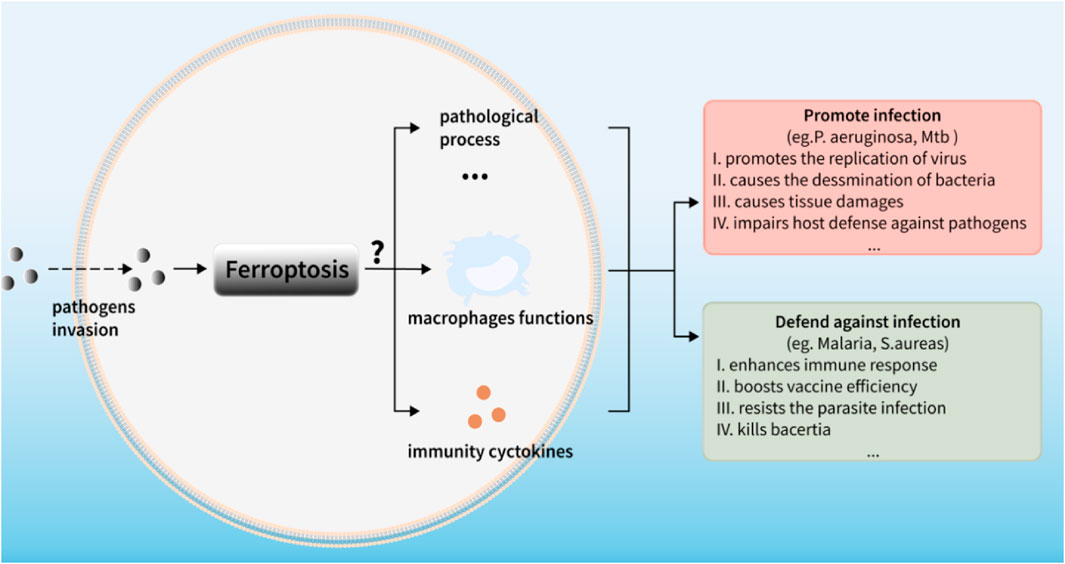
FIGURE 5. Ferroptosis: a mixed blessing for infectious diseases.
Iron is of great significance both to the invading of bacteria and fungi in human body. Patients with clinical liver transplantation are prone to fungal infection, which seriously threatens the patients. A retrospective study suggested that iron overload was an independent risk factor for fungal infection after liver transplantation, and controlling iron content before transplantation could reduce the risk of fungal infection (Alexander et al., 2006). Also, unlike ferroptosis regulated by Fenton response mediated by LOXs or Fe2+, NOXs can regulate ferroptosis induced by exogenous Fe2+ in Aspergillus flavus (Yao L. et al., 2021). However, there are few published researches that uncover the links between ferroptosis and fungal infections, such as Candida, Aspergillus, and Saccharomyces infections. Obviously, more works are needed to done to further explore this terra incognita.
Infection of GPX4-deficient mice with Leishmania could lead to a reduction in the number of CD4+ T cells, which contributes to the maintenance of Leishmania in vivo (Matsushita et al., 2015). Malaria caused by Plasmodium infection in humans is prevalent all over the world, especially in Africa. Hence, the elimination of malaria is particularly important for the public health (Cotter et al., 2013). Once infected with malaria parasites, the host-owned pattern recognition receptors recognize the PAMPs and DAMPs of the parasite and activate immune cells to secrete inflammatory factors, causing oxidative stress to generate a large amount of ROS, which may eventually lead to cell death (Cotter et al., 2013). What’s more, Singh et al. reported that the mutation of amino acid 47 in human TP53 caused defects in p53-induced ferroptosis, resulting in massive intracellular iron accumulation in macrophages, which would lead to M2-polarization of macrophages, eventually helping resist Plasmodium infection but also promoting bacterial infections such as Listeria (Singh et al., 2020). In addition, malaria parasites can obtain sufficient iron from hemoglobin by attacking red blood cells, but can also lead to excess ferrous iron, exacerbating the Fenton reaction (Sena-Dos-Santos et al., 2021). Obviously, Plasmodium infection is closely related to iron homeostasis and ROS, and intracellular iron metabolism is important for resistance to Plasmodium. Notably, the SLC7A11-GPX4 pathway helps resist the infection of liver Plasmodium (Kain et al., 2020). And p53 indirectly suppresses the activity of GPX4 via inhibiting SLC7A11, causing lipid peroxidation to kill Plasmodium. Apparently, Plasmodium is sensitive to ferroptosis, and induction of ferroptosis can protect against Plasmodium infection to a certain extent.
Nowadays, glucocorticoids are used as clinical routine to treat COVID-19, such as dexamethasone. However, this kind of treatment comes along with dose-dependent side effects, which might limit the therapy efficiency in clinical. Mässenhausen et al. firstly discovered that dexamethasone sensitized ferroptosis by GSH depletion (Von Mässenhausen et al., 2022). Dialectically, it is crucial to understand the mechanisms of the side effects brought by high dose of glucocorticoids. And importantly, how can they minimize the ferroptosis damage of side effects and optimize the clinical treatment effects of dexamethasone? Meanwhile, hallmarks of COVID-19 are also associated with iron overload and problematic ROS scavenging. Due to the research we mentioned above (Meyer, 2006; Wang et al., 2021a; Kung et al., 2022), we hypothesized that ferroptosis inhibitors had potential to be a kind of adjuvant therapy, for example, could ameliorate the side effects of ferroptosis damage from the use of glucocorticoids, and impaire viral replication via iron chelation. Suggestively, multiple interactions between SARS-Cov-2 and ferroptosis may provide diverse perspectives in the exploration of strategy for COVID-19 treatment.
For the recent years, the therapeutic potential of ferroptosis was widely discussed in cancer or other diseases, excepted for infectious diseases. Although the concept of ferroptosis was officially proposed until 2012, but its relevant mechanisms had existed in many diseases for a long time. Table 1 and 2 may give us some hints or clues for the exploration of therapeutic methods. These inhibitors or inducers are involved in many basic biological processes, such as iron metabolism, redox reaction, and protein synthesis. And these processes always appear to be dysregulated in diseased conditions. And it is reasonable to seek therapy targets via three aspects: iron overload, LPO and GPX4 antioxidant system. Obviously, it is of worth to discuss the possibility of therapy for these drugs in infectious diseases, especially when close interrelationships between ferroptosis and the diseases are showed. Crucially, what can iron accumulation, lipid peroxidation damage and disrupted cellular redox contribute to the treatment of infectious diseases? The understanding of these works would further benefit the use of ferroptosis for therapies.
Additionally, a summary table of the potential therapeutic target or treatment of ferroptosis has been made up (Table 3). Notably, among these possible options discussed here are a combination of hypothesis stemming from in vivo or in vitro researches.
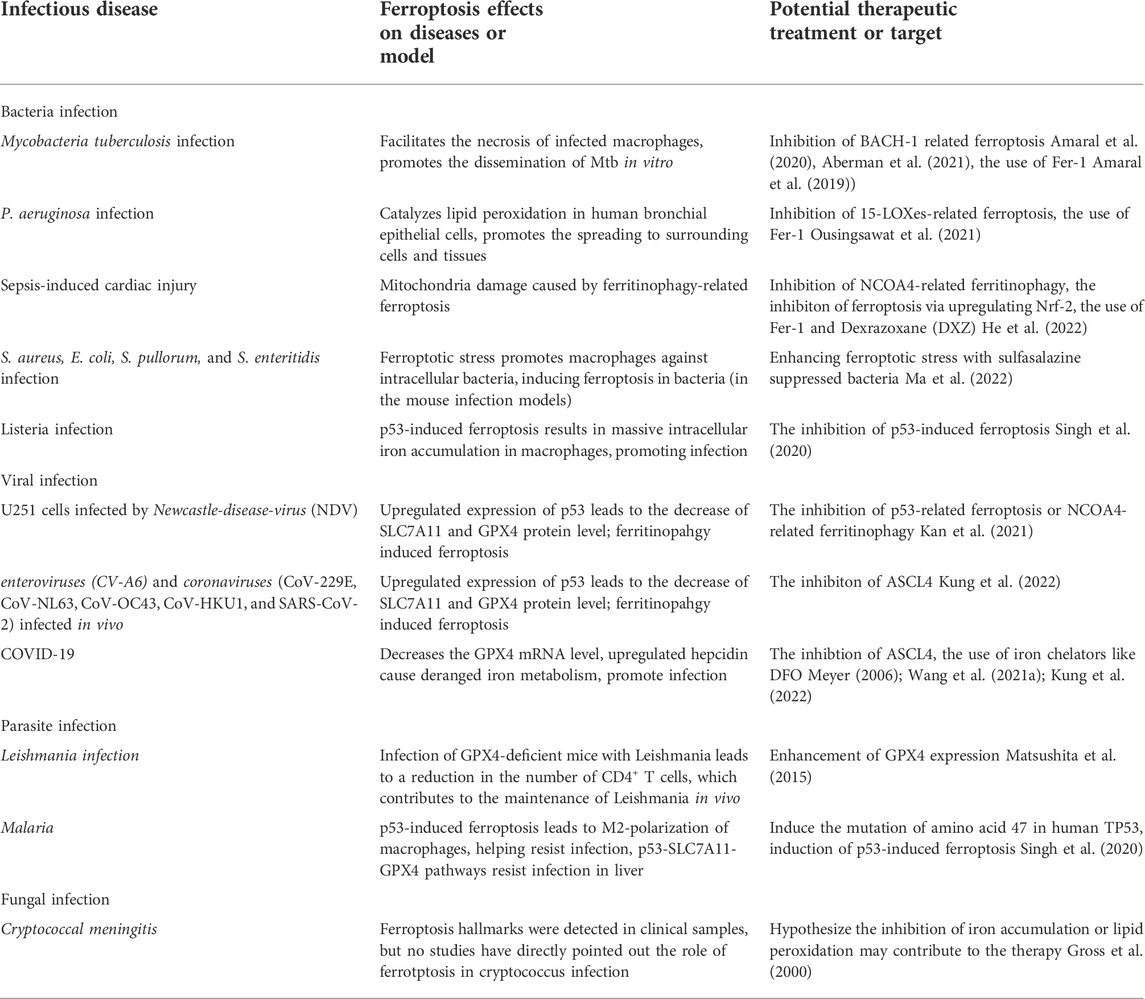
TABLE 3. Summary of the relationships between ferroptosis and infectious diseases and potential therapeutic target or treatment.
For many years, soaring progresses have been made in delineating the mechanisms that modulate ferroptosis and its occurrence in different diseases (i.e., Parkinson’s, kidney diseases, cancers), however, only a visble crack can be seen in the door of infectious diseases. Therefore, bridging ferroptosis with infectious diseases is particularly on the map. As we reviewed above, multiple functions of ferroptosis in infectious diseases can be generally considered as a double-edged sword (Figure 5). On the one hand, ferroptosis poses threats to immune cells like perturbing the normal immunity performance against infections in macrophages. On the other hand, some kinds of “pro-immunity” ways of ferroptosis, such as its occurrence in pathogens inside defensing against the infection, causing the release of DAMPs as PAMPs and activating the immune responses, can facilitate the host control of infections. For example, some intracellular pathogens evade the killing of macrophages by immune escape while ferroptosis promotes macrophages to kill intracellular bacteria, indicating that ferroptosis may be used as a means of treating intracellular bacteria. However, this effect might be inversed to show impaired killing effects of host cells to the intracellular bacteria due to the dysfunction of host cell ferroptosis. Such fascinating facts undoubtedly point out the engaging connections between ferroptosis and host immunity, especially in host cells, such as macrophages. Obviously, more thorough clarifications about the modulations and functions of ferroptosis in infectious diseases are needed to be done.
Furthermore, relationships between ferroptosis-related pathways, metabolism, and host immunity against infections are also striking enough in the discovery tour. As proof, the “selenium-GPX4-ferroptosis” pathway provides a novel insight into the development and enhancement of vaccination. Meanwhile, iron homeostasis, one of the key factors of ferroptosis, to some degree is a tiger that host riding with. Excess iron-induced ferroptosis causes the damage, but on the bright side, NCOA4-mediated ferrtinophagy can possibly be utilized by macrophages to kill intracellular bacteria via inducing ferroptosis. Also, iron homeostasis is maintained by various proteins and transcription factors, such as iron chaperone proteins PCBP1, a negative regulator of ferroptosis. Although the connections between PCBP1 and infectious diseases haven’t been elucidated, these kinds of functional molecules may provide more inspiration for the future work in this field. Simultaneously, lipid metabolism involved in infectious diseases, such as 15-LOXes in P. aeruginosa catalyzed lipid peroxidation in human bronchial epithelial cells to induce ferroptosis with increased damage, is also worth to be mentioned. This means that ferroptosis-driven lipid metabolism might be a target for the development of infectious diseases. Similarly, it is reported that ROS, which induces ferroptosis, mainly comes from mitochondria (Li et al., 2021). Given the importance of mitochondrial as a multifunctional unit in managing basic physiological events and diverse host responses against infections, it makes sense to extrapolate that modulation mitochondrial functions might be a way to control ferroptosis pathways upon infections. In summary, compelling evidences indicate the complex connections among physiological and pathological metabolism, ferroptosis, and pathogen infections, which might be a new entry points of therapy in infectious diseases.
Many researchers have proposed their perspectives about the role of ferroptosis or illustrated the mechanisms involved in infectious diseases. For example, While Amaral et al. suggested the inhibition of ferroptosis might ameliorate the Mtb infection, they also considered that simultaneously lessen tissue damage while reducing pathogen burden and dissemination is an attractive aspect of this strategy (Amaral et al., 2019). And Yao et al. proposed that regulation of ferroptosis as a strategy to boost humoral immunity in infection and following vaccination (Yao Y. et al., 2021). Summarily, these diverse perspectives elicit public an open mindedness in the exploration of ferroptosis for infectious diseases treatment.
However, plenty of questions remain unknown, such as whether it is possible to develop ferroptosis as target into a new therapeutic method for infectious diseases or not. To what extent can ferroptosis assist the host killing of pathogens, and how can we minimize the damage of ferroptosis to host physiological functions? How does ferroptosis mediate the pathological progresses during the infections? How do ferroptosis-related regulations affect the innate and adaptive immunity against the infections? What are the similarities and complementarities between ferroptosis and cell death such as autophagy and necrosis in infectious diseases? Undoubtedly, tons of puzzles sitting right there need us to solve in the future. Delving into and thoroughly settling the mechanisms of ferroptosis in host upon infections is a promising, yet albeit challenging strategy to help conquer these diseases. And we believe that with the increasing understanding of the relations and underlying mechanisms between ferroptosis and infectious diseases, the regulation of ferroptosis might be developed into novel therapeutic strategy, which would further benefit the control of the threatening infectious diseases.
LX drafted the manuscript, HH, SF, BZ, JW and JZ helped to revise the manuscript, JP and JX were responsible for leading this work and revising the manuscript.
This work was supported by National Natural Science Foundation of China (81870016), Natural Science Foundation of Guangdong Province (2022A1515011223 and 2022A1515010525), Project of Educational Commission of Guangdong Province of China (2021KTSCX038), Guangdong Basic and Applied Basic Research Foundation (2020A1515010283 and 2021B1515140068), Open Research Fund of Songshan Lake Materials Laboratory (2021SLABFN10), Medical Scientific Research Foundation of Guangdong Province (A2018434), Science and Technology Project of Dongguan (20211800904782, 202110571025), Key project of Science Foundation of Guangdong Medical university (GDMUZ2019005), Funds for Ph.D. researchers of Guangdong Medical University in 2021 (4SG21236G) and Hospital Fund for The First Dongguan Affiliated Hospital of Guangdong Medical University (PU2022002) and Discipline Construction Project of Guangdong Medical University (4SG21229GDGFY01).
We tender our apologies to those authors whose deserving research was not cited in this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aberman, K., Fisher, L., Oland, S., Sher, A., and Amaral, E. P. (2021). The transcription factor Bach1 plays an important role in regulating macrophage ferroptosis upon iron supplementation. J. Immunol. 206, 111.
Alexander, J., Limaye, A. P., Ko, C. W., Bronner, M. P., and Kowdley, K. V. (2006). Association of hepatic iron overload with invasive fungal infection in liver transplant recipients. Liver Transpl. 12, 1799–1804. doi:10.1002/lt.20827
Amaral, E. P., Costa, D. L., Namasivayam, S., Riteau, N., Kamenyeva, O., Mittereder, L., et al. (2019). Die another way: Ferroptosis drives tuberculosis pathology. J. Exp. Med. 216, 556–570. doi:10.1084/jem.20181776
Amaral, E. P., Namasivayam, S., Costa, D. L., Fisher, L., Bomfim, C. C. B., Andrade, B. B., et al. (2020). The transcription factor BACH1 promotes tissue damage and host susceptibility in Mycobacterium tuberculosis infection by reducing expression of Gpx4, a major negative regulator of ferroptosis. J. Immunol. 204, 227.
Anderson, G. J., and Vulpe, C. D. (2009). Mammalian iron transport. Cell. Mol. Life Sci. 66, 3241–3261. doi:10.1007/s00018-009-0051-1
Barluzzi, R., Saleppico, S., Nocentini, A., Boelaert, J. R., Neglia, R., Bistoni, F., et al. (2002). Iron overload exacerbates experimental meningoencephalitis by Cryptococcus neoformans. J. Neuroimmunol. 132, 140–146. doi:10.1016/s0165-5728(02)00324-7
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi:10.1038/s41586-019-1705-2
Brigelius-Flohe, R., and Maiorino, M. (2013). Glutathione peroxidases. Biochim. Biophys. Acta 1830, 3289–3303. doi:10.1016/j.bbagen.2012.11.020
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. doi:10.1126/science.1092385
Byrd, T. F. (1997). Tumor necrosis factor alpha (TNFalpha) promotes growth of virulent Mycobacterium tuberculosis in human monocytes iron-mediated growth suppression is correlated with decreased release of TNFalpha from iron-treated infected monocytes. J. Clin. Invest. 99, 2518–2529. doi:10.1172/JCI119436
Cairo, G., Recalcati, S., Mantovani, A., and Locati, M. (2011). Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 32, 241–247. doi:10.1016/j.it.2011.03.007
Campbell, D. R., Skikne, B. S., and Cook, J. D. (1986). Cerebrospinal fluid ferritin levels in screening for meningism. Arch. Neurol. 43, 1257–1260. doi:10.1001/archneur.1986.00520120041014
Castanheira, F. V. S., and Kubes, P. (2019). Neutrophils and NETs in modulating acute and chronic inflammation. Blood 133, 2178–2185. doi:10.1182/blood-2018-11-844530
Chen, D., Chu, B., Yang, X., Liu, Z., Jin, Y., Kon, N., et al. (2021). iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 12, 3644. doi:10.1038/s41467-021-23902-6
Chen, Y., and Chen, Y. (2021). Clinical challenges with hypervirulent Klebsiella pneumoniae (hvKP) in China. J. Transl. Int. Med. 9, 71–75. doi:10.2478/jtim-2021-0004
Cheng, Z., and Li, Y. (2007). What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: An update. Chem. Rev. 107, 748–766. doi:10.1021/cr040077w
Chu, B., Kon, N., Chen, D., Li, T., Liu, T., Jiang, L., et al. (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591. doi:10.1038/s41556-019-0305-6
Cotter, C., Sturrock, H. J. W., Hsiang, M. S., Liu, J., Phillips, A. A., Hwang, J., et al. (2013). The changing epidemiology of malaria elimination: New strategies for new challenges. Lancet 382, 900–911. doi:10.1016/S0140-6736(13)60310-4
Dai, E., Han, L., Liu, J., Xie, Y., Kroemer, G., Klionsky, D. J., et al. (2020). Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 16, 2069–2083. doi:10.1080/15548627.2020.1714209
Dar, H. H., Tyurina, Y. Y., Mikulska-Ruminska, K., Shrivastava, I., Ting, H. C., Tyurin, V. A., et al. (2018). Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Invest. 128, 4639–4653. doi:10.1172/JCI99490
Delehouze, C., Comte, A., Leon-Icaza, S. A., Cougoule, C., Hauteville, M., Goekjian, P., et al. (2022). Nigratine as dual inhibitor of necroptosis and ferroptosis regulated cell death. Sci. Rep. 12, 5118. doi:10.1038/s41598-022-09019-w
Ding, W., Ma, Y., Ma, C., Malone, D. C., Ma, A., Tang, W., et al. (2021). The lifetime cost estimation of human papillomavirus-related diseases in China: A modeling study. J. Transl. Int. Med. 9, 200–211. doi:10.2478/jtim-2021-0039
Dixon, Scott j., Lemberg, Kathryn m., Lamprecht, Michael r., Skouta, R., Zaitsev, Eleina m., Gleason, Caroline e., et al. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi:10.1016/j.cell.2012.03.042
Dixon, S. J., Patel, D. N., Welsch, M., Skouta, R., Lee, E. D., Hayano, M., et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. doi:10.7554/eLife.02523
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. doi:10.1038/nchembio.2239
Eaton, J. K., Furst, L., Ruberto, R. A., Moosmayer, D., Hilpmann, A., Ryan, M. J., et al. (2020). Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 16, 497–506. doi:10.1038/s41589-020-0501-5
Fang, S., Yu, X., Ding, H., Han, J., and Feng, J. (2018). Effects of intracellular iron overload on cell death and identification of potent cell death inhibitors. Biochem. Biophys. Res. Commun. 503, 297–303. doi:10.1016/j.bbrc.2018.06.019
Fuhrmann, D. C., Mondorf, A., Beifuss, J., Jung, M., and Brune, B. (2020). Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 36, 101670. doi:10.1016/j.redox.2020.101670
Gangaidzo, I. T., Moyo, V. M., Mvundura, E., Aggrey, G., Murphree, N. L., Khumalo, H., et al. (2001). Association of pulmonary tuberculosis with increased dietary iron. J. Infect. Dis. 184, 936–939. doi:10.1086/323203
Ganz, T. (2009). Iron in innate immunity: Starve the invaders. Curr. Opin. Immunol. 21, 63–67. doi:10.1016/j.coi.2009.01.011
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308. doi:10.1016/j.molcel.2015.06.011
Gaschler, M. M., Andia, A. A., Liu, H., Csuka, J. M., Hurlocker, B., Vaiana, C. A., et al. (2018). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 14, 507–515. doi:10.1038/s41589-018-0031-6
Gross, N. T., Hultenby, K., Mengarelli, S., Camner, P., and Jarstrand, C. (2000). Lipid peroxidation by alveolar macrophages challenged with Cryptococcus neoformans, Candida albicans or Aspergillus fumigatus. Med. Mycol. 38, 443–449. doi:10.1080/mmy.38.6.443.449
Guo, J., Xu, B., Han, Q., Zhou, H., Xia, Y., Gong, C., et al. (2018). Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res. Treat. 50, 445–460. doi:10.4143/crt.2016.572
Halliwell, B., and Chirico, S. (1993). Lipid peroxidation: Its mechanism, measurement, and significance. Am. J. Clin. Nutr. 57, 715S–724S; discussion 724S-725S. ; discussion 724S-725S. doi:10.1093/ajcn/57.5.715S
He, R., Liu, B., Xiong, R., Geng, B., Meng, H., Lin, W., et al. (2022). Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. 8, 43. doi:10.1038/s41420-021-00807-3
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi:10.1080/15548627.2016.1187366
Hu, Q., Zhang, Y., Lou, H., Ou, Z., Liu, J., Duan, W., et al. (2021). GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 12, 706. doi:10.1038/s41419-021-04008-9
Imlay, J. A., Chin, S. M., and Linn, S. (1988). Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240, 640–642. doi:10.1126/science.2834821
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2018). Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422. doi:10.1016/j.cell.2017.11.048
Jarvis, J. N., Meintjes, G., Bicanic, T., Buffa, V., Hogan, L., Mo, S., et al. (2015). Cerebrospinal fluid cytokine profiles predict risk of early mortality and immune reconstitution inflammatory syndrome in HIV-associated cryptococcal meningitis. PLoS Pathog. 11, e1004754. doi:10.1371/journal.ppat.1004754
Jha, A. K., Huang, S. C., Sergushichev, A., Lampropoulou, V., Ivanova, Y., Loginicheva, E., et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. doi:10.1016/j.immuni.2015.02.005
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. doi:10.1038/nature14344
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P., Doll, S., Croix, C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi:10.1038/nchembio.2238
Kain, H. S., Glennon, E. K. K., Vijayan, K., Arang, N., Douglass, A. N., Fortin, C. L., et al. (2020). Liver stage malaria infection is controlled by host regulators of lipid peroxidation. Cell Death Differ. 27, 44–54. doi:10.1038/s41418-019-0338-1
Kalra, A., and Chawla, N. (2020). Quandary surrounding COVID19 transmission. J. Transl. Int. Med. 8, 274–275. doi:10.2478/jtim-2020-0041
Kan, X., Yin, Y., Song, C., Tan, L., Qiu, X., Liao, Y., et al. (2021). Newcastle-disease-virus-induced ferroptosis through nutrient deprivation and ferritinophagy in tumor cells. iScience 24, 102837. doi:10.1016/j.isci.2021.102837
Kang, Y. P., Mockabee-Macias, A., Jiang, C., Falzone, A., Prieto-Farigua, N., Stone, E., et al. (2021). Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 33, 174–189.e7. doi:10.1016/j.cmet.2020.12.007
Kapralov, A. A., Yang, Q., Dar, H. H., Tyurina, Y. Y., Anthonymuthu, T. S., Kim, R., et al. (2020). Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 16, 278–290. doi:10.1038/s41589-019-0462-8
Karuppagounder, S. S., Alin, L., Chen, Y., Brand, D., Bourassa, M. W., Dietrich, K., et al. (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 84, 854–872. doi:10.1002/ana.25356
Kloditz, K., and Fadeel, B. (2019). Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 5, 65. doi:10.1038/s41420-019-0146-x
Koppula, P., Zhuang, L., and Gan, B. (2021). Cystine transporter slc7a11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12, 599–620. doi:10.1007/s13238-020-00789-5
Kung, Y. A., Chiang, H. J., Li, M. L., Gong, Y. N., Chiu, H. P., Hung, C. T., et al. (2022). Acyl-coenzyme A synthetase long-chain family member 4 is involved in viral replication organelle formation and facilitates virus replication via ferroptosis. mBio 13, e0271721. doi:10.1128/mbio.02717-21
Lachowicz, J. I., Pichiri, G., Piludu, M., Fais, S., Orru, G., Congiu, T., et al. (2022). Thymosin β4 is an endogenous iron chelator and molecular switcher of ferroptosis. Int. J. Mol. Sci. 23, 551. doi:10.3390/ijms23010551
Lee, H., Zandkarimi, F., Zhang, Y., Meena, J. K., Kim, J., Zhuang, L., et al. (2020). Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22, 225–234. doi:10.1038/s41556-020-0461-8
Lee, J., You, J. H., and Roh, J. L. (2022). Poly(rC)-binding protein 1 represses ferritinophagy-mediated ferroptosis in head and neck cancer. Redox Biol. 51, 102276. doi:10.1016/j.redox.2022.102276
Li, C., Zhang, Y., Liu, J., Kang, R., Klionsky, D. J., and Tang, D. (2021). Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 17, 948–960. doi:10.1080/15548627.2020.1739447
Li, N., Wang, W., Zhou, H., Wu, Q., Duan, M., Liu, C., et al. (2020). Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 160, 303–318. doi:10.1016/j.freeradbiomed.2020.08.009
Li, W., Feng, G., Gauthier, J. M., Lokshina, I., Higashikubo, R., Evans, S., et al. (2019). Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Invest. 129, 2293–2304. doi:10.1172/JCI126428
Liu, Y., and Gu, W. (2022). p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 29, 895–910. doi:10.1038/s41418-022-00943-y
Lorincz, T., Jemnitz, K., Kardon, T., Mandl, J., and Szarka, A. (2015). Ferroptosis is involved in acetaminophen induced cell death. Pathol. Oncol. Res. 21, 1115–1121. doi:10.1007/s12253-015-9946-3
Luo, X., Gong, H. B., Gao, H. Y., Wu, Y. P., Sun, W. Y., Li, Z. Q., et al. (2021). Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 28, 1971–1989. doi:10.1038/s41418-020-00719-2
Ma, R., Fang, L., Chen, L., Wang, X., Jiang, J., and Gao, L. (2022). Ferroptotic stress promotes macrophages against intracellular bacteria. Theranostics 12, 2266–2289. doi:10.7150/thno.66663
Ma, S., Dielschneider, R. F., Henson, E. S., Xiao, W., Choquette, T. R., Blankstein, A. R., et al. (2017). Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One 12, e0182921. doi:10.1371/journal.pone.0182921
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W., and Kimmelman, A. C. (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. doi:10.1038/nature13148
Matsushita, M., Freigang, S., Schneider, C., Conrad, M., Bornkamm, G. W., and Kopf, M. (2015). T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212, 555–568. doi:10.1084/jem.20140857
Meyer, D. (2006). Iron chelation as therapy for HIV and Mycobacterium tuberculosis co-infection under conditions of iron overload. Curr. Pharm. Des. 12, 1943–1947. doi:10.2174/138161206777442164
Minotti, G., and Aust, S. D. (1989). The role of iron in oxygen radical mediated lipid peroxidation. Chem. Biol. Interact. 71, 1–19. doi:10.1016/0009-2797(89)90087-2
Moosmayer, D., Hilpmann, A., Hoffmann, J., Schnirch, L., Zimmermann, K., Badock, V., et al. (2021). Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallogr. D. Struct. Biol. 77, 237–248. doi:10.1107/S2059798320016125
Murray, P. J. (2017). Macrophage polarization. Annu. Rev. Physiol. 79, 541–566. doi:10.1146/annurev-physiol-022516-034339
Nairz, M., Fritsche, G., Brunner, P., Talasz, H., Hantke, K., and Weiss, G. (2008). Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur. J. Immunol. 38, 1923–1936. doi:10.1002/eji.200738056
Nishizawa, H., Matsumoto, M., Shindo, T., Saigusa, D., Kato, H., Suzuki, K., et al. (2020). Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J. Biol. Chem. 295, 69–82. doi:10.1074/jbc.RA119.009548
Ou, Y., Wang, S.-J., Li, D., Chu, B., and Gu, W. (2016). Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. 113 (44), E6806–E6812. doi:10.1073/pnas.1607152113
Ousingsawat, J., Schreiber, R., Gulbins, E., Kamler, M., and Kunzelmann, K. (2021). P. aeruginosa induced lipid peroxidation causes ferroptotic cell death in airways. Cell. Physiol. biochem. 55, 590–604. doi:10.33594/000000437
Paradkar, P. N., De Domenico, I., Durchfort, N., Zohn, I., Kaplan, J., and Ward, D. M. (2008). Iron depletion limits intracellular bacterial growth in macrophages. Blood 112, 866–874. doi:10.1182/blood-2007-12-126854
Park, E., and Chung, S. W. (2019). ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 10, 822. doi:10.1038/s41419-019-2064-5
Protchenko, O., Baratz, E., Jadhav, S., Li, F., Shakoury-Elizeh, M., Gavrilova, O., et al. (2021). Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology 73, 1176–1193. doi:10.1002/hep.31328
Recalcati, S., and Cairo, G. (2021). Macrophages and iron: A special relationship. Biomedicines 9, 1585. doi:10.3390/biomedicines9111585
Rodriguez, G. M. (2006). Control of iron metabolism in Mycobacterium tuberculosis. Trends Microbiol. 14, 320–327. doi:10.1016/j.tim.2006.05.006
Schaible, U. E., and Kaufmann, S. H. (2004). Iron and microbial infection. Nat. Rev. Microbiol. 2, 946–953. doi:10.1038/nrmicro1046
Sena-Dos-Santos, C., Braga-Da-Silva, C., Marques, D., Azevedo Dos Santos Pinheiro, J., Ribeiro-Dos-Santos, A., and Cavalcante, G. C. (2021). Unraveling cell death pathways during malaria infection: What do we know so far? Cells 10, 479. doi:10.3390/cells10020479
Shah, R., Shchepinov, M. S., and Pratt, D. A. (2018). Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 4, 387–396. doi:10.1021/acscentsci.7b00589
Shi, H., Bencze, K. Z., Stemmler, T. L., and Philpott, C. C. (2008). A cytosolic iron chaperone that delivers iron to ferritin. Science 320, 1207–1210. doi:10.1126/science.1157643
Shimada, K., Skouta, R., Kaplan, A., Yang, W. S., Hayano, M., Dixon, S. J., et al. (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503. doi:10.1038/nchembio.2079
Singh, K. S., Leu, J. I., Barnoud, T., Vonteddu, P., Gnanapradeepan, K., Lin, C., et al. (2020). Author Correction: African-centric TP53 variant increases iron accumulation and bacterial pathogenesis but improves response to malaria toxin. Nat. Commun. 11, 1541. doi:10.1038/s41467-020-15366-x
Skouta, R., Dixon, S. J., Wang, J., Dunn, D. E., Orman, M., Shimada, K., et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 136, 4551–4556. doi:10.1021/ja411006a
Song, X., Zhu, S., Chen, P., Hou, W., Wen, Q., Liu, J., et al. (2018). AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system xc(-) activity. Curr. Biol. 28, 2388–2399. e2385. doi:10.1016/j.cub.2018.05.094
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi:10.1016/j.cell.2017.09.021
Stockwell, B. R., Jiang, X., and Gu, W. (2020). Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 30, 478–490. doi:10.1016/j.tcb.2020.02.009
Sun, W. Y., Tyurin, V. A., Mikulska-Ruminska, K., Shrivastava, I. H., Anthonymuthu, T. S., Zhai, Y. J., et al. (2021). Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 17, 465–476. doi:10.1038/s41589-020-00734-x
Tarangelo, A., Magtanong, L., Bieging-Rolett, K. T., Li, Y., Ye, J., Attardi, L. D., et al. (2018). p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 22, 569–575. doi:10.1016/j.celrep.2017.12.077
Thaiss, C. A., Zmora, N., Levy, M., and Elinav, E. (2016). The microbiome and innate immunity. Nature 535, 65–74. doi:10.1038/nature18847
Theil, E. C. (2004). Iron, ferritin, and nutrition. Annu. Rev. Nutr. 24, 327–343. doi:10.1146/annurev.nutr.24.012003.132212
Urrutia, P., Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. (2013). Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549. doi:10.1111/jnc.12244
Vance, R. E., Hong, S., Gronert, K., Serhan, C. N., and Mekalanos, J. J. (2004). The opportunistic pathogen Pseudomonas aeruginosa carries a secretable arachidonate 15-lipoxygenase. Proc. Natl. Acad. Sci. U. S. A. 101, 2135–2139. doi:10.1073/pnas.0307308101
Vilcheze, C., Hartman, T., Weinrick, B., and Jacobs, W. R. (2013). Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nat. Commun. 4, 1881. doi:10.1038/ncomms2898
Von Mässenhausen, A., Zamora Gonzalez, N., Maremonti, F., Belavgeni, A., Tonnus, W., Meyer, C., et al. (2022). Dexamethasone sensitizes to ferroptosis by glucocorticoid receptor-induced dipeptidase-1 expression and glutathione depletion. Sci. Adv. 8, eabl8920. doi:10.1126/sciadv.abl8920
Wang, S. J., Li, D., Ou, Y., Jiang, L., Chen, Y., Zhao, Y., et al. (2016). Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 17, 366–373. doi:10.1016/j.celrep.2016.09.022
Wang, Y., Huang, J., Sun, Y., Stubbs, D., He, J., Li, W., et al. (2021a). SARS-CoV-2 suppresses mRNA expression of selenoproteins associated with ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food Chem. Toxicol. 153, 112286. doi:10.1016/j.fct.2021.112286
Wang, Y., Quan, F., Cao, Q., Lin, Y., Yue, C., Bi, R., et al. (2021b). Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 28, 231–243. doi:10.1016/j.jare.2020.07.007
Wang, Y., Tian, Q., Hao, Y., Yao, W., Lu, J., Chen, C., et al. (2022). The kinase complex mTORC2 promotes the longevity of virus-specific memory CD4(+) T cells by preventing ferroptosis. Nat. Immunol. 23, 303–317. doi:10.1038/s41590-021-01090-1
Wen, Q., Liu, J., Kang, R., Zhou, B., and Tang, D. (2019). The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 510, 278–283. doi:10.1016/j.bbrc.2019.01.090
Wu, C. Y., Yang, Y. H., Lin, Y. S., Chang, G. H., Tsai, M. S., Hsu, C. M., et al. (2021a). Dihydroisotanshinone I induced ferroptosis and apoptosis of lung cancer cells. Biomed. Pharmacother. 139, 111585. doi:10.1016/j.biopha.2021.111585
Wu, W., Geng, Z., Bai, H., Liu, T., and Zhang, B. (2021b). Ammonium ferric citrate induced ferroptosis in non-small-cell lung carcinoma through the inhibition of GPX4-GSS/GSR-GGT axis activity. Int. J. Med. Sci. 18, 1899–1909. doi:10.7150/ijms.54860
Xie, Y., Zhu, S., Song, X., Sun, X., Fan, Y., Liu, J., et al. (2017). The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 20, 1692–1704. doi:10.1016/j.celrep.2017.07.055
Xu, C., Xiao, Z., Wang, J., Lai, H., Zhang, T., Guan, Z., et al. (2021a). Discovery of a potent glutathione peroxidase 4 inhibitor as a selective ferroptosis inducer. J. Med. Chem. 64, 13312–13326. doi:10.1021/acs.jmedchem.1c00569
Xu, X., Lin, D., Tu, S., Gao, S., Shao, A., and Sheng, J. (2021b). Is ferroptosis a future direction in exploring cryptococcal meningitis? Front. Immunol. 12, 598601. doi:10.3389/fimmu.2021.598601
Yang, H., and Lu, S. (2020). COVID-19 and tuberculosis. J. Transl. Int. Med. 8, 59–65. doi:10.2478/jtim-2020-0010
Yang, W. S., Sriramaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi:10.1016/j.cell.2013.12.010
Yao, L., Ban, F., Peng, S., Xu, D., Li, H., Mo, H., et al. (2021a). Exogenous iron induces NADPH oxidases-dependent ferroptosis in the conidia of Aspergillus flavus. J. Agric. Food Chem. 69, 13608–13617. doi:10.1021/acs.jafc.1c04411
Yao, Y., Chen, Z., Zhang, H., Chen, C., Zeng, M., Yunis, J., et al. (2021b). Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat. Immunol. 22, 1127–1139. doi:10.1038/s41590-021-00996-0
Ye, F., Chai, W., Xie, M., Yang, M., Yu, Y., Cao, L., et al. (2019). HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am. J. Cancer Res. 9, 730–739.
Yee, P. P., Wei, Y., Kim, S. Y., Lu, T., Chih, S. Y., Lawson, C., et al. (2020). Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 11, 5424. doi:10.1038/s41467-020-19193-y
Yu, Y., Jiang, L., Wang, H., Shen, Z., Cheng, Q., Zhang, P., et al. (2020). Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 136, 726–739. doi:10.1182/blood.2019002907
Zhang, H. L., Hu, B. X., Li, Z. L., Du, T., Shan, J. L., Ye, Z. P., et al. (2022b). PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 24, 88–98. doi:10.1038/s41556-021-00818-3
Zhang, H., Liu, J., Zhou, Y., Qu, M., Wang, Y., Guo, K., et al. (2022a). Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int. J. Biol. Sci. 18, 3337–3357. doi:10.7150/ijbs.69141
Zhang, Y., Swanda, R. V., Nie, L., Liu, X., Wang, C., Lee, H., et al. (2021). mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 12, 1589. doi:10.1038/s41467-021-21841-w
Zhang, Z., Guo, M., Shen, M., Kong, D., Zhang, F., Shao, J., et al. (2020). The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 36, 101619. doi:10.1016/j.redox.2020.101619
Zhou, B., Liu, J., Kang, R., Klionsky, D. J., Kroemer, G., and Tang, D. (2020a). Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 66, 89–100. doi:10.1016/j.semcancer.2019.03.002
Zhou, C., Chen, Y., Ji, Y., He, X., and Xue, D. (2020b). Increased serum levels of hepcidin and ferritin are associated with severity of COVID-19. Med. Sci. Monit. 26, e926178. doi:10.12659/MSM.926178
Zilka, O., Shah, R., Li, B., Friedmann Angeli, J. P., Griesser, M., Conrad, M., et al. (2017). On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 3, 232–243. doi:10.1021/acscentsci.7b00028
Zindel, J., and Kubes, P. (2020). DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu. Rev. Pathol. 15, 493–518. doi:10.1146/annurev-pathmechdis-012419-032847
Keywords: ferroptosis, mechanism, host immunity, infectious diseases, therapy strategy
Citation: Xiao L, Huang H, Fan S, Zheng B, Wu J, Zhang J, Pi J and Xu J-F (2022) Ferroptosis: A mixed blessing for infectious diseases. Front. Pharmacol. 13:992734. doi: 10.3389/fphar.2022.992734
Received: 12 July 2022; Accepted: 04 August 2022;
Published: 07 September 2022.
Edited by:
Shibiao Wan, St. Jude Children’s Research Hospital, United StatesReviewed by:
Si-Yuan Song, Baylor College of Medicine, United StatesCopyright © 2022 Xiao, Huang, Fan, Zheng, Wu, Zhang, Pi and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiang Pi, amlhbmdwaUBnZG11LmVkdS5jbg==; Jun-Fa Xu, eHVqdW5mYUBnZG11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.