 Sukanya Bhunia
Sukanya Bhunia Nagesh Kolishetti
Nagesh Kolishetti Adriana Yndart Arias
Adriana Yndart Arias Arti Vashist
Arti Vashist Madhavan Nair
Madhavan Nair
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 25 October 2022
Sec. Neuropharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.989717
This article is part of the Research TopicNeuropharmacology of Neurodegenerative diseases: Exploration of Pathways that involved in Neurodegenerative disordersView all 4 articles
Despite the significant advances in neurology, the cure for neurodegenerative conditions remains a formidable task to date. Among various factors arising from the complex etiology of neurodegenerative diseases, neuroinflammation and oxidative stress play a major role in pathogenesis. To this end, some phytocannabinoids isolated from Cannabis sativa (widely known as marijuana) have attracted significant attention as potential neurotherapeutics. The profound effect of ∆9-tetrahydrocannabinol (THC), the major psychoactive component of cannabis, has led to the discovery of the endocannabinoid system as a molecular target in the central nervous system (CNS). Cannabidiol (CBD), the major non-psychoactive component of cannabis, has recently emerged as a potential prototype for neuroprotective drug development due to its antioxidant and anti-inflammatory properties and its well-tolerated pharmacological behavior. This review briefly discusses the role of inflammation and oxidative stress in neurodegeneration and demonstrates the neuroprotective effect of cannabidiol, highlighting its general mechanism of action and disease-specific pathways in Parkinson’s disease (PD) and Alzheimer’s disease (AD). Furthermore, we have summarized the preclinical and clinical findings on the therapeutic promise of CBD in PD and AD, shed light on the importance of determining its therapeutic window, and provide insights into identifying promising new research directions.
With the increase in life expectancy worldwide, the prevalence of neurodegenerative diseases (NDs) has become a concern due to their socio-economic impact. NDs are characterized by progressive loss of structure and function of nerve cells in the central and/or peripheral nervous systems (Katsnelson et al., 2016). The structural loss of neurons and synapses including demyelination and/or neuronal death causes gradual loss of motor and/or cognitive skills, eventually leading to functional loss. Aging is found to be a major risk for NDs. Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the most common NDs, while the other forms include amyotrophic lateral sclerosis (ALS), Huntington’s disease, spinal muscular atrophy, motor neuron diseases, prion diseases, and spinocerebellar ataxia. However, irrespective of their differences in pathophysiology, which are determined by observing the aggregated characteristic protein via neuropathological evaluation at autopsy (Dugger and Dickson 2017), chronic neuroinflammation is the fundamental pathological mechanism driving both onsets and the progression of the NDs (Kinney et al., 2018).
Oxidative stress (OS) arises from redox imbalance in the cell when the cellular antioxidant systems fail to counter the excess of reactive species. Excess accumulation of reactive species causes structural alteration in protein, DNA, and cell membrane which leads to neuronal death. Persistent inflammation also plays a major role in neurodegeneration (Glass et al., 2010). The insoluble protein aggregates and/or oxidized lipid molecules cause microglial activation, a key factor in the pathogenesis of NDs. Activated microglia release inflammatory mediators that again activate astrocytes to produce reactive species and inflammatory cytokines, which in turn further activate microglia, and thereby a feed-forward loop is established favoring chronic oxidative stress and persistent neuroinflammation. Neuronal cells, especially those present in the CNS, are more susceptible to oxidative stress due to their high metabolic rate (Salim, 2017). In aging with poor self-renewal capacity of neuronal cells and a gradual decline in activity of the antioxidant enzymes to counterbalance the excessive reactive species, neuronal death becomes prevalent (Hou et al., 2019). In addition, in aging with a retarded rate of autophagy, clearance of dysfunctional mitochondria or aggregated protein becomes difficult, further favoring the environment for chronic oxidative stress and neuroinflammation. Though the gold standards for treating such diseases, for example, l-DOPA for Parkinson’s disease and small molecule acetylcholinesterase inhibitors for AD, provide symptomatic relief (Rizek et al., 2016; Marucci et al., 2021), the discovery of druggable targets and drugs that can mitigate disease progression still remains an unmet challenge.
For the past few decades, cannabinoids, secondary plant metabolites extracted from Cannabis sativa, especially ∆9-tetrahydrocannabinol (THC) and cannabidiol (CBD), have garnered significant attention (Cooray et al., 2020). Despite the anecdotal history of cannabis’s medicinal use since the medieval era, systematic medicinal exploration started in 1970 after its widespread use as a “recreational drug.” The profound behavioral effect of cannabis including euphoria and analgesia led to investigating the potential receptors in humans and rodents, leading to the discovery of the endocannabinoid system (ECS), which regulates several physiological processes such as cognition and analgesia. ECS consists of two primary receptors of the G-protein-coupled receptor (GPR) family: cannabinoid receptors 1 and 2 (CB1R and CB2R), two primary ligands known as endocannabinoids (namely, anandamide (AEA) and 2-arachidonoylglycerol (2AG)) and enzymes (namely, fatty acid amide hydrolase (FAAH) and monoglyceride lipase (MAGL)) that control the release and degradation of endocannabinoids (Alexander, 2016). CB1R is predominantly found in the nerve terminals of the CNS and in peripheral neural and other tissue (e.g., gastrointestinal tract) with lower density. The two endocannabinoids, AEA and 2AG (Figure 1A), act as retrograde synaptic messengers. In response to a transient intracellular Ca2+ elevation, endocannabinoids are released from post synapses in the synaptic space, travel to the presynaptic cleft, and activate the CB1 receptor (CB1R). Activation of CB1R blocks the release of neurotransmitters such as GABA (an inhibitory neurotransmitter) and glutamate (an excitatory neurotransmitter). AEA and 2AG are finally degraded by FAAH and MAGL, respectively. On the other hand, CB2R is mostly found in immune-associated cells and acts as an immunomodulator that can inhibit immune cell migration.
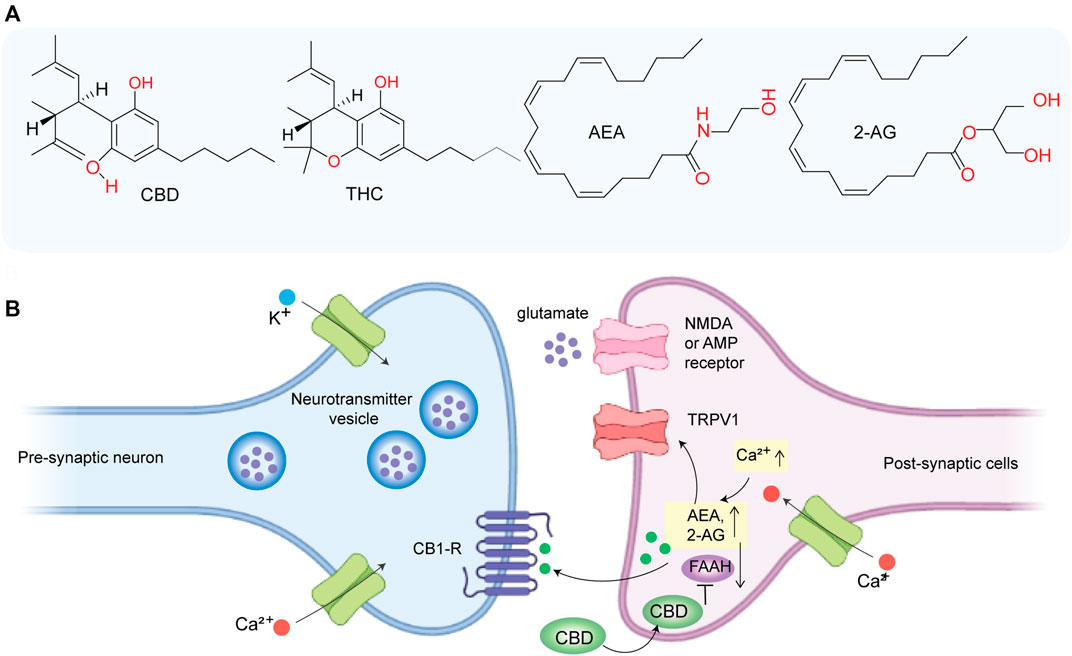
FIGURE 1. (A) Chemical structure of CBD, THC, anandamide (AEA), and 2-arachidonoylglycerol (2AG). (B) CBD indirectly activates CB1 receptor and TRPV1 by inhibiting FAAH and thereby promoting the accumulation of AEA at synapse which may play a role in inhibiting glutamate-based excitotoxicity.
Some cannabinoids can mimic the effect of endocannabinoids and can regulate nerve impulses in certain pathways (Piomelli, 2003). For instance, THC acts as a partial agonist of the CB1 receptor (Ki = ∼10 nM) and exerts its psychoactive effect by activating the CB1 receptor (Kimura et al., 2019). Previously, major research on ECS has focused on THC, the major psychoactive component of W extract. However, recently, CBD has attracted major attention due to its neuroprotective activity (Cassano et al., 2020), its non-psychoactive nature, and well-tolerated pharmacological behavior (Bergamaschi et al., 2011). Although CBD has a low affinity for CB1 (Ki = ∼2,000 nM) (Kimura et al., 2019), it indirectly activates CB1 by inhibiting FAAH which aids in the accumulation of AEA (Figure 1B) and prevents glutamate-induced excitotoxicity on neurons (Peres et al., 2018). CBD acts as an inverse agonist of CB2R. Importantly, much of the recent evidence suggested that CBD has high anti-inflammatory and antioxidant activities which play a major role in its neuroprotective behavior, and it is mediated via multiple molecular targets other than CB1R and CB2R. In this review article, we have briefly discussed the role of oxidative stress and neuroinflammation in neurodegeneration and provided an overview of the general mechanism of neuroprotective behavior of CBD along with molecular targets. In addition, we have summarized clinical and preclinical findings on the therapeutic promise of CBD in PD and AD.
Persistent neuroinflammation and chronic oxidative stress play a crucial role in the pathogenesis of neurodegenerative diseases. Microglia cells, the immune cells of the brain, act as a “double-edged sword” (Hickman et al., 2018). It normally acts as basic housekeeping, which maintains regular neuronal function, tissue repair, and brain homeostasis. However, in the presence of persistent inflammatory stimulus, microglia shift to their activated forms, which release inflammatory mediators and reactive species that can directly cause neuronal death and activate astrocytes and neurons to express more inflammatory mediators and RS. Once the feed-forward loops of inflammatory responses are established, the normal resolution mechanisms are overwhelmed, leading to neurodegeneration (Glass et al., 2010).
The inflammatory response represents a highly regulated cascade of events containing three major types of agents: 1) inducers and sensors, 2) transduction systems, and 3) amplifiers and effectors (Glass et al., 2010). In the initiation phase, inflammation stimuli are recognized by some specific cell surface receptors of microglia. For instance, pathogen-associated molecular patterns from an infectious agent (e.g., lipopolysaccharide (LPS) from Gram-negative bacteria) are recognized by pattern recognition receptors (PRRs) as “stranger” signals, while components released from necrotic cells are recognized as “danger signals.” ATP released following traumatic injury and neuronal death is sensed by purinergic receptors on microglia and astrocytes, while the “scavenger receptors” respond to oxidized lipids, proteins, and apoptotic cells. In the second phase, activation of such cell surface-sensing receptors leads to the activation of some signal transduction pathways (e.g., IkB kinase and mitogen-activated protein kinase, i.e., MAPK), which further control the activities of multiple transcription factors such as nuclear factor-κB (NF-κB) and activator protein 1 (AP-1). These transcription factors work together to regulate the expression of cytokines (e.g., tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β)) which propagate the inflammation (amplifier) and chemokines (e.g., COX-2) that recruit additional immune cells (effector cells) depending on the target cell. In addition, reactive species (e.g., ROS and inducible nitric oxide synthase (iNOS)) are produced as an anti-microbial defense mechanism of cells (Glass et al., 2010).
Amyloid-β (Aβ) aggregates and neurofibrillary tangles of tau protein (NFT), the proteins involved in AD, create a stress response in neurons. These aggregates also interact with microglia and astrocytes via cell surface receptors (such as TLRs and the receptor for advanced glycation endproducts (RAGE)) to produce several factors, such as pro-inflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6), chemokines (e.g., COX-2), ROS, NO, prostaglandins (e.g., PGE2), and caspase that promote the death of cholinergic neurons (Figure 2A). Genetic, environmental, and age-related factors contribute to the initiation of inflammation (Migliore and Coppedè, 2009; Glass et al., 2010). For instance, the expression of ApoE4, the strongest genetic factor of AD (Liu et al., 2013), causes defects in the microglial clearance of Aβ. ApoE is a lipid transporter protein that participates in the clearance of Aβ by microglia via a low-density lipoprotein receptor (LDL-R) or LDL-R-related protein 1 (LRP1) receptor, and the binding of ApoE to LDL-R or LRP1 suppresses expression of the stress-responsive kinase JNK pathway. However, the efficiency of ApoE in clearing Aβ is isoform-specific. The ApoE4/Aβ complex is the least preferred compared with the other isoforms of ApoE for microglial uptake by the LRP1 receptor, which results in the accumulation of Aβ with the high expression of ApoE4 (Pocivavsek et al., 2009). Other than genetic factors, environmental factors such as obesity due to a high-calorie diet also induce a low-grade but persistent inflammation. Activated microglia release TNF-α and IL-1β that activate astrocytes, which in turn activate microglia (Liu et al., 2020). Furthermore, the cholinergic neuron itself has NF-κB sites in the promoters of amyloid precursor protein (APP), presenilin, and BACE1 (beta-secretase 1) (Sastre et al., 2008) which produces more Aβ upon inflammatory stimuli and further amplifies microglia-mediated inflammation. Thus, neurons and glia act in combination to amplify the production of neurotoxic factors in AD pathology. Similarly, in PD, the process of aggregation of α-synuclein, the major factor involved in PD, from monomers to fibrils via oligomeric intermediate is considered to be neurotoxic, causing neuronal death (Danzer et al., 2007). Neuronal death itself and subsequent extracellularly released aggregated α-synuclein can induce microglial activation via the purinergic receptor (Figure 2B). Aggregated α-synuclein is sensed and internalized by microglia via cell surface gangliosides (Park et al., 2009), resulting in microglial activation which produces pro-inflammatory cytokines (e.g., TNF-α and IL-1β) and mediators of oxidative stress including ROS and NO (Reynolds et al., 2008). Dopaminergic neurons in the substantia nigra are vulnerable to oxidative stress (Kastner et al., 1992) due to their enhanced intracellular oxidative processes. ROS production via the activation of NADPH oxidase upon internalization of α-synuclein is a crucial factor for microglia activation in PD pathology.
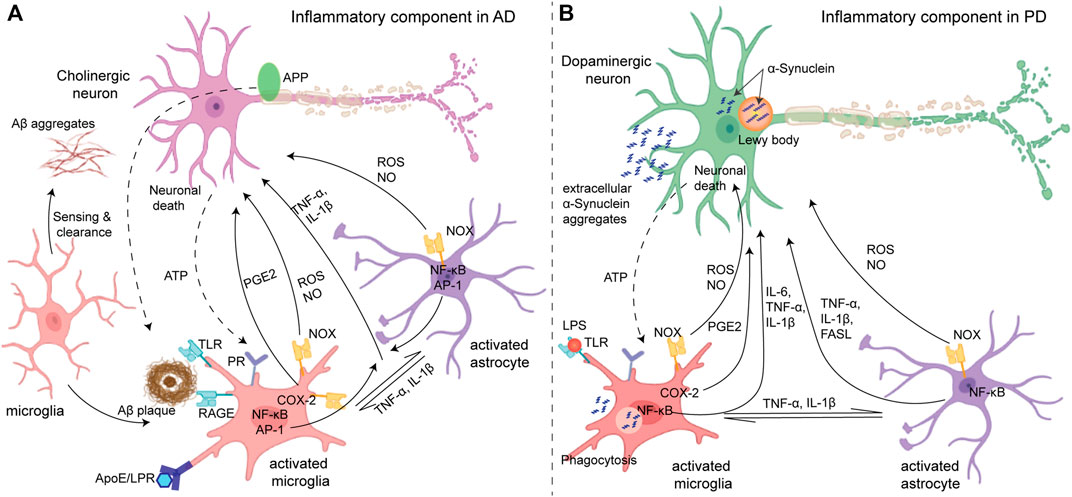
FIGURE 2. (A) Inflammatory components in Alzheimer’s disease (left): initially, microglia act as neuroprotectants that can sense and migrate to the Aβ aggregates. The Aβ aggregates interact with different cell surface receptors (TLR and RAGE) of microglia which induce activation of some transcription factors such as NF-kB and AP-1 and produce reactive species (ROS and NO) and cytokines (microglial activation). However, under sustained inflammation, activated microglia exhibit compromised Aβ clearance ability due to impaired ApoE/LRP uptake, while the production of ROS and inflammatory mediators remains unaltered which directly causes apoptosis/necrosis in neurons. Activated microglia can also activate astrocytes by releasing cytokines which in return can further activate the microglia. In addition, ATP released from neuronal death activates microglia via the purinergic receptors (PR), and collectively, a feed-forward loop is established. (B) Parkinson’s disease (right): intracellular deposition of α-synuclein known as Lewy bodies caused stress in dopaminergic neurons, while the oligomeric α-synuclein in the extracellular space causes activation of glia cells (microglia and astrocytes) in a similar manner to amplify the neuroinflammation and oxidative stress causing the death of dopaminergic neurons. Adapted with permission from (Glass et al., 2010).
OS plays a major role in neurodegeneration, especially in the CNS, which is vulnerable to OS due to its high metabolic rate and limited cell renewal capacity. In CNS inflammation, NADPH oxidases (NOX), NOS, and dysfunctional mitochondria are major sources of reactive species (Fischer and Maier, 2015). ROS can directly damage neurons, facilitate protein aggregates via impaired proteosomal degradation, and initiate inflammation via lipid peroxidation which causes microglial activation. In aging with a reduced rate of autophagy, the removal of dysfunctional mitochondria and aggregated proteins becomes compromised, contributing to the risk of age-related neurodegeneration. In AD, oxidative stress activates some kinases (e.g., JNK, p38 MAPK, and GSK-3β) which promote the generation of Aβ (Tamagno et al., 2005) and NFT (Lovell et al., 2004). Sustained activation of kinases also causes neuronal apoptosis. In PD, α-synuclein can directly impair the mitochondrial complex I in dopaminergic neurons which are vulnerable to oxidative stress due to their high rate of internal dopamine metabolism (Devi et al., 2008). Thus, prevention of oxidative stress and neuroinflammation may hold promise in mitigating the progression of neurodegenerative disease.
Phytocannabinoids have garnered significant attention lately due to their inherent neuroactive and strong antioxidant properties. Approval of Sativex, a combination of THC and CBD (1:1), in treating spasticity and/or neuropathic pain in multiple sclerosis (MS) in 2005, became a breakthrough in cannabis research. CBD, beyond reducing the adverse effect of THC when used in combination (Russo and Guy 2006), can individually exhibit a broad spectrum of therapeutic benefits. CBD alone has been FDA-approved for treating treatment-resistant pediatric epilepsy in 2018 (Devinsky et al., 2016). In addition, its anticonvulsant, antiemetic, and sleep-inducing properties have been explored to treat epilepsy and sleep disorders, as well as for treating psychiatric disorders such as schizophrenia, anxiety, and depression (Fernández-Ruiz et al., 2013). CBD showed a better safety profile and tolerability in patients (1,500 mg/day) than THC (Bergamaschi et al., 2011). Unlike THC, CBD did not alter cardiovascular parameters, psychomotor, and psychological functions, presumably because CBD does not directly target the CB1 receptor (Pertwee, 2006).
Emerging preclinical research studies demonstrate that CBD can exhibit neuroprotective effects due to its antioxidant and anti-inflammatory properties, which may potentially treat different neurodegenerative diseases (Hampson et al., 1998; Esposito et al., 2006; Esposito, Scuderi et al., 2007). For instance, CBD can act as a scavenger of reactive oxygen species (ROS) and inhibit lipid peroxidation, which inhibits caspase-mediated apoptosis of neurons (Iuvone et al., 2004). Interestingly, the neuroprotective effect of CBD is stronger than that of common antioxidants such as α-tocopherol or ascorbate (Atalay et al., 2019), which indicates that CBD may interact with some molecular targets. CBD has a low affinity for CB1 and CB2 receptors; however, it can indirectly activate CB1R via inhibition of FAAH and thereby promote the accumulation of endocannabinoids, which may prevent glutamate-mediated neurotoxicity (Pertwee 2008). In addition, CBD can also prevent glutamate-induced neurotoxicity occurring via the N-methyl-d-aspartate (NMDA) receptor, 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), or kainate receptor in the cannabinoid receptor-independent pathway as demonstrated by Hampson et al. (1998).
CBD can also prevent neuroinflammation by acting on multiple molecular targets (Figure 3). CBD can reduce the expression of iNOS in neurons via the downregulation of NF-κB and phosphorylated p38 MAPK (Esposito et al., 2006). CBD, being an inverse agonist of the CB2 receptor (Thomas et al., 2007), can inhibit microglial activation in a CB2 receptor-associated pathway (Ramírez et al., 2005). Peroxisome proliferation-activated receptor gamma (PPARγ) (Hegde et al., 2015; O'Sullivan, 2016) is an important target of CBD. PPARγ is a member of the ligand-activated transcriptional factor of the nuclear hormone receptor superfamily, which is a key mediator of energy homeostasis and associated with many biological functions including inflammation (Daynes and Jones, 2002). Several studies have reported that PPAR-γ agonists exhibit neuroprotective effects via regulating gene transcription related to the pathogenesis of neurodegeneration (Kumar et al., 2020). CBD also exhibits neuroprotective and anti-inflammatory effects via the A2A receptor where it can stimulate A2A-mediated signaling in immune cells by inhibiting the uptake of adenosine (Carrier Erica et al., 2006). Activation of the A2A receptor by CBD decreases microglial activation in an ATP-dependent pathway (Martín-Moreno et al., 2011) and can improve cognitive and motor function via inhibition of TNF-α expression and increasing expression of brain-derived neurotrophic factor (BDNF) (Magen et al., 2009). In addition, CBD can also target the TRPV1 receptor to exhibit its neuroprotective effect (Giuliano et al., 2021).
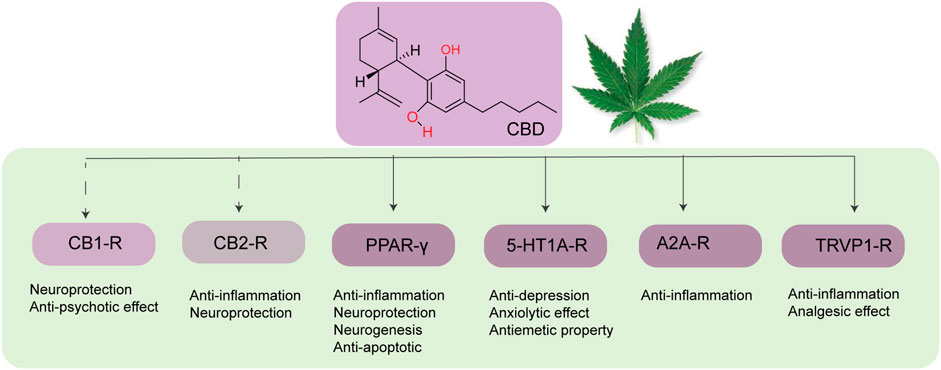
FIGURE 3. Different molecular targets of CBD in the brain.
CBD also has a significant effect on improving mitochondrial dysfunction. Emerging evidence suggests that mitochondrial dysfunction via both mitochondrial iron accumulation and ROS production is directly related to neurodegenerative disorders. To this end, da Silva et al. have demonstrated that CBD can reverse iron-induced mitochondrial dysfunction by rescuing mitochondrial ferritin and epigenetic modulation of mitochondrial DNA (da Silva et al., 2018). CBD can also act as a modest affinity agonist toward the 5-HT1A (serotonin) receptor (Russo et al., 2005), which is present in the synaptic membrane of several regions of the brain (Hoyer et al., 1986) and exhibits its anti-depression property.
PD is the second most prevalent neurodegenerative disease (after AD) associated with motor deficits such as tremors at rest, rigidity, and postural instability. Progressive loss of dopaminergic neurons in the midbrain substantia nigra is the pathological hallmark of PD, which results from the intracellular accumulation of Lewy bodies containing misfolded α-synuclein protein. The genetic studies reveal that the mutations in α-synuclein, PARK7, PINK1, ubiquitin genes, and population-specific gene mutations such as the glucocerebrosidase gene in Ashkenazi Jews are directly related to the onset of PD. Although the current standard of care including treatment with l-DOPA and deep brain stimulation can alleviate the symptoms, there is no therapeutic solution to date to cease the progression of neurodegeneration in PD. To this end, mitochondrial dysfunction and oxidative stress are presumed to play key roles in the pathogenesis of PD, and recent studies indicate that antioxidant phytocannabinoids such as CBD can exhibit potential therapeutic benefits due to their neuroprotective effect (Table 1). CBD is also demonstrated to improve the antioxidant defense of striatal neurons projected toward the substantia nigra (Sagredo et al., 2007). Using a rat striatum lesion, Sagredo et al. have demonstrated that CBD can completely rescue the endogenous antioxidant defense diminished by treatment with 3-nitropropionic acid, an inhibitor of mitochondrial complex II. Particularly, CBD can enhance mRNA levels for SOD-II and partially for SOD-I. Using agonists of CB1, CB2, TRPV1, and A2A, authors have shown that this neuroprotective effect is mediated via the TRPV1 receptor. Similarly, using a 1-methyl-4-phenylpyridinium (MPP(+))-induced cellular model of PD in SH-SY5Y and PC12 cells, Santos et al. have reported that CBD can enhance neuronal cell viability, differentiation, and the expression of synaptic (synaptophysin and synapsin I) and axonal (growth-associated protein-43) proteins (Santos et al., 2015). This neuritogenic effect of CBD against MPP(+)-induced PD is independent of nerve growth factor (NGF) but associated with the involvement of trkA receptors. A similar observation has also been reported by Gugliandolo et al. using MPP(+)-induced PD in SH-SY5Y cells (Gugliandolo et al., 2020). In this study, the authors have demonstrated that in addition to the trkA receptors, presumably ERK and AKT/mTOR pathways are also involved in the neuroprotection, which can be correlated with the interaction of CBD molecules with CB2 and TRVP1 receptors.
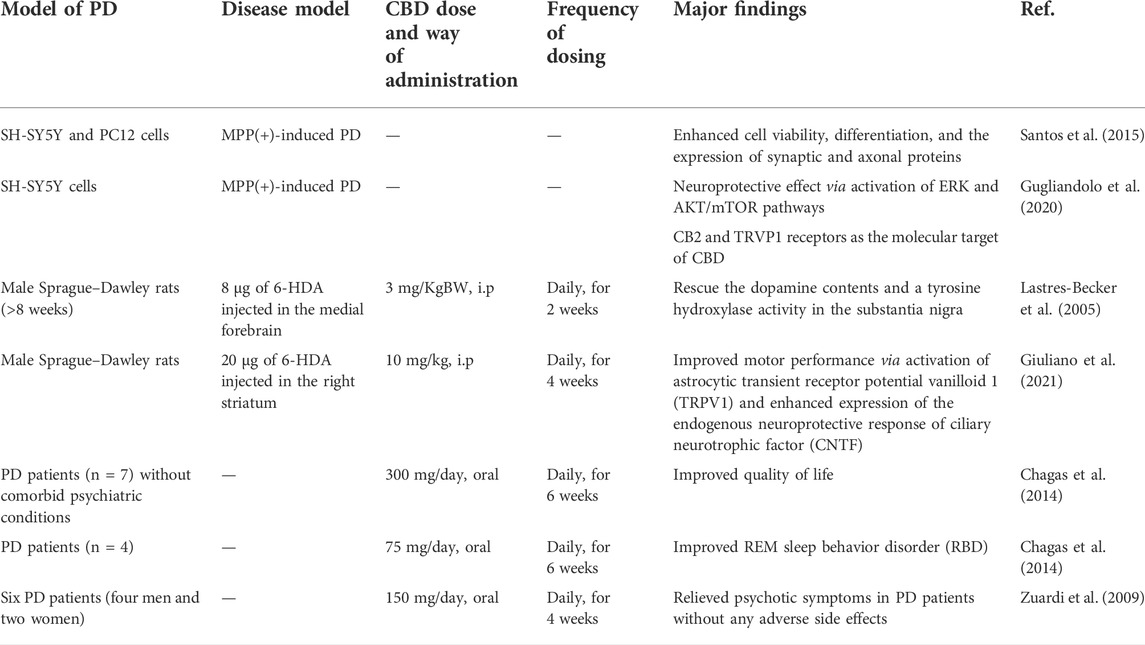
TABLE 1. CBD on PD, in vitro, in vivo, and clinical study.
The neuroprotective effect of CBD has been investigated in animal models of PD. Using a 6-hydroxydopamine (6-HDA) neurotoxin-induced PD model of rats, Ruiz and co-workers have demonstrated that a daily dose of 3 mg/KgBW of CBD over the course of 2 weeks can rescue the dopamine contents and tyrosine hydroxylase activity in the substantia nigra (Lastres-Becker et al., 2005). Interestingly, this effect is even more than that observed for the same dose of THC and is independent of the CB1 receptor. It is worth mentioning that the MPP(+)-induced animal model of PD is related to PD induced by neurotoxins, while 6-HDA-induced PD mimics the oxidative stress model of PD. To gain mechanistic insight, the authors have examined the direct effect of CBD on neuronal cultures, and neuronal cultures conditioned with media from CBD-treated glial cell culture. Authors have observed that neuroprotection is more pronounced with the conditioned media, indicating a neuroprotective role of CBD-treated glial cells. To further investigate if it is the involvement of CB receptors or the antioxidant properties of phytocannabinoids that play a key role in the observed neuroprotective behavior, PD-bearing rats were treated with small molecule inhibitors of the endocannabinoid system with and without antioxidant properties, namely, AM404 and UCM707, respectively (García-Arencibia et al., 2007). Recovery of dopamine depletion is observed when rats were treated with AM404 but not with UCM707, indicating antioxidant properties of phytocannabinoid molecules/analogue behind the neuroprotective role against PD which is further confirmed by upregulation of superoxide dismutase, a key endogenous enzyme against oxidative stress. It is worth mentioning that a recent study by the same group using a similar 6-HDA-induced cellular and murine model of PD demonstrates that the quinone derivative of CBD exhibits its neuroprotective behavior via PPAR-γ receptors (Burgaz et al., 2021). Furthermore, recently, Giuliano et al. have demonstrated that a higher dose of CBD (10 mg/KgBW) can reduce nigrostriatal degeneration and improve motor function in rats bearing 6-HDA-induced PD (Giuliano et al., 2021). Authors have reported that this effect is mediated via the activation of TRPV1 in astrocytes which further enhances expression of the endogenous neuroprotective response of ciliary neurotrophic factor (CNTF).
The effect of CBD on PD has been investigated in clinical settings too. For instance, in an exploratory double-blind clinical trial with PD patients (n = 7) without dementia or comorbid psychiatric conditions, CBD (300 mg/day) does not improve motor function significantly but has been found to improve the quality of life among the PD patients (Chagas et al., 2014). In addition, CBD also significantly improves REM sleep behavior disorder (RBD) in PD patients (n = 4) without side effects (Chagas et al., 2014). Importantly, in another open-label pilot study in six PD patients (four men and two women), CBD was found to relieve psychotic symptoms in PD patients without any adverse side effects or negative effects on motor neurons (Zuardi et al., 2009). Taken together, CBD is well tolerated in humans and effective in relieving some PD-related symptoms, indicating its potential as a complementary treatment for PD. In-depth preclinical and clinical studies with a large cohort of patients, however, are needed to further confirm this possibility.
AD is the most prevalent neurodegenerative disease which is characterized by progressive cognitive decline and memory dysfunction prior to the normal aging process, affecting ∼40 million people worldwide (Tiwari et al., 2019). Progression of AD is associated with a gradual loss of cholinergic neurons in the hippocampus and neocortex of the brain. The pathological hallmarks for both late-onset sporadic AD (>95% of cases) and early-onset familial AD (<5% cases) are deposition of extracellular Aβ-senile plaques, intracellular NFT of hyperphosphorylated tau protein, and loss of cholinergic neurons. Familial AD results from a genetic mutation in the APP gene or in the presenilin 1 and 2 (PS1 and PS2) genes that lead to an altered ratio of amyloid β42 to amyloid β40 and eventually rapid aggregation of Aβ peptide to form Aβ plaque (Bettens et al., 2013). The cause of sporadic AD is still unclear but it is majorly influenced by environmental and lifestyle factors, with the APOE4 gene being the primary genetic risk factor (Kamboh, 2004). Irrespective of the origin of AD, Aβ accumulation initiates a cascade of events mainly including excitotoxicity, neuroinflammation, and oxidative stress that ultimately led to neurodegeneration and cognitive impairment. Aβ accumulation enhances glutamate release from astrocytes (Talantova et al., 2013) and neuronal cells (de Paula Faria et al., 2022). Excess glutamate stimulates the extrasynaptic N-methyl-d-aspartate (NMDA) receptors and causes an intense influx of Ca2+, which results in excitotoxicity, mitochondrial dysfunction, and neuronal cell death (Hardingham and Bading, 2010). In addition, Aβ accumulation suppresses synaptic transmission via promoting endocytosis of NMDA receptors at synapses (Hardingham and Bading, 2010) and alters synaptic plasticity (Figure 4) (He et al., 2019). Microglia initially clear Aβ and have neuroprotective effects. Upon persistent microglial activation, it facilitates Aβ aggregation and neurodegeneration (Cai et al., 2014).
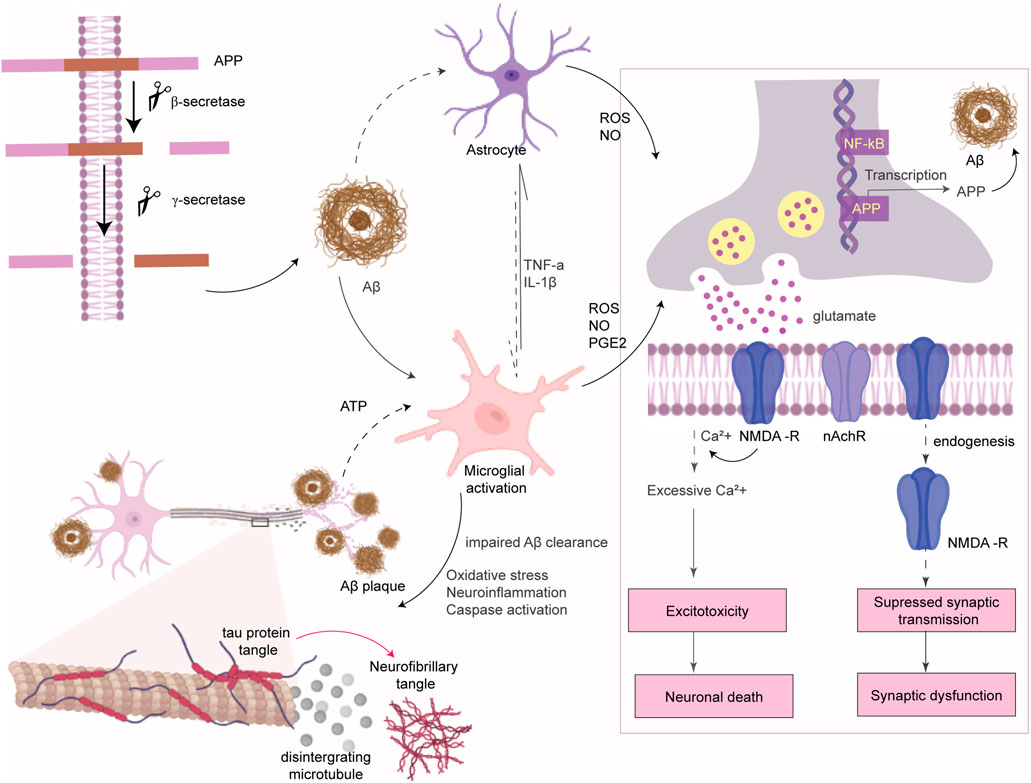
FIGURE 4. Amyloid β pathogenesis of AD. APP protein is processed by secretase enzymes to produce Aβ peptides. Aggregated Aβ causes microglial activation to produce ROS and inflammatory cytokines which further activate astrocytes. ROS binds to NF-κB sites in the promoters of APP in neurons to activate transcription of the Aβ peptide. Aβ plaques enhance glutamate release from neurons and astrocytes. Prolonged activation of extrasynaptic NMDA receptors by excess glutamate leads to intense transient Ca2+ influx which alters some signaling pathways and causes neuronal death (excitotoxicity). Aβ also inhibits the activity of acetylcholine esterase and lowers the expression of nicotinic acetylcholine receptor (nAchR). In addition, Aβ accumulation promotes endocytosis of NMDA receptors at synapses which suppresses synaptic transmission and alters synaptic plasticity.
Cholinergic neurons are majorly affected in AD with a severely diminished transcription for acetylcholine esterase enzyme (ChAT) transcription, which is closely associated with cognitive decline in AD patients (Wilcock et al., 1982). The FDA-approved drugs (three acetylcholinesterase inhibitors such as donepezil, rivastigmine, and galantamine and one NMDA receptor antagonist, namely, memantine) till date mostly target to improve the cognitive deficit via facilitating cholinergic neurotransmission or via mitigating the excitotoxicity by antagonizing the NMDA receptor. However, these therapies offer only temporary symptomatic benefits (Husna Ibrahim et al., 2020). To this end, considering the crucial role of oxidative stress and neuroinflammation in AD progression, antioxidants and nonsteroidal anti-inflammatory drugs have attracted significant attention in exploring their potential benefit to mitigate Aβ-initiated events of AD progression. In this context, cannabinoids such as CBD have garnered tremendous attention.
Since the observance of the neuroprotective effect of CBD (Hampson et al., 1998) in 1998, Esposito et al. first (in 2004) reported that pre-exposure to CBD can protect rat neuronal cells PC12 from Aβ peptide-induced neurotoxicity (Iuvone et al., 2004). CBD acts as a scavenger of ROS and dose-dependently inhibits lipid peroxidation followed by inhibition of caspase 3-mediated apoptosis of neuronal cells. To further investigate the molecular pathway of neuroprotection, the same group reported that CBD can inhibit the phosphorylation of GSK-3β (Esposito et al., 2006). GSK-3β, a kinase enzyme, plays a significant role in the pathogenesis of AD (Lauretti et al., 2020). It promotes the formation of Aβ plaque by altering the PS1/γ-secretase complex during APP cleavage and hyper phosphorylates tau promoting neurofibrillary tangle formation. It is hyperactive in the brain of AD patients and can act as a therapeutic target (Ly et al., 2013). Importantly, inhibition of GSK-3β stabilizes β-catenin, a protein present at both pre- and postsynaptic terminals, and influences synaptic size and strength (Maguschak and Ressler, 2008), which improves learning and memory deficits (Onishi et al., 2011). To this end, Esposito et al. have reported that the reduction of the phosphorylation of GSK-3β by CBD inhibits neurofibrillary tangle formation and rescue of the Wnt/β-catenin pathway (Esposito et al., 2006).
In addition to the antioxidant effect of CBD in AD, the same group has reported that CBD can also exhibit anti-inflammatory effects. iNOS and its enzymatic product NO are the major neurotoxic effectors of AD found both in Aβ-stimulated neuronal cells (Iuvone et al., 2004) and in post-mortem AD brains (Haas et al., 2002). NF-κB, a redox-sensitive transcription factor that is activated by a family of stress-activated kinases including p38 MAP kinase, regulates the expression of genes involved in oxidative, inflammatory, and immune responses. Esposito et al. have found that CBD can inhibit both NO production and iNOS protein expression at a low concentration (10–6 to 10–4 M) and significantly prevent NF-κB activation and phosphorylation of p38 MAPK in PC12 cells against Aβ insult (Esposito et al., 2006). To further investigate such anti-inflammatory effects of CBD in an animal model, the same group has also reported that early administration of CBD (i.p., 2.5 or 10 mg kg−1) for 7 days in C57 mice inoculated with Aβ-peptide in the hippocampus can markedly mitigate reactive gliosis by suppressing the expression of IL-1β and iNOS (Esposito et al., 2007).
Interestingly, while there is a quest for the molecular mechanism of CBD’s anti-inflammatory and neuroprotective action in AD, recent studies indicate that PPARγ plays a crucial role. An elevated expression level of PPARγ has been found in AD brain tissues (Kitamura et al., 1999). However, PPARγ agonists have been found to induce beneficial neuroprotective and anti-inflammatory effects in cultured cortical neurons (Gray et al., 2012) and improve learning and memory deficits in transgenic mice models of AD (Escribano et al., 2010). To this end, O'Sullivan et al. have reported that being a small molecule, CBD can transport to the nucleus and bind to activate the transcriptional activity of PPARγ (O'Sullivan, Sun et al., 2009). Using a rat model of AD injected with fibrillar Aβ peptide at the hippocampus, Esposito et al. have reported that CBD can mitigate Aβ-induced neuroinflammation and promote neurogenesis via activation of PPARγ which is diminished when CBD is used in combination with PPARγ antagonists. To further investigate the molecular pathway, authors have demonstrated that activating PPARγ by CBD diminishes Aβ-induced inflammation in cultured astrocytes via inhibition of NF-κB (Kozela et al., 2010; Esposito et al., 2011) and promotes ubiquitination of APP to reduce expression of Aβ peptide in neuronal SHSY5YAPP+ cells (Scuderi et al., 2014).
CBD can also influence other cells including microglia and mesenchymal stem cell toward neuroprotection. For instance, CBD can inhibit microglia activation in response to Aβ insult in cultured N13 microglial cells and in primary microglia cells (BV-2) presumably via cannabinoid and adenosine A2A receptors (Martín-Moreno et al., 2011). Libro et al. have examined the regenerative potential of CBD in conditioning human mesenchymal stem cells (hMSCs) toward recovering from the neurodegeneration of AD (Libro et al., 2016). Using a transcriptomic analysis with the next-generation sequencing (NGS) study, authors have demonstrated that hMSCs pre-treated with CBD show downregulation of kinase genes responsible for tau phosphorylation, specifically the PI3K/Akt/GSK-3β pathway and secretase genes responsible for Aβ production. Using small molecule antagonists, authors have demonstrated that this effect of CDB is exhibited via the TRVP1 receptor and is independent of the cannabinoid receptor. CBD is also reported to enhance the migration of adipose-derived MSCs in a dose- and time-dependent manner, and this migration of MSCs is mediated via the CB2 receptor and GPR55 receptor by activation of the p42/44 MAPK pathway (Schmuhl et al., 2014). Cannabidiol (CBD) can act as a preventive agent to protect synaptic plasticity against AD (Hughes and Herron, 2019). Using hippocampal slices of C57BL/6J mice perfused with oligomeric beta-amyloid peptide (Aβ1–42) in the presence or absence of CBD treatment, Huges et al. have reported that pretreatment with CBD (not post-treatment) can recover the synaptic transmission attenuated by Aβ. The in vitro findings on CBD’s neuroprotective properties are summarized in Figure 5 and Table 2.
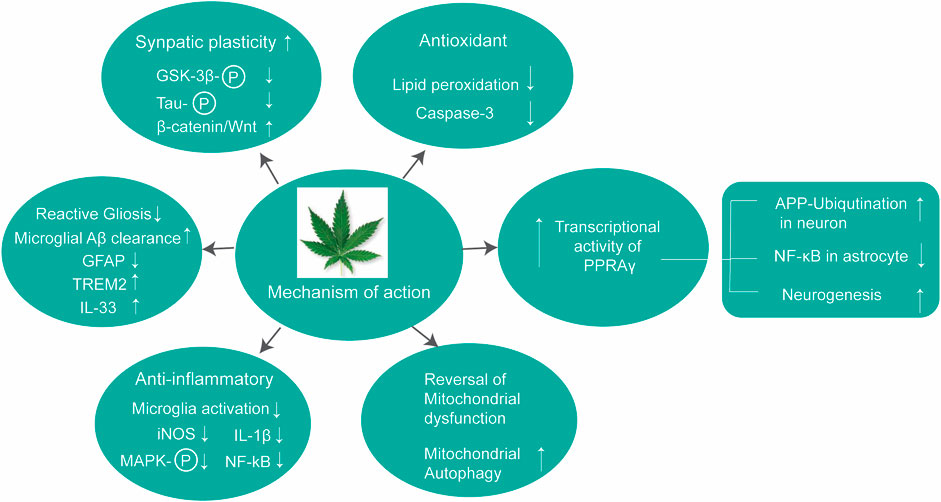
FIGURE 5. Molecular pathways of contributing neuroprotective role of CBD.

TABLE 2. CBD on AD, in vitro study.
The neuroprotective efficacy of CBD has also been evaluated using different animal models of AD including pharmacological rodent models of partial AD in C57BL/6J and Wistar rats where the disease is induced by injecting Aβ peptide on hippocampus and in established transgenic mice (Table 3). Cheng et al. have first evaluated the efficacy of CBD in a double transgenic mouse model of familial AD (APPxPS1 mice) containing mutations in both the APP and PS1 genes. Notably, such mice start developing Aβ plaques as early as 4–6 months of age which further increases with age and exhibits a sexual dimorphism profile with female APPxPS1 mice showing higher pathological levels of AD markers compared to males. Authors have demonstrated that daily treatment of CBD (20 mg/kg) for 3 weeks rescued social recognition memory and object recognition deficits in 6-month-old male mice with no effect on fear-associated memory or anxiety measures (Cheng et al., 2014). A similar behavioral outcome has also been observed when APPxPS1 mice are treated in prophylactic settings with the same dose of CBD for 8 months (Cheng et al., 2014). However, 20 mg/kg CBD does not alter amyloid load or oxidative damage, although it impacts neuroinflammation and cholesterol. A recent study demonstrates that 50 mg/kg CBD can moderately reduce Aβ load in the hippocampus of a 12-month-old APPxPS1 male along with restoration of social recognition memory and spatial learning deficit (Watt et al., 2020). In contrast, some other studies reported that small to medium doses of CBD, for example, at a dose of 0.75 mg/kg of CBD (Aso et al., 2015) or 5 mg/kg of CBD (Coles et al., 2020) can improve the cognitive deficit in APPxPS1 mice. Some cannabinoids exhibit biphasic dose responses (Rey et al., 2012); therefore, it is crucial to investigate a range of dosages to estimate the window of the therapeutic effectiveness of the drug.
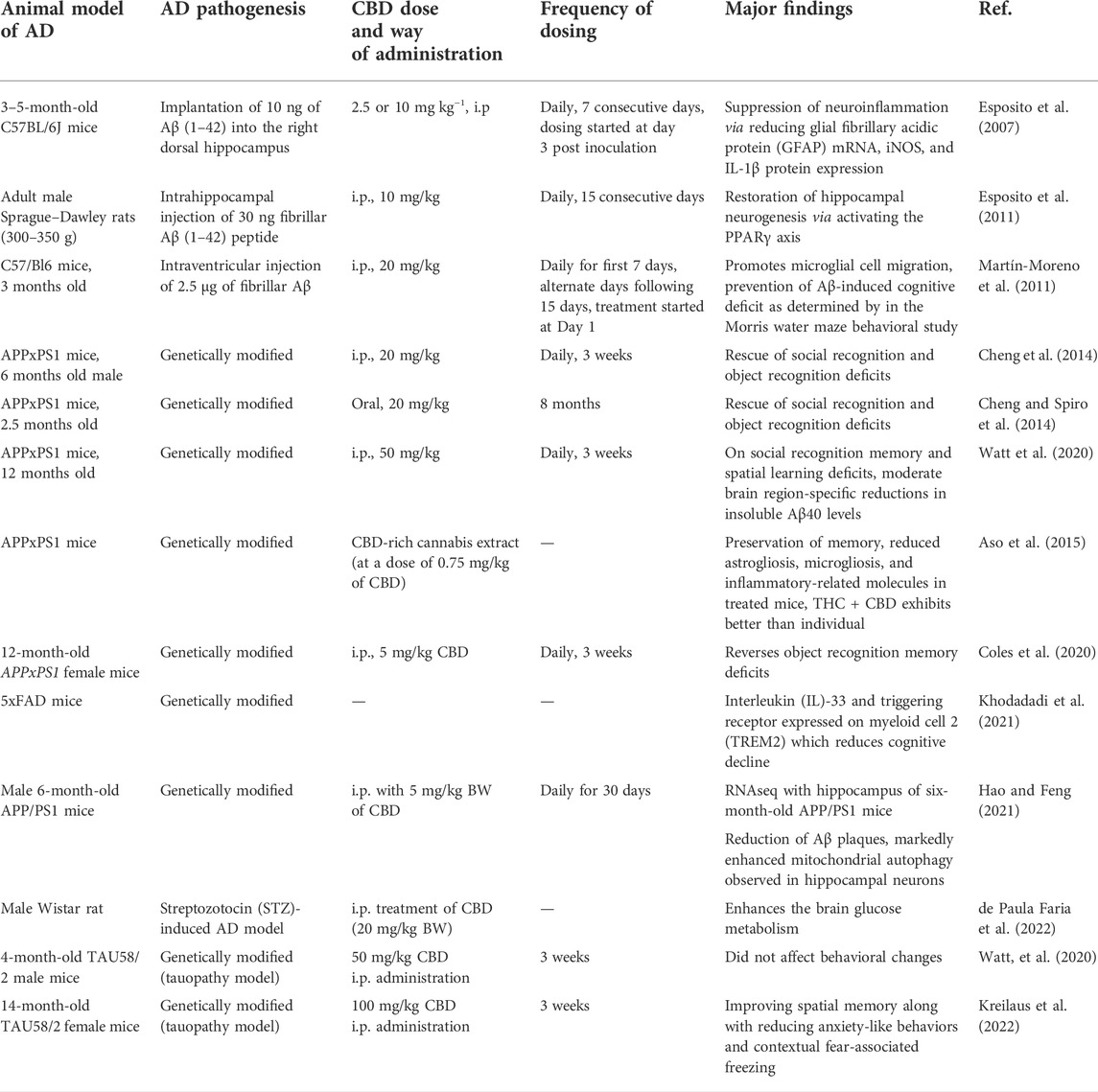
TABLE 3. CBD on AD, in vivo study.
To have a mechanistic insight into CBD’s various functions on AD-bearing animals, Hao et al. have used transcriptomic approaches. Authors have performed RNASeq using the hippocampus of six-month-old APPxPS1 mice treated (i.p.) with 5 mg/kg BW of CBD daily for 30 days. Using bioinformatics-based prediction tools (Gene Set Enrichment Analysis (GSEA) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis), authors have reported the reduction of Aβ plaques in CBD-treated mice which is related to improved immune response and enhanced mitochondrial autophagy in hippocampal neurons (Hao and Feng 2021). In another recent study using 5xFAD mice (which mimic early-onset familial AD), Hesam et al. reported that CBD treatment enhances the expression of IL-33 and triggers receptors expressed on myeloid cells 2 (TREM2). IL-33 is a cytokine and TREM2 is a microglial surface receptor, both of which skew microglia in enhancing Aβ clearance, and CBD treatment enhances the expression of such mediators to prevent the cognitive decline in mice (Khodadadi É et al., 2021). More recently, Faria et al. have investigated the effect of CBD on preventing the hypometabolism observed in AD, and have found that 20 mg/kg BW of CBD enhances the brain glucose metabolism in a streptozotocin (STZ)-induced AD model of the male Wistar rat (de Paula Faria et al., 2022).
In addition to the amyloid β model, researchers have also investigated the effect of CBD in the tauopathy model of AD. Recently, Alali et al. have reported that CBD can inhibit aggregation of tau protein (Alali et al., 2021). Using in vitro and in silico biochemical methods such as circular dichroism (CD) and atomic force microscopy (AFM), the author has demonstrated that CBD molecules bind to recombinant human tau protein and inhibit its aggregation. TAU58/2 transgenic mice, containing a P301S mutation in human tau which emphasizes tau pathogenesis of AD, are used as tauopathy rodent models of AD. Behavioral characteristics for anxiety, cognition, and motor functions have been investigated in the presence and absence of CBD in TAU58/2 transgenic mouse. Although chronic CBD treatment of 50 mg/KgBW for 3 weeks (i.p.) did not affect behavioral changes in such mice (Watt et al., 2020), a higher dose (100 mg/kg) of CBD is reported to be effective in improving spatial memory along with reducing anxiety-like behaviors and contextual fear-associated freezing in all mice (Kreilaus et al., 2022).
CBD has also been reported to combine with THC or other drugs. Sativex, the mixed cannabis (THC + CBD), reduces learning impairment and Aβ-42 peptide levels in AβPP/PS1 transgenic mice when chronically administered during the early symptomatic stage (Aso et al., 2015). Authors have demonstrated that this anti-Alzheimer efficacy of the combined cannabis (THC + CBD) is via reduction of astrogliosis, microgliosis, and inflammatory-related molecules in APPxPS1 mice. Using a genome-wide gene expression study, the author has identified that the redox protein thioredoxin-2 and the signaling protein Wnt16 are the significant targets for the (THC + CBD)-induced effects. The same group has also reported that the combined cannabinoids (THC + CBD) can improve memory impairment in APPxPS1 mice with an advanced stage of AD, which is not associated with APP processing or reducing glial activity, leading to Aβ deposition as observed in the early stages but via reduced GluR2/3 and increased levels of GABA-A Rα1 (Aso et al., 2016). GluR2/3 is a subtype of an AMPA glutamate receptor, and GABA-A Rα1 is a receptor subunit of the GABAergic system which plays a role in preventing neurodegeneration in both AD and in aging. Reduced GluR2/3 can prevent glutamate toxicity, while enhanced GABA-A Rα1 expression plays a role in preventing neuronal dysfunction. Authors have demonstrated that this effect of combined cannabinoids, however, is AD-specific and does not affect the cognitive impairment in healthy aging as observed in wild-type mice. The same group has also investigated if there is any role of the CB2 receptor in this combined anti-AD effect of (THC + CBD) using CB2 knockout APPxPS1 mice. They have found that although the CB2 receptor has no significant role in the therapeutic benefit of (THC + CBD), the lack of CB2 receptor interferes with the APP processing to reduce the cortical Aβ deposition by enhancing the levels of soluble Aβ40 (Aso et al., 2016).
To further enhance the therapeutic efficacy of CBD, it is chemically conjugated to a fragment of an AChE inhibitor. The new cannabidiol−carbamate hybrid lead compound (namely, C16) exhibits a much stronger inhibition of butyrylcholinesterase (BuChE) (IC50 = 5.3 nM) than that of CBD (IC50 = 0.67 μM) and crosses the BBB. Authors have demonstrated that C16 is able to improve scopolamine-induced cognition impairment in mice, and the treated mice exhibit better behavioral activity than donepezil (Jiang et al., 2021). Contrary to other observations, Amini et al. have reported that CBD, when delivered using chitosan nanoparticles, reduces Aβ plaque formation and learning and memory in AD-bearing rats via enhancing the expression levels of CB1 and CB2, though the effect of empty chitosan NPs on CB1 and CB2 has not been investigated in this study (Amini and Abdolmaleki, 2021).
A clinical trial with THC-free CBD oil has just started (Leszko and Meenrajan, 2021). In Poland, 63 caregivers out of 73 reported CBD to be effective in managing behavioral symptoms of AD, which indicates the initial promise of CBD oil in the clinical study. Collectively, the non-psychoactive CBD holds significant promises in managing behavioral deficit of AD due to its antioxidant and anti-inflammatory properties with a note that its dose and frequency of treatment should be thoroughly investigated in detail in near future.
Despite the significant advancement in neurology, the cure of neurodegenerative diseases such as AD and PD continues to remain an unmet challenge. Emerging evidence indicates that oxidative stress and inflammation are common amplifiers of disease progression, and the inhibition of neuroinflammation may result in clinical benefit in treating NDs. However, care should be taken when choosing anti-inflammatory drugs that may have a potential adverse effect due to activation of the innate immune system. In addition, as neurodegenerative diseases require long-term therapy, the high safety profile of the drug is a crucial factor. Furthermore, to be clinically effective, the therapeutics should gain access to the CNS and specifically target the cells or pathways. To this end, non-psychoactive phytocannabinoid CBD holds significant promises due to their antioxidant and anti-inflammatory properties, high safety profile, and good tolerability. Specifically, neuroprotection via PPARγ activation by CBD may hold significant promise as rosiglitazone, an agonist of PPARγ, exhibited prominent clinical benefit either alone (phase 2 clinical trials) or in combination with donepezil (Phase 3) in treating AD patients. However, a thorough investigation of the required dosages and treatment windows in different transgenic mouse models is necessary. Importantly, rodent age and sex are crucial factors when evaluating the therapeutic effectiveness of CBD in some neurodegenerative diseases.
Clinical application of CBD has just started with FDA approval of Epidiolex (CBD in oil formulation) in 2018 for treating pediatric epilepsy. However, despite its multi-faceted therapeutic potential, its clinical approval is impeded by intrinsic characteristics such as poor bioavailability and variable pharmacokinetics profile (Millar et al., 2020). Potential ways to overcome this including usage of drug delivery systems and/or different routes of drug administration are in the preclinical and clinical stages of development. For example, to avoid the precipitation of CBD in the gastrointestinal tract, CBD is used in formulation with oils, surfactants, solvents, or cyclodextrin carriers which are being investigated while the i.p. route of administration exhibits a better brain accumulation than the oral route. Considering the general trend of PK study regarding a small percentage of brain accumulation of therapeutics, nano- or micro-sized droplets of such formulations have gained significant importance. In addition, oxidative degradation and drug–drug interaction (when used in combination) of CBD are crucial factors that should be taken care of. To this end, investigation of lipid-based nanocarriers that can solubilize CBD in lipid core has just started (Aparicio-Blanco et al., 2019). Herein, the brain-targeting lipophilic nanocarrier which can solubilize the CBD in its lipid core and deliver a payload to the brain holds significant promise. Collectively, CBD is harmless at a low dose, and preclinical studies indicate its beneficial neuroprotective effect. Clinical trials for CBD in conventional neurodegenerative diseases have started showing its initial promise, especially in improving behavioral health. Thus, CBD may find its clinical application beyond pediatric epilepsy not only to conventional neurodegenerative diseases but also to neurodegenerative conditions secondary to other CNS complications either alone or as an adjuvant in the near future.
SB and MN have conceptualized the content, NK, AA, AV have provided constructive feedback to improve quality of the manuscript.
SB, AA, and MN are grateful for support from the National Institutes of Health (NIH) grants DA042706, DA052271, DA040537, DA037838, and DA034547 and Florida Department of Health grant 8AZ04, NK thanks the H&N Wertheim Research Pilot Project from Florida International University (FIU) for their support, and AV is thankful for support from the FIU Foundation via grant number AWD0011094.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alali, S., Riazi, G., Ashrafi-Kooshk, M. R., Meknatkhah, S., Ahmadian, S., Hooshyari Ardakani, M., et al. (2021). Cannabidiol inhibits tau aggregation in vitro. Cells 10 (12), 3521. doi:10.3390/cells10123521
Alexander, S. P. (2016). Therapeutic potential of cannabis-related drugs. Prog. Neuropsychopharmacol. Biol. Psychiatry 64, 157–166. doi:10.1016/j.pnpbp.2015.07.001
Amini, M., and Abdolmaleki, Z. (2021). The effect of cannabidiol coated by nano-chitosan on learning and memory, hippocampal CB1 and CB2 levels, and amyloid plaques in an alzheimer's disease rat model. Neuropsychobiology 81, 171–183. doi:10.1159/000519534
Aparicio-Blanco, J., Romero, I. A., Male, D. K., Slowing, K., García-García, L., and Torres-Suárez, A. I. (2019). Cannabidiol enhances the passage of lipid nanocapsules across the blood–brain barrier both in vitro and in vivo. Mol. Pharm. 16 (5), 1999–2010. doi:10.1021/acs.molpharmaceut.8b01344
Aso, E., Andrés-Benito, P., Carmona, M., Maldonado, R., and Ferrer, I. (2016). Cannabinoid receptor 2 participates in amyloid-β processing in a mouse model of alzheimer's disease but plays a minor role in the therapeutic properties of a cannabis-based medicine. J. Alzheimers Dis. 51 (2), 489–500. doi:10.3233/JAD-150913
Aso, E., Andrés-Benito, P., and Ferrer, I. (2016). Delineating the efficacy of a cannabis-based medicine at advanced stages of dementia in a murine model. J. Alzheimers Dis. 54 (3), 903–912. doi:10.3233/JAD-160533
Aso, E., Sánchez-Pla, A., Vegas-Lozano, E., Maldonado, R., and Ferrer, I. (2015). Cannabis-based medicine reduces multiple pathological processes in AβPP/PS1 mice. J. Alzheimers Dis. 43 (3), 977–991. doi:10.3233/JAD-141014
Atalay, S., Jarocka-Karpowicz, I., and Skrzydlewska, E. (2019). Antioxidative and anti-inflammatory properties of cannabidiol. Antioxidants (Basel, Switz. 9 (1), 21. doi:10.3390/antiox9010021
Bergamaschi, M. M., Queiroz, R. H., Zuardi, A. W., and Crippa, J. A. (2011). Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr. Drug Saf. 6 (4), 237–249. doi:10.2174/157488611798280924
Bettens, K., Sleegers, K., and Van Broeckhoven, C. (2013). Genetic insights in Alzheimer's disease. Lancet. Neurol. 12 (1), 92–104. doi:10.1016/S1474-4422(12)70259-4
Burgaz, S., García, C., Gómez-Cañas, M., Rolland, A., Muñoz, E., Fernández-Ruiz, J., et al. (2021). Preclinical investigation in neuroprotective effects of the GPR55 ligand VCE-006.1 in experimental models of Parkinson's disease and amyotrophic lateral sclerosis. Molecules 26 (11), 7643. doi:10.3390/molecules26247643
Cai, Z., Hussain, M. D., and Yan, L. J. (2014). Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease. Int. J. Neurosci. 124 (5), 307–321. doi:10.3109/00207454.2013.833510
Carrier Erica, J., Auchampach John, A., and Hillard Cecilia, J. (2006). Inhibition of an equilibrative nucleoside transporter by cannabidiol: A mechanism of cannabinoid immunosuppression. Proc. Natl. Acad. Sci. U. S. A. 103 (20), 7895–7900. doi:10.1073/pnas.0511232103
Cassano, T., Villani, R., Pace, L., Carbone, A., Bukke, V. N., Orkisz, S., et al. (2020). From cannabis sativa to cannabidiol: Promising therapeutic candidate for the treatment of neurodegenerative diseases. Front. Pharmacol. 11, 124. doi:10.3389/fphar.2020.00124
Chagas, M. H., Eckeli, A. L., Zuardi, A. W., Pena-Pereira, M. A., Sobreira-Neto, M. A., Sobreira, E. T., et al. (2014). Cannabidiol can improve complex sleep-related behaviours associated with rapid eye movement sleep behaviour disorder in Parkinson's disease patients: A case series. J. Clin. Pharm. Ther. 39 (5), 564–566. doi:10.1111/jcpt.12179
Chagas, M. H., Zuardi, A. W., Tumas, V., Pena-Pereira, M. A., Sobreira, E. T., Bergamaschi, M. M., et al. (2014). Effects of cannabidiol in the treatment of patients with Parkinson's disease: An exploratory double-blind trial. J. Psychopharmacol. 28 (11), 1088–1098. doi:10.1177/0269881114550355
Cheng, D., Low, J. K., Logge, W., Garner, B., and Karl, T. (2014). Chronic cannabidiol treatment improves social and object recognition in double transgenic APPswe/PS1∆E9 mice. Psychopharmacology 231 (15), 3009–3017. doi:10.1007/s00213-014-3478-5
Cheng, D., Spiro, A. S., Jenner, A. M., Garner, B., and Karl, T. (2014). Long-term cannabidiol treatment prevents the development of social recognition memory deficits in alzheimer's disease transgenic mice. J. Alzheimers Dis. 42, 1383–1396. doi:10.3233/JAD-140921
Coles, M., Watt, G., Kreilaus, F., and Karl, T. (2020). Medium-dose chronic cannabidiol treatment reverses object recognition memory deficits of APPSwe/PS1ΔE9 transgenic female mice. Front. Pharmacol. 11, 587604. doi:10.3389/fphar.2020.587604
Cooray, R., Gupta, V., and Suphioglu, C. (2020). Current aspects of the endocannabinoid system and targeted THC and CBD phytocannabinoids as potential therapeutics for Parkinson's and alzheimer's diseases: A review. Mol. Neurobiol. 57 (11), 4878–4890. doi:10.1007/s12035-020-02054-6
da Silva, V. K., de Freitas, B. S., Dornelles, V. C., Kist, L. W., Bogo, M. R., Silva, M. C., et al. (2018). Novel insights into mitochondrial molecular targets of iron-induced neurodegeneration: Reversal by cannabidiol. Brain Res. Bull. 139, 1–8. doi:10.1016/j.brainresbull.2018.01.014
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of α-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27 (34), 9220–9232. doi:10.1523/JNEUROSCI.2617-07.2007
Daynes, R. A., and Jones, D. C. (2002). Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2 (10), 748–759. doi:10.1038/nri912
de Paula Faria, D., Estessi de Souza, L., Duran, F. L. S., Buchpiguel, C. A., Britto, L. R., Crippa, J. A. S., et al. (2022). Cannabidiol treatment improves glucose metabolism and memory in streptozotocin-induced alzheimer's disease rat model: A proof-of-concept study. Int. J. Mol. Sci. 23 (3), 1076. doi:10.3390/ijms23031076
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283 (14), 9089–9100. doi:10.1074/jbc.M710012200
Devinsky, O., Marsh, E., Friedman, D., Thiele, E., Laux, L., Sullivan, J., et al. (2016). Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. Lancet. Neurol. 15 (3), 270–278. doi:10.1016/S1474-4422(15)00379-8
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9 (7), a028035. doi:10.1101/cshperspect.a028035
Escribano, L., Simón, A.-M., Gimeno, E., Cuadrado-Tejedor, M., López de Maturana, R., García-Osta, A., et al. (2010). Rosiglitazone rescues memory impairment in alzheimer's transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 35 (7), 1593–1604. doi:10.1038/npp.2010.32
Esposito, G., De Filippis, D., Carnuccio, R., Izzo, A. A., and Iuvone, T. (2006). The marijuana component cannabidiol inhibits beta-amyloid-induced tau protein hyperphosphorylation through Wnt/beta-catenin pathway rescue in PC12 cells. J. Mol. Med. 84 (3), 253–258. doi:10.1007/s00109-005-0025-1
Esposito, G., De Filippis, D., Maiuri, M. C., De Stefano, D., Carnuccio, R., and Iuvone, T. (2006). Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett. 399 (1-2), 91–95. doi:10.1016/j.neulet.2006.01.047
Esposito, G., Scuderi, C., Savani, C., Steardo, L., De Filippis, D., Cottone, P., et al. (2007). Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br. J. Pharmacol. 151 (8), 1272–1279. doi:10.1038/sj.bjp.0707337
Esposito, G., Scuderi, C., Valenza, M., Togna, G. I., Latina, V., De Filippis, D., et al. (2011). Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS One 6 (12), e28668. doi:10.1371/journal.pone.0028668
Fernández-Ruiz, J., Sagredo, O., Pazos, M. R., García, C., Pertwee, R., Mechoulam, R., et al. (2013). Cannabidiol for neurodegenerative disorders: Important new clinical applications for this phytocannabinoid? Br. J. Clin. Pharmacol. 75 (2), 323–333. doi:10.1111/j.1365-2125.2012.04341.x
Fischer, R., and Maier, O. (2015). Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 610813. doi:10.1155/2015/610813
García-Arencibia, M., González, S., de Lago, E., Ramos, J. A., Mechoulam, R., and Fernández-Ruiz, J. (2007). Evaluation of the neuroprotective effect of cannabinoids in a rat model of Parkinson's disease: Importance of antioxidant and cannabinoid receptor-independent properties. Brain Res. 1134 (1), 162–170. doi:10.1016/j.brainres.2006.11.063
Giuliano, C., Francavilla, M., Ongari, G., Petese, A., Ghezzi, C., Rossini, N., et al. (2021). Neuroprotective and symptomatic effects of cannabidiol in an animal model of Parkinson's disease. Int. J. Mol. Sci. 22 (16), 8920. doi:10.3390/ijms22168920
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140 (6), 918–934. doi:10.1016/j.cell.2010.02.016
Gray, E., Ginty, M., Kemp, K., Scolding, N., and Wilkins, A. (2012). The PPAR-gamma agonist pioglitazone protects cortical neurons from inflammatory mediators via improvement in peroxisomal function. J. Neuroinflammation 9 (1), 63. doi:10.1186/1742-2094-9-63
Gugliandolo, A., Pollastro, F., Bramanti, P., and Mazzon, E. (2020). Cannabidiol exerts protective effects in an in vitro model of Parkinson's disease activating AKT/mTOR pathway. Fitoterapia 143, 104553. doi:10.1016/j.fitote.2020.104553
Haas, J., Storch-Hagenlocher, B., Biessmann, A., and Wildemann, B. (2002). Inducible nitric oxide synthase and argininosuccinate synthetase: Co-induction in brain tissue of patients with alzheimer's dementia and following stimulation with β-amyloid 1–42 in vitro. Neurosci. Lett. 322 (2), 121–125. doi:10.1016/s0304-3940(02)00095-2
Hampson, A. J., Grimaldi, M., Axelrod, J., and Wink, D. (1998). Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. U. S. A. 95 (14), 8268–8273. doi:10.1073/pnas.95.14.8268
Hao, F., and Feng, Y. (2021). Cannabidiol (CBD) enhanced the hippocampal immune response and autophagy of APP/PS1 Alzheimer's mice uncovered by RNA-seq. Life Sci. 264, 118624. doi:10.1016/j.lfs.2020.118624
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11 (10), 682–696. doi:10.1038/nrn2911
He, Y., Wei, M., Wu, Y., Qin, H., Li, W., Ma, X., et al. (2019). Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4, 5-bisphosphate. Nat. Commun. 10 (1), 1193. doi:10.1038/s41467-019-09114-z
Hegde, V. L., Singh, U. P., Nagarkatti, P. S., and Nagarkatti, M. (2015)., 194. Baltimore, Md, 5211–5222. Critical role of mast cells and peroxisome proliferator-activated receptor γ in the induction of myeloid-derived suppressor cells by marijuana cannabidiol in vivoJ. Immunol.11. doi:10.4049/jimmunol.1401844
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21 (10), 1359–1369. doi:10.1038/s41593-018-0242-x
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15 (10), 565–581. doi:10.1038/s41582-019-0244-7
Hoyer, D., Pazos, A., Probst, A., and Palacios, J. M. (1986). Serotonin receptors in the human brain. I. Characterization and autoradiographic localization of 5-HT1A recognition sites. Apparent absence of 5-HT1B recognition sites. Brain Res. 376 (1), 85–96. doi:10.1016/0006-8993(86)90902-9
Hughes, B., and Herron, C. E. (2019). Cannabidiol reverses deficits in hippocampal LTP in a model of Alzheimer’s disease. Neurochem. Res. 44 (3), 703–713. doi:10.1007/s11064-018-2513-z
Husna Ibrahim, N., Yahaya, M. F., Mohamed, W., Teoh, S. L., Hui, C. K., and Kumar, J. (2020). Pharmacotherapy of alzheimer's disease: Seeking clarity in a time of uncertainty. Front. Pharmacol. 11, 261. doi:10.3389/fphar.2020.00261
Iuvone, T., Esposito, G., Esposito, R., Santamaria, R., Di Rosa, M., and Izzo, A. A. (2004). Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 89 (1), 134–141. doi:10.1111/j.1471-4159.2003.02327.x
Jiang, X., Zhang, Z., Zuo, J., Wu, C., Zha, L., Xu, Y., et al. (2021). Novel cannabidiol−carbamate hybrids as selective BuChE inhibitors: Docking-based fragment reassembly for the development of potential therapeutic agents against Alzheimer's disease. Eur. J. Med. Chem. 223, 113735. doi:10.1016/j.ejmech.2021.113735
Kamboh, M. I. (2004). Molecular genetics of late-onset alzheimer's disease. Ann. Hum. Genet. 68 (4), 381–404. doi:10.1046/j.1529-8817.2004.00110.x
Kastner, A., Hirsch, E. C., Lejeune, O., Javoy-Agid, F., Rascol, O., and Agid, Y. (1992). Is the vulnerability of neurons in the substantia nigra of patients with Parkinson's disease related to their neuromelanin content? J. Neurochem. 59 (3), 1080–1089. doi:10.1111/j.1471-4159.1992.tb08350.x
Katsnelson, A., De Strooper, B., and Zoghbi Huda, Y. (2016). Neurodegeneration: From cellular concepts to clinical applications. Sci. Transl. Med. 8 (364), 364ps18–364ps318. doi:10.1126/scitranslmed.aal2074
Khodadadi, H., Salles É, L., Jarrahi, A., Costigliola, V., Khan, M. B., Yu, J. C., et al. (2021). Cannabidiol ameliorates cognitive function via regulation of IL-33 and TREM2 upregulation in a murine model of alzheimer's disease. J. Alzheimers Dis. 80 (3), 973–977. doi:10.3233/JAD-210026
Kimura, T., Takaya, M., Usami, N., Watanabe, K., and Yamamoto, I. (2019). ∆9-Tetrahydrocannabinol, a major marijuana component, enhances the anesthetic effect of pentobarbital through the CB1 receptor. Forensic Toxicol. 37 (1), 207–214. doi:10.1007/s11419-018-0457-2
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. 4, 575–590. doi:10.1016/j.trci.2018.06.014
Kitamura, Y., Shimohama, S., Koike, H., Kakimura, J.-i., Matsuoka, Y., Nomura, Y., et al. (1999). Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-γ in alzheimer's disease brains. Biochem. Biophys. Res. Commun. 254 (3), 582–586. doi:10.1006/bbrc.1998.9981
Kozela, E., Pietr, M., Juknat, A., Rimmerman, N., Levy, R., and Vogel, Z. (2010). Cannabinoids Delta(9)-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2 microglial cells. J. Biol. Chem. 285 (3), 1616–1626. doi:10.1074/jbc.M109.069294
Kreilaus, F., Przybyla, M., Ittner, L., and Karl, T. (2022). Cannabidiol (CBD) treatment improves spatial memory in 14-month-old female TAU58/2 transgenic mice. Behav. Brain Res. 425, 113812. doi:10.1016/j.bbr.2022.113812
Kumar, P. B. R., Kumar, A. P., Jose, J. A., Prabitha, P., Yuvaraj, S., Chipurupalli, S., et al. (2020). Minutes of PPAR-γ agonism and neuroprotection. Neurochem. Int. 140, 104814. doi:10.1016/j.neuint.2020.104814
Lastres-Becker, I., Molina-Holgado, F., Ramos, J. A., Mechoulam, R., and Fernández-Ruiz, J. (2005). Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: Relevance to Parkinson's disease. Neurobiol. Dis. 19 (1), 96–107. doi:10.1016/j.nbd.2004.11.009
Lauretti, E., Dincer, O., and Praticò, D. (2020). Glycogen synthase kinase-3 signaling in Alzheimer's disease. Biochim. Biophys. Acta. Mol. Cell Res. 1867 (5), 118664. doi:10.1016/j.bbamcr.2020.118664
Leszko, M., and Meenrajan, S. (2021). Attitudes, beliefs, and changing trends of cannabidiol (CBD) oil use among caregivers of individuals with Alzheimer's disease. Complement. Ther. Med. 57, 102660. doi:10.1016/j.ctim.2021.102660
Libro, R., Diomede, F., Scionti, D., Piattelli, A., Grassi, G., Pollastro, F., et al. (2016). Cannabidiol modulates the expression of alzheimer's disease-related genes in mesenchymal stem cells. Int. J. Mol. Sci. 18 (1), E26. doi:10.3390/ijms18010026
Liu, C.-C., Liu, C.-C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 9 (2), 106–118. doi:10.1038/nrneurol.2012.263
Liu, L.-R., Liu, J.-C., Bao, J.-S., Bai, Q.-Q., and Wang, G.-Q. (2020). Interaction of microglia and astrocytes in the neurovascular unit. Front. Immunol. 11, 1024. doi:10.3389/fimmu.2020.01024
Lovell, M. A., Xiong, S., Xie, C., Davies, P., and Markesbery, W. R. (2004). Induction of hyperphosphorylated tau in primary rat cortical neuron cultures mediated by oxidative stress and glycogen synthase kinase-3. J. Alzheimers Dis. 6, 659–671. doi:10.3233/jad-2004-6610
Ly, P. T., Wu, Y., Zou, H., Wang, R., Zhou, W., Kinoshita, A., et al. (2013). Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 123 (1), 224–235. doi:10.1172/JCI64516
Magen, I., Avraham, Y., Ackerman, Z., Vorobiev, L., Mechoulam, R., and Berry, E. M. (2009). Cannabidiol ameliorates cognitive and motor impairments in mice with bile duct ligation. J. Hepatol. 51 (3), 528–534. doi:10.1016/j.jhep.2009.04.021
Maguschak, K. A., and Ressler, K. J. (2008). Beta-catenin is required for memory consolidation. Nat. Neurosci. 11 (11), 1319–1326. doi:10.1038/nn.2198
Martín-Moreno, A. M., Reigada, D., Ramírez, B. G., Mechoulam, R., Innamorato, N., Cuadrado, A., et al. (2011). Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to alzheimer's disease. Mol. Pharmacol. 79 (6), 964.
Marucci, G., Buccioni, M., Ben, D. D., Lambertucci, C., Volpini, R., and Amenta, F. (2021). Efficacy of acetylcholinesterase inhibitors in Alzheimer's disease. Neuropharmacology 190, 108352. doi:10.1016/j.neuropharm.2020.108352
Migliore, L., and Coppedè, F. (2009). Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 667 (1), 82–97. doi:10.1016/j.mrfmmm.2008.10.011
Millar, S. A., Maguire, R. F., Yates, A. S., and O'Sullivan, S. E. (2020). Towards better delivery of cannabidiol (CBD). Pharm. (Basel) 13 (9), E219. doi:10.3390/ph13090219
O'Sullivan, S. E. (2016). An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 173 (12), 1899–1910. doi:10.1111/bph.13497
O'Sullivan, S. E., Sun, Y., Bennett, A. J., Randall, M. D., and Kendall, D. A. (2009). Time-dependent vascular actions of cannabidiol in the rat aorta. Eur. J. Pharmacol. 612 (1-3), 61–68. doi:10.1016/j.ejphar.2009.03.010
Onishi, T., Iwashita, H., Uno, Y., Kunitomo, J., Saitoh, M., Kimura, E., et al. (2011). A novel glycogen synthase kinase-3 inhibitor 2-methyl-5-(3-{4-[(S )-methylsulfinyl]phenyl}-1-benzofuran-5-yl)-1, 3, 4-oxadiazole decreases tau phosphorylation and ameliorates cognitive deficits in a transgenic model of Alzheimer’s disease. J. Neurochem. 119 (6), 1330–1340. doi:10.1111/j.1471-4159.2011.07532.x
Park, J.-Y., Kim, K. S., Lee, S.-B., Ryu, J.-S., Chung, K. C., Choo, Y.-K., et al. (2009). On the mechanism of internalization of α-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 110 (1), 400–411. doi:10.1111/j.1471-4159.2009.06150.x
Peres, F. F., Lima, A. C., Hallak, J. E. C., Crippa, J. A., Silva, R. H., and Abílio, V. C. (2018). "Cannabidiol as a promising strategy to treat and prevent movement disorders?". Frontiers in Pharmacology. 9. 482. doi:10.3389/fphar.2018.00482
Pertwee, R. G. (2006). Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 147, S163–S171. doi:10.1038/sj.bjp.0706406
Pertwee, R. G. (2008). The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 153 (2), 199–215. doi:10.1038/sj.bjp.0707442
Piomelli, D. (2003). The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 4 (11), 873–884. doi:10.1038/nrn1247
Pocivavsek, A., Burns, M. P., and Rebeck, G. W. (2009). Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia 57 (4), 444–453. doi:10.1002/glia.20772
Ramírez, B. G., Blázquez, C., Pulgar, T. G. d., Guzmán, M., and Ceballos, M. L. de (2005). Prevention of alzheimer's disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 25 (8), 1904–1913. doi:10.1523/JNEUROSCI.4540-04.2005
Rey, A. A., Purrio, M., Viveros, M.-P., and Lutz, B. (2012). Biphasic effects of cannabinoids in anxiety responses: CB1 and GABA(B) receptors in the balance of GABAergic and glutamatergic neurotransmission. Neuropsychopharmacology 37 (12), 2624–2634. doi:10.1038/npp.2012.123
Reynolds, A. D., Kadiu, I., Garg, S. K., Glanzer, J. G., Nordgren, T., Ciborowski, P., et al. (2008). Nitrated alpha-synuclein and microglial neuroregulatory activities. J. Neuroimmune Pharmacol. 3 (2), 59–74. doi:10.1007/s11481-008-9100-z
Rizek, P., Kumar, N., and Jog, M. S. (2016). An update on the diagnosis and treatment of Parkinson disease. CMAJ Can. Med. Assoc. J. = J. de l'Association medicale Can. 188 (16), 1157–1165. doi:10.1503/cmaj.151179
Russo, E. B., Burnett, A., Hall, B., and Parker, K. K. (2005). Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res. 30 (8), 1037–1043. doi:10.1007/s11064-005-6978-1
Russo, E., and Guy, G. W. (2006). A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypotheses 66 (2), 234–246. doi:10.1016/j.mehy.2005.08.026
Sagredo, O., Ramos, J. A., Decio, A., Mechoulam, R., and Fernández-Ruiz, J. (2007). Cannabidiol reduced the striatal atrophy caused 3-nitropropionic acid in vivo by mechanisms independent of the activation of cannabinoid, vanilloid TRPV1 and adenosine A2A receptors. Eur. J. Neurosci. 26 (4), 843–851. doi:10.1111/j.1460-9568.2007.05717.x
Salim, S. (2017). Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 360 (1), 201–205. doi:10.1124/jpet.116.237503
Santos, N. A. G., Martins, N. M., Sisti, F. M., Fernandes, L. S., Ferreira, R. S., Queiroz, R. H. C., et al. (2015). The neuroprotection of cannabidiol against MPP⁺-induced toxicity in PC12 cells involves trkA receptors, upregulation of axonal and synaptic proteins, neuritogenesis, and might be relevant to Parkinson's disease. Toxicol. Vitro 30 (1), 231–240. Part B). doi:10.1016/j.tiv.2015.11.004
Sastre, M., Walter, J., and Gentleman, S. M. (2008). Interactions between APP secretases and inflammatory mediators. J. Neuroinflammation 5 (1), 25. doi:10.1186/1742-2094-5-25
Schmuhl, E., Ramer, R., Salamon, A., Peters, K., and Hinz, B. (2014). Increase of mesenchymal stem cell migration by cannabidiol via activation of p42/44 MAPK. Biochem. Pharmacol. 87 (3), 489–501. doi:10.1016/j.bcp.2013.11.016
Scuderi, C., Steardo, L., and Esposito, G. (2014). Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARγ involvement. Phytother. Res. 28 (7), 1007–1013. doi:10.1002/ptr.5095
Talantova, M., Sanz-Blasco, S., Zhang, X., Xia, P., Akhtar, M. W., Okamoto, S.-i., et al. (2013). Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. U. S. A. 110 (27), E2518–E2527. doi:10.1073/pnas.1306832110
Tamagno, E., Parola, M., Bardini, P., Piccini, A., Borghi, R., Guglielmotto, M., et al. (2005). Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 92 (3), 628–636. doi:10.1111/j.1471-4159.2004.02895.x
Thomas, A., Baillie, G. L., Phillips, A. M., Razdan, R. K., Ross, R. A., and Pertwee, R. G. (2007). Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 150 (5), 613–623. doi:10.1038/sj.bjp.0707133
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer's disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomedicine 14, 5541–5554. doi:10.2147/IJN.S200490
Watt, G., Chesworth, R., Przybyla, M., Ittner, A., Garner, B., Ittner, L. M., et al. (2020). Chronic cannabidiol (CBD) treatment did not exhibit beneficial effects in 4-month-old male TAU58/2 transgenic mice. Pharmacol. Biochem. Behav. 196, 172970. doi:10.1016/j.pbb.2020.172970
Watt, G., Shang, K., Zieba, J., Olaya, J., Li, H., Garner, B., et al. (2020). Chronic treatment with 50 mg/kg cannabidiol improves cognition and moderately reduces Aβ40 levels in 12-month-old male AβPPswe/PS1ΔE9 transgenic mice. J. Alzheimers Dis. 74 (3), 937–950. doi:10.3233/JAD-191242
Wilcock, G. K., Esiri, M. M., Bowen, D. M., and Smith, C. C. (1982). Alzheimer's disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J. Neurol. Sci. 57 (2-3), 407–417. doi:10.1016/0022-510x(82)90045-4
Keywords: cannabidiol, neuroinflammation, oxidative stress, neurodegenerative diseases, Alzheimer’s disease, Parkinson’s disease
Citation: Bhunia S, Kolishetti N, Arias AY, Vashist A and Nair M (2022) Cannabidiol for neurodegenerative disorders: A comprehensive review. Front. Pharmacol. 13:989717. doi: 10.3389/fphar.2022.989717
Received: 08 July 2022; Accepted: 27 September 2022;
Published: 25 October 2022.
Edited by:
Luca Ferraro, University of Ferrara, ItalyReviewed by:
Md. Siddiqul Islam, Southeast University, BangladeshCopyright © 2022 Bhunia, Kolishetti, Arias, Vashist and Nair. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sukanya Bhunia, c2JodW5pYUBmaXUuZWR1; Madhavan Nair, bmFpcm1AZml1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.