
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 14 October 2022
Sec. Cardiovascular and Smooth Muscle Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.988408
 Genlong Xue1†Xiaolei Yang1,2†Ge Zhan1†Xin Wang2,3Jinghan Gao1Yong Zhao1Xinying Wang1Jiatian Li1Zhenwei Pan4*Yunlong Xia1,2*
Genlong Xue1†Xiaolei Yang1,2†Ge Zhan1†Xin Wang2,3Jinghan Gao1Yong Zhao1Xinying Wang1Jiatian Li1Zhenwei Pan4*Yunlong Xia1,2*Background: Recent clinical trials indicate that sodium–glucose cotransporter 2 (SGLT2) inhibitors improve cardiovascular outcomes in myocardial infarction (MI) patients, but the underlying mechanisms remain unknown. As arrhythmia often occurs during myocardial infarction, it is the main cause of death.
Objective: The purpose of this study was to investigate the influence of empagliflozin (EMPA), an SGLT2 inhibitor, on cardiac electrophysiological remodeling and arrhythmia susceptibility of myocardial infarction mice.
Methods: ECG was obtained from mice 1 week after MI to determine the QT interval. In an electrophysiological study and optical mapping was performed to evaluate the function of EMPA and underlying mechanisms of post-myocardial-infarction in mice.
Results: EMPA treatment significantly reduced the QT interval of MI mice (MI + EMPA 50.24 ms vs. MI 64.68 ms). The membrane potential and intracellular Ca [Cai] were mapped from 13 MI hearts and five normal hearts using an optical mapping technique. A dynamic pacing protocol was used to determine action potential duration and [Cai] at baseline and after EMPA (10 umol/L) infusion. EMPA perfusion did not change the APD80 and CaT80 in normal ventricles while shortening them in an infarct zone, bordering zone, and remote zone of MI hearts at 200 ms, 150 ms, 120 ms, and 100 ms pacing cycle length. The conduction velocity of infarcted ventricles was 0.278 m/s and 0.533 m/s in normal ventricles at baseline (p < 0.05). After EMPA administration, the conduction velocity of infarcted ventricles increased to 0.363 m/s, whereas no significant changes were observed in normal ventricles. The action potential rise time, CaT rise time, and CaT tau time were improved after EMPA perfusion in infarcted ventricles, whereas no significant changes were observed in normal ventricles. EMPA decreases early afterdepolarizations premature ventricular beats, and ventricular fibrillation (VF) in infarcted ventricles. The number of phase singularities (baseline versus EMPA, 6.26 versus 3.25), dominant frequency (20.52 versus 10.675 Hz), and ventricular fibrillation duration (1.072 versus 0.361 s) during ventricular fibrillation in infarcted ventricles were all significantly decreased by EMPA.
Conclusion: Treatment with EMPA improved post-MI electrophysiological remodeling and decreased substrate for VF of MI mice. The inhibitors of SGLT2 may be a new class of agents for the prevention of ventricle arrhythmia after chronic MI.
Ventricular arrhythmias are the main cause of mortality in patients suffering from myocardial infarction (MI) (Behnes et al., 2018; Frontera et al., 2020). Myocardial infarction is associated with significant electrophysiological remodeling including aberrant cardiac conduction and altered action potential duration (APD) in post-MI hearts (Jiang et al., 2007; Mendonca Costa et al., 2018; Chowdhury et al., 2021). The inhomogeneous prolongation of APD and slowing of cardiac conduction velocity in the infarct zone (IZ), bordering zone (BZ), and remote zone (RZ) are prone to induce arrhythmias in infarcted hearts (Mendonca Costa et al., 2018; Wen et al., 2018). Post-MI malignant ventricular arrhythmias were also related to abnormal Ca2+ handling (Belevych et al., 2012). The functional disorder of calcium transient dynamics leads to Ca2+ cycling disturbance in infarcted ventricles, which induces secondary rises of [Cai] and triggers early afterdepolarizations (EADs) and ventricular arrhythmias (Chang et al., 2013). Irregularity of APD and calcium transient has been taken as a hallmark of cardiac electrical disturbances after MI.
Sodium–glucose cotransporter 2 (SGLT2) inhibitors such as empagliflozin (EMPA) comprise a group of anti-hyperglycemic drugs that promote urinary excretion of glucose by inhibiting glucose reabsorption through SGLT2 blockade (Kondo et al., 2021). Surprisingly, recent large-scale cardiovascular safety trials demonstrate that SGLT2 inhibitors exert not only antidiabetic actions but also cardioprotective effects (Zelniker and Braunwald, 2020)and the use of SGLT2 inhibitors in patients with diabetes mellitus (DM), chronic kidney disease (CKD), and heart failure (HF) to reduce related cardiac complications and comorbidities of cardiac arrhythmias (Li et al., 2021). Clinical research findings showed that dapagliflozin proved to be clinically effective in patients with heart failure (HF) with reduced ejection fraction regardless of diabetes, suggesting its robust benefits in some specific patients with HF (Tanaka and Node, 2020). Kondo et al. (2021) discovered that canagliflozin suppressed myocardial NADPH oxidase activity and improved NOS coupling to anti-inflammatory and ant-apoptotic effects in the human myocardium, indicating that the cardioprotective effect was not related to their systemic glucose-lowering function.
SGLT2 inhibitor, empagliflozin, was shown to attenuate cardiac fibrosis (Li et al., 2019a) and pressure overload-induced heart failure (Yurista et al., 2019). Lately, Philippaert et al. (2021) demonstrated that empagliflozin inhibits late-INa in a transaortic constriction (TAC)-induced mouse heart failure model, prevents the activation of the NLRP3 inflammasome, and improves functional recovery in an ex vivo cardiac ischemia/reperfusion injury model. However, the action of empagliflozin on electrophysiological remodeling and arrhythmia susceptibility during chronic myocardial infarction remains unclear.
Therefore, in this study, we explored the effects of empagliflozin on action potential repolarization and dynamics of calcium transient in chronic infarcted ventricles of mice. We found that empagliflozin reduced ventricular arrhythmia susceptibility by shortening APD80 and CaT80 of infarcted ventricles, improving the dynamics of AP rise time, CaT rise time, and Tau and decreasing the occurrences of EADs, PVBs, and secondary rises of [Cai]. The study highlights a novel mechanism for the regulation of empagliflozin on ventricular arrhythmia and reveals its therapeutic potential for post-MI-related cardiac arrhythmias.
This study was approved and monitored by the Laboratory Animal Resource Center at Dalian Medical University and conformed to the Guide for the care and use of laboratory animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Adult C57BL/6 mice (8 weeks old) were provided by the animal center at Liaoning Changsheng Biotechnology Co., Ltd.
C57BL/6 mice underwent MI, as described previously (Alibhai et al., 2014). Mice were anesthetized by spontaneous inhalation and maintained under general anesthesia with 2% isoflurane. Animals were intubated and ventilated with air using a small-animal respirator. The heart was exposed by a left-side limited thoracotomy, and the left ventricle was visualized and the pericardial sac was ruptured to expose the left anterior descending (LAD) coronary artery. The LAD was ligated with a 7–0 prolene suture 2 mm below the tip of the left auricle. We studied the EMPA (10 umol) impact on MI hearts that followed up 1 week after MI.
Mice were anesthetized as previously described (Si et al., 2020). Intervals (HR, QRS, QT, and RR) were measured and analyzed using a BL-420 Biological Signal Acquisition System (Chengdu Techman Software Co. Ltd., Chengdu, China). Surface electrocardiograph (ECG) was recorded. QTc interval was calculated as QT interval (ms) divided by the square root of the RR interval (ms) divided by 100 (Mitchell et al., 1998). The maximum slope-–intercept method was used to define the end of the T-wave.
Mice were heparinized and euthanized with 2,2,2-tribromoethanol (200 mg/kg, intraperitoneal injection; Sigma, St Louis, MO, United States). The heart was isolated and Langendorff perfused with Tyrode’s solution (NaCl 128.2 mM, CaCl2•2H2O 1.3 mM, KCl 4.7 mM, MgCl2•6H2O 1.85 mM, NaH2PO4•2H2O 1.19 mM, Na2CO3 20 mM, and glucose 11.1 mM; pH 7.35) at 37°C. After 10 min of stabilization, the hearts were stained with Rhod-2 AM (1.48 μM) for [Cai] and RH237 (10 μM) for membrane voltage (Vm) mapping. The double-stained hearts were excited at 710 nm wavelength for Vm and 580 nm wavelength for [Cai] using a two-wavelength light-emitting device. The fluorescence was filtered and recorded simultaneously with an OMS-PCIE-2002 camera (MappingLab, United Kingdom) at 1 ms/frame and 100 × 100 pixels with a spatial resolution of 0.35 × 0.35 mm2 per pixel. Optical signals were processed with both spatial (3 × 3 pixels Gaussian filter). Phase mapping was performed to evaluate the location and evolution of phase singularities (PSs). Blebbistatin (10–20 μmol/L, Tocris, Ellisville, and MO) was used to inhibit motion artifacts during optical mapping.
A pair of hook bipolar electrodes was inserted into the bottom of the heart for pacing. A pseudo-electrocardiograph was obtained with widely spaced bipolar electrodes to determine the ventricular rhythm. The ventricles were initially paced at a constant pacing cycle length (PCL) of 200 ms. The PCLs were progressively shortened (200, 150, 120, and 100 ms) with a duration of 3 s to record Vm and [Cai] mapping. Pacing was performed with S1–S2 using a basic PCL of 100 ms and with S2 decreased by 2 ms until the ventricular effective refractory period was reached. Finally, optical recordings were then performed during ventricular tachycardia (VT)/VF inducibility.
Data are expressed as mean ± SEM. Statistical analysis was performed using an unpaired Student’s t-test or one-way analysis of variance (ANOVA), followed by Tukey’s post hoc analysis. p < 0.05 was considered statistically different.
A surface ECG was measured to determine the QT interval from anesthetized WT, WT + EMPA, MI, and MI + EMPA mice. In infarcted hearts, EMPA treatment significantly shortened the QT interval (MI vs. MI + EMPA: 64.68 vs. 50.24 ms; p < 0.05) (Figures 1A and B) and QTc interval (MI vs. MI + EMPA: 57.39 vs. 45.80 ms; p < 0.05) (Figure 1C). The QT intervals were not changed by EMPA in sham control (WT) mice. EMPA significantly increased the heart rate of MI hearts (MI vs. MI + EMPA: 434.98 bpm vs. 479.84 bpm; p < 0.05), while there was no change of heart rate in WT mice (Figure 1D). Furthermore, QRS intervals were shortened in MI + EMPA mice compared to MI (MI vs. MI + EMPA: 18.20 vs. 14.48 ms; p < 0.05) (Figure 1D). EMPA treatment led to reduced development of PVBs and VF in MI mice (Figure 1F).
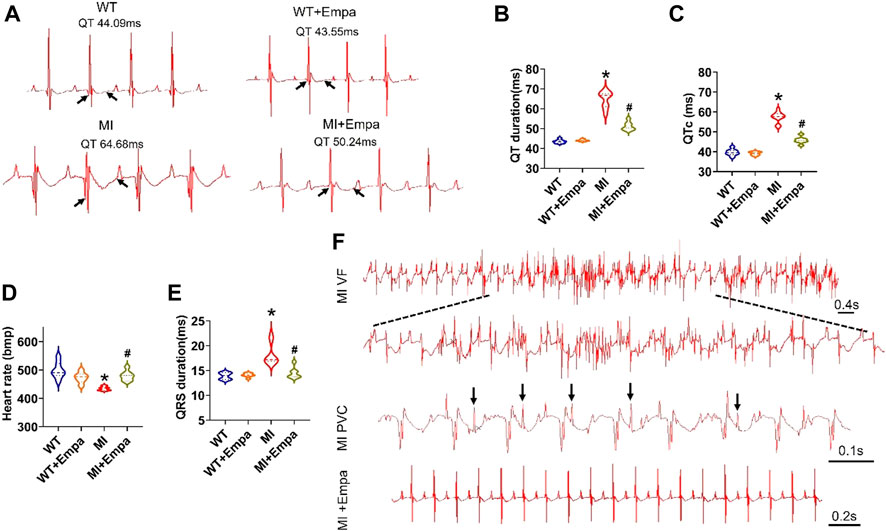
FIGURE 1. Effects of EMPA on QT interval and ventricular arrhythmias in chronic MI mice. (A,B) Representative ECG traces of the QT interval in WT, WT + EMPA, MI, and MI + EMPA. N = 6–10. *p < 0.05 vs. WT; #p < 0.05 vs. MI. p-values were determined using the unpaired t test. (C) QTc of WT, WT + EMPA, MI, and MI + EMPA mice. N = WT: 8; WT + EMPA: 6; MI: 10; MI + EMPA: 7; *p < 0.05 vs. WT; and #p < 0.05 vs. MI. (D,E) heart rate and QRS of WT, WT + EMPA, MI, and MI + EMPA mice. N = 6–9; *p < 0.05 vs. WT; and #p < 0.05 vs. MI. (F) Representative ECG traces of the VF and PVCs in MI and MI + EMPA mice.
We further evaluated the effects of EMPA on APD of ex vivo perfused MI hearts by optical mapping. At baseline, the mean APD at 80% repolarization (APD80) in the IZ, BZ, and RZ of MI ventricles was longer than that in the normal ventricles at all PCLs (Figures 2A,D). EMPA treatment did not produce a significant influence on APD80 in normal ventricles, and the ΔAPD80 is very small (Figures 2B,C). In the infarcted ventricles, EMPA shortened APD80 at all pacing cycle lengths (PCLs) in IZ, BZ, and RZ; however, the effects were more apparent at longer PCLs, i.e., in the IZ of infarcted ventricles, at PCL200 ms, baseline: 105 ms, EMPA: 72.75 ms, p < 0.05; at PCL 150 ms, baseline: 90.75 ms, EMPA: 63.75 ms, p < 0.05 (Figures 2D,E); in the BZ of infarcted ventricles, at PCL200 ms, baseline:92.91 ms, EMPA: 69.28 ms, p < 0.05; at PCL 150 ms, baseline: 77.25 ms, EMPA: 64.14 ms, p < 0.05 (Figures 2D,G); and in the RZ of infarcted ventricles, at PCL200 ms, baseline: 81.24 ms, EMPA: 63.29 ms, p < 0.05; at PCL 150 ms, baseline: 75.21 ms, EMPA: 56.81 ms, p < 0.05; p < 0.05, Figures 2D,I). EMPA also shortened ΔAPD80 at all pacing cycle lengths (PCLs) in the infarcted ventricles IZ, BZ, and RZ (Figures 2F,H,J).
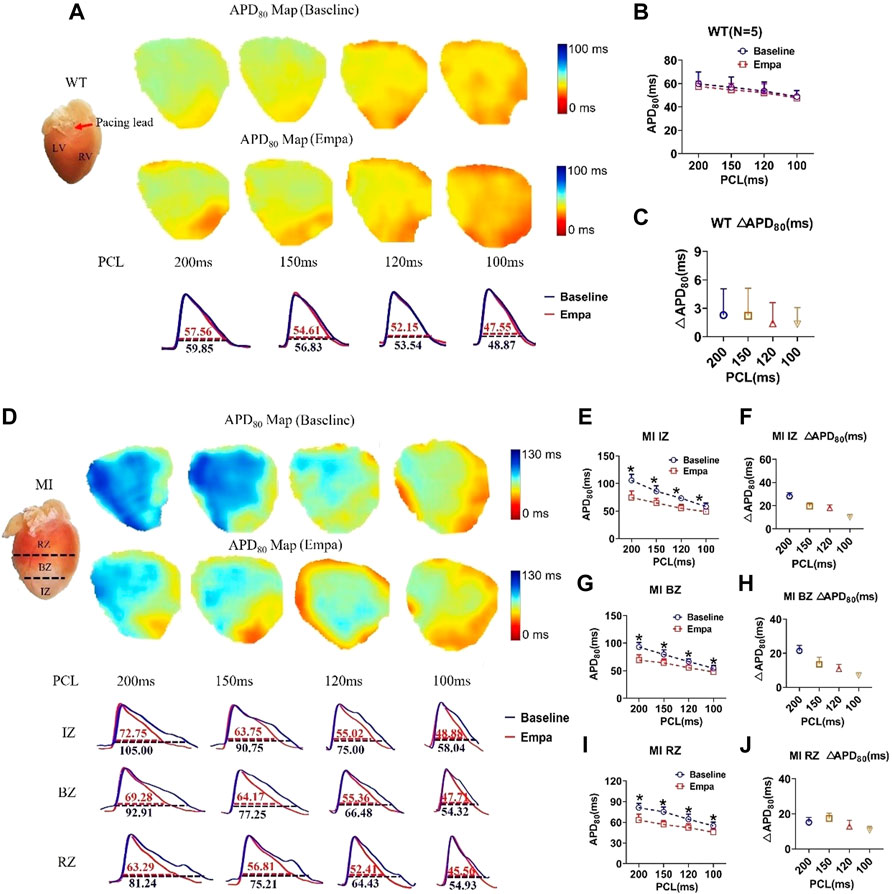
FIGURE 2. Effects of EMPA on action potential duration (APD) at different pacing cycle lengths (PCLs) in normal and infarcted hearts. (A) Representative membrane potential traces and APD at 80% repolarization (APD80) maps at baseline and in the presence of EMPA (10 nmol/L) in normal ventricles. (B,C) APD80 and ΔAPD80 associated with different PCLs at baseline and after EMPA infusion in normal ventricles. N = 5. (D) Representative membrane potential traces and APD at 80% repolarization (APD80) maps at baseline and in the presence of EMPA (10 nmol/L) in IZ, BZ, and RZ zones of infarcted hearts (E,F) APD80 and ΔAPD80 associated with different PCLs at baseline and after EMPA infusion in infarcted ventricles of IZ. N = 9–12. *p < 0.05 vs. baseline, p-values were determined by unpaired t test. (G,H) APD80 and ΔAPD80 associated with different PCLs at baseline and after EMPA infusion in infarcted ventricles of BZ. N = 10–12. *p < 0.05 vs. baseline. (I,J) APD80 and ΔAPD80 associated with different PCLs at baseline and after EMPA infusion in infarcted ventricles of RZ. N = 9–11. *p < 0.05 vs. baseline.
In the infarcted ventricles, the conduction velocity map showed that EMPA treatment did not significantly change conduction velocity in normal ventricles (Figures 3A,B), while it significantly increased the conduction velocity of infarcted hearts (Figures 3C,D). AP rise maps at baseline and after EMPA infusion did not change in all PCLs from recordings in the WT mice hearts (Figures 3E,F). In the infarcted ventricles, EMPA shortened AP rise time at all PCLs in IZ, BZ, and RZ zones, and the effects were more apparent at longer PCLs (Figures 3G–J).
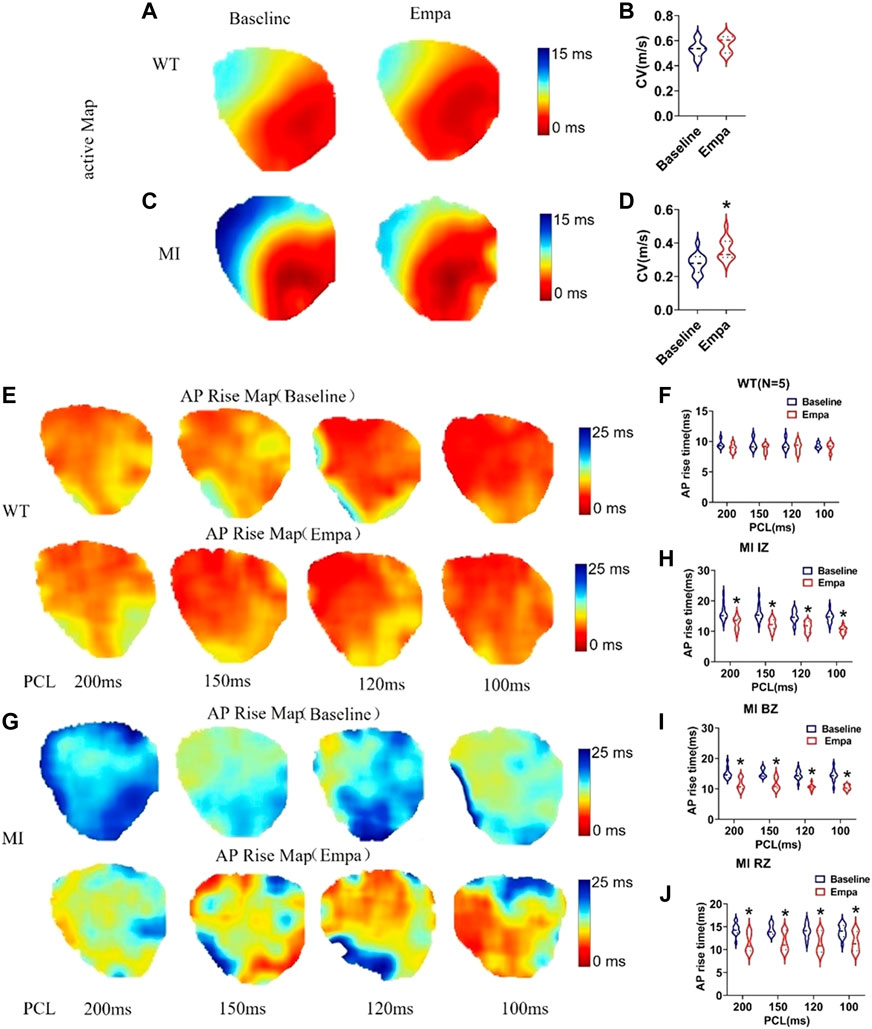
FIGURE 3. Effects of EMPA on conduction velocity (CV) and AP rise time at different pacing cycle lengths (PCLs) in normal and infarcted ventricles. (A,C) Maps of activation at baseline and in the presence of EMPA (10 umol/L) in normal and infarcted ventricles. (B,D) conduction velocity at baseline and after EMPA infusion in normal and infarcted ventricles. N = 5 for WT; N = 13 for MI. *p < 0.05 versus baseline. p-values were determined using the unpaired t test. (E) Action potential (AP) rise time (TRise) maps associated with different PCLs at baseline and in the presence of EMPA (10 umol/L) in normal ventricles. (F) TRise associated with different PCLs at baseline and after EMPA infusion in normal ventricles. N = 5. (G) TRise maps associated with different PCLs at baseline and in the presence of EMPA in infarcted ventricles. (H–J) TRise associated with different PCLs at baseline and after EMPA infusion in infarcted ventricles of IZ, BZ, and RZ. N = 11–13. *p < 0.05 versus baseline. p-values were determined using the unpaired t test.
We investigated the effects of EMPA on Ca2+ cycling characteristics by comparing the 80% Ca2+ transient duration (CaTD80). CaT80 maps at baseline and after EMPA infusion were not changed in the hearts of WT mice. EMPA did not significantly change CaT80 in normal ventricles, and ΔCaT80 is very small (Figures 4A–C). In the infarcted ventricles, EMPA shortened CaT80 at all PCLs in IZ, BZ, and RZ zones, and the effects were more apparent at longer PCLs (Figures 4D,E,J,I). EMPA also shortened ΔCaT80 at all PCLs in the IZ, BZ, and RZ zones of infarcted ventricles (Figures 4F,H,J).
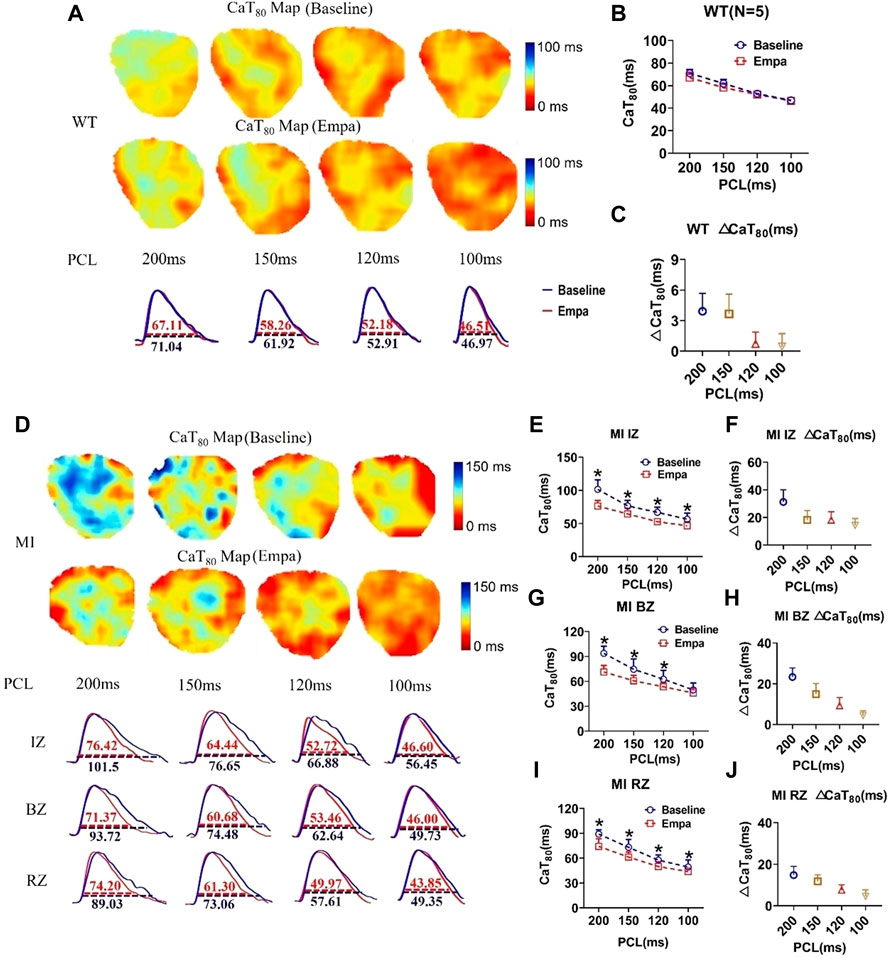
FIGURE 4. Effects of EMPA on Ca2+ transient duration (CaTD) at different pacing cycle lengths (PCLs) in normal and infarcted ventricles. (A) Representative Ca2+ transient traces and CaT at 80% repolarization (CaT80) maps at baseline and in the presence of EMPA (10 umol/L) in normal ventricles. (B,C) CaT80 and ΔCaT80 associated with different PCLs at baseline and after EMPA infusion in normal ventricles. N = 5. (D) Representative Ca2+ transient traces and CaT80 maps at baseline and in the presence of EMPA in infarcted ventricles of IZ, BZ, and RZ. (E,F) CaT80 and ΔCaT80 associated with different PCLs at baseline and after EMPA infusion in infarcted ventricles of IZ. N = 9. *p < 0.05 vs. baseline, p-values were determined by unpaired t test. (G,H) CaT80 and ΔCaT80 associated with different PCLs at baseline and after EMPA infusion in infarcted ventricles of BZ. N = 10–11. *p < 0.05 vs. baseline. (I,J) CaT80 and ΔCaT80 associated with different PCLs at baseline and after EMPA infusion in infarcted ventricles of RZ. N = 9. *p < 0.05 vs. baseline.
In the normal ventricles, the CaT rise map did not change at baseline and after EMPA infusion at all PCLs (Figures 5A,B). However, in the infarcted ventricles, after EMPA infusion, the CaT rise time significantly shortened at all PCLs in IZ, BZ, and RZ zones of MI hearts (Figures 5C–F). [Cai] transient decay constant Tau maps were not changed in all PCLs at baseline and after EMPA infusion in the hearts of WT mice (Figures 5G,H). In infarcted ventricles, EMPA decreased Tau at all PCLs in IZ, BZ, and RZ zones (Figures 5I–L).
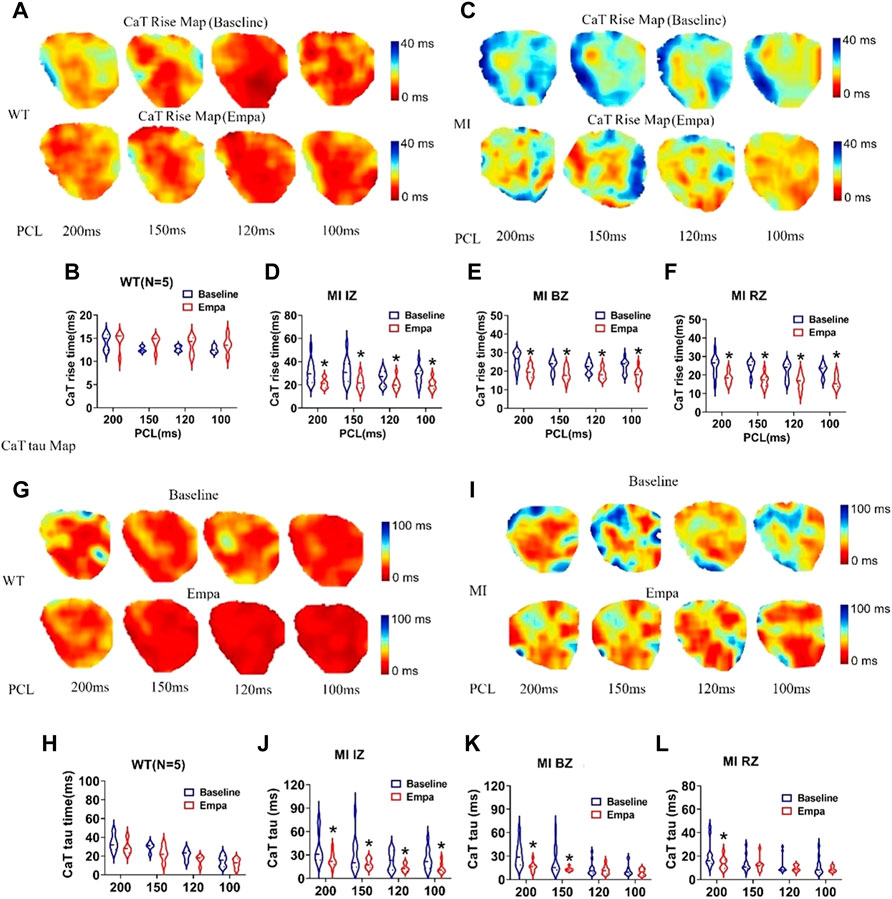
FIGURE 5. Effects of EMPA on Ca2+ transient rise time (CaTRise) and Tau at different pacing cycle lengths (PCLs) in normal and infarcted ventricles. (A) Ca2+ transient rise time (CaTRise) maps associated with different PCLs at baseline and in the presence of EMPA (10 umol/L) in normal ventricles. (B) CaTRise associated with different PCLs at baseline and after EMPA infusion in normal ventricles. N = 5. (C) CaTRise maps associated with different PCLs at baseline and in the presence of EMPA in infarcted ventricles. (D–F) CaTRise associated with different PCLs at baseline and after EMPA infusion in infarcted ventricles of IZ, BZ, and RZ. N = 9–12. *p < 0.05 versus baseline. p-values were determined using an unpaired t test. (G,H) Ca2+ transient decay time (Tau) maps and Tau associated with different PCLs at baseline and in the presence of EMPA normal ventricles. N = 5. (I–L) Tau maps and Tau associated with different PCLs at baseline and in the presence of EMPA in infarcted ventricles of IZ, BZ, and RZ. N = 10–13. *p < 0.05 versus baseline. p-values were determined using an unpaired t test.
We observed a secondary rise of [Cai] in infarcted ventricles both at baseline and after EMPA administration (Figures 6A,B). The secondary rise of [Cai] was decreased in the presence of EMPA, along with reduced EADs (red arrows at site). At baseline, the spontaneous secondary rise of [Cai] was detected, and EADs was increased in infarcted ventricles. The activation sites of EADs beats colocalized with the highest secondary rises of [Cai] regions. We analyzed the total EADs episodes in infarcted hearts and found that it was inhibited by EMPA (baseline: 15.625, EMPA: 5.125 episodes per heart). The activation site of EADs always occurred at the site with longer APD and larger amplitude of the secondary rises of [Cai]. EMPA administration reduced secondary [Cai] rises, [Cai] transient duration, EAD induction rate, and EAD episodes (Figures 6C,D).
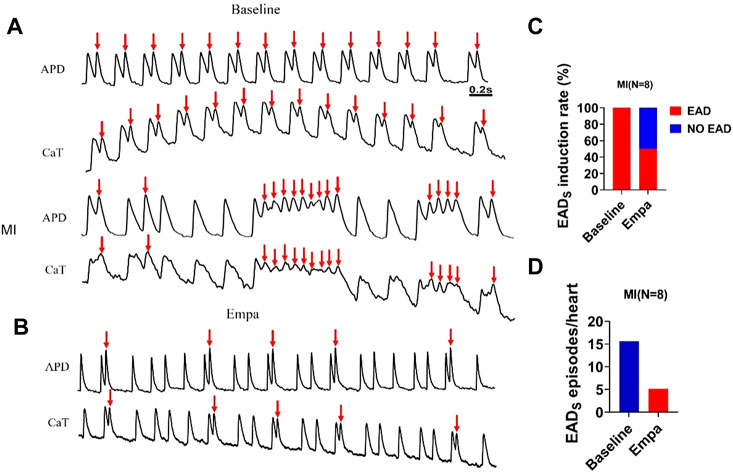
FIGURE 6. EMPA reduced secondary rises of [Cai] and EADs in infarcted ventricles. (A,B) EAD and Secondary rises of [Cai] traces at baseline and in the presence of EMPA in infarcted ventricles. (C,D) EAD induction rate and EADs episodes at baseline and in the presence of EMPA. N = 8.
VF was rarely induced in the normal ventricles at a PCL of 100 ms at baseline or after EMPA with an S1/S2 pacing protocol (Figure 7A). In infarcted ventricles, at baseline, there was significantly induced VF at a PCL of 100 ms. We performed a specific focus on how VFs are initiated in the infarcted ventricles. An example of VF induced with an S1/S2 pacing protocol is shown in Figure 7B Active maps of the 1–7 wave showed the earliest activation of the VF occurred at the sites. After EMPA infusion, either VF induction rate, VF episodes, or VF duration was decreased in infarcted ventricles (Figures 7C–E).
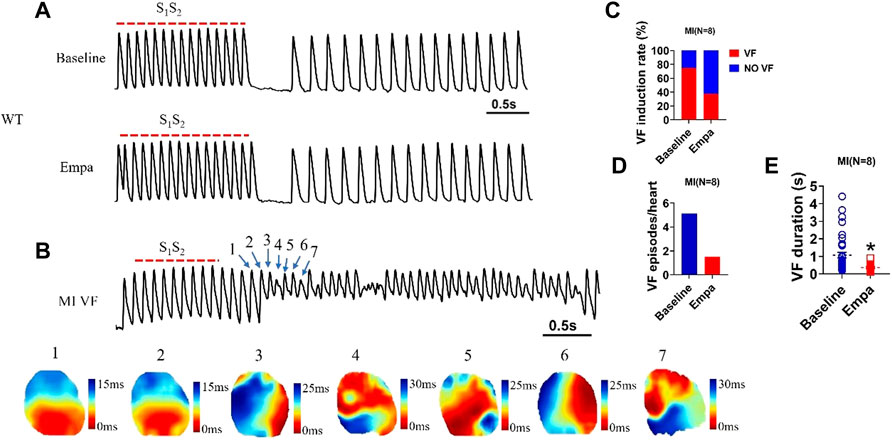
FIGURE 7. EMPA reduced the VF vulnerability in infarcted ventricles. (A) Ventricular fibrillation (VF) induced by S1S2 pacing at baseline and in the presence of EMPA in normal ventricles. N = 5 mice. (B) Ventricular fibrillation (VF) and active map by S1S2 pacing at baseline in infarcted ventricles. N = 8 mice. (C–E) VF induction rate and duration at baseline and in the presence of EMPA in infarcted ventricles. N = 8 mice. *p < 0.05 versus baseline. p-values were determined using an unpaired t test.
We analyzed the VF dynamics in infarcted ventricles, and consecutive phase maps sampled at 10 ms intervals during VF were analyzed for phase singularities (PSs). EMPA decreased the number of phase singularities compare to baseline (p < 0.05; Figures 8A,E). The dominant frequency of VF was decreased from 20.52 Hz at baseline to 10.675 Hz after EMPA infusion (p < 0.05; Figures 7B–D).
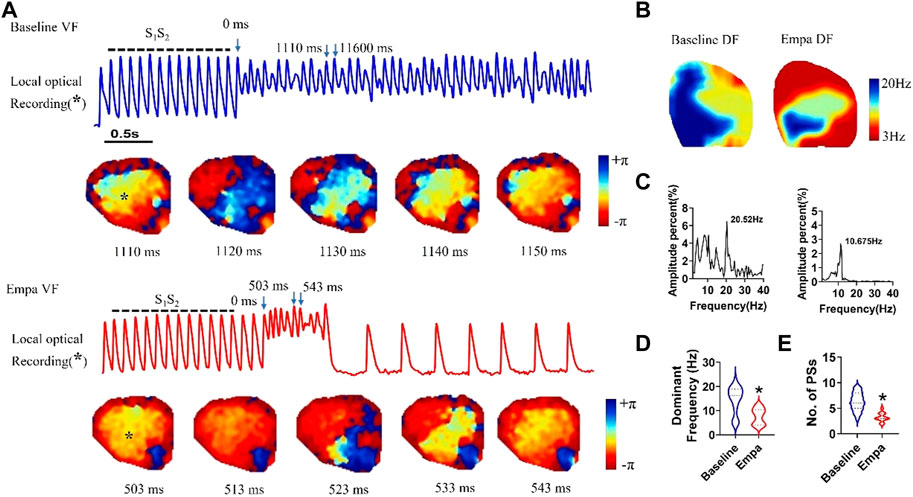
FIGURE 8. Effects of EMPA on the wave breaks and dominant frequency of ventricular fibrillation in infarcted ventricles. (A) Recordings of S1S2 pacing-induced sustained VF episodes at baseline and after EMPA infusion in an infarcted ventricle. Top, corresponding optical recording of VF at the asterisk site. Consecutive phase maps sampled at 10 ms intervals during VF at baseline and after EMPA infusion. (B–D) DF distribution of VF at baseline and after EMPA infusion. N = 10 mice. *p < 0.05 versus baseline. (E) Effects of EMPA on the number of phase singularities before and after EMPA infusion in infarcted ventricles. Note that the numbers of phase singularity are decreased by EMPA. N = 10 mice. *p < 0.05 versus baseline. p-values were determined using an unpaired t test.
In this paper, we identified the following key observations (Figure 9): 1) EMPA inhibited the prolongation of QT interval and induction of ventricular arrhythmias in infarcted ventricles; 2) the APD80 and AP rise time were shortened by EMPA in the IZ, BZ, and RZ zones of infarcted ventricles; 3) the CaT80, CaT rise time, and Tau were shortened by EMPA in the IZ, BZ, and RZ zones of infarcted ventricles; 4) EMPA reversibly reduced the incidence of calcium disturbances-induced EADs and VF in infarcted ventricles; and 5) EMPA prevented activation of the PSs and DF and improved functional recovery in post-MI hearts.
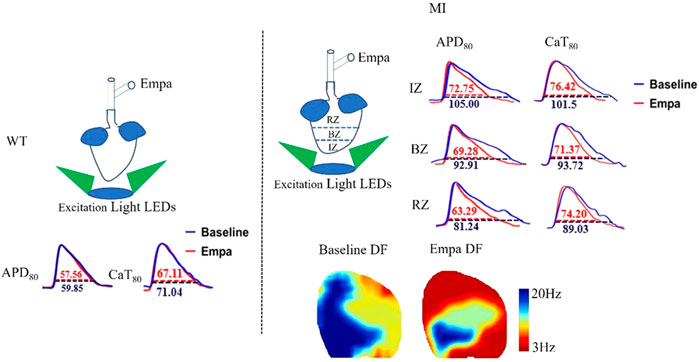
FIGURE 9. Schematic summary of the signaling pathway of EMPA and arrhythmia in post-MI.
The role of SGLT isoforms in myocardial physiology is controversial (Di Franco et al., 2017; Ng et al., 2018). However, studies have shown that SGLT1 is abundantly expressed in the heart, whereas SGLT2 is barely detectable (Banerjee et al., 2009; Li et al., 2019b; Kondo et al., 2021). SGLT2 inhibitors improve heart failure process independent of hypoglycemic mechanisms (Raz and Cahn, 2016; Shah and Fang, 2022). Empagliflozin is the most selective SGLT2 inhibitor (Anker and Butler, 2018), it suppresses cardiomyocyte autophagic cell death to confer cardioprotective effects. In myocardial infarction (MI) mouse models with and without diabetes mellitus, EMPA treatment significantly reduced the infarct size and myocardial fibrosis (Jiang et al., 2022). In addition to participating in the process of heart failure, EMPA also plays a role in cardiac arrhythmia. EMPA reduced late-INa in cardiomyocytes from mice with heart failure and cardiac Nav1.5 sodium channels containing the long QT syndrome 3 mutations R1623Q or ΔKPQ (Philippaert et al., 2021). In this study, we demonstrated the effect of EMPA on arrhythmia of a chronic MI mouse model.
Induction of arrhythmias after myocardial infarction is associated with an increased risk of sudden death (Zaman and Kovoor, 2014; Chatterjee and Levy, 2020). Following MI, the risk for ventricular tachyarrhythmia and sudden cardiac death in patients is increased (Hegyi et al., 2018; Li et al., 2020; Liu et al., 2021). QT interval and changes in the corrected QT (QTc) were important indicators of APD, which is mainly determined by ventricular repolarization. QT interval and QTc were significantly increased after MI (Tao et al., 2020). We discovered that QT and QTc were obviously recovered after EMPA treatment in post-MI mice. In chronic ischemic cardiomyopathy, VF is most often attributable to APD repolarization heterogeneity and conduction abnormalities in the IZ, BZ, and RZ zones (Saffitz and Kleber, 2012; Arbustini et al., 2018). We found that acute perfusion of EMPA shortened APD80 in the infarcted ventricles. This indicates that EMPA may transiently affect channel currents rather than channel proteins. EMPA was shown to directly decrease late-INa current in the mouse heart failure model (Philippaert et al., 2021). In this study, we found EMPA increased conduction velocity and reduced the rise time of action potential, which may be related to its influence on the sodium channel in infarcted ventricles. The Ca2+ influx triggers Ca2+ release by activating ryanodine receptors (RyRs) in the SR membrane from intracellular sarcoplasmic reticulum (SR) Ca2+ stores (Shen, 2006; Yang et al., 2020). Resting calcium may increase when [Cai] is taken up again into the SR by the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA), and extrusion from the cell primarily via sarcolemmal sodium–calcium exchange (NCX) was suppressed (Zhang et al., 2018; Gorski et al., 2019). The contractile dysfunction occurring in MI or hypoxia was primarily caused by impairment of intracellular Ca2+ homeostasis due to the disturbance of Ca2+ handling (McCrink et al., 2017; Zhang et al., 2018). The known Ca2+ handling dysregulation in MI includes calcium transient duration increase (Maczewski and Mackiewicz, 2008), intracellular Ca2+ overload (Morciano et al., 2015), and Ca2+ leak via RYR2 channels from SR (Valverde et al., 2019). In infarcted hearts, EMPA reduced CaT80 duration, increased CaT rise time, and shortened calcium transient decay constant Tau, indicating that EMPA improved calcium cycling during MI. The secondary rise of [Cai] during late action potential plateau is commonly observed in MI but not in normal hearts (Chang et al., 2015). The mechanism of the secondary rise of [Cai] is attributed to Ca2+ entry increase, which further promotes greater Ca2+ entry and additional sarcoplasmic reticulum Ca2+ release during phases 2 and 3 of the action potential (Chang et al., 2013; Chang et al., 2018). Under these conditions, EAD take-off potentials were observed to occur in action potential repolarization, suggesting a secondary rise of [Cai], and calcium cycling contributes to EAD formation, possibly including other ionic mechanisms. Our data demonstrated that EMPA reduced the secondary rise of [Cai] numbers and triggers; decreased EADs, PVBs, and VF occurrence; and improved the PS and DF in infarcted ventricles.
In conclusion, EMPA is a novel regulator of APD and calcium transient remodeling after chronic MI, which suppresses the prolongation of APD, the disturbance of calcium handling, and reduced arrhythmia susceptibility. The current study enriched our understanding of the electrophysiological action of SGLT2 inhibitors and indicated that EMPA possesses the potential to become therapeutic agents for ventricular arrhythmias.
The raw data supporting the conclusions of this article will be made available by the authors without undue reservation.
The animal study was reviewed and approved by Dalian Medical University Laboratory Animal Ethics Committee.
GX, XY, and GZ performed experiments, analyzed data, and prepared the manuscript. GZ, XW, YZ, JG, XW, and JL helped perform experiments and collect data. ZP oversaw the project and proofread the manuscript. YX designed the project, oversaw the experiments, and prepared the manuscript.
This study was supported by Chang Jiang Scholars Program (grant number T2017124), the National Natural Science Foundation of China (grant number 81970286), the Program of Liao Ning Distinguished Professor, and the Liaoning Revitalization Talents Program (grant number XLYC2002096).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alibhai, F. J., Tsimakouridze, E. V., Chinnappareddy, N., Wright, D. C., Billia, F., O'Sullivan, M. L., et al. (2014). Short-term disruption of diurnal rhythms after murine myocardial infarction adversely affects long-term myocardial structure and function. Circ. Res. 114, 1713–1722. doi:10.1161/CIRCRESAHA.114.302995
Anker, S. D., and Butler, J. (2018). Empagliflozin, calcium, and SGLT1/2 receptor affinity: Another piece of the puzzle. Esc. Heart Fail. 5, 549–551. doi:10.1002/ehf2.12345
Arbustini, E., Kramer, C. M., and Narula, J. (2018). Arrhythmogenic potential of border zone after myocardial infarction: Scar is more than just a healed wound. JACC. Cardiovasc. Imaging 11, 573–576. doi:10.1016/j.jcmg.2017.07.003
Banerjee, S. K., McGaffin, K. R., Pastor-Soler, N. M., and Ahmad, F. (2009). SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 84, 111–118. doi:10.1093/cvr/cvp190
Behnes, M., Mashayekhi, K., Weiss, C., Nienaber, C., Lang, S., Reiser, L., et al. (2018). Prognostic impact of acute myocardial infarction in patients presenting with ventricular tachyarrhythmias and aborted cardiac arrest. J. Am. Heart Assoc. 7, e010004. doi:10.1161/JAHA.118.010004
Belevych, A. E., Terentyev, D., Terentyeva, R., Ho, H. T., Gyorke, I., Bonilla, I. M., et al. (2012). Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 110, 569–577. doi:10.1161/CIRCRESAHA.111.260455
Chang, P. C., Hsieh, Y. C., Hsueh, C. H., Weiss, J. N., Lin, S. F., and Chen, P. S. (2013). Apamin induces early afterdepolarizations and torsades de pointes ventricular arrhythmia from failing rabbit ventricles exhibiting secondary rises in intracellular calcium. Heart rhythm. 10, 1516–1524. doi:10.1016/j.hrthm.2013.07.003
Chang, P. C., Lu, Y. Y., Lee, H. L., Lin, S. F., Chu, Y., Wen, M. S., et al. (2018). Paradoxical effects of sodium-calcium exchanger inhibition on torsade de Pointes and early afterdepolarization in a heart failure rabbit model. J. Cardiovasc. Pharmacol. 72, 97–105. doi:10.1097/FJC.0000000000000598
Chang, P. C., Wo, H. T., Lee, H. L., Lin, S. F., Wen, M. S., Chu, Y., et al. (2015). Role of sarcoplasmic reticulum calcium in development of secondary calcium rise and early afterdepolarizations in long QT syndrome rabbit model. PLoS One 10, e0123868. doi:10.1371/journal.pone.0123868
Chatterjee, N. A., and Levy, W. C. (2020). Sudden cardiac death after myocardial infarction. Eur. J. Heart Fail. 22, 856–858. doi:10.1002/ejhf.1744
Chowdhury, R. A., Debney, M. T., Protti, A., Handa, B. S., Patel, K. H. K., Lyon, A. R., et al. (2021). Rotigaptide infusion for the first 7 Days after myocardial infarction-reperfusion reduced late complexity of myocardial architecture of the healing border-zone and arrhythmia inducibility. J. Am. Heart Assoc. 10, e020006. doi:10.1161/JAHA.120.020006
Di Franco, A., Cantini, G., Tani, A., Coppini, R., Zecchi-Orlandini, S., Raimondi, L., et al. (2017). Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 243, 86–90. doi:10.1016/j.ijcard.2017.05.032
Frontera, A., Melillo, F., Baldetti, L., Radinovic, A., Bisceglia, C., D'Angelo, G., et al. (2020). High-density characterization of the ventricular electrical substrate during sinus rhythm in post-myocardial infarction patients. JACC. Clin. Electrophysiol. 6, 799–811. doi:10.1016/j.jacep.2020.04.008
Gorski, P. A., Jang, S. P., Jeong, D., Lee, A., Lee, P., Oh, J. G., et al. (2019). Role of SIRT1 in modulating acetylation of the sarco-endoplasmic reticulum Ca(2+)-ATPase in heart failure. Circ. Res. 124, e63–e80. doi:10.1161/CIRCRESAHA.118.313865
Hegyi, B., Bossuyt, J., Griffiths, L. G., Shimkunas, R., Coulibaly, Z., Jian, Z., et al. (2018). Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc. Natl. Acad. Sci. U. S. A. 115, E3036–E3044. doi:10.1073/pnas.1718211115
Jiang, H., Lu, Z., Yu, Y., Zhao, D., Jian, X., Yang, B., et al. (2007). Effects of metoprolol on sympathetic remodeling and electrical remodeling at infarcted border zone after myocardial infarction in rabbits. Cardiology 108, 176–182. doi:10.1159/000096647
Jiang, K., Xu, Y., Wang, D., Chen, F., Tu, Z., Qian, J., et al. (2022). Cardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis. Protein Cell 13, 336–359. doi:10.1007/s13238-020-00809-4
Kondo, H., Akoumianakis, I., Badi, I., Akawi, N., Kotanidis, C. P., Polkinghorne, M., et al. (2021). Effects of canagliflozin on human myocardial redox signalling: Clinical implications. Eur. Heart J. 42, 4947–4960. doi:10.1093/eurheartj/ehab420
Li, C., Zhang, J., Xue, M., Li, X., Han, F., Liu, X., et al. (2019). SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 18, 15. doi:10.1186/s12933-019-0816-2
Li, H. L., Lip, G. Y. H., Feng, Q., Fei, Y., Tse, Y. K., Wu, M. Z., et al. (2021). Sodium-glucose cotransporter 2 inhibitors (SGLT2i) and cardiac arrhythmias: A systematic review and meta-analysis. Cardiovasc. Diabetol. 20, 100. doi:10.1186/s12933-021-01293-8
Li, J., Xu, C., Liu, Y., Li, Y., Du, S., Zhang, R., et al. (2020). Fibroblast growth factor 21 inhibited ischemic arrhythmias via targeting miR-143/EGR1 axis. Basic Res. Cardiol. 115, 9. doi:10.1007/s00395-019-0768-4
Li, Z., Agrawal, V., Ramratnam, M., Sharma, R. K., D'Auria, S., Sincoular, A., et al. (2019). Cardiac sodium-dependent glucose cotransporter 1 is a novel mediator of ischaemia/reperfusion injury. Cardiovasc. Res. 115, 1646–1658. doi:10.1093/cvr/cvz037
Liu, M., Liu, H., Parthiban, P., Kang, G. J., Shi, G., Feng, F., et al. (2021). Inhibition of the unfolded protein response reduces arrhythmia risk after myocardial infarction. J. Clin. Invest. 15, 147836. doi:10.1172/JCI147836
Maczewski, M., and Mackiewicz, U. (2008). Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovasc. Res. 79, 42–51. doi:10.1093/cvr/cvn057
McCrink, K. A., Maning, J., Vu, A., Jafferjee, M., Marrero, C., Brill, A., et al. (2017). β-Arrestin2 improves post-myocardial infarction heart failure via sarco(endo)plasmic reticulum Ca2+-ATPase-dependent positive inotropy in cardiomyocytes. Hypertension 70, 972–981. doi:10.1161/HYPERTENSIONAHA.117.09817
Mendonca Costa, C., Plank, G., Rinaldi, C. A., Niederer, S. A., and Bishop, M. J. (2018). Modeling the electrophysiological properties of the infarct border zone. Front. Physiol. 9, 356. doi:10.3389/fphys.2018.00356
Mitchell, G. F., Jeron, A., and Koren, G. (1998). Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 274, H747–H751. doi:10.1152/ajpheart.1998.274.3.H747
Morciano, G., Giorgi, C., Bonora, M., Punzetti, S., Pavasini, R., Wieckowski, M. R., et al. (2015). Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 78, 142–153. doi:10.1016/j.yjmcc.2014.08.015
Ng, K. M., Lau, Y. M., Dhandhania, V., Cai, Z. J., Lee, Y. K., Lai, W. H., et al. (2018). Empagliflozin ammeliorates high glucose induced-cardiac dysfuntion in human iPSC-derived cardiomyocytes. Sci. Rep. 8, 14872. doi:10.1038/s41598-018-33293-2
Philippaert, K., Kalyaanamoorthy, S., Fatehi, M., Long, W., Soni, S., Byrne, N. J., et al. (2021). Cardiac late sodium channel current is a molecular target for the sodium/glucose cotransporter 2 inhibitor empagliflozin. Circulation 143, 2188–2204. doi:10.1161/CIRCULATIONAHA.121.053350
Raz, I., and Cahn, A. (2016). Heart failure: SGLT2 inhibitors and heart failure -- clinical implications. Nat. Rev. Cardiol. 13, 185–186. doi:10.1038/nrcardio.2016.35
Saffitz, J. E., and Kleber, A. G. (2012). Gap junctions, slow conduction, and ventricular tachycardia after myocardial infarction. J. Am. Coll. Cardiol. 60, 1111–1113. doi:10.1016/j.jacc.2012.05.020
Shah, K. S., and Fang, J. C. (2022). Sodium-glucose cotransporter 2 inhibitors in heart failure. Annu. Rev. Pharmacol. Toxicol. 62, 109–120. doi:10.1146/annurev-pharmtox-052120-014725
Shen, J. X. (2006). Isoprenaline enhances local Ca2+ release in cardiac myocytes. Acta Pharmacol. Sin. 27, 927–932. doi:10.1111/j.1745-7254.2006.00383.x
Si, R., Zhang, Q., Tsuji-Hosokawa, A., Watanabe, M., Willson, C., Lai, N., et al. (2020). Overexpression of p53 due to excess protein O-GlcNAcylation is associated with coronary microvascular disease in type 2 diabetes. Cardiovasc. Res. 116, 1186–1198. doi:10.1093/cvr/cvz216
Tanaka, A., and Node, K. (2020). Clinical application of sodium-glucose cotransporter 2 inhibitor into a real-world setting of heart failure care. Cardiovasc. Diabetol. 19, 132. doi:10.1186/s12933-020-01113-5
Tao, B., Liu, Z., Wei, F., Fan, S., Cui, S., Xia, H., et al. (2020). Over-expression of Kv4.3 gene reverses cardiac remodeling and transient-outward K(+) current (Ito) reduction via CaMKII inhibition in myocardial infarction. Biomed. Pharmacother. Dec 132, 110896. doi:10.1016/j.biopha.2020.110896
Valverde, C. A., Mazzocchi, G., Di Carlo, M. N., Ciocci Pardo, A., Salas, N., Ragone, M. I., et al. (2019). Ablation of phospholamban rescues reperfusion arrhythmias but exacerbates myocardium infarction in hearts with Ca2+/calmodulin kinase II constitutive phosphorylation of ryanodine receptors. Cardiovasc. Res. 115, 556–569. doi:10.1093/cvr/cvy213
Wen, H. Z., Xie, P., Zhang, F., Ma, Y., Li, Y. L., and Xu, S. K. (2018). Neuropilin 1 ameliorates electrical remodeling at infarct border zones in rats after myocardial infarction. Auton. Neurosci. 214, 19–23. doi:10.1016/j.autneu.2018.08.001
Yang, H. Q., Zhou, P., Wang, L. P., Zhao, Y. T., Ren, Y. J., Guo, Y. B., et al. (2020). Compartmentalized β1-adrenergic signalling synchronizes excitation-contraction coupling without modulating individual Ca2+ sparks in healthy and hypertrophied cardiomyocytes. Cardiovasc. Res. 116, 2069–2080. doi:10.1093/cvr/cvaa013
Yurista, S. R., Sillje, H. H. W., Oberdorf-Maass, S. U., Schouten, E. M., Pavez Giani, M. G., Hillebrands, J. L., et al. (2019). Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 21, 862–873. doi:10.1002/ejhf.1473
Zaman, S., and Kovoor, P. (2014). Sudden cardiac death early after myocardial infarction: Pathogenesis, risk stratification, and primary prevention. Circulation 129, 2426–2435. doi:10.1161/CIRCULATIONAHA.113.007497
Zelniker, T. A., and Braunwald, E. (2020). Clinical benefit of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J. Am. Coll. Cardiol. 75, 435–447. doi:10.1016/j.jacc.2019.11.036
Keywords: sodium–glucose cotransporter 2, empagliflozin, myocardial infarction, membrane potential, intracellular Ca2+
Citation: Xue G, Yang X, Zhan G, Wang X, Gao J, Zhao Y, Wang X, Li J, Pan Z and Xia Y (2022) Sodium–Glucose cotransporter 2 inhibitor empagliflozin decreases ventricular arrhythmia susceptibility by alleviating electrophysiological remodeling post-myocardial-infarction in mice. Front. Pharmacol. 13:988408. doi: 10.3389/fphar.2022.988408
Received: 07 July 2022; Accepted: 25 August 2022;
Published: 14 October 2022.
Edited by:
Pasquale Paolisso, University of Naples Federico II, ItalyCopyright © 2022 Xue, Yang, Zhan, Wang, Gao, Zhao, Wang, Li, Pan and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunlong Xia, yunlong_xia@126.com; Zhenwei Pan, panzw@ems.hrbmu.edu.cn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.