 Oval Yadav1
Oval Yadav1 Manjeet Kumar
Manjeet Kumar Azaj Ansari
Azaj Ansari
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 03 October 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.982484
This article is part of the Research TopicMetal-Complexed Molecules of Natural and Synthetic Origin: Pharmacological Advances and Therapeutic ApplicationsView all 5 articles
Recent years have witnessed a growing interest in the biological activity of metal complexes of α-aminophosphonates. Here for the first time, a detailed DFT study on five α-aminophosphonate ligated mononuclear/dinuclear CuII complexes is reported using the dispersion corrected density functional (B3LYP-D2) method. The electronic structures spin densities, FMO analysis, energetic description of spin states, and theoretical reactivity behaviour using molecular electrostatic potential (MEP) maps of all five species are reported. All possible spin states of the dinuclear species were computed and their ground state S values were determined along with the computation of their magnetic coupling constants. NBO analysis was also performed to provide details on stabilization energies. A molecular docking study was performed for the five complexes against two SARS-CoV-2 coronavirus protein targets (PDB ID: 6LU7 and 7T9K). The docking results indicated that the mononuclear species had a higher binding affinity for the targets compared to the dinuclear species. Among the species investigated, species I showed the highest binding affinity with the SARS-CoV-2 Omicron protease. NPA charge analysis showed that the heteroatoms of model species III had a more nucleophilic nature. A comparative study was performed to observe any variations and/or correlations in properties among all species.
Metal-based complexes containing organic ligands play crucial roles in biochemical systems including as biocatalysts, metal ions conductors across cell membranes, and new potential drugs for the treatment of various diseases (Congradyova and Jomova, 2013; Baek et al., 2018; Ali et al., 2021; Kiwaan et al., 2021). Along with the advancement of medicinal inorganic chemistry, such complexes have gained considerable interest as therapeutic agents (Ali and van Lier, 1999; Louie and Meade, 1999; Sakurai et al., 2002; Rafique et al., 2010). Complexation with metal ions has been shown to alter the pharmacological and therapeutic effects of ligands, providing new opportunities for the design of metal-based drugs in medicinal chemistry (Weder et al., 2002).
α-Aminophosphonates are an important class of amino acid analogues (Mucha et al., 2011) with diverse applications, in medicinal chemistry, agriculture and other industries, as synthetic intermediates with interesting pharmacological properties (Gemmill et al., 2005; Dinér and Amedjkouh, 2006; Juribasic et al., 2011; Tusek-Bozic, 2013; Boshta et al., 2018; Imam et al., 2018). They have been reported to inhibit enzymes involved in amino acid metabolism, thus having a direct influence on cell functions (Naydenova et al., 2010). Many studies have reported the remarkable activity of α-aminophosphonates as pharmacophores (Reddy et al., 2012; Chinthaparthi et al., 2013). These compounds exhibit potent biological properties including as plant growth regulators (Kafarski et al., 1989), antiviral (Long et al., 2008; Chen et al., 2009; Singh et al., 2020), anti-inflammatory agents (Sujatha et al., 2017; Romero-Estudillo et al., 2019), herbicides (Chen et al., 2015), protein inhibitors (Sienczyk and Oleksyszyn, 2009; Wang et al., 2012; Moreno-Cinos et al., 2019), antitumor/antiproliferative agents (Gu and Jin, 2012; Kraicheva et al., 2012; Liu et al., 2017; López-Francés et al., 2021), and antimicrobials (Atherton et al., 1986; El Sayed et al., 2011; Rezaei et al., 2011; Sudileti et al., 2019a). The biological activity of α-aminophosphonates has been linked with the presence of heterocyclic motifs in their structures (Sudileti et al., 2019b; Poola et al., 2019; Mohan et al., 2020). Diphenyl ester derivatives of α-aminophosphonates and their complexes had remarkable activity against serine proteases which play a crucial role in cancer cell disruption. Wang et al. reported that some cyclic α-aminophosphonates could mimic the antitumor activity of doxorubicin drug (Ma et al., 2011).
Platinum group metal-based aminophosphonate complexes have been reported to exhibit potent activity against tumour cells (Aranowska et al., 2006). A few other transition metals, like gadolinium, technetium, and rhenium have been previously used in metal-based complexes with ligands as therapeutic agents and for diagnostic purposes (Tusek-Bozic, 2013). Several α-aminophosphonates ligated metal complexes have been reported (Azzam et al., 2020). Derivatives of metal ligated aminophosphonates have gained huge attention due to their wide-range spectrum of bioactivities and tremendous application in the medical field (Azzam et al., 2020). Comprehensive studies have previously been carried out using copper to form copper (II) complexes with predominant applications as antiviral, anticancer and antimicrobial agents (Ranford et al., 1993; Marzano et al., 2006; Tardito et al., 2007; Repicka et al., 2009; Creaven et al., 2010; Saeed et al., 2010; Konarikova et al., 2013; Santini et al., 2014; Wehbe et al., 2017; Massoud et al., 2018). To the best of our knowledge, no computational research has been carried out on carbocyclic ring containing α-aminophosphonate ligands containing copper ions.
Over the past 2 years, the COVID-19 pandemic has caused a significant number of deaths worldwide. Immense efforts have been made to develop anti-COVID vaccines and antiviral drugs, but there is no effective therapeutic approach that can effectively cure this disease to date (Xu et al., 2006). α-Aminophosphonates have been reported to exhibit antiviral activity (Mohapatra et al., 2022). The growing interest in the biological activity of α-aminophosphonates metal complexes, and the reported feasibility of their large scale synthesis, motivated us to carry out an extensive computational study on these compounds with a particular focus on the interaction of metal ligation with the α-aminophosphonates. Our study also aimed to explore the potential of CuII α-aminophosphonates complexes to interact with key proteins of the COVID-19-causing SARS-CoV-2 coronavirus. Herein we report a DFT study on five α-aminophosphonate (L) ligated mononuclear/dinuclear Copper (II) complexes, namely species I [CuII(L) (Br) (H2O)], species II [CuII(L) (Br)2], species III [CuII(L) (Cl)2], species IV [Cu2(HL) (Cl)4(H2O)] and species V [Cu2(HL) (Br)2(Cl)2(H2O)]. Among these species, I and IV have been experimentally synthesized and well characterized (Azzam et al., 2020). Species II and III are model species of I whereas species V is a model species of IV. Here we have addressed and discussed: electronic structures, spin state energetics, Mulliken spin densities, FMO analysis and electrostatic potentials, NBO analysis and molecular docking towards two SARS-CoV-2 protein targets (PDB ID: 6LU7 and 7T9K) with all five species followed by cross comparison among them.
Gaussian 16 was used to perform all geometry optimizations employing a dispersion corrected density functional B3LYP-D2 method (Frisch et al., 2016). The accuracy of the used functional was confirmed in previous studies (Ansari et al., 2013; Ansari et al., 2014; Ansari et al., 2015; Ansari et al., 2017; Yadav et al., 2021; Ansari and Monika, 2022). The LACVP basis set comprising LanL2DZ (Los Alamos effective core potential) for the copper metal centre (Dunning et al., 1977; Hay and Wadt, 1985a; Hay and Wadt, 1985b; Wadt and Hay, 1985) and a 6-31G (Ditchfield et al., 1971) basis set for the other atoms (P, H, C, N, O, Cl and Br) were employed. Frequency calculations were done on the optimized structure to confirm the minima on the potential-energy surface. The single point energy calculations were performed on optimized geometry using TZVP basis set with acetonitrile as a solvent using the polarizable continuum solvent (PCM) model (Schafer et al., 1992; Schafer et al., 1994; Tomasi et al., 2005). The quoted DFT energies are B3LYP-D2 solvation including free-energy corrections with TZVP basis set at room temperature.
Initially, we optimized α-aminophosphonate (L; see Supplementary Figure S1) and coordinated with the copper centre to generate copper [CuII(L) (Br) (H2O)] species I. The latter was synthesized by the reaction of CuII salt with ligand (L) and characterized by various techniques like FT-IR and EI-Mass (Azzam et al., 2020). This species was reported recently to show activity against HT-29 cancer cell lines. The higher bioactivity of species I compared to the corresponding parent ligand suggested that it could be further exploited for other pharmacological applications (Azzam et al., 2020). The optimized geometry and spin density plot of species I are shown in Figure 1. The Cu-Br, Cu-N and Cu-O4 bond lengths were computed to be 2.466 Å, 2.153 Å and 1.917 Å, respectively. The phenyl rings of the phosphonate moiety were cis to each other and in the same plane. Our computed results showed that species I found as a distorted square planar, leaning towards tetrahedral, geometry in good agreement with experimental observations (Azzam et al., 2020). A spin density of ρ = 0.408 was located at the copper centre (see Figure 1B and Supplementary Table S1) which corresponds to unpaired electron nature at the metal centre with strong electron delocalization present in the system. The eigenvalue plot is shown in Supplementary Figure S2 and the electronic configuration at the metal centre was found to be (dxz)2 (dyz)2 (dz2)2 (dxy)2 (dx2-y2)1. The HOMO-LUMO gap for species I was computed to be 4.770 eV.
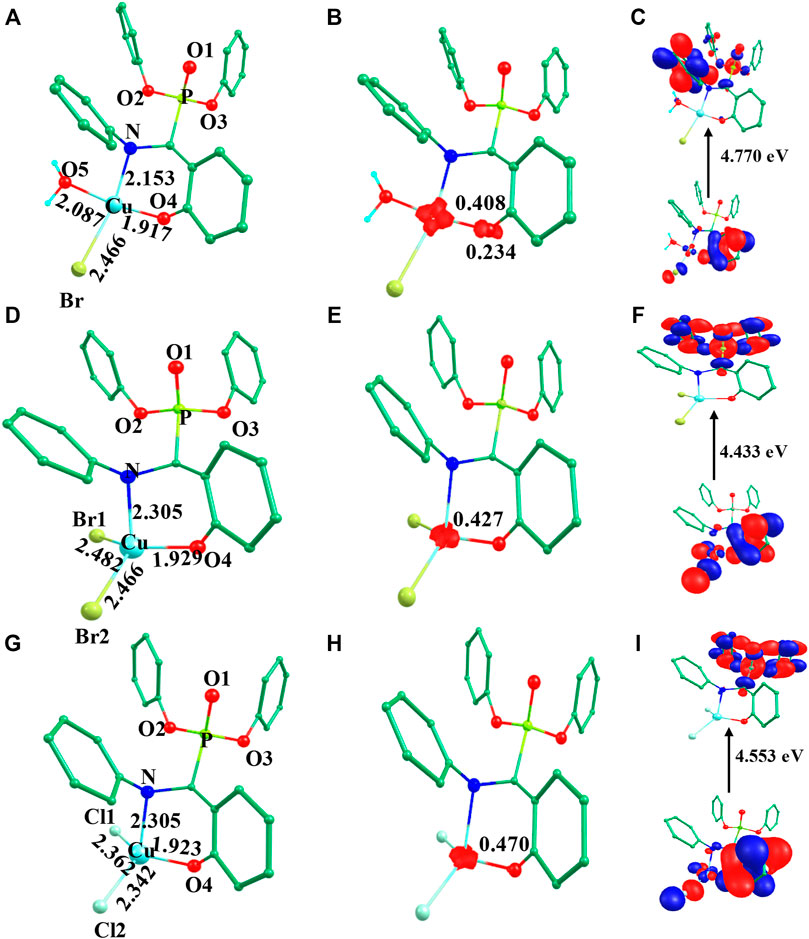
FIGURE 1. B3LYP-D2 (A) optimized structure of species I and its (B) spin density plot and (C) HOMO-LUMO gap; (D) optimized structure of species II and its (E) spin density plot and (F) HOMO-LUMO gap; (G) optimized structure of species III and its (H) spin density plot and (I) HOMO-LUMO gap. (Bond lengths are in Å).
The previously reported remarkable bioactivity of species I (Azzam et al., 2020) prompted us to further study its models (species II and III). In species II, the water molecule was replaced by a bromide and in species III, both bromides were replaced by chloride ligands. The optimized geometry and spin density plot of species II is shown in Figures 1D,E. Here again, a distortion of the square planar geometry, leaning towards tetrahedral, was witnessed. The distortion was expressed by bond angles of 116.4° and 100.0° for ∠Br1-Cu-Br2 and ∠Br2-Cu-O. The computed Cu-Bravg, Cu-N and Cu-O4 bond lengths were found to be 2.474 Å, 2.305 Å and 1.929 Å, respectively. The computed spin density of ρ = 0.427 was located at the copper centre (see Figure 1E and Supplementary Table S1) which corresponded to the unpaired electron character at the metal centre. The eigenvalue plot is shown in Supplementary Figure S3 and the electronic configuration at the metal centre was found to be (dxz)2 (dyz)2 (dz2)2 (dxy)2 (dx2-y2)1. The HOMO-LUMO gap for species II was computed to be 4.433 eV.
The optimized geometry and spin density plot of species III is shown in Figures 1G,H. The computed Cu-Clavg, Cu-N and Cu-O4 bond lengths were found to be 2.352 Å, 2.305 Å and 1.923 Å, respectively. The bond angles, 87.8° and 119.5° for ∠Cl1-Cu-N and ∠Cl1-Cu-Cl2, expressed distortion in the square planar geometry of the system towards tetrahedral. The computed data suggested that both model species exhibited nearly the same distortion in their geometry and that the substitution with a halide had little effect on the geometry of the complexes. A spin density ρ = 0.470 located at the copper centre corresponding to the presence of an unpaired electron. The electronic configuration at the metal centre was found to be (dyz)2 (dxz)2 (dz2)2 (dxy)2 (dx2-y2)1 (see Supplementary Figure S4). The HOMO-LUMO gap for species III was computed to be 4.553 eV. This is larger than that obtained for species II and smaller than for species I.
Here, species IV is the dinuclear derivative of species I where halogen and water substituents are coordinated to the metal centre. Species IV has previously been well characterized by analytical, spectral and thermal data (Azzam et al., 2020). For this species, two spin states 3IV and 1IV are possible due to ferro and antiferromagnetic interactions between the metal centres (see Table 1). Following optimization of both spin states, our computed data revealed 3IV as the ground state with 1IV lying 2.5 kJ/mol high in energy (see Figure 2 and Table 1). The magnetic exchange J value was calculated as 23.9 cm−1, in accordance with experimental data (Azzam et al., 2020).
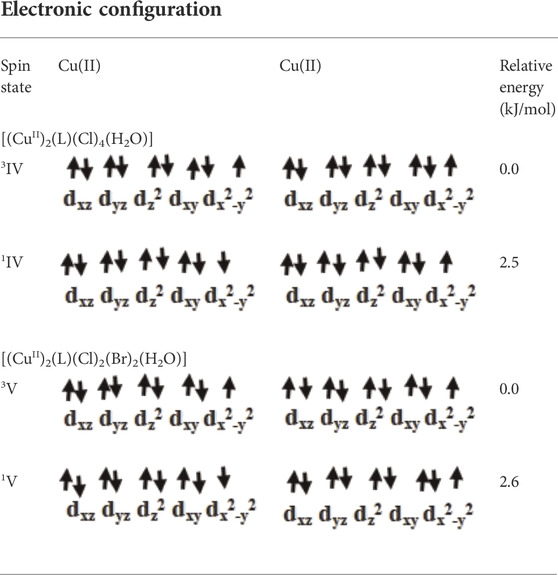
TABLE 1. Possible spin states of species IV and V.
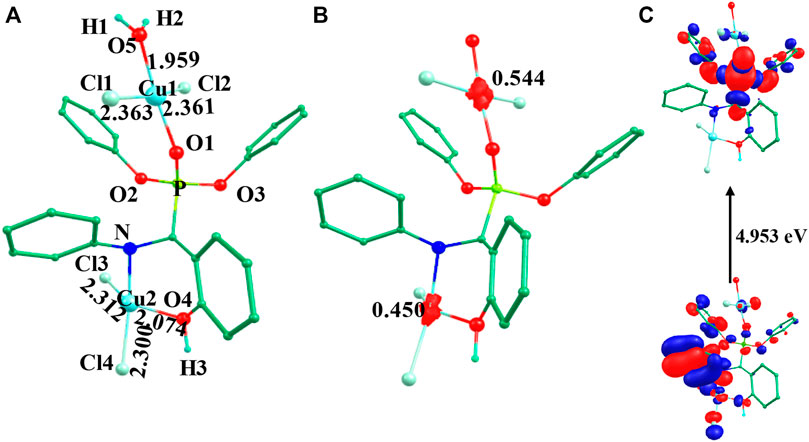
FIGURE 2. B3LYP-D2 (A) optimized structure, (B) spin density plot and (C) HOMO-LUMO gap of the species 3IV (Bond lengths are in Å).
The optimized geometry and spin density plot for 3IV are shown in Figure 2. The computed Cu1-Clavg and Cu2-Clavg bond lengths were found to be 2.362 Å and 2.306 Å, respectively. Similarly, Cu1-O5, Cu1-O1, Cu2-N and Cu2-O4 bond lengths were computed to be 1.959 Å, 1.945 Å, 2.117 Å and 2.074 Å, respectively. Similar to species I-III, both copper centres adopted a distorted square planar geometry towards tetrahedral which was evident from the bond angles 173.2°, 87.1°, 148.9°, 104.9° and 160.2° for ∠Cl1-Cu1-Cl2, ∠Cl1-Cu1-O5, ∠Cl3-Cu2-O4, ∠Cl3-Cu2-Cl4, ∠N-Cu2-Cl4, respectively. The two phenyl rings of the phosphonate moiety were coplanar to each other and the remaining two moved out of the plane to reduce strain in the system. The Cu1 and Cu2 centres had a spin density value of ρ = 0.544 and ρ = 0.450, respectively, corresponding to the presence of unpaired electron at both the metal centres (see Figure 2B). The HOMO-LUMO gap was computed to be 4.953 eV and was higher than those computed for species I-III. The electronic configuration at both copper centres was found to be (dxz)2 (dyz)2 (dxy)2 (dz2)2 (dx2-y2)1 (see Supplementary Figure S5). The optimized structure, spin density plot and HOMO-LUMO gap of 1IV is shown in Supplementary Figure S6.
A DFT study was carried out on species V (model species of IV) where the chloride ligand at Cu2 was replaced by the bromide ligand. Following optimization of both possible spin interactions, our results revealed that 3V was computed to be the ground state with 1V lying 2.6 kJ/mol high in energy (see Table 1). The magnetic exchange J value was calculated to be 11.5 cm−1 which suggested the ferromagnetic nature of this species (Ansari et al., 2017; Azzam et al., 2020; Yadav et al., 2021).
The optimized geometry and spin density plot of 3V are shown in Figure 3. The computed Cu1-Clavg and Cu2-Bravg bond lengths were found to be 2.363 Å and 2.434 Å, respectively. The main structural parameters including bond lengths Cu1-O5, Cu1-O1, Cu2-N and Cu2-O4 were computed to be 1.958 Å, 1.945 Å, 2.129 Å and 2.117 Å, respectively. A distorted square planar geometry towards tetrahedral was predicted at both the copper centres which was apparent from the bond angle values of 103.8°, 141.4° and 157.9° for ∠Br1-Cu2-Br2, ∠Br1-Cu2-O4, ∠Br2-Cu2-N, respectively. The distortion in geometry was more pronounced at Cu2 compared to Cu1. The spin density plot suggested that more delocalization was favored at the Cu2 centre (ρ = 0.544(Cu1)/0.376(Cu2); see Figure 3B. The electronic configuration at both copper centres was found to be (dxz)2 (dyz)2 (dxy)2 (dz2)2 (dx2-y2)1. The HOMO-LUMO gap of 3V was computed to be 4.776 eV. The optimized structure, spin density plot and HOMO-LUMO gap of 1V is shown in Supplementary Figure S9.
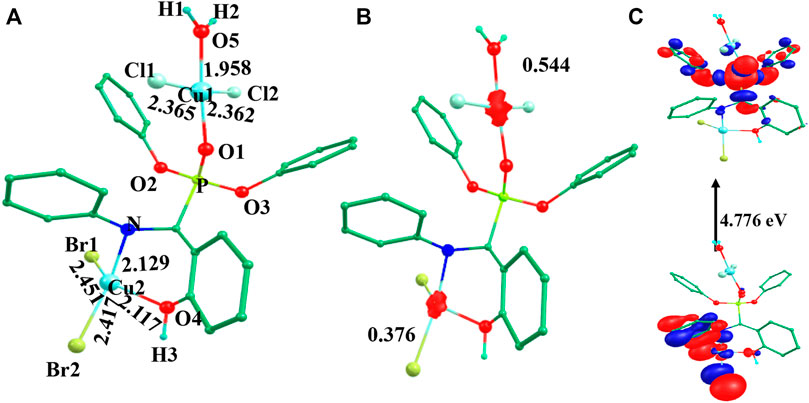
FIGURE 3. B3LYP-D2 (A) optimized structure, (B) spin density plot and (C) HOMO-LUMO gap of the species 3V (Bond lengths are in Å).
To exploit the positive and negative potential regions, molecular electrostatic surfaces were mapped for all five species. Different regions of electrostatic potential are commonly described with a colour grading, using red for the most negative potential surface, blue for the most positive potential, and green and yellow for a potential between the two extremes (Drissi et al., 2015). The electrostatic potential maps of species I to V are shown in Figure 4. The electrostatic potential distribution for the molecule in the crystal was calculated from Eq. 1 (Takayanagi et al., 1996):
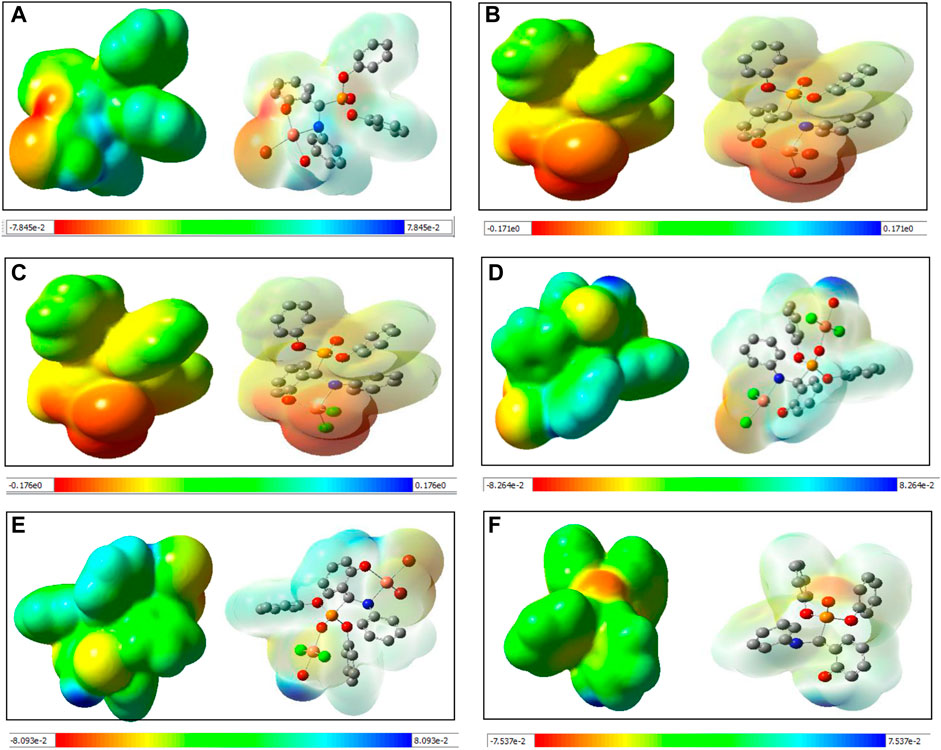
FIGURE 4. Computed molecular electrostatic potential maps (solid/transparent) of (A) species I, (B) species II, (C) species III, (D) species IV, (E) species V, and (F) Ligand.
A close inspection of the MEP maps revealed that the presence of localized negative potential regions were located at the O atom of the phosphonate moiety in the mononuclear species I-III while the negative potential was dispersed to neighbouring atoms in both dinuclear species (IV and V). Within the mononuclear species, a more negative potential was located on species II and III compared to species I. Thus, it may be predicted that the model species II and III are more prone to electrophilic attack. It is also noteworthy that the region around the aromatic ring witnessed a gradual depletion in the red and blue colours and an increase in green. On the other hand, dinuclear species showed blue and green regions that possessed positive potentials.
NBO analysis of all species was performed to gain in depth knowledge about the bonding aspects and charge transfer occurring in all five species. The NBO approach makes use of a set of localized bond, anti-bond and Rydberg extra valence orbitals. NBO analysis provides the representations of orbitals and the highest viable percentage of electron density based on mathematically accurate results (Monika et al., 2021). All five species showed a common backbone skeleton comprising of α-aminophosphoante ligated to the metal core. The only differences were in the substituents present at the metal centre. Thus, the study of the interaction of substituents with the copper centre helped to better understand the bonding aspects of all species under investigation. The NBO plots of substituents with the metal of species I-III are shown in Figure 5. The examination of the NBO plot of species I revealed that the Cu-O5 bond was quite polar in nature with 28.58% orbital contribution from copper and 71.42% from oxygen (O5). Similarly, the orbital contributions of the Cu-Br bond was computed to be 74.28% by bromine and 25.72% by copper metal. The NBO plots were also computed for the model species II and III. Among the mononuclear species, the metal-substituent bond was more polar in the case of model species III. In the dinuclear species 3IV, the NBO plots were computed for both metal centres. The orbital contribution for the Cu1-Cl1 bond was computed to be 23.10% by Cu1 and 76.90% by Cl1. The orbital contribution for the highly polar Cu1-Cl2 bond was computed to be 21.94% by Cu1 and 78.06% by Cl2. The orbital contribution for the Cu2-Cl3 bond was computed to be 29.47% by Cu2 and 70.53% by Cl3. The orbital contribution for the Cu2-Cl4 bond was computed to be 27.04% by Cu2 and 72.96% by Cl4. Similarly, Cu-Clavg orbital contribution for 3IV was computed to be 22.5% by Cu1 and 77.5% by chloride. Cu-Bravg orbital contribution of 3IV was computed to be 32.81% by Cu2 and 67.19% by bromide (Supplementary Figure S11). The NBO plots for 1IV and 1V are shown in Supplementary Figure S12.
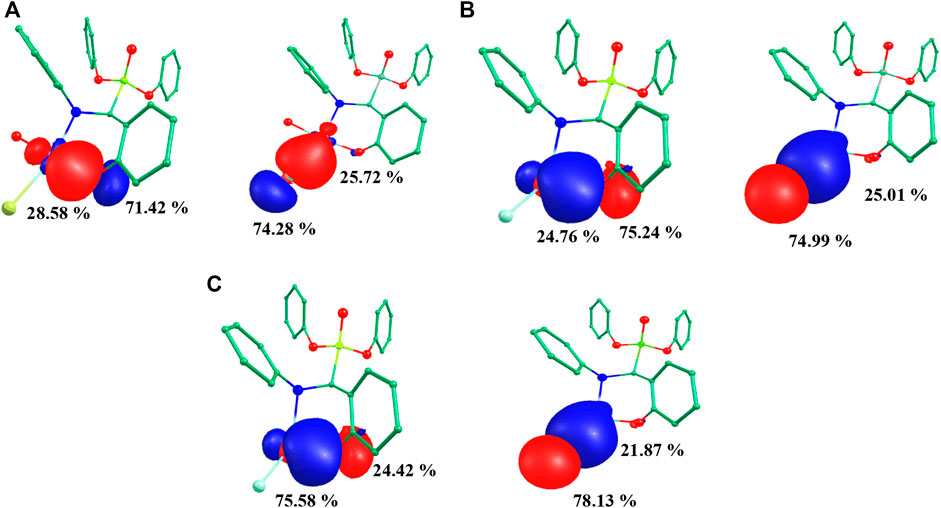
FIGURE 5. Computed NBO plots of (A) species I, (B) species II and (C) species III.
The structure of species I exhibited a type of Lewis structure (99.70%), 0.18% of valence non-Lewis, and 0.10% of Rydberg non-Lewis (Figure 6). Both Species II and III exhibited a type of Lewis structure (99.99%) and a negligible percentage of valence non-Lewis and of Rydberg non-Lewis. The data for the remaining are shown Supplementary Figures S13–18.
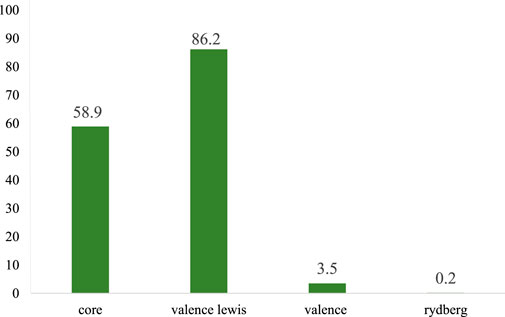
FIGURE 6. Bar diagram representing the natural population analysis of species I.
NBO analysis allows to explore donor-acceptor interactions by making use of the second-order perturbation approach (Fock matrix). The strength of interaction stabilization energy E(2) related with electron delocalisation among the donor (i) and acceptor (j) was calculated using the following equation:
where qi is the donor orbital occupancy, εj and εi depict diagonal elements and F (i, j) denotes off diagonal NBO Fock matrix element. The more intensive the interaction between the acceptor-donor electron, the greater the value for stabilization energy E(2) is, indicative of greater conjugation in the whole system (Sumrra et al., 2018). The feasible dominant intensive interactions of species I are listed in Table 2. The computed data showed that strong intramolecular hyper-conjugative interactions stabilized by pi electrons were present in species I. The system was mainly stabilized by hyper-conjugative interactions of π(C31-C34) → π*(C29-C30), π(C29-C30) → π*(C32-C36), π(C32-C36) → π*(C31-C34), π(C44-C47) → π*(C42-C43) responsible for conjugation in the aromatic ring (see Supplementary Figure S19). Further conjugation was due to interactions π(C45-C49) → π*(C44-C47), π (C6-C10) → π*(C5-C8), π(C6-C10) → π*(C3-C4), π(C5-C8) → π*(C3-C4). The most important (n → π*) interactions were n (C19) → π*(C15-C17), n (C19) → π*(C16-C18), and n (O28) → π*(C29-C30) with a stabilization energy of 33.79, 28.99 and 7.75 kcal/mol, respectively. Further stabilization was achieved by interactions n (O26) → σ*(P25-O28) and n (O26) → σ*(P25-O27) having a stabilization energy of 9.39 and 12.40 kcal/mol, respectively. A similar trend was computed in the model species II (see Supplementary Table S2 and Supplementary Figure S20) and III (see Supplementary Table S3 and Supplementary Figure S21).

TABLE 2. Perturbation theory energy analysis of species I.
In the dinuclear species 3IV, the system was mainly stabilized by π→ π* transitions, including π(C6-C10) → π*(C3-C4), π(C6-C10) → π*(C5-C8), π(C16-C18) → π*(C14-C15), π(C17-C19) → π*(C16-C18), π(C31-C34) → π*(C29-C30), π(C44-C47) → π*(C42-C43), π(C45-C49) → π*(C44-C47) (see Supplementary Table S4 and Supplementary Figure S22) with respective stabilization energies of 10.53, 10.01, 12.22, 11.29, 11.17, 11.13 and 10.71 kcal/mol (see Supplementary Table S4 for remaining transitions). Other important intra-molecular hyper-conjugative interactions were also present, providing extra stability to the system. These interactions included n → n* transitions such as n (O1) → n*(Cu53) with a stabilization energy 9.95 kcal/mol and n (N2) → n*(Cu53) with a stabilization energy of 14.76 kcal/mol. The dominant donor-acceptor interactions of 1IV are shown in Supplementary Table S5 and Supplementary Figure S22. The Fock-matrix based second order perturbation theory transitions for the remaining species (3IV and 1IV) are listed in Supplementary Tables S6,7 and Supplementary Figure S23.
The computation of NPA charges helps to predict the reactivity of a system by analysing the presence of electrophilic and nucleophilic centres (Biswas et al., 2021). The NPA charges of heteroatoms of all five species are listed in Table 3. The NPA charges were computed as −0.506 (O1) and −0.246 (O4) for species I, −0.516(O1), −0.268(O4), −0.217(Cl1), −0.277 (Cl2) for species II and −0.517 (O1), −0.268(O4), −0.248 (Br1), −0.304 (Br2) for species II. Among the mononuclear species, the NPA charges were more negative in the case of model species (II and III) compared to previously reported species (species I). Among the model species, species III was computed to have more negative NPA charges, thus predicting its comparatively higher nucleophilic character (see Table 3). In the case of dinuclear species, the computed NPA charges were −0.499 (O1), −0.324 (O4), −0.236(Cl1), −0.221 (Cl2), −0.169 (Cl3), −0.190 (Cl4) for species 3IV and −0.499 (O1), −0.327 (O4), −0.236 (Cl1), −0.221 (Cl2), −0.121 (Br1), −0.141 (Br2) for species 1V. These NPA charges were comparable for both species except for the halogen atoms which were more negative for species IV. The NPA charge analysis predicted that the heteroatoms of the mononuclear species were more nucleophilic than those of the dinuclear species. Among the mononuclear species, the heteroatoms of the model species III were predicted to be more nucleophilic.
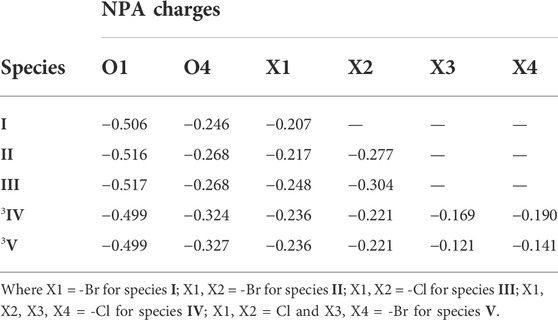
TABLE 3. Computed NPA charges of all the reported species.
Several α-aminophosphonate derivatives, exhibiting interesting biological properties, have previously been synthesized (El-Boraey et al., 2015; Gundluru et al., 2021; Tian et al., 2022). Many studies have reported their potential as antiviral agents (Hu et al., 2008; Song et al., 2010; Amira et al., 2021). Here we performed a molecular docking study of ligand L and species I-V against the target proteases PDB ID: 6LU7 (SARS-CoV-2) and PDB ID: 7T9K (SARS-CoV-2 Omicron). The high-resolution 3D structures of both SARS-CoV-2 proteases were downloaded from the protein data bank (https://www.rcsb.org/). Docking calculations were carried out using the AutoDock Vina software and AutoDockTools (ADT) (Trott et al., 2010). Prior to the docking calculations, all water molecules were removed. It was ensured that the residues of the active site of each protein were included when assigning both grid box sizes (30 × 30 × 30 Å centered at X = -10.773, Y = 12.066, Z = 68.545 for 6LU7 and 27 × 27 × 27 Å centered at X = 178.412393, Y = 147.822071 and Z = 222.093571 for 7T9K). All ligands were geometrically optimized geometries and the docked poses and ligand-protein interactions were visualized using the Discover Studio Visualizer 4.0 software (BIOVIA Systemes, Dassault, 2021). The analysis of the docked models was performed to investigate the binding affinity and the nature of the intermolecular bonding interactions between each species and the target proteins. The modes of interactions of the ligand with proteins were determined by investigating their favourable orientations of binding.
The molecular docking of all species (I-V) with both SARS-CoV-2 proteases revealed that the mononuclear species (I-III) had more binding affinity than the dinuclear species (IV-V) for the protein targets (see Table 4). Among all species, species I had the highest binding affinity (binding energy -5.9 kcal/mol) with the SARS-CoV-2 Omicron protease. The favourable orientation and important interactions of species I and the Omicron protease are shown in Figure 7. Species I showed two types of significant interactions, one was H-bonding and the other was Pi-Sigma interactions. H-bonding occurred at a distance of 2.367 Å and Pi-Sigma interactions with VAL83 and LEU110 occurring at a distance of 3.590 Å and 3.451 Å, respectively. The molecular docking results obtained for the remaining species are shown in Supplementary Figures S24–S34.
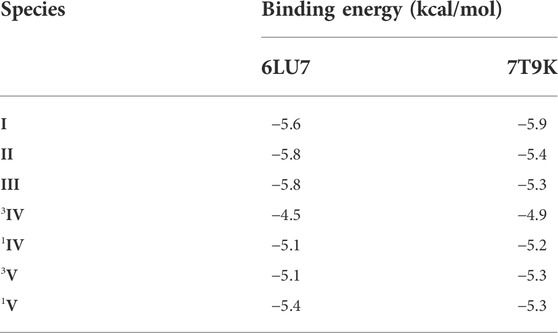
TABLE 4. Favourable binding energies (docking scores) for the docked poses for species I-V with the selected SARS-CoV-2 proteases.

FIGURE 7. Molecular docking study of species I with the SARS-CoV-2 Omicron variant protease (7T9K).
Species I-III are mononuclear whereas species IV-V are dinuclear. Species II, III and V are model species. All species showed a distortion in geometry from square planar towards tetrahedral. Among the mononuclear species, I was computed to have the highest HOMO-LUMO gap. In the case of the dinuclear species, low spin states had a comparatively lower HOMO-LUMO gap compared to high spin states. The computed MEP maps predicted that the mononuclear species had more negative potential, and thus may show be more electrophilic than the dinuclear species which had a comparatively more positive potential. Further theoretical reactivity predictions based on MEP analysis suggested that the model species II and III had the most electrophilic regions. NPA charge analysis predicted that the heteroatoms of the mononuclear species were more nucleophilic than those of the dinuclear species and among them, the heteroatoms of species III were more nucleophilic. NBO analysis suggested that all systems were stabilized by intramolecular hyper-conjugative interactions. The stabilization energy value (E(2a)) values indicated that the dinuclear species E(2a) were comparatively more stabilized with these interactions compared to the mononuclear species. Among the mononuclear species, the model species gained more stabilization as evident from their higher values of (E(2a)). Molecular docking studies indicated that all species showed interactions with various amino acid residues of two SARS-CoV-2 proteases. The mononuclear species were predicted to exhibit a higher binding affinity for the protein targets compared to the parent ligand as evident by their higher docking scores and number of hydrogen and hydrophobic bonds with the targets.
CuII α-aminophosphonate complexes have gained considerable interest due to their valuable biological properties. Here, we have carried out calculations on five species using DFT method using a dispersion corrected functional B3LYP-D2 method to predict the correct ground state of the dinuclear species; high spin (S = 3) for species IV and species V. Spin density values confirmed the presence of unpaired electron nature at the copper centre of species I, II and III and also on each copper centre of dinuclear species. Species 1V was computed to have the smallest transient energy gap (2.233 eV) whereas species 3IV was computed to have the highest energy gap (4.953 eV). Computed MEP maps predicted that the mononuclear species were more electrophilic than the dinuclear species. Among the mononuclear species, the most negative potential was located on model species II and III. This was also evident from NPA charges analysis which suggested more negative charges present at heteroatoms in species II and III. Among these two, NPA charges were found to be more negative for species III. NBO analysis suggested that all species were stabilized by hyper conjugative interactions. Molecular docking predicted that the metal complexed ligands showed better affinity for the SARS-CoV-2 targets than the parent ligand in agreement with experimental findings. Among the metal complexes, the mononuclear species have comparatively higher interactions than the dinuclear species. Further studies, including in vitro assays, are warranted to confirm the potential of CuII α-aminophosphonate complexes as new therapeutic agents against COVID-19.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
OY: Conceptualization, Methodology, Validation, Visualization, Writing—original draft, Writing—review and Editing. MK: Validation, Editing, HM: Visualization, Writing—review. KY: Visualization, Writing—review. VS: Validation, Editing, Funding Acquisition, AA: Supervision, Resources, Software, Editing.
OY thanks CSIR New Delhi for SRF fellowship. MK sincerely thanks to the Central University of Haryana for financial assistance and department of Chemistry, Central University of Haryana. VS. thanks Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde for the support. AA would like to thanks the Central University of Haryana for computing facility.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.982484/full#supplementary-material
Ali, A., Sepay, N., Afzal, M., Sepay, N., Alarifi, A., Shahid, M., et al. (2021). Molecular designing, crystal structure determination and in silico screening of copper(II) complexes bearing 8-hydroxyquinoline derivatives as anti-COVID-19. Bioorg. Chem. 110, 104772. doi:10.1016/j.bioorg.2021.104772
Ali, H., and van Lier, J. E. (1999). Metal complexes as photo- and radiosensitizers. Chem. Rev. 99, 2379–2450. doi:10.1021/cr980439y
Amira, A., Aouf, Z., K'tir, H., Chemam, Y., Ghodbane, R., Zerrouki, R., et al. (2021). Recent advances in the synthesis of α‐aminophosphonates: A review. ChemistrySelect 6, 6137–6149. doi:10.1002/slct.202101360
Ansari, A., Ansari, M., Singha, A., and Rajaraman, G. (2017). Interplay of electronic cooperativity and exchange coupling in regulating the reactivity of diiron(IV)-oxo complexes towards C-H and O-H bond activation. Chemistry 23, 10110–10125. doi:10.1002/chem.201701059
Ansari, A., Jayapal, P., and Rajaraman, G. (2014). C-H bond activation by metal–superoxo species: what derives higher reactivity?. Angew. Chem. Int. Ed. 54, 564–568. doi:10.1002/anie.201409844
Ansari, A., Kaushik, A., and Rajaraman, G. (2013). Mechanistic insights on the ortho-hydroxylation of aromatic compounds by non-heme iron complex: A computational case study on the comparative oxidative ability of ferric-hydroperoxo and high-valent Fe(IV)═O and Fe(V)═O intermediates. J. Am. Chem. Soc. 135, 4235–4249. doi:10.1021/ja307077f
Ansari, M., Vyas, N., Ansari, A., and Rajaraman, G. (2015). Oxidation of methane by an N-bridged high-valent diiron-oxo species: Electronic structure implications on the reactivity. Dalton Trans. 44, 15232–15243. doi:10.1039/c5dt01060h
Ansari, A., and Monika, (2022). Effect of ring size of TMC ligands in controlling C-H bond activation by metal-superoxo. Dalton Trans. 51, 5878–5889. doi:10.1039/d2dt00491g
Aranowska, K., Graczyk, J., Checinska, L., Pakulska, W., and Ochocki, J. (2006). Antitumor effect of Pt(II) amine phosphonate complexes on sarcoma Sa-180 in mice. Crystal structure of cis-dichlorobis (diethyl-4-Pyridylmethylphosphonate-kN) platinum(II)hydrate, cis-[PtCl2(4-Pmpe)2].H2O. Pharmazie 61, 457
Atherton, F. R., Hassall, C. H., and Lambert, R. W. (1986). Synthesis and structure-activity relationships of antibacterial phosphonopeptides incorporating (1-aminoethyl) phosphonic acid and (aminomethyl) phosphonic acid. J. Med. Chem. 29, 29
Azzam, M. A., El-Boraey, H. A. L., and El-Sayed, I. E. T. (2020). Transition metal complexes of α-aminophosphonates part II: Synthesis, spectroscopic characterization, and in vitro anticancer activity of copper(II) complexes of α-aminophosphonates. Phosphorus, Sulfur, Silicon Relat. Elem. 195, 339–347. doi:10.1080/10426507.2019.1700258
Baek, J., Rungtaweevoranit, B., Pei, X., Park, M., Fakra, S. C., Liu, Y. S., et al. (2018). Bioinspired metal-organic framework catalysts for selective methane oxidation to methanol. J. Am. Chem. Soc. 140, 18208–18216. doi:10.1021/jacs.8b11525
Biswas, J. P., Ansari, M., Paik, A., Sasmal, S., Paul, S., Rana, S., et al. (2021). Effect of the ligand backbone on the reactivity and mechanistic paradigm of non-heme iron(IV)-oxo during olefin epoxidation. Angew. Chem. Int. Ed. Engl. 60, 14030–14039. doi:10.1002/anie.202102484
Boshta, N. M., Elgamal, E. A., and El-Sayed, I. E. (2018). Bioactive amide and α-aminophosphonate inhibitors for methicillin-resistant Staphylococcus aureus (MRSA). Monatsh Chem. 149, 2349–2358. doi:10.1007/s00706-018-2303-y
Chen, J.-L., Tang, W., Che, J.-Y., Chen, K., Yan, G., Gu, Y.-C., et al. (2015). Synthesis and biological activity evaluation of novel α-amino phosphonate derivatives containing a pyrimidinyl moiety as potential herbicidal agents. J. Agric. Food Chem. 63, 7219–7229. doi:10.1021/acs.jafc.5b02335
Chen, M. H., Chen, Z., Song, B.-A., Bhadury, P. S., Yang, S., Cai, X.-J., et al. (2009). Synthesis and antiviral activities of chiral thiourea derivatives containing an alpha-aminophosphonate moiety. J. Agric. Food Chem. 57, 1383–1388. doi:10.1021/jf803215t
Chinthaparthi, R. R., Bhatnagar, I., Gangireddy, C. S. R., Syama, S. C., and Cirandur, S. R. (2013). Green synthesis of α-aminophosphonate derivatives on a solid supported TiO2-SiO2 Catalyst and their anticancer activity. Arch. Pharm. Chem. Life Sci. 346, 667–676. doi:10.1002/ardp.201300214
Congradyova, A., and Jomova, K. (2013). Antifungal effect of copper (II)-phenanthroline complex with 5-chlorosalicyclic acid. J. Microbiol. Biotechnol. Food Sci. 2, 1377–1383.
Creaven, B. S., Duff, B., Egan, D. A., Kavanagh, K., Rosair, G., Thangella, V. R., et al. (2010). Anticancer and antifungal activity of copper(II) complexes of quinolin-2(1H)-one-derived Schiff bases. Inorganica Chim. Acta 363, 4048–4058. doi:10.1016/j.ica.2010.08.009
Dinér, P., and Amedjkouh, M. (2006). Aminophosphonates as organocatalysts in the direct asymmetric aldol reaction: Towards syn selectivity in the presence of Lewis bases. Org. Biomol. Chem. 4, 2091–2096. doi:10.1039/b605091c
Ditchfield, R., Hehre, W. J., and Pople, J. A. (1971). Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 54, 724–728. doi:10.1063/1.1674902
Drissi, M., Benhalima, N., Megrouss, Y., Rachida, R., Chouaih, A., and Hamzaoui, F. (2015). Theoretical and experimental electrostatic potential around the m-nitrophenol molecule. Molecules 20, 4042–4054. doi:10.3390/molecules20034042
Dunning, T. H., and Hay, P. J. (1977). “Gaussian basis sets for molecular calculations,” in Methods of electronic structure theory. Modern theoretical chemistry. Editor H. F. Schaefer (Boston, MA: Springer), Vol. 3. doi:10.1007/978-1-4757-0887-5_1
El Sayed, I., El Kosy, S. M., Hawata, M. A., El Gokha, A. A.-A., Tolan, A., and Abd Ei–Sattar, M. M. (2011). Synthesis and antibacterial activity of α-aminophosphonates bearing neocryptolepine moiety. J. Am. Sci. 7, 357.
El-Boraey, H. A. L., El-Gokha, A. A. A., El-Sayed, I. E. T., and Azzam, M. A. (2015). Transition metal complexes of α-aminophosphonates Part I: Synthesis, spectroscopic characterization, and in vitro anticancer activity of copper (II) complexes of α-aminophosphonates. Med. Chem. Res. 24, 2142–2153. doi:10.1007/s00044-014-1282-8
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 revision (A.01. Wallingford CT: Gaussian, Inc..
Gemmill, W. R., Smith, M. D., and Reisner, B. A. (2005). A tetrahedrally coordinated cobalt(II) aminophosphonate containing one-dimensional channels. J. Solid State Chem. 178, 2658–2662. doi:10.1016/j.jssc.2005.05.035
Gu, L., and Jin, C. (2012). Synthesis and antitumor activity of α-aminophosphonates containing thiazole[5,4-b]pyridine moiety. Org. Biomol. Chem. 10, 7098–7102. doi:10.1039/c2ob25875g
Gundluru, M., Badavath, V. N., Shaik, H. Y., Sudileti, M., Nemallapudi, B. R., Gundala, S., et al. (2021). Design, synthesis, cytotoxic evaluation and molecular docking studies of novel thiazolyl α-aminophosphonates. Res. Chem. Intermed. 47, 1139–1160. doi:10.1007/s11164-020-04321-6
Hay, P. J., and Wadt, W. R. (1985). Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310. doi:10.1063/1.448975
Hay, P. J., and Wadt, W. R. (1985). Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 82, 270–283. doi:10.1063/1.448799
Hu, D.-Y., Wan, Q.-Q., Yang, S., Song, B.-A., Bhadury, P. S., Jin, L.-H., et al. (2008). Synthesis and antiviral activities of amide derivatives containing the α-aminophosphonate moiety. J. Agric. Food Chem. 56, 998–1001. doi:10.1021/jf072394k
Imam, E. A., El-Sayed, I. E. T., Mahfouz, M. G., Tolba, A. A., Akashi, T., Galhoum, A. A., et al. (2018). Synthesis of α-aminophosphonate functionalized chitosan sorbents: Effect of methyl vs phenyl group on uranium sorption. Chem. Eng. J. 352, 1022–1034. doi:10.1016/j.cej.2018.06.003
Juribasic, M., Molcanov, K., Kojic-Prodic, B., Bellotto, L., Kralj, M., Zani, F., et al. (2011). Palladium(II) complexes of quinolinylaminophosphonates: Synthesis, structural characterization, antitumor and antimicrobial activity. J. Inorg. Biochem. 105, 867. doi:10.1016/j.jinorgbio.2011.03.011
Kafarski, P., Lejczak, B., Slesak, E., and Przetocki, J. (1989). Plant growth regulating activity of aromatic aminophosphonates and their short peptides. Pestic. Sci. 25, 137–143. doi:10.1002/ps.2780250205
Kiwaan, H. A., El-Mowafy, A. S., and El-Bindary, A. A.. (2021). Synthesis, spectral characterization, DNA binding, catalytic and in vitro cytotoxicity of some metal complexes, J. Mol. Liq 326, 115381. doi:10.1016/j.molliq.2021.115381
Konarikova, K., Andrezalova, L., Rapta, P., Slovakova, M., Durackova, Z., Laubertova, L., et al. (2013). Effect of the Schiff base complex diaqua-(N-salicylidene-l-glutamato)copper(II) monohydrate on human tumor cells. Eur. J. Pharmacol. 721, 178–184. doi:10.1016/j.ejphar.2013.09.038
Kraicheva, I., Tsacheva, I., Vodenicharova, E., Tashev, E., Tosheva, T., Kril, A., et al. (2012). Synthesis, antiproliferative activity and genotoxicity of novel anthracene-containing aminophosphonates and a new anthracene-derived Schiff base. Bioorg. Med. Chem. 20, 117–124. doi:10.1016/j.bmc.2011.11.024
Long, N., Cai, X.-J., Song, B.-A., Yang, S., Chen, Z., Bhadury, P. S., et al. (2008). Synthesis and antiviral activities of cyanoacrylate derivatives containing an alpha-aminophosphonate moiety. J. Agric. Food Chem. 56, 5242–5246. doi:10.1021/jf800405m
López-Francés, A., del Corte, X. D., Martínez de Marigorta, E., Palacios, F., and Vicario, J. (2021). Ugi reaction on α-phosphorated ketimines for the synthesis of tetrasubstituted α-aminophosphonates and their applications as antiproliferative agents. Molecules 26, 1654. doi:10.3390/molecules26061654
Liu, J., Liao, P., Hu, J., Zhu, H., Wang, Y., Li, Y., et al. Synthesis and antitumor activities of chiral dipeptide thioureas containing an alpha-aminophosphonate moiety, Molecules, 22 (2017) 238. doi:10.3390/molecules22020238
Louie, A. Y., and Meade, T. J. (1999). Metal complexes as enzyme inhibitors. Chem. Rev. 99, 2711–2734. doi:10.1021/cr9804285
Ma, Y., Wang, J. G., Wang, B., and Li, Z. M. (2011). Integrating molecular docking, DFT and CoMFA/CoMSIA approaches for a series of naphthoquinone fused cyclic α-aminophosphonates that act as novel topoisomerase II inhibitors. J. Mol. Model. 17, 1899–1909. doi:10.1007/s00894-010-0898-y
Marzano, C., Pellei, M., Alidori, S., Brossa, A., Lobbia, G. G., Tisato, F., et al. (2006). New copper(I) phosphane complexes of dihydridobis(3-nitro-1,2,4-triazolyl)borate ligand showing cytotoxic activity. J. Inorg. Biochem. 100, 299–304. doi:10.1016/j.jinorgbio.2005.11.014
Massoud, S. S., Louka, F. R., Ducharme, G. T., Fischer, R. C., Mautner, F. A., Vančo, J., et al. (2018). Copper(II) complexes based on tripodal pyrazolyl amines: Synthesis, structure, magnetic properties and anticancer activity. J. Inorg. Biochem. 180, 39–46. doi:10.1016/j.jinorgbio.2017.11.023
Mohan, G., Kuma, S., Sudileti, M., Sridevi, C., Venkatesu, P., and Reddy, C. S. (2020). Excellency of pyrimidinyl moieties containing α-aminophosphonates over benzthiazolyl moieties for thermal and structural stability of stem bromelain. Int. J. Biol. Macromol. 165, 2010–2021. doi:10.1016/j.ijbiomac.2020.10.065
Mohapatra, R. K., Azam, M., Mohapatra, P. K., Sarangi, A. K., Abdalla, M., Perekhoda, L., et al. (2022). Computational studies on potential new anti-Covid-19 agents with a multi-target mode of action. J. King Saud Univ. - Sci. 34, 102086. doi:10.1016/j.jksus.2022.102086
Monika, , , Yadav, O., Chauhan, H., and Ansari, A. (2021). Electronic structures, bonding, and spin state energetics of biomimetic mononuclear and bridged dinuclear iron complexes: A computational examination. Struct. Chem. 32, 1473–1488. doi:10.1007/s11224-020-01690-x
Moreno-Cinos, C., Sassetti, E., Salado, I. G., Witt, G., Benramdane, S., Reinhardt, L., et al. (2019). α-Amino diphenyl phosphonates as novel inhibitors of Escherichia coli ClpP protease. J. Med. Chem. 62, 774–797. doi:10.1021/acs.jmedchem.8b01466
Mucha, A., Kafarski, P., and Berlicki, L. (2011). Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 54, 5955–5980. doi:10.1021/jm200587f
Naydenova, E. D., Todorov, P. T., and Troev, K. D. (2010). Recent synthesis of aminophosphonic acids as potential biological importance. Amino Acids 38, 23–30. doi:10.1007/s00726-009-0254-7
Poola, S., Nadiveedhi, M. R., Sarva, S., Gundluru, M., Nagaripati, S., Shaik, M. S., et al. (2019). Nano Sb2O3 catalyzed green synthesis, cytotoxic activity, and molecular docking study of novel α-aminophosphonates. Med. Chem. Res. 28, 528–544. doi:10.1007/s00044-019-02302-y
Rafique, S., Idrees, M., Nasim, A., Akbar, H., and Athar, A. (2010). Transition metal complexes as potential therapeutic agents. Biotechnol. Mol. Biol. Rev. 5, 38–45.
Ranford, J. D., Sadler, P. J., and Tocher, D. A. (1993). Cytotoxicity and antiviral activity of transition-metal salicylato complexes and crystal structure of Bis(diisopropylsalicylato)(1,10-phenanthroline)copper(II). J. Chem. Soc. Dalton Trans. 22, 3393–3399. doi:10.1039/dt9930003393
Reddy, C. B., Kumar, K. S., Kumar, M. A., Narayana Reddy, M. V. N., Krishna, B. S., Naveen, M., et al. (2012). PEG-SO(3)H catalyzed synthesis and cytotoxicity of α-aminophosphonates-aminophosphonates. Eur. J. Med. Chem. 47, 553–559. doi:10.1016/j.ejmech.2011.11.026
Repicka, Z., Krupkova, L., Maria, K., Moncol, J., Hudecova, D., and Valigura, D. (2009). Bis(5-Me(O)salicylato) copper (II) complexes with/without diethylnicotinamide – preparation, structure and properties. Acta Chim. Slov. 2, 52
Rezaei, Z., Khabnadideh, S., Zomorodian, K., Pakshir, K., Nadali, S., Mohtashami, N., et al. (2011). Design, synthesis, and antifungal activity of new α-aminophosphonates. Int. J. Med. Chem. 2011, 678101. doi:10.1155/2011/678101
Romero-Estudillo, I., Viveros-Ceballos, J. L., Cazares-Carreño, O., González-Morales, A., de Jesús, B., López-Castillo, M., et al. (2019). 000Synthesis of new α-aminophosphonates: Evaluation as anti-inflammatory agents and QSAR studies. Bioorg. Med. Chem. 27, 2376–2386. doi:10.1016/j.bmc.2018.12.041
Saeed, S., Rashid, N., Ali, M., and Hussain, R. (2010). Synthesis, characterization and antibacterial activity of nickel (II) and copper (II) complexes of N-(alkyl(aryl)carbamothioyl)-4-nitrobenzamide. Eur. J. Chem. 1, 200–205. doi:10.5155/eurjchem.1.3.200-205.120
Sakurai, H., Kojima, Y., Yoshikawa, Y., Kawabe, K., and Yasui, H. (2002). Antidiabetic vanadium(IV) and zinc(II) complexes. Coord. Chem. Rev. 226, 187–198. doi:10.1016/s0010-8545(01)00447-7
Santini, C., Pellei, M., Gandin, V., Porchia, M., Tisato, F., and Marzano, C. (2014). Advances in copper complexes as anticancer agents. Chem. Rev. 114, 815–862. doi:10.1021/cr400135x
Schafer, A., Horn, H., and Ahlrichs, R. (1992). Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577. doi:10.1063/1.463096
Schafer, A., Huber, C., and Ahlrichs, R. (1994). Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835. doi:10.1063/1.467146
Sienczyk, M., and Oleksyszyn, J. (2009). Irreversible inhibition of serine proteases - design and in vivo activity of diaryl α-Aminophosphonate derivatives. Curr. Med. Chem. 16, 1673–1687. doi:10.2174/092986709788186246
Singh, I. P., Gupta, S., and Kumar, S. (2020). Thiazole compounds as antiviral agents: An update. Med. Chem. 16, 4–23. doi:10.2174/1573406415666190614101253
Song, B., Jin, L., Yang, S., and Bhadury, P. S. (2010). Studies on α-aminophosphonates with antiviral activity, environment-friendly antiviral agents for plants. Berlin, Heidelberg: Springer, 7–93. doi:10.1007/978-3-642-03692-7_2
Sudileti, M., Chintha, V., Nagaripati, S., Gundluru, M., Yasmin, S. H., Wudayagiri, R., et al. (2019). Green synthesis, molecular docking, anti-oxidant and anti-inflammatory activities of α-aminophosphonates. Med. Chem. Res. 28, 1740–1754. doi:10.1007/s00044-019-02411-8
Sudileti, M., Gundluru, M., Sarva, S., Tellamekala, S., Hari, B., Meriga, B., et al. (2019). Tungstosulfonic acid-catalyzed green synthesis and bioassay of α-aminophosphonates. Monatsh. Chem. 150, 1101–1109. doi:10.1007/s00706-019-2385-1
Sujatha, B., Mohan, S., Subramanyam, C., and Rao, K. P. (2017). Microwave-assisted synthesis and anti-inflammatory activity evaluation of some novel α-aminophosphonates. Phosphorus, Sulfur, Silicon Relat. Elem. 192, 1110–1113. doi:10.1080/10426507.2017.1331233
Sumrra, S. H., Atif, A. H., Zafar, M. N., Khalid, M., Tahir, M. N., Nazar, M. F., et al. (2018). Synthesis, Crystal Structure, Spectral and DFT studies of potent isatin derived metal complexes. J. Mol. Struct. 1166, 110–120. doi:10.1016/j.molstruc.2018.03.132
Takayanagi, H., Kai, T., Yamaguchi, S.-I., Takeda, K., and Goto, M. (1996). Studies on picrate. VIII. Crystal and molecular structures of aromatic amine picrates: Aniline, N-methylaniline, N, N-dimethylaniline and o-, m- and p-phenylenediamine picrates. Chem. Pharm. Bull. 44, 2199–2204. doi:10.1248/cpb.44.2199
Tardito, S., Bussolati, O., Maffini, M., Tegoni, M., Giannetto, M., Dall'asta, V., et al. (2007). Thioamido coordination in a thioxo-1,2,4-triazole copper(II) complex enhances nonapoptotic programmed cell death associated with copper accumulation and oxidative stress in human cancer cells. J. Med. Chem. 50, 1916–1924. doi:10.1021/jm061174f
Tian, J., Ji, R., Wang, H., Li, S., and Zhang, G. (2022). Discovery of novel α-aminophosphonates with hydrazone as potential antiviral agents combined with active fragment and molecular docking. Front. Chem. 10, 911453. doi:10.3389/fchem.2022.911453
Tomasi, J., Mennucci, B., and Cammi, R. (2005). Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094. doi:10.1021/cr9904009
Trott, O., Olson, A. J., and Vina, A. (2010). Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem. 31, 455–461. doi:10.1002/jcc.21334
Tusek-Bozic, L. J. (2013). Aminophosphonate metal complexes of biomedical potential. Cmc 20, 2096–2117. doi:10.2174/0929867311320160004
Wadt, W. R., and Hay, P. J. (1985). Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 82, 284–298. doi:10.1063/1.448800
Wang, Q., Zhu, M., Zhu, R., Lu, L., Yuan, C., Xing, S., et al. (2012). Exploration of α-aminophosphonate N-derivatives as novel, potent and selective inhibitors of protein tyrosine phosphatases. Eur. J. Med. Chem. 49, 354–364. doi:10.1016/j.ejmech.2012.01.038
Weder, J. E., Dillon, C. T., Hambley, T. W., Kennedy, B. J., Lay, P. A., Biffin, J. R., et al. (2002). Copper complexes of non-steroidal anti-inflammatory drugs: An opportunity yet to be realized. Coord. Chem. Rev. 232, 95–126. doi:10.1016/s0010-8545(02)00086-3
Wehbe, M., Lo, C., Leung, A. W. Y., Dragowska, W. H., Ryan, G. M., and Bally, M. B. (2017). Copper (II) complexes of bidentate ligands exhibit potent anti-cancer activity regardless of platinum sensitivity status. Invest.. New Drugs 35, 682–690. doi:10.1007/s10637-017-0488-2
Xu, Y., Yan, K., Song, B., Xu, G., Yang, S., Xue, W., et al. (2006). Synthesis and antiviral bioactivities of alpha-aminophosphonates containing alkoxyethyl moieties. Molecules 11, 666–676. doi:10.3390/11090666
Keywords: DFT, molecular docking, NBO, MEP map, copper species
Citation: Yadav O, Kumar M, Mittal H, Yadav K, Seidel V and Ansari A (2022) Theoretical exploration on structures, bonding aspects and molecular docking of α-aminophosphonate ligated copper complexes against SARS-CoV-2 proteases. Front. Pharmacol. 13:982484. doi: 10.3389/fphar.2022.982484
Received: 30 June 2022; Accepted: 24 August 2022;
Published: 03 October 2022.
Edited by:
Ranjan K. Mohapatra, Government College of Engineering, IndiaReviewed by:
Tulika Gupta, Banaras Hindu University, IndiaCopyright © 2022 Yadav, Kumar, Mittal, Yadav, Seidel and Ansari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Veronique Seidel, dmVyb25pcXVlLnNlaWRlbEBzdHJhdGguYWMudWs=; Azaj Ansari, YWphei5hbGFtMkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.