
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 23 August 2022
Sec. Neuropharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.976799
This article is part of the Research Topic Current Status of Natural Products Targeting Alzheimer’s Disease View all 11 articles
 Tapan Behl1*
Tapan Behl1* Dapinder Kaur2Aayush Sehgal2
Dapinder Kaur2Aayush Sehgal2 Rajeev K. Singla3,4
Rajeev K. Singla3,4 Hafiz A. Makeen5Mohammed Albratty6Hassan A. Alhazmi6,7Abdulkarim M. Meraya8
Hafiz A. Makeen5Mohammed Albratty6Hassan A. Alhazmi6,7Abdulkarim M. Meraya8 Simona Bungau9,10*
Simona Bungau9,10*Alzheimer’s disease (AD) is perceived with various pathophysiological characteristics such oxidative stress, senile plaques, neuroinflammation, altered neurotransmission immunological changes, neurodegenerative pathways, and age-linked alterations. A great deal of studies even now are carried out for comprehensive understanding of pathological processes of AD, though many agents are in clinical trials for the treatment of AD. Retinoids and retinoic acid receptors (RARs) are pertinent to such attributes of the disease. Retinoids support the proper functioning of the immunological pathways, and are very potent immunomodulators. The nervous system relies heavily on retinoic acid signaling. The disruption of retinoid signaling relates to several pathogenic mechanisms in the normal brain. Retinoids play critical functions in the neuronal organization, differentiation, and axonal growth in the normal functioning of the brain. Disturbed retinoic acid signaling causes inflammatory responses, mitochondrial impairment, oxidative stress, and neurodegeneration, leading to Alzheimer’s disease (AD) progression. Retinoids interfere with the production and release of neuroinflammatory chemokines and cytokines which are located to be activated in the pathogenesis of AD. Also, stimulating nuclear retinoid receptors reduces amyloid aggregation, lowers neurodegeneration, and thus restricts Alzheimer’s disease progression in preclinical studies. We outlined the physiology of retinoids in this review, focusing on their possible neuroprotective actions, which will aid in elucidating the critical function of such receptors in AD pathogenesis.
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder marked with personality changes, memory and cognitive deficits due to loss of the neurons in the frontal cortex and hippocampus. The histological indicator of AD is amyloid plaques, comprising of insoluble amyloid-ß (Aß) peptides (Kabir et al., 2021). Overactive inflammatory astrocytes and microglia localized with the senile plaques are considered to be linked with the pathological lesions (Ayaz et al., 2019a). It is now commonly accepted that mostly affects older individuals over the age of 65 as a geing is a major trigger for Alzheimer’s disease. AD is presently the most common neurological condition affecting over 15 million individuals globally (Andreeva et al., 2017). The world population of individuals with AD is constantly increasing. Clinical evidence links Alzheimer’s disease to dementia and memory loss. AD is characterized by extra-neuronal Aß plaques deposition and intracellular neurofibrillary tangles in the temporal lobe. Aß plaques are made up of accumulated amyloid-beta peptides, whereas neurofibrillary tangles are made up of tau protein that has been hyperphosphorylated (Querfurth and LaFerla, 2010). Oxidative stress, neuroinflammation, and mitochondrial dysfunction are all triggered by the development of these aggregates, resulting in the loss of not only neurons but also white matter in the brain. New research reveals that the pathogenesis of Alzheimer’s disease may be caused by a complicated interaction involving aberrant Aß and tau proteins. The amyloid hypothesis of AD claims that the buildup of Aß plaques in the temporal lobe of the brain is the basic pillar of neurodegeneration and memory deficits in AD patients (Musiek and Holtzman, 2015; Behl et al., 2020). The amyloid hypothesis’s fundamental weakness is its failure to definitively establish the molecular mechanisms that link amyloidosis to NFTsfor neurodegeneration in Alzheimer’s disease (Eriksen and Janus, 2007). There are numerous different possibilities concerning AD pathophysiology, and several natural compounds such as flavanoids, retinoids, lipoic acids have been developed to treat AD based on these beliefs (Ayaz et al., 2019b). The majority of Alzheimer’s models are based on a single theory for the causation of AD, which is a serious flaw in the research. To create viable therapies that will treat the majority of instances, a full understanding of the condition is required. Researchers used different genetically comparable transgenic knock-in animals of AD and tau dysfunction associated with AD and dementia to investigate changes in retinoid signaling at the transcriptional levels in such models. Female rat hippocampal and frontal combined primary cultures were also used to undertake an early assessment of the therapeutic potential of a new family of synthetic retinoids (RAR-M1) that target both biological and non-biological receptors (Endres et al., 2014a; Khatib et al., 2020).
Vitamin-A analogs, both natural and synthetic, are known as retinoids. These chemicals are significant in memory because they are believed to play important functions in adult brain development (Das et al., 2014a). As a result, there is a surge of attention in discovering more about the biology and chemistry of known and new retinoids, as well as their therapeutic potential in the treatment of acute and chronic disorders like AD. Since retinoic acid (RA), a vitamin A derivative, can bind to nuclear receptors and regulate the expression of multiple genes in cells, it executes the majority of biological mechanisms (Lerner et al., 2012). Retinoids activate their target genes by interacting with nuclear receptors including retinoic acid receptors (RAR) and retinoid X receptors (RXR), which are transcriptional modifiers that are reported to be expressed in the prefrontal cortex, amygdala, and hippocampus regions of the brain (Goodman and Pardee, 2003a). These receptors bind to a specific DNA sequence and either suppress or promote target gene expression (Khorasanizadeh and Rastinejad, 2001). In animals, retinoid deprivation or mutations in the RAR and RXR genes have been linked to the suppression of spatial memory and learning, as well as the emergence of depression (Nomoto et al., 2012). In retinoid-deficit rats, suppressing RAR expression resulted in the accumulation of amyloid-beta (Aß) polypeptide in the vasculature, according to research (Shudo et al., 2009). Retinoids play a vital function in neuroprotection by preventing neuroinflammatory processes (Lee et al., 2009a). Microglia, have been shown to suppress the production of neuroinflammatory cytokines and chemokines (Goncalves et al., 2013a). To the improvement of cholinergic neurotransmission, retinoid receptor agonists were known to stimulate the expression of the choline acetyltransferase (ChAT) and acetylcholine transporter genes (Mufson et al., 2008). Retinoids are conventionally accepted as antioxidants, have significant role in the maintenance of brain activity during advanced age. AD patients have been observed with moderate levels of serum vitamin A. Clinically, it was reported that the cognitive abilities in the group of 442 patients of AD were improved with increased serum Vitamin-A levels (Mohammadzadeh Honarvar et al., 2017a). It has been observed that vitamin A and beta-carotene restrain the generation of amyloid-ß fibrils from amyloid precursor protein (APP) and induce variation in the fibrillar anatomy of amyloid beta proteins (Fahrenholz et al., 2010). Retinoic acid (RA), a dynamic metabolite of vitamin A, has been observed to regulate the gene expression relying on APP processing in nuclear receptors including RAR and RXR. Vitamin A deficiency potentiaiates the depoisition of Aß peptides and decreases the long term potentiation of the hippocampus in animals. Researches has revealed that RA potentaite the appearance of the MNSOD2 gene in neuroblast cells, thus decreasing oxidative stress, an essential pathological factor in AD. Studies revealed that memory and cognitive impairments, and reduced neuroplasticity, are due to RXR and RAR mutations (Connor and Sidell, 1997a; Etchamendy et al., 2003a; La Fata et al., 2014; Pierzchalski, 2015).In this review, we discussed the chemistry and biochemistry of various natural and synthetic retinoids, as well as their efficacy in preventing neurodegeneration in Alzheimer’s disease.
The importance of retinoids in the growth and differentiation of the prenatal and postnatal brain has been recognised long ago (Jiang et al., 2012; Cunningham and Duester, 2015; Bonney et al., 2018). Yet, a substantial size of the data suggests that retinoid signalling is important in adult brain activity as well (Kour and Rath, 2016; Mishra et al., 2018). Vitamin A is the most widely occuring retinoid, regulating a number of physiolgical functions including embryogenesis, cellular proliferation, cell growth, and cell death, as well as proper brain functioning (Khillan, 2014). It is primarily synthesized from pro-vitamin A carotenoids, which may be found in a variety of colourful vegetables and fruits as well as in animal sources like egg yolks, and dairy products. Vitamin A carotenoids are synthesized by some microorganisms and photosynthetic plants which are metabolised to retinol in the small intestines of animals (Green and Fascetti, 2016). Some noteworthy scientific papers have already been published outlining the role of natural and synthetic retinoids in drug development and signaling cascades (Das et al., 2014b; Haffez et al., 2018; Chisholm et al., 2019). At present, there are number of major synthetic retinoids for their therapeutic use, even though some differences in haematological parameters occur following isotretinoin therapy, yet is the most excellent therapy for treating acne vulgaris in clinical studies (Gencoglan et al., 2018). Since, retinoids have conjugated double bonds, they are rapidly oxidised or isomerized by the action of oxidants, light, or intense heat. Increased ingestion of pro-vitamin A carotenoids is linked to a decreased risk of various brain disorders, including Alzheimer’s disease, according to epidemiological research (Lakey-Beitia et al., 2017; Yang et al., 2017). Retinoids also improves visual acuity and protects against age-linked macular degeneration (Harrison, 2019). Therefore, natural and synthetic retinoids are now being explored intensively for effective neuroprotection in neuronal injuries and disorders (Chakrabarti et al., 2016a). The biosynthetic pathway of RA begins either with conversion of retinol to retinal or the manufacture of retinal (Figure 1) as provided by different food sources, often known as vitamin A (Sommer and Vyas, 2012). Two oxidation processes are required to synthesize retinoic acid from retinol. The activity of retinol dehydrogenase and the transition of NAD+ to NADH converts retinol to retinal (Hong et al., 2015). As in the following oxidation reaction, RA is synthesized from retinal with the activity of retinaldehyde dehydrogenases (RALHDs or ALDHs), which come in a variety of types (Kedishvili, 2016). Retinol attach to cellular retinol-binding proteins (CRBPs), whereas RA binds to cellular RA-binding proteins (CRABPs) in the cytoplasm (Napoli, 2017). CRBPs are classified into two classes (CRBP type I and type II); likewise, CRABPs are categorised into two classes (CRABP type I and type II), and they transport RA to RARs and RXRs which are identical but differ in amino acid sequences. CRBPs are essential for the absorption and metabolism of retinol, whereas CRABPs are involved in the control of several RA signalling pathways and the availability of retinol to its receptors (Zhang et al., 2012). All RA isomers activate RARs, which function as heterodimers with RXR nuclear subtypes. Generally, retinoids occur naturally and are chemical conjugates of vitamin A. The RAR-RXR dimer regulates transcriptional activity by interacting with a retinoic acid response element (RARE) found in the gene promoter. Both α and Γ types of RARs are largely found in the adult brain, with higher expression in both the cortex and the hippocampus, while RARß, RXRΓ, RXRß, and RXR have limited distribution. Although levels might be significant in such confined locations; for example, RAR is expressed primarily in the hypothalamus and striatum. RAR protein’s localized activity does not resemble one of their messenger RNA (mRNA) transcripts, suggesting that post-translational regulation of their representation is important. RA seems to have the ability to control the number of genes actively or passively through RARs (Hong et al., 2015; Kedishvili, 2016). The cytochrome P450 enzymes of the CYP26 family (Napoli, 2017), especially CYP26B1, are principally accountable for shutting down the network (Zhang et al., 2012).
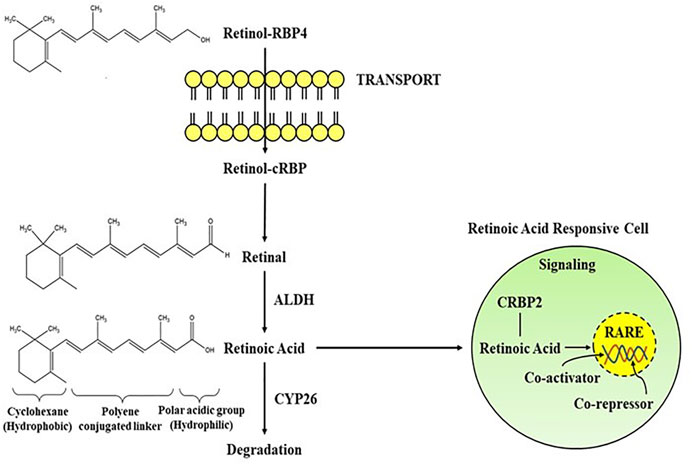
FIGURE 1. Schematic representation of the transport and signaling pathways of retinoic acid from retinol via the activity of alcohol dehydrogenase (ALDH) and degradation through CYP26. Retinoic acid mediates its activity by interacting with the RA receptors in RA responsive cell. RBP, Retinol binding protein; RARE, Retinoic acid response element.
Although the epigenetic roles of retinoids (regulation of transcription) are well-known, the non-genomic function is also important in its physiological activities. The non-genomic activity of retinoids is mostly assumed to be in intracellular pathways, such as stimulation of various kinase pathways, which permits faster indirect effects than the comparatively slow transcriptional processes. RAR is frequently involved in such cytosolic pathways, but retinoids can also interact with some other receptors like CRABP1 or protein kinase-Cα (Ochoa et al., 2003). The modulation of neuroplasticity through stimulation of mammalian target of rapamycin (mTOR) and ERK1/2 phosphorylation-induced mitogen-activated protein kinase (MAPK) is believed to include non-genomic processes (Zhong et al., 2018). RAR’s interaction with the RNA-binding protein PurA is also being hypothesized to prevent axonal protein translation via encouraging mRNA transport to suppress ribonucleoprotein molecules. This inhibition is reversed by retinoids, enabling PurA-mediated axonal RNA transfer to recommence, resulting in neuronal growth stimulation. Aside from intracellular functions, Napoli and Chen found membrane-linked RAR-α in cholinergic neuronal cells, which increases neurogenesis through rapid ATRA-mediated neuronal translation (Chen and Napoli, 2008). Surprisingly, synapto genesis requires the integration of genomic and non-genomic mechanisms (Khatib et al., 2019). Furthermore, specific RAR agonists have different abilities to stimulate genomic and non-genomic systems, which can be engaged separately. RAR-α impacts the cholinergic as well as the GABAnergic and glutamatergic pathways. When RA acts on RAR, it increases the mRNA expression of Choline acetyltransferase (ChAT) and vesicular ACh transporter (VAChT), according to research done on mouse cell lines. As ChAT is necessary for memory encoding and the production of acetylcholine (ACh). Memory encoding and working memory are impaired when muscarinic ACh receptors are blocked, but additional information is enhanced when nicotinic ACh receptors are stimulated (Hasselmo, 2006) (Figure 1).
Dietary supplementation of carotenoids has been shown to play a crucial role in preventing several neu-rodegenerative diseases, including AD (Obulesu et al., 2011a). Retinoids are involved in neuronal patterning, differentiation, and axon outgrowth. Retinoid deprivation leads to impairment of normal brain development and function, resulting in the appearance of symptoms of different neurodegenerative diseases, including AD. Recent investigations indicate that retinoids can induce generation of specific neuronal cell types and also regenerate axons after damage (Maden, 2007). In addition, retinoids are involved in the maintenance of the differentiated state of adult neurons and neural stem cells as well as altered RA signaling levels. Many studies involving genetic analysis of AD have confirmed direct correlation between the genes that encode molecules involved in the RA signaling pathway and those that are considered to be involved in the pathogenesis of AD (Goodman, 2006a).
Retinoids are thought to perform through a variety of processes, according to a substantial amount of research. These chemicals have anti-inflammatory and antioxidant properties. Retinoids have also been discovered to play an important function as antioxidant enzymes as they contribute to the modulation of cytotoxic effects of ROS by eliminating free radicals from cells (Lushchak, 2011). ROS generation and deposition are enhanced in cells during oxidative stress environments like as metal exposure. Retinoids offer protection from this unstable state through a variety of methods, such as inhibition of ROS generation, free radicals clearance, subsequent activation of antioxidant enzymes, and modulation of defence signalling pathways including Nrf2 signalling (Alpsoy et al., 2009). In this view, clinical data suggests that retinoid levels in the liver have been reported as a defence mechanism towards ROS-mediated metal exposure (Defo et al., 2012). Retinoic acid (RA) has been shown to protect neurons from oxidative stress and apoptotic death by lowering glutathione levels (Lee et al., 2009b). In hippocampus cells, it also improves superoxide dimustase (SOD1 and SOD-2) activity (Obulesu et al., 2011b). Furthermore, retinoids are involved in the development and proper functioning of several cells and organs of the immune system, indicating that they play an important regulatory role on various inflammatory responses. The impaired generation of RA-dependent tolerogenic macrophages and dendritic cells in Vit A-deficient cells has been shown to exacerbate inflammatory response (Saurer et al., 2007). Study found that appropriate retinoid levels are required for proper functioning of epithelial barrier stability (Filteau et al., 2001). Retinoids, when consumed in sufficient amounts, have been shown to reduce inflammation in animals (Kang et al., 2007). Also, the anti-inflammatory actions of retinoids involves the inhibition of the synthesis of inflammatory cytokines by reducing the translocation of the NF-kappaB transcription factor (Horton et al., 2001). Additionally, RA has a profound suppressive effect on T-cells (Th cells) which are involved in neuroinflammation whereas RA stimulates Th2, a subtype of T-cell that has anti-inflammatory properties (Nozaki et al., 2006). Furthermore, RA has been demonstrated to increase FoxP3 T-cells, which decrease the inflammation (Kim, 2011). These findings suggest that retinoids are anti-inflammatory agents which may have clinical efficacy in neuroinflammatory disorders including AD (Mohammadzadeh Honarvar et al., 2017b).
Microglia maintain brain homeostasis in normal circumstances. When pathogenic triggers, such as proinflammatory cytokines, pH changes, or hypoxia, are detected, the cells undergo a metamorphosis known as “activation,” which can lead to a chronic prolonged, vicious cycle of “sub-threshold” neuroinflammation. This moderate, yet continually sustained pro-inflammatory condition is thought to represent the neuronal component supporting neurodegenerative pathophysiology (Dantzer et al., 2008). Additionally, cytokines activate indoleamine-2,3-dioxygenase (IDO), a major enzyme in the kynurenine pathway (KP), which destroys the serotonin precursor tryptophan (Hochstrasser et al., 2011) culminating in a microglial-mediated rise in the neurotoxic NMDA-receptor agonist quinolinic acid (Schwarcz et al., 2012). The kynurenine pathway not only serves as a marker for inflammatory activation but also serves as a link between neuro-inflammation and transmitter imbalances, which are linked to a variety of neurodegenerative diseases. As a result, microglial stimulation is a viable therapeutic target for a variety of neurological diseases. Also, Aß causes activation of microglia and austrocytes to produce proinflammatory mediators, while retinoic acid can prevent the development of these proinflammatory cytokines by interacting with RARs, which are found in astrocytic and microglial cells. Retinoids have been shown to stimulate RAR and RXR, allowing these cells to adjust actions and limit production. Among the most fundamental tasks in the therapy of AD is to inhibit neuroinflammatory reactions. Previous research has found that retinoic acid can help in reducing neuroinflammation in neurodegenerative disorders. By reducing the transcriptional activity of the NFK-B, RA was believed to decrease the Aß-mediated generation of TNF-α and suppress the activity of inducible nitric oxide synthase (iNOS) in microglial cells (Kaur et al., 2006). Retinoids also reduce Aß-mediated neuroinflammation, amyloidogenesis, and cognitive impairments in animal models according to the latest research (Behairi et al., 2016). In transgenic mice, RA stimulates neural stem cell growth while suppressing microglial reactivity, resulting in hippocampal neurogenesis (Takamura et al., 2017). In an animal model with Aß-induced neuroinflammation, anti-inflammatory functions of a RAR ligand Am80 (Tamibarotene) (Table 1) were examined, and the analysis indicated that Am80 might endorse the generation of brain-derived neurotrophic factor (BDNF), thus providing cytoprotective outcomes in disease states (Katsuki et al., 2009). Upon administration of Am580 suppressed inflammation-mediated neuronal loss in cultured neurons (Jarvis et al., 2010). In numerous neurological diseases, namely Alzheimer’s disease, RA implies a vital function in suppressing neuroinflammatory reactions and encouraging clearance of Aß additional study is presently being taken for a better comprehension of the microscopic basis for the mechanism of actions of retinoids and carotenoids in the reduction of neuroinflammation in AD. Both retinoic acid and carotenoids provide effective anti-oxidative and anti-inflammatory actions and thus can be used in neuroprotection. They can slow the production and buildup of amyloid plaques, reduce the peroxidation of lipids, and inhibit the release of pro-inflammatory factors, all of which enhance mental abilities (Mohammadzadeh Honarvar et al., 2017c). All such studies have suggested that retinoic acid is performed via different routes to provide highly effective neuroprotection in AD. A recent study showed that RA reduced neurotoxicity in the rat brain via regulating the activity of Sirtuin 1, a class 3 histone deacetylase belonging to the Sirtuin protein family, as well as NFκ-B (Priyanka et al., 2018).

TABLE 1. The schematic data revealing the potential role of synthetic and natural retinoids as well as vitamin A derivatives in ameliorating the pathogenesis of Alzheimer’s disease in various experimental models through distinct mechanisms/pathways.
RA is a CNS morphogen that is recognized to prevent neuroinflammation (Hellmann-Regen et al., 2013) and microglial activation and also, it is important for neuronal growth. RA is a powerful neuroprotective drug that has been linked to neuroplasticity (Maghsoodi et al., 2008). In neurological diseases, each of these pathways is reported to be disturbed. Furthermore, there is a considerable amount of direct evidence supporting retinoid signaling’s role in the pathophysiology of AD (Bremner and McCaffery, 2008) and other neurological diseases (van Neerven et al., 2008), implying that local brain RA might act as an “endogenous antidepressant.” Moreover, the commonly prescribed and well known antidepressant fluoxetine inhibits RA degradation (Wietrzych-Schindler et al., 2011), implying that fluoxetine’s neuroprotective, anti-inflammatory, and perhaps anti-depressant effects are all achieved via RA signaling (Hellmann-Regen et al., 2015).
Lower intrinsic levels of retinoids have been linked to cognitive loss in the elderly (Huang et al., 2018), and have been found to decrease in the ageing mouse brain (Touyarot et al., 2013). This reduction affects cognition since RA promotes neurogenesis and neuroplasticity, both of which are necessary for memory and learning (Shearer et al., 2012). If low RA levels induce AD, it is reasonable to expect that enhancing them would be beneficial, and multiple in-vitro investigations have shown that RA lowers amyloid-beta toxicity (Sahin et al., 2005). Moreover, a vitamin A-deficient diet in mice causes disturbance in RA signalling pathway and Aß accumulation in the blood vessels of frontal brain neurons, which can be restored by RA treatment (Husson et al., 2006a). Increasing the RA signalling through its receptor agonists promotes cognition in transgenic mice models of AD, clears Aß in microglia and neurons, and has a powerful anti-inflammatory effect (Goncalves et al., 2013b). As a result, synthetic retinoids might be used to treat Alzheimer’s disease and other neurodegenerative diseases. Because of its multiple therapeutic benefits, synthetic retinoid tamibarotene (Am80) is being explored intensively as a possible medication for Alzheimer’s disease which tends to reduce the level of insoluble Aß42 in APP23 AD model mice, according (Kawahara et al., 2009) As such acitretin is under investigation, that has been shown to raise the levels of the α-secretase of amyloid-protein precursor (APP), promoting the non-amyloidogenic process in neuroblastoma cells and lowering Aß levels in APP/PS-1 AD model (Tippmann et al., 2009). Acitretin has also been shown to pass the blood-brain barrier in rodents (Holthoewer et al., 2012). Endres et al. (2014b) studied the effects of oral acitretin treatment on α-secretase-acquired APP levels in the cerebrospinal fluid (CSF) of patients with AD. Additionally, acitretin raised APPs- levels and increased non-amyloidogenic processing of APP in humans, according to the studies (Endres et al., 2014b). Although some experimental data on the retinoids utilization in clinical trials is available, research on their possible therapeutic efficacy in AD is still in its initial stages. The data from a clinical trial of bexarotene (RXR ligand) in patients of AD showed that it may decrease cerebral amyloid and enhance plasma Aß1–42 in ApoE4 noncarriers (Cummings et al., 2016). Furthermore, acitretin entered Phase 2 trials in 2010, with preliminary findings showing a 25% rise in APPs-α levels in the CSF in the treatment group of AD patients (Endres et al., 2014c). Additionally, acitretin modulates certain genes connected to the pathogenesis of Alzheimer’s disease, including choline acetyltransferase (chAT). Gonçalves and others tested the effectiveness of numerous retinoid receptor agonists, namely AM 580, CD 2019, and CD437, that are specific RARα, RAR, and RAR Γ agonists, accordingly, on the AD experimental model. They showed that activating the RAR receptor signaling pathway promotes Aß removal by increasing the expression of NEP and IDE enzymes and modifying glial cell production of the pro-inflammatory cytokine TNF-α (Kobayashi et al., 1994) (Figure 2).

FIGURE 2. The overactivation of the ß- and Γ-secretases complexes may lead to the formation of Aß fibrils which successively leads to the generation of amyloid-ß plaques and neuroinflammation by activating the pro-inflammatory cytokines like TNF and IL. Such type of overactivation can be inhibited by the activity of retinoids like retinoic acid (RA). TNF-α, Tumour necrosis factor-alpha; IL, interleukins; Aß, amyloid beta; APP, Amyloid precursor protein; C99, carboxyl fragment 99.
As a result, a clear link between both reduced IDE mRNA levels and increased Aß deposition was already reported in the AD brain. Furthermore, reducing IDE expression may increase the likelihood of developing AD. Additionally, NEP-associated proteolytic processing of Aß has been shown to activate the signaling of RAR in brain cells (Goncalves et al., 2013a). Therefore, preliminary studies clearly show that natural and artificial retinoids can control Aß production and accumulation, suggesting that retinoids could be employed as a promising therapy for AD pathology as shown in Figure 2. Acitretin, a biosynthetic retinoid, is now being tested in humans under Phase 2 clinical trials. Although, there seems to be no experimental evidence supporting this treatment in Alzheimer’s patients. AD is related to impaired removal of Aß from the brain which is modulated by APOE. IDE is a protein that dissolves Aß deposits and is found in the brain (Manzine et al., 2019). RA regulates IDE transcription, and the gene’s promoter contains a RAR-α response element (Vekrellis et al., 2000). APP is a protein that aids in the production of Aß but is not cytotoxic and stimulates nerve growth (Melino et al., 1996). Also, the MAPT promoter has been demonstrated to be regulated by RA. The examination of MAPT’s (König et al., 1990) nucleotide coding region discovered that it includes a RAR-α response element (Goedert et al., 1989), starting at 5′ -16. The genotype has four distinct TGACC domains, each of which potentially provides retinoid responsiveness (Duester et al., 1991). Variants of one or more RARs may alter the transcriptional activity of such components, according to the latest findings. In contrast to RA, transforming growth factor- ß (TGF-ß), a mediator crucially engaged in AD, Aß plaque generation (Hall et al., 1992), neurological damage, and neuroinflammation, favorably regulates APP translation (Burton et al., 2002). The treatment of TGF-2ß, which decreases amyloid load in mouse models, thus appears to shield terminally differentiated cells encoding TGF-B2 targets against Aß toxicity (Grammas and Ovase, 2002). TGF-2 processes are increased by RA and decreased by their insufficiency (Wyss-Coray et al., 2001). In retinoid-deficit rats, RA restores TGF-ß2 in a tissue-specific manner. As a result, RA may enhance APP synthesis in healthy neurons through the TGF-ß signaling, including SMAD4, which is highly labeled in the AD brain. Transthyretin (TTR) may eliminate or inhibit the production of Aß, according to studies, and greater degrees of this seems to reduce amyloid-induced toxicity (Freemantle et al., 2002). TTR contents were found to be decreased in AD (White and Kelly, 2001), but no TTR alterations were found in an AD sample. Enhanced TTR does have other effects, including stabilizing RARG2 and decreasing the receptor’s responsiveness to RA. The MAPK pathway is inhibited to accomplish this goal. All of the foregoing empirical evidence reinforces the notion that retinoid dysfunction is a critical component tin the emergence of Aß toxicity. This is especially important given the age-related reduction in retinoid availability seen in both healthy and AD humans (Serot et al., 1997). It will be important to analyze these individuals in the coding, 5′, 3′, and promoters to see whether these genetic changes in the retinoid receptors at regions are associated to increases AD susceptibility (Figure 2).
Multiple neurotransmitter channels, particularly the catecholaminergic and cholinergic pathways, are disrupted in AD (Mecocci et al., 2002). Cholinergic neurons in the frontal cortex which extends to the neocortex, amygdala, and hippocampus degenerate, which is a characteristic of AD. The depletion of cholinergic neurons causes cognitive issues in experimental animals (Veng et al., 2003; Trillo et al., 2013), and impaired cholinergic signaling is associated with the initial stages of dementia. Treatment of cholinesterase inhibitors (drugs that stop the Ach metabolism and extend its activity in the cortex) was shown to enhance cognition in AD models, and these medications are now being used to address the symptoms of cognitive impairment in patients of neurological diseases (Hunter et al., 2004). Retinoids, which have neurotrophic actions on cholinergic neuronal cells and are markedly reduced in AD patients (Santucci et al., 1989), may serve as a complementary therapy for AD characteristics. RAR stimulation can increase the production of choline acetyltransferase (ChAT) and the VAChT protein, both of these aid in the transportation of Ach into presynaptic vesicles for their release (Goodman, 2006b). Retinoids have been shown to boost Ach and ChAT mRNA levels (Figure 3) (Berse and Blusztajn, 1995a). Dysfunction of monoaminergic pathways, such as the dopaminergic and noradrenergic systems, has also been linked to AD. As a result, the locus coeruleus, a brain’s primary noradrenergic center with considerable distribution in the cortical regions, shows severe deterioration in AD. In AD, tyrosine hydroxylase (a flow-restricting protein for noradrenaline and dopamine production) and dopamine-beta-hydroxylase (DBH, the enzyme essential for noradrenaline generation) levels are also diminished (Ayaz et al., 2021). Furthermore, AD is linked to a lower extent of norepinephrine in the cortex (Mann et al., 1980), and dopamine in the cortex, striatum, and amygdala (Iversen et al., 1983). The trophic effects of RA on the monoaminergic system may help in the mitigation of AD signs because they govern the activity of tyrosine and dopamine hydroxylase (Reinikainen et al., 1988) and could also explicitly regulate the articulation of dopamine D2 receptors by interacting with the RARE promoter site (Pinessi et al., 1987). Some researchers used a delayed recall training test to illustrate the potential usefulness of RA in improving learning and memory. As previously described in regards to RA and AD, this highly utilized scenario for memory is believed to necessitate stimulation of receptors in the 3 neuromodulatory systems: 1) muscarinic cholinergic receptors, 2) dopamine receptors, 3) and adrenergic receptors. The cholinergic receptor antagonist scopolamine was shown to impair memory as predicted, while RAR and RXR agonists were discovered to restore cognitive performance. While the specific process for this impact has yet to be discovered, Shudo and others hypothesized that a retinoid-mediated increase of D2 receptor activation might be one possibility (Kim et al., 2001). Retinoids and forskolin, an adenylate cyclase agonist, had already been demonstrated to enhance the quantity of ChAT mRNA in murine cells. Others have previously found that drugs that elevate cytoplasmic cAMP amount influence ChAT activity transcriptionally in a range of experimental conditions (Samad et al., 1997a; Ledesma and Dotti, 2012; Chakrabarti et al., 2016b). By analyzing the identical blots in sequence with ChAT and VAChT cDNA after a 48-h exposure of the cells with optimum amounts of RA, studies were able to effectively examine the effects of retinoids on ChAT and VAChT mRNA (Walsh and Selkoe, 2007).
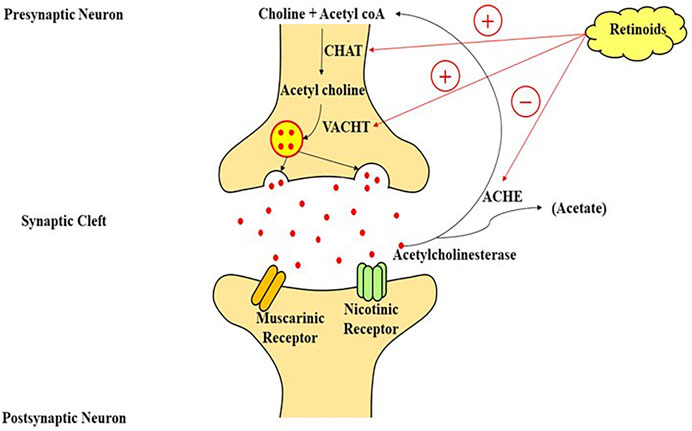
FIGURE 3. A model of retinoic acid signaling in the central nervous system, which plays an important role in the stimulation of transcriptional machinery to modulate the cholinergic neurotransmission through the production of choline-acetyl transferase mRNA and acetylcholine on the interaction of retinoic acid with its nuclear receptors. CHAT, choline acetyltransferase; VACHT, vesicular acetylcholine transporter; acetyl coA, acetyl coenzyme A; ACHE, acetylcholinesterase enzyme.
Alteration in cholinergic system, which result in a reduction in Ach, are one of the characteristics of AD. The abnormal scopolamine-induced release of Ach due to blocked presynaptic autoreceptor in vitamin A deficient rats, results in cognition loss. Lower synthesis of ChAT, as well as brain cell death induced by RA and the gene promoter, are likely to be the causes of ACh release impairment (Gessel et al., 2012; Saleem et al., 2021). Increased ChAT appears to be favourable, and it has been proposed that RA regulates the vascular ACh transporter as well (Pedersen et al., 1995). In a passive avoidance experiment, retinoid restored scopolamine-derived cognitive loss (Berse and Blusztajn, 1995b). Retinoids also have a role in the expression of tyrosine hydroxylase, dopamine hydroxylase, and the dopamine D2 receptor. Dopamine receptors activity is regulated primarily by RA via interaction with a RA response element (RARE) at the promoter (Shudo et al., 2004).
Hence, retinoid signalling appears to have a function in regulating both acetylcholine production (through ChAT) and cholinergic availability at synapses (via AChE) which might serve a function in the pathogenesis and treatment of Alzheimer’s disease. Yet, further research is required (Samad et al., 1997b) (Figure 3).
Retinol/Vitamin A is a fat-soluble vitamin whose major metabolite, retinoic acid, performs several biological functions comparable to hormones. The hippocampus and adjacent regions produce and metabolise RA, as well as express retinol-binding protein (Lane and Bailey, 2005). The majority of studies on the involvement of retinoids in memory and learning have used Vitamin A deficient (VAD) rats, RA receptor (RAR) transgenic mice, and ageing mouse models, etc. Etchamendy showed that VAD diet in mice model had dramatically decreased spatial memory and learning after 31 weeks, but that after a few days of RA therapy, the behaviour reverted to normal (Misner et al, 2001). VAD-mediated learning and spatial cognition deficits in rats may be facilitated by RA and RARs, according to some investigations (Etchamendy et al., 2003b). RA activates the RAR, a nuclear receptor that controls genes regulating the neuronal proliferation and differentiation, neurite outgrowth, synaptic plasticity, and other activities (Hernández-Pinto et al., 2006; McCaffery et al., 2006). The downstream pathways in RA activation, however, are unknown. Long-term potentiation (LTP) is the most common form of neuroplasticity, which is thought to be the cornerstone of memory and learning. RA levels and RARs, as well as neurogranin (RC3) and neuromodulin (GAP43), which together play a key role in modulating the uptake of Ca2+ and Ca/CaMK in neuroplasticity, have all been investigated in analyses of LTP and long-term depression (LTD) and the influence of VAD (Mark et al., 2006). Various in-vitro investigations have revealed that NMDA-NR1 may be a promising target for RA in regards to glutamate receptors. In neural development, RA might stimulate NMDA-NR1 subunit expression in human pluripotent adipose tissue stromal cells (Krucker et al., 2002). During growth, the gene expression of NMDA-NR1 can alter dynamically, such that certain quantity of NR1 subunits can be observed throughout all regions of the newborn rats brain, but 3 weeks after birth, this expression appears to reach its peak, before progressively declining to adult human levels (Kulikov et al., 2007). Such arrangement was observed to be comparable with RAR expression in neural tissue in some previous studies (Luo et al., 1996). The link between Ca2+, NMDA receptors, and RA signalling in vivo, on the other hand, is yet unknown. LTP and LTD, which are the most fundamental processes of cognitive performance, are initiated by Ca2+ influx (Jiang et al., 2011). The LTP and LTD were observed to be impaired in RAR or RAR-RXR knockout mice (Bliss et al., 2006). (Figure 4) Furthermore, in cultured hippocampus, acute RA treatment enhances small excitatory post-synaptic activity magnitude (Chiang et al., 1998). The NMDA receptor, as well as other glutamate and non-glutamate receptors (such as TrkB), can control Ca2+ influx in neurons (Aoto et al., 2008). Many types of LTP and LTD have been linked to NMDA receptors (Lynch, 2004). The major mechanism for Ca2+ influx into neurons through the post-synaptic membrane is thought to be interaction with NMDA receptors (Figure 4) (Isaac et al., 2009). Complex cognitive activities, including memory generation, necessitate RAR activation via all-trans-RA (ATRA) to promote neuroplasticity, according to some studies (Fukumori et al., 2010).
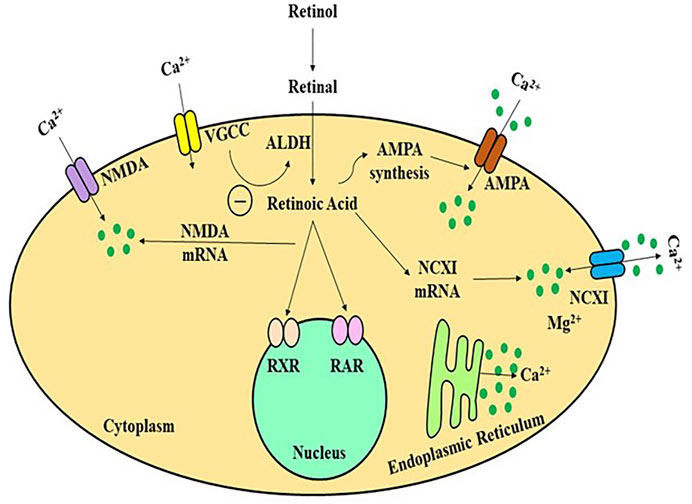
FIGURE 4. A schematic framework of activity-dependent retinoid generation in neurons. Appropriate synaptic transmission inhibits RA production in a Ca2+-dependent manner by activating glutamate receptors and L-type Ca2+channels. Limiting glutamate receptors or L-type Ca2+ channels reduces dendritic L-type Ca2+ intake and de-represses retinoid production, allowing AMPARs to be translated and inserted synaptically neural cells to increase the calcium influx. ALDH, alcohol dehydrogenase; AMPA, amino-hydroxy-methyl-isoxazolepropionic acid receptor; NMDA, N-methyl-D-aspartate receptor; VGCC, volatge gated calcium channel; RAR/RXR, retinoic acid receptors.
Calcineurin is a calcium-binding enzyme that is important for the link between calcium and RA in neuronal cells. Calcineurin suppression promotes the generation of RA. These alterations occur by stimulating the RA generation and RAR stimulation (Etchamendy et al., 2001). RAR is a protein that stimulates the production of calcium-permeable channels in this network. It is nongenomic because it does not involve its traditional job of controlling transcriptional activity, but rather the fast activity of RAR in the cytosol to control protein expression. RAR is continually carried to the nerve cells, where this functions as an mRNA-binding protein, preventing the production of mRNA like glutamate receptor 1 (GluR1), a critical component of the AMPA receptor calcium receptor. When RA interacts with RAR, it causes the receptor to alter the shape and the detachment of GluR1 mRNA, allowing it to be translated (Arendt et al., 2015). The postsynaptic surface is injected with newly translated GluR1 subunit-containing AMPA channels, permitting calcium uptake into the neuron, thus reinforcing the excitatory contact and turning off RA generation, disrupting the neuroplasticity. Thus the loss of neuroplasticity can be cured by shrinking inhibitory synapses as well as the stimulation of excitatory nerve terminals.
The results of this investigation revealed that RA-mediated morphological and physiological neuroplasticity in adult human cortical lines requires mRNA transcription, as evidenced by recent studies on the importance of protein synthesis in neuroplasticity (Poon and Chen, 2008). Synaptopodin was found as a target and regulator of RA-mediated neuroplasticity at the molecular level. Synaptopodin is an actin-regulating protein related to the spinal machinery (Biever et al., 1979), a membrane expansion of the endoplasmic reticulum observed in a subset of the telencephalic dendritic spine. The findings of such studies showed that 1) synaptopodin complexes are noticed in roughly 70% of nerve cells in the cortical region 2) synaptopodin is an indicator of the human spine system; 3) synaptopodin complexes and spine systems are observed in large nerve cells; and 4) a plasticity-mediating signal, such as RA treatment, stimulates the transformation of synaptopodin complexes, spine system. The significance of synaptopodin and the spine system in neuroplasticity is uncertain; although, it has been suggested that they play a role in the local synthesis of protein and regulation of cytosolic calcium dynamics (Deller et al., 2003). Synaptopodin interacts with actin and -actinin and has been proposed to control spinal mobility and long-term spinal integrity via Rho-A signaling (Lenz et al., 2021). Synaptopodin and myosin V was discovered recently to be associated with this mechanism (Yap et al., 2020). Synaptopodin might thus be involved in the structural and functional alterations seen at synaptic vesicles undergoing NMDA receptor-induced neuroplasticity (Jedlicka and Deller, 2017). Both retinoids and synaptopodin have already been related to the homeostatic neuroplasticity in mouse neural tissue, as well as the production and transport of Ca2+-dependent AMPA receptors. In diseased states of the brain, changes in retinoid signaling and synaptopodin activity have been linked to neuroplasticity abnormalities (Soden and Chen, 2010). As a result, in the neural tissue of patients with AD and cognitive impairment, changes in synaptopodin activity and retinoid signaling have now been found. Considering that retinoids have been suggested as a promising medicinal route for AD-linked cognitive impairment (Endres et al., 2014a). RA may work by altering the expression of synaptopodin and hence enhancing the capability of adult neural cells to enhance neuroplasticity. Eventually, VAD can harm cognitive function throughout postnatal life. One mechanism behind this phenomenon might be VAD-mediated downregulation of neural RAR, which results in lower production of NMDA-NR1 through no direct transcription, impairing neuronal Ca2+ excitability and decreasing LTP (Figure 4; Table 1).
Retinoids, vitamin A including carotenoids and beta-carotene, are lipophilic molecules produced by plants and animals which support various processes for cell growth, development, and differentiation (Duester, 2008; Shannon et al., 2017). Vitamin A is ingested in the form of pro-vitamin A carotenoids from plants and preformed vitamin A from animal-derived food which are modified to all-trans-retinol through a reactive pathway in the intestine. Through diet, carotenoids directly acts as ROS scavengers via energy transfer (Muller and Bohm, 2011). The deficiency of vitamin A and its metabolites were studied for their coalition with cognitive impairment in adult animals, thus highlighting the importance of sufficient vitamin A levels. Carotenoids are categorized as- 1) Non-pro-vitamin A carotenoids like lutein and 2) pro-vitamin A carotenoids including β-carotene and retinal, and non-pro-vitamin A carotenoids such as lycopene and lutein (Mohammadzadeh Honarvar et al., 2017d). Retinoids perform by various mechanisms and reach out the free radicals in mitochondria, plasma and cell membranes through electron transfer, physical scavenging, and hydrogen abstraction (Woodall et al., 1997). Also, they can react indirectly with several signaling pathways, such as the nuclear factor elytroid-2-related factor 2 (Nrf2), mitogen-activated protein kinase (MAPK), and NF-κB (Palozza et al., 2003; Ben-Dor et al., 2005). The antioxidative effects of retinoids includes quenching of singlet oxygen molecules and scavenging peroxyl radicals. Centrally located retinol-binding proteins (RBPs) are known to control the transport of retinol through the BBB into the brain (MacDonald et al., 1990). Higher levels of retinoids were implicated in the frontal lobe cortex of the postmortem human brain (Craft et al., 2004). ß-carotene, a precursor of retinoic acid, has also been observed in the singlet oxygen scavenging, free radical quenching, and lipid anti-oxidation in plasma. Thus, ß-carotene is considered to be localized in the lipid core of the membranes and implied as one of the favourable antioxidant to scavenge hydrophobic radicals in the membrane for clinical utilization (de Oliveira et al., 2012). Retinoids are structural derivatives of vitamin A which are reported to be involved in normal brain functioning including neuronal growth, development, and differentiation. Retinoids are known to regulate the glucocorticosteroids abundance in the brain which is an essential physiological process that can be observed in various stress-linked conditions to preserve the neuroplasticity within the hippocampus (Bonhomme et al., 2014). Under the oxidative stress conditions including metal injury and ROS accumulation, retinoids preserve the cells against this imbalance through several pathways, such as Nrf-2 and other defensive mechanisms like inhibition of ROS generation, free radical scavenging, induction of antioxidant enzymes. It has also been reported that RA has a neuroprotective role against apoptosis and oxidative stress via decreasing glutathione and restoring SOD-1 and SOD-2 in the hippocampus (Ahlemeyer et al., 2001). The role of retinoid signal transduction in the control of dopaminergic neurotransmission was noticed during the high levels of RA-synthesizing enzymes and RAR, which potentially has a significant action on regulation of cell survival, adaptation, and homeostatic regulation of the dopaminergic system (Lévesque and Rouillard, 2007). Retinoid expression plays a significant function in memory and cognitive performance, and synaptic plasticity as well (Crandall et al., 2004). RA supplementation upregulated μ-type opioid receptor 1 (MOR1) and its cascade and alleviated dyskinetic movements, which is a known result of long-term therapy of L-DOPA, in animal model (Pan et al., 2019). Additionally, RA induced the neuroprotective action on dopamine neurons in MPTP-treated mice model of AD. Administration of RA-loaded polymeric nanoparticle largely curbs the dopamine neuron loss in the substantia nigra and axonal innervations in the striatum (Esteves et al., 2015). Moreover, a research on synthetic retinoid revealed the reduction in neuroinflammation, Aß load, and oxidative stress with age-linked cognitive improved in AD patients (Bitarafan et al., 2016).
Growing data suggests that retinoid insufficiency may have a role in the pathology of AD, including enhanced Aß accumulation and memory loss (dos Santos Guilherme et al., 2019). Retinoid serum levels were found to be lower in AD patients in a meta-analysis (Das et al., 2019). RA transmission, on the other hand, have been shown to reduce the development of AD in rats (Bonnet et al., 2008; da Silva et al., 2014) and might be used as treatment strategy in patients with AD (Reinhardt et al., 2016). In developing countries, VAD is significantly more common among pregnant women and school—age children (Kitaoka et al., 2013). Vitamin A deficiency may contribute to AD development by disrupting APP processing and resulting in Aß buildup. In AD mouse models, it was discovered that VAD raised Aß levels. Retinoids were implicated in the transcriptional control of the APP and BACE1 genes via NF-B and RARs in earlier studies (West, 2002; Endres et al., 2014d). In the mouse model of AD, retinoid deficiency promoted BACE1-induced APP cleavage and Aß production, facilitating senile plaque accumulation as well as cognitive impairments. Additionally, the VAD-induced Aß rise may impede RA production, aggravating AD pathogenesis (Koryakina et al., 2009). A production and clearance are both important factors in controlling Aß levels in the brain. Microglia, a phagocyte and a innate immune cell in the brain, play critical role in removal of Aβ peptides (Husson et al., 2006b). It was found that inhibiting RAR reduced Aß clearance by microglia in some investigation, implying that RAR-dependent Aß breakdown is assisted by microglia (Goncalves et al., 2013c).
In vitro data imply that all-trans-retinoic acid may also affect tau protein production, particularly the amount of phosphorylated forms of tau (Lee and Landreth, 2010). The utilization of retinoids with the goal of rectifying or decreasing neurodegenerative effects may also include the regulation of neuroinflammation, another pathological mechanism that causes neuronal and synaptic loss in diseases like AD. In vitro, amyloid-ß increases the production and release of the inflammatory cytokines such as tumour necrosis factor- and inducible NOS in microglial cells of AD model. Studies on the clinical benefits of omega-3 fatty acids may actively support the neuroprotective effects of retinoids in dementia. A nutritional investigation of patients with various kinds of dementia revealed that a diet high in antioxidants and polyunsaturated fats, especially vitamin A, may have a preventive role (Zeng et al., 2017). Retinoid signalling has already been proposed as a possible target for developing new Alzheimer’s therapy (Charpentier et al., 1995). Because there is contradictory data in the field, it is difficult to say if AD patients have a dysfunction of RA expression. Rinaldi et al. (2003), Larrieu and Layé (2018) found that serum levels of retinols are lower in AD patients, while Connor and Sidell (Goodman and Pardee, 2003b) found that hippocampus retinoid content is equivalent in AD and control groups. Corcoran et al. (2004), Rinaldi et al. (2003) found lower levels of RAR and RALDH2 in AD brains, indicating that RA signalling is likely to be impaired in patients. Studies in aged mice indicate that RAR levels are reduced which may eventually causes impaired RA-mediated actions. Likewise, biological stimulation of RA signalling can restore the memory and learning as reported in aged animal models (Connor and Sidell, 1997b). However, the pathogenesis of Alzheimer’s disease is complicated, involving many biological activities, and retinoids can influence these systems by influencing expression of genes and serving as an antioxidant. Particular data like “the retinoid activity in AD” should be interpreted with caution. To investigate retinoid treatment for Alzheimer’s disease, researchers must first determine the specific involvement of retinoid and its receptors in the many mechanisms that regulate plaque generation, neurofibrillary tangles, cholinergic signaling, and ApoE activity in the adult brain in vivo (Corcoran et al., 2004). It has been proposed that retinoid signalling is a strong, plausible avenue for future Alzheimer’s treatments. It is uncertain if Alzheimer’s patients have retinoid signalling given that RA has been utilised in the treatment of AD and there are contradicting results. For example, GL Fata and others observed the serum level of vitamin A in Alzheimer’s decreased While Connor and Seidel suggested that hippocampal levels of retinoids in Alzheimer’s is similar to that of controls, A decline in RARα concentration and retinal dehydrogenase 2 in Alzheimer’s brain, indicating that retinoid signaling may be impaired in patients, In older mice showed that the frequency of RAR decreased, which may eventually lead to a reduction in retinoic acid-dependent effects. In fact, perceptive deficiencies seen in older mice can be inverted by activating retinoic acid. However, the pathogenesis of Alzheimer’s disease includes numerous and intricate signalling pathways, and retinoids may alter these processes by controlling gene expression in addition to having antioxidant characteristic consideration of vitamin A and its receptors’ effects on numerous pathways, including plaque formation, neuroinflammation, and neurotransmission, is important in order to treat AD with retinoids (Eftekhari et al., 2021; Ayaz et al., 2022).
Retinoid signaling is clearly involved in neurodegenerative diseases like AD, as well fdas memory and learning. The relevance of sufficient Vitamin A status for such cognitive activities has been highlighted in adult mice and rats, but the exact targeting of retinoid signalling pathways behind these pathology have yet to be identified. Remarkably little is focused on the impact of ATRA signalling on other brain functions, given the enormous number of neuronal functions that could potentially be controlled by retinoids in the adult brain. Transgenic mouse models have revealed a role for retinoids in cholinnergic signaling, gene transcription, neuroplasticity, amyloid plaque formation. Nevertheless, such data may not clearly distinguish between the well-described protective effects of retinoids and neurodegeneration. Future study in this new subject is needed to enhance our knowledge of the role of retinoids in the brain. Retinoids, retinoid ligands, and inhibitors have a lot of promise as treatments for Alzheimer’s, and probably other neurodegenerative disorders. However, we need to learn more about the neuronal genes and molecular pathways that are precisely regulated by retinoids, as well as the physiological consequences in the brain, before such interventions may be explored.
Conceptualization, TB and DK; methodology, TB and AS; investigation, TB, DK, and RKS; resources, TB and SB.; data curation, TB, HM, and MA; writing-original draft preparation, TB and DK; writing-review and editing, HA and AM; visualization, AS and TB; supervision, TB and SB.
The fundings for publication of this paper are provided by University of Oradea, Oradea, Romania, by an Internal project.
RKS has an honorary-based association with the iGlobal Research and Publishing Foundation (iGRPF), New Delhi, India. All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahlemeyer, B., Bauerbach, E., Plath, M., Steuber, M., Heers, C., Tegtmeier, F., et al. (2001). Retinoic acid reduces apoptosis and oxidative stress by preservation of SOD protein level. Free Radic. Biol. Med. 30 (10), 1067–1077. doi:10.1016/s0891-5849(01)00495-6
Alpsoy, L., Yildirim, A., and Agar, G. (2009). The antioxidant effects of vitamin A, C, and E on aflatoxin B1-induced oxidative stress in human lymphocytes. Toxicol. Ind. Health 25 (2), 121–127. doi:10.1177/0748233709103413
Andreeva, T. v., Lukiw, W. J., and Rogaev, E. I. (2017). Biological basis for amyloidogenesis in alzheimer’S disease. Moscow: Biochemistry, 82.
Aoto, J., Nam, C. I., Poon, M. M., Ting, P., and Chen, L. (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60 (2), 308–320. doi:10.1016/j.neuron.2008.08.012
Arendt, T., Brückner, M. K., Morawski, M., Jäger, C., and Gertz, H. J. (2015). Early neurone loss in Alzheimer’s disease: Cortical or subcortical? Acta Neuropathol. Commun. 3, 10. doi:10.1186/s40478-015-0187-1
Ayaz, M., Ali, T., Sadiq, A., Ullah, F., and Naseer, M. I. (2022). Current trends in medicinal plant research and neurodegenerative disorders. Front. Pharmacol., 2522.
Ayaz, M., Sadiq, A., Junaid, M., Ullah, F., Ovais, M., Ullah, I., et al. (2019). Flavonoids as prospective neuroprotectants and their therapeutic propensity in aging associated neurological disorders. Front. Aging Neurosci. 11, 155. doi:10.3389/fnagi.2019.00155
Ayaz, M., Ullah, F., Sadiq, A., Kim, M. O., and Ali, T. (2019). Editorial: Natural products-based drugs: Potential therapeutics against Alzheimer's disease and other neurological disorders. Front. Pharmacol. 10, 1417. doi:10.3389/fphar.2019.01417
Ayaz, M., Wadood, A., Sadiq, A., Ullah, F., Anichkina, O., and Ghufran, M. (2021). In-silico evaluations of the isolated phytosterols from polygonum hydropiper L against BACE1 and MAO drug targets. J. Biomol. Struct. Dyn. 8, 1–9. doi:10.1080/07391102.2021.1940286
Behairi, N., Belkhelfa, M., Rafa, H., Labsi, M., Deghbar, N., Bouzid, N., et al. (2016). All-trans retinoic acid (ATRA) prevents lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment in aged rats. J. Neuroimmunol. 300, 21–29. doi:10.1016/j.jneuroim.2016.10.004
Behl, T., Kaur, I., Sehgal, A., Kumar, A., Uddin, M. S., and Bungau, S. (2020). The interplay of ABC transporters in Aβ translocation and cholesterol metabolism: implicating their roles in Alzheimer’s disease. Molec. Neurobiol.. doi:10.1007/s12035-020-02211-x
Ben-Dor, A., Steiner, M., Gheber, L., Danilenko, M., Dubi, N., Linnewiel, K., et al. (2005). Carotenoids activate the antioxidant response element transcription system. Mol. Cancer Ther. 4 (1), 177–186. doi:10.1158/1535-7163.177.4.1
Berse, B., and Blusztajn, J. K. (1995). Coordinated up-regulation of choline acetyltransferase and vesicular acetylcholine transporter gene expression by the retinoic acid receptor α, cAMP, and leukemia inhibitory factor/ciliary neurotrophic factor signaling pathways in a murine septal cell line (∗). J. Biol. Chem. 270 (38), 22101–22104. doi:10.1074/jbc.270.38.22101
Berse, B., and Blusztajn, J. K. (1995). Coordinated up-regulation of choline acetyltransferase and vesicular acetylcholine transporter gene expression by the retinoic acid receptor α, CAMP, and leukemia inhibitory factor/ciliary neurotrophic factor signaling pathways in a murine septal cell line. J. Biol. Chem. 270, 22101–22104. doi:10.1074/jbc.270.38.22101
Biever, A., Glock, C., Tushev, G., Ciirdaeva, E., Dalmay, T., Langer, J. D., et al. (1979). Monosomes actively translate synaptic MRNAs in neuronal processes. Science 2020, eaay4991. doi:10.1126/science.aay4991
Bitarafan, S., Saboor-Yaraghi, A., Sahraian, M. A., Soltani, D., Nafissi, S., Togha, M., et al. (2016). Effect of vitamin A supplementation on fatigue and depression in multiple sclerosis patients: A double-blind placebo-controlled clinical trial. Iran. J. Allergy Asthma Immunol. 31, 13–19.
Bliss, T. V., Collingridge, G. L., and Laroche, S. (2006). Neuroscience. ZAP and ZIP, a story to forget. Science 313 (5790), 1058–1059. doi:10.1126/science.1132538
Bonhomme, D., Minni, A. M., Alfos, S., Roux, P., Richard, E., Higueret, P., et al. (2014). Vitamin A status regulates glucocorticoid availability in wistar rats: Consequences on cognitive functions and hippocampal neurogenesis? Front. Behav. Neurosci. 8, 20. doi:10.3389/fnbeh.2014.00020
Bonnet, E., Touyarot, K., Alfos, S., Pallet, V., Higueret, P., and Abrous, D. N. (2008). Retinoic acid restores adult hippocampal neurogenesis and reverses spatial memory deficit in vitamin A deprived rats. PloS one 3 (10), e3487. doi:10.1371/journal.pone.0003487
Bonney, S., Dennison, B. J., Wendlandt, M., and Siegenthaler, J. A. (2018). Retinoic acid regulates endothelial β-catenin expression and pericyte numbers in the developing brain vasculature. Front. Cell. Neurosci. 12, 476. doi:10.3389/fncel.2018.00476
Bremner, J. D., and McCaffery, P. (2008). The neurobiology of retinoic acid in affective disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 315–331. doi:10.1016/J.PNPBP.2007.07.001
Burton, T., Liang, B., Dibrov, A., and Amara, F. (2002). Transcriptional activation and increase in expression of Alzheimer’s beta-amyloid precursor protein gene is mediated by TGF-beta in normal human astrocytes. Biochem. Biophys. Res. Commun. 295, 702–712. doi:10.1016/S0006-291X(02)00724-6
Chakrabarti, M., McDonald, A. J., Will Reed, J., Moss, M. A., Das, B. C., and Ray, S. K. (2016). Molecular signaling mechanisms of natural and synthetic retinoids for inhibition of pathogenesis in Alzheimer’s disease. J. Alzheimers Dis. 50 (2), 335–352. doi:10.3233/JAD-150450
Chakrabarti, M., Mcdonald, A. J., Will Reed, J., Moss, M. A., Das, B. C., and Ray, S. K. (2016). Molecular signaling mechanisms of natural and synthetic retinoids for inhibition of pathogenesis in Alzheimer’s disease. J. Alzheimers Dis. 50, 335–352. doi:10.3233/JAD-150450
Charpentier, B., Bernardon, J. M., Eustache, J., Millois, C., Martin, B., Michel, S., et al. (1995). Synthesis, structure-affinity relationships, and biological activities of ligands binding to retinoic acid receptor subtypes. J. Med. Chem. 38 (26), 4993–5006. doi:10.1021/jm00026a006
Chen, N., and Napoli, J. L. (2008). All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. FASEB J. 22, 236–245. doi:10.1096/fj.07-8739com
Chiang, M. Y., Misner, D., Kempermann, G., Schikorski, T., Giguère, V., Sucov, H. M., et al. (1998). An essential role for retinoid receptors RARbeta and RXRgamma in long-term potentiation and depression. Neuron 21 (6), 1353–1361. doi:10.1016/s0896-6273(00)80654-6
Chisholm, D. R., Tomlinson, C. W., Zhou, G. L., Holden, C., Affleck, V., Lamb, R., et al. (2019). Fluorescent retinoic acid analogues as probes for biochemical and intracellular characterization of retinoid signaling pathways. ACS Chem. Biol. 14 (3), 369–377. doi:10.1021/acschembio.8b00916
Connor, M. J., and Sidell, N. (1997). Retinoic acid synthesis in normal and Alzheimer diseased brain and human neural cells. Mol. Chem. Neuropathol. 30 (3), 239–252. doi:10.1007/BF02815101
Connor, M. J., and Sidell, N. (1997). Retinoic acid synthesis in normal and Alzheimer diseased brain and human neural cells. Mol. Chem. Neuropathol. 30 (3), 239–252. doi:10.1007/BF02815101
Corcoran, J. P., So, P. L., and Maden, M. (2004). Disruption of the retinoid signalling pathway causes a deposition of amyloid β in the adult rat brain. Eur. J. Neurosci. 20 (4), 896–902. doi:10.1111/j.1460-9568.2004.03563.x
Craft, N. E., Haitema, T. B., Garnett, K. M., Fitch, K. A., and Dorey, C. K. (2004). Carotenoid, tocopherol, and retinol concentrations in elderly human brain. Exp. Anim. 21, 22.
Crandall, J., Sakai, Y., Zhang, J., Koul, O., Mineur, Y., Crusio, W. E., et al. (2004). 13-cis-retinoic acid suppresses hippocampal cell division and hippocampal-dependent learning in mice. Proc. Natl. Acad. Sci. U. S. A. 101 (14), 5111–5116. doi:10.1073/pnas.0306336101
Cummings, J. L., Zhong, K., Kinney, J. W., Heaney, C., Moll-Tudla, J., Joshi, A., et al. (2016). Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer’s disease. Alz. Res. Ther. 8 (1), 4–9. doi:10.1186/s13195-016-0173-2
Cunningham, T. J., and Duester, G. (2015). Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 16 (2), 110–123. doi:10.1038/nrm3932
da Silva, S. L., Vellas, B., Elemans, S., Luchsinger, J., Kamphuis, P., Yaffe, K., et al. (2014). Plasma nutrient status of patients with Alzheimer's disease: Systematic review and meta-analysis. Alzheimers Dement. 10 (4), 485–502. doi:10.1016/j.jalz.2013.05.1771
Dantzer, R., O’Connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. doi:10.1038/nrn2297
Das, B. C., Dasgupta, S., and Ray, S. K. (2019). Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer’s disease. Neural Regen. Res. 14 (11), 1880–1892. doi:10.4103/1673-5374.259604
Das, B. C., Thapa, P., Karki, R., Das, S., Mahapatra, S., Liu, T. C., et al. (2014). Retinoic acid signaling pathways in development and diseases. Bioorg. Med. Chem. 22 (2), 673–683. doi:10.1016/j.bmc.2013.11.025
Das, B. C., Thapa, P., Karki, R., Das, S., Mahapatra, S., Liu, T. C., et al. (2014). Retinoic acid signaling pathways in development and diseases. Bioorg. Med. Chem. 22, 673–683. doi:10.1016/j.bmc.2013.11.025
de Oliveira, B. F., Veloso, C. A., Nogueira-Machado, J. A., de Moraes, E. N., dos Santos, R. R., Cintra, M. T., et al. (2012). Ascorbic acid, alpha-tocopherol, and beta-carotene reduce oxidative stress and proinflammatory cytokines in mononuclear cells of Alzheimer's disease patients. Nutr. Neurosci. 15 (6), 244–251. doi:10.1179/1476830512Y.0000000019
Defo, M. A., Pierron, F., Spear, P. A., Bernatchez, L., Campbell, P. G., and Couture, P. (2012). Evidence for metabolic imbalance of vitamin A2 in wild fish chronically exposed to metals. Ecotoxicol. Environ. Saf. 85, 88–95. doi:10.1016/j.ecoenv.2012.08.017
Deller, T., Korte, M., Chabanis, S., Drakew, A., Schwegler, H., Stefani, G. G., et al. (2003). Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc. Natl. Acad. Sci. U. S. A. 100, 10494–10499. doi:10.1073/pnas.1832384100
dos Santos Guilherme, M., Stoye, N. M., Rose-John, S., Garbers, C., Fellgiebel, A., and Endres, K. (2019). The synthetic retinoid acitretin increases IL-6 in the central nervous system of Alzheimer disease model mice and human patients. Front. Aging Neurosci. 11, 182. doi:10.3389/fnagi.2019.00182
Duester, G. (2008). Retinoic acid synthesis and signaling during early organogenesis. Cell 134 (6), 921–931. doi:10.1016/j.cell.2008.09.002
Duester, G., Shean, M. L., McBride, M. S., and Stewart, M. J. (1991). Retinoic acid response element in the human alcohol dehydrogenase gene ADH3: Implications for regulation of retinoic acid synthesis. Mol. Cell. Biol. 11, 1638–1646. doi:10.1128/mcb.11.3.1638
Duester, G. (2022). Synaptic plasticity is altered by treatment with pharmacological levels of retinoic acid acting nongenomically however endogenous retinoic acid has not been shown to have nongenomic activity. J. Neurol. Disord., 10.
Eftekhari, A., Shanehb, D., Hoseinnejhad, S., Ceferov, Z., and Asefy, Z. (2021). Retinoids as potential therapeutic approach in prevention of Alzheimer's disease. Ann. Clin. Laboratory Res. 9 (9), 0.
Endres, K., Fahrenholz, F., Lotz, J., Hiemke, C., Teipel, S., Lieb, K., et al. (2014). Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 83 (21), 1930–1935. doi:10.1212/WNL.0000000000001017
Endres, K., Fahrenholz, F., Lotz, J., Hiemke, C., Teipel, S., Lieb, K., et al. (2014). Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 83 (21), 1930–1935. doi:10.1212/WNL.0000000000001017
Endres, K., Fahrenholz, F., Lotz, J., Hiemke, C., Teipel, S., Lieb, K., et al. (2014). Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 83 (21), 1930–1935. doi:10.1212/WNL.0000000000001017
Endres, K., Fahrenholz, F., Lotz, J., Hiemke, C., Teipel, S., Lieb, K., et al. (2014). Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 83, 1930–1935. doi:10.1212/WNL.0000000000001017
Eriksen, J. L., and Janus, C. G. (2007). Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav. Genet. 37, 79–100. doi:10.1007/s10519-006-9118-z
Esteves, M., Cristóvão, A. C., Saraiva, T., Rocha, S. M., Baltazar, G., Ferreira, L., et al. (2015). Retinoic acid-loaded polymeric nanoparticles induce neuroprotection in a mouse model for Parkinson's disease. Front. Aging Neurosci. 7, 20. doi:10.3389/fnagi.2015.00020
Etchamendy, N., Enderlin, V., Marighetto, A., Pallet, V., Higueret, P., and Jaffard, R. (2003). Vitamin A deficiency and relational memory deficit in adult mice: Relationships with changes in brain retinoid signalling. Behav. Brain Res. 145 (1-2), 37–49. doi:10.1016/s0166-4328(03)00099-8
Etchamendy, N., Enderlin, V., Marighetto, A., Pallet, V., Higueret, P., and Jaffard, R. (2003). Vitamin A deficiency and relational memory deficit in adult mice: Relationships with changes in brain retinoid signalling. Behav. Brain Res. 145 (1-2), 37–49. doi:10.1016/s0166-4328(03)00099-8
Etchamendy, N., Enderlin, V., Marighetto, A., Vouimba, R. M., Pallet, V., Jaffard, R., et al. (2001). Alleviation of a selective age-related relational memory deficit in mice by pharmacologically induced normalization of brain retinoid signaling. J. Neurosci. 21 (16), 6423–6429. doi:10.1523/jneurosci.21-16-06423.2001
Fahrenholz, F., Tippmann, F., and Endres, K. (2010). Retinoids as a perspective in treatment of Alzheimer’s disease. Neurodegener. Dis. 7 (1-3), 190–192. doi:10.1159/000295662
Filteau, S. M., Rollins, N. C., Coutsoudis, A., Sullivan, K. R., Willumsen, J. F., and Tomkins, A. M. (2001). The effect of antenatal vitamin A and β-carotene supplementation on gut integrity of infants of HIV-infected South African women. J. Pediatr. Gastroenterol. Nutr. 32 (4), 464–470. doi:10.1097/00005176-200104000-00014
Freemantle, S. J., Kerley, J. S., Olsen, S. L., Gross, R. H., and Spinella, M. J. (2002). Developmentally-related candidate retinoic acid target genes regulated early during neuronal differentiation of human embryonal carcinoma. Oncogene 21, 2880–2889. doi:10.1038/sj.onc.1205408
Fukasawa, H., Nakagomi, M., Yamagata, N., Katsuki, H., Kawahara, K., Kitaoka, K., et al. (2012). Tamibarotene: A candidate retinoid drug for Alzheimer’s disease. Biol. Pharm. Bull. 35 (8), 1206–1212. doi:10.1248/bpb.b12-00314
Fukasawa, H., Nakagomi, M., Yamagata, N., Katsuki, H., Kawahara, K., Kitaoka, K., et al. (2012). Tamibarotene: A candidate retinoid drug for Alzheimer’s disease. Biol. Pharm. Bull. 35 (8), 1206–1212. doi:10.1248/bpb.b12-00314
Fukumori, R., Nakamichi, N., Takarada, T., Kambe, Y., Matsushima, N., Moriguchi, N., et al. (2010). Inhibition by 2-Methoxy-4-ethylphenol of Ca2+ influx through acquired and native N-Methyl-D-aspartate–Receptor channels. J. Pharmacol. Sci. 112, 273–281. doi:10.1254/jphs.09294fp
Gencoglan, G., Inanir, I., Miskioglu, M., and Gunduz, K. (2018). Evaluation of sequential effect of isotretinoin on the haematological parameters in patients with acne vulgaris. Cutan. Ocul. Toxicol. 37 (2), 139–142. doi:10.1080/15569527.2017.1359837
Gessel, M. M., Bernstein, S., Kemper, M., Teplow, D. B., and Bowers, M. T. (2012). Familial alzheimers disease mutations differentially alter amyloid β-protein oligomerization. ACS Chem. Neurosci. 3, 909–918. doi:10.1021/CN300050D/SUPPL_FILE/CN300050D_SI_001
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526. doi:10.1016/0896-6273(89)90210-9
Goncalves, M. B., Clarke, E., Hobbs, C., Malmqvist, T., Deacon, R., Jack, J., et al. (2013). Amyloid β inhibits retinoic acid synthesis exacerbating A lzheimer disease pathology which can be attenuated by an retinoic acid receptor α agonist. Eur. J. Neurosci. 37 (7), 1182–1192. doi:10.1111/ejn.12142
Goncalves, M. B., Clarke, E., Hobbs, C., Malmqvist, T., Deacon, R., Jack, J., et al. (2013). Amyloid β inhibits retinoic acid synthesis exacerbating A lzheimer disease pathology which can be attenuated by an retinoic acid receptor α agonist. Eur. J. Neurosci. 37 (7), 1182–1192. doi:10.1111/ejn.12142
Goncalves, M. B., Clarke, E., Hobbs, C., Malmqvist, T., Deacon, R., Jack, J., et al. (2013). Amyloid β inhibits retinoic acid synthesis exacerbating alzheimer disease pathology which can Be attenuated by an retinoic acid receptor α agonist. Eur. J. Neurosci. 37, 1182–1192. doi:10.1111/EJN.12142
Goodman, A. B. (2006). Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset alzheimer disease. J. Cell. Physiol. 209, 598–603. doi:10.1002/jcp.20784
Goodman, A. B., and Pardee, A. B. (2003). Evidence for defective retinoid transport and function in late onset Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 100 (5), 2901–2905. doi:10.1073/pnas.0437937100
Goodman, A. B. (2006). Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset Alzheimer disease. J. Cell. Physiol. 209 (3), 598–603. doi:10.1002/jcp.20784
Goodman, A. B., and Pardee, A. B. (2003). Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 100, 2901–2905. doi:10.1073/pnas.0437937100
Grammas, P., and Ovase, R. (2002). Cerebrovascular transforming growth factor-β contributes to inflammation in the Alzheimer’s disease brain. Am. J. Pathol. 160, 1583–1587. doi:10.1016/S0002-9440(10)61105-4
Green, A. S., and Fascetti, A. J. (2016). Meeting the vitamin A requirement: The efficacy and importance of β-carotene in animal species. ScientificWorldJournal., 7393620. doi:10.1155/2016/7393620
Haffez, H., Chisholm, D. R., Tatum, N. J., Valentine, R., Redfern, C., Pohl, E., et al. (2018). Probing biological activity through structural modelling of ligand-receptor interactions of 2, 4-disubstituted thiazole retinoids. Bioorg. Med. Chem. 26 (8), 1560–1572. doi:10.1016/j.bmc.2018.02.002
Hall, R. K., Scott, D. K., Noisin, E. L., Lucas, P. C., and Granner, D. K. (1992). Activation of the phosphoenolpyruvate carboxykinase gene retinoic acid response element is dependent on a retinoic acid receptor/coregulator complex. Mol. Cell. Biol. 12, 5527–5535. doi:10.1128/mcb.12.12.5527
Harrison, E. H. (2019). Mechanisms of transport and delivery of vitamin A and carotenoids to the retinal pigment epithelium. Mol. Nutr. Food Res. 63 (15), 1801046. doi:10.1002/mnfr.201801046
Hasselmo, M. E. (2006). The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol. 16, 710–715. doi:10.1016/j.conb.2006.09.002
Hellmann-Regen, J., Kronenberg, G., Uhlemann, R., Freyer, D., Endres, M., and Gertz, K. (2013). Accelerated degradation of retinoic acid by activated microglia. J. Neuroimmunol. 256, 1–6. doi:10.1016/J.JNEUROIM.2012.11.005
Hellmann-Regen, J., Uhlemann, R., Regen, F., Heuser, I., Otte, C., Endres, M., et al. (2015). Direct inhibition of retinoic acid catabolism by fluoxetine. J. Neural Transm. 122, 1329–1338. doi:10.1007/s00702-015-1407-3
Hernández-Pinto, A. M., Puebla-Jiménez, L., and Arilla-Ferreiro, E. (2006). A vitamin A-free diet results in impairment of the rat hippocampal somatostatinergic system. Neuroscience 141 (2), 851–861. doi:10.1016/j.neuroscience.2006.04.034
Hochstrasser, T., Ullrich, C., Sperner-Unterweger, B., and Humpel, C. (2011). Inflammatory stimuli reduce survival of serotonergic neurons and induce neuronal expression of indoleamine 2, 3-dioxygenase in rat dorsal raphe nucleus organotypic brain slices. Neuroscience 184, 128–138. doi:10.1016/j.neuroscience.2011.03.070
Holthoewer, D., Endres, K., Schuck, F., Hiemke, C., Schmitt, U., and Fahrenholz, F. (2012). Acitretin, an enhancer of alpha-secretase expression, crosses the blood-brain barrier and is not eliminated by P-glycoprotein. Neurodegener. Dis. 10 (1-4), 224–228. doi:10.1159/000334300
Hong, S. H., Kim, K. R., and Oh, D. K. (2015). Biochemical properties of retinoid-converting enzymes and biotechnological production of retinoids. Appl. Microbiol. Biotechnol. 99 (19), 7813–7826. doi:10.1007/s00253-015-6830-8
Horton, J. W., White, D. J., Maass, D. L., Hybki, D. P., Haudek, S., and Giroir, B. (2001). Antioxidant vitamin therapy alters burn trauma-mediated cardiac NF-kappaB activation and cardiomyocyte cytokine secretion. J. Trauma 50 (3), 397–406. doi:10.1097/00005373-200103000-00002
Huang, X., Zhang, H., Zhen, J., Dong, S., Guo, Y., Van Halm-Lutterodt, N., et al. (2018). Diminished circulating retinol and elevated α-TOH/retinol ratio predict an increased risk of cognitive decline in aging Chinese adults, especially in subjects with ApoE2 or ApoE4 genotype. Aging (Albany NY) 10 (12), 4066–4083. doi:10.18632/aging.101694
Hunter, C. L., Bimonte-Nelson, H. A., Nelson, M., Eckman, C. B., and Granholm, A. C. (2004). Behavioral and neurobiological markers of Alzheimer’s disease in Ts65Dn mice: Effects of estrogen. Neurobiol. Aging 25, 873–884. doi:10.1016/j.neurobiolaging.2003.10.010
Husson, M., Enderlin, V., Delacourte, A., Ghenimi, N., Alfos, S., Pallet, V., et al. (2006). Retinoic acid normalizes nuclear receptor mediated hypo-expression of proteins involved in β-amyloid deposits in the cerebral cortex of vitamin A deprived rats. Neurobiol. Dis. 23 (1), 1–10. doi:10.1016/j.nbd.2006.01.008
Husson, M., Enderlin, V., Delacourte, A., Ghenimi, N., Alfos, S., Pallet, V., et al. (2006). Retinoic acid normalizes nuclear receptor mediated hypo-expression of proteins involved in β-amyloid deposits in the cerebral cortex of vitamin A deprived rats. Neurobiol. Dis. 23 (1), 1–10. doi:10.1016/j.nbd.2006.01.008
Isaac, J. T., Buchanan, K. A., Muller, R. U., and Mellor, J. R. (2009). Hippocampal place cell firing patterns can induce long-term synaptic plasticity in vitro. J. Neurosci. 29 (21), 6840–6850. doi:10.1523/JNEUROSCI.0731-09.2009
Iversen, L. L., Rossor, M. N., Reynolds, G. P., Hills, R., Roth, M., Mountjoy, C. Q., et al. (1983). Loss of pigmented dopamine-β-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer’s type. Neurosci. Lett. 39, 95–100. doi:10.1016/0304-3940(83)90171-4
Jarvis, C. I., Goncalves, M. B., Clarke, E., Dogruel, M., Kalindjian, S. B., Thomas, S. A., et al. (2010). Retinoic acid receptor-α signalling antagonizes both intracellular and extracellular amyloid-β production and prevents neuronal cell death caused by amyloid-β. Eur. J. Neurosci. 32, 1246–1255. doi:10.1111/j.1460-9568.2010.07426.x
Jedlicka, P., and Deller, T. (2017). Understanding the role of synaptopodin and the spine apparatus in hebbian synaptic plasticity – new perspectives and the need for computational modeling. Neurobiol. Learn. Mem. 138, 21–30. doi:10.1016/j.nlm.2016.07.023
Jiang, W., Wen, E. Y., Gong, M., Shi, Y., Chen, L., Bi, Y., et al. (2011). The pattern of retinoic acid receptor expression and subcellular, anatomic and functional area translocation during the postnatal development of the rat cerebral cortex and white matter. Brain Res. 1382, 77–87. doi:10.1016/j.brainres.2011.01.027
Jiang, W., Yu, Q., Gong, M., Chen, L., Wen, E. Y., Bi, Y., et al. (2012). Vitamin A deficiency impairs postnatal cognitive function via inhibition of neuronal calcium excitability in hippocampus. J. Neurochem. 121 (6), 932–943. doi:10.1111/j.1471-4159.2012.07697.x
Kabir, M. T., Uddin, M. S., Zamman, S., Begum, Y., Ashraf, G. M., Bin-Jumah, M. N., et al. (2021). Molecular mechanisms of metal toxicity in the pathogenesis of Alzheimer’s disease. Molec. Neurobiol. 58, 1–20. doi:10.1007/s12035-020-02096-w
Kang, S. G., Lim, H. W., Andrisani, O. M., Broxmeyer, H. E., and Kim, C. H. (2007). Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J. Immunol. 179, 3724–3733. doi:10.4049/jimmunol.179.6.3724
Katsuki, H., Kurimoto, E., Takemori, S., Kurauchi, Y., Hisatsune, A., Isohama, Y., et al. (2009). Retinoic acid receptor stimulation protects midbrain dopaminergic neurons from inflammatory degeneration via BDNF-mediated signaling. J. Neurochem. 110, 707–718. doi:10.1111/j.1471-4159.2009.06171.x
Kaur, C., Sivakumar, V., Dheen, S. T., and Ling, E. A. (2006). Insulin-like growth factor I and II expression and modulation in amoeboid microglial cells by lipopolysaccharide and retinoic acid. Neuroscience 138, 1233–1244. doi:10.1016/j.neuroscience.2005.12.025
Kawahara, K., Nishi, K., Suenobu, M., Ohtsuka, H., Maeda, A., Nagatomo, K., et al. (2009). Oral administration of synthetic retinoid Am80 (tamibarotene) decreases brain β-amyloid peptides in APP23 mice. Biol. Pharm. Bull. 32 (7), 1307–1309. doi:10.1248/bpb.32.1307
Kawahara, K., Suenobu, M., Ohtsuka, H., Kuniyasu, A., Sugimoto, Y., Nakagomi, M., et al. (2014). Cooperative therapeutic action of retinoic acid receptor and retinoid x receptor agonists in a mouse model of Alzheimer's disease. J. Alzheimers Dis. 42 (2), 587–605. doi:10.3233/JAD-132720
Kedishvili, N. Y. (2016). Retinoic acid synthesis and degradation. Biochem. Retin. Signal. II 81, 127–161. doi:10.1007/978-94-024-0945-1_5
Khatib, T., Chisholm, D. R., Whiting, A., Platt, B., and McCaffery, P. (2020). Decay in retinoic acid signaling in varied models of Alzheimer’s disease and in-vitro test of novel retinoic acid receptor ligands (RAR-MS) to regulate protective genes. J. Alzheimers Dis. 73, 935–954. doi:10.3233/JAD-190931
Khatib, T., Marini, P., Nunna, S., Chisholm, D. R., Whiting, A., Redfern, C., et al. (2019). Genomic and non-genomic pathways are both crucial for peak induction of neurite outgrowth by retinoids. Cell Commun. Signal. 17, 40. doi:10.1186/s12964-019-0352-4
Khillan, J. S. (2014). Vitamin A/retinol and maintenance of pluripotency of stem cells. Nutrients 6 (3), 1209–1222. doi:10.3390/nu6031209
Khorasanizadeh, S., and Rastinejad, F. (2001). Nuclear-receptor interactions on DNA-response elements. Trends biochem. Sci. 26, 384–390. doi:10.1016/S0968-0004(01)01800-X
Kim, C. H. (2011). Retinoic acid, immunity, and inflammation. Vitam. Horm. 86, 83–101. doi:10.1016/B978-0-12-386960-9.00004-6
Kim, H. S., Hong, S. J., Ledoux, M. S., and Kim, K. S. (2001). Regulation of the tyrosine hydroxylase and dopamine β-hydroxylase genes by the transcription factor AP-2. J. Neurochem. 76, 280–294. doi:10.1046/j.1471-4159.2001.00044.x
Kitaoka, K., Shimizu, N., Ono, K., Chikahisa, S., Nakagomi, M., Shudo, K., et al. (2013). The retinoic acid receptor agonist Am80 increases hippocampal ADAM10 in aged SAMP8 mice. Neuropharmacology 72, 58–65. doi:10.1016/j.neuropharm.2013.04.009
Kobayashi, M., Matsuoka, I., and Kurihara, K. (1994). Cholinergic differentiation of cultured sympathetic neurons induced by retinoic acid. Induction of choline acetyltransferase-MRNA and suppression of tyrosine hydroxylase-MRNA levels. FEBS Lett. 337, 259–264. doi:10.1016/0014-5793(94)80204-1
König, G., Masters, C. L., and Beyreuther, K. (1990). Retinoic acid induced differentiated neuroblastoma cells show increased expression of the ΒA4 amyloid gene of Alzheimer’s disease and an altered splicing pattern. FEBS Lett. 269, 305–310. doi:10.1016/0014-5793(90)81181-M
Koryakina, A., Aeberhard, J., Kiefer, S., Hamburger, M., and Küenzi, P. (2009). Regulation of secretases by all‐trans‐retinoic acid. FEBS J. 276 (9), 2645–2655. doi:10.1111/j.1742-4658.2009.06992.x
Kour, S., and Rath, P. C. (2016). All-trans retinoic acid induces expression of a novel intergenic long noncoding RNA in adult rat primary hippocampal neurons. J. Mol. Neurosci. 58 (2), 266–276. doi:10.1007/s12031-015-0671-x
Krucker, T., Siggins, G. R., McNamara, R. K., Lindsley, K. A., Dao, A., Allison, D. W., et al. (2002). Targeted disruption of RC3 reveals a calmodulin-based mechanism for regulating metaplasticity in the hippocampus. J. Neurosci. 22 (13), 5525–5535.
Kulikov, A. V., Rzhaninova, A. A., Goldshtein, D. V., and Boldyrev, A. A. (2007). Expression of NMDA receptors in multipotent stromal cells of human adipose tissue under conditions of retinoic acid-induced differentiation. Bull. Exp. Biol. Med. 144 (4), 626–629. doi:10.1007/s10517-007-0390-6
La Fata, G., Weber, P., and Mohajeri, M. H. (2014). Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 6 (12), 5453–5472. doi:10.3390/nu6125453
Lakey-Beitia, J., Doens, D., Kumar, D. J., Murillo, E., Fernandez, P. L., Rao, K. S., et al. (2017). Anti-amyloid aggregation activity of novel carotenoids: Implications for Alzheimer’s drug discovery. Clin. Interv. Aging 12, 815–822. doi:10.2147/CIA.S134605
Lane, M. A., and Bailey, S. J. (2005). Role of retinoid signalling in the adult brain. Prog. Neurobiol. 75 (4), 275–293. doi:10.1016/j.pneurobio.2005.03.002
Larrieu, T., and Layé, S. (2018). Food for mood: Relevance of nutritional omega-3 fatty acids for depression and anxiety. Front. Physiol. 9, 1047. doi:10.3389/fphys.2018.01047
Ledesma, M. D., and Dotti, C. G. (2012). Peripheral cholesterol, metabolic disorders and Alzheimer’s disease. Front. Biosci. 4, 181–194. doi:10.2741/368
Lee, C. Y., and Landreth, G. E. (2010). The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 117 (8), 949–960. doi:10.1007/s00702-010-0433-4
Lee, H. P., Casadesus, G., Zhu, X., Lee, H. G., Perry, G., Smith, M. A., et al. (2009). All-trans retinoic acid as a novel therapeutic strategy for Alzheimer’s disease. Expert Rev. Neurother. 9, 1615–1621. doi:10.1586/ern.09.86
Lee, S. A., Belyaeva, O. V., and Kedishvili, N. Y. (2009). Biochemical characterization of human epidermal retinol dehydrogenase 2. Chem. Biol. Interact. 178 (1-3), 182–187. doi:10.1016/j.cbi.2008.09.019
Lenz, M., Eichler, A., Kruse, P., Muellerleile, J., Deller, T., Jedlicka, P., et al. (2021). All-trans retinoic acid induces synaptopodin-dependent metaplasticity in mouse dentate granule cells. Elife 10, e71983. doi:10.7554/eLife.71983
Lerner, A. J., Gustaw-Rothenberg, K., Smyth, S., and Casadesus, G. (2012). Retinoids for treatment of Alzheimer’s disease. BioFactors 38, 84–89. doi:10.1002/biof.196
Lévesque, D., and Rouillard, C. (2007). Nur77 and retinoid X receptors: Crucial factors in dopamine-related neuroadaptation. Trends Neurosci. 30 (1), 22–30. doi:10.1016/j.tins.2006.11.006
Liu, Z. M., Wang, K. P., Ma, J., and Guo Zheng, S. (2015). The role of all-trans retinoic acid in the biology of Foxp3+ regulatory T cells. Cell. Mol. Immunol. 12 (5), 553–557. doi:10.1038/cmi.2014.133
Luo, J., Bosy, T. Z., Wang, Y., Yasuda, R. P., and Wolfe, B. B. (1996). Ontogeny of NMDA R1 subunit protein expression in five regions of rat brain. Brain Res. Dev. Brain Res. 92 (1), 10–17. doi:10.1016/0165-3806(95)00191-3
Lushchak, V. I. (2011). Environmentally induced oxidative stress in aquatic animals. Aquat. Toxicol. 101 (1), 13–30. doi:10.1016/j.aquatox.2010.10.006
Lynch, M. A. (2004). Long-term potentiation and memory. Physiol. Rev. 84 (1), 87–136. doi:10.1152/physrev.00014.2003
Maggio, N., and Vlachos, A. (2014). Synaptic plasticity at the interface of health and disease: New insights on the role of endoplasmic reticulum intracellular calcium stores. Neuroscience, 135–146. doi:10.1016/j.neuroscience.2014.09.041
MacDonald, P. N., Bok, D., and Ong, D. E. (1990). Localization of cellular retinol-binding protein and retinol-binding protein in cells comprising the blood-brain barrier of rat and human. Proc. Natl. Acad. Sci. U. S. A. 87 (11), 4265–4269. doi:10.1073/pnas.87.11.4265
Maden, M. (2007). Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 8 (10), 755–765. doi:10.1038/nrn2212
Maghsoodi, B., Poon, M. M., Nam, C. I., Aoto, J., Ting, P., and Chen, L. (2008). Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. U. S. A. 105, 16015–16020. doi:10.1073/pnas.0804801105
Mann, D. M. A., Lincoln, J., Yates, P. O., Stamp, J. E., and Toper, S. (1980). Changes in the monoamine containing neurones of the human CNS in senile dementia. Br. J. Psychiatry 136, 533–541. doi:10.1192/bjp.136.6.533
Manzine, P. R., Ettcheto, M., Cano, A., Busquets, O., Marcello, E., Pelucchi, S., et al. (2019). ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 113, 108661. doi:10.1016/j.biopha.2019.108661
Mark, M., Ghyselinck, N. B., and Chambon, P. (2006). Function of retinoid nuclear receptors: Lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 46, 451–480. doi:10.1146/annurev.pharmtox.46.120604.141156
McCaffery, P., Zhang, J., and Crandall, J. E. (2006). Retinoic acid signaling and function in the adult hippocampus. J. Neurobiol. 66 (7), 780–791. doi:10.1002/neu.20237
Mecocci, P., Cristina Polidori, M., Cherubini, A., Ingegni, T., Mattioli, P., Catani, M., et al. (2002). Lymphocyte oxidative DNA damage and plasma antioxidants in alzheimer disease. Arch. Neurol. 59, 794–798. doi:10.1001/archneur.59.5.794
Melino, G., Draoui, M., Bernardini, S., Bellincampi, L., Reichert, U., and Cohen, P. (1996). Regulation by retinoic acid of insulin-degrading enzyme and of a related endoprotease in human neuroblastoma cell lines. Cell Growth Differ. 7, 787–796.
Mishra, S., Kelly, K. K., Rumian, N. L., and Siegenthaler, J. A. (2018). Retinoic acid is required for neural stem and progenitor cell proliferation in the adult hippocampus. Stem Cell Rep. 10 (6), 1705–1720. doi:10.1016/j.stemcr.2018.04.024
Misner, D. L., Jacobs, S., Shimizu, Y., De Urquiza, A. M., Solomin, L., Perlmann, T., et al. (2001). Vitamin A deprivation results in reversible loss of hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. U. S. A. 98 (20), 11714–11719. doi:10.1073/pnas.191369798
Mohammadzadeh Honarvar, N., Saedisomeolia, A., Abdolahi, M., Shayeganrad, A., Taheri Sangsari, G., Hassanzadeh Rad, B., et al. (2017). Molecular anti-inflammatory mechanisms of retinoids and carotenoids in Alzheimer’s disease: A review of current evidence. J. Mol. Neurosci. 61 (3), 289–304. doi:10.1007/s12031-016-0857-x
Mohammadzadeh Honarvar, N., Saedisomeolia, A., Abdolahi, M., Shayeganrad, A., Taheri Sangsari, G., Hassanzadeh Rad, B., et al. (2017). Molecular anti-inflammatory mechanisms of retinoids and carotenoids in Alzheimer’s disease: A review of current evidence. J. Mol. Neurosci. 61 (3), 289–304. doi:10.1007/s12031-016-0857-x
Mohammadzadeh Honarvar, N., Saedisomeolia, A., Abdolahi, M., Shayeganrad, A., Taheri Sangsari, G., Hassanzadeh Rad, B., et al. (2017). Molecular anti-inflammatory mechanisms of retinoids and carotenoids in Alzheimer’s disease: A review of current evidence. J. Mol. Neurosci. 61 (3), 289–304. doi:10.1007/s12031-016-0857-x
Mohammadzadeh Honarvar, N., Saedisomeolia, A., Abdolahi, M., Shayeganrad, A., Taheri Sangsari, G., Hassanzadeh Rad, B., et al. (2017). Molecular anti-inflammatory mechanisms of retinoids and carotenoids in Alzheimer’s disease: A review of current evidence. J. Mol. Neurosci. 61, 289–304. doi:10.1007/s12031-016-0857-x
Mufson, E. J., Counts, S. E., Perez, S. E., and Ginsberg, S. D. (2008). Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 8, 1703–1718. doi:10.1586/14737175.8.11.1703
Muller, L., and Bohm, V. (2011). Antioxidant activity of β-carotene compounds in different in vitro assays. Molecules 16 (2), 1055–1069. doi:10.3390/molecules16021055
Musiek, E. S., and Holtzman, D. M. (2015). Three dimensions of the amyloid hypothesis: Time, space and “wingmen. Nat. Neurosci. 18, 800–806. doi:10.1038/nn.4018
Napoli, J. L. (2017). Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol. Ther. 173, 19–33. doi:10.1016/j.pharmthera.2017.01.004
Nomoto, M., Takeda, Y., Uchida, S., Mitsuda, K., Enomoto, H., Saito, K., et al. (2012). Dysfunction of the RAR/RXR signaling pathway in the forebrain impairs hippocampal memory and synaptic plasticity. Mol. Brain 5, 8. doi:10.1186/1756-6606-5-8
Nozaki, Y., Yamagata, T., Sugiyama, M., Ikoma, S., Kinoshita, K., and Funauchi, M. (2006). Anti-inflammatory effect of all-trans-retinoic acid in inflammatory arthritis. Clin. Immunol. 119, 272–279. doi:10.1016/j.clim.2005.11.012
Obulesu, M., Dowlathabad, M. R., and Bramhachari, P. V. (2011). Carotenoids and Alzheimer’s disease: An insight into therapeutic role of retinoids in animal models. Neurochem. Int. 59 (5), 535–541. doi:10.1016/j.neuint.2011.04.004
Obulesu, M., Dowlathabad, M. R., and Bramhachari, P. V. (2011). Carotenoids and Alzheimer’s disease: An insight into therapeutic role of retinoids in animal models. Neurochem. Int. 59 (5), 535–541. doi:10.1016/j.neuint.2011.04.004
Ochoa, W. F., Torrecillas, A., Fita, I., Verdaguer, N., Corbalán-García, S., and Gomez-Fernandez, J. C. (2003). Retinoic acid binds to the C2-domain of protein kinase C(alpha). Biochemistry 42, 8774–8779. doi:10.1021/bi034713g
Palozza, P., Serini, S., Torsello, A., Di Nicuolo, F., Piccioni, E., Ubaldi, V., et al. (2003). Beta-carotene regulates NF-kappaB DNA-binding activity by a redox mechanism in human leukemia and colon adenocarcinoma cells. J. Nutr. 133 (2), 381–388. doi:10.1093/jn/133.2.381
Pan, J., Yu, J., Sun, L., Xie, C., Chang, L., Wu, J., et al. (2019). ALDH1A1 regulates postsynaptic μ–opioid receptor expression in dorsal striatal projection neurons and mitigates dyskinesia through transsynaptic retinoic acid signaling. Sci. Rep. 9 (1), 3602–3604. doi:10.1038/s41598-019-40326-x
Pedersen, W. A., Berse, B., Schüler, U., Wainer, B. H., Blusztajn, J. K., and Schuler, U. (1995). All-trans- and 9-cis-retinoic acid enhance the cholinergic properties of a murine septal cell line: Evidence that the effects are mediated by activation of retinoic acid receptor-alpha. J. Neurochem. 65 (1), 50–58. doi:10.1046/j.1471-4159.1995.65010050.x
Pierzchalski, K. A. (2015). Impact of cellular retinol binding protein, type I on retinoic acid biosynthesis and homeostasis. Baltimore): Doctoral dissertation, University of Maryland.
Pinessi, L., Rainero, I., de Gennaro, T., Gentile, S., Portaleone, P., and Bergamasco, B. (1987). Biogenic amines in cerebrospinal fluid and plasma of patients with dementia of alzheimer type. Funct. Neurol. 2, 51–58.
Poon, M. M., and Chen, L. (2008). Retinoic acid-gated sequence-specific translational control by RARalpha. Proc. Natl. Acad. Sci. U. S. A. 105, 20303–20308. doi:10.1073/pnas.0807740105
Priyanka, S. H., Syam Das, S., Thushara, A. J., Rauf, A. A., and Indira, M. (2018). All trans retinoic acid attenuates markers of neuroinflammation in rat brain by modulation of SIRT1 and NFκB. Neurochem. Res. 43, 1791–1801. doi:10.1007/s11064-018-2595-7
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer's disease. N. Engl. J. Med. 362, 329–344. doi:10.1056/NEJMRA0909142
Reinhardt, S., Ow Grimm, M., Stahlmann, C., Hartmann, T., Shudo, K., Tomita, T., et al. (2016). Rescue of hypovitaminosis a induces non-amyloidogenic amyloid precursor protein (APP) processing. Curr. Alzheimer Res. 13 (11), 1277–1289. doi:10.2174/1567205013666160603002105
Reinikainen, K. J., Paljärvi, L., Huuskonen, M., Soininen, H., Laakso, M., and Riekkinen, P. J. (1988). A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer’s disease. J. Neurol. Sci. 84, 101–116. doi:10.1016/0022-510X(88)90179-7
Rinaldi, P., Polidori, M. C., Metastasio, A., Mariani, E., Mattioli, P., Cherubini, A., et al. (2003). Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 24 (7), 915–919. doi:10.1016/s0197-4580(03)00031-9
Sahin, M., Karaüzüm, S. B., Perry, G., Smith, M. A., and Alicigüzel, Y. (2005). Retinoic acid isomers protect hippocampal neurons from amyloid-β induced neurodegeneration. Neurotox. Res. 7 (3), 243–250. doi:10.1007/BF03036453
Saleem, U., Akhtar, R., Anwar, F., Shah, M. A., Chaudary, Z., Ayaz, M., et al. (2021). Neuroprotective potential of Malva neglecta is mediated via down-regulation of cholinesterase and modulation of oxidative stress markers. Metab. Brain Dis. 36 (5), 889–900. doi:10.1007/s11011-021-00683-x
Samad, T. A., Krezel, W., Chambon, P., and Borrelli, E. (1997). Regulation of dopaminergic pathways by retinoids: Activation of the D2 receptor promoter by members of the retinoic acid receptor–retinoid X receptor family. Proc. Natl. Acad. Sci. U. S. A. 94 (26), 14349–14354. doi:10.1073/pnas.94.26.14349
Samad, T. A., Krezel, W., Chambon, P., and Borrelli, E. (1997). Regulation of dopaminergic pathways by retinoids: Activation of the D2 receptor promoter by members of the retinoic acid receptor-retinoid X receptor family. Proc. Natl. Acad. Sci. U. S. A. 94, 14349–14354. doi:10.1073/pnas.94.26.14349
Santucci, A. C., Kanof, P. D., and Haroutunian, V. (1989). Effect of physostigmine on memory consolidation and retrieval processes in intact and nucleus basalis-lesioned rats. Psychopharmacol. Berl. 99, 70–74. doi:10.1007/BF00634455
Saurer, L., McCullough, K. C., and Summerfield, A. (2007). In vitro induction of mucosa-type dendritic cells by all-trans retinoic acid. J. Immunol. 179 (6), 3504–3514. doi:10.4049/jimmunol.179.6.3504
Schwarcz, R., Bruno, J. P., Muchowski, P. J., and Wu, H. Q. (2012). Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477. doi:10.1038/nrn3257
Serot, J. M., Christmann, D., Dubost, T., and Couturier, M. (1997). Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 63, 506–508. doi:10.1136/JNNP.63.4.506
Shannon, S. R., Moise, A. R., and Trainor, P. A. (2017). New insights and changing paradigms in the regulation of vitamin A metabolism in development. WIREs Dev. Biol. 6 (3), e264. doi:10.1002/wdev.264
Shearer, K. D., Stoney, P. N., Morgan, P. J., and McCaffery, P. J. (2012). A vitamin for the brain. Trends Neurosci. 35 (12), 733–741. doi:10.1016/j.tins.2012.08.005
Shudo, K., Kagechika, H., Yamazaki, N., Igarashi, M., and Tateda, C. (2004). A synthetic retinoid Am80 (tamibarotene) rescues the memory deficit caused by scopolamine in a passive avoidance paradigm. Biol. Pharm. Bull. 27 (11), 1887–1889. doi:10.1248/bpb.27.1887
Shudo, K., Fukasawa, H., Nakagomi, M., and Yamagata, N. (2009). Towards retinoid therapy for alzheimers disease. Curr. Alzheimer Res. 6, 302–311. doi:10.2174/156720509788486581
Soden, M. E., and Chen, L. (2010). Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J. Neurosci. 30, 16910–16921. doi:10.1523/JNEUROSCI.3660-10.2010
Sodhi, R. K., and Singh, N. (2014). Retinoids as potential targets for Alzheimer's disease. Pharmacol. Biochem. Behav. 120, 117–123. doi:10.1016/j.pbb.2014.02.016
Sommer, A., and Vyas, K. S. (2012). A global clinical view on vitamin A and carotenoids. Am. J. Clin. Nutr. 96 (5), 1204S–6S. doi:10.3945/ajcn.112.034868
Takamura, R., Watamura, N., Nikkuni, M., and Ohshima, T. (2017). All-trans retinoic acid improved impaired proliferation of neural stem cells and suppressed microglial activation in the Hippocampus in an Alzheimer’s mouse model. J. Neurosci. Res. 95, 897–906. doi:10.1002/jnr.23843
Tippmann, F., Hundt, J., Schneider, A., Endres, K., and Fahrenholz, F. (2009). Up‐regulation of the α‐secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 23 (6), 1643–1654. doi:10.1096/fj.08-121392
Tousi, B. (2015). The emerging role of bexarotene in the treatment of Alzheimer’s disease: Current evidence. Neuropsychiatr. Dis. Treat. 11, 311–315. doi:10.2147/NDT.S61309
Touyarot, K., Bonhomme, D., Roux, P., Alfos, S., Lafenêtre, P., Richard, E., et al. (2013). A mid-life vitamin A supplementation prevents age-related spatial memory deficits and hippocampal neurogenesis alterations through CRABP-I. PLoS One 8 (8), e72101. doi:10.1371/journal.pone.0072101
Trillo, L., Das, D., Hsieh, W., Medina, B., Moghadam, S., Lin, B., et al. (2013). Ascending monoaminergic systems alterations in Alzheimer’s disease. Translating basic science into clinical care. Neurosci. Biobehav. Rev. 37, 1363–1379. doi:10.1016/j.neubiorev.2013.05.008
van Neerven, S., Kampmann, E., and Mey, J. (2008). RAR/RXR and PPAR/RXR signaling in neurological and psychiatric diseases. Prog. Neurobiol. 85, 433–451. doi:10.1016/j.pneurobio.2008.04.006
Vekrellis, K., Ye, Z., Qiu, W. Q., Walsh, D., Hartley, D., Chesneau, V., et al. (2000). Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 20, 1657–1665. doi:10.1523/jneurosci.20-05-01657.2000
Veng, L. M., Granholm, A. C., and Rose, G. M. (2003). Age-related sex differences in spatial learning and basal forebrain cholinergic neurons in F344 rats. Physiol. Behav. 80, 27–36. doi:10.1016/S0031-9384(03)00219-1
Walsh, D. M., and Selkoe, D. J. (2007). Aβ oligomers - a decade of discovery. J. Neurochem., 1172–1184. doi:10.1111/j.1471-4159.2006.04426.x
West, K. P. (2002). Extent of vitamin A deficiency among preschool children and women of reproductive age. J. Nutr. 132 (9), 2857S–66S. doi:10.1093/jn/132.9.2857S
White, J. T., and Kelly, J. W. (2001). Support for the multigenic hypothesis of amyloidosis: The binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc. Natl. Acad. Sci. U. S. A. 98, 13019–13024. doi:10.1073/pnas.241406698
Wietrzych-Schindler, M., Szyszka-Niagolov, M., Ohta, K., Endo, Y., Pérez, E., de Lera, A. R., et al. (2011). Retinoid X receptor gamma is implicated in docosahexaenoic acid modulation of despair behaviors and working memory in mice. Biol. Psychiatry 69, 788–794. doi:10.1016/j.biopsych.2010.12.017
Woodall, A. A., Lee, S. W., Weesie, R. J., Jackson, M. J., and Britton, G. (1997). Oxidation of carotenoids by free radicals: Relationship between structure and reactivity. Biochim. Biophys. Acta 1336 (1), 33–42. doi:10.1016/s0304-4165(97)00006-8
Wyss-Coray, T., Lin, C., Yan, F., Yu, G. Q., Rohde, M., Mcconlogue, L., et al. (2001). TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 7, 612–618. doi:10.1038/87945
Yang, F., Wolk, A., Håkansson, N., Pedersen, N. L., and Wirdefeldt, K. (2017). Dietary antioxidants and risk of Parkinson's disease in two population‐based cohorts. Mov. Disord. 32 (11), 1631–1636. doi:10.1002/mds.27120
Yap, K., Drakew, A., Smilovic, D., Rietsche, M., Paul, M. H., Vuksic, M., et al. (2020). The actin-modulating protein synaptopodin mediates long-term survival of dendritic spines. Elife 9, e62944. doi:10.7554/eLife.62944
Zeng, J., Chen, L., Wang, Z., Chen, Q., Fan, Z., Jiang, H., et al. (2017). Marginal vitamin A deficiency facilitates Alzheimer’s pathogenesis. Acta Neuropathol. 133 (6), 967–982. doi:10.1007/s00401-017-1669-y
Zhang, Y. R., Zhao, Y. Q., and Huang, J. F. (2012). Retinoid-binding proteins: Similar protein architectures bind similar ligands via completely different ways. PLoS One 7 (5), e36772. doi:10.1371/journal.pone.0036772
Keywords: retinoids, neuroinflammation, RXRS, RARS, neurotransmission, neuroplasticity
Citation: Behl T, Kaur D, Sehgal A, Singla RK, Makeen HA, Albratty M, Alhazmi HA, Meraya AM and Bungau S (2022) Therapeutic insights elaborating the potential of retinoids in Alzheimer’s disease. Front. Pharmacol. 13:976799. doi: 10.3389/fphar.2022.976799
Received: 23 June 2022; Accepted: 18 July 2022;
Published: 23 August 2022.
Edited by:
Prof. Dr. Haroon Khan, Abdul Wali Khan University Mardan, PakistanReviewed by:
Muhammad Ayaz, University of Malakand, PakistanCopyright © 2022 Behl, Kaur, Sehgal, Singla, Makeen, Albratty, Alhazmi, Meraya and Bungau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tapan Behl, dGFwYW5iZWhsMzFAZ21haWwuY29t; Simona Bungau, c2J1bmdhdUB1b3JhZGVhLnJv
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.