
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 23 August 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.936758
 Rui Xuan Huang1
Rui Xuan Huang1 Damrongrat Siriwanna2
Damrongrat Siriwanna2 William C. Cho3
William C. Cho3 Tsz Kin Wan1
Tsz Kin Wan1 Yan Rong Du4
Yan Rong Du4 Adam N. Bennett2
Adam N. Bennett2 Qian Echo He2
Qian Echo He2 Jun Dong Liu2
Jun Dong Liu2 Xiao Tai Huang5
Xiao Tai Huang5 Kei Hang Katie Chan1,2,6*
Kei Hang Katie Chan1,2,6*Lung cancer is the leading cause of cancer deaths globally, and lung adenocarcinoma (LUAD) is the most common type of lung cancer. Gene dysregulation plays an essential role in the development of LUAD. Drug repositioning based on associations between drug target genes and LUAD target genes are useful to discover potential new drugs for the treatment of LUAD, while also reducing the monetary and time costs of new drug discovery and development. Here, we developed a pipeline based on machine learning to predict potential LUAD-related target genes through established graph attention networks (GATs). We then predicted potential drugs for the treatment of LUAD through gene coincidence-based and gene network distance-based methods. Using data from 535 LUAD tissue samples and 59 precancerous tissue samples from The Cancer Genome Atlas, 48,597 genes were identified and used for the prediction model building of the GAT. The GAT model achieved good predictive performance, with an area under the receiver operating characteristic curve of 0.90. 1,597 potential LUAD-related genes were identified from the GAT model. These LUAD-related genes were then used for drug repositioning. The gene overlap and network distance with the target genes were calculated for 3,070 drugs and 672 preclinical compounds approved by the US Food and Drug Administration. At which, bromoethylamine was predicted as a novel potential preclinical compound for the treatment of LUAD, and cimetidine and benzbromarone were predicted as potential therapeutic drugs for LUAD. The pipeline established in this study presents new approach for developing targeted therapies for LUAD.
Lung cancer is one of the most common cancers globally. In 2020, approximately 2.21 million people had lung cancer, and there were 1.8 million lung cancer-related deaths, which was the highest cancer mortality rate in that year (World Health Organization, 2021). Lung adenocarcinoma (LUAD) belongs to the non-small cell lung cancer (NSCLC) family, which accounts for 40% of all lung cancer types. LUAD is the most common type of lung cancer (Zappa and Mousa, 2016). It has unique cellular and molecular characteristics compared with other lung cancer subtypes. Previous studies have shown that high or low expression levels of certain genes in cancer tissues play a vital role in the development of LUAD (Chen et al., 2019). The dysregulation of these genes provides an opportunity for targeted therapy. Targeted therapy is currently one of the options for the postoperative treatment of patients with LUAD. For example, the epithelial growth factor receptor (EGFR) inhibitors, gefitinib, and erlotinib are already used to treat LUAD patients (Janku et al., 2010; Pao and Chmielecki, 2010). Due to the large number of genes involved in the development of LUAD, the therapeutic effects of inhibiting only a single gene may be limited. Therefore, it is necessary to discover potential genes related to LUAD and identify drugs that target these genes (Cho, 2013).
Targeted therapy refers to a treatment targeting previously defined carcinogenic sites at the cellular and molecular levels. The site may be a protein, a gene, or a gene fragment inside the tumor cell (American Cancer Society, 2021). Targeted therapies interfere with cancer-causing or tumor growth-promoting molecules or genes to treat cancer. The development of new drugs based on cancer-related genes is time-consuming and labor-intensive, and it takes an average of 13 years and $870 million from development to market (Paul et al., 2010; Sertkaya et al., 2016). Therefore, repurposing drugs that have already been approved is a more cost-effective approach. Hundreds of drugs have been retargeted, and new uses have been found using algorithms that integrate genomic and phenotypic data (Cheng et al., 2019; Kenneth and Cho, 2022). Most existing drug repositioning methods are limited to drug similarity networks and disease similarity networks, which do not account for information about cancer-related genes themselves or topological information between different biological networks (Cheng et al., 2019; Kenneth and Cho, 2022).
With the recent development of artificial intelligence and big data disciplines, an increasing number of machine learning (ML) and deep learning (DL) networks are being developed and used to analyze highly nonlinear network structures, such as gene, protein, disease, and drug networks (Angermueller et al., 2016; Ching et al., 2018). Using a network analysis model based on ML and DL, the combined analysis of genomics and systems biology networks makes it possible to identify druggable targets and perform drug repositioning while predicting cancer-related genes.
In this study, a target gene prediction–drug repositioning pipeline was constructed for LUAD. To more accurately predict potential drugs for the treatment of LUAD, target genes associated with LUAD were identified before drug prediction. Due to the complex network relationship between genes and the wide range of characteristics of the genes themselves, a graph neural network (GNN)-based graph attention network (GAT) model was built to be a gene classifier, which can predict genes that currently have unconfirmed relationship with LUAD are in fact associated with LUAD or not. Other models including a GNN-based graph convolutional network (GCN) model, a DL-based TabNet model, an ML-based random forest (RF) model (Ho, 1995), an Adaptive Boosting (AdaBoost) model (Freund et al., 1999), and an XGBoost classifier model (Chen and Guestrin, 2016) were built to compare their performances with the GAT model. The drug prediction performed in this study was based on the assumption that the target genes of a drug for the treatment of LUAD are related to the genes associated with LUAD. Under this assumption, two drug prediction methods, network overlap-based, and network distance-based methods were used to predict potential drugs for the treatment of LUAD.
Gene expression data were collected for 594 samples from The Cancer Genome Atlas (TCGA, https://www.cancer.gov/tcga) (Cancer Genome Atlas Research Network et al., 2013), of which 59 were precancerous tissue (normal) samples, and 535 were tumor samples. Genes were extracted from TCGA. To define LUAD-related target genes, the fragments per kilobase of exon per million mapped fragments (FPKM) values from the transcriptome profiling data were downloaded from TCGA. To further group genes and to identify genes that have statistically significant differences in expression in cancer tissue compared to normal tissue, differential expression analysis was performed. Specifically, fold change was calculated by dividing the mean FKPM in cancer tissue by the mean FPKM from normal tissue, thereby providing the fold change value for each gene. To facilitate differential expression analysis, log fold change was converted to log2fold change (log2FC), and adjusted p-value (false discovery rate, FDR) was calculated by limma package (v3.38.3) (Ritchie et al., 2015). Genes with an FDR ≥0.05 were excluded from the study. Genes with log2FC > 2 were classified as up-regulated genes, whereas those with log2FC < −2 were classified as down-regulated genes. Both up-regulated, and down-regulated genes were considered to be associated with LUAD. These LUAD-related genes were added to ‘‘positive’’ gene lists to facilitate subsequent model building. On the contrary, genes with log2FC > −0.3 and log2FC < 0.3 were considered not to be statistically differentially expressed in cancer tissue compared to normal tissue and were added to the “negative” genes list in the study. A Bayesian information criterion (BIC) method-based sensitivity analysis was performed to illustrate the rationale for the Log2FC threshold used in this study (Supplementary Material SB). The remaining genes were having an unconfirmed relationship with LAUD, and thus were added to the “ambiguous” genes list in the study. Positive and negative gene lists were used for GAT model training, validation, and testing, and the ambiguous gene list was used for potential LUAD-related target prediction. The model was designed to predict the association of genes with ambiguous relationships to LUAD (classified as LUAD-related or not).
To verify the predictive ability and validity of the model, a subset of genes was obtained from the Comparative Toxicogenomics Database (CTD) (Davis et al., 2020). The criteria of LUAD-related genes were: 1) Reported to have direct evidence of “marker/mechanism; ” 2) Had an Inference Score greater than 11.09 (75% quantile) or a Reference Count greater than 6 (75% quantile). The same number of genes that were reported to other diseases from CTD were randomly picked with the following criteria: 1) Not included in the training set or validation set of GAT; 2) Not included in LAUD-related gene list from CTD.
Gene function annotation analysis was performed using Gene Analytics (https://ga.genecards.org/) (Ben-Ari Fuchs et al., 2016) and VarElect (https://varelect.genecards.org/). p-values, matching scores, and average disease-causing likelihoods of gene-related tissues, systems, diseases, and pathways; gene ontology information; and human phenotype ontology information were collected, with a threshold p-value of 0.05. For convenient data processing, the original p-values were replaced by −log10 (p-values). Missing values were imputed with zeros to indicate that there was no association.
Gene function annotation features with greater than 40% of values missing, those with absolute Pearson’s correlation coefficient values greater than 0.98, and those that did not contribute to cumulative importance of 0.99 were excluded from the analysis. Samples were divided into training (80%), validation (10%), and testing sets (10%) using a stratified sampling method. The imputation of missing values and data normalization were performed on the training, validation, and testing sets separately after dataset division to avoid data leakage.
Gene-gene interaction information was collected from the Human Reference Interactome (http://www.interactome-atlas.org/) (Luck et al., 2020) ,RNAInter v4.0 (http://www.rnainter.org/) (Kang et al., 2022), and the human protein-coding Gene Functional Association Network. The high-confidence gene associations from different databases were merged, and gene-gene interaction subgraphs were constructed according to the genes required for the study. Functionally related protein-coding genes, RNA-RNA associations, and RNA-protein associations were recorded as the background gene-gene interaction network of this study. As the interaction edges from the Human Reference Interactome were un-weighted, the unweighted interactions were replaced by the average edge weight of the subgraph. The weights of all edges are input into the network as initial values. During the learning process of the network, the weights of each edge are iteratively updated. Drug target genes were collected from PharmGKB (https://www.pharmgkb.org/) (Whirl-Carrillo et al., 2021), the Binding Database (https://www.bindingdb.org/) (Liu et al., 2007), and DrugBank (https://go.drugbank.com/) (Wishart et al., 2018). Data from human subjects collected and used in this research project have been approved by the University’s Research Ethics Subcommittee. Data is available at: https://github.com/Clement-HUANG/LUAD-Drug-Repositioning. The hyperparameters and sought ranges of trained models in this study are available in the Supplementary Table S1.
A GAT model was developed for LUAD-related target gene prediction. During the construction of the graph, genes were input as nodes, and the features of the nodes were high-dimensional gene function annotation features. The edges of the graph were the gene-gene associations, and the weights of the edges were the weights of the association. The three-fold cross-validation method was used to obtain optimal model parameters, and “auc” was used as the model metric. The random search method was applied for the approximate range of hyperparameters, and the grid search method was applied afterward to search for the best hyperparameters in a small range. The trained GAT model was used to analyze the “ambiguous” gene list for the final LUAD-related target gene prediction. ML-based models, including RF, AdaBoost, and XGBoost classifier models; a DL-based TabNet model; and a GNN-based GCN model were built to compare their performance with the GAT model. A support vector machine classifier and a logistic regression model were also built but were excluded from the comparison as they were time-consuming to execute, and they showed low precision for processing high-dimensional data. Gene enrichment analysis was applied for LUAD-related genes identified by differential gene expression analysis and LUAD-related genes predicted by the GAT model. To extend the GAT model established in this study, additional experiments were applied to test the ability of the GAT model to predict genes associated with enriched GO terms and KEGG pathways. In addition, the gene interaction networks of BioGRID (Stark et al., 2006), BioSNAP (PP-Pathways), and BioSNAP (TFG-Ohmnet) (Leskovec and Sosič, 2016) were collected to test the sensitivity of the GAT model to different gene interaction networks. The GAT-trained gene-gene interaction network was derived, and the genes were clustered using the K-mean clustering method. This method’s optimal K value (the number of clusters) was determined by the silhouette index (Rousseeuw, 1987), which described the node’s association and its corresponding cluster. To better interpret this network, gene enrichment analysis using the DAVID online database (Huang Da et al., 2009; Sherman et al., 2022) was applied to the genes of each cluster.
The drug repositioning module was based on the LUAD-related gene interaction network and drug-gene interaction network. Among them, the LUAD-related gene network was built according to LUAD-related genes and their associated genes from the original gene-gene interaction network; and the drug-gene network was constructed according to drug target genes and their directly related genes from the original gene-gene interaction network. The module was mainly composed of the network overlap-based drug prediction model, the network distance-based drug prediction model, and the summary model. The Jaccard score is a statistic that measures the similarity between two sample sets. In the network overlap-based drug prediction model, the similarity between the LUAD-related target gene networks and the drug target gene networks was calculated using the Jaccard score as the measure (Bass et al., 2013). The Jaccard score between the LUAD-related target gene networks and lists (Ng) and the ith drug target gene network (Ndi) was calculated according to Eq. 1.
The network distance-based drug prediction model calculated the length of the shortest path (d) from the genes (g) in the LUAD-related target gene network (Ng) to the ith drug (Ndi) in the target gene network (Nd) according to Eq. 2.
The z-score was used to standardize the distance between Ng and Ndi. It was calculated based on the shortest path length (d) and it’s mean (
Jaccard scores and z-scores for the drugs were calculated using the online tool PharmOmics (http://mergeomics.research.idre.ucla.edu/) (Chen et al., 2022) and were included in the final summary model. Drugs with a Jaccard score more than the mean value of all the drugs, and a z-score less than −2 were output as potential therapeutics for LUAD (one side p-value < 0.05). Based on the difference between the values for Ng and Ndi, drugs with an overlap (overlapped genes/Ndi) < 50% were excluded.
To validate the affinities of potential drugs, and preclinical compounds predicted from this study with their target proteins, the half-maximal inhibitory concentration (IC50) values were collected from Binding Database. The drug-target protein pairs with median IC50 values less than 10,000 nM were considered as having physical drug-target interactions (Cheng et al., 2019).
The target gene prediction–drug repositioning pipeline is shown in Figure 1. LUAD target gene prediction was based on genes, gene interaction networks, and functional annotation data. Through the established GAT, potential LUAD-related target genes were predicted. The target genes were passed into the drug repositioning module as the input of the drug redirection network. The module was a double-layer structure. The first layer was composed of a network overlap-based drug prediction model and a network distance-based drug prediction model, and the second layer was a summary of the results of the two models.
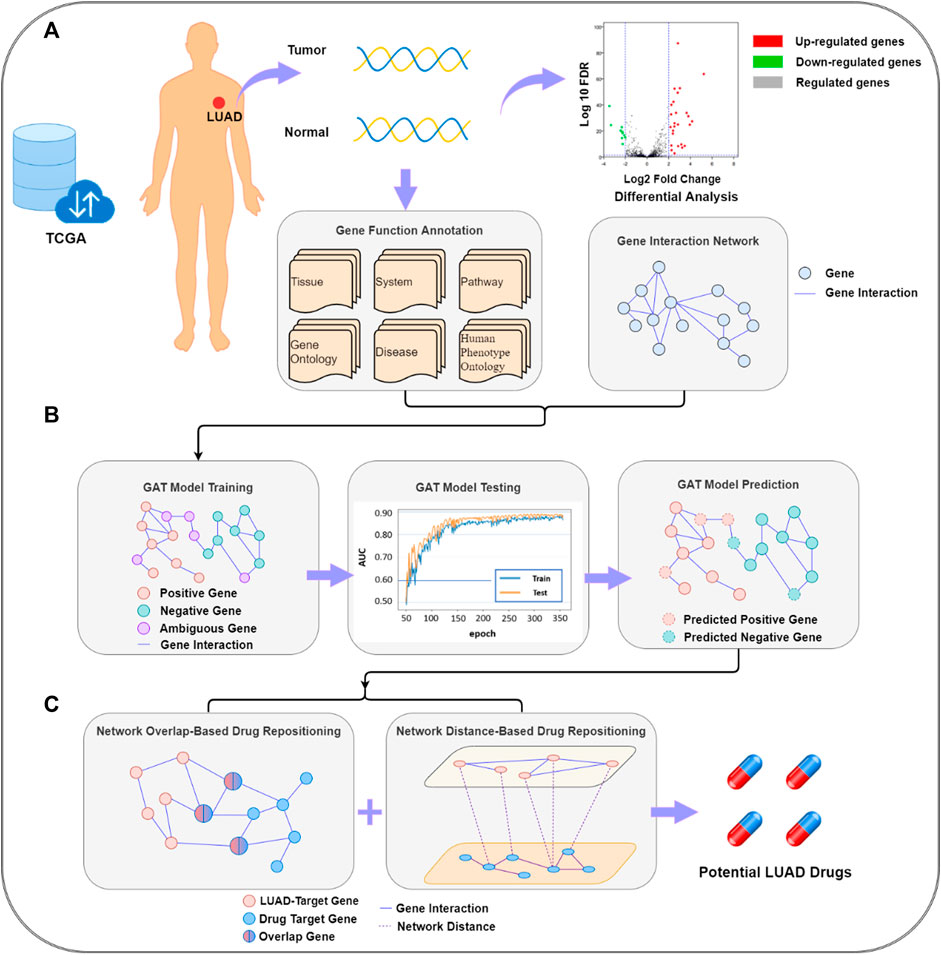
FIGURE 1. Lung adenocarcinoma target gene prediction–drug repositioning pipeline. (A) data collection and pre-processing; (B) graph attention network (GAT) model building and lung adenocarcinoma (LUAD)-related target gene prediction; (C) potential LUAD drug prediction. FDR, false discovery rate; TCGA, The Cancer Gene Atlas.
Genes were extracted from TCGA. The FPKM data for the genes were used for differential gene expression analysis (Figure 2). Of the 48,597 genes (19,054 protein-coding genes; 29,543 non-coding genes) included in the analysis, 19,730 had an FDR <0.05, 1,119 were upregulated, and 1,064 were downregulated. After excluding pseudogenes, the remaining 1,946 dysregulated genes were classified as positive genes (LUAD-related target genes) for model training. The absolute value of log2 fold change (log2FC) was <0.3 for 1,379 genes, and these genes were considered to be irrelevant to LUAD and thus were used as negative genes for training. The remaining 13,062 genes were classified as having an unclear relationship with LUAD, awaiting prediction by the model.
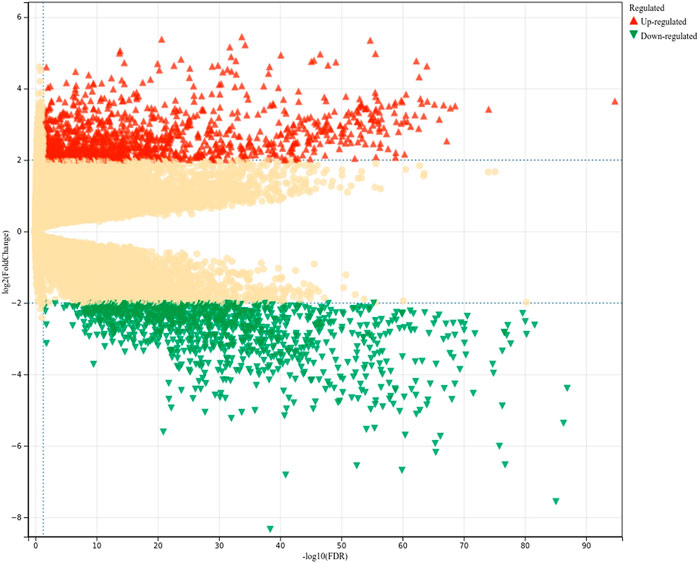
FIGURE 2. Analysis of differentially expressed LUAD-related genes. FDR, false discovery rate.
One hundred and fifty functional annotation features (tissues, systems, diseases, and pathways; gene ontology information; and human phenotype ontology information) of the genes were collected, and suitable features were selected to train the model. Features with greater than 40% of values missing and those with absolute Pearson’s correlation coefficient values greater than 0.98 were removed from the analysis due to low quality and high redundancy, respectively. Sixty-eight features were selected and used for subsequent model training. The 15 most significant prediction features with their normalized importance scores and the number of features with a cumulative importance score greater than 0.99 are shown in Supplementary Figure S1. Of all the gene functional annotation features, the importance score of gene enrichment significance was highest in the nervous system (normalized importance coefficient >0.05). The enrichment significance of genes in lung, brain, blood, kidney, and epithelial cells also had a high ranking (>0.03). In terms of disease enrichment features, genes were more significantly enriched in breast cancer than in lung cancer. The enrichment significance of pathways and gene ontologies related to lung cancer did not rank within the top 15.
The models’ power to predict LUAD-related target genes was determined based on the area under the receiver operating characteristic curve (AUROC), as shown in Figure 3. The GAT model achieved the best prediction performance in the internal data tests, reaching the highest AUROC value (0.90). The GNN-based GCN, ML-based RF, XGBoost, and Adaboost models also achieved good prediction performance (AUROC = 0.87, 0.81, 0.83, and 0.85, respectively). However, the DL-based TabNet model did not show good performance, with an AUROC value of less than 0.8. The external validation dataset from CTD achieved an AUROC of 0.82. In addition to AUROC, model evaluation metrics such as precision, recall, and F1-score were also used to measure the predictive ability of the model (Table 1). GNN-based GAT and GCN models achieved above 0.80 in precision, recall, and F1-score, which were better than DL-based TabNet, ML-based XGBoost, RF, and Adaboost. GAT achieved the highest precision, recall, and F1-score (precision = 0.85, recall = 0.85, F1-score = 0.85), indicating its strong classification ability for positive and negative samples. The precision, recall, and F1-score of the external validation dataset from CTD all achieved 0.78. 134 GO terms and KEGG pathways were enriched by LUAD-related genes (FDR<0.05), including 69 GO biological process terms, 27 GO cellular component terms, 28 GO molecular function terms, and 10 KEGG pathways. The top 5 terms of each category, together with the prediction AUROC and AUPRC, were reported according to the order of FDR (Supplementary Table S2).

FIGURE 3. Areas under the receiver operating characteristic curves of the models. AUROC, area under the receiver operating characteristic curve; GAT, graph attention networks; GCN, graph convolutional network.
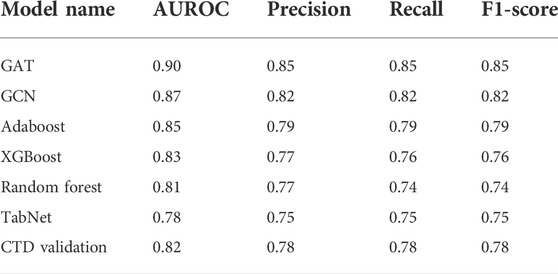
TABLE 1. Model evaluation metrics: AUROC, precision, recall, and F1 score.
The prediction of LUAD-related genes in this study was sensitive to the gene interaction network. The results and prediction performance of loading different gene interaction networks into the GAT model were not completely consistent, which might be caused by various gene interaction network sources. In addition to the results reported in Table 1, the GAT model achieved an AUROC of 0.85 using the gene interaction network from the Biogrid database; an AUROC of 0.87 using the gene interaction network from the BioSNAP database; and in particular, an AUROC of 0.85 using the lung tissue-related gene interaction network from the BioSNAP database (Supplementary Table S3). Although good LUAD-related gene prediction ability can be achieved using lung tissue-related gene interaction networks, it might be a risk to be applied in drug repositioning analysis, because drug genes are not necessarily associated with lungs only and vice versa.
After training the GAT model, genes with ambiguous relationships with LUAD were input into the trained GAT model to identify more LUAD-related genes. The output of the GAT model was the likelihood (probability) that a gene was a LUAD-related target gene, with values ranging from 0 to 1. Genes with an output probability value closer to 1 had a stronger association with LUAD. The Youden index of AUROC was calculated to find the optimal threshold for the model. The genes with an output higher than the optimal threshold can be considered to be associated with LUAD. The optimal threshold of the GAT model was 0.664, and genes with scores >0.664 were all potential LUAD-related genes originally. To simplify the complexity of the drug prediction network and improve the computational speed, the genes with output scores higher than 0.9 were used as the predicted LUAD-related genes for subsequent drug prediction. Using a threshold between 0.664 and 0.9 increased the complexity of the drug prediction network and the computation time of the Jaccard score and z-score, but not able to identify more drugs that reached a statistically significant level. Of the 10,858 genes identified as having an ambiguous relationship with LUAD, 1,597 genes had an output score >0.9 and were considered to be potential target genes for LUAD treatment.
Based on the results of GAT model training, the interaction network between genes was drawn (Figure 4). Among them, the blue nodes are LUAD-related genes identified by the gene differential expression analysis, the red nodes are LUAD-related genes predicted by the GAT model, and the grey nodes are LUAD-unrelated genes. The distance between nodes is determined by the strength of the interaction between genes, and two nodes with close distances indicate strong interactions between the two genes. In the gene-gene interaction network, 8 clusters of genes were found. The number of clusters is determined from the optimal silhouette index of the K-mean clustering method. Among the 8 clusters, the 7th cluster contained more LUAD-related genes (>50%). To further investigate the genes network, gene enrichment analysis using the DAVID online tool was applied to the genes of each cluster. The enrichment of genes in each cluster was assessed according to the top 5 GO terms from low to high FDR (threshold FDR <0.05), and the enriched KEGG pathways are reported (Supplementary Table S4).
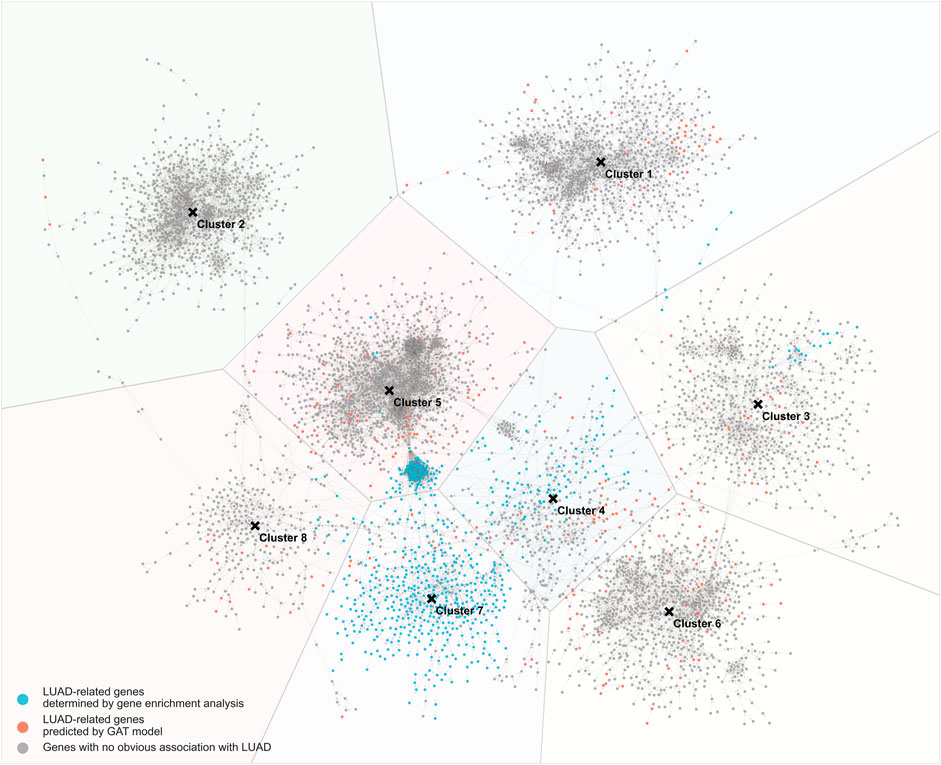
FIGURE 4. Gene-gene interaction network trained by GAT model. Blue nodes: LUAD-related genes determined by gene enrichment analysis; red nodes: LUAD-related genes predicted by GAT model; grey nodes: genes with no obvious association with LUAD.
The original positive samples and the potential target genes for LUAD treatment predicted by the GAT model were integrated and input into the drug prediction module, along with 3,070 drugs approved by the Food and Drug Administration and 672 preclinical compounds for use in humans. In the overlap-based drug prediction network, Jaccard scores were calculated to represent the degree of overlap between drug target genes and LUAD-related genes. The Jaccard scores for all drugs were generally normally distributed (Supplementary Figure S2A), with a mean of 0.036 and a standard deviation of 0.011. In the network distance-based drug prediction network, the z-score of each drug was calculated to represent the distance between the drug target gene network and the LUAD target gene network. A smaller z-score represented a closer distance. The z-score distribution of the individual drugs was not normal (Supplementary Figure S2B), with a mean of -3.848 and a standard deviation of 2.951. In total, 1,855 drugs and preclinical compounds from the overlap-based network had Jaccard scores higher than the mean Jaccard score, and 35 drugs or preclinical compounds had z-scores less than -2 and were thus considered to be potentially associated with LUAD.
The prediction results of the two networks were summarized, and drugs with a drug-gene overlap rate of less than 50% were eliminated. Ten drugs or compounds met all the requirements and were predicted to be potential drugs for preclinical studies of LUAD (Table 2). The predicted results were compared with data from the DrugBank database, the CTD, and literature searches of PubMed. Of the ten predicted drugs and preclinical compounds, three were new findings and had not previously been reported. Of the remaining seven, five were in the laboratory stage (in vitro), one was entering the clinical trial stage (phase 2), and one was reported to be discovered (reported) with experiments under preparation.
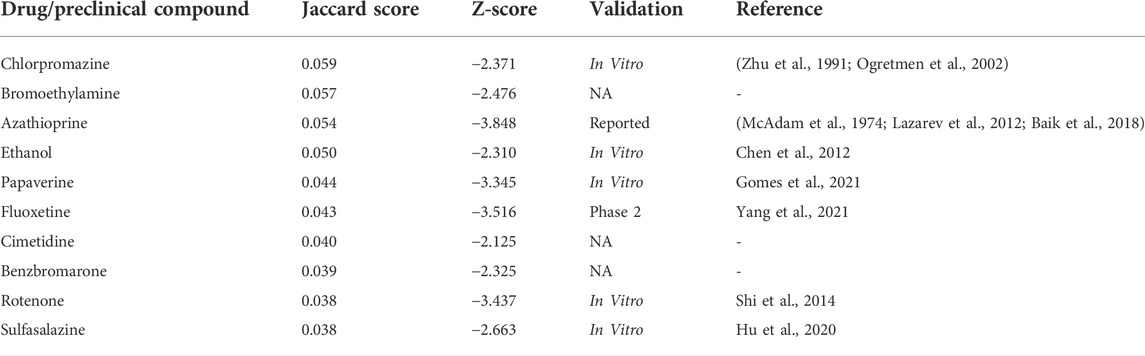
TABLE 2. Potential drugs or preclinical compounds for lung adenocarcinoma.
There were 47 binding target proteins from the three potential drugs predicted from this study, and 14 of them were identified from GAT and with an IC50 value less than 10,000 nM, which presents the physical interaction between drugs and targets (Table 3). The proteins and genes that were not found by GAT or IC50 larger than 10,000 nM were listed in (Supplementary Table S5).
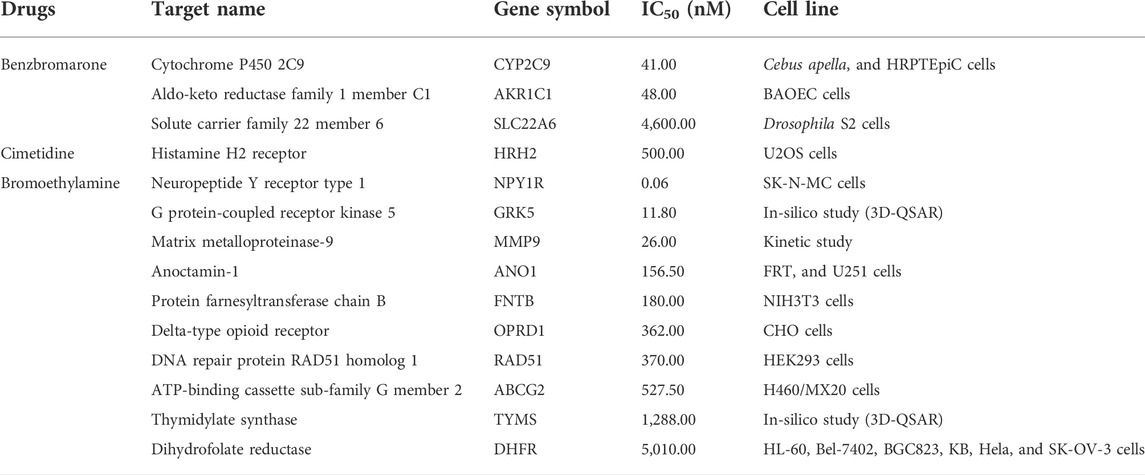
TABLE 3. Physical drug-target interaction pairs and IC50 values.
In this study, a LUAD-related target genes and drug prediction pipeline was developed. A GAT model was used for target gene prediction, and an overlap-based network and a network distance-based methods were used for LUAD drug prediction. Using this pipeline, together with FPKM data from TCGA, a gene interaction network, gene function annotation features, and drug target gene information, 1,597 potential LUAD-related target genes and 10 potential drugs or preclinical compounds for the treatment of LUAD were predicted. The AUROC of the GAT model developed in this study reached 0.89, which indicates the robust prediction of LUAD-related target genes.
Due to the absence of standard datasets of LUAD-related and -unrelated genes (Schulte-Sasse et al., 2021), we used a more stringent log2FC criterion than conventional studies. An absolute value of log2FC greater than 2 was required for a gene to be considered to be related to LUAD, while traditionally, an absolute value of log2FC greater than 1 is used as the threshold. A stricter threshold selection is beneficial to reduce the error of the sample data in the training set and thus achieve more accurate predictions. As few datasets reported genes unrelated to cancer, genes with an absolute value of log2FC close to 0 were classified as unrelated genes.
We also compared the prediction performance of the GAT model with the prediction performance of traditional ML and DL models. Compared with the TabNET model, the ML-based decision tree model had better prediction performance. There are two possible reasons for this. First, the training sample size and the number of features were small (Kotsiantis, 2007). There were only approximately 3,000 genes and less than 100 features in the training set. Within the scope of this analysis, ML-based decision trees had obvious advantages. Second, the TabNet model is not a GNN-based model, and therefore, it is not conducive to summarizing topological information from gene interaction networks. Compared with ML-based decision trees, deep learning models (GAT and GCN) based on GNN had better prediction performance. GNNs are applied to graphs in the non-Euclidean space. Non-Euclidean spaces represent more arbitrary spaces than Euclidean spaces due to arbitrary connections between nodes, thus allowing GNNs to obtain topological information more effectively. For feature extraction, the analytical ability of the GNNs was close to that of the ML-based decision tree. Both GCN and GAT are types of GNNs, and both methods aggregate the features of neighboring vertices to the central vertex and use the local stationary points on the graph to learn new vertex feature expressions. The difference is that GCN uses the Laplacian matrix, whereas GAT uses the attention coefficient. To a certain extent, GAT models are more precise as the correlation between vertex features is better incorporated into the model (Li et al., 2015). Due to the difference in the vertex operation mode, changing the structure of the graph in the test task has little impact on the GAT. In the training process of most graph neural networks, the training set is only a subgraph of the whole graph, and the testing set is a new subgraph for the model. GCN is a calculation method for the entire graph, and the node features of the entire graph are updated in one calculation, which makes the GCN model inefficient and poor in handling unknown subgraphs (Kipf and Welling, 2016). The learned parameters in GCNs are primarily related to the graph structure. On the other hand, for the GAT model, the new subgraph only affects the relationship matrix of nodes to feature mapping. The dimension of this matrix is determined at the beginning of the model establishment and is only related to the number of features of each node. Therefore, GATs are more effective than GCN at inductive tasks, such as supervised learning (Hamilton et al., 2017).
In the drug prediction module, overlap-based and network distance-based networks are commonly used prediction methods, and several drug prediction models have been built using similar theories (Tu, 1996; Huang et al., 2016; Li et al., 2020; Jain et al., 2021). As there are no data on which type of predictive model is more reliable, both predictive models were used in this study and then summarized to predict potential drugs for the treatment of LUAD.
Among the ten predicted drugs or preclinical compounds, three were newly discovered using this prediction pipeline. The remaining eight drugs have been previously identified as potential drugs for the treatment of LUAD, and some are already in the laboratory testing stage or clinical trial stage. Bromoethylamine is a new potential preclinical compound for LUAD treatment identified in this study (Jaccard score = 0.057, z-score = -2.476). An association between bromoethylamine and LUAD has not been reported in DrugBank, the CTD, or in previous studies. Notably, in the CTD, bromoethylamine is reported to interact with hepatitis A virus cellular receptor 1 and natriuretic peptide receptor 1, which coincide with the genes predicted in this study. Additionally, both genes are associated with fibrosis (Cherngwelling et al., 2021; Priego et al., 2021), which is an important phenotype that occurs throughout the progression of LUAD (Angel et al., 2020). Moreover, these two genes also show strong associations with kidney-related diseases (Kanki et al., 2014).
Bromoethylamine is used to synthesize anti-radon, a brain drug. Anti-radon has a central excitatory effect. It promotes the release of active sulfhydryl groups in the body and participates in the redox process of brain cells, thereby promoting and restoring the metabolism of brain cells to enable the rapid restoration of brain function in traumatic coma patients. In addition, there are anti-central nervous system depressant drugs that are clinically suitable for traumatic coma, coma caused by other reasons, the sequelae of traumatic brain injury, carbon monoxide poisoning, cerebral hypoxia, and sleeping pill poisoning (Sun and Li, 2008). Cimetidine (Jaccard score = 0.040, z-score = −2.125) was also predicted to be a potential treatment for LUAD. Although the use of cimetidine for the treatment of NSCLC has been assessed in clinical trials, it did not appear directly as a LUAD-related drug. However, it was used as an allergy prevention drug in a clinical trial of paclitaxel for the treatment of NSCLC (Hainsworth and Greco, 1994). Currently, cimetidine is used to treat gastroesophageal reflux disease, peptic ulcer disease, and dyspepsia. It is also being tested in clinical trials for the treatment of chronic obstructive pulmonary disease and community-acquired pneumonia. The target genes of cimetidine include Epidermal Growth Factor Receptor (EGFR) and Aryl Hydrocarbon Receptor (AHR), both of which are LUAD-related genes. Benzbromarone is another drug newly identified in this study as having potential for the treatment of LUAD (Jaccard score = 0.039, z-score = −2.235). Benzbromarone has been studied for its effects on the development of hepatoma and metabolic pathways of lipids, proteins, amino acids, and their derivatives (Calvisi et al., 2011). It has also been used in trials studying the basic science and treatment of heart failure, hyperuricemia, chronic kidney disease, abnormal renal function, gout, and asymptomatic hyperuricemia. Benzbromarone is currently being assessed in the clinical trials for the treatment of heart failure/hyperuricemia, type 2 diabetes, and pulmonary idiopathic arterial hypertension. No previous studies have linked benzbromarone with LUAD.
The seven remaining drugs have previously been reported to be LUAD-related or have entered clinical trials for LUAD treatment. For example, chlorpromazine, which was predicted in this study (Jaccard score = 0.059, z-score = −2.371), is a phenothiazine anti-psychotic used to treat nausea, vomiting, preoperative anxiety, schizophrenia, bipolar disorder, and severe behavioral problems in children. Chlorpromazine has been reported to modulate the metabolism of papillary lung adenocarcinoma cells by targeting c-Myc (Ciribilli et al., 2016). In addition, in cell line experiments, chlorpromazine has been shown to be effective at inhibiting the growth of lung tumor cell lines (the human small cell carcinoma-derived cell line IRSC-10M and the adenocarcinoma-derived cell line A549) (Zhu et al., 1991). Fluoxetine (Jaccard score = 0.043, z-score = −3.516) was originally developed as an anti-depressant in the selective serotonin reuptake inhibitor class (March et al., 2004). It is commonly used to treat depression and sometimes obsessive-compulsive disorder and bulimia (Husted et al., 2007). In addition, fluoxetine is also a promising drug for treating patients with depression and NSCLC (Yang et al., 2021). The use of this drug for the treatment of a variety of disorders may be related to the significant expression levels of LUAD-related genes in the nervous system (Hsu et al., 2020), which was also partially confirmed by the feature selection in this study. This finding of nervous system-related drugs, including selective serotonin reuptake inhibitors, being potentially suitable for treating lung cancer is not an independent event, as aducanumab, a drug for Alzheimer’s disease (Schneider, 2020), has also been experimentally tested for lung cancer and encouraging results were observed (Morrison, 2016).
Azathioprine has previously been reported to be used for treating patients with LUAD, but its effect and mechanism of action are unknown (McAdam et al., 1974; Lazarev et al., 2012; Baik et al., 2018). Ethanol has the potential to suppress CL1-5 human LUAD cell migration, and it inhibits matrix metalloproteinase-2/9 via the extracellular signal-regulated kinase, c-Jun N-terminal kinase, p38, and phosphatidylinositol 3-kinase/Akt signalling pathways (Chen et al., 2012). A clinical trial of ethanol for the treatment of LUAD is currently in the recruiting stage. Papaverine may be used to treat various types of smooth muscle spasms, including vasospasm and visceral spasm associated with acute myocardial infarction and angina pectoris. It is currently being assessed in a clinical trial for the treatment of NSCLC. Rotenone has the potential to sensitize NSCLC cell lines to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis (Shi et al., 2014), but it has not been assessed in a clinical trial, possibly because of its potent toxicity. Sulfasalazine is an anti-inflammatory drug used to treat Crohn’s disease and rheumatoid arthritis. Previous studies have shown that sulfasalazine abolishes the phosphorylation of AXL and other receptor tyrosine kinases, thereby reducing LUAD metastasis and drug resistance (Lay et al., 2007). No clinical trials using sulfasalazine for the treatment of LUAD have been conducted.
The present study has some limitations. The drug prediction pipeline is not able to distinguish between drugs that are used to relieve the symptoms of LUAD and those that eradicate LUAD. The reason is that the GAT model established in this study only screens out the genes related to LUAD, and it cannot determine whether the abnormal expression of genes occurs before or after the onset of LUAD. Therefore, it cannot be determined whether the predicted drug targets a gene involved in the development of LUAD, a gene that is abnormally expressed after the development of LUAD, or both. However, if LUAD-related oncogenes can be identified using statistical-based Mendelian randomization analysis or cellular or biological assays, potential drugs for the treatment of LUAD may be more targeted. In addition, the node attention coefficients in the GAT model of this study and the weights of edges between nodes may have potential implications for the prediction of new gene associations.
In this study, a GAT-based target gene prediction–drug repositioning pipeline was constructed for LUAD. Using this pipeline, 1,597 genes were predicted as potential target genes for LUAD, bromoethylamine was predicted as a novel potential compound for the treatment of LUAD, cimetidine and benzbromarone were predicted as potential therapeutic drugs for LUAD. In the future, it is a potential research direction to use the attention coefficients and edge weights obtained in the GAT model to predict new gene-gene interaction relationships. Introducing cell line experiments and animal experiments to verify the efficacy of treating LUAD with bromoethylamine, cimetidine, and benzbromarone will also be a key research direction.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by College Research Ethics Sub-committee, College of Engineering, City University of Hong Kong. The patients/participants provided their written informed consent to participate in this study.
RH: Conceptualization, Methodology, Data Curation, Software, Writing Original Draft, Editing; DS: Data Curation, Software, Writing, Review, and Editing; WC: Advice, Writing, Review and Editing, Supervision; TW: Validation; YD: Visualization; AB: Writing, Review and Editing; QH: Writing, Review, and Editing; JL: Writing Review and Editing; XH: Data Curation, Writing, Review and Editing, Supervision; KC: Conceptualization, Writing, Review and Editing, Supervision, Project administration.
This work was supported by the City University of Hong Kong New Research Initiatives/Infrastructure Support from Central (APRC; grant number 9610401). This funding body was not involved in the study design, data analysis, writing, or submission of the manuscript.
The authors would like to thank the following organizations for providing useful database in this study: The Cancer Genome Atlas for gene expression level; Gene Analytics and VarElect for gene features; Human Reference Interactome for gene-gene interactions; and PharmGKB, Binding Database, and DrugBank for drug target genes and drug database. This study might not be possible without the support from them.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.936758/full#supplementary-material
American Cancer Society (2021). American cancer society. E. coli [Online]. [Accessed]. Available: https://www.cancer.org/treatment.
Angel, P. M., Bruner, E., Bethard, J., Clift, C. L., Ball, L., Drake, R. R., et al. (2020). Extracellular matrix alterations in low-grade lung adenocarcinoma compared with normal lung tissue by imaging mass spectrometry. J. Mass Spectrom. 55, e4450. doi:10.1002/jms.4450
Angermueller, C., Parnamaa, T., Parts, L., and Stegle, O. (2016). Deep learning for computational biology. Mol. Syst. Biol. 12, 878. doi:10.15252/msb.20156651
Baik, K. W., Kim, S. H., and Shin, H. Y. (2018). Paraneoplastic neuromyelitis optica associated with lung adenocarcinoma in a young woman. J. Clin. Neurol. 14, 246–247. doi:10.3988/jcn.2018.14.2.246
Bass, J. I. F., Diallo, A., Nelson, J., Soto, J. M., Myers, C. L., and Walhout, A. J. M. (2013). Using networks to measure similarity between genes: Association index selection. Nat. Methods 10, 1169–1176. doi:10.1038/nmeth.2728
Ben-Ari Fuchs, S., Lieder, I., Stelzer, G., Mazor, Y., Buzhor, E., Kaplan, S., et al. (2016). GeneAnalytics: An integrative gene set analysis tool for next generation sequencing, RNAseq and microarray data. Omics 20, 139–151. doi:10.1089/omi.2015.0168
Calvisi, D. F., Wang, C., Ho, C., Ladu, S., Lee, S. A., Mattu, S., et al. (2011). Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 140, 1071–1083. doi:10.1053/j.gastro.2010.12.006
Chen, B., Gao, T., Yuan, W., Zhao, W., Wang, T.-H., and Wu, J. (2019). Prognostic value of survival of MicroRNAs signatures in non-small cell lung cancer. J. Cancer 10, 5793–5804. doi:10.7150/jca.30336
Chen, T., and Guestrin, C. (2016). “Xgboost: A scalable tree boosting system,” in Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining, 785–794.
Chen, Y.-W., Diamante, G., Ding, J., Nghiem, T. X., Yang, J., Ha, S.-M., et al. (2022). PharmOmics: A species- and tissue-specific drug signature database and gene-network-based drug repositioning tool. iScience 25, 104052. doi:10.1016/j.isci.2022.104052
Chen, Y. Y., Liu, F. C., Chou, P. Y., Chien, Y. C., Chang, W. S., Huang, G. J., et al. (2012). Ethanol extracts of fruiting bodies of Antrodia cinnamomea suppress CL1-5 human lung adenocarcinoma cells migration by inhibiting matrix metalloproteinase-2/9 through ERK, JNK, p38, and PI3K/Akt signaling pathways. Evid. Based. Complement. Altern. Med. 2012, 378415. doi:10.1155/2012/378415
Cheng, F. X., Lu, W. Q., Liu, C., Fang, J. S., Hou, Y., Handy, D. N. E., et al. (2019). A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun. 10, 3476. doi:10.1038/s41467-019-10744-6
Cherngwelling, R., Pengrattanachot, N., Swe, M. T., Thongnak, L., Promsan, S., Phengpol, N., et al. (2021). Agomelatine protects against obesity-induced renal injury by inhibiting endoplasmic reticulum stress/apoptosis pathway in rats. Toxicol. Appl. Pharmacol. 425, 115601. doi:10.1016/j.taap.2021.115601
Ching, T., Himmelstein, D. S., Beaulieu-Jones, B. K., Kalinin, A. A., Do, B. T., Way, G. P., et al. (2018). Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 15, 20170387. doi:10.1098/rsif.2017.0387
Cho, W. C. (2013). Targeting signaling pathways in lung cancer therapy. Expert Opin. Ther. Targets 17, 107–111. doi:10.1517/14728222.2013.729043
Ciribilli, Y., Singh, P., Inga, A., and Borlak, J. (2016). c-Myc targeted regulators of cell metabolism in a transgenic mouse model of papillary lung adenocarcinoma. Oncotarget 7, 65514–65539. doi:10.18632/oncotarget.11804
Davis, A. P., Grondin, C. J., Johnson, R. J., Sciaky, D., Wiegers, J., Wiegers, T. C., et al. (2020). Comparative Toxicogenomics database (CTD): Update 2021. Nucleic Acids Res. 49, D1138–D1143. doi:10.1093/nar/gkaa891
Freund, Y., Schapire, R., and Abe, N. (1999). A short introduction to boosting. Journal-Japanese Soc. Artif. Intell. 14, 1612.
Gomes, D. A., Joubert, A. M., and Visagie, M. H. (2021). In vitro effects of papaverine on cell proliferation, reactive oxygen species, and cell cycle progression in cancer cells. Molecules 26, 6388. doi:10.3390/molecules26216388
Hainsworth, J. D., and Greco, F. A. (1994). Paclitaxel administered by 1-hour infusion. Preliminary results of a phase I/II trial comparing two schedules. Cancer 74, 1377–1382. doi:10.1002/1097-0142(19940815)74:4<1377::aid-cncr2820740431>3.0.co;2-u
Hamilton, W. L., Ying, R., and Leskovec, J. (2017). Inductive representation learning on large graphs. Adv. Neural Inf. Process. Syst. 30, 30.2017
Ho, T. K. (1995). “Random decision forests,” in Proceedings of 3rd international conference on document analysis and recognition (IEEE), 278–282.
Hsu, L. C., Tu, H. F., Hsu, F. T., Yueh, P. F., and Chiang, I. T. (2020). Beneficial effect of fluoxetine on anti-tumor progression on hepatocellular carcinoma and non-small cell lung cancer bearing animal model. Biomed. Pharmacother. 126, 110054. doi:10.1016/j.biopha.2020.110054
Hu, K., Li, K., Lv, J., Feng, J., Chen, J., Wu, H., et al. (2020). Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Invest. 130, 1752–1766. doi:10.1172/JCI124049
Huang, C. H., Chang, P. M. H., Hsu, C. W., Huang, C. Y. F., and Ng, K. L. (2016). Drug repositioning for non-small cell lung cancer by using machine learning algorithms and topological graph theory. Bmc Bioinforma. 17, 2. doi:10.1186/s12859-015-0845-0
Huang Da, W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi:10.1038/nprot.2008.211
Husted, D. S., Shapira, N. A., Murphy, T. K., Mann, G. D., Ward, H. E., and Goodman, W. K. (2007). Effect of comorbid tics on a clinically meaningful response to 8-week open-label trial of fluoxetine in obsessive compulsive disorder. J. Psychiatr. Res. 41, 332–337. doi:10.1016/j.jpsychires.2006.05.007
Jain, A. S., Prasad, A., Pradeep, S., Dharmashekar, C., Achar, R. R., Ekaterina, S., et al. (2021). Corrigendum: Everything old is new again: Drug repurposing approach for non-small cell lung cancer targeting MAPK signaling pathway. Front. Oncol. 11, 822865. doi:10.3389/fonc.2021.822865
Janku, F., Stewart, D. J., and Kurzrock, R. (2010). Targeted therapy in non-small-cell lung cancer--is it becoming a reality? Nat. Rev. Clin. Oncol. 7, 401–414. doi:10.1038/nrclinonc.2010.64
Kang, J., Tang, Q., He, J., Li, L., Yang, N., Yu, S., et al. (2022). RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. 50, D326–D332. doi:10.1093/nar/gkab997
Kanki, M., Moriguchi, A., Sasaki, D., Mitori, H., Yamada, A., Unami, A., et al. (2014). Identification of urinary miRNA biomarkers for detecting cisplatin-induced proximal tubular injury in rats. Toxicology 324, 158–168. doi:10.1016/j.tox.2014.05.004
Kenneth, K. W. T., and Cho, W. C. S. (2022). Drug repurposing for cancer therapy in the era of precision medicine. Curr. Mol. Pharmacol. 15, 895–903. doi:10.2174/1874467215666220214104530
Kipf, T. N., and Welling, M. (2016). Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907.
Kotsiantis, S. B. (2007). Supervised machine learning: A Review of classification techniques. Emerg. Artif. Intell. Appl. Comput. Eng. 160, 3–24.
Lay, J. D., Hong, C. C., Huang, J. S., Yang, Y. Y., Pao, C. Y., Liu, C. H., et al. (2007). Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res. 67, 3878–3887. doi:10.1158/0008-5472.CAN-06-3191
Lazarev, I., Sion-Vardy, N., and Ariad, S. (2012). EML4-ALK-positive non-small cell lung cancer in a patient treated with azathioprine for ulcerative colitis. Tumori 98, e98–101. doi:10.1700/1146.12652
Leskovec, J., and Sosič, R. (2016). Snap: A general-purpose network analysis and graph-mining library. ACM Trans. Intell. Syst. Technol. 8, 1–20. doi:10.1145/2898361
Li, B. R., Dai, C., Wang, L. J., Deng, H. L., Li, Y. Y., Guan, Z., et al. (2020). A novel drug repurposing approach for non-small cell lung cancer using deep learning. Plos One 15, e0233112. doi:10.1371/journal.pone.0233112
Li, Y., Tarlow, D., Brockschmidt, M., and Zemel, R. (2015). Gated graph sequence neural networks. arXiv preprint arXiv:1511.05493.
Liu, T., Lin, Y., Wen, X., Jorissen, R. N., and Gilson, M. K. (2007). BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 35, D198–D201. doi:10.1093/nar/gkl999
Luck, K., Kim, D.-K., Lambourne, L., Spirohn, K., Begg, B. E., Bian, W., et al. (2020). A reference map of the human binary protein interactome. Nature 580, 402–408. doi:10.1038/s41586-020-2188-x
March, J., Silva, S., Petrycki, S., Curry, J., Wells, K., Fairbank, J., et al. (2004). Fluoxetine, cognitive-behavioral therapy, and their combination for adolescents with depression - treatment for adolescents with depression study (TADS) randomized controlled trial. Jama-Journal Am. Med. Assoc. 292, 807–820. doi:10.1001/jama.292.7.807
Mcadam, L., Paulus, H. E., and Peter, J. B. (1974). Adenocarcinoma of the lung during azathioprine therapy. Arthritis Rheum. 17, 92–94. doi:10.1002/art.1780170114
Morrison, C. (2016). Hope for anti-amyloid antibodies surges, yet again. Nat. Biotechnol. 34, 1082–1083. doi:10.1038/nbt1116-1082b
Ogretmen, B., Pettus, B. J., Rossi, M. J., Wood, R., Usta, J., Szulc, Z., et al. (2002). Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. Role for endogenous ceramide in mediating the action of exogenous ceramide. J. Biol. Chem. 277, 12960–12969. doi:10.1074/jbc.M110699200
Pao, W., and Chmielecki, J. (2010). Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 10, 760–774. doi:10.1038/nrc2947
Paul, S. M., Mytelka, D. S., Dunwiddie, C. T., Persinger, C. C., Munos, B. H., Lindborg, S. R., et al. (2010). How to improve R&D productivity: The pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov. 9, 203–214. doi:10.1038/nrd3078
Priego, A. R., Parra, E. G., Mas, S., Morgado-Pascual, J. L., Ruiz-Ortega, M., and Rayego-Mateos, S. (2021). Bisphenol A modulates autophagy and exacerbates chronic kidney damage in mice. Int. J. Mol. Sci. 22, 7189. doi:10.3390/ijms22137189
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. doi:10.1093/nar/gkv007
Rousseeuw, P. J. (1987). Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65. doi:10.1016/0377-0427(87)90125-7
Schneider, L. (2020). A resurrection of aducanumab for Alzheimer's disease. Lancet. Neurol. 19, 111–112. doi:10.1016/S1474-4422(19)30480-6
Schulte-Sasse, R., Budach, S., Hnisz, D., and Marsico, A. (2021). Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 3, 513–526. doi:10.1038/s42256-021-00325-y
Sertkaya, A., Wong, H. H., Jessup, A., and Beleche, T. (2016). Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials 13, 117–126. doi:10.1177/1740774515625964
Sherman, B. T., Hao, M., Qiu, J., Jiao, X., Baseler, M. W., Lane, H. C., et al. (2022). David: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. doi:10.1093/nar/gkac194
Shi, Y. L., Feng, S., Chen, W., Hua, Z. C., Bian, J. J., and Yin, W. (2014). Mitochondrial inhibitor sensitizes non-small-cell lung carcinoma cells to TRAIL-induced apoptosis by reactive oxygen species and Bcl-X(L)/p53-mediated amplification mechanisms. Cell Death Dis. 5, e1579. doi:10.1038/cddis.2014.547
Stark, C., Breitkreutz, B.-J., Reguly, T., Boucher, L., Breitkreutz, A., and Tyers, M. (2006). BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539. doi:10.1093/nar/gkj109
Sun, Q. H., and Li, H. F. (2008). Observation on the effect of Yadongke cerebral maze in the treatment of neurological combined neuroencephalitis in children. Chin. J. Pract. Pediatr. 23, 3.
Tu, J. V. (1996). Advantages and disadvantages of using artificial neural networks versus logistic regression for predicting medical outcomes. J. Clin. Epidemiol. 49, 1225–1231. doi:10.1016/s0895-4356(96)00002-9
Cancer Genome Atlas Research Network Weinstein, J. N., Collisson, E. A., Mills, G. B., Shaw, K. R. M., Ozenberger, B. A., Ellrott, K., et al. (2013). The cancer genome Atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. doi:10.1038/ng.2764
Whirl-Carrillo, M. A.-O., Huddart, R. A.-O., Gong, L., Sangkuhl, K. A.-O., Thorn, C. F., Whaley, R. A.-O., et al. (2021). An evidence-based framework for evaluating pharmacogenomics knowledge for personalized medicine.
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–d1082. doi:10.1093/nar/gkx1037
World Health Organization (2021). World Health organization. E. coli [Online]. [Accessed]. Available: https://www.who.int/news-room/fact-sheets/detail/cancer.
Yang, Z., Li, Z. M., Guo, Z. J., Ren, Y., Zhou, T., Xiao, Z. J., et al. (2021). Antitumor effect of fluoxetine on chronic stress-promoted lung cancer growth via suppressing kynurenine pathway and enhancing cellular immunity. Front. Pharmacol. 12, 685898. doi:10.3389/fphar.2021.685898
Zappa, C., and Mousa, S. A. (2016). Non-small cell lung cancer: current treatment and future advances. Transl. Lung Cancer Res. 5, 288–300. doi:10.21037/tlcr.2016.06.07
Keywords: lung adenocarcinoma, drug repositioning, gene prediction, graph attention networks, machine learning, deep learning
Citation: Huang RX, Siriwanna D, Cho WC, Wan TK, Du YR, Bennett AN, He QE, Liu JD, Huang XT and Chan KHK (2022) Lung adenocarcinoma-related target gene prediction and drug repositioning. Front. Pharmacol. 13:936758. doi: 10.3389/fphar.2022.936758
Received: 05 May 2022; Accepted: 01 August 2022;
Published: 23 August 2022.
Edited by:
Yuhei Nishimura, Mie University, JapanReviewed by:
Kazim Yalcin Arga, Marmara University, TurkeyCopyright © 2022 Huang, Siriwanna, Cho, Wan, Du, Bennett, He, Liu, Huang and Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kei Hang Katie Chan, a2toY2hhbkBjaXR5dS5lZHUuaGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.