
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 23 August 2022
Sec. Renal Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.935937
This article is part of the Research TopicApplications of Herbal Medicine to Control Chronic Kidney Disease: Volume IIView all 30 articles
 Mengying Yao1,2†
Mengying Yao1,2† Shaozong Qin2†
Shaozong Qin2† Jiachuan Xiong2
Jiachuan Xiong2 Wang Xin2Xu Guan2Shuiqin Gong2Jing Chen2Yong Liu2Bo Zhang2
Wang Xin2Xu Guan2Shuiqin Gong2Jing Chen2Yong Liu2Bo Zhang2 Jinghong Zhao2*
Jinghong Zhao2* Yinghui Huang2,*
Yinghui Huang2,*Background: Acute kidney injury (AKI) occurs in approximately 7–18% of all hospitalizations, but there are currently no effective drug therapy for preventing AKI or delaying its progression to chronic kidney disease (CKD). Recent studies have shown that Scutellaria baicalensis, a traditional Chinese herb, could attenuate cisplatin-induced AKI, although the mechanism remains elusive. Further, it is unknown whether its major active component, Oroxylin A (OA), can alleviate kidney injury.
Methods: The therapeutic effect of OA was evaluated by using ischemia-reperfusion (IR) and cisplatin mediated-AKI mice and HK-2 cells under hypoxia-reoxygenation (HR) conditions. HE staining, transmission electron microscopy, flow cytometry, immunofluorescence, qPCR, Western blot, PPARα inhibitor, BNIP3 siRNA and ChIP assay were used to explore the role and mechanism of OA in AKI.
Results: OA ameliorated tubular damage and dramatically decreased serum creatinine (Scr) and urea nitrogen (BUN), and the expressions of renal injury markers (Kim-1, Ngal) in AKI mice induced by both IR injury and cisplatin, as well as attenuating AKI-to-CKD transition. In vitro experiments showed that OA alleviated HR-induced mitochondrial homeostasis imbalance in renal tubular epithelial cells. Mechanistically, OA dose-dependently induced the expression of Bcl-2/adenovirus E1B 19-kDa interacting protein (BNIP3), while knockdown of BNIP3 expression reversed the protection of OA against HR-mediated mitochondrial injury. Network pharmacological analysis and experimental validation suggested that OA enhanced BNIP3 expression via upregulating the expression of peroxisome proliferator activated receptor alpha (PPARα), which induced the transcription of BNIP3 via directly binding to its promoter region. Both in vitro and in vivo experiments confirmed that the renoprotective effect of OA was dramatically reduced by GW6471, a PPARα antagonist.
Conclusion: Our findings revealed that OA ameliorates AKI-to-CKD transition by maintaining mitochondrial homeostasis through inducing PPARα-BNIP3 signaling pathway, indicating that OA may serve as a candidate therapeutic strategy for alleviating AKI and CKD.
Acute kidney injury (AKI) is a common critical illness, characterized by a sharp decrease of glomerular filtration rate, accompanied by retention of nitrogen products (such as creatinine and urea nitrogen) (Bellomo et al., 2012; Levey and James, 2017; Ronco et al., 2019). AKI is also one of the significant risk factors for progressive chronic kidney disease (CKD) and end-stage renal disease (ESRD) (Chawla et al., 2014; Kramann et al., 2014; Chen et al., 2019). Although the kidney can gradually recover from AKI through regenerative capacity and repair, accumulating evidence suggests that there is a high risk of developing CKD after AKI (Bucaloiu et al., 2012; Coca et al., 2012; Wu et al., 2014). Yet, no effective drug is clinically applicable for AKI or AKI-to-CKD transition. Therefore, novel therapeutic strategies to alleviate AKI are highly desired.
Traditional Chinese medicine reveals that chronic renal failure is located in the spleen and kidney, and the pathogenesis can be summarized as deficiency of the yang qi of spleen, while kidney is the cause and the internal stagnation of dampness, turbidity, stasis and toxin is the symptom (Liu et al., 2014). Recently, traditional Chinese medicine suggests that herbal medicines that are capable of invigorating the spleen and kidney, promoting blood circulation, and removing turbidity may be effective in the treatment of CKD and renal fibrosis (Zhao et al., 2022). Scutellaria baicalensis Georgi (SB), a traditional Chinese medicine, has been prescribed to treat diseases in China for thousands of years, such as inflammation (Guan et al., 2020) and fibrosis (Kong et al., 2011; Pan et al., 2012), even though the mechanism remains elusive. SB can alleviate chemotherapy-induced AKI and protect against CKD (Huang et al., 2019; Guan et al., 2020). Simultaneously, baicalenin (a component of SB) can protect the kidney injury induced by renal ischemia-reperfusion injury (IRI) (Lin et al., 2014) or myocardial IR (Lai et al., 2016). Oroxylin A (OA), the main active component of SB, can alleviate lipopolysaccharide (LPS)-mediated acute injury of liver and lung (Tseng et al., 2012; Huang et al., 2015). However, the role and mechanism of OA in the process of AKI-to-CKD transition remain unclear.
Given the function of the kidney to remove waste from the blood by filtration and regulate fluid and electrolyte balance, it is destined to consume a large amount of energy, which is mainly provided by mitochondrial fatty acid β-oxidation (FAO) (Bhargava and Schnellmann, 2017; Sun et al., 2019). Mitochondrial energy metabolism is one of the important components of mitochondrial homeostasis, and maintaining mitochondrial homeostasis will benefit for the restoration of renal function (Emma et al., 2016; Bhargava and Schnellmann, 2017; Sun et al., 2019; Tang et al., 2021). Mitochondrial damage in proximal tubule cells has gradually evolved as a feature of various forms of AKI (Parikh et al., 2015; Sun et al., 2019). Accumulating evidences, including ours, suggested mitochondrial injury as a major contributor to acute and chronic kidney disease (Huang et al., 2020a; Huang et al., 2020b; Yu et al., 2020; Huang et al., 2022). Therefore, exploring therapeutic strategies aimed at restoring mitochondrial homeostasis may hold great potential for improving renal function (Emma et al., 2016; Bhargava and Schnellmann, 2017; Sun et al., 2019; Tang et al., 2021).
In the present study, based on the effect of OA in alleviating acute liver injury and acute lung injury (Tseng et al., 2012; Huang et al., 2015), the therapeutic potential of OA in AKI was evaluated and its molecular mechanism was explored. Our findings suggest that OA maintains mitochondrial homeostasis via inducing peroxisome proliferator activated receptor alpha (PPARα)-Bcl-2/adenovirus E1B 19-kDa interacting protein (BNIP3) signaling pathway, thereby alleviating AKI and its progression to CKD.
Additional details for all methods are provided in the Supplementary Methods.
Oroxylin A (OA) and Cisplatin were obtained from MedChemExpress (MCE, Monmouth Junction, NJ, United States). GW6471 (PPARα antagonist) and WY-14643 (PPARα agonist) were bought from Selleckchem (Houston, TX, United States).
Male C57BL/6J mice (8-week-old) were purchased from Chongqing Tengxin Bioscience (Chongqing, China). The construction of IRI-induced AKI mouse model was previously described (Hu et al., 2017).
For the hypoxia-reoxygenation (HR) model, HK-2 cells were incubated in a hypoxic incubator at 1% O2 for 24 h, and then the cells were put back into the normal incubator and reoxygenated for 6 h. After treatment with OA, the cells were harvested for the subsequent analysis.
The collected HK-2 cells and mouse kidney tissue were immersed in a transmission electron microscopy solution containing 2.5% glutaraldehyde and 4% paraformaldehyde, and the samples were treated in the transmission electron microscopy room of the Central Laboratory of Army Medical University. The microstructure of mitochondria was observed using TEM (JEM-1400PLUS, Japan).
All data were presented as mean ± SEM. Unpaired t test or one-way analysis of variance (ANOVA) with Tukey’s test were used for statistical analysis. Statistical significance was defined as P < 0.05.
The chemical formula of OA was shown in Figure 1A. To determine the therapeutic effect of OA, OA (20 mg/kg) was intravenously injected into sham and AKI mice. HE staining showed that OA improved renal damage in AKI mice (Figures 1B,C). In AKI mice, the levels of Scr, BUN and the expressions of kidney injury markers (Kidney injury molecule-1, Kim-1; and Neutrophil gelatinase-associated lipocalin, Ngal) and apoptosis-related proteins, including B-cell lymphoma-2 (Bcl-2) and Bcl-2-associated x (Bax), were markedly elevated, which were significantly ameliorated by OA treatment (Figures 1D–H). Meanwhile, we also found that OA alleviated cisplatin-induced renal tubular damage, increased levels of Scr and BUN, and upregulated expressions of Kim-1 and Ngal (Supplementary Figures 1A–E). These data suggest that OA can ameliorate IR and Cisplatin-induced AKI.
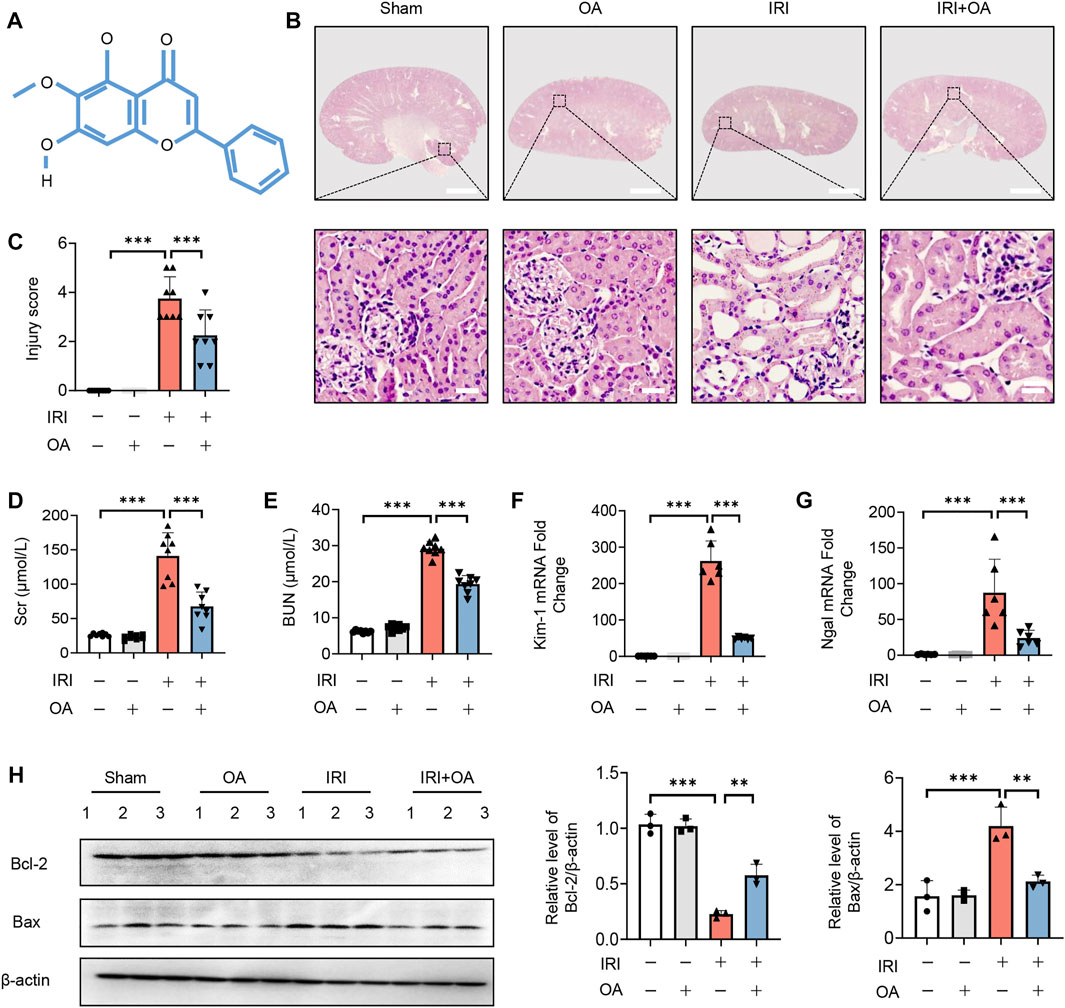
FIGURE 1. OA alleviates IRI-induced AKI in vivo. (A) The chemical structure of OA. (B) Representative micrographs of HE staining of kidney sections from sham and IRI-induced AKI mice intravenously injected with control or OA. Scale bars, 1.25 mm (top) and 50 μm (bottom). (C) Scoring of injury score according to HE staining of kidney sections from mice in (A) (n = 8). Effects of OA on Scr (D) and BUN (E) of mice in (A) (n = 8). The mRNA levels of Kim-1 (F) and Ngal (G) were analyzed by qPCR (n = 8). (H) The expressions of Bcl-2 and Bax in kidney tissues from mice in (A) were analyzed by Western blot (n = 3). Data are expressed as means ± SEM. ∗∗P < 0.01, ∗∗∗P < 0.001.
Subsequently, we analyzed the kidney tissues by TEM observation, and found that mitochondrial morphological damage, including mitochondrial swelling and vacuolation with disorganized or fragmented cristae, and the accumulation of lipid droplets in AKI mice were significantly improved after OA treatment (Figure 2A). Meanwhile, the decreased expressions of mitochondrial oxidative phosphorylation (OXPHO)-related genes (synthase H+ transporting mitochondrial F1 complex alpha subunit 1, Atp5a1; NADH ubiquinone oxidoreductase alpha subunit, Ndufa; NADH ubiquinone oxidoreductase flavoprotein, Ndufv and Cytochrome c, Cytc) and fatty acid oxidation (FAO)-related genes (Carnitine palmityl transferase 1b, Cpt1b; Long Chain Acyl Coenzyme A Synthetase 1, ACSL-1; O-octanoyltransferase, CROT; Acyl Coenzyme A Dehydrogenase Medium chain, ACADM) and lipid deposition in AKI mice were also restored by OA (Supplementary Figures 2A–C). Further, CCK-8 assays demonstrated that OA did not inhibit the viability of HK-2 cells, when its concentration was less than 100 μM (Figure 2B). To verify the effect of OA in vitro, HK-2 cells were incubated with OA (10 μM) after HR treatment. It was found that OA could alleviate mitochondrial damage, as evidenced by reducing apoptosis and ROS production, as well as restoring mitochondrial membrane potential, ATP level and the expression levels of OXPHO- and FAO-related gene (Figures 2C–I). Further, OA attenuated HR-mediated disorder of mitochondrial biogenesis, as evidenced by downregulated transcription factor A, mitochondrial (TFAM) expression and reduced mtDNA copy number (Figures 2J,K). These findings reveal that OA improves HR-induced mitochondrial morphological and functional damage.
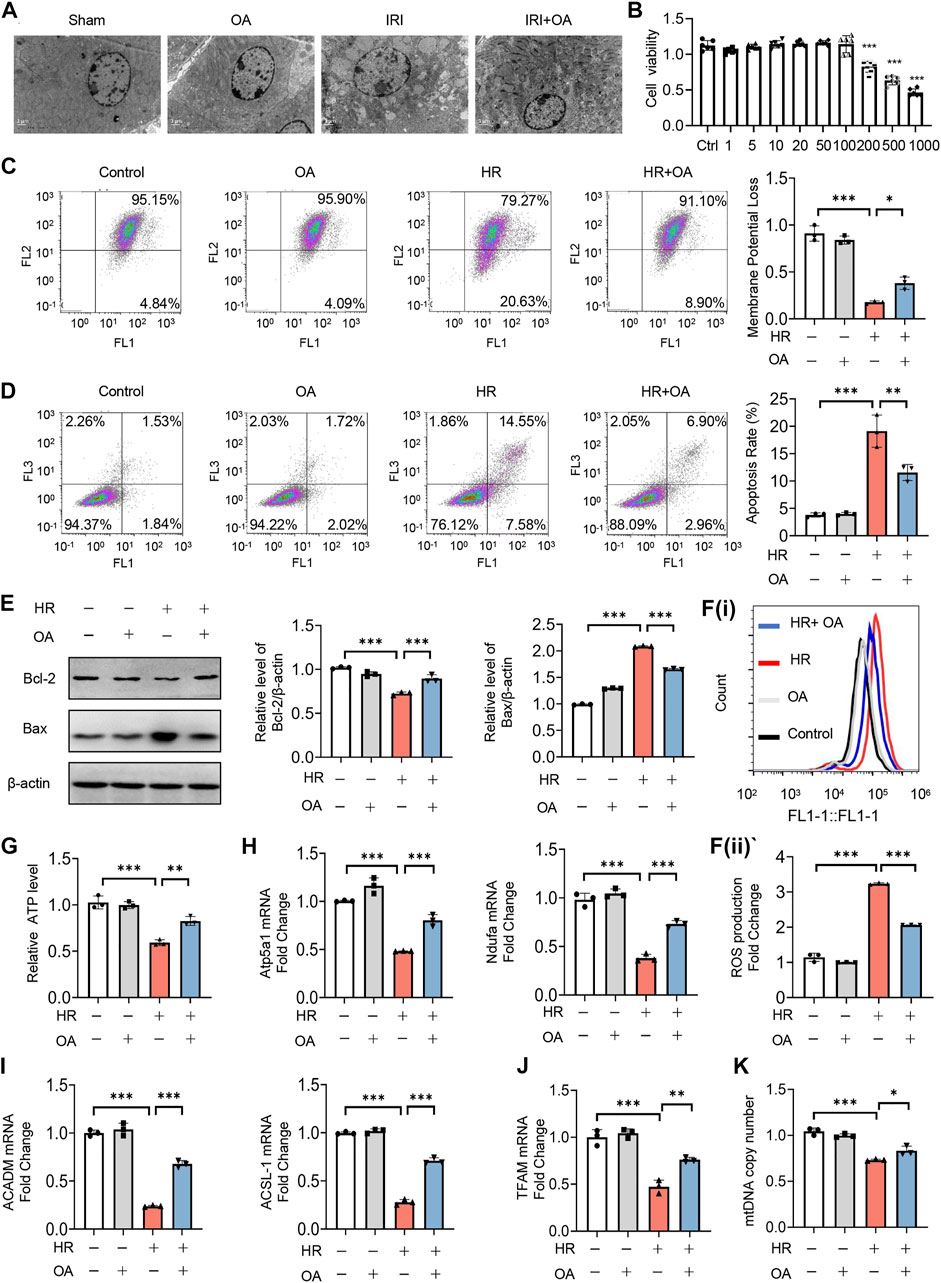
FIGURE 2. OA ameliorates hypoxia-reoxygenation (HR)-mediated mitochondrial homeostasis imbalance in vitro. (A) Representative TEM images of kidney tissues from sham and AKI mice treated with control or OA (20 mg/kg) for 24 h. Scale bar, 1 μm. (B) Cell viability of HK-2 cells incubated with indicated concentrations of OA for 48 h was determined using CCK-8. (C–I) HK-2 cells under HR exposure were treated with control or OA (10 μM), then the mitochondrial membrane potential (C), apoptosis (D,E), ROS (F), ATP (G), the expressions of Atp5a1, Ndufa, ACADM and ACSL-1 (H,I) were analyzed (n = 3). (J,K) HK-2 cells under HR exposure were treated with control or OA (10 μM), the expression of TFAM (J) and relative mitochondrial DNA copy number (K) were analyzed by qPCR (n = 3). Data are expressed as means ± SEM. ns: no significance. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Since recent studies reported that BNIP3 was essential for mitochondrial quality control and bioenergetics (Choi et al., 2016), we evaluated its role in OA-mediated effects. We found that OA dose-dependently induced BNIP3 expression at both transcriptional and translational level (Figures 3A,B). Simultaneously, BNIP3 expression was further enhanced in both OA-treated cells exposed to HR and kidney tissues from AKI mice (Figures 3C–F).
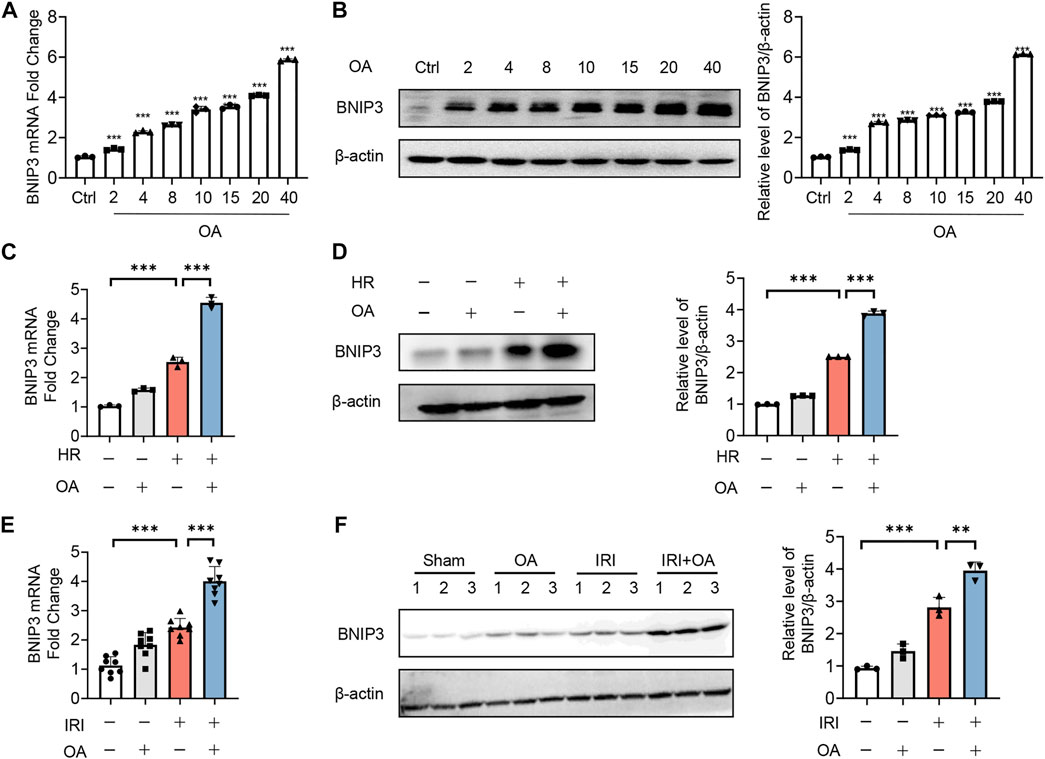
FIGURE 3. OA enhances BNIP3 expression in vitro and in vivo. (A,B) qPCR (A) and Western blot (B) analysis of the expression of BNIP3 in HK-2 cells treated with OA for 24 h (n = 3). (C,D) The mRNA and protein expression of BNIP3 in HK-2 cells exposed to HR followed by incubation with control or OA were analyzed by qPCR (C) and Western blot (D) (n = 3). (E,F) qPCR (E, n = 8) and Western blot (F, n = 3) analysis of the expression of BNIP3 in kidney tissues from sham and AKI mice treated with control or OA. Data are expressed as means ± SEM. ∗∗P < 0.01, ∗∗∗P < 0.001.
To verify whether OA maintains mitochondrial homeostasis by inducing BNIP3 expression, we first transfected siRNAs against BNIP3 (siBNIP3) into HK-2 cells, and then treated the cells with HR model (Figures 4A,B). We found that OA cannot improve mitochondrial injury when BNIP3 was silenced, as evidenced by increased ROS production and apoptosis, decreased ATP levels, and downregulated expressions of OXPHO-related gene Atp5a1 and FAO-related gene ACADM (Figures 4C–I). As BNIP3 is well known for mitophagy induction that can protect against AKI (Fu et al., 2020), we also investigated the role of mitophagy after OA treatment. We treated HK-2 cells exposed to normoxia or HR injury with control or OA. However, the Western blot assay and immunofluorescence observations showed that OA neither induced mitophagy under normoxia conditions nor aggravated HR-mediated mitophagy (Supplementary Figures 3A–C). Further, OA could not regulate mitophagy in AKI mice either (Supplementary Figure 3D). These results collectively indicate that OA maintains mitochondrial homeostasis through inducing the expression of BNIP3 without regulating mitophagy in renal tubular epithelial cells.
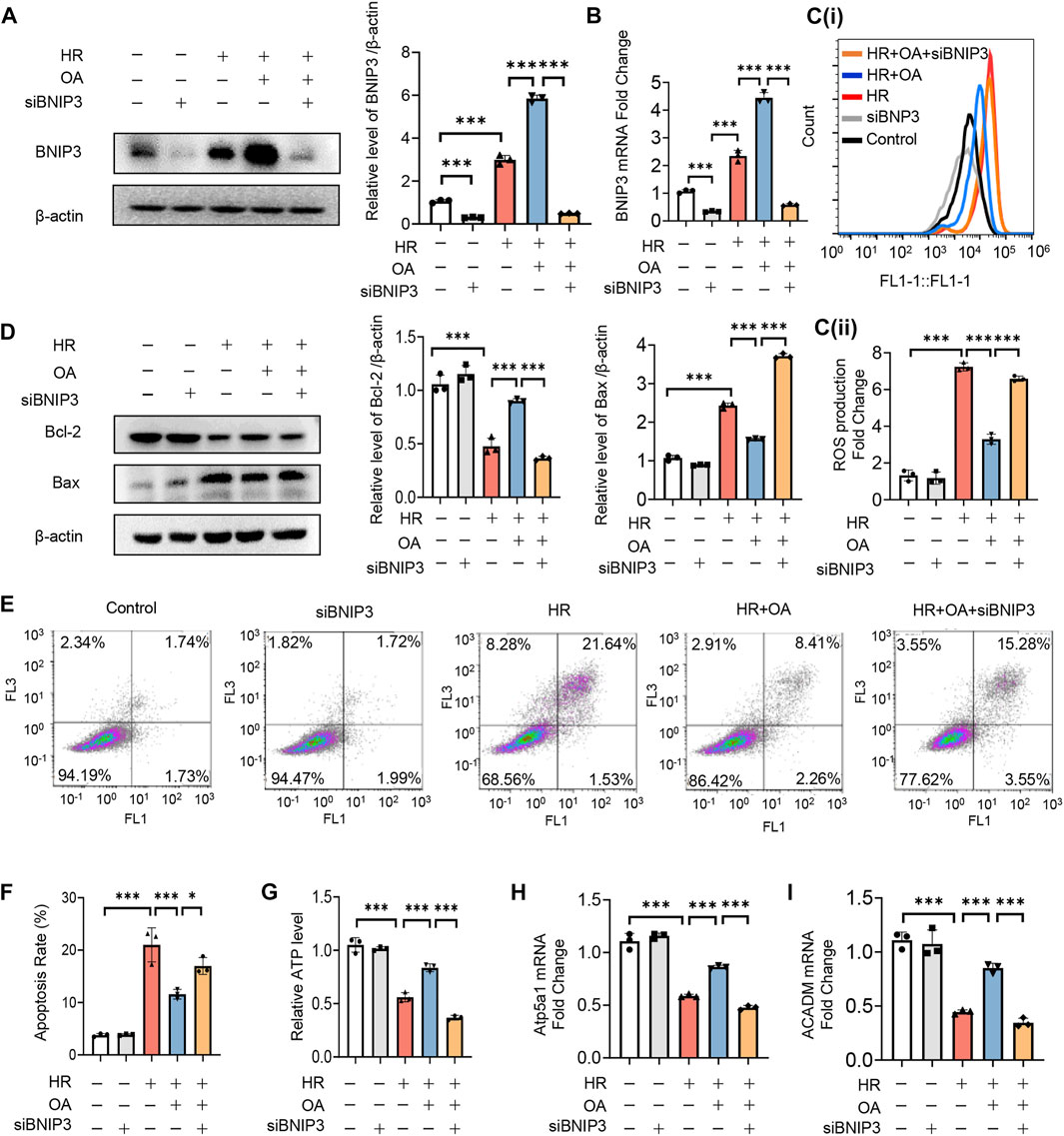
FIGURE 4. OA improves mitochondrial homeostasis imbalance through inducing BNIP3 expression. (A–I) After 24 h transfection with siBNIP3, HK-2 cells were exposed to HR and treated with control or OA. The protein expression level of BNIP3 (A), the levels of BNIP3 mRNA (B), ROS (C), apoptosis (D–F), ATP (G), the expression levels of OXPHO-related gene (H) and FAO-related gene (I) were detected (n = 3). Data are expressed as means ± SEM. ∗P < 0.05, ∗∗∗P < 0.001.
To investigate the upstream transcription factors of BNIP3, JASPAR, Genecard and SwissTarget prediction (network pharmacology analysis) were used, which predicted three potential transcription factors, including PPARα, signal transducer and transcription 3 (STAT3) and hypoxia-inducible factor 1 activator of subunit alpha (HIF-1α) (Figure 5A). After screening, we found that only the expression of PPARα was restored by OA in HK-2 cells exposed to HR and kidney tissues from AKI mice (Figures 5B–D). The expressions of PPARα and BNIP3 were also enhanced by a PPARα agonist, WY14163 (Figure 5E). Further, ChIP assay demonstrated that PPARα could induce BNIP3 transcription via directly binding to its promoter region (Figures 5F,G).
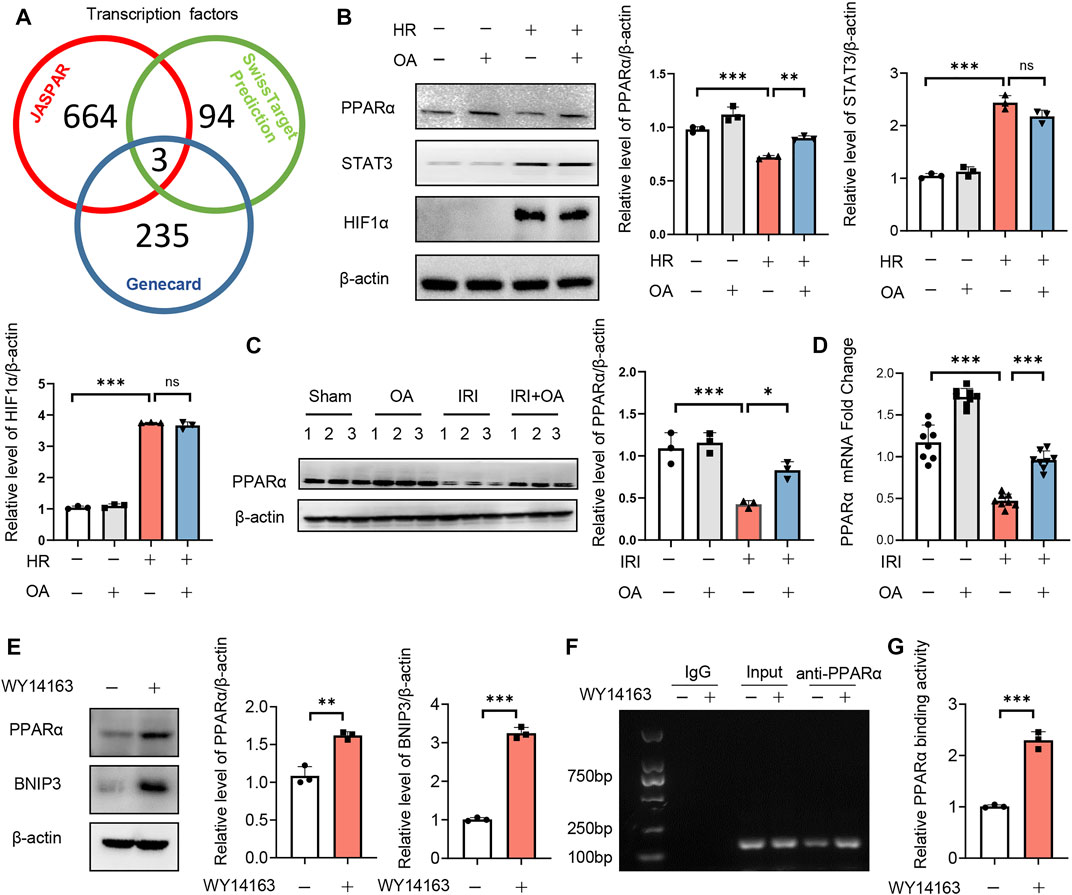
FIGURE 5. Transcription factor PPARα induces BNIP3 expression via directly binding to its promoter region. (A) JASPAR, Genecard and SwissTarget (network pharmacology analysis) were employed to predict the upstream transcription factors of BNIP3, and three potential transcription factors were obtained, including PPARα, STAT3 and HIF-1α. (B) Expression of PPARα, STAT3, and HIF-1α in HK-2 cells incubated with control or OA after HR treatment (n = 3). (C,D) Expression of PPARα in kidney tissue from sham and AKI mice treated with control or OA (n = 3–8). (E) Expressions of PPARα and BNIP3 in HK-2 cells treated with Control or PPARα agonist (n = 3). (F,G) The binding ability of PPARα to BNIP3 promoter region was assayed by ChIP (n = 3). Data are expressed as means ± SEM. ns: no significance. ∗∗P < 0.01, ∗∗∗P < 0.001.
To further verify the role of PPARα, we treated HK-2 cells under normal or HR injury with OA in combined with or without GW6471, a PPARα antagonist. We found that OA cannot upregulate the expressions of PPARα and BNIP3 after the co-incubation with GW6471 (Figure 6A). Meanwhile, OA did not alleviate mitochondrial damage when PPARα expression was inhibited, as evidenced by elevated ROS production and apoptosis, repressed mitochondrial membrane potential and ATP levels, and suppressed expressions of Atp5a1 and ACADM (Figures 6B–H). These results collectively suggest that OA restores mitochondrial homeostasis through enhancing PPARα-BNIP3 signaling pathway.
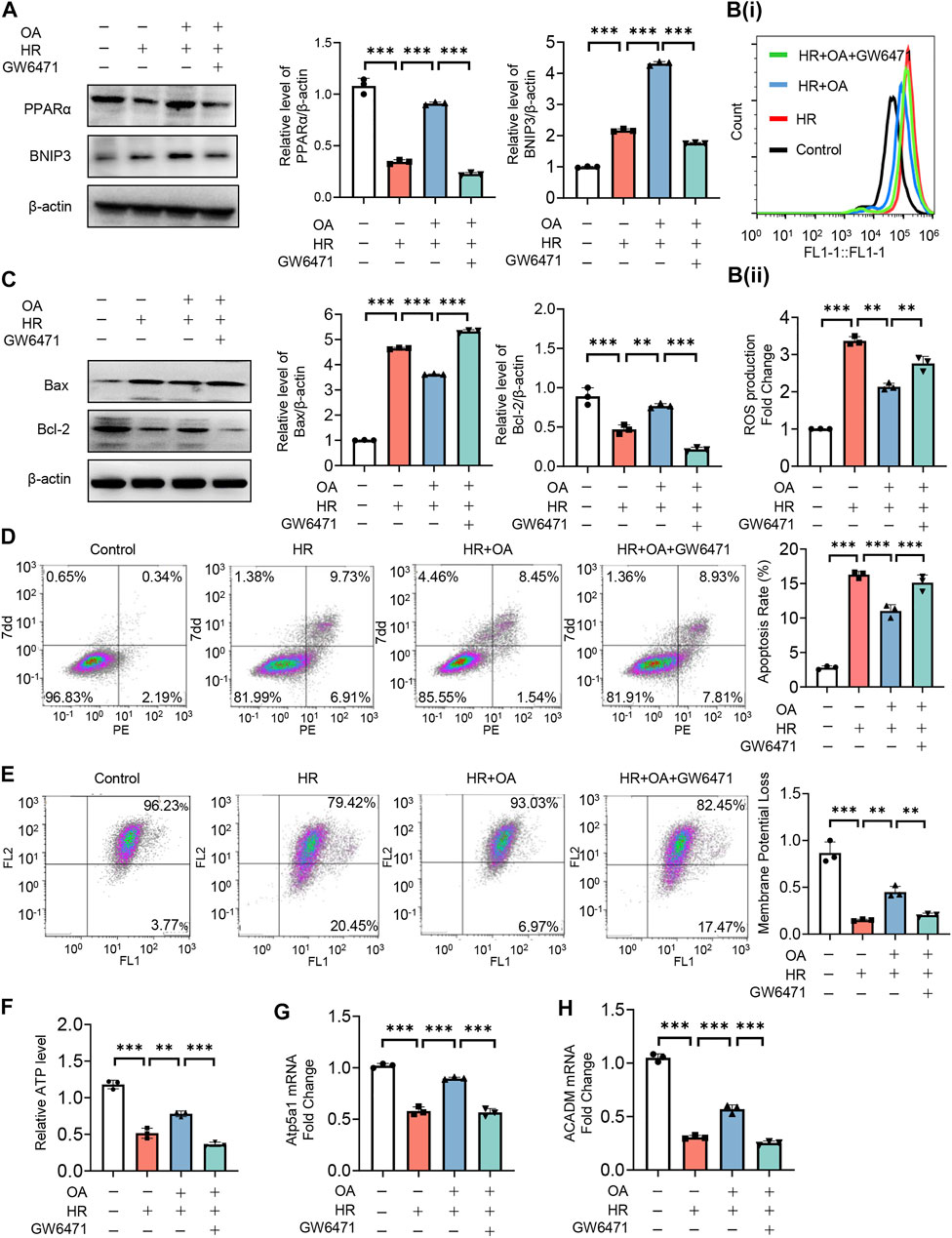
FIGURE 6. OA alleviates mitochondrial homeostasis imbalance by enhancing PPARα-BNIP3 signaling pathway. (A–H) HK-2 cells were treated with OA and control or GW6471 (PPARα antagonist) under normal or HR injury conditions, and then the levels of PPARα and BNIP3 expressions (A), ROS (B), apoptosis (C,D), mitochondrial membrane potential (E), ATP (F), OXPHO-related (G) and FAO-related gene expressions (H) were determined (n = 3). Data are expressed as means ± SEM. ∗∗P < 0.01, ∗∗∗P < 0.001.
To analyze the in vivo effect of PPARα, OA-treated AKI mice were injected with control or GW6471. HE staining exhibited that OA did not ameliorate renal tubular injury in AKI mice, when PPARα expression was inhibited (Figures 7A,B). Consistent with the histological findings, OA could not restore the serum levels of in Scr, BUN, the expressions of kidney injury markers (Kim-1 and Ngal), apoptosis-related genes (Bax and Bcl-2), OXPHO-related genes (Atp5a1 and Ndufa) and FAO-related genes (ACSL-1 and ACADM), or BNIP3 in AKI mice (Figures 7C–J).
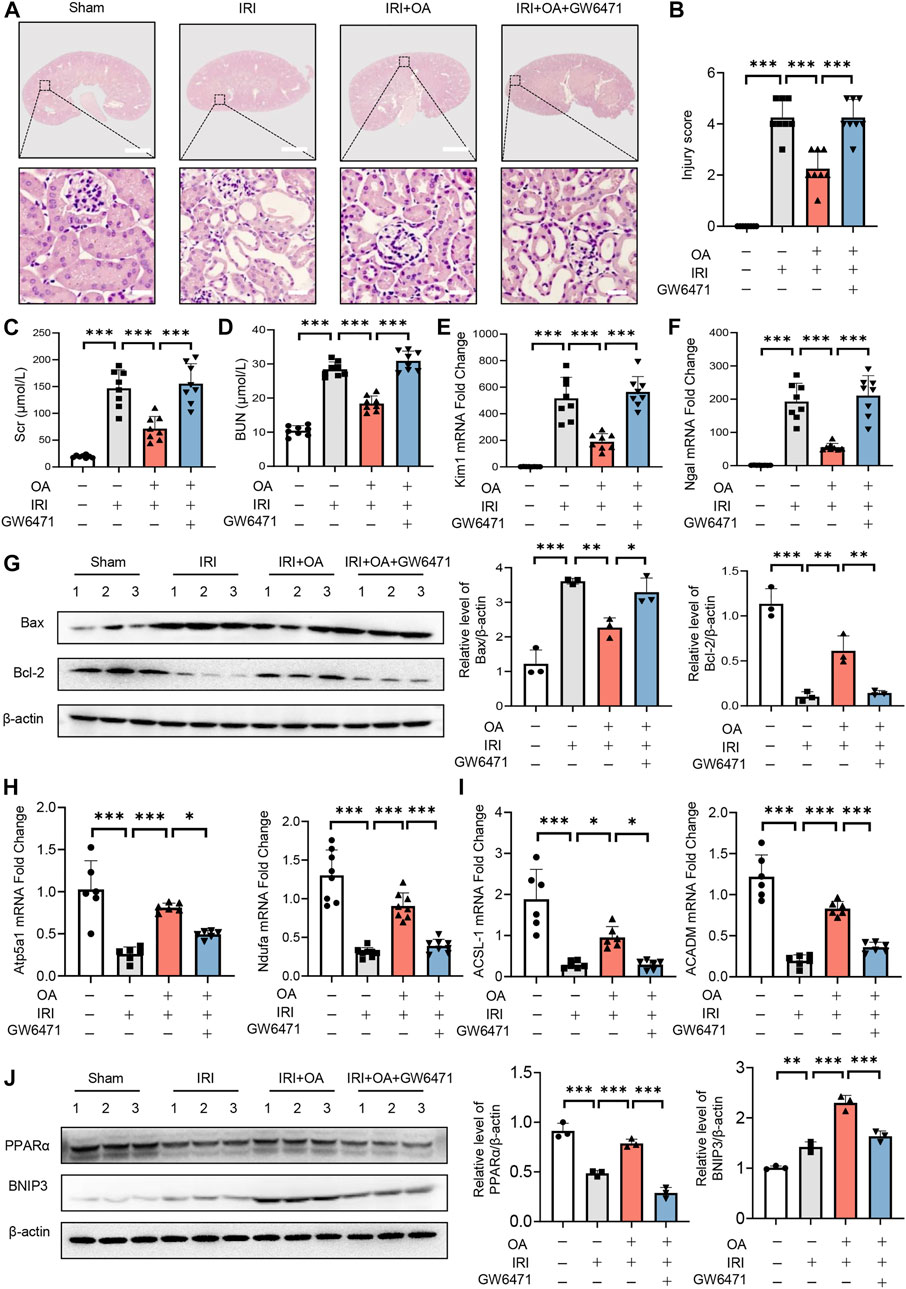
FIGURE 7. PPARα inhibition reversed the protective effect of OA in AKI mice. (A) OA-treated Sham and AKI mice were injected with control or PPARα antagonist. Representative HE staining images of kidney sections from sham and AKI mice treated with control or OA in combination with or without PPARα antagonist. Scale bars, 1.25 mm (top) and 50 μm (bottom). (B) Quantitative assessment of tubular damage (n = 8). Effects of OA on serum Scr (C) and BUN (D) (n = 8). mRNA levels of Kim-1 (E) and Ngal (F) by qPCR (n = 8). (G) The expressions of Bcl-2 and Bax in kidney tissues were analyzed by Western blot analysis (n = 3). (H,I) The expression levels of OXPHO-related gene (H) and FAO-related gene (I) (n = 8) were detected by qPCR. (J) The expression levels of PPARα and BNIP3 in kidney tissues were measured by Western blot (n = 3). Data are expressed as means ± SEM. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
To further confirm the role of OA, AKI-to-CKD transition mouse model was constructed. HE and Masson staining showed that tubulointerstitial injury and fibrosis were significantly improved after OA treatment (Figures 8A,B). Besides, we also found that the expressions of fibrosis markers, including fibronectin and α-smooth muscle actin (α-SMA), were markedly decreased by OA in the kidney tissues from IRI mice (Figure 8C). These findings reveal that OA could ameliorate the progression of AKI to CKD.
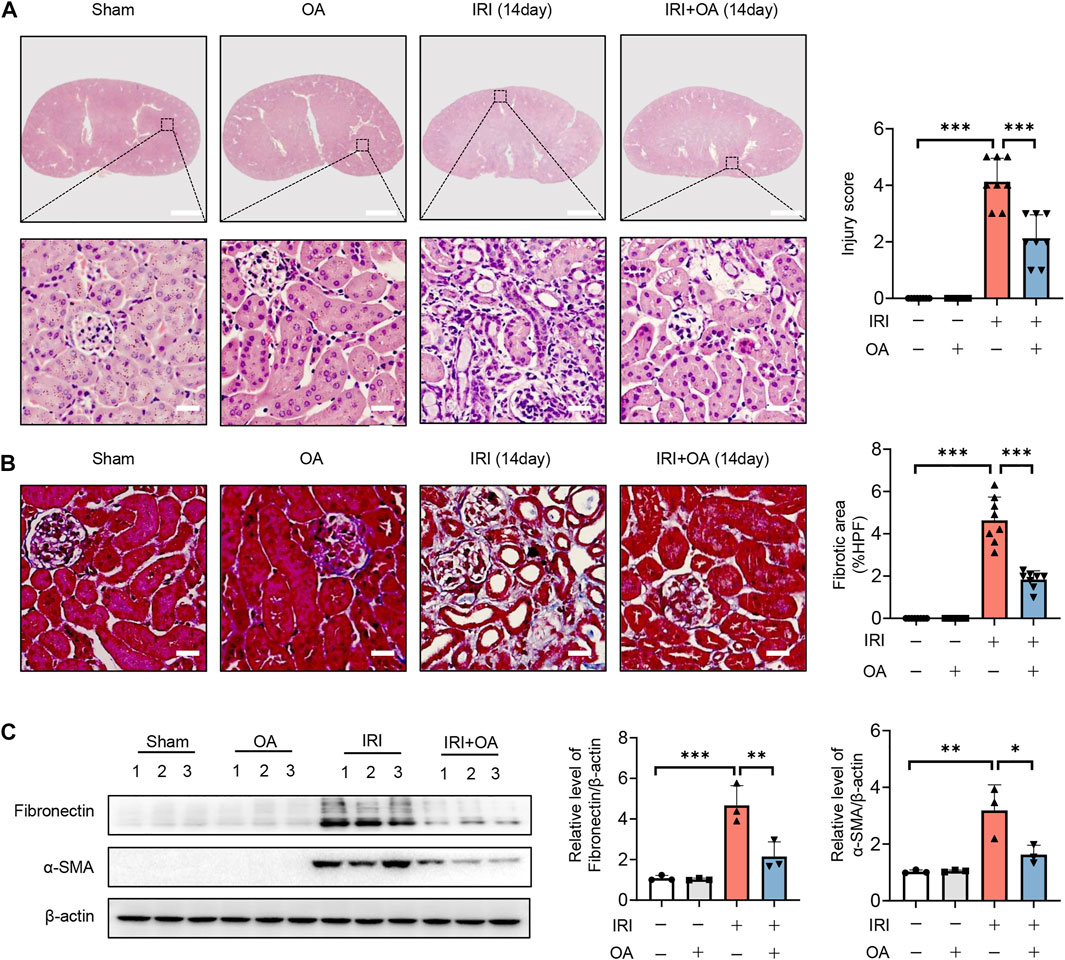
FIGURE 8. OA attenuates the transition of AKI-to-CKD. A 35-min bilateral IRI-induced AKI-to-CKD mouse model was constructed, which was intravenously injected with control or OA (20 mg/kg). Mice were euthanized 2 weeks later. (A) Representative images of HE staining. Scale bars, 1.25 mm (top) and 50 μm (bottom). (B) Representative images of Masson staining. Scale bar, 50 μm. (C) Western blot analysis of the expressions of fibrosis markers (Fibronectin and α-SMA) in the kidney tissues (n = 3). Data are expressed as means ± SEM. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
AKI remains a difficult clinically critical disease, with high morbidity and mortality (Bellomo et al., 2012; Levey and James, 2017; Ronco et al., 2019). Although significant progression has been made in the pathogenesis, there is still no drugs applicable for curing AKI in clinical practice (Hong et al., 2021; Kim et al., 2021), which highlights the urgent necessity to develop novel therapeutic strategies for AKI.
Although emerging studies support the beneficial role of OA in the pathogenesis of LPS-mediated acute injury of liver and lung (Tseng et al., 2012; Huang et al., 2015), the therapeutic role of OA in ischemic AKI is unclear. This study has identified the ability of OA in ameliorating AKI and delaying its transition to CKD. It was found that OA alleviated IRI and Cisplatin-induced renal tubular damage. In addition, at 14 days post-35 min bilateral renal IRI, mouse kidneys developed fibrosis and progressively progressed to CKD, which was prevented by the administration of OA. Further, OA significantly ameliorated HR-mediated mitochondrial injury via inducing PPARα-BNIP3 signaling pathway, which may shed new light on the treatment of AKI and its progression to CKD.
The kidney is a human organ with the second highest mitochondrial abundance, which plays a vital role in maintaining kidney health (Wang et al., 2020). Increasing evidence demonstrated mitochondrial injury as a crucial cause of kidney diseases (Huang et al., 2020a; Huang et al., 2020b; Scholz et al., 2021), while maintaining mitochondrial homeostasis may improve kidney injury (Szeto et al., 2016; Bhargava and Schnellmann, 2017). In this study, we found that OA alleviated mitochondrial damage through inducing the expression of BNIP3. BNIP3 is localized in the mitochondrial outer membrane, playing a vital role in mitochondrial quality control (Chourasia et al., 2015; Lee et al., 2017; Tang et al., 2019; Fu et al., 2020). Recent studies have reported that the loss of BNIP3 aggravated IR-induced AKI, while overexpression of BNIP3 not only reversed the reduction of mitophagy, but also alleviated renal injury, indicating that BNIP3-mediated mitophagy plays a protective role in AKI (Tang et al., 2019; Fu et al., 2020). However, in the present study, we found that OA attenuated AKI without regulating mitophagy in the kidney, which coincided with other reports that mitophagy can also be induced independent of BNIP3 (Choi et al., 2016, Yuan and Pan, 2018). In addition, BNIP3 ensured mitochondrial quality and integrity via regulating FAO in the liver (Glick et al., 2012; Choi et al., 2016), reflecting a new role of BNIP3 in mitochondrial quality control. We demonstrated that OA attenuated HR-mediated mitochondrial injury, including mitochondrial energy metabolism remodeling, through upregulating BNIP3 expression in renal tubular epithelial cells. In-depth in vivo studies using kidney-specific BNIP3 knockout mice may help validate the in vitro findings and overcome this limitation. Despite of this, our results together with previous reports collectively suggest that therapeutic approaches targeting BNIP3-mediated mitochondrial homeostasis may develop as a promising strategy for the treatment of AKI.
Further network pharmacological analysis, transcription factor prediction and experimental validations suggested that OA enhanced BNIP3 expression via upregulating the expression of PPARα, which induced BNIP3 transcription via directly binding to its promoter region. PPARα is a subtype of ligand-activated transcription factor, which has been shown mainly distributed in tissues with a high FAO, such as kidney and brown fat (Pawlak et al., 2015). PPARα plays a crucial role in lipid metabolism by regulating various target genes involved in FAO and lipoprotein metabolism (Nan et al., 2014; Pawlak et al., 2015; Yu et al., 2015). 90% of the ATP required by proximal tubule cells in the renal cortex is produced by mitochondrial FAO (Console et al., 2020), which is an essential part of mitochondrial energy metabolism. As reported, FAO decreases during hypoxia, exhibiting the same phenotype as renal fibrosis, which gradually leads to CKD and ESRD (Console et al., 2020). In this study, we observed that PPARα expression was significantly upregulated in OA-treated cells under HR conditions and in kidney tissues from AKI mice. Of note, when PPARα expression was inhibited, OA could neither ameliorate HR-induced mitochondrial homeostasis imbalance in vitro, nor IRI-mediated AKI in vivo, further supporting the regulatory effect of OA on PPARα. Therefore, OA-induced PPARα-BNIP3 axis-mediated mitochondrial homeostasis may serve as a novel target for AKI treatment.
As reported, OA possesses multi-pharmacological activities not only restricted in anti-tumor effects, but also in anti-inflammation and neuro-protection (Lu et al., 2016). In carcinoma, OA could increase ROS level, induce apoptosis, suppress angiogenesis and enhance mitochondrial dysfunction via inactivating sirtuin 1 (SIRT1)-forkhead box O3 (FOXO3)-BNIP3 axis (Lu et al., 2016; Yao et al., 2021). However, in normal tissues, OA plays a protective effect through inhibiting mitochondrial ROS, repressing inflammatory response, and promoting angiogenesis (Lu et al., 2016; Kai et al., 2020; Zhang et al., 2021). Recently, a literature review compared the inconsistent roles of OA in tumors and normal tissues, and found the high selectivity of OA due to the unfolded protein response and protein kinase B pathway, since OA (100–200 μM) selectively killed many more hepatocellular carcinoma cells (HepG2) than normal hepatocytes (L02) (Mu et al., 2009; Xu et al., 2012; Lu et al., 2016). Further, the concentrations of OA used in tumor treatment were mainly 40–200 μM (Lu et al., 2016), while the therapeutic dose of OA was 10 μM in the present study. As shown in Figure 2B, OA could not inhibit the viability of HK-2 cells, when its concentration was less than 100 μM. Therefore, the different roles and underlying mechanisms of OA may be attributed to tissue specificity and various dosages, and low doses of OA may improve tumor progression while protecting normal cells.
The present study demonstrated the therapeutic effect of OA, the main active component of a traditional Chinese medicine SB, on AKI and its progression to CKD. Mechanistically, OA significantly improved HR-induced mitochondrial injury via enhancing PPARα-BNIP3 signaling pathway. Therefore, therapeutic approaches using OA or targeting PPARα-BNIP3 axis to modulate mitochondrial homeostasis may provide novel strategies for the treatment of AKI-to-CKD transition.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by The Animal Ethics Review Board of the Army Medical University.
YH and JZ designed the study and revised the manuscript. MY and SQ performed the experiments and drafted the manuscript. JX, WX, XG, SG, and JC carried out the collation and analysis of the experimental data. YL and BZ took part in the animal studies. All authors approved the final version of the paper.
This work was supported by research grants from Key program of the Natural Science Foundation of China (No. 82030023), the Natural Science Foundation of China (Nos. 81873605, 82170705, 81800621), Chongqing Science and Technology Talent Program (cstc2021ycjh-bgzxm0145), Natural Science Foundation of Chongqing Science & Technology Commission (cstc2021jcyj-msxmX0672), Frontier specific projects of Xinqiao Hospital (No. 2018YQYLY004), and Personal training Program for Clinical Medicine Research of Army Medical University (No. 2018XLC1007).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.935937/full#supplementary-material
Bellomo, R., Kellum, J. A., and Ronco, C. (2012). Acute kidney injury. Lancet 9843, 756–766. doi:10.1016/S0140-6736(11)61454-2
Bhargava, P., and Schnellmann, R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 10, 629–646. doi:10.1038/nrneph.2017.107
Bucaloiu, I. D., Kirchner, H. L., Norfolk, E. R., Hartle, J. E., and Perkins, R. M. (2012). Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 5, 477–485. doi:10.1038/ki.2011.405
Chawla, L. S., Eggers, P. W., Star, R. A., and Kimmel, P. L. (2014). Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 1, 58–66. doi:10.1056/NEJMra1214243
Chen, D. Q., Feng, Y. L., Chen, L., Liu, J. R., Wang, M., Vaziri, N. D., et al. (2019). Poricoic acid a enhances melatonin inhibition of AKI-to-CKD transition by regulating Gas6/AxlNFκB/Nrf2 axis. Free Radic. Biol. Med. 484-97, 484–497. doi:10.1016/j.freeradbiomed.2019.01.046
Choi, J. W., Jo, A., Kim, M., Park, H. S., Chung, S. S., Kang, S., et al. (2016). BNIP3 is essential for mitochondrial bioenergetics during adipocyte remodelling in mice. Diabetologia 3, 571–581. doi:10.1007/s00125-015-3836-9
Chourasia, A. H., Tracy, K., Frankenberger, C., Boland, M. L., Sharifi, M. N., Drake, L. E., et al. (2015). Mitophagy defects arising from BNIP3 loss promote mammary tumor progression to metastasis. EMBO Rep. 9, 1145–1163. doi:10.15252/embr.201540759
Coca, S. G., Singanamala, S., and Parikh, C. R. (2012). Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 5, 442–448. doi:10.1038/ki.2011.379
Console, L., Scalise, M., Giangregorio, N., Tonazzi, A., Barile, M., Indiveri, C., et al. (2020). The link between the mitochondrial fatty acid oxidation derangement and kidney injury. Front. Physiol. 794. doi:10.3389/fphys.2020.00794
Emma, F., Montini, G., Parikh, S. M., and Salviati, L. (2016). Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 5, 267–280. doi:10.1038/nrneph.2015.214
Fu, Z. J., Wang, Z. Y., Xu, L., Chen, X. H., Li, X. X., Liao, W. T., et al. (2020). HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 36, 101671. doi:10.1016/j.redox.2020.101671
Glick, D., Zhang, W., Beaton, M., Marsboom, G., Gruber, M., Simon, M. C., et al. (2012). BNIP3 regulates mitochondrial function and lipid metabolism in the liver. Mol. Cell. Biol. 13, 2570–2584. doi:10.1128/MCB.00167-12
Guan, Y., Chen, K., Quan, D., Kang, L., Yang, D., Wu, H., et al. (2020). The combination of scutellaria baicalensis Georgi and Sophora japonica L. ameliorate renal function by regulating gut microbiota in spontaneously hypertensive rats. Front. Pharmacol. 11, 575294. doi:10.3389/fphar.2020.575294
Hong, X., Zhou, Y., Wang, D., Lyu, F., Guan, T., Liu, Y., et al. (2021). Exogenous Wnt1 prevents acute kidney injury and its subsequent progression to chronic kidney disease. Front. Physiol. 12, 745816. doi:10.3389/fphys.2021.745816
Hu, M. C., Shi, M., Gillings, N., Flores, B., Takahashi, M., Kuro, O. M., et al. (2017). Recombinant alpha-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 5, 1104–1114. doi:10.1016/j.kint.2016.10.034
Huang, H., Zhang, X., and Li, J. (2015). Protective effect of oroxylin a against lipopolysaccharide and/or D-galactosamine-induced acute liver injury in mice. J. Surg. Res. 2, 522–528. doi:10.1016/j.jss.2015.01.047
Huang, T. H., Wu, T. H., Guo, Y. H., Li, T. L., Chan, Y. L., Wu, C. J., et al. (2019). The concurrent treatment of scutellaria baicalensis Georgi enhances the therapeutic efficacy of cisplatin but also attenuates chemotherapy-induced cachexia and acute kidney injury. J. Ethnopharmacol. 243, 112075. doi:10.1016/j.jep.2019.112075
Huang, Y., Wang, S., Zhou, J., Liu, Y., Du, C., Yang, K., et al. (2020a). IRF1-mediated downregulation of PGC1α contributes to cardiorenal syndrome type 4. Nat. Commun. 1, 4664. doi:10.1038/s41467-020-18519-0
Huang, Y., Zhou, J., Wang, S., Xiong, J., Chen, Y., Liu, Y., et al. (2020b). Indoxyl sulfate induces intestinal barrier injury through IRF1-DRP1 axis-mediated mitophagy impairment. Theranostics 16, 7384–7400. doi:10.7150/thno.45455
Huang, Y., Xin, W., Xiong, J., Yao, M., Zhang, B., Zhao, J., et al. (2022). The intestinal microbiota and metabolites in the Gut-Kidney-Heart axis of chronic kidney disease. Front. Pharmacol. 13, 837500. doi:10.3389/fphar.2022.837500
Kai, J., Yang, X., Wang, Z., Wang, F., Jia, Y., Wang, S., et al. (2020). Oroxylin a promotes PGC-1α/Mfn2 signaling to attenuate hepatocyte pyroptosis via blocking mitochondrial ROS in alcoholic liver disease. Free Radic. Biol. Med. 89–102. doi:10.1016/j.freeradbiomed.2020.03.031
Kim, S., Lee, S. A., Yoon, H., Kim, M. Y., Yoo, J. K., Ahn, S. H., et al. (2021). Exosome-based delivery of super-repressor IκBα ameliorates kidney ischemia-reperfusion injury. Kidney Int. 3, 570–584. doi:10.1016/j.kint.2021.04.039
Kong, E. K., Yu, S., Sanderson, J. E., Chen, K. B., Huang, Y., Yu, C. M., et al. (2011). A novel anti-fibrotic agent, baicalein, for the treatment of myocardial fibrosis in spontaneously hypertensive rats. Eur. J. Pharmacol. 2-3, 175–181. doi:10.1016/j.ejphar.2011.02.033
Kramann, R., Tanaka, M., and Humphreys, B. D. (2014). Fluorescence microangiography for quantitative assessment of peritubular capillary changes after AKI in mice. J. Am. Soc. Nephrol. 9, 1924–1931. doi:10.1681/ASN.2013101121
Lai, C. C., Huang, P. H., Yang, A. H., Chiang, S. C., Tang, C. Y., Tseng, K. W., et al. (2016). Baicalein, a component of scutellaria baicalensis, attenuates kidney injury induced by myocardial ischemia and reperfusion. Planta Med. 3, 181–189. doi:10.1055/s-0035-1558114
Lee, H. J., Jung, Y. H., Choi, G. E., Ko, S. H., Lee, S. J., Lee, S. H., et al. (2017). BNIP3 induction by hypoxia stimulates FASN-dependent free fatty acid production enhancing therapeutic potential of umbilical cord blood-derived human mesenchymal stem cells. Redox Biol. 13, 426–443. doi:10.1016/j.redox.2017.07.004
Levey, A. S., and James, M. T. (2017). Acute kidney injury. Ann. Intern. Med. 9, Itc66–itc80. doi:10.7326/aitc201711070
Lin, M., Li, L., Li, L., Pokhrel, G., Qi, G., Rong, R., et al. (2014). The protective effect of baicalin against renal ischemia-reperfusion injury through inhibition of inflammation and apoptosis. BMC Complement. Altern. Med. 14, 19. doi:10.1186/1472-6882-14-19
Liu, X., Liu, L., Chen, P., Zhou, L., Zhang, Y., Wu, Y., et al. (2014). Clinical trials of traditional Chinese medicine in the treatment of diabetic nephropathy--a systematic review based on a subgroup analysis. J. Ethnopharmacol. 2, 810–819. doi:10.1016/j.jep.2013.11.028
Lu, L., Guo, Q., and Zhao, L. (2016). Overview of oroxylin A: A promising flavonoid compound. Phytother. Res. 11, 1765–1774. doi:10.1002/ptr.5694
Mu, R., Qi, Q., Gu, H., Wang, J., Yang, Y., Rong, J., et al. (2009). Involvement of p53 in oroxylin A-induced apoptosis in cancer cells. Mol. Carcinog. 12, 1159–1169. doi:10.1002/mc.20570
Nan, Y. M., Wang, R. Q., and Fu, N. (2014). Peroxisome proliferator-activated receptor alpha, a potential therapeutic target for alcoholic liver disease. World J. Gastroenterol. 25, 8055–8060. doi:10.3748/wjg.v20.i25.8055
Pan, T. L., Wang, P. W., Leu, Y. L., Wu, T. H., and Wu, T. S. (2012). Inhibitory effects of Scutellaria baicalensis extract on hepatic stellate cells through inducing G2/M cell cycle arrest and activating ERK-dependent apoptosis via Bax and caspase pathway. J. Ethnopharmacol. 3, 829–837. doi:10.1016/j.jep.2011.12.028
Parikh, S. M., Yang, Y., He, L., Tang, C., Zhan, M., Dong, Z., et al. (2015). Mitochondrial function and disturbances in the septic kidney. Semin. Nephrol. 1, 108–119. doi:10.1016/j.semnephrol.2015.01.011
Pawlak, M., Lefebvre, P., and Staels, B. (2015). Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 3, 720–733. doi:10.1016/j.jhep.2014.10.039
Ronco, C., Bellomo, R., and Kellum, J. A. (2019). Acute kidney injury. Lancet 10212, 1949–1964. doi:10.1016/s0140-6736(19)32563-2
Scholz, H., Boivin, F. J., Schmidt-Ott, K. M., Bachmann, S., Eckardt, K. U., Scholl, U. I., et al. (2021). Kidney physiology and susceptibility to acute kidney injury: implications for renoprotection. Nat. Rev. Nephrol. 5, 335–349. doi:10.1038/s41581-021-00394-7
Sun, J., Zhang, J., Tian, J., Virzi, G. M., Digvijay, K., Cueto, L., et al. (2019). Mitochondria in sepsis-induced AKI. J. Am. Soc. Nephrol. 7, 1151–1161. doi:10.1681/ASN.2018111126
Szeto, H. H., Liu, S., Soong, Y., Alam, N., Prusky, G. T., Seshan, S. V., et al. (2016). Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 5, 997–1011. doi:10.1016/j.kint.2016.06.013
Tang, C., Han, H., Liu, Z., Liu, Y., Yin, L., Cai, J., et al. (2019). Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 9, 677. doi:10.1038/s41419-019-1899-0
Tang, C., Cai, J., Yin, X. M., Weinberg, J. M., Venkatachalam, M. A., Dong, Z., et al. (2021). Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 5, 299–318. doi:10.1038/s41581-020-00369-0
Tseng, T. L., Chen, M. F., Tsai, M. J., Hsu, Y. H., Chen, C. P., Lee, T. J., et al. (2012). Oroxylin-A rescues LPS-induced acute lung injury via regulation of NF-κB signaling pathway in rodents. PLoS One 10, e47403. doi:10.1371/journal.pone.0047403
Wang, Y., Cai, J., Tang, C., and Dong, Z. (2020). Mitophagy in acute kidney injury and kidney repair. Cells 2, E338. doi:10.3390/cells9020338
Wu, V. C., Wu, C. H., Huang, T. M., Wang, C. Y., Lai, C. F., Shiao, C. C., et al. (2014). Long-term risk of coronary events after AKI. J. Am. Soc. Nephrol. 3, 595–605. doi:10.1681/ASN.2013060610
Xu, M., Lu, N., Sun, Z., Zhang, H., Dai, Q., Wei, L., et al. (2012). Activation of the unfolded protein response contributed to the selective cytotoxicity of oroxylin A in human hepatocellular carcinoma HepG2 cells. Toxicol. Lett. 2, 113–125. doi:10.1016/j.toxlet.2012.05.008
Yao, J., Wang, J., Xu, Y., Guo, Q., Sun, Y., Liu, J., et al. (2021). CDK9 inhibition blocks the initiation of PINK1-PRKN-mediated mitophagy by regulating the SIRT1-FOXO3-BNIP3 axis and enhances the therapeutic effects involving mitochondrial dysfunction in hepatocellular carcinoma. Autophagy. 18, 1–19. doi:10.1080/15548627.2021.2007027
Yuan, Y., and Pan, S. S. (2018). Parkin mediates mitophagy to participate in cardioprotection induced by late exercise preconditioning but Bnip3 does not. J. Cardiovasc Pharmacol. 5, 303–316. doi:10.1097/FJC.0000000000000572
Yu, X. H., Zheng, X. L., and Tang, C. K. (2015). Peroxisome proliferator-activated receptor alpha in lipid metabolism and atherosclerosis. Adv. Clin. Chem. 71, 171–203. doi:10.1016/bs.acc.2015.06.005
Yu, X., Xu, M., Meng, X., Li, S., Liu, Q., Bai, M., et al. (2020). Nuclear receptor PXR targets AKR1B7 to protect mitochondrial metabolism and renal function in AKI. Sci. Transl. Med. 543, eaay7591. doi:10.1126/scitranslmed.aay7591
Zhang, L., Chen, L., Li, C., Shi, H., Wang, Q., Yang, W., et al. (2021). Oroxylin a attenuates limb ischemia by promoting angiogenesis via modulation of endothelial cell migration. Front. Pharmacol. 12, 705617. doi:10.3389/fphar.2021.705617
Keywords: oroxylin A, acute kidney injury, mitochondrial energy metabolism, BNIP3, PPARα
Citation: Yao M, Qin S, Xiong J, Xin W, Guan X, Gong S, Chen J, Liu Y, Zhang B, Zhao J and Huang Y (2022) Oroxylin A ameliorates AKI-to-CKD transition through maintaining PPARα-BNIP3 signaling-mediated mitochondrial homeostasis. Front. Pharmacol. 13:935937. doi: 10.3389/fphar.2022.935937
Received: 04 May 2022; Accepted: 12 July 2022;
Published: 23 August 2022.
Edited by:
Jianping Chen, Shenzhen Traditional Chinese Medicine Hospital, ChinaReviewed by:
Wenju Li, National Renal Disease Clinical Medicine Research Center, ChinaCopyright © 2022 Yao, Qin, Xiong, Xin, Guan, Gong, Chen, Liu, Zhang, Zhao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinghong Zhao, emhhb2poQHRtbXUuZWR1LmNu; Yinghui Huang, aWtreWh1YW5nQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.