 Olena Zaitseva
Olena Zaitseva Annett Hoffmann
Annett Hoffmann Christoph Otto2
Christoph Otto2 Harald Wajant
Harald Wajant
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 21 October 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.935086
This article is part of the Research Topic Targeting TNF/TNFR Signaling Pathways View all 6 articles
Fibroblast growth factor-inducible 14 (Fn14) is a member of the tumor necrosis factor (TNF) receptor superfamily (TNFRSF) and is activated by its ligand TNF-like weak inducer of apoptosis (TWEAK). The latter occurs as a homotrimeric molecule in a soluble and a membrane-bound form. Soluble TWEAK (sTWEAK) activates the weakly inflammatory alternative NF-κB pathway and sensitizes for TNF-induced cell death while membrane TWEAK (memTWEAK) triggers additionally robust activation of the classical NF-κB pathway and various MAP kinase cascades. Fn14 expression is limited in adult organisms but becomes strongly induced in non-hematopoietic cells by a variety of growth factors, cytokines and physical stressors (e.g., hypoxia, irradiation). Since all these Fn14-inducing factors are frequently also present in the tumor microenvironment, Fn14 is regularly found to be expressed by non-hematopoietic cells of the tumor microenvironment and most solid tumor cells. In general, there are three possibilities how the tumor-Fn14 linkage could be taken into consideration for tumor therapy. First, by exploitation of the cancer associated expression of Fn14 to direct cytotoxic activities (antibody-dependent cell-mediated cytotoxicity (ADCC), cytotoxic payloads, CAR T-cells) to the tumor, second by blockade of potential protumoral activities of the TWEAK/Fn14 system, and third, by stimulation of Fn14 which not only triggers proinflammtory activities but also sensitizes cells for apoptotic and necroptotic cell death. Based on a brief description of the biology of the TWEAK/Fn14 system and Fn14 signaling, we discuss the features of the most relevant Fn14-targeting biologicals and review the preclinical data obtained with these reagents. In particular, we address problems and limitations which became evident in the preclinical studies with Fn14-targeting biologicals and debate possibilities how they could be overcome.
Fibroblast growth factor (FGF)-inducible 14 (Fn14) is an unusual small member of the tumor necrosis factor (TNF) receptor superfamily (TNFRSF) of 14 kDa (Wiley et al., 2001). TNFRSF receptors (TNFRs) are characterized by a cysteine rich domain (CRD) which is found in one to six copies in their N-terminal extracellular parts. Structurally three TNFR subgroups can be distinguished: 1) death receptors (DRs), which harbor a death domain (DD) in their cytoplasmic part mediating the assembly of receptor-associated or cytoplasmic caspase activating protein complexes (Dickens et al., 2012) 2) TRAF-interacting TNFRs, which have no DD but instead contain one or more binding motifs for adapter proteins of the TNF receptor associated factor (TRAF) family in their cytoplasmic domain and 3) decoy TNFRs, which lack an intracellular domain. Fn14 belongs to the second subgroup of TNFRs and binds TRAF1, TRAF2, TRAF3, and TRAF5 (Wiley et al., 2001).
Fn14 is dynamically and highly expressed during development. In contrast, in the healthy adult organism Fn14 expression is low and mainly limited to mesenchymal and epithelial progenitor cells. Importantly, however, rapid and strong upregulation of Fn14 expression takes place in practically any non-hematological cell type upon tissue injury (Winkles, 2008; Burkly et al., 2011). The injury-associated pattern of Fn14 expression is in good accordance with the fact that growth factors (e.g., EGF, FGF), various cytokines (e.g., IFNγ, TNF, TGFβ) but also microbial (e.g., LPS) and physical stressors (e.g., hypoxia, irradiation) are potent Fn14 inducers. In accordance with the expression pattern of Fn14, manifold and complex functions of Fn14 have been described in tissue repair and regeneration. For example, it has been found that regenerative responses after the injury of muscles, pancreas and the liver are delayed in Fn14 knockout mice (Girgenrath et al., 2006; Mittal et al., 2010a; Mittal et al., 2010b). Furthermore, excessive or chronic engagement of Fn14 may also promote repair-related adverse effects, such as fibrosis and chronic inflammation (Mittal et al., 2010a; Mittal et al., 2010b; Kuramitsu et al., 2013).
The tissue damage that is unavoidably associated with tumor growth typically comes along with sterile inflammation and triggering of repair processes. As a consequence, the tumor microenvironment contains a variety of the aforementioned Fn14-inducing factors and expression of Fn14 has been accordingly described for a huge variety of tumors of non-hematopoietic origin (Table 1).
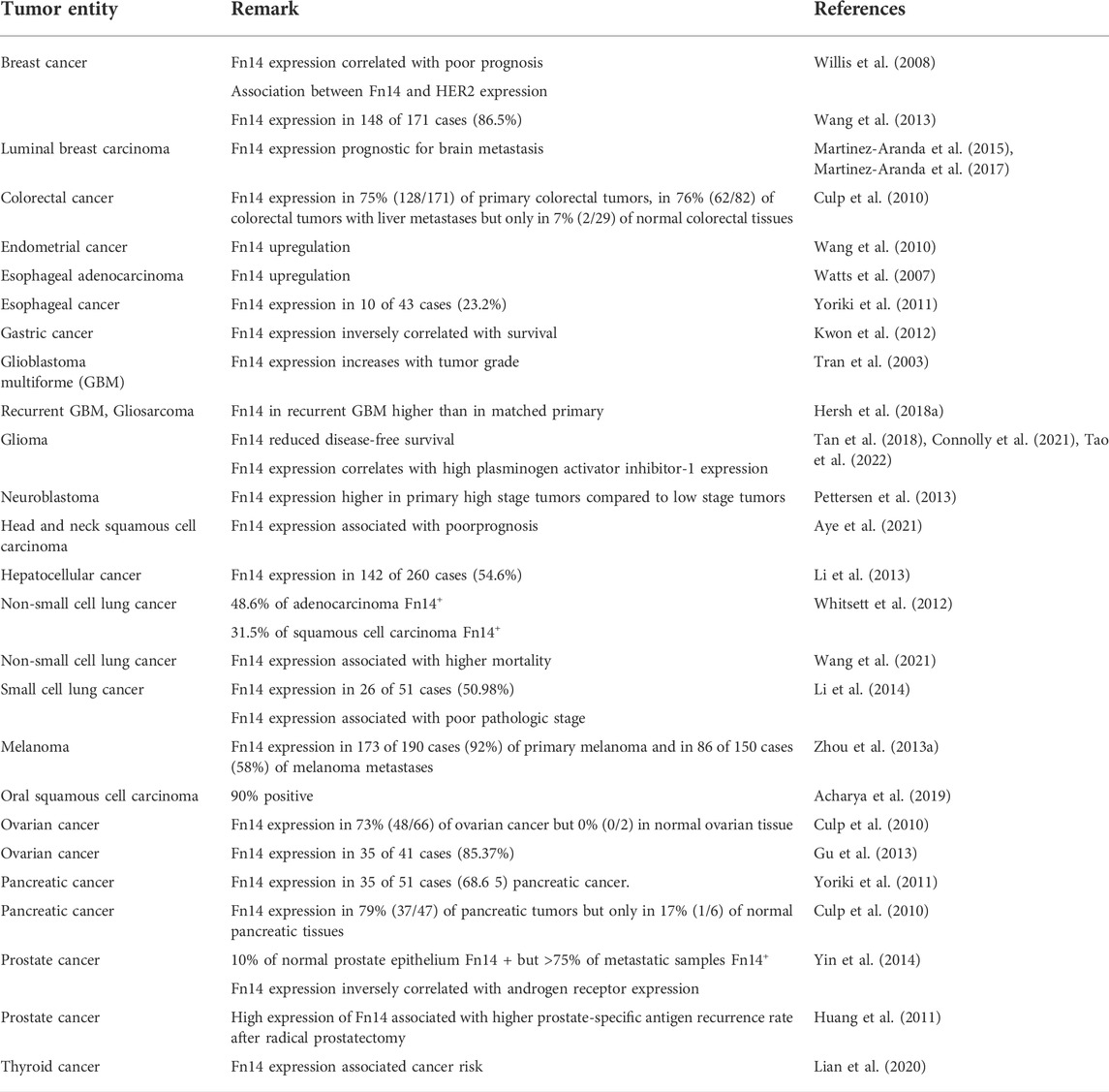
TABLE 1. Cancer-associated expression of Fn14.
Fn14 signaling is activated by two variants of tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), membrane-bound TWEAK (memTWEAK) and soluble TWEAK (sTWEAK). TWEAK is a typical ligand of the TNF superfamily (TNFSF). Accordingly, it is a type II homotrimeric transmembrane protein (memTWEAK) from which a soluble, likewise trimeric, molecule (sTWEAK) can be released by furin protease-mediated proteolytic processing (Bodmer et al., 2002). An early study claimed that death receptor (DR3) is also a TWEAK receptor (Marsters et al., 1998), but we and others could not confirm this interaction (Schneider et al., 1999; Kaptein et al., 2000). The scavenger receptor CD163 has also been identified as a TWEAK receptor (Bover et al., 2007), but we did not observe this interaction in highly sensitive cellular binding assays (Fick et al., 2012). TWEAK expression has been described in a variety of cell lines and cell types by immunohistochemistry and RT-PCR. Doubtless detection of memTWEAK, however, has only been reported for monocytes, dendritic cells, NK cells and T-cells and a very few tumor cell lines (Nakayama et al., 2000; Kaplan et al., 2002; Kawakita et al., 2004; Felli et al., 2005; Maecker et al., 2005). Thus, it appears that sTWEAK is more abundantly expressed, compared to memTWEAK.
TWEAK received its name due to the initial observation that it induces apoptosis in a very few cell lines (Chicheportiche et al., 1997). As mentioned before, TNF receptor 1 (TNFR1) and other apoptosis inducing receptors of the TNFRSF trigger cell death signaling by means of a characteristic protein-protein interaction domain present in their cytoplasmic part, the so called death domain (DD). Fn14 does not contain a DD (Chicheportiche et al., 1997). Accordingly, early on we could show in the Kym-1 cell line that Fn14 has no own authentic ability to directly instruct the assembly of a caspase-8 activating protein complex and instead triggers apoptosis indirectly by stimulating the production of TNF which subsequently kills the Kym-1 cells via TNFR1 (Schneider et al., 1999). A similar indirect mechanism was later on reported for some of the other few cell lines undergoing TWEAK-induced apoptosis (Vince et al., 2008). Stimulation of TNF production is not sufficient for TWEAK-induced apoptosis. The latter furthermore requires a second Fn14-mediated process, which sensitizes for TNFR1-induced activation of caspase-8 and apoptosis (Schneider et al., 1999; Vince et al., 2008; Wicovsky et al., 2009). This sensitizing mechanism is based on the reduction of the cytoplasmic TNFR1-available pool of TRAF2 and the TRAF2-associated molecules cIAP1 and cIAP2 by receptor recruitment. Originally this process has been identified as a mechanism by which TNFR2 enhances TNFR1-induced apoptosis (Duckett and Thompson, 1997; Weiss et al., 1997; Weiss et al., 1998). The concerted action of TRAF1, TRAF2 and the cIAPs attenuates the ability of TNFR1 to activate caspase-8 (Wang et al., 1998) Therefore, the sequestration of these antiapoptotic molecules by Fn14 enhances TNF-induced apoptosis. Efficient recruitment of TRAF2, and under some circumstances TRAF2 degradation, is the most receptor proximal event triggered by Fn14 activation and thus operates also in cells which do not produce TNF in response to Fn14 stimulation (Wicovsky et al., 2009). Consequently, TWEAK enhances the apoptotic effect of exogenous TNF also in cells in which TWEAK alone is unable to trigger cell death. Membrane TWEAK as well as soluble TWEAK trigger TRAF2 recruitment to Fn14, accordingly both TWEAK variants enhance TNF-induced apoptosis.
TNFR1, by help of the redundant involvement of the TNFR1-associated signaling molecules TRADD and RIPK1 (Fullsack et al., 2019), stimulates apoptotic cell death by triggering the formation of a cytosolic caspase-8 activating complex, called complex II (Micheau and Tschopp, 2003; Ting and Bertrand, 2016; Siegmund et al., 2017). Both, TRADD and RIPK1, can independently recruit TRAF2 and the cIAPs to the plasma membrane-associated TNFR1 signaling complex but also to the cytosolic caspase-8 activating complex. This way, the antiapoptotic TRAF2-cIAP1/2 complexes gain physically access to the caspase-8 activating protein machinery. Noteworthy, caspase-8 activation in context of apoptosis-induction by the TNFR1-related death receptors TRAILR1, TRAILR2 and CD95 occurs directly at the initially assembled plasma membrane associated receptor complexes. Since in these cases TRAF2 and cIAPs have less efficient access to caspase-8, apoptosis induction by these death receptors remains largely unaffected by TWEAK/Fn14-dependent TRAF2-cIAP1/2 sequestration. Intriguingly, we and others could show that TWEAK and Fn14 also enhance necroptosis triggered by various insults (Wicovsky et al., 2009; Karl et al., 2014; Anany et al., 2018; Martin-Sanchez et al., 2018). Necroptosis is a form of cell death that can be triggered in RIPK3-expressing cells especially when caspase-8 activation is suppressed. Necroptosis induction depends on the formation of a cytosolic RIPK1- and RIPK3-containing protein complex and deubiquitination of linearly- and K63-ubiquitinated RIPK1. Noteworthy, K63-ubiquitination of RIPK1 in context of TNFR1 signaling is performed by cIAP1 and cIAP2. Therefore, it is not surprising that TWEAK/Fn14-mediated sequestration of TRAF2-cIAP1/2 complexes enhances TNF-induced necroptosis. Other than in the case of caspase-8 activation, TNFR1, CD95 and the TRAIL death receptors do not differ in the use of the RIPK1-RIPK3 core complex for necroptosis triggering and this also applies to necroptosis induction by double-stranded RNA and TLR3 (Karl et al., 2014; Anany et al., 2018). Accordingly, TWEAK has been found to enhance necroptosis induction by all these triggers (Karl et al., 2014; Anany et al., 2018). The physiological and/or pathophysiological relevance of the apoptosis- and necroptosis-enhancing activity of the TWEAK-Fn14 system has been poorly investigated yet in vivo. There is, however, evidence that it contributes in vivo to TNF-driven cell death of intestinal epithelial cells and the delayed phase of cell death in acute kidney injury (Chopra et al., 2015; Grabinger et al., 2017; Martin-Sanchez et al., 2018). Moreover, TRAF2 and the cIAPs have recently been identified as major targets facilitating CD8+ T-cell elimination of tumor cells in a TNF-dependent manner in vivo (Vredevoogd et al., 2019). Therefore, Fn14-dependent TRAF2-cIAP1/2 sequestration could also be a process that could be exploited for tumor therapy (see also below).
TRAF2 along with cIAP1 and cIAP2 are not only of relevance in the regulation of DR-induced cell death signaling pathways but play also a complex role in the activation of signaling pathways culminating in nuclear translocation of transcription factors of the NF-κB family and transcription of NF-κB-regulated genes. The transcription factors of the NF-κB family are dimeric and are sequestered in the cytosol (Baeuerle and Baltimore, 1988a; Baeuerle and Baltimore, 1988b; Henkel et al., 1992; Rice et al., 1992; Huang et al., 1997; Sun, 2017; Zhang et al., 2017). Cytosolic sequestration of NF-κB dimers is largely based on masking of their nuclear localization sequence (NLS). The latter can be achieved either by intramolecular interaction with an ankyrin-repeat containing inhibitory domain which is present in two NF-κB proteins that are initially expressed as immature precursor molecules (p100, p105) or by intermolecular interaction with ankyrin-repeat containing IκB proteins (Baeuerle and Baltimore, 1988a; Baeuerle and Baltimore, 1988b; Henkel et al., 1992; Rice et al., 1992; Sun, 2017; Zhang et al., 2017) According to the signaling mechanisms that result in demasking of the NLS two fundamentally different NF-κB signaling pathways can be distinguished, the classical or canonical NF-κB pathway and the alternative or noncanonical NF-κB pathway (Sun, 2017; Zhang et al., 2017). Intriguingly, TRAF2, cIAP1 and cIAP2 play a crucial role in both signaling pathways but with a qualitatively opposing effect. In the classical NF-κB pathway, TRAF2 and the cIAPs are crucially involved in the receptor/signal-induced activation of the so called IKK complex which phosphorylates IκB proteins to trigger their degradation and p105 to accelerate its constitutive processing to mature p50 (Sun, 2017; Zhang et al., 2017). In contrast, TRAF2 and the cIAPs in concert with TRAF3 inhibit in unstimulated cells the alternative NF-κB pathway by K48-ubiquitination and proteasomal degradation of the NF-κB inducing kinase (NIK), a constitutively active kinase of the MAP3K family. NIK is able to stimulate the phosphorylation and activation of IKK1, which in turn triggers proteolytic processing of the inactive p100 NF-κB precursor protein to the NF-κB subunit p52 (Sun, 2017; Zhang et al., 2017).
Sequestration of TRAF2-cIAP1/2 complexes and TRAF3 to the TWEAK-induced Fn14 signaling complex, as described above in context of the death receptor-Fn14 crosstalk, results therefore in reduced NIK degradation, NIK accumulation, p100 to p52 processing and eventually enhanced nuclear translocation of p52-containing NF-κB dimers (Wajant, 2013). Again, sTWEAK as well as membrane TWEAK efficiently trigger this signaling pathway (Roos et al., 2010). Fn14 also activates the classical NF-κB pathway by crucial help of TRAF2 and cIAPs (Figure 1). The trimeric TRAF2 molecule interacts with a single molecule of cIAP1 or cIAP2 (Mace et al., 2010; Zheng et al., 2010). Activation of cIAP1 and cIAP2 requires homotypic interaction/dimerization of their RING domain (Feltham et al., 2011). In cIAP1/2 monomers, however, the RING domain is intramolecularly inhibited by shielding by the BIR3, UBA and CARD domains (Dueber et al., 2011; Feltham et al., 2011; Lopez et al., 2011). The need of RING domain dimerization for cIAP1/2 activation suggests that TRAF23-cIAP1 and TRAF23-cIAP2 complexes have no or only a low E3 ligase activity. Accordingly, clustering of trimeric TWEAK-Fn14 complexes seems necessary to promote the proximity of two or more TRAF23-cIAP1/2 complexes to favor RING domain dimerization and activation of the classical NF-κB pathway (Figure 1). Additionally, we observed that soluble TWEAK trimers which bind three Fn14 molecules largely fail to trigger the classical NF-κB pathway despite robust TRAF2 recruitment while oligomerized soluble TWEAK trimers robustly activate this response (Roos et al., 2010). The involvement of the IKK complex, TRAF2, cIAPs and the IKK-stimulating kinase TAK1 in the activation of the classical NF-κB pathway by Fn14 has been proven by using Fn14 knockout cells, siRNA experiments and pharmacological inhibition but is poorly understood in detail (Saitoh et al., 2003; Kumar et al., 2009; Varfolomeev et al., 2012). Intriguingly, the E3 ligase activity of the TRAF2-cIAP1/2 complexes in classical and alternative NF-κB signaling are qualitatively different, promoting K48 ubiquitination to instruct proteasomal degradation of ubiquitinated proteins versus K63 ubiquitination to create docking sides for downstream signaling molecules. How the decision is made for the K48 or K63 type of ubiquitination by TRAF2 and the cIAPs is still largely unknown.
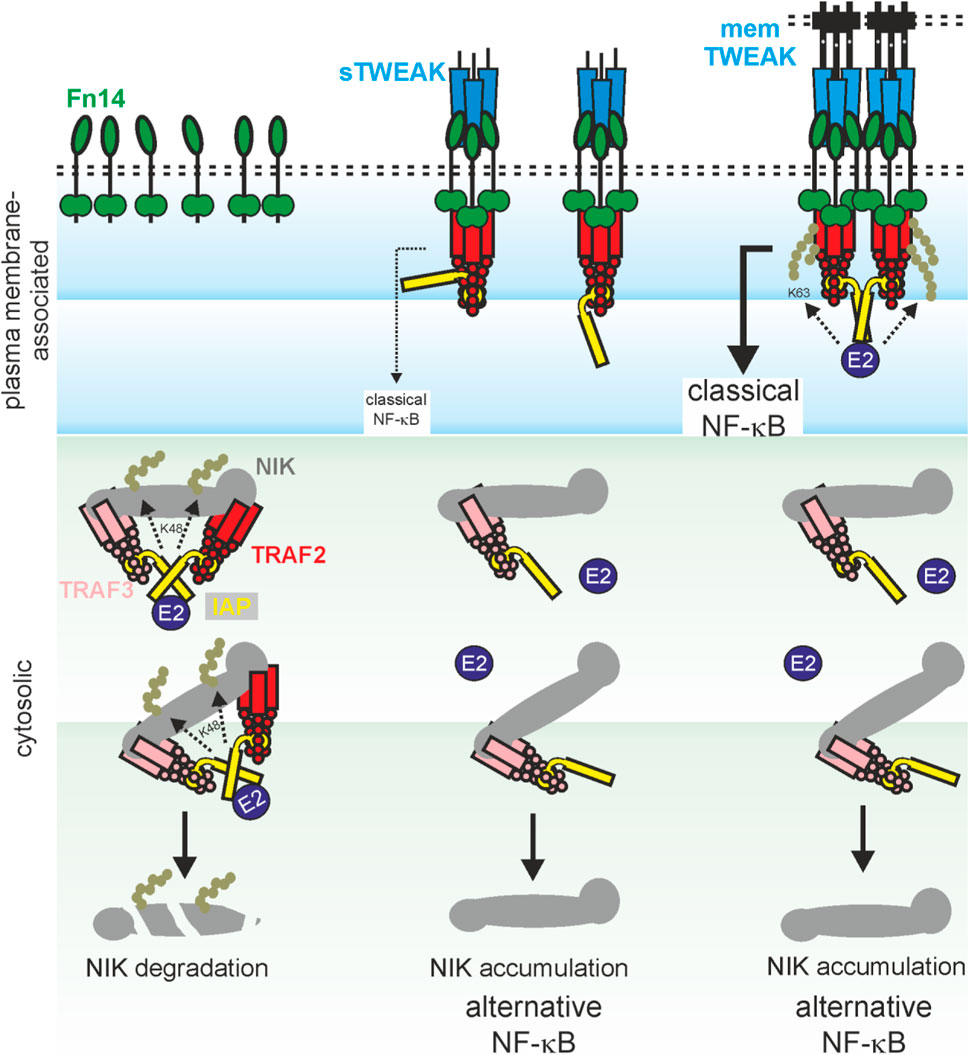
FIGURE 1. Model of TWEAK-induced NF-κB signaling. Dependent on the interplay of sTWEAK, memTWEAK, Fn14, TRAF-cIAP1/2 and TRAF3-cIAP1/2-NIK complexes, three different states of NFκB signaling can be distinguished: i) In the absence of TWEAK, TRAF3-cIAP1/2-NIK complexes interact with TRAF2-cIAP1/2 complexes in the cytoplasm. This results in cIAP1/2 transactivation and by help of E2 proteins to K48 ubiquitination of NIK by the cIAPs, proteasomal degradation of NIK and thus in constitutive inhibition of the alternative NF-κB pathway (left panel). ii) In the presence of sTWEAK, ligated trimeric Fn14 complexes are formed which recruit a single TRAF2-cIAP1/2 complex and reduces so the availability of the latter for NIK degradation. As a consequence, there is reduced NIK degradation leading to accumulation of NIK and eventually to signal-induced alternative NF-κB signaling. Since the Fn14-bound single TRAF2-cIAP1 and TRAF2-cIAP2 complexes remain largely inactive due to missing cIAP1/2 transactivation, there is no/poor classical NFκB signaling (middle panel). iii) In the presence of memTWEAK, ligated trimeric Fn14 complexes are formed, too but due to their high concentrations in the cell-to-cell contact zone these complexes cluster secondarily leading to the recruitment of several TRAF2-cIAP1/2 complexes to the Fn14 clusters. Again, this reduces the availability of TRAF2-cIAPs complexes for NIK degradation and triggers the alternative NF-κB pathway. However, since the Fn14-clusters bind several TRAF2-cIAP1/2 complexes, transactivation of cIAPs becomes possible leading eventually in classical NFκB signaling (right panel).
Noteworthy, TRAF2 and the structurally related protein TRAF1 form heterotrimers which recruit cIAP1 and cIAP2 even better than TRAF2 trimers (Zheng et al., 2010) and act in many aspects similar to the TRAF2-cIAP1 and TRAF2-cIAP2 complexes. TRAF1 is poorly expressed in most cells and is instead induced by NF-κB transcription factors (Wajant, 2013). Moreover, TRAF1, in contrast to many other NF-κB-regulated proinflammatory factors, such as IL8 and IL6, is not only robustly induced via the classical NF-κB pathway but also via the alternative NF-κB pathway. Thus, TRAF1 in contrast to most other NF-κB-regulated factors is already induced by sTWEAK (Carmona Arana et al., 2014). Interestingly, we furthermore observed that TRAF1 antagonizes TRAF2 degradation and thus maintains TRAF2-dependent signaling events (Wicovsky et al., 2009). It is therefore tempting to speculate that TRAF1 gains relevance in the adjustment of Fn14 activities when cells are chronically exposed to TWEAK.
Based on the ability to sensitize cells for TNF-induced cell death, the TWEAK/Fn14 system has obviously the potential to suppress together with TNF tumor development. Indeed, Vredevoogd et al. (2019) found a TNF response signature in patients responding to an immune checkpoint blockade and identified TRAF2 as a major factor preventing tumor cell death-induction by CD8+ T-cell produced TNF in animal models. Moreover, they mimicked the TNF-sensitizing effect of TRAF2 knockout in their models by Fn14 stimulation (Vredevoogd et al., 2019). Increased activity of the TWEAK/Fn14 system has also been associated with better outcome (overall-/disease-free survival) in patients suffering on colorectal cancer and was furthermore correlated with TWEAK-induced inhibition of in vitro invasiveness of colon cancer cell lines (Lin et al., 2012).
Most cellular activities engaged by the Fn14/TWEAK system, however, have the potential to promote cancer development. This in particular accounts for its ability to stimulate cell migration. TWEAK/Fn14-promoted cell migration and/or invasiveness have been reported for various tumor cell lines in vitro, including glioblastoma cells, NSCLC, ovarian cancer cells, breast cancer cell lines and androgen-independent prostate cancer cells (Tran et al., 2003; Willis et al., 2008; Dai et al., 2009; Huang et al., 2011; Lin et al., 2012; Whitsett et al., 2012; Asrani et al., 2013; Whitsett et al., 2014; Cheng et al., 2015; Jandova et al., 2015; Armstrong et al., 2016; Wang et al., 2018; Hu et al., 2019; Li et al., 2019). Moreover, there is also evidence from in vivo models that the TWEAK/Fn14 system can act as a crucial driver of tumor cell migration and metastasis. A549 cells overexpressing Fn14 showed enhanced lung metastasis upon tail vein injection in mice (Whitsett et al., 2012) and H460 tumor cells over-expressing Fn14 showed more metastases in lung, liver and lymph nodes compared to mice challenged with H460 cells with low Fn14 levels (Jandova et al., 2015). Similarly, in a prostate cancer xenograft model with bone metastatic DU145 RasG37 tumor cells, knockdown of Fn14 resulted in significantly reduced numbers of liver and brain tumors (Yin et al., 2014). The relevance of the TWEAK/Fn14 system for tumor cell migration and invasiveness is particular well understood in glioblastoma. Overexpression of Fn14 in the glioblastoma cell lines SF767 and T98G resulted in enhanced cell motility, membrane ruffling and lamellipodia formation (Tran et al., 2003). Moreover, TWEAK induces in glioblastoma cell lines sequential activation of the Rho GTPase Cdc42 and Rac1 along with Rac1-dependent cell migration (Tran et al., 2003; Fortin et al., 2012). In line with these findings, the guanine nucleotide exchange factors (GEFs) Ect2 and Trio, which promote the conversion of Cdc42 and Rac1 to their GTP-bound active state, have been crucially involved in TWEAK-induced cell migration in the T98G glioma cell line (Fortin et al., 2012). Similarly, it has been demonstrated that RhoA and the RhoA-specific GEF Src homology 3 domain-containing guanine nucleotide exchange factor (SGEF) mediate TWEAK-induced cell migration in the glioma cell lines U87 and U118 (Fortin Ensign et al., 2013). RhoA and SGEF but also the Cdc42/Ect2-Rac1/Trio axis furthermore mediate TWEAK-induced lamellipodia formation in T98G cells (Fortin et al., 2012; Fortin Ensign et al., 2013). In overexpression experiments co-immunoprecipitation of Fn14 and the aforementioned Rho-GTPases and GEFs have been observed (Fortin et al., 2012; Fortin Ensign et al., 2013) opening the possibility that these factors are part of the TWEAK-induced Fn14 signaling complex. In line with this idea, it has been found that TRAF2 and SGEF interact constitutively and recruited to Fn14 in a TWEAK-dependent manner. Furthermore, it has been described that TRAF2 knockdown inhibits TWEAK-induced cell migration in U87 and U118 cells (Fortin Ensign et al., 2013). Later studies revealed additionally that TWEAK not only generally stimulates cell motility in glioma cells but also chemotaxis. Accordingly, it has been reported that TWEAK induces activation of the non-receptor tyrosine kinase Lyn and that the knockdown of the latter abrogates TWEAK-induced chemotactic migration in T98A and A172 glioma cells as well as Rac1 activation (Dhruv et al., 2014). It is worth mentioning that SGEF expression is upregulated in glioma cell lines in response to Fn14 activation and is not only required for cell migration but also seems to contribute to resistance to DNA damaging chemotherapeutic drugs, such as temozolomide (TMZ), which is used to treat glioblastoma (Ensign et al., 2016) In fact, patient derived glioblastoma cells with acquired TMZ resistance display higher Fn14 expression and enhanced migratory activity compared to TMZ-sensitive cells (Hersh et al., 2018a; Hersh et al., 2018b). In accordance with a central role of the TWEAK/Fn14 system for the motility and aggressiveness of glioblastoma cells, Fn14 expression is strongly increased in gliosarcoma, a particularly aggressive form of high grade glioma, isocitrate dehydrogenase 1 (IDH1) wild-type glioblastoma, which have a poorer prognosis than IDH1 mutated glioblastoma, mesenchymal type glioblastomas and recurrent glioblastoma (Tran et al., 2006; Fortin et al., 2009; Perez et al., 2016; Hersh et al., 2018a). Moreover, experimentally generated proneural-like gliomas (PDGF-A expression, p53 knockdown) in rats convert to highly invasive brain cancer with constitutive classical and alternative NF-κB signaling upon introduction of Fn14 (Connolly et al., 2021).
Fn14 activities, however, also contribute to tumor development beyond improving cell migration. For example, Fn14 is induced by proangiogenic proteins such as fibroblast growth factor (FGF)-2 and vascular endothelial growth factor (VEGF)-A and vice versa TWEAK and Fn14 can promote the expression of FGF-2 and VEGF (Donohue et al., 2003; Dai et al., 2009). In line with this reciprocal regulation, it has been observed that TWEAK-expressing Hek293 cells elicit enhanced angiogenesis in nude mice (Ho et al., 2004). There is furthermore evidence from an orthotopic model of urothelial carcinoma that ALKBH3 in cooperation with an autocrine TWEAK-Fn14 loop maintains expression of Fn14 and VEGF in tumor vessels to promote angiogenesis and tumor progression (Shimada et al., 2012).
Considering the complex biology of Fn14 both the inhibition of TWEAK or Fn14 and the activation of Fn14 can have beneficial therapeutic effects (Aggarwal et al., 2012). The interference with Fn14 activity is straightforwardly possible by the help of anti-TWEAK or anti-Fn14 antibodies preventing TWEAK-Fn14 interaction or by soluble fusion proteins containing the extracellular domain of Fn14. The efficacy of TWEAK- and Fn14-blocking antibodies and a Fn14–Fc decoy receptor has been demonstrated in various preclinical disease models (Table 2).
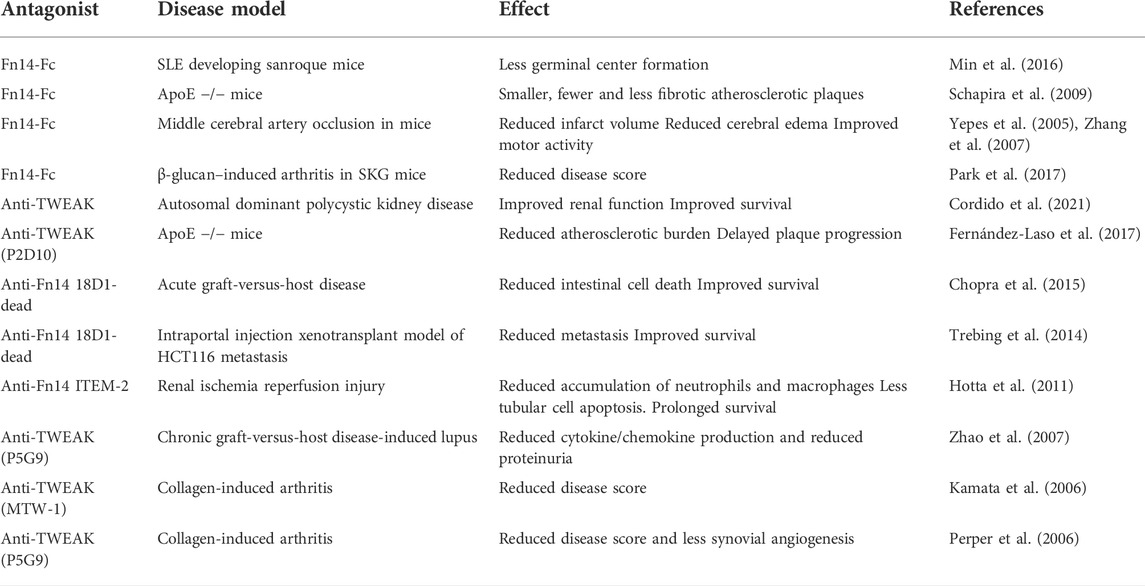
TABLE 2. Examples of the use of TWEAK- and Fn14 antagonists in preclinical disease models.
The usefulness of recombinant soluble TWEAK is limited in several ways. Soluble TWEAK trimers are rapidly cleared from the circulation in mice with a half-life of less than 1 h (Muller et al., 2010). Furthermore, as already discussed, it does not comprehensively stimulate Fn14. Both limitations of sTWEAK can be overcome by genetic fusion with heterologous protein domains. For example, the serum retention of sTWEAK can be strongly enhanced by genetic fusion with serum albumin (Muller et al., 2010). Connecting two or more sTWEAK trimers or enabling cell surface anchoring of sTWEAK furthermore confer membrane TWEAK-like activity (Figure 2). Thus, a Fc-TWEAK fusion protein forms hexamers, has memTWEAK-like activity and presumably act by enforcing close proximity of two liganded Fn14 trimers (Roos et al., 2010). The Fc domain furthermore greatly improves serum retention. It has been furthermore demonstrated that a cell surface antigen-recognizing trimeric scFv-sTWEAK fusion protein acts as sTWEAK in the absence of the cell surface antigen but convert to a molecule with memTWEAK activity when bound to the cell surface exposed target of its scFv domain (Roos et al., 2010). Recombinant TWEAK variants have been used in some preclinical models. However, expression of rec. TWEAK seems to be less efficient than that of antibodies and in general, there is less experience with the translational development and approval of recombinant TNFLs.
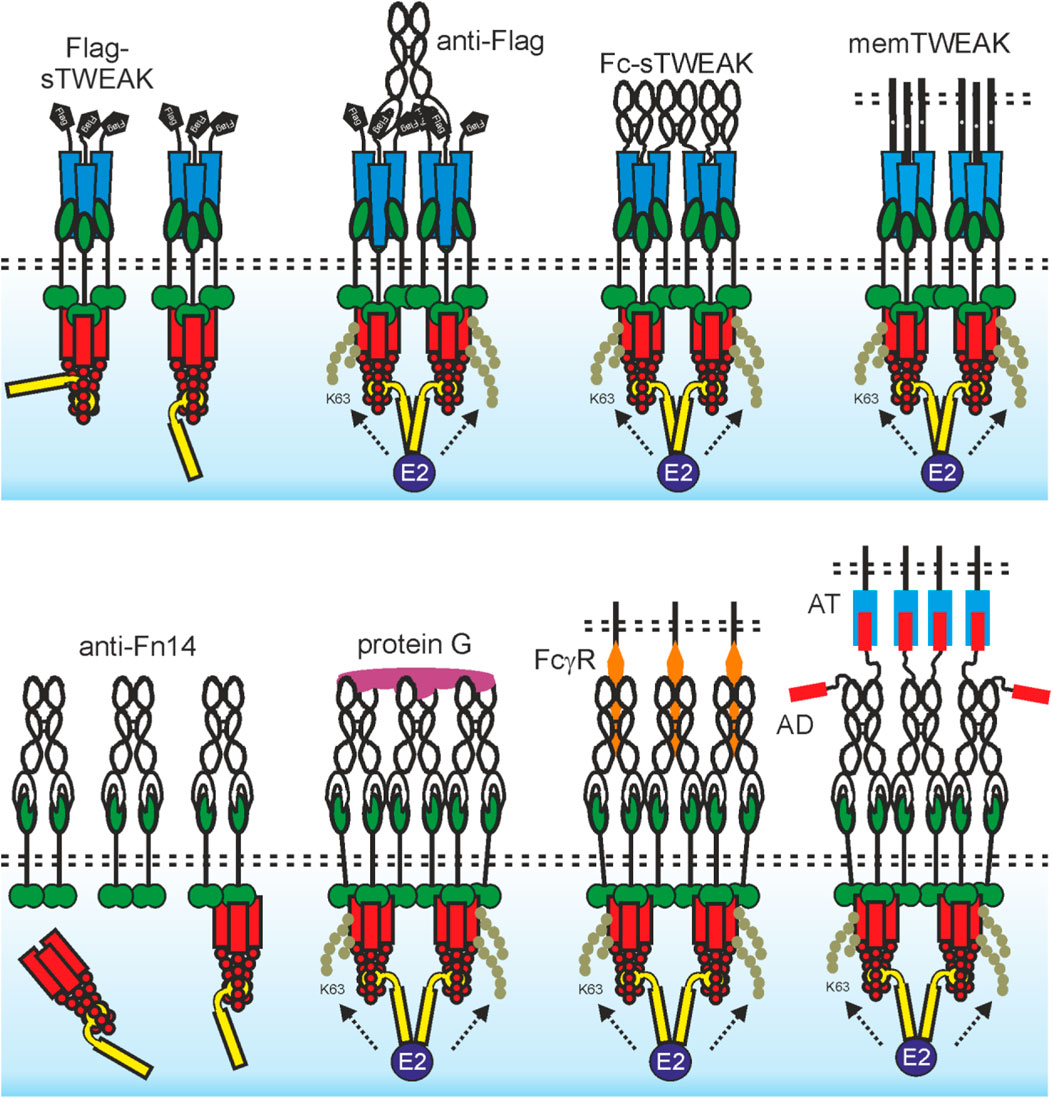
FIGURE 2. The agonistic quality of TWEAK trimers and anti-Fn14 antibodies is determined by oligomerization and membrane-association. Upper panel: An sTWEAK trimer recruits three Fn14 molecules. The Fn14 trimers assembled by sTWEAK in turn recruit complexes of a TRAF2 trimer with one cIAP1 or cIAP2 molecule. This results in reduced availability of these molecules for other binding partners but does not trigger efficient cIAP transactivation. If the proximity of two (or more) of these sTWEAK-associated Fn14 trimers is enforced, e.g., by use of antibody crosslinking (e.g., Flag-tagged sTWEAK with anti-Flag antibody) or by genetic fusion with an oligomerization domain (e.g., Fc domain), this results in close neighborhood of two (or more) TRAF2-cIAP1/2 complexes and thus in cIAP transactivation and classical NF-κB signaling (see also Figure 1). Due to the high concentrations of trimeric Fn14 complexes in the cell-to-cell contact zone with memTWEAK expressing cells, there is spontaneous clustering of the Fn14 complexes and again cIAP transactivation due to the tight neighborhood of TRAF2-cIAP1/2 complexes. Lower panel: An anti-Fn14 antibody recruits two Fn14 molecules. The anti-Fn14-bound Fn14 dimers recruit one TRAF2-cIAP1/2 complex but less efficient than an sTWEAK trimer. Thus, there is some reduction in the cytosolic availability of TRAF2-cIAPs and therefore alternative NF-κB signaling despite weaker as with sTWEAK (see upper panel and Figure 1). If the linkage of three (or more) of these anti-Fn14-associated Fn14 dimers is enforced, e.g. by protein G crosslinking this also induces neighborhood of several TRAF2-cIAP1/2 complexes and thus cIAP transactivation and classical NF-κB signaling. Due to the high concentrations of dimeric Fn14 complexes in the cell-to-cell contact zone with cells decorated with FcγR-bound anti-Fn14 antibodies (or anchoring target (AT)-bound anti-Fn14 antibody fusion proteins with an anchoring domain (AD), there is again spontaneous clustering of the Fn14 complexes and cIAP transactivation.
Anti-Fn14 antibodies alone have no or only limited agonism (Figure 2). However, anti-Fn14 antibodies regularly gain comprehensive and strong agonism upon cross-linking with secondary antibodies or protein G (Salzmann et al., 2013; Trebing et al., 2014). This resembles the situation of sTWEAK which gains maximal and comprehensive memTWEAK-like activity after oligomerization (see above). The idiotype of an anti-TNFR antibody and thus the recognized epitope is typically considered as the main factor determining agonistic activity. However, there is now broad evidence that antibodies specific for a subgroup of the TNFRSF, including Fn14, regularly gain high agonistic activity by FcγR-binding (Wajant, 2015) (Figure 2). Indeed, the non-competitive anti-Fn14 antibodies PDL192 and 5B6 and the blocking antibodies P4A8 and 18D1 recognize different epitopes within the extracellular domain of Fn14. All these antibodies show, however, strong classical NF-κB activation after FcγR-binding (Trebing et al., 2014). Moreover, all combinations of antibody isotype and FcγR type which show binding also result in significant Fn14 activation (Medler et al., 2019). The isotype is therefore obviously only of importance for agonistic activity of an anti-Fn14 antibody in so far that it determines the ability to bind FcγRs (Medler et al., 2019). Importantly, the maximal Fn14 activity achieved with FcγR-anchored antibodies is comparable to those elicited by memTWEAK (Medler et al., 2019). In sum, it can be asserted that oligomerization or FcγR-binding is regularly required, but also sufficient, for the agonism of antibodies targeting Fn14 and that it is largely the sheer cell surface anchoring that constitutes the agonism of FcγR-bound antibodies while FcγR-specific activities are largely irrelevant. With respect to the potential translational exploitation of anti-Fn14 antibodies as agonists, it is important to realize that the required FcγR binding comes along with several limitations. First, due to poor availability of FcγR-expressing cells and/or low cellular FcγR expression levels, an only suboptimal agonistic activity might be reached in vivo. Second, FcγR-mediated effects triggered by the antibody-FcγR interaction can counteract the anticipated therapeutic effects which are actually aspired by the anti-Fn14 antibody treatment. For example, the aim of triggering Fn14-mediated inflammatory activity in tumors might be antagonized by stimulation of the anti-inflammatory FcγRIIB or FcγRIIIA-mediated ADCC of non-transformed Fn14-expressing cells. Third, considerable antibody doses are typically required to overcome competition with serum IgGs for FcγR binding. Worth mentioning the FcγR-binding related limitations of anti-Fn14 antibodies can be overcome by modification in a way enabling them to anchor to cells in a FcγR-independent fashion. Thus, anti-Fn14 antibody fusion proteins having an anchoring domain (e.g., a scFv domain) enabling binding to a cell surface-exposed anchoring target (AT) distinct from FcγRs display strong Fn14 stimulation when presented bound to the AT but not in presence of anchoring target-negative cells (Figure 2). Anchoring dependent Fn14 agonism was also achieved with FcγR-binding defective antibody mutants and Fab2-fragments (Medler et al., 2019; WO2019129644 A1). Worth mentioning, anti-Fn14 immobilized on gold nanoparticles are also highly agonistic (Aido et al., 2021). In this case, it is, however, unclear whether the agonism is due to oligomerization of several anti-Fn14 antibody molecules on a single particle or the surface-associated mode of presentation.
The high tumor-associated expression of Fn14 (Table 1) along with its proliferation-, cell migration- and angiogenesis promoting activities lead to the consideration of the TWEAK/Fn14 system as a promising target for tumor therapy. In this context, two major approaches have been discussed and evaluated. First, the exploitation of Fn14 expression to direct anti-cancer effector activities to the tumor and to destroy Fn14-expressing tumor cells. Second, blockade of Fn14 or TWEAK to inhibit protumoral activities of these molecules. A third, yet not or only poorly investigated possibility is to activate Fn14 to exploit its proinflammatory and cell death sensitizing properties to destroy tumor cells and/or the tumor microenvironment.
The principle usefulness of targeting Fn14 as a tumor-associated tumor antigen has been successfully demonstrated in preclinical studies with antibodies, antibody drug conjugates and CAR T-cells specific for Fn14 (Table 3).
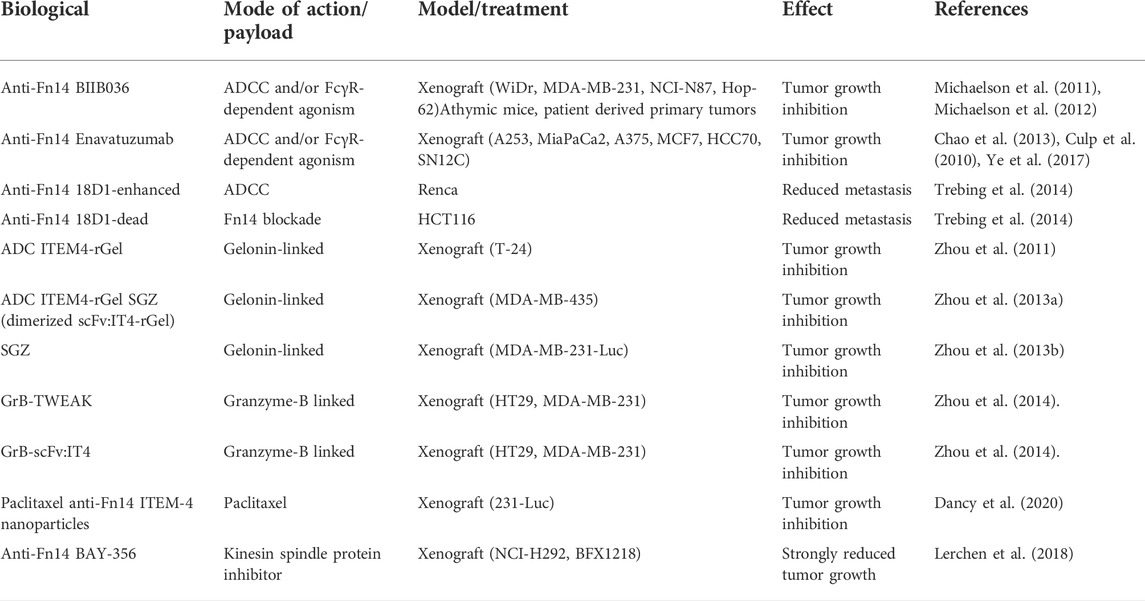
TABLE 3. Anti-tumor activity of Fn14-targeting biologicals.
The anti-Fn14 antibody BIIB036 (humanized version of anti-Fn14 P4A8) showed a strong anti-tumor effect on various xenografted tumor cell lines (Table 3) and patient derived primary tumors (Michaelson et al., 2011; Michaelson et al., 2012). In vitro studies proved efficient ADCC induction by BIIB036 (Michaelson et al., 2011). Similarly, the anti-Fn14 antibody enavatuzumab (humanized version of anti-Fn14 PDL192) displayed anti-tumor efficacy on several xenografted tumor cell lines and again this correlated in most cases with the ability of the antibody to interact with FcγRs and ADCC induction (Culp et al., 2010; Chao et al., 2013; Ye et al., 2017). The anti-Fn14 antibody 18D1 furthermore showed a FcγR-dependent antitumoral effect in the syngeneic Renca lung metastasis model (Trebing et al., 2014). As discussed already above, anti-Fn14 antibodies regularly acquire high memTWEAK-mimicking agonistic activity upon FcγR-binding. The FcγR-dependent antitumoral effects of BIIB036, enavatuzumab and 18D1 are thus not necessarily indicative for an exclusive and obligate relevance of FcγR effector mechanisms but could, at least in some cases, also reflect the stimulation of Fn14 signaling. In fact, enavatuzumab inhibited xenotransplanted BT549 tumor cells and this correlated with the ability of these tumor cells to respond to enavatuzumab with activation of the classical and alternative NF-κB pathway (Purcell et al., 2014). In sum, Fc-FcγR-interaction dependent antitumor activity of anti-Fn14 antibodies has been demonstrated in various models in vivo, however, it has, not been clarified yet to which extend this antitumor activity is due to stimulation of FcγR-induced effector functions, e.g., ADCC, and/or the enhanced agonism of FcγR-bound anti-Fn14 antibodies and Fn14-mediated signaling. In fact, the two mechanisms not necessarily act mutually exclusive. As discussed before, Fn14 can also elicit protumoral activities. Such activities could be stimulated by FcγR-interacting anti-Fn14 antibodies, too, but should be of transient nature in the case of antibodies triggering destruction of the Fn14-expressing cells. The enthusiasm about the potential of Fn14-targeting with FcγR-interacting antibodies, however, is dampened through a phase I clinical study with enavatuzumab in patients with solid malignancies revealing dose-limiting liver and pancreatic toxicities (Lam et al., 2018).
Immunotoxins composed of geolin chemically linked to the anti-Fn14 antibody ITEM-4 or genetically fused to an ITEM-4 derived scFv were also found to show strong antitumor effects to xenotransplanted cancer cells (T-24, MDA-MB4-432 melanoma cells, MDA-MB-231 breast cancer cells) which were otherwise poorly sensitive for ITEM-4 (Zhou et al., 2011; Zhou et al., 2013a; Zhou et al., 2013b). Likewise, a Fn14-specific antibody-drug conjugate with an aglycosylated variant of the anti-Fn14 antibody Bay-365 and a kinesin spindle protein inhibitor showed antitumor efficacy against xenotransplanted NCI-H292 cells (Lerchen et al., 2018). Granzyme B fusion proteins of sTWEAK or an anti-Fn14 scFv domain showed selective cytotoxicity on Fn14-expressing cells and inhibited tumor growth of xenografted HT29 and MDA-MB-231 tumor cells, too (Zhou et al., 2014). The usefulness of Fn14 as a target to direct cytotoxic drugs to tumors has also been reported with ITEM-4 conjugated paclitaxel-loaded nanoparticles which outperformed nontargeted nanoparticles in preclinical models of primary and brain metastatic triple-negative breast cancer (Dancy et al., 2020). In view of the fact that anti-Fn14 antibodies immobilized on gold nanoparticles acquire agonistic activity (Aido et al., 2021), it appears possible that engagement of Fn14 signaling contributed to the superior antitumoral activity of Fn14-targeted paclitaxel-loaded nanoparticles.
Considering the protumoral effects of Fn14, especially its cell migration-stimulating activity (see 3.), there is also evidence for the idea that blockade of the Fn14/TWEAK system can elicit antitumoral effects. It has been observed in an intraportal injection xenotransplant model of hepatic metastasis with HCT116 colon cancer cells that a FcγR-binding defective variant of the blocking anti-Fn14 antibody 18D1 reduces metastasis (Trebing et al., 2014). Regression of tumor development has also been reported for the TWEAK-neutralizing antibody RG7212 in several xenograft tumor models (ACHN, MDA-MB-231, Caki-1, Calu-3) with high Fn14 expression (Yin et al., 2014). Moreover, RG7212 showed very good tolerability and evidence for tumor regression in heavily pretreated patients with solid tumors in a phase I monotherapy study (Lassen et al., 2015). Since there is yet no reliable possibility to distinguish between endogenous sTWEAK- and memTWEAK-induced effects, it is fully unclear whether the beneficial effects of the inhibition of the TWEAK-Fn14 interaction in these studies reflect protumoral activities of soluble or membrane TWEAK or of both forms of the ligand. It is also unclear whether the antitumoral effects of TWEAK/Fn14 blockade in the aforementioned models mirror general protumoral activities of Fn14 or whether the net effect of the Fn14 activation on tumor development changes if a certain threshold of Fn14 activity is exceeded. Indeed, there is evidence for different thresholds of Fn14 stimulation for enhancement of TNFR1-induced cell death and activation of the alternative NF-κB pathway (Salzmann et al., 2013). In the absence of FcγR-binding, anti-Fn14 antibodies, similar to sTWEAK, significantly trigger p100 processing, the hallmark of the alternative NF-κB pathway. However, while sTWEAK is also sufficient to sensitize for TNF receptor 1-induced cell death, free anti-Fn14 antibodies fail to do so or even block the corresponding sTWEAK response (Salzmann et al., 2013). Therefore, it appears that oligomerized or FcγR-bound Fn14 antibodies comprehensively mimic memTWEAK, while free bivalent anti-Fn14 antibodies only resemble sTWEAK in a response selective manner. One have to consider that in vivo sTWEAK is presumably dominant over memTWEAK due to the efficient processing of memTWEAK to sTWEAK by furin proteases. Therefore, even in cases where the endogenous activity of the TWEAK/Fn14 system elicits protumoral effects, strong exogenous Fn14 stimulation, especially with memTWEAK mimicking Fn14 agonists, might nevertheless display antitumoral activities.
Since FcγR-binding regularly converts anti-Fn14 antibodies into strong membrane TWEAK-mimicking agonists one could mean that the potential of triggering antitumoral effector functions should be evident from the various studies with anti-Fn14 antibodies described above. Indeed, there is initial evidence from in vivo studies with enavatuzumab that anti-Fn14-induced “agonistic” chemokine production result in enhanced migration of myeloid cells into the tumor microenvironment where then the same anti-Fn14 antibody acts antitunoral by its effector activity (Ye et al., 2017). However, the full antitumoral potential of anti-Fn14 agonism is presumably underestimated from studies with anti-Fn14 antibodies. Besides the fact that antibody-linked effector activities might mask antitumoral activities of Fn14 molecules stimulated by FcγR-bound anti-Fn14 antibodies, one have to consider that most anti-Fn14 antibodies have been primarily studied in xenograft models (Table 3) without a functional adaptive immune system. It is thus tempting to speculate that antitumor activities related to proinflammatory Fn14 signaling engaged by FcγR-bound anti-Fn14 antibodies have been underestimated, yet. The antitumoral activity of pure Fn14 agonists, however, has been poorly addressed yet in vivo. This is not at least due to the fact that there are yet no agonistic Fn14 antibodies displaying FcγR-independent agonism. More elaborated studies with recombinant TWEAK variants are therefore necessary in the future to evaluate the therapeutic potential of agonistic Fn14 targeting. A first promising study in this respect revealed strong inhibition of tumor development from MDA-MB-231 tumor cells injected in nude mice after treatment with Fc-TWEAK or adenoviral administration of TWEAK (Michaelson et al., 2011). In view of very recent results, which identified TRAF2 and the cIAPs as major targets facilitating CD8+ T-cell elimination of tumor cells in a TNF-dependent manner in patients responding to immune checkpoint blockade (Vredevoogd et al., 2019), it appears furthermore promising to evaluate the potential of Fn14 agonists in combination therapies with checkpoint inhibitors.
As already briefly discussed, Fn14-related activities crucially contribute to complications arising from overshooting and/or chronic repair processes. In view of the concept of cancer as a wound that does not heal, it appears possible to target the TWEAK/Fn14 system in cancer not with the aim to attack the tumor development process itself but rather to attenuate detrimental effects of cancer development and cancer treatments on healthy parts of the body. Indeed, there is evidence from animal models that Fn14 blockade can reduce tumor-associated cachexia or therapy-induced diarrhea (Johnston et al., 2015; Sezaki et al., 2017). Furthermore, in good accordance with its sensitizing effect for cytotoxic TNFR1 signaling and the well established high sensitivity of the intestinal epithelium for TNF, it was found that a blocking anti-Fn14 antibody has a strong protective effect on graft-versus host disease in the intestine (Chopra et al., 2015). Notably, the GvHD protective effect of Fn14 blockade allowed efficient treatment of syngeneic tumors by adoptive transfer of allogeneic T-cells (Chopra et al., 2015).
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by a grant of the Deutsche Krebshilfe to CO and HW (project number 70114009).
The University of Wuerzburg filed a patent describing novel Fn14 agonists with OZ, CO, and HW as co-inventors.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acharya, S., Prabhu, P., Patil, V. S., Acharya, A. B., and Nikhil, K. (2019). Immunohistochemical expression of tumor necrosis factor-like weak inducer of apoptosis and fibroblast growth factor-inducible immediate early response protein 14 in oral squamous cell carcinoma and its implications. J. Investig. Clin. Dent. 10, e12469. doi:10.1111/jicd.12469
Aggarwal, B. B., Gupta, S. C., and Kim, J. H. (2012). Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 119, 651–665. doi:10.1182/blood-2011-04-325225
Aido, A., Zaitseva, O., Wajant, H., Buzgo, M., and Simaite, A. (2021). Anti-Fn14 antibody-conjugated nanoparticles display membrane TWEAK-like agonism. Pharmaceutics 13, 1072. doi:10.3390/pharmaceutics13071072
Anany, M. A., Kreckel, J., Fullsack, S., Rosenthal, A., Otto, C., Siegmund, D., et al. (2018). Soluble TNF-like weak inducer of apoptosis (TWEAK) enhances poly(I:C)-induced RIPK1-mediated necroptosis. Cell. Death Dis. 9, 1084. doi:10.1038/s41419-018-1137-1
Armstrong, C. L., Galisteo, R., Brown, S. A., and Winkles, J. A. (2016). TWEAK activation of the non-canonical NF-κB signaling pathway differentially regulates melanoma and prostate cancer cell invasion. Oncotarget 7, 81474–81492. doi:10.18632/oncotarget.13034
Asrani, K., Keri, R. A., Galisteo, R., Brown, S. A., Morgan, S. J., Ghosh, A., et al. (2013). The HER2- and heregulin β1 (HRG)-inducible TNFR superfamily member Fn14 promotes HRG-driven breast cancer cell migration, invasion, and MMP9 expression. Mol. Cancer Res. 11, 393–404. doi:10.1158/1541-7786.MCR-12-0542
Aye, L., Song, X., Yang, J., Hu, L., Sun, X., Zhou, J., et al. (2021). Identification of a costimulatory molecule gene signature to predict survival and immunotherapy response in head and neck squamous cell carcinoma. Front. Cell. Dev. Biol. 9, 695533. doi:10.3389/fcell.2021.695533
Baeuerle, P. A., and Baltimore, D. (1988a). Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-kappa B transcription factor. Cell. 53, 211–217. doi:10.1016/0092-8674(88)90382-0
Baeuerle, P. A., and Baltimore, D. (1988b). I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science 242, 540–546. doi:10.1126/science.3140380
Bodmer, J. L., Schneider, P., and Tschopp, J. (2002). The molecular architecture of the TNF superfamily. Trends biochem. Sci. 27, 19–26. doi:10.1016/s0968-0004(01)01995-8
Bover, L. C., Cardo-Vila, M., Kuniyasu, A., Sun, J., Rangel, R., Takeya, M., et al. (2007). A previously unrecognized protein-protein interaction between TWEAK and CD163: Potential biological implications. J. Immunol. 178, 8183–8194. doi:10.4049/jimmunol.178.12.8183
Burkly, L. C., Michaelson, J. S., and Zheng, T. S. (2011). TWEAK/Fn14 pathway: An immunological switch for shaping tissue responses. Immunol. Rev. 244, 99–114. doi:10.1111/j.1600-065X.2011.01054.x
Carmona Arana, J. A., Seher, A., Neumann, M., Lang, I., Siegmund, D., and Wajant, H. (2014). TNF receptor-associated factor 1 is a major target of soluble TWEAK. Front. Immunol. 5, 63. doi:10.3389/fimmu.2014.00063
Chao, D. T., Su, M., Tanlimco, S., Sho, M., Choi, D., Fox, M., et al. (2013). Expression of TweakR in breast cancer and preclinical activity of enavatuzumab, a humanized anti-TweakR mAb. J. Cancer Res. Clin. Oncol. 139, 315–325. doi:10.1007/s00432-012-1332-x
Cheng, E., Whitsett, T. G., Tran, N. L., and Winkles, J. A. (2015). The TWEAK receptor Fn14 is an src-inducible protein and a positive regulator of src-driven cell invasion. Mol. Cancer Res. 13, 575–583. doi:10.1158/1541-7786.MCR-14-0411
Chicheportiche, Y., Bourdon, P. R., Xu, H., Hsu, Y. M., Scott, H., Hession, C., et al. (1997). TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 272, 32401–32410. doi:10.1074/jbc.272.51.32401
Chopra, M., Brandl, A., Siegmund, D., Mottok, A., Schafer, V., Biehl, M., et al. (2015). Blocking TWEAK-Fn14 interaction inhibits hematopoietic stem cell transplantation-induced intestinal cell death and reduces GVHD. Blood 126, 437–444. doi:10.1182/blood-2015-01-620583
Connolly, N. P., Galisteo, R., Xu, S., Bar, E. E., Peng, S., Tran, N. L., et al. (2021). Elevated fibroblast growth factor-inducible 14 expression transforms proneural-like gliomas into more aggressive and lethal brain cancer. Glia 69, 2199–2214. doi:10.1002/glia.24018
Cordido, A., Nuñez-Gonzalez, L., Martinez-Moreno, J. M., Lamas-Gonzalez, O., Rodriguez-Osorio, L., Perez-Gomez, M. V., et al. (2021). TWEAK signaling pathway blockade slows cyst growth and disease progression in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 32, 1913–1932. doi:10.1681/ASN.2020071094
Culp, P. A., Choi, D., Zhang, Y., Yin, J., Seto, P., Ybarra, S. E., et al. (2010). Antibodies to TWEAK receptor inhibit human tumor growth through dual mechanisms. Clin. Cancer Res. 16, 497–508. doi:10.1158/1078-0432.CCR-09-1929
Dai, L., Gu, L., Ding, C., Qiu, L., and Di, W. (2009). TWEAK promotes ovarian cancer cell metastasis via NF-kappaB pathway activation and VEGF expression. Cancer Lett. 283, 159–167. doi:10.1016/j.canlet.2009.03.036
Dancy, J. G., Wadajkar, A. S., Connolly, N. P., Galisteo, R., Ames, H. M., Peng, S., et al. (2020). Decreased nonspecific adhesivity, receptor-targeted therapeutic nanoparticles for primary and metastatic breast cancer. Sci. Adv. 6, eaax3931. doi:10.1126/sciadv.aax3931
Dhruv, H. D., Whitsett, T. G., Jameson, N. M., Patel, F., Winkles, J. A., Berens, M. E., et al. (2014). Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) promotes glioblastoma cell chemotaxis via Lyn activation. Carcinogenesis 35, 218–226. doi:10.1093/carcin/bgt289
Dickens, L. S., Powley, I. R., Hughes, M. A., and Macfarlane, M. (2012). The 'complexities' of life and death: Death receptor signalling platforms. Exp. Cell. Res. 318, 1269–1277. doi:10.1016/j.yexcr.2012.04.005
Donohue, P. J., Richards, C. M., Brown, S. A., Hanscom, H. N., Buschman, J., Thangada, S., et al. (2003). TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arterioscler. Thromb. Vasc. Biol. 23, 594–600. doi:10.1161/01.ATV.0000062883.93715.37
Duckett, C. S., and Thompson, C. B. (1997). CD30-dependent degradation of TRAF2: Implications for negative regulation of TRAF signaling and the control of cell survival. Genes. Dev. 11, 2810–2821. doi:10.1101/gad.11.21.2810
Dueber, E. C., Schoeffler, A. J., Lingel, A., Elliott, J. M., Fedorova, A. V., Giannetti, A. M., et al. (2011). Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 334, 376–380. doi:10.1126/science.1207862
Ensign, S. P., Roos, A., Mathews, I. T., Dhruv, H. D., Tuncali, S., Sarkaria, J. N., et al. (2016). SGEF is regulated via TWEAK/Fn14/NF-κB signaling and promotes survival by modulation of the DNA repair response to temozolomide. Mol. Cancer Res. 14, 302–312. doi:10.1158/1541-7786.MCR-15-0183
Felli, N., Pedini, F., Zeuner, A., Petrucci, E., Testa, U., Conticello, C., et al. (2005). Multiple members of the TNF superfamily contribute to IFN-gamma-mediated inhibition of erythropoiesis. J. Immunol. 175, 1464–1472. doi:10.4049/jimmunol.175.3.1464
Feltham, R., Bettjeman, B., Budhidarmo, R., Mace, P. D., Shirley, S., Condon, S. M., et al. (2011). Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J. Biol. Chem. 286, 17015–17028. doi:10.1074/jbc.M111.222919
Fernández-Laso, V., Sastre, C., Méndez-Barbero, N., Egido, J., Martín-Ventura, J. L., Gómez-Guerrero, C., et al. (2017). TWEAK blockade decreases atherosclerotic lesion size and progression through suppression of STAT1 signaling in diabetic mice. Sci. Rep. 7, 46679. doi:10.1038/srep46679
Fick, A., Lang, I., Schafer, V., Seher, A., Trebing, J., Weisenberger, D., et al. (2012). Studies of binding of tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) to fibroblast growth factor inducible 14 (Fn14). J. Biol. Chem. 287, 484–495. doi:10.1074/jbc.M111.287656
Fortin Ensign, S. P., Mathews, I. T., Eschbacher, J. M., Loftus, J. C., Symons, M. H., and Tran, N. L. (2013). The Src homology 3 domain-containing guanine nucleotide exchange factor is overexpressed in high-grade gliomas and promotes tumor necrosis factor-like weak inducer of apoptosis-fibroblast growth factor-inducible 14-induced cell migration and invasion via tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 288, 21887–21897. doi:10.1074/jbc.M113.468686
Fortin, S. P., Ennis, M. J., Savitch, B. A., Carpentieri, D., Mcdonough, W. S., Winkles, J. A., et al. (2009). Tumor necrosis factor-like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol. Cancer Res. 7, 1871–1881. doi:10.1158/1541-7786.MCR-09-0194
Fortin, S. P., Ennis, M. J., Schumacher, C. A., Zylstra-Diegel, C. R., Williams, B. O., Ross, J. T., et al. (2012). Cdc42 and the guanine nucleotide exchange factors Ect2 and trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol. Cancer Res. 10, 958–968. doi:10.1158/1541-7786.MCR-11-0616
Fullsack, S., Rosenthal, A., Wajant, H., and Siegmund, D. (2019). Redundant and receptor-specific activities of TRADD, RIPK1 and FADD in death receptor signaling. Cell. Death Dis. 10, 122. doi:10.1038/s41419-019-1396-5
Girgenrath, M., Weng, S., Kostek, C. A., Browning, B., Wang, M., Brown, S. A., et al. (2006). TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J. 25, 5826–5839. doi:10.1038/sj.emboj.7601441
Grabinger, T., Bode, K. J., Demgenski, J., Seitz, C., Delgado, M. E., Kostadinova, F., et al. (2017). Inhibitor of apoptosis protein-1 regulates tumor necrosis factor-mediated destruction of intestinal epithelial cells. Gastroenterology 152, 867–879. doi:10.1053/j.gastro.2016.11.019
Gu, L., Dai, L., Cao, C., Zhu, J., Ding, C., Xu, H. B., et al. (2013). Functional expression of TWEAK and the receptor Fn14 in human malignant ovarian tumors: Possible implication for ovarian tumor intervention. PLoS One 8, e57436. doi:10.1371/journal.pone.0057436
Henkel, T., Zabel, U., van Zee, K., Muller, J. M., Fanning, E., and Baeuerle, P. A. (1992). Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-kappa B subunit. Cell. 68, 1121–1133. doi:10.1016/0092-8674(92)90083-o
Hersh, D. S., Harder, B. G., Roos, A., Peng, S., Heath, J. E., Legesse, T., et al. (2018a). The TNF receptor family member Fn14 is highly expressed in recurrent glioblastoma and in GBM patient-derived xenografts with acquired temozolomide resistance. Neuro. Oncol. 20, 1321–1330. doi:10.1093/neuonc/noy063
Hersh, D. S., Peng, S., Dancy, J. G., Galisteo, R., Eschbacher, J. M., Castellani, R. J., et al. (2018b). Differential expression of the TWEAK receptor Fn14 in IDH1 wild-type and mutant gliomas. J. Neurooncol. 138, 241–250. doi:10.1007/s11060-018-2799-3
Ho, D. H., Vu, H., Brown, S. A., Donohue, P. J., Hanscom, H. N., and Winkles, J. A. (2004). Soluble tumor necrosis factor-like weak inducer of apoptosis overexpression in HEK293 cells promotes tumor growth and angiogenesis in athymic nude mice. Cancer Res. 64, 8968–8972. doi:10.1158/0008-5472.CAN-04-1879
Hotta, K., Sho, M., Yamato, I., Shimada, K., Harada, H., Akahori, T., et al. (2011). Direct targeting of fibroblast growth factor-inducible 14 protein protects against renal ischemia reperfusion injury. Kidney Int. 79, 179–188. doi:10.1038/ki.2010.379
Hu, G., Liang, L., Liu, Y., Liu, J., Tan, X., Xu, M., et al. (2019). TWEAK/Fn14 interaction confers aggressive properties to cutaneous squamous cell carcinoma. J. Invest. Dermatol. 139, 796–806. doi:10.1016/j.jid.2018.09.035
Huang, D. B., Huxford, T., Chen, Y. Q., and Ghosh, G. (1997). The role of DNA in the mechanism of NFkappaB dimer formation: Crystal structures of the dimerization domains of the p50 and p65 subunits. Structure 5, 1427–1436. doi:10.1016/s0969-2126(97)00293-1
Huang, M., Narita, S., Tsuchiya, N., Ma, Z., Numakura, K., Obara, T., et al. (2011). Overexpression of Fn14 promotes androgen-independent prostate cancer progression through MMP-9 and correlates with poor treatment outcome. Carcinogenesis 32, 1589–1596. doi:10.1093/carcin/bgr182
Jandova, J., Mason, C. J., Pawar, S. C., and Watts, G. S. (2015). Fn14 receptor promotes invasive potential and metastatic capacity of non-small lung adenocarcinoma cells through the up-regulation of integrin α6. Neoplasma 62, 41–52. doi:10.4149/neo_2015_006
Johnston, A. J., Murphy, K. T., Jenkinson, L., Laine, D., Emmrich, K., Faou, P., et al. (2015). Targeting of Fn14 prevents cancer-induced cachexia and prolongs survival. Cell. 162, 1365–1378. doi:10.1016/j.cell.2015.08.031
Kamata, K., Kamijo, S., Nakajima, A., Koyanagi, A., Kurosawa, H., Yagita, H., et al. (2006). Involvement of TNF-like weak inducer of apoptosis in the pathogenesis of collagen-induced arthritis. J. Immunol. 177, 6433–6439. doi:10.4049/jimmunol.177.9.6433
Kaplan, M. J., Lewis, E. E., Shelden, E. A., Somers, E., Pavlic, R., Mccune, W. J., et al. (2002). The apoptotic ligands TRAIL, TWEAK, and Fas ligand mediate monocyte death induced by autologous lupus T cells. J. Immunol. 169, 6020–6029. doi:10.4049/jimmunol.169.10.6020
Kaptein, A., Jansen, M., Dilaver, G., Kitson, J., Dash, L., Wang, E., et al. (2000). Studies on the interaction between TWEAK and the death receptor WSL-1/TRAMP (DR3). FEBS Lett. 485, 135–141. doi:10.1016/s0014-5793(00)02219-5
Karl, I., Jossberger-Werner, M., Schmidt, N., Horn, S., Goebeler, M., Leverkus, M., et al. (2014). TRAF2 inhibits TRAIL- and CD95L-induced apoptosis and necroptosis. Cell. Death Dis. 5, e1444. doi:10.1038/cddis.2014.404
Kawakita, T., Shiraki, K., Yamanaka, Y., Yamaguchi, Y., Saitou, Y., Enokimura, N., et al. (2004). Functional expression of TWEAK in human hepatocellular carcinoma: Possible implication in cell proliferation and tumor angiogenesis. Biochem. Biophys. Res. Commun. 318, 726–733. doi:10.1016/j.bbrc.2004.04.084
Kumar, M., Makonchuk, D. Y., Li, H., Mittal, A., and Kumar, A. (2009). TNF-like weak inducer of apoptosis (TWEAK) activates proinflammatory signaling pathways and gene expression through the activation of TGF-beta-activated kinase 1. J. Immunol. 182, 2439–2448. doi:10.4049/jimmunol.0803357
Kuramitsu, K., Sverdlov, D. Y., Liu, S. B., Csizmadia, E., Burkly, L., Schuppan, D., et al. (2013). Failure of fibrotic liver regeneration in mice is linked to a severe fibrogenic response driven by hepatic progenitor cell activation. Am. J. Pathol. 183, 182–194. doi:10.1016/j.ajpath.2013.03.018
Kwon, O. H., Park, S. J., Kang, T. W., Kim, M., Kim, J. H., Noh, S. M., et al. (2012). Elevated fibroblast growth factor-inducible 14 expression promotes gastric cancer growth via nuclear factor-κB and is associated with poor patient outcome. Cancer Lett. 314, 73–81. doi:10.1016/j.canlet.2011.09.016
Lam, E. T., Eckhardt, S. G., Messersmith, W., Jimeno, A., O'Bryant, C. L., Ramanathan, R. K., et al. (2018). Phase I study of enavatuzumab, a first-in-class humanized monoclonal antibody targeting the TWEAK receptor, in patients with advanced solid tumors. Mol. Cancer Ther. 17, 215–221. doi:10.1158/1535-7163.MCT-17-0330
Lassen, U. N., Meulendijks, D., Siu, L. L., Karanikas, V., Mau-Sorensen, M., Schellens, J. H., et al. (2015). A phase I monotherapy study of RG7212, a first-in-class monoclonal antibody targeting TWEAK signaling in patients with advanced cancers. Clin. Cancer Res. 21, 258–266. doi:10.1158/1078-0432.CCR-14-1334
Lerchen, H. G., Wittrock, S., Stelte-Ludwig, B., Sommer, A., Berndt, S., Griebenow, N., et al. (2018). Antibody-drug conjugates with pyrrole-based KSP inhibitors as the payload class. Angew. Chem. Int. Ed. Engl. 57, 15243–15247. doi:10.1002/anie.201807619
Li, L., Ameri, A. H., Wang, S., Jansson, K. H., Casey, O. M., Yang, Q., et al. (2019). EGR1 regulates angiogenic and osteoclastogenic factors in prostate cancer and promotes metastasis. Oncogene 38, 6241–6255. doi:10.1038/s41388-019-0873-8
Li, N., Hu, W. J., Shi, J., Xue, J., Guo, W. X., Zhang, Y., et al. (2013). Roles of fibroblast growth factor-inducible 14 in hepatocellular carcinoma. Asian pac. J. Cancer Prev. 14, 3509–3514. doi:10.7314/apjcp.2013.14.6.3509
Li, X., Zhu, W., Chen, Z., Luo, L., Huang, J., Zhang, F., et al. (2014). Fibroblast growth factor-inducible 14 regulates cell growth and multidrug resistance of small-cell lung cancer through the nuclear factor-κB pathway. Anticancer. Drugs 25, 1152–1164. doi:10.1097/CAD.0000000000000153
Lian, M., Cao, H., Baranova, A., Kural, K. C., Hou, L., He, S., et al. (2020). Aging-associated genes TNFRSF12A and CHI3L1 contribute to thyroid cancer: An evidence for the involvement of hypoxia as a driver. Oncol. Lett. 19, 3634–3642. doi:10.3892/ol.2020.11530
Lin, B. R., Huang, M. T., Chen, S. T., Jeng, Y. M., Li, Y. J., Liang, J. T., et al. (2012). Prognostic significance of TWEAK expression in colorectal cancer and effect of its inhibition on invasion. Ann. Surg. Oncol. 19, S385–S394. doi:10.1245/s10434-011-1825-x
Lopez, J., John, S. W., Tenev, T., Rautureau, G. J., Hinds, M. G., Francalanci, F., et al. (2011). CARD-mediated autoinhibition of cIAP1's E3 ligase activity suppresses cell proliferation and migration. Mol. Cell. 42, 569–583. doi:10.1016/j.molcel.2011.04.008
Mace, P. D., Smits, C., Vaux, D. L., Silke, J., and Day, C. L. (2010). Asymmetric recruitment of cIAPs by TRAF2. J. Mol. Biol. 400, 8–15. doi:10.1016/j.jmb.2010.04.055
Maecker, H., Varfolomeev, E., Kischkel, F., Lawrence, D., Leblanc, H., Lee, W., et al. (2005). TWEAK attenuates the transition from innate to adaptive immunity. Cell. 123, 931–944. doi:10.1016/j.cell.2005.09.022
Marsters, S. A., Sheridan, J. P., Pitti, R. M., Brush, J., Goddard, A., and Ashkenazi, A. (1998). Identification of a ligand for the death-domain-containing receptor Apo3. Curr. Biol. 8, 525–528. doi:10.1016/s0960-9822(98)70204-0
Martin-Sanchez, D., Fontecha-Barriuso, M., Carrasco, S., Sanchez-Nino, M. D., Massenhausen, A. V., Linkermann, A., et al. (2018). TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl. Acad. Sci. U. S. A. 115, 4182–4187. doi:10.1073/pnas.1716578115
Martinez-Aranda, A., Hernandez, V., Guney, E., Muixi, L., Foj, R., Baixeras, N., et al. (2015). FN14 and GRP94 expression are prognostic/predictive biomarkers of brain metastasis outcome that open up new therapeutic strategies. Oncotarget 6, 44254–44273. doi:10.18632/oncotarget.5471
Martinez-Aranda, A., Hernandez, V., Moreno, F., Baixeras, N., Cuadras, D., Urruticoechea, A., et al. (2017). Predictive and prognostic brain metastases assessment in luminal breast cancer patients: FN14 and GRP94 from diagnosis to prophylaxis. Front. Oncol. 7, 283. doi:10.3389/fonc.2017.00283
Medler, J., Nelke, J., Weisenberger, D., Steinfatt, T., Rothaug, M., Berr, S., et al. (2019). TNFRSF receptor-specific antibody fusion proteins with targeting controlled FcγR-independent agonistic activity. Cell. Death Dis. 10, 224. doi:10.1038/s41419-019-1456-x
Michaelson, J. S., Amatucci, A., Kelly, R., Su, L., Garber, E., Day, E. S., et al. (2011). Development of an Fn14 agonistic antibody as an anti-tumor agent. MAbs 3, 362–375. doi:10.4161/mabs.3.4.16090
Michaelson, J. S., Kelly, R., Yang, L., Zhang, X., Wortham, K., and Joseph, I. B. (2012). The anti-Fn14 antibody BIIB036 inhibits tumor growth in xenografts and patient derived primary tumor models and enhances efficacy of chemotherapeutic agents in multiple xenograft models. Cancer Biol. Ther. 13, 812–821. doi:10.4161/cbt.20564
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 114, 181–190. doi:10.1016/s0092-8674(03)00521-x
Min, H. K., Kim, S. M., Park, J. S., Byun, J. K., Lee, J., Kwok, S. K., et al. (2016). Fn14-Fc suppresses germinal center formation and pathogenic B cells in a lupus mouse model via inhibition of the TWEAK/Fn14 Pathway. J. Transl. Med. 14, 98. doi:10.1186/s12967-016-0846-4
Mittal, A., Bhatnagar, S., Kumar, A., Lach-Trifilieff, E., Wauters, S., Li, H., et al. (2010a). The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J. Cell. Biol. 188, 833–849. doi:10.1083/jcb.200909117
Mittal, A., Bhatnagar, S., Kumar, A., Paul, P. K., Kuang, S., and Kumar, A. (2010b). Genetic ablation of TWEAK augments regeneration and post-injury growth of skeletal muscle in mice. Am. J. Pathol. 177, 1732–1742. doi:10.2353/ajpath.2010.100335
Muller, N., Schneider, B., Pfizenmaier, K., and Wajant, H. (2010). Superior serum half life of albumin tagged TNF ligands. Biochem. Biophys. Res. Commun. 396, 793–799. doi:10.1016/j.bbrc.2010.04.134
Nakayama, M., Kayagaki, N., Yamaguchi, N., Okumura, K., and Yagita, H. (2000). Involvement of TWEAK in interferon gamma-stimulated monocyte cytotoxicity. J. Exp. Med. 192, 1373–1380. doi:10.1084/jem.192.9.1373
Park, J. S., Kim, S. M., Jung, K. A., Lee, J., Kwok, S. K., Cho, M. L., et al. (2017). Inhibition of the TWEAK/Fn14 pathway attenuates autoimmune arthritis in a SKG mouse model. Histol. Histopathol. 32, 481–490. doi:10.14670/HH-11-813
Perez, J. G., Tran, N. L., Rosenblum, M. G., Schneider, C. S., Connolly, N. P., Kim, A. J., et al. (2016). The TWEAK receptor Fn14 is a potential cell surface portal for targeted delivery of glioblastoma therapeutics. Oncogene 35, 2145–2155. doi:10.1038/onc.2015.310
Perper, S. J., Browning, B., Burkly, L. C., Weng, S., Gao, C., Giza, K., et al. (2006). TWEAK is a novel arthritogenic mediator. J. Immunol. 177, 2610–2620. doi:10.4049/jimmunol.177.4.2610
Pettersen, I., Baryawno, N., Abel, F., Bakkelund, W. H., Zykova, S. N., Winberg, J. O., et al. (2013). Expression of TWEAK/Fn14 in neuroblastoma: Implications in tumorigenesis. Int. J. Oncol. 42, 1239–1248. doi:10.3892/ijo.2013.1800
Purcell, J. W., Kim, H. K., Tanlimco, S. G., Doan, M., Fox, M., Lambert, P., et al. (2014). Nuclear factor κB is required for tumor growth inhibition mediated by enavatuzumab (PDL192), a humanized monoclonal antibody to TweakR. Front. Immunol. 4, 505. doi:10.3389/fimmu.2013.00505
Rice, N. R., Mackichan, M. L., and Israel, A. (1992). The precursor of NF-kappa B p50 has I kappa B-like functions. Cell. 71, 243–253. doi:10.1016/0092-8674(92)90353-e
Roos, C., Wicovsky, A., Muller, N., Salzmann, S., Rosenthal, T., Kalthoff, H., et al. (2010). Soluble and transmembrane TNF-like weak inducer of apoptosis differentially activate the classical and noncanonical NF-kappa B pathway. J. Immunol. 185, 1593–1605. doi:10.4049/jimmunol.0903555
Saitoh, T., Nakayama, M., Nakano, H., Yagita, H., Yamamoto, N., and Yamaoka, S. (2003). TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J. Biol. Chem. 278, 36005–36012. doi:10.1074/jbc.M304266200
Salzmann, S., Seher, A., Trebing, J., Weisenberger, D., Rosenthal, A., Siegmund, D., et al. (2013). Fibroblast growth factor inducible (Fn14)-specific antibodies concomitantly display signaling pathway-specific agonistic and antagonistic activity. J. Biol. Chem. 288, 13455–13466. doi:10.1074/jbc.M112.435917
Schapira, K., Burkly, L. C., Zheng, T. S., Wu, P., Groeneweg, M., Rousch, M., et al. (2009). Fn14-Fc fusion protein regulates atherosclerosis in ApoE-/- mice and inhibits macrophage lipid uptake in vitro. Arterioscler. Thromb. Vasc. Biol. 29, 2021–2027. doi:10.1161/ATVBAHA.109.195040
Schneider, P., Schwenzer, R., Haas, E., Muhlenbeck, F., Schubert, G., Scheurich, P., et al. (1999). TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur. J. Immunol. 29, 1785–1792. doi:10.1002/(SICI)1521-4141(199906)29:06<1785::AID-IMMU1785>3.0.CO;2-U
Sezaki, T., Hirata, Y., Hagiwara, T., Kawamura, Y. I., Okamura, T., Takanashi, R., et al. (2017). Disruption of the TWEAK/Fn14 pathway prevents 5-fluorouracil-induced diarrhea in mice. World J. Gastroenterol. 23, 2294–2307. doi:10.3748/wjg.v23.i13.2294
Shimada, K., Fujii, T., Tsujikawa, K., Anai, S., Fujimoto, K., and Konishi, N. (2012). ALKBH3 contributes to survival and angiogenesis of human urothelial carcinoma cells through NADPH oxidase and tweak/Fn14/VEGF signals. Clin. Cancer Res. 18, 5247–5255. doi:10.1158/1078-0432.CCR-12-0955
Siegmund, D., Lang, I., and Wajant, H. (2017). Cell death-independent activities of the death receptors CD95, TRAILR1, and TRAILR2. FEBS J. 284, 1131–1159. doi:10.1111/febs.13968
Sun, S. C. (2017). The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17, 545–558. doi:10.1038/nri.2017.52
Tan, D., Pang, F. M., Li, D., Zhang, L., Wu, J., Liu, Z. Q., et al. (2018). Overexpression of Fn14 in gliomas: Tumor progression and poor prognosis. Future Oncol. 14, 1273–1284. doi:10.2217/fon-2017-0598
Tao, R., Liu, Q., Huang, R., Wang, K., Sun, Z., Yang, P., et al. (2022). A novel TNFSF-based signature predicts the prognosis and immunosuppressive status of lower-grade glioma. Biomed. Res. Int. 2022, 3194996. doi:10.1155/2022/3194996
Ting, A. T., and Bertrand, M. J. M. (2016). More to life than NF-κB in TNFR1 signaling. Trends Immunol. 37, 535–545. doi:10.1016/j.it.2016.06.002
Tran, N. L., Mcdonough, W. S., Donohue, P. J., Winkles, J. A., Berens, T. J., Ross, K. R., et al. (2003). The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am. J. Pathol. 162, 1313–1321. doi:10.1016/S0002-9440(10)63927-2
Tran, N. L., Mcdonough, W. S., Savitch, B. A., Fortin, S. P., Winkles, J. A., Symons, M., et al. (2006). Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-kappaB and correlate with poor patient outcome. Cancer Res. 66, 9535–9542. doi:10.1158/0008-5472.CAN-06-0418
Trebing, J., Lang, I., Chopra, M., Salzmann, S., Moshir, M., Silence, K., et al. (2014). A novel llama antibody targeting Fn14 exhibits anti-metastatic activity in vivo. MAbs 6, 297–308. doi:10.4161/mabs.26709
Varfolomeev, E., Goncharov, T., Maecker, H., Zobel, K., Komuves, L. G., Deshayes, K., et al. (2012). Cellular inhibitors of apoptosis are global regulators of NF-κB and MAPK activation by members of the TNF family of receptors. Sci. Signal. 5, ra22. doi:10.1126/scisignal.2001878
Vince, J. E., Chau, D., Callus, B., Wong, W. W., Hawkins, C. J., Schneider, P., et al. (2008). TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J. Cell. Biol. 182, 171–184. doi:10.1083/jcb.200801010
Vredevoogd, D. W., Kuilman, T., Ligtenberg, M. A., Boshuizen, J., Stecker, K. E., de Bruijn, B., et al. (2019). Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell. 178, 585–599. doi:10.1016/j.cell.2019.06.014
Wajant, H. (2015). Principles of antibody-mediated TNF receptor activation. Cell. Death Differ. 22, 1727–1741. doi:10.1038/cdd.2015.109
Wajant, H. (2013). The TWEAK-Fn14 system as a potential drug target. Br. J. Pharmacol. 170, 748–764. doi:10.1111/bph.12337
Wang, C. Y., Mayo, M. W., Korneluk, R. G., Goeddel, D. V., and Baldwin, A. S. (1998). NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281, 1680–1683. doi:10.1126/science.281.5383.1680
Wang, D., Fung, J. N., Tuo, Y., Hu, L., and Chen, C. (2010). TWEAK/Fn14 promotes apoptosis of human endometrial cancer cells via caspase pathway. Cancer Lett. 294, 91–100. doi:10.1016/j.canlet.2010.01.027
Wang, J., Liu, Y., Wei, M. J., Mi, X. Y., and Wang, E. H. (2013). Clinical correlations and prognostic relevance of Fn14 expression in breast carcinoma. Histol. Histopathol. 28, 859–864. doi:10.14670/HH-28.859
Wang, W., Liu, F., Wang, C., Wang, C., Tang, Y., and Jiang, Z. (2018). Src promotes metastasis of human non-small cell lung cancer cells through fn14-mediated NF-κB signaling. Med. Sci. Monit. 24, 1282–1294. doi:10.12659/msm.906266
Wang, Y., Zhou, P., Cui, C., He, X., Bian, Y., and Wang, X. (2021). The expression of Nanog protein and fibroblast growth factor-inducible molecule 14 in patients with non-small cell lung cancer and their relationship with pathological characteristics and prognosis. Transl. Cancer Res. 10, 2470–2477. doi:10.21037/tcr-21-724
Watts, G. S., Tran, N. L., Berens, M. E., Bhattacharyya, A. K., Nelson, M. A., Montgomery, E. A., et al. (2007). Identification of Fn14/TWEAK receptor as a potential therapeutic target in esophageal adenocarcinoma. Int. J. Cancer 121, 2132–2139. doi:10.1002/ijc.22898
Weiss, T., Grell, M., Hessabi, B., Bourteele, S., Muller, G., Scheurich, P., et al. (1997). Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80: Requirement of the TNF receptor-associated factor-2 binding site. J. Immunol. 158, 2398–2404.
Weiss, T., Grell, M., Siemienski, K., Muhlenbeck, F., Durkop, H., Pfizenmaier, K., et al. (1998). TNFR80-dependent enhancement of TNFR60-induced cell death is mediated by TNFR-associated factor 2 and is specific for TNFR60. J. Immunol. 161, 3136–3142.
Whitsett, T. G., Cheng, E., Inge, L., Asrani, K., Jameson, N. M., Hostetter, G., et al. (2012). Elevated expression of Fn14 in non-small cell lung cancer correlates with activated EGFR and promotes tumor cell migration and invasion. Am. J. Pathol. 181, 111–120. doi:10.1016/j.ajpath.2012.03.026
Whitsett, T. G., Fortin Ensign, S. P., Dhruv, H. D., Inge, L. J., Kurywchak, P., Wolf, K. K., et al. (2014). FN14 expression correlates with MET in NSCLC and promotes MET-driven cell invasion. Clin. Exp. Metastasis 31, 613–623. doi:10.1007/s10585-014-9653-6
Wicovsky, A., Salzmann, S., Roos, C., Ehrenschwender, M., Rosenthal, T., Siegmund, D., et al. (2009). TNF-like weak inducer of apoptosis inhibits proinflammatory TNF receptor-1 signaling. Cell. Death Differ. 16, 1445–1459. doi:10.1038/cdd.2009.80
Wiley, S. R., Cassiano, L., Lofton, T., Davis-Smith, T., Winkles, J. A., Lindner, V., et al. (2001). A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 15, 837–846. doi:10.1016/s1074-7613(01)00232-1
Willis, A. L., Tran, N. L., Chatigny, J. M., Charlton, N., Vu, H., Brown, S. A., et al. (2008). The fibroblast growth factor-inducible 14 receptor is highly expressed in HER2-positive breast tumors and regulates breast cancer cell invasive capacity. Mol. Cancer Res. 6, 725–734. doi:10.1158/1541-7786.MCR-08-0005
Winkles, J. A. (2008). The TWEAK-fn14 cytokine-receptor axis: Discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 7, 411–425. doi:10.1038/nrd2488
Ye, S., Fox, M. I., Belmar, N. A., Sho, M., Chao, D. T., Choi, D., et al. (2017). Enavatuzumab, a humanized anti-TWEAK receptor monoclonal antibody, exerts antitumor activity through attracting and activating innate immune effector cells. J. Immunol. Res. 2017, 5737159. doi:10.1155/2017/5737159
Yepes, M., Brown, S. A., Moore, E. G., Smith, E. P., Lawrence, D. A., and Winkles, J. A. (2005). A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am. J. Pathol. 166, 511–520. doi:10.1016/S0002-9440(10)62273-0
Yin, J., Liu, Y. N., Tillman, H., Barrett, B., Hewitt, S., Ylaya, K., et al. (2014). AR-regulated TWEAK-FN14 pathway promotes prostate cancer bone metastasis. Cancer Res. 74, 4306–4317. doi:10.1158/0008-5472.CAN-13-3233
Yoriki, R., Akashi, S., Sho, M., Nomi, T., Yamato, I., Hotta, K., et al. (2011). Therapeutic potential of the TWEAK/Fn14 pathway in intractable gastrointestinal cancer. Exp. Ther. Med. 2, 103–108. doi:10.3892/etm.2010.181
Zhang, Q., Lenardo, M. J., and Baltimore, D. (2017). 30 Years of NF-κB: A blossoming of relevance to human pathobiology. Cell. 168, 37–57. doi:10.1016/j.cell.2016.12.012
Zhang, X., Winkles, J. A., Gongora, M. C., Polavarapu, R., Michaelson, J. S., Hahm, K., et al. (2007). TWEAK-Fn14 pathway inhibition protects the integrity of the neurovascular unit during cerebral ischemia. J. Cereb. Blood Flow. Metab. 27, 534–544. doi:10.1038/sj.jcbfm.9600368
Zhao, Z., Burkly, L. C., Campbell, S., Schwartz, N., Molano, A., Choudhury, A., et al. (2007). TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J. Immunol. 179, 7949–7958. doi:10.4049/jimmunol.179.11.7949
Zheng, C., Kabaleeswaran, V., Wang, Y., Cheng, G., and Wu, H. (2010). Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: Affinity, specificity, and regulation. Mol. Cell. 38, 101–113. doi:10.1016/j.molcel.2010.03.009
Zhou, H., Ekmekcioglu, S., Marks, J. W., Mohamedali, K. A., Asrani, K., Phillips, K. K., et al. (2013a). The TWEAK receptor Fn14 is a therapeutic target in melanoma: Immunotoxins targeting Fn14 receptor for malignant melanoma treatment. J. Invest. Dermatol. 133, 1052–1062. doi:10.1038/jid.2012.402
Zhou, H., Hittelman, W. N., Yagita, H., Cheung, L. H., Martin, S. S., Winkles, J. A., et al. (2013b). Antitumor activity of a humanized, bivalent immunotoxin targeting fn14-positive solid tumors. Cancer Res. 73, 4439–4450. doi:10.1158/0008-5472.CAN-13-0187
Zhou, H., Marks, J. W., Hittelman, W. N., Yagita, H., Cheung, L. H., Rosenblum, M. G., et al. (2011). Development and characterization of a potent immunoconjugate targeting the Fn14 receptor on solid tumor cells. Mol. Cancer Ther. 10, 1276–1288. doi:10.1158/1535-7163.MCT-11-0161
Zhou, H., Mohamedali, K. A., Gonzalez-Angulo, A. M., Cao, Y., Migliorini, M., Cheung, L. H., et al. (2014). Development of human serine protease-based therapeutics targeting Fn14 and identification of Fn14 as a new target overexpressed in TNBC. Mol. Cancer Ther. 13, 2688–2705. doi:10.1158/1535-7163.MCT-14-0346
Keywords: agonistic antibodies, cell death, Fn14, NFκB, TNF, TWEAK
Citation: Zaitseva O, Hoffmann A, Otto C and Wajant H (2022) Targeting fibroblast growth factor (FGF)-inducible 14 (Fn14) for tumor therapy. Front. Pharmacol. 13:935086. doi: 10.3389/fphar.2022.935086
Received: 03 May 2022; Accepted: 10 October 2022;
Published: 21 October 2022.
Edited by:
James Jeiwen Chou, Harvard Medical School, United StatesReviewed by:
Matthew S. Hayden, Dartmouth College, United StatesCopyright © 2022 Zaitseva, Hoffmann, Otto and Wajant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harald Wajant, aGFyYWxkLndhamFudEBtYWlsLnVuaS13dWVyemJ1cmcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.