 Yuanying Wang
Yuanying Wang Ziyun Guo
Ziyun Guo Ruimin Ma1
Ruimin Ma1 Jingwei Wang
Jingwei Wang Qiao Ye
Qiao Ye
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 01 July 2022
Sec. Respiratory Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.924754
This article is part of the Research Topic Pulmonary Fibrosis: one manifestation, various diseases View all 12 articles
Background: Limited data are available regarding the entire spectrum of interstitial lung disease with a progressive fibrosing feature. We investigated the prevalence and prognostic predictive characteristics in patients with PF-ILD.
Methods: This retrospective cohort study included patients with fibrosing ILD who were investigated between 1 January 2015 and 30 April 2021. We recorded clinical features and outcomes to identify the possible risk factors for fibrosing progression as well as mortality.
Results: Of the 579 patients with fibrosing ILD, 227 (39.21%) met the criteria for progression. Clubbing of fingers [odds ratio (OR) 1.52, 95% confidence interval (CI) 1.03 to 2.24, p = 0.035] and a high-resolution computed tomography (HRCT)-documented usual interstitial pneumonia (UIP)-like fibrotic pattern (OR 1.95, 95% CI 1.33 to 2.86, p = 0.001) were risk factors for fibrosis progression. The mortality was worse in patients with PF with hypoxemia [hazard ratio (HR) 2.08, 95% CI 1.31 to 3.32, p = 0.002], in those with baseline diffusion capacity of the lung for carbon monoxide (DLCO) % predicted <50% (HR 2.25, 95% CI 1.45 to 3.50, p < 0.001), or in those with UIP-like fibrotic pattern (HR 1.68, 95% CI 1.04 to 2.71, p < 0.001).
Conclusion: Clubbing of fingers and an HRCT-documented UIP-like fibrotic pattern were more likely to be associated with progressive fibrosing with varied prevalence based on the specific diagnosis. Among patients with progressive fibrosing, those with hypoxemia, lower baseline DLCO% predicted, or UIP-like fibrotic pattern showed poor mortality.
The disease course in fibrosing interstitial lung disease (ILD) is highly heterogeneous and difficult to predict. Some patients experience a stable course or slowly functional decline while other patients may experience a rapidly progressive change (Cottin et al., 2018). Progressive fibrosing interstitial lung disease (PF-ILD) is a term referring to a group of diseases characterized by a high-resolution computed tomography (HRCT)-documented increase in the extent of pulmonary fibrosis, a decline in lung function, worsening respiratory symptoms and quality of life, and a high risk of early mortality despite available treatments, with a clinical course similar to that of idiopathic pulmonary fibrosis (IPF) (Cottin et al., 2018). PF-ILDs include a heterogeneous group of disorders associated with multifactorial etiology, including IPF, connective tissue disease-associated ILD (CTD-ILD), fibrotic hypersensitivity pneumonitis (FHP), unclassifiable idiopathic interstitial pneumonia (uIIP), and idiopathic non-specific interstitial pneumonia (Cottin et al., 2019). Reportedly, PF-ILDs may develop in approximately 33.0% of patients with fibrosing ILDs other than IPF (Olson et al., 2018; Nasser et al., 2021a; Ruaro et al., 2021; Lee et al., 2022). The estimated incidence of PF-ILD ranged from 4.0 to 4.7/100,000 person-years and the estimated prevalence from 6.6 to 19.4/100,000 individuals (Nasser et al., 2021a; Nasser et al., 2021b).
Several clinical trials have encompassed patients with progressive fibrosing, with the eligibility criteria for these studies helping to guide a standardized diagnosis of PF-ILD (Wongkarnjana et al., 2020). Although no uniform criteria were established, guidelines recommended monitoring of fibrosis progression based on evaluation of multiple components, such as a decline in lung function, increase in chest imaging-documented fibrosis, symptomatic worsening, and composite measures of these variables (George et al., 2020; Wongkarnjana et al., 2020; Raghu et al., 2022).
In view of the clinical and underlying physiological similarities between IPF and other PF-ILDs, anti-fibrotic agents, such as nintedanib or pirfenidone, are considered to be effective against progressive fibrosis (Flaherty et al., 2019; Gibson et al., 2020). However, watchful observation until the onset of HRCT-documented lung function decline and extensive fibrosis delays initiation of early anti-fibrotic therapy, and patients invariably present with clinically significant and irreversible injury. Several studies have investigated the clinical features and likely predictors in patients at high risk of PF-ILD at baseline (Kolb and Vašáková, 2019). Certain conditions, such as FHP (which is not associated with any specific antigen) and increasing age predispose to PF-ILD (Sharma et al., 2021; Churg, 2022). Smoking status was a risk factor for rheumatoid arthritis-ILD (RA-ILD) or primary Sjögren syndrome associated with ILD (Johnson, 2017; Wang et al., 2018). Male sex and a high baseline modified Rodnan skin score were strong predictors of forced vital capacity (FVC) decline in patients with systemic sclerosis-ILD (SSc-ILD) (Hoffmann-Vold et al., 2021). Risk factors for mortality have also been identified. Regardless of a specific diagnosis of ILD, patients with an HRCT-documented usual interstitial pneumonia (UIP) pattern or extensive traction bronchiectasis, FVC decline, or older age showed the highest mortality risk (Walsh et al., 2018; Yang et al., 2022). In addition, peripheral blood monocyte counts have been investigated in IPF and other ILDs as a predictor of prognosis (Misharin et al., 2017; Barratt et al., 2021). Although studies have reported findings for individual types of PF-ILDs (for example, SSc-ILD, RA-ILD, other CTD-ILDs, and FHP), limited data are available regarding the PF-ILDs as a whole group (Khanna et al., 2020; Skibba et al., 2020; Orlandi et al., 2022). Early accurate diagnosis of most ILDs was difficult. Studying clinical courses for the heterogeneous group of fibrosing ILDs as a whole group was essential to identify patients with the most rapidly progressive feature or poor prognosis and initiate early anti-fibrotic treatment.
The aims of this study were: 1) to investigate the prevalence and clinical features of the PF-ILDs and 2) to explore the potential factors associated with progression or mortality.
This observational retrospective cohort study was performed at Beijing Chao-Yang Hospital, China, a regional tertiary referral center specialized in the management of ILDs. We retrospectively screened all patients aged ≥18 years with a multidisciplinary diagnosis of fibrosing ILDs between 1 January 2015 and 30 April 2021. Multidisciplinary diagnoses were conducted between pulmonologists (QY, NW, and YW), radiologists (YF and JW), a rheumatologist (ZG), and a pathologist (RM) experienced in the diagnosis of ILD based on clinical characteristics, HRCT, and lung biopsy if appropriate. Patients whose results of baseline pulmonary function tests (PFTs) and HRCT were available were enrolled in the study. Following were the exclusion criteria: 1) <10% fibrosis documented on baseline HRCT, 2) diagnosis of pulmonary embolism, decompensated heart failure, or lower respiratory tract infections associated with disease progression, 3) lung cancer at baseline, 4) withdrawal of consent for study participation, and 5) loss to follow-up (Figure 1). The study was registered at www.chictr.org.cn ChiCTR2100049247 and was approved by the Ethics Committee of Beijing Chao-Yang Hospital (2020-KY-437). All procedures were performed in accordance with the principles of the Declaration of Helsinki.
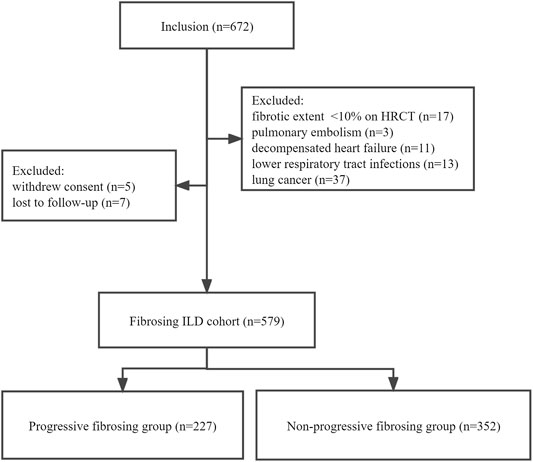
FIGURE 1. Patient flowchart. ILD, interstitial lung disease; PFT, pulmonary function test; HRCT, high-resolution computed tomography.
We reviewed patients’ medical records to uniformly extract clinical data at the first clinical visit, including demographic information, physical examination, and routine clinical laboratory test results. Clinical and survival data were obtained from medical records, outpatient follow-up records (usually every 6–12 months), hospitalization details, and telephone interviews. Hypoxemia was defined as the partial pressure of oxygen in the arterial blood (PaO2) of less than 80 mm Hg obtained from the arterial blood gas (ABG) test at rest. The derivative blood cell count inflammation indexes included the neutrophil-to-lymphocyte ratio (NLR: neutrophils/lymphocytes), monocyte-to-lymphocyte ratio (MLR: monocytes/lymphocytes), platelet-to-lymphocyte ratio (PLR: platelet/lymphocyte), systemic inflammatory index (SII: neutrophils × platelets/lymphocytes), systemic inflammatory response index (SIRI: neutrophils × monocytes/lymphocytes), and aggregate index of systemic inflammation (AISI: neutrophils × platelets × monocytes/lymphocytes). The HRCT-documented UIP-like pattern included definitive UIP or a probable UIP pattern according to the Clinical Practice Guideline of IPF (Raghu et al., 2011; Raghu et al., 2018). Two thoracic radiologists (YF and JW) blinded to the clinical data independently determined whether the fibrosis involved >10% of the total lung and reviewed the HRCT scans by visual assessment. The disagreements were resolved via consensus. The kappa value for the interobserver correlation was 0.84.
The follow-up period ended on 30 April 2021. The primary outcome was progressive fibrosing. Patients who fulfilled any of the following criteria within 24 months despite administration of standard treatment were considered experiencing progressive fibrosis (Cottin et al., 2018; Flaherty et al., 2019): 1) a relative decline of ≥10% in FVC, 2) relative decline of ≥15% in diffusion capacity of the lung for carbon monoxide (DLCO), and 3) worsening symptoms and/or worsening radiological findings accompanied by a ≥5% to <10% relative decrease in FVC. The secondary outcome was all-cause mortality during the follow-up period. Survival was calculated from the time of the first visit to the outcome or the end of follow-up.
PASS software, version 11.0 (NCSS, LLC. Kaysville, Utah, United States) was used to calculate the sample size. The sample allocation ratio was 1: 2 (PF: non-PF), given the estimated prevalence of PF-ILD among patients with fibrosing ILDs was 33.0% (Nasser et al., 2021a). We used a two-sided log-rank test for the 6-year all-cause mortality with 1-year accrual periods and an estimated 10.0% proportion of dropping out for both groups. An overall sample size of 260 patients (174 in the non-PF-ILD group and 86 in the PF-ILD group) was deemed sufficient to achieve >90% power at a 0.05% significance level to detect a hazard ratio (HR) of death with the 6-year mortality rate considered to be 40.0% for the PF group and 18.0% for the non-PF group (Adegunsoye et al., 2018). However, we attempted to enroll as many patients as possible within the study period. The final cohort included 579 patients, of which 227 patients were diagnosed with PF-ILD (85 deaths), fulfilling the 10-events-per-variable criterion of multivariable logistic regression and Cox analysis.
Statistical analyses were performed using SPSS Statistics software, version 26 (IBM, Inc., Chicago, IL, United States). Missing data were imputed using mean/median or removed for a complete case analysis. Data were expressed as means (SD) or medians (range), depending on the distribution. The Mann–Whitney U or t-test was used for continuous variables for comparison of intergroup differences and the chi-squared test or Fisher’s exact test for categorical variables. Linear mixed-effects analysis was applied to analyze disease behavior over time, with random terms for intercept and slope (for time-from-diagnosis). The covariance for the repeated measures was left autoregressive 1: heterogeneous, which yielded the best fit. Multicollinearity diagnostic tests were performed. Logistic regression analyses were used to investigate risk factors for the PF-ILD, and continuous variables were converted into dichotomous variables mainly through the median cut-off. The cut-off value was 70.0% for FVC% predicted and 50.0% for DLCO% predicted (Abe et al., 2021; Lacedonia et al., 2022). Survival curves were obtained using the Kaplan–Meier method, and Cox proportional analyses were performed to identify prognostic factors for mortality. Proportional hazards in the Cox analysis were checked using the Schoenfeld residual test. A p-value < 0.05 was considered statistically significant.
In this study, 277 (39.21%) out of 579 ILD patients met the criteria for PF-ILD; 64, 88, and 108 patients met criteria 1, 2, and 3, respectively; 28 patients met both the criteria 1 and 2, and 16 patients met both the criteria 2 and 3 (Supplementary Figure S1). Statistically significant baseline differences were observed in age (p = 0.001), underlying diagnosis (p < 0.001), clubbing of fingers (p = 0.005), and HRCT-documented UIP-like fibrotic pattern (p < 0.001) between the PF-ILD and non-PF-ILD groups (Table 1). Baseline blood count values did not differ significantly between patients with PF and non-PF. Within the PF-ILD group, gender, age, smoking status, UIP-like fibrotic pattern, FVC, GAP stage distribution, and anti-fibrotic treatment were statistically significant between IPF and non-IPF patients (Table 2). Comorbidity information for the whole cohort is shown in Supplementary Figure S2. The prevalence for pulmonary hypertension was 8.81% in the PF group and 5.11% in the non-PF group.
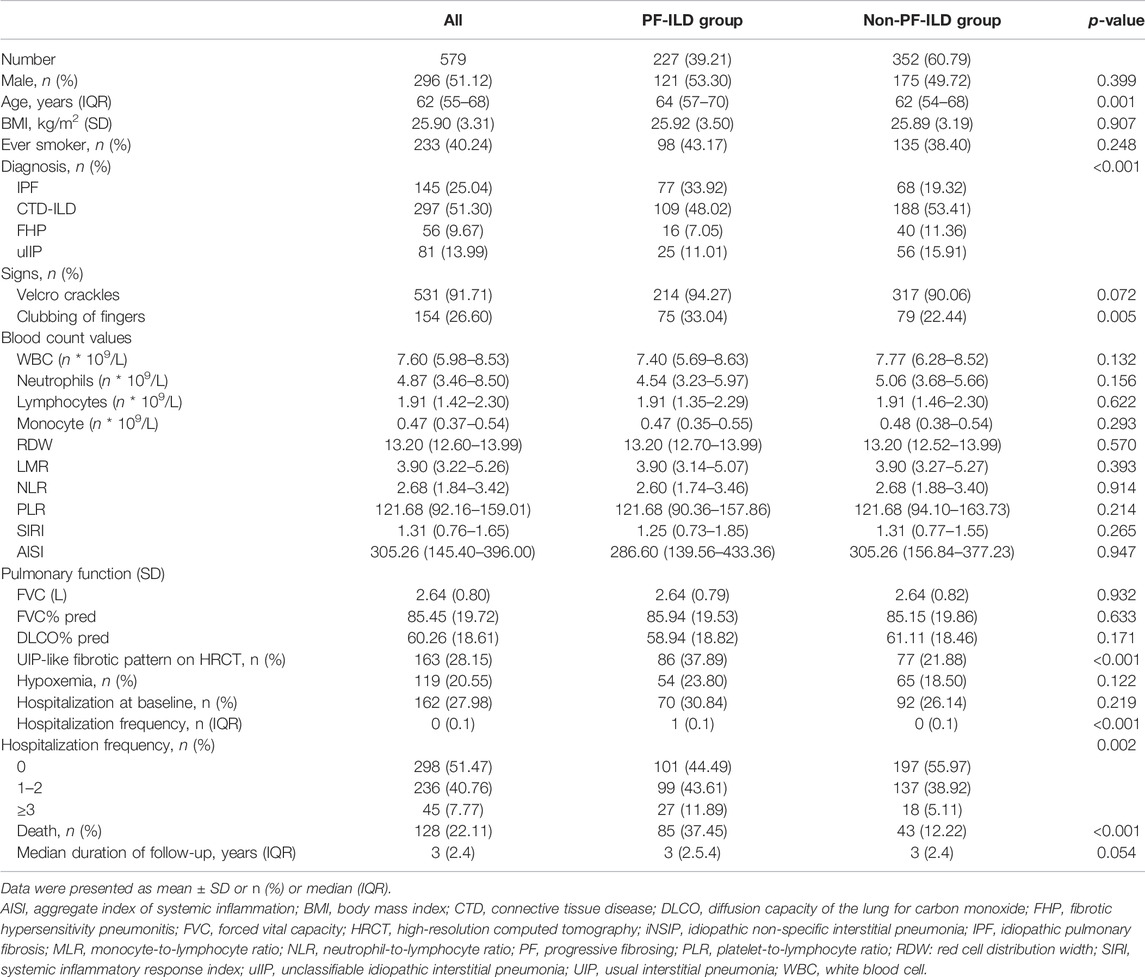
TABLE 1. Demographics and clinical characteristics of the cohort.
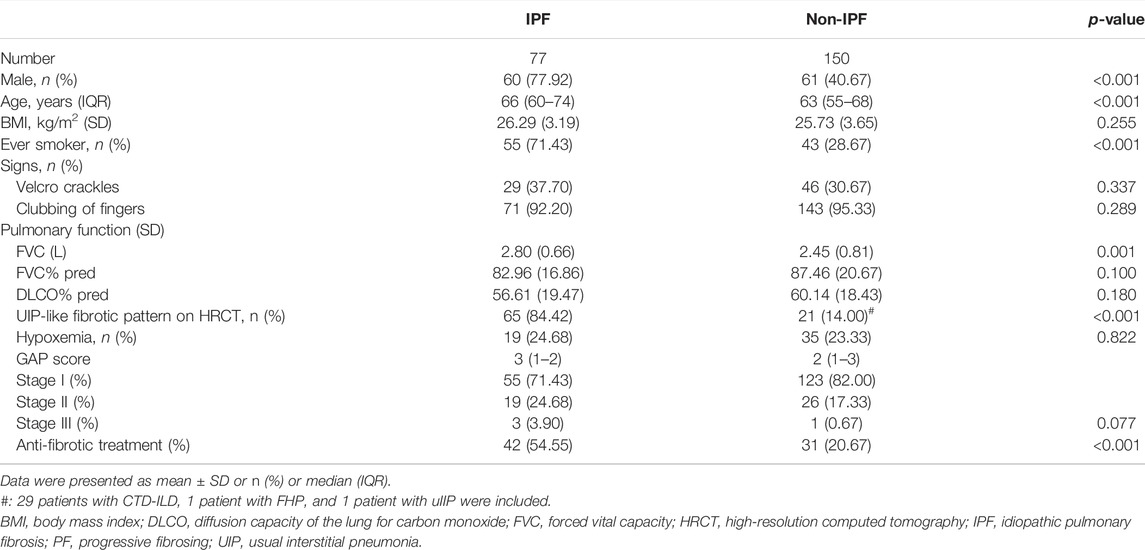
TABLE 2. Demographics and clinical characteristics of patients within the PF group.
The most frequent diagnoses included CTD-ILD (n = 297%, 51.30%), IPF (n = 145%, 25.04%), uIIP (n = 81%, 13.99%), and FHP (n = 56%, 9.67%) (Table 1). The prevalence of PF-ILD in patients with ILDs, including those with IPF (n = 77), CTD-ILD (n = 109), FHP (n = 16), and uIIP (n = 25) was 53.10%, 36.70%, 28.57%, and 30.86%, respectively (Supplementary Figure S3).
In the entire cohort, 74.96% of patients (n = 434) received at least one of the following treatments: administration of glucocorticoids, immunosuppressive agents, anti-fibrotic treatment, and oxygen therapy. Anti-fibrotic treatment was administered to 32.16% of patients with PF-ILD (n = 73) (Supplementary Table S1). In the PF group, 42 (54.50%) of IPF patients and 31 (20.70%) of non-IPF patients (29 CTD-ILD, 1 FHP, and 1 uIIP) received anti-fibrotic treatment (Table 2). Patients hospitalized within 1 month of the first visit were considered hospitalized at baseline. We observed that 298 (51.47%) patients underwent hospitalization at least once during follow-up, and 27 (11.89%) of 227 patients in the PF-ILD group vs. 18 (5.11%) of the 352 patients in the non-PF group underwent hospitalization at least thrice (Table 1).
Figure 2 showed the mean lung function measures across different time intervals. The slope of FVC differed between the PF-ILD (−0.07 L/6 months, 95% CI −0.09 to −0.05) and non-PF-ILD (0.01 L/6 months, 95% CI 0.01–0.05) subgroups (p = 0.001) (Figure 2D). The slope of FVC% predicted in the PF-ILD group was −1.42%/6 months (95% CI −1.99 to −0.86), whereas that in the non-PF-ILD group was 2.80%/6 months (95% CI 2.12–3.49) (p = 0.04) (Figure 2E). The slope of DLCO% predicted also differed between subgroups (−1.84%/6 months, 95% CI −2.43 to −1.25 in the PF-ILD group vs. 1.93%/6 months, 95% CI 1.35 to 2.51 in the non-PF-ILD group, p = 0.003) (Figure 2F).
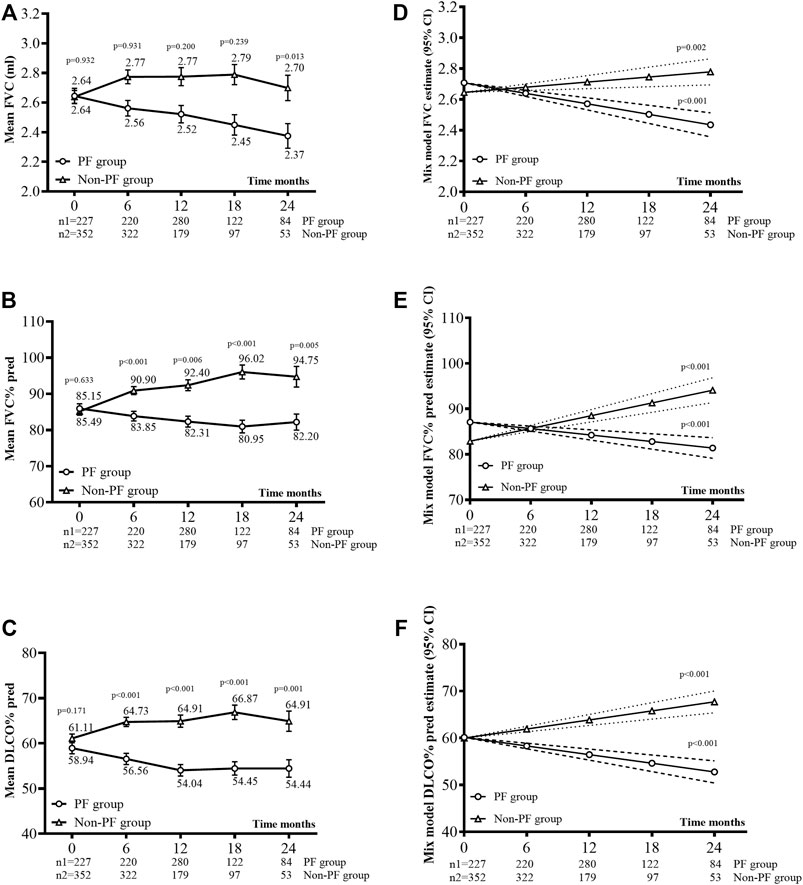
FIGURE 2. Lung function changes. (A), (B), and (C) Mean FVC ml (±SE), mean FVC% predicted (±SE), and mean DLCO% predicted (±SE) during observation. (D), (E), and (F) Disease behavior (FVC, FVC% pred, and DLCO% pred) over time was analyzed using a linear mixed model for each group, with a random effect for the intercept and slope. The covariance for the repeated measures was left AR (autoregressive)1: heterogeneous, as this yielded the best fit.
Table 3 shows the results of logistic regression analysis for predictors of progressive fibrosing. Multivariable analysis was performed on significant factors obtained from univariate analysis with a p-value < 0.2. Although the NLR (OR 0.69, 95% CI 0.55 to 1.08, p = 0.039) was significantly associated with PF, the OR included the value 1. Patients with clubbing of fingers (OR 1.52, 95% CI 1.03 to 2.24, p = 0.035) and an HRCT-documented UIP-like fibrotic pattern (OR 1.95, 95% CI 1.33 to 2.86, p = 0.001) showed a high risk of fibrosis progression.
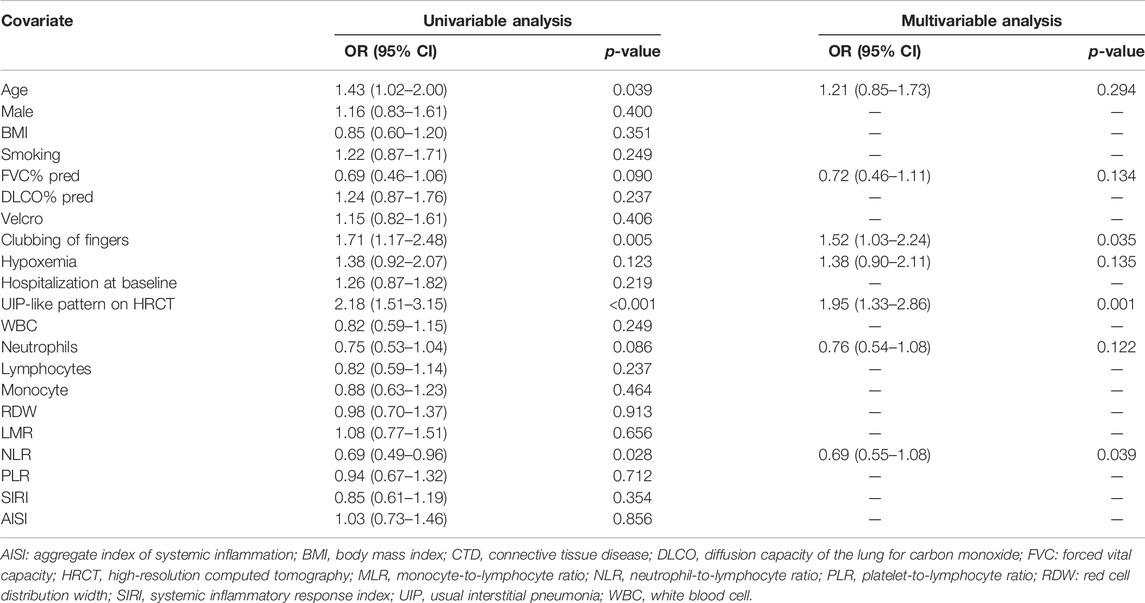
TABLE 3. Factors associated with progressive fibrosing.
Patients were followed up for a median duration of 3 (2–4) years with 128 (22.1%) deaths during follow-up, and the median overall survival (OS) time for whole patients with fibrosing ILDs was 6 years. In the non-PF group, the median overall survival was not reached, while in the PF-ILD group, 85 of 227 (37.45%) patients died, with a median OS of 5 years (Supplementary Figure S4). The median survival time was shorter in patients with hypoxemia (OS: 4 years), baseline DLCO% predicted <50% (OS: 4 years), and UIP-like pattern (OS: 4.5 years). Subgroup analysis based on anti-fibrotic treatment using the log-rank test showed no significant intergroup difference (p = 0.435) (Figure 3). Within patients in the PF group who received anti-fibrotic treatment, IPF patients showed worse OS (OS: 4.5 years) than non-IPF patients (OS: 6 years) (log-rank test, p = 0.003, Supplementary Figure S5). Table 4 shows the results of univariate and multivariate Cox analyses of predictors for all-cause mortality. By multivariate Cox analysis, lower DLCO% predicted (HR 2.25, 95% CI 1.45 to 3.50, p < 0.001), hypoxemia (HR 2.08, 95% CI 1.31 to 3.32, p = 0.002), and UIP-like fibrosis pattern on HRCT (HR 1.68, 95% CI 1.04 to 2.71, p = 0.034) were associated with an increased risk of mortality.
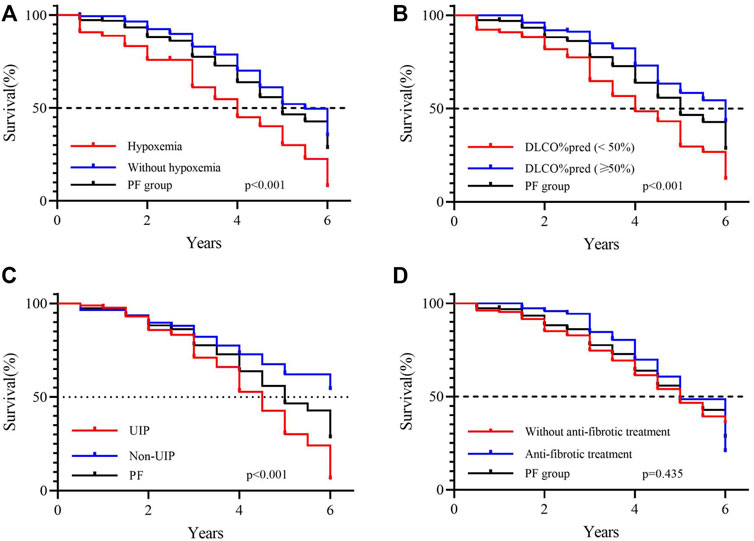
FIGURE 3. Survival in patients with progressive fibrosing ILD. (A) Survival according to with or without hypoxemia at baseline (log-rank test, p < 0.001); (B) survival according to DLCO% pred at baseline with a 50% threshold (log-rank test, p < 0.001); (C) survival according to the UIP-like fibrotic pattern on HRCT (log-rank test, p < 0.001); (D) survival according to with or without anti-fibrotic treatment during observation (log-rank test, p = 0.435).
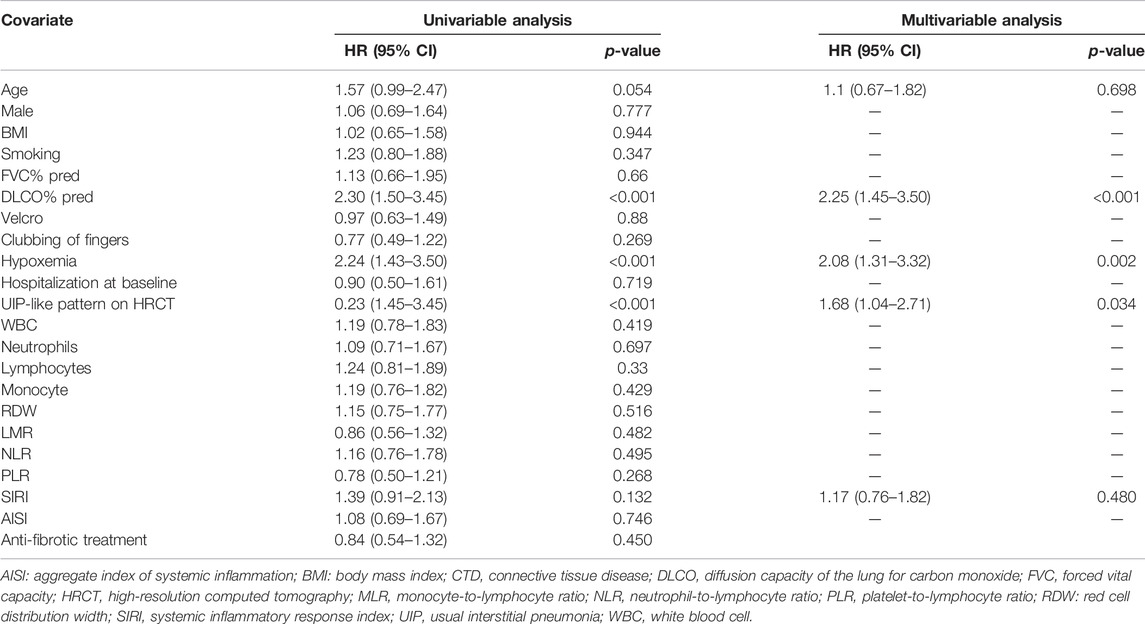
TABLE 4. Factors associated with 6-year all-cause mortality in the PF-group.
We provided a clinical cohort profile of patients with fibrosing ILD in China. A significant percentage of patients with fibrosing ILD tend to develop progressive fibrosing, which is associated with accelerated decline in lung function secondary to progressive fibrosis and symptomatic worsening (Nasser et al., 2021b). Prevalence of non-IPF PF-ILD varied from 28.57% to 39.07% owing to various diagnosis subgroups in our data, consistent with the findings reported by previous studies (approximately 33.0%) (Flaherty et al., 2019; Nasser et al., 2021b). A large-scale survey estimated that 18.0%–32.0% of patients diagnosed with non-IPF ILDs developed progressive fibrosis with overall survival of 61–80 months in the US, France, Germany, Italy, Spain, the United Kingdom, and Japan (Wijsenbeek et al., 2019). PF-ILD was identified in 135 of 396 (34.1%) patients with non-IPF ILDs in a cohort of Korean patients (Kwon et al., 2021). In another Chinese single-center cohort of 608 patients, 132 patients (21.7%) with ILD met the criteria for PF-ILD (Chen et al., 2021). The difference in findings between previous studies and our study may be attributable to the older age, poorer DLCO% predicted, or various disease classifications used for non-IPF ILDs in our patients. In our study, half of the patients with IPF showed rapid disease progression within 24 months. The clinical course of IPF is variable and difficult to predict, with a median survival time of 2–3 years after diagnosis without anti-fibrotic therapy. However, approximately 25.0% of patients survive over 5 years after the initial diagnosis (Kärkkäinen et al., 2019). Therefore, it is possible that lung function in patients with IPF remained stable or declined only slightly during the first 24 months but eventually led to poor outcomes.
Healthcare utilization is expected to be higher in patients with PF-ILD (Holtze et al., 2018; Wuyts et al., 2020). Hospitalization frequency was higher in the PF-ILD group than in the non-PF-ILD group during follow-up. Patients with PF-ILD received glucocorticoids for primary therapy, which emphasizes the need for alternative treatment. Many clinical trials have confirmed the efficacy of anti-fibrotic drugs to slow disease progression in patients with PF-ILD (Behr et al., 2021; Nambiar et al., 2021). Our data showed a trend of better survival in patients with PF-ILD, though not statistically significant, who received anti-fibrotic treatment; nintedanib and pirfenidone are known to prolong life expectancy in patients with IPF (Costabel et al., 2017; Lancaster et al., 2019). In the INBUILD trial, the annual rate of decline in the FVC in patients with PF-ILD was significantly lower among patients who received nintedanib (−80.8 ml/year) than among those who received a placebo (−187.8 ml/year) (Flaherty et al., 2019). However, only 32.16% of patients receive anti-fibrotic treatment in our real-world clinical settings, which indicates the possible financial burden associated with these drugs.
Overall, the mean lung function measures remained stable during follow-up in the non-PF-ILD group but showed a decline in the PF-ILD group. In the PF-ILD group, the change in the estimated mean annual FVC was −140 ml. In our study, the decline in the FVC in patients with PF-ILD was similar to that reported by the PROGRESS clinical cohort after 52 weeks (−136 ml) (Nasser et al., 2021b). Another study reported that the mean annual FVC change was −69.9 ml in a United States cohort and −50.0 ml in a United Kingdom cohort of patients with PF-ILD (Oldham et al., 2021). These differences may be attributable to the heterogeneous lung function trajectory across the ILD subgroups. Analysis of diagnosis subgroups showed a mean annual FVC change of −37.2 ml in the CTD-ILD, −92.0 ml in the FHP, and −69.5 ml in the IIP groups (Oldham et al., 2021). We did not perform diagnosis stratification owing to the limited sample size. A greater number of studies that perform the stratification are warranted to investigate the lung function trajectory.
We observed that the HRCT-documented UIP-like fibrotic pattern in patients with fibrosing ILDs was associated with more rapid disease progression, which suggests that morphological patterns may be prognostically significant; our findings are consistent with those reported by previous studies (Nasser et al., 2021b). Moreover, our study showed that clubbing of fingers (enlargement of the ends of the finger accompanied by a downward sloping of the nails) significantly predicted PF-ILD. A previous study reported that the prevalence of clubbing of fingers in cases of fibrosing ILDs ranged from 7.0% to 42.0% (van Manen et al., 2017). Clubbing of fingers is considered an essential clinical finding in patients with fibrosing ILDs and represents decreased oxygen levels and poor prognosis (Kanematsu et al., 1994; van Manen et al., 2017).
Hypoxemia, low DLCO% predicted, and HRCT-documented UIP pattern were associated with worse mortality in patients with PF. Reportedly, male sex, older age, lower FVC or DLCO at baseline, decline in lung function compared with the baseline, subgroups based on diagnosis, HRCT-documented UIP-like fibrotic pattern, or honeycombing were indicators of poor prognosis (Wongkarnjana et al., 2020). Cluster analysis was used to classify patients with ILD into clinical subgroups based on their clinical courses. Patients with the most rapid decline in lung function or fibrosis progression and poor survival were usually clustered together in ILDs or IP with myositis-specific autoantibodies (Adegunsoye et al., 2018; Lia et al., 2022). These findings suggest that evidence of progressive pulmonary fibrosis may be useful to define a group of patients with fibrosing ILD of multifactorial etiologies and poor prognoses. Although accurate and prompt diagnosis is important to enable initiation of optimal management, it is possible to identify patients at high risk of progression through observation of disease behavior despite the specific diagnosis. Validation cohorts are required to confirm mortality predictors.
Given the possible relationship between prognosis and biomarkers obtained by the routinely provided blood cell count test in IPF, we sought to explore the prognostic capacity of the blood cell count and the combined indexes in PF-ILDs. A pooled retrospective analysis of 2,067 IPF patients derived from the clinical trials showed that patients with higher monocyte count had a higher 1-year risk of progression, all-cause mortality, and all-cause hospitalization (Kreuter et al., 2021). On the contrary, we did not observe the relationship between disease progression or mortality and blood cell count and the combined index. According to a multicenter retrospective study, higher baseline NLR and absolute monocyte counts predict worse survival in IPF but not in fHP (Barratt et al., 2021), highlighting the potential divergence in the underlying mechanisms of these diseases. However, our sample size is smaller than the previous reports for the prognostic role of blood cell counts. Second, single peripheral blood cell measures may be influenced by many short-term factors. Further multicenter studies with a larger population would be informative about the applicability of blood cell counts in predicting prognosis.
We provided data on longitudinal clinical outcomes in patients with fibrosing ILD from China. Patients evaluated in a tertiary referral center through a multidisciplinary discussion were routinely followed up every 6–12 months with pulmonary function tests, HRCT, and visits. Unlike previous studies for FP-ILDs (Flaherty et al., 2019; Wijsenbeek et al., 2019; Nasser et al., 2021b) which generally emphasized non-IPF patients, our cohort included both IPF and non-IPF patients as a whole group and also provided separate data for both groups. In this way, the exploration between clinical characteristics and prognosis of patients with PF-ILD is of more clinically utility because it is hard to make specific diagnosis for some ILDs at an early stage, especially for IPF. In addition, we tried to study the relationship between the patient’s blood count values and prognosis, which was not explored in PF-ILD patients as a whole group before. The negative results were supported by some previous research studies while opposed by others studying specific diagnosis group of diseases, highlighting cautious use of routine blood test values as prognosis predictors.
Several limitations should be considered: 1) the retrospective and monocentric design of this study may have led to a selection bias. 2) This was a small-scale study in which we observed a reduction in available data on lung function over time, which resulted in an inaccurate estimation of the lung function trajectory. 3) Acute exacerbations, as one of the leading causes of death in patients with IPF (annual incidence of up to 20.0%) (Richeldi, 2015; Kolb et al., 2018; Ruaro et al., 2021; Yoon et al., 2021), were not investigated because some exacerbations were not recorded in the medical system of our hospital. The annual incidence of acute exacerbations in patients with non-IPF PF-ILD was 13.9% (Nasser et al., 2021b; Petnak et al., 2021; Yoon et al., 2021). Further studies are warranted to thoroughly investigate the effects of acute exacerbations. 4) We used all-cause mortality rather than ILD-related mortality as the outcome because all deaths did not occur at the hospital, and some causes of death were not reported reliably.
In summary, we presented a hospital population-based cohort profile of patients with PF-ILDs in China. We reported that 28.57%–53.10% of patients with fibrosing ILD developed a progressive fibrosing feature. Patients with clubbing of fingers or an HRCT-documented UIP pattern showed a high potential risk for PF-ILD. The risk factors associated with mortality in patients with PF-ILD included hypoxemia, low DLCO% predicted, or an HRCT-documented UIP pattern. The relationship between prognosis and blood cell count and the combined index was not observed, highlighting the divergence in the underlying mechanisms of PF-ILDs.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Beijing Chao-Yang Hospital. The patients/participants provided their written informed consent to participate in this study.
The authors were responsible for all content of the manuscript. YW performed data collection, analyzed the data, contributed to interpretation, and wrote the manuscript. ZG, RM, JW, and YF were responsible for recruiting the patients and collecting the data. NW helped in the drafting and revision. QY contributed as primary investigator and was responsible for designing the study, recruiting the patients, analyzing and interpreting the data, and writing the manuscript. All authors read and approved the final manuscript.
The work was supported by the National Natural Science Foundation of China (81970061).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all patients who were involved in this study. We express our thanks to Miss Moyang Xu of the University of Michigan and Ann Arbor for polishing the language and grammar of the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.924754/full#supplementary-material
AISI, aggregate index of systemic inflammation; BMI, body mass index; CI, confidence interval; CTD, connective tissue disease; DLCO, diffusion capacity of the lung for carbon monoxide; FHP, fibrotic hypersensitivity pneumonitis; FVC, forced vital capacity; HR, hazard ratio; HRCT, high-resolution computed tomography; iNSIP, idiopathic non-specific interstitial pneumonia; IIP, idiopathic interstitial pneumonia; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; IQR, interquartile range; MLR, monocyte-to-lymphocyte ratio; NLR, neutrophil-to-lymphocyte ratio; OR, hazard ratio; PF, progressive fibrosing; PFT, pulmonary function testing; PLR, platelet-to-lymphocyte ratio; pSS-ILD, primary Sjögren syndrome-associated ILD; SD, standard deviation; RDW: red cell distribution width; SIRI, systemic inflammatory response index; SSc-ILD, sclerosis-associated ILD; UIP, usual interstitial pneumonia; WBC, white blood cell.
Abe, M., Tsushima, K., Ishii, D., Shikano, K., Yoshioka, K., Sakayori, M., et al. (2021). Risk Factors for Acute Exacerbation Following Bronchoalveolar Lavage in Patients with Suspected Idiopathic Pulmonary Fibrosis: A Retrospective Cohort Study. Adv. Respir. Med. 89 (2), 101–109. doi:10.5603/ARM.a2021.0012
Adegunsoye, A., Oldham, J. M., Chung, J. H., Montner, S. M., Lee, C., Witt, L. J., et al. (2018). Phenotypic Clusters Predict Outcomes in a Longitudinal Interstitial Lung Disease Cohort. Chest 153 (2), 349–360. doi:10.1016/j.chest.2017.09.026
Barratt, S. L., Creamer, A. W., Adamali, H. I., Duckworth, A., Fallon, J., Fidan, S., et al. (2021). Use of Peripheral Neutrophil to Lymphocyte Ratio and Peripheral Monocyte Levels to Predict Survival in Fibrotic Hypersensitivity Pneumonitis (fHP): A Multicentre Retrospective Cohort Study. BMJ Open Respir. Res. 8 (1), e001063. doi:10.1136/bmjresp-2021-001063
Behr, J., Prasse, A., Kreuter, M., Johow, J., Rabe, K. F., Bonella, F., et al. (2021). Pirfenidone in Patients with Progressive Fibrotic Interstitial Lung Diseases Other Than Idiopathic Pulmonary Fibrosis (RELIEF): a Double-Blind, Randomised, Placebo-Controlled, Phase 2b Trial. Lancet Respir. Med. 9 (5), 476–486. doi:10.1016/s2213-2600(20)30554-3
Chen, X., Guo, J., Yu, D., Jie, B., and Zhou, Y. (2021). Predictors of Mortality in Progressive Fibrosing Interstitial Lung Diseases. Front. Pharmacol. 12, 754851. doi:10.3389/fphar.2021.754851
Churg, A. (2022). Hypersensitivity Pneumonitis: New Concepts and Classifications. Mod. Pathol. 35 (Suppl. 1), 15–27. doi:10.1038/s41379-021-00866-y
Costabel, U., Albera, C., Lancaster, L. H., Lin, C. Y., Hormel, P., Hulter, H. N., et al. (2017). An Open-Label Study of the Long-Term Safety of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (RECAP). Respiration 94 (5), 408–415. doi:10.1159/000479976
Cottin, V., Hirani, N. A., Hotchkin, D. L., Nambiar, A. M., Ogura, T., Otaola, M., et al. (2018). Presentation, Diagnosis and Clinical Course of the Spectrum of Progressive-Fibrosing Interstitial Lung Diseases. Eur. Respir. Rev. 27 (150), 180076. doi:10.1183/16000617.0076-2018
Cottin, V., Wollin, L., Fischer, A., Quaresma, M., Stowasser, S., and Harari, S. (2019). Fibrosing Interstitial Lung Diseases: Knowns and Unknowns. Eur. Respir. Rev. 28 (151), 180100. doi:10.1183/16000617.0100-2018
Flaherty, K. R., Wells, A. U., Cottin, V., Devaraj, A., Walsh, S. L. F., Inoue, Y., et al. (2019). Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 381 (18), 1718–1727. doi:10.1056/NEJMoa1908681
George, P. M., Spagnolo, P., Kreuter, M., Altinisik, G., Bonifazi, M., Martinez, F. J., et al. (2020). Progressive Fibrosing Interstitial Lung Disease: Clinical Uncertainties, Consensus Recommendations, and Research Priorities. Lancet Respir. Med. 8 (9), 925–934. doi:10.1016/s2213-2600(20)30355-6
Gibson, C. D., Kugler, M. C., Deshwal, H., Munger, J. S., and Condos, R. (2020). Advances in Targeted Therapy for Progressive Fibrosing Interstitial Lung Disease. Lung 198 (4), 597–608. doi:10.1007/s00408-020-00370-1
Hoffmann-Vold, A. M., Allanore, Y., Alves, M., Brunborg, C., Airó, P., Ananieva, L. P., et al. (2021). Progressive Interstitial Lung Disease in Patients with Systemic Sclerosis-Associated Interstitial Lung Disease in the EUSTAR Database. Ann. Rheum. Dis. 80 (2), 219–227. doi:10.1136/annrheumdis-2020-217455
Holtze, C., Flaherty, K., Kreuter, M., Luppi, F., Moua, T., Vancheri, C., et al. (2018). Healthcare Utilisation and Costs in the Diagnosis and Treatment of Progressive-Fibrosing Interstitial Lung Diseases. Eur. Respir. Rev. 27 (150), 180078. doi:10.1183/16000617.0078-2018
Johnson, C. (2017). Recent Advances in the Pathogenesis, Prediction, and Management of Rheumatoid Arthritis-Associated Interstitial Lung Disease. Curr. Opin. Rheumatol. 29 (3), 254–259. doi:10.1097/bor.0000000000000380
Kanematsu, T., Kitaichi, M., Nishimura, K., Nagai, S., and Izumi, T. (1994). Clubbing of the Fingers and Smooth-Muscle Proliferation in Fibrotic Changes in the Lung in Patients with Idiopathic Pulmonary Fibrosis. Chest 105 (2), 339–342. doi:10.1378/chest.105.2.339
Kärkkäinen, M., Kettunen, H. P., Nurmi, H., Selander, T., Purokivi, M., and Kaarteenaho, R. (2019). Comparison of Disease Progression Subgroups in Idiopathic Pulmonary Fibrosis. BMC Pulm. Med. 19 (1), 228. doi:10.1186/s12890-019-0996-2
Khanna, D., Tashkin, D. P., Denton, C. P., Renzoni, E. A., Desai, S. R., and Varga, J. (2020). Etiology, Risk Factors, and Biomarkers in Systemic Sclerosis with Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 201 (6), 650–660. doi:10.1164/rccm.201903-0563CI
Kolb, M., Bondue, B., Pesci, A., Miyazaki, Y., Song, J. W., Bhatt, N. Y., et al. (2018). Acute Exacerbations of Progressive-Fibrosing Interstitial Lung Diseases. Eur. Respir. Rev. 27 (150), 180071. doi:10.1183/16000617.0071-2018
Kolb, M., and Vašáková, M. (2019). The Natural History of Progressive Fibrosing Interstitial Lung Diseases. Respir. Res. 20 (1), 57. doi:10.1186/s12931-019-1022-1
Kreuter, M., Lee, J. S., Tzouvelekis, A., Oldham, J. M., Molyneaux, P. L., Weycker, D., et al. (2021). Monocyte Count as a Prognostic Biomarker in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 204 (1), 74–81. doi:10.1164/rccm.202003-0669OC
Kwon, B. S., Choe, J., Chae, E. J., Hwang, H. S., Kim, Y. G., and Song, J. W. (2021). Progressive Fibrosing Interstitial Lung Disease: Prevalence and Clinical Outcome. Respir. Res. 22 (1), 282. doi:10.1186/s12931-021-01879-6
Lacedonia, D., Correale, M., Tricarico, L., Scioscia, G., Stornelli, S. R., Simone, F., et al. (2022). Survival of Patients with Idiopathic Pulmonary Fibrosis and Pulmonary Hypertension under Therapy with Nintedanib or Pirfenidone. Intern Emerg. Med. 17 (3), 815–822. doi:10.1007/s11739-021-02883-w
Lancaster, L., Crestani, B., Hernandez, P., Inoue, Y., Wachtlin, D., Loaiza, L., et al. (2019). Safety and Survival Data in Patients with Idiopathic Pulmonary Fibrosis Treated with Nintedanib: Pooled Data from Six Clinical Trials. BMJ Open Respir. Res. 6 (1), e000397. doi:10.1136/bmjresp-2018-000397
Lee, C. T., Ventura, I. B., Phillips, E. K., Leahy, A., Jablonski, R., Montner, S., et al. (2022). Interstitial Lung Disease in Firefighters: An Emerging Occupational Hazard. Front. Med. (Lausanne) 9, 864658. doi:10.3389/fmed.2022.864658
Lia, Y., Fana, Y., Wanga, Y., Yanga, S., Dua, X., and Yea, Q. (2022). Phenotypic Clusters and Survival Analyses in Interstitial Pneumonia with Myositis-specific Autoantibodies. Sarcoidosis Vasc. Diffuse Lung Dis. 38 (4), e2021047. doi:10.36141/svdld.v38i4.11368
Misharin, A. V., Morales-Nebreda, L., Reyfman, P. A., Cuda, C. M., Walter, J. M., McQuattie-Pimentel, A. C., et al. (2017). Monocyte-derived Alveolar Macrophages Drive Lung Fibrosis and Persist in the Lung over the Life Span. J. Exp. Med. 214 (8), 2387–2404. doi:10.1084/jem.20162152
Nambiar, A. M., Walker, C. M., and Sparks, J. A. (2021). Monitoring and Management of Fibrosing Interstitial Lung Diseases: a Narrative Review for Practicing Clinicians. Ther. Adv. Respir. Dis. 15, 17534666211039771. doi:10.1177/17534666211039771
Nasser, M., Larrieu, S., Boussel, L., Si-Mohamed, S., Bazin, F., Marque, S., et al. (2021a). Estimates of Epidemiology, Mortality and Disease Burden Associated with Progressive Fibrosing Interstitial Lung Disease in France (The PROGRESS Study). Respir. Res. 22 (1), 162. doi:10.1186/s12931-021-01749-1
Nasser, M., Larrieu, S., Si-Mohamed, S., Ahmad, K., Boussel, L., Brevet, M., et al. (2021b). Progressive Fibrosing Interstitial Lung Disease: a Clinical Cohort (The PROGRESS Study). Eur. Respir. J. 57 (2), 2002718. doi:10.1183/13993003.02718-2020
Oldham, J. M., Lee, C. T., Wu, Z., Bowman, W. S., Vu Pugashetti, J., Dao, N., et al. (2021). Lung Function Trajectory in Progressive Fibrosing Interstitial Lung Disease. Eur. Respir. J. 59 (6), 2101396. doi:10.1183/13993003.01396-2021
Olson, A. L., Gifford, A. H., Inase, N., Fernández Pérez, E. R., and Suda, T. (2018). The Epidemiology of Idiopathic Pulmonary Fibrosis and Interstitial Lung Diseases at Risk of a Progressive-Fibrosing Phenotype. Eur. Respir. Rev. 27 (150), 180077. doi:10.1183/16000617.0077-2018
Orlandi, M., Landini, N., Sambataro, G., Nardi, C., Tofani, L., Bruni, C., et al. (2022). The Role of Chest CT in Deciphering Interstitial Lung Involvement: Systemic Sclerosis versus COVID-19. Rheumatol. Oxf. 61 (4), 1600–1609. doi:10.1093/rheumatology/keab615
Petnak, T., Lertjitbanjong, P., Thongprayoon, C., and Moua, T. (2021). Impact of Antifibrotic Therapy on Mortality and Acute Exacerbation in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Chest 160 (5), 1751–1763. doi:10.1016/j.chest.2021.06.049
Raghu, G., Collard, H. R., Egan, J. J., Martinez, F. J., Behr, J., Brown, K. K., et al. (2011). An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-Based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 183 (6), 788–824. doi:10.1164/rccm.2009-040GL
Raghu, G., Remy-Jardin, M., Myers, J. L., Richeldi, L., Ryerson, C. J., Lederer, D. J., et al. (2018). Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 198 (5), e44–e68. doi:10.1164/rccm.201807-1255ST
Raghu, G., Remy-Jardin, M., Richeldi, L., Thomson, C. C., Inoue, Y., Johkoh, T., et al. (2022). Idiopathic Pulmonary Fibrosis (An Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 205 (9), e18–e47. doi:10.1164/rccm.202202-0399ST
Richeldi, L. (2015). Time for Prevention of Idiopathic Pulmonary Fibrosis Exacerbation. Ann. Am. Thorac. Soc. 12 Suppl 2, S181–S185. doi:10.1513/AnnalsATS.201504-210AW
Ruaro, B., Baratella, E., Confalonieri, P., Wade, B., Marrocchio, C., Geri, P., et al. (2021). High-Resolution Computed Tomography: Lights and Shadows in Improving Care for SSc-ILD Patients. Diagnostics 11 (11), 1960. doi:10.3390/diagnostics11111960
Sharma, A., Ferraro, V., Renzoni, E. A., and Morisset, J. (2021). Chronic Hypersensitivity Pneumonitis: Real World Diagnostic Criteria. Curr. Opin. Pulm. Med. 27 (5), 414–421. doi:10.1097/mcp.0000000000000799
Skibba, M., Drelich, A., Poellmann, M., Hong, S., and Brasier, A. R. (2020). Nanoapproaches to Modifying Epigenetics of Epithelial Mesenchymal Transition for Treatment of Pulmonary Fibrosis. Front. Pharmacol. 11, 607689. doi:10.3389/fphar.2020.607689
van Manen, M. J. G., Vermeer, L. C., Moor, C. C., Vrijenhoeff, R., Grutters, J. C., Veltkamp, M., et al. (2017). Clubbing in Patients with Fibrotic Interstitial Lung Diseases. Respir. Med. 132, 226–231. doi:10.1016/j.rmed.2017.10.021
Walsh, S. L. F., Devaraj, A., Enghelmayer, J. I., Kishi, K., Silva, R. S., Patel, N., et al. (2018). Role of Imaging in Progressive-Fibrosing Interstitial Lung Diseases. Eur. Respir. Rev. 27 (150), 180073. doi:10.1183/16000617.0073-2018
Wang, Y., Hou, Z., Qiu, M., and Ye, Q. (2018). Risk Factors for Primary Sjögren Syndrome-Associated Interstitial Lung Disease. J. Thorac. Dis. 10 (4), 2108–2117. doi:10.21037/jtd.2018.03.120
Wijsenbeek, M., Kreuter, M., Olson, A., Fischer, A., Bendstrup, E., Wells, C. D., et al. (2019). Progressive Fibrosing Interstitial Lung Diseases: Current Practice in Diagnosis and Management. Curr. Med. Res. Opin. 35 (11), 2015–2024. doi:10.1080/03007995.2019.1647040
Wongkarnjana, A., Scallan, C., and Kolb, M. R. J. (2020). Progressive Fibrosing Interstitial Lung Disease: Treatable Traits and Therapeutic Strategies. Curr. Opin. Pulm. Med. 26 (5), 436–442. doi:10.1097/mcp.0000000000000712
Wuyts, W. A., Papiris, S., Manali, E., Kilpeläinen, M., Davidsen, J. R., Miedema, J., et al. (2020). The Burden of Progressive Fibrosing Interstitial Lung Disease: A DELPHI Approach. Adv. Ther. 37 (7), 3246–3264. doi:10.1007/s12325-020-01384-0
Yang, S., Chai, D., Li, Y., Wang, Y., Zhan, X., Zhang, L., et al. (2022). Patterns of Lung Diseases Predict Survival in Patients with MPO-ANCA-Associated Vasculitis: a Single-Center Retrospective Study. Clin. Rheumatol. 41 (3), 783–793. doi:10.1007/s10067-021-05964-5
Keywords: progressive fibrosing interstitial lung disease, prognosis, lung function test, blood count values, anti-fibrotic treatment
Citation: Wang Y, Guo Z, Ma R, Wang J, Wu N, Fan Y and Ye Q (2022) Prognostic Predictive Characteristics in Patients With Fibrosing Interstitial Lung Disease: A Retrospective Cohort Study. Front. Pharmacol. 13:924754. doi: 10.3389/fphar.2022.924754
Received: 20 April 2022; Accepted: 23 May 2022;
Published: 01 July 2022.
Edited by:
Barbara Ruaro, University of Trieste, ItalyReviewed by:
Stefano Tavano, University of Trieste, ItalyCopyright © 2022 Wang, Guo, Ma, Wang, Wu, Fan and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiao Ye, eWVxaWFvX2NoYW95YW5nQHNpbmEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.