 Luyan Liu
Luyan Liu Yi Wu
Yi Wu Kaiyan Ye
Kaiyan Ye Meichun Cai
Meichun Cai Guanglei Zhuang
Guanglei Zhuang Jieyi Wang
Jieyi Wang- 1Lyvgen Biopharma, Shanghai, China
- 2State Key Laboratory of Oncogenes and Related Genes, Department of Obstetrics and Gynecology, Ren Ji Hospital, Shanghai Cancer Institute, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 3Shanghai Key Laboratory of Gynecologic Oncology, Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Co-stimulation signaling in various types of immune cells modulates immune responses in physiology and disease. Tumor necrosis factor receptor superfamily (TNFRSF) members such as CD40, OX40 and CD137/4-1BB are expressed on myeloid cells and/or lymphocytes, and they regulate antigen presentation and adaptive immune activities. TNFRSF agonistic antibodies have been evaluated extensively in preclinical models, and the robust antitumor immune responses and efficacy have encouraged continued clinical investigations for the last two decades. However, balancing the toxicities and efficacy of TNFRSF agonistic antibodies remains a major challenge in the clinical development. Insights into the co-stimulation signaling biology, antibody structural roles and their functionality in immuno-oncology are guiding new advancement of this field. Leveraging the interactions between antibodies and the inhibitory Fc receptor FcγRIIB to optimize co-stimulation agonistic activities dependent on FcγRIIB cross-linking selectively in tumor microenvironment represents the current frontier, which also includes cross-linking through tumor antigen binding with bispecific antibodies. In this review, we will summarize the immunological roles of TNFRSF members and current clinical studies of TNFRSF agonistic antibodies. We will also cover the contribution of different IgG structure domains to these agonistic activities, with a focus on the role of FcγRIIB in TNFRSF cross-linking and clustering bridged by agonistic antibodies. We will review and discuss several Fc-engineering approaches to optimize Fc binding ability to FcγRIIB in the context of proper Fab and the epitope, including a cross-linking antibody (xLinkAb) model and its application in developing TNFRSF agonistic antibodies with improved efficacy and safety for cancer immunotherapy.
1 Introduction
Immunotherapy has become an important cancer treatment option, and the antagonistic antibodies of immune checkpoint inhibitors PD-(L)1 have led a cancer treatment revolution in the last decade. Many studies have revealed the roles of adaptive immunities in tumor elimination and the mechanisms by which cancers evade immune response (Wei, Duffy et al. 2018, Morad, Helmink et al. 2021). CD8 T-cell dysfunction was identified as one mechanism of tumor escape, sometimes caused by overactivation of immune checkpoint receptors, such as PD-1. Since the response to PD-(L)1 treatment varies among patients and cancer types, with a limited response rate of around 10%–30%, it is important to identify new immuno-therapeutic targets and optimize the corresponding drug design with improved therapeutic efficacy and safety.
Co-stimulatory receptors have been considered as important as and complementary to the checkpoint inhibitors to promote immune antitumor activities. Many tumor necrosis factor (TNF) receptor superfamily (TNFRSF) members have been identified as the co-stimulatory receptors in antitumor immunity (Croft et al. 2013). For example, the TNFRSF members CD137 (4-1BB) and CD40 showed their potential as immunotherapy targets by stimulating the proliferation and cytotoxic activities of tumor-reactive CD8 T cells directly and indirectly, respectively (Jeong and Park, 2020). Effective TNFRSF agonistic antibodies may contribute to the therapeutic repertoires against cancers.
TNFRSF receptors are normally activated by molecular clustering induced by cognate cell membrane TNFSF ligands. Antibodies have bivalency in antigen binding and can induce cell membrane receptor clustering and activation. The binding epitope of the TNFRSF target is one of the key factors determining the intrinsic agonistic activities of the antibody. Antibodies are also multi-domain immunoglobulins possessing Fc receptor interaction in addition to antigen binding functionality. Thus, the agonistic activity of an antibody can vary drastically in different environments resulting from potential multiple molecular interactions. In fact, Fc and Fc gamma receptor (FcγR) interaction has been identified as the most dominant factor in determining overall agonistic activities of an antibody. Binding to FcγRs via Fc provides the antibody an opportunity for multivalent interactions beyond its antigen target. Thus, Fc/FcγR interaction can facilitate target receptor cross-linking and clustering by TNFRSF agonistic antibodies (Medler et al. 2019). As receptor molecule clustering is the fundamental mechanism for TNFRSF members to trigger their downstream signaling, Fc/FcγR mediated cross-linking can dramatically affect the agonistic activity of TNFRSF antibodies, generating super TNFRSF agonistic antibodies in certain cases.
Here, we review recent advances in our understanding of agonistic antibodies targeting TNFRSF in cancers. We focus on the structural and functional correlations of these TNFRSF agonistic antibodies, more specifically the impact of Fc-gamma receptor IIB (FcγRIIB) on their antitumor activities and safety. We will discuss the principles of how FcγRIIB-dependent TNFRSF agonistic antibodies can be engineered to induce receptor clustering and activation optimally in the tumor microenvironment while minimizing systemic toxicity.
2 TNFSF-TNFRSF System
2.1 TNFSF-TNFRSF Expression Profile and Biological Function
The tumor necrosis factor superfamily (TNFSF) and their receptor superfamily (TNFRSF), composed of 19 ligands and 29 receptors respectively, play diversified roles in inflammation, apoptosis, proliferation and morphogenesis (Figure 1; Table 1) (Dostert et al. 2019). The genes of TNFSF and TNFRSF are clustered in several distinct genetic regions within the major histocompatibility complex (MHC). The human MHC is a large genetic region containing a dense proportion of genes implicated in immunity. Phylogenetic analyses clearly indicate that TNFSF and TNFRSF emerged and diverged from the common ancestors within proto-MHC region before the appearance of vertebrates. TNFSF and TNFRSF co-evolve together with the adaptive immune system (Collette et al., 2003, Glenney and Wiens, 2007, Wiens and Glenney, 2011). That evolutionary history might explain the highly conserved structural and functional properties among TNFSF and TNFRSF members. They have an important role in adaptive immunity and control of immune homeostasis. Structurally, they share a similar ligand-receptor trimeric structure for signaling activation. Functionally, TNFRSF members show some overlapping expression profiles and biological functions such as promoting the proliferation of T and B cells or maturation of dendritic cells and macrophages. At the same time, each TNFSF or TNFRSF member is assumed to have its distinct spatiotemporal expression profile, signaling network, and functional impact on the immune system.
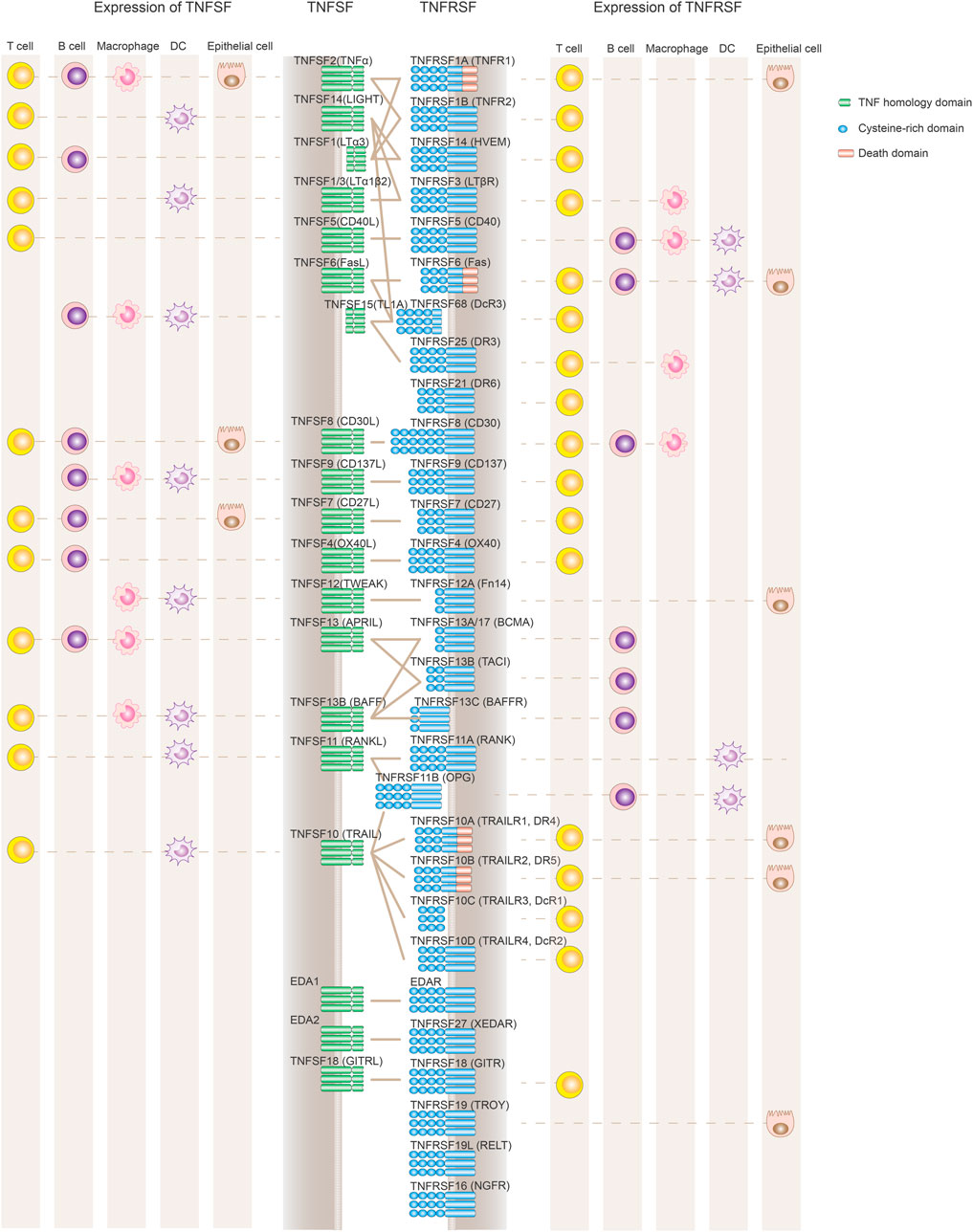
FIGURE 1. Expression and interaction of TNFSF and TNFRSF members. TNFRSF members (depicted in blue on the right) contain variable numbers of cysteine-rich domains (CRD) in their ligand-binding extracellular regions. TNFSF ligands (left side shown in green) are active primarily as non-covalently associated homotrimers or homodimers to facilitate the formation of TNFRSF trimer clustering and its downstream signaling activation. TNFRSF members regulate the immune system mainly through either stimulating cell proliferation and maturation or promoting apoptotic cell death via a death domain. Also depicted are the primary cell types expressing TNFRSF and TNFSF, although the list is not comprehensive to represent the complex expression profile of each molecule, and some cell populations such as NK cells or monocytes are not included. Both TNFSF and TNFRSF members are widely and dynamically expressed in different immune cell populations.
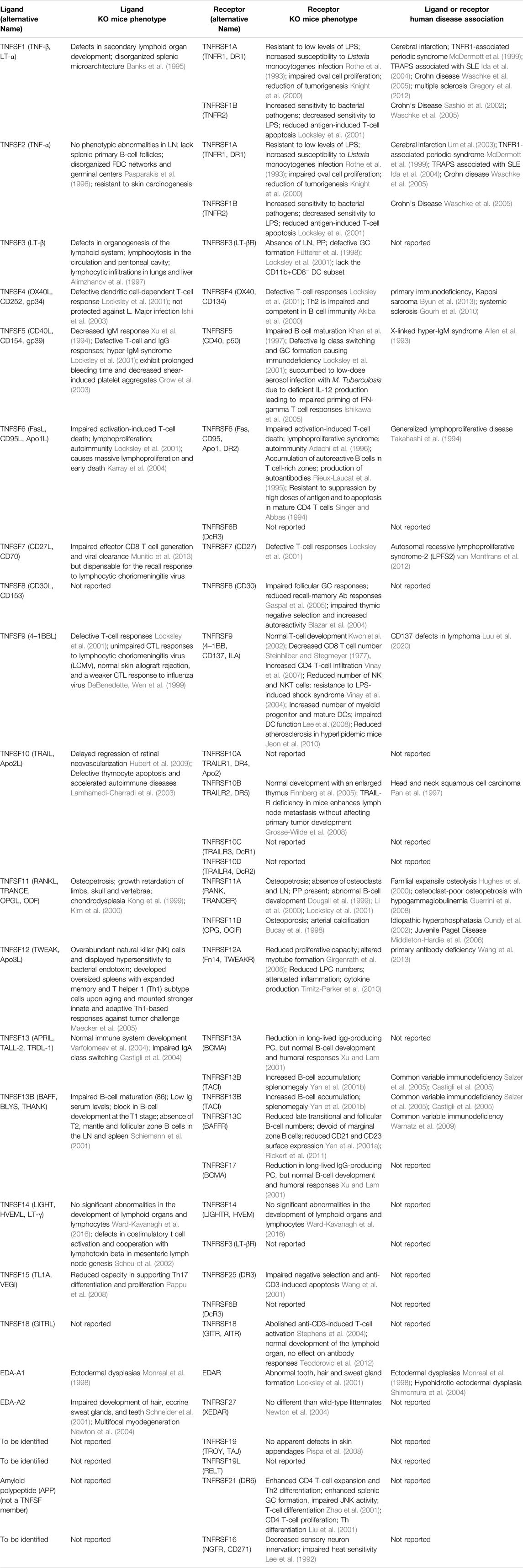
TABLE 1. Tumor necrosis factor superfamily ligands and receptors and their functions.
Although there is still a lot to be learned about the nuanced divergence among the TNFSF and TNFRSF members, our knowledge has been enriched gradually during the last three decades (Aggarwal et al., 2012, Croft and Siegel, 2017, Dostert et al., 2019) (Table 1). From mouse gene knock-out studies and human genetics analyses, TNFRSF and TNFSF members have been proven to play critical roles in immune system development and immune modulation. In the case of the stimulation of T cell antitumor activities, the known T cell co-stimulators including CD27, CD137 and OX40 were demonstrated to have distinct effects. For example, CD27 and HVEM expressed on resting T cells function early after the initial activation of T cells, while OX40 and CD137 signals on T cells are induced at a later stage after antigen-activation, and OX40 and CD137 showed distinct preferential effects on CD4 and CD8 T cells respectively (Watts, 2005). In another experiment to characterize the impact of CD27, CD137, OX40, and GITR on the CD8 T-cell cytokine response, although both CD137 and CD27 increased the sensitivity of the stimulation, only CD137 was able to prolong the response duration and had the strongest amplification effect on cytokine production. In contrast, GITR and OX40 had almost no effect shown in this study (Nguyen et al., 2021). In an ex vivo tumor-infiltrating lymphocytes (TILs) stimulation assay with autologous tumor cells, CD137+ TILs exhibited the greatest frequency among the effector cells expressing IFN-γ, TNF-α, Granzyme B, perforin, and IL-2, compared to OX40+, PD-1+, CD25+, and CD69+ TILs, suggesting that CD137+ TILs might have the most effective antitumor activities.
2.2 TNSRSF Signaling Activation Driven by Receptor Clustering
TNFRSF members are naturally activated by their corresponding TNFSF ligands in a highly regulated manner. As above mentioned above, the structural principles of TNFRSF-TNFSF interactions and downstream signaling activation are well conserved among the TNFRSF and TNFSF members. TNFRSF receptors are type 1 transmembrane proteins that adopt elongated structures containing mainly 3-4 highly conserved “cysteine-rich domains” (CRDs). TNFSF ligands are type II transmembrane proteins non-covalently linked as homotrimer or homodimers via a conserved carboxy-terminal homology domain called the TNF homology domain (THD). TNFSF trimer mainly binds to the 2nd and 3rd CRDs (CRD 2–3) of TNFRSF members and facilitates the formation of an active TNFSF3–TNFRSF3 complex (Figure 2) (Banner et al., 1993; Locksley et al., 2001; Compaan and Hymowitz, 2006). This complex is the minimal unit for TNFRSF signaling activation. In fact, a significant fraction of TNFRSF members need further oligomerization of two or more TNFSF3–TNFRSF3 complexes to be able to fully stimulate TNFRSF downstream signaling cascades (Figure 2) (Wajant, 2015). In natural physiological conditions, the ligands of this category of TNFRSF work as membrane- bound, while soluble TNFSF does not show secondary receptor clustering ability. The molecular and cellular regulation mechanisms of TNFSF and TNFRSF enable a precise control of TNFRSF signaling with spatial-temporal specificity.
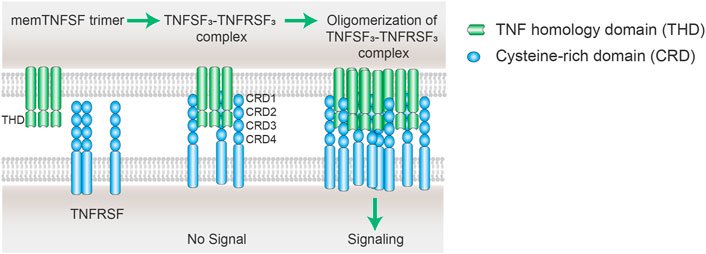
FIGURE 2. Scheme of oligomerization of TNFSF3–TNFRSF3 complexes. The cell membrane TNFSF (mem TNFSF, often associated as trimer) binds to TNFRSF via the interactions between the TNF homology domain (THD) and cysteine rich domain (CRD), resulting in the formation of TNFSF3-TNFRSF3 complex. However, the minimum TNFSF-TNFRSF complex is insufficient to trigger signaling of some TNFRSF members including CD40, CD137, OX40, and GITR. Secondary interaction of the initial trimeric complex leads to oligomerization of the ligand-receptor complexes. The clustering of multiple TNFRSFs is necessary to activate receptor intracellular signaling pathways.
3 Therapeutic Potential of TNFRSF Agonistic Antibodies
Many TNFRSF members have been evaluated as potential novel immunotherapy targets, given their role in activation, clonal expansion, and survival of immune cells including cytotoxic T cells, the same population regulated by PD-1. In fact, the interest of targeting TNFRSF in medicine emerged by the end of 1990s, even earlier than the development of PD-1 antagonists. Besides T cell co-stimulators such as CD137, CD27, OX40, GITR or TNFR2, for antigen presentation and innate immune activation, CD40 is also an important target for cancer immunotherapy. Nowadays, there are dozens if not hundreds of TNFRSF agonist candidates in the format of monoclonal or multi-specific antibodies being tested in clinical trials. They are being investigated or developed as monotherapy or in combination with immune checkpoint inhibitors, such as anti-CTLA4 or anti PD-1/PD-L1 antibodies, or standard treatment like chemotherapy. The clinical development status of representative TNFRSF agonistic antibodies (Croft et al., 2013; Assal et al. 2015; Burugu et al., 2018; Lee, 2021) are summarized in Table 2.
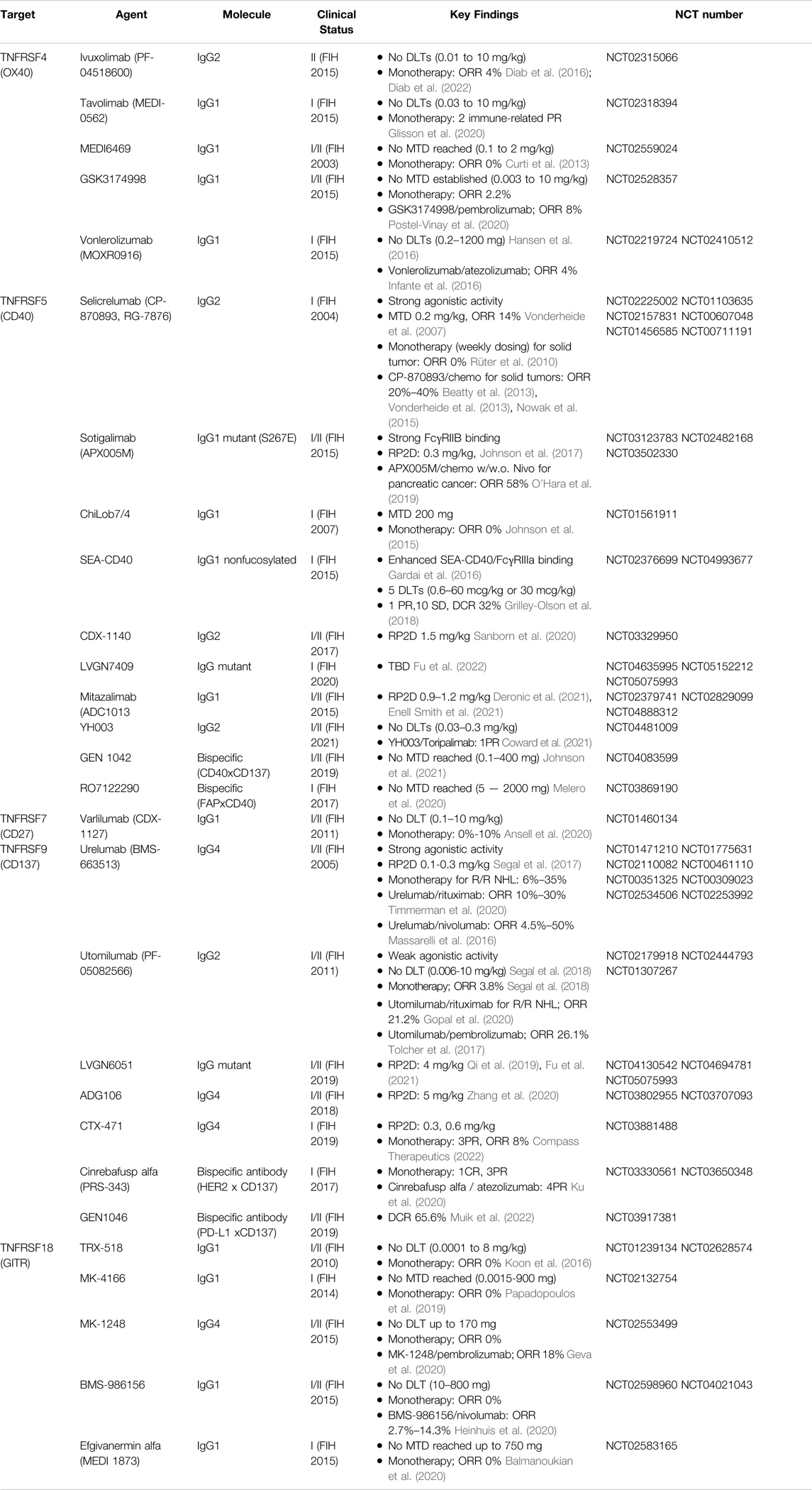
TABLE 2. Clinical studies of anti-TNFRSF agonistic antibodies for cancer treatment, alone or in combination.
Although showing promising antitumor activities in preclinical tests, the clinical performance of TNFRSF agonists remains unsatisfactory. Dose-limiting toxicity or insufficient agonistic potency has been hampering the clinical development. For example, selicrelumab (CP-870893) and urelumab (BMS-663513) are two well-studied TNFRSF potent agonistic monoclonal antibodies undergone in clinical trials. Selicrelumab was an anti-CD40 agonistic antibody developed as human IgG2 showing potent activity without cross-linking. Urelumab was the first clinical strong anti-CD137 agonist developed as human IgG4 capable of binding to FcγRs and cross-linking, yielding a super agonistic antibody in vivo. Both agents showed treatment-related adverse events such as liver toxicity or cytokine release syndrome at low doses had maximal tolerated dose (MTD) at 0.2–0.3 mg/kg, which severely limited their full clinical evaluation.
Various efforts are being applied to optimize TNFRSF agonistic antibodies to overcome toxicity by improving their local agonism. Fc-engineering of anti-CD40 has been tested to enhance efficacy in the case of sotigalimab (APX005M), which uses IgG1 with S267E variation that has high affinity for FcγRIIB (Filbert et al., 2021). Newer agonistic antibodies leveraging FcγRIIB cross-linking with IgG4 such as ADG106 or CTX-471, or with designed IgG Fc mutant selective for binding to FcγRIIB such as LVGN6051 or LVGN7409, have also entered clinical development. Additionally, bi-specific antibodies have been developed by engineering an additional binding site to tumor antigen, such as HER2, fibroblast activation protein (FAP), or PD-L1 for targeted agonism towards tumors (Table 2). It remains to be seen if these attempts will resolve the clinical challenges for TNFRSF agonistic antibodies.
The rich literature on TNFRSF agonistic antibody development provides essential and excellent learnings for continued improvement in the approaches to optimize the design and development of TNFRSF agonistic antibodies. In this review, we focus on monoclonal antibody agonists with regular IgG structure.
4 Mechanism of Action of TNFRSF Agonistic Antibodies
4.1 Background on IgG and FcγR
4.1.1 Human and Mouse Immunoglobulins
Among the five classes of human immunoglobulins (IgA, IgD, IgE, IgG, and IgM), IgG is most frequently chosen for therapeutic antibody development (Table 2). Antibodies possess regions that recognize antigens, the fragment antigen binding (Fab) region, and the effector function region, the fragment crystallizable (Fc) region that recognize Fc receptors and complement proteins. The second constant domain (CH2) in Fc and the first constant domain (CH1) in Fab are linked via a hinge. The hinge contains disulfide bonds from conserved cysteine residues that stabilize the structure of IgG, while allowing Fab and Fc conformational flexibility.
There are four IgG subclasses in human known as IgG1, IgG2, IgG3, and IgG4, and in mouse named as IgG1, IgG2a, IgG2b, and IgG3. They share more than 95% homology in the amino acid sequences of the Fc regions but show major differences in the amino acid composition and structure of the hinge region. IgGs participate in many important immune protection functions including the clearance of pathogens and toxins, and lysis and removal of infected or malignant cells. Detection and presentation of abnormal antigens are often the first steps for immune protection. This process involves the cellular uptake and intracellular processing of those antigens assisted by IgGs and FcγRs expressed on immune cells including antigen presenting cells (APCs).
4.1.2 Human and Mouse Fc Gamma Receptors
There are three major groups of human activating FcγRs (including FcγRI, FcγRIIA, FcγRIIC, FcγRIIIA, and FcγRIIIB). All activating FcγRs, except FcγRIIIB, are associated with an immunoreceptor tyrosine-based activation motif (ITAM) either in the intracellular domain (FcγRIIA and FcγRIIC) or associated with the common FcRγ chain (FcγRI and FcγRIIIA) (Vidarsson and van de Winkel, 1998; Bruhns, Iannascoli et al. 2009; Bruhns and Jönsson 2015). Similarly, there are three activating FcγRs in mouse (FcγRI, FcγRIII, and FcγRIV) with an ITAM motif. In contrast, there is only one inhibitory FcγR (FcγRIIB) in humans or the mouse. FcγRIIB contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) in the intracellular domain (Nimmerjahn et al. 2005; Hogarth and Pietersz, 2012) (Figure 3A).
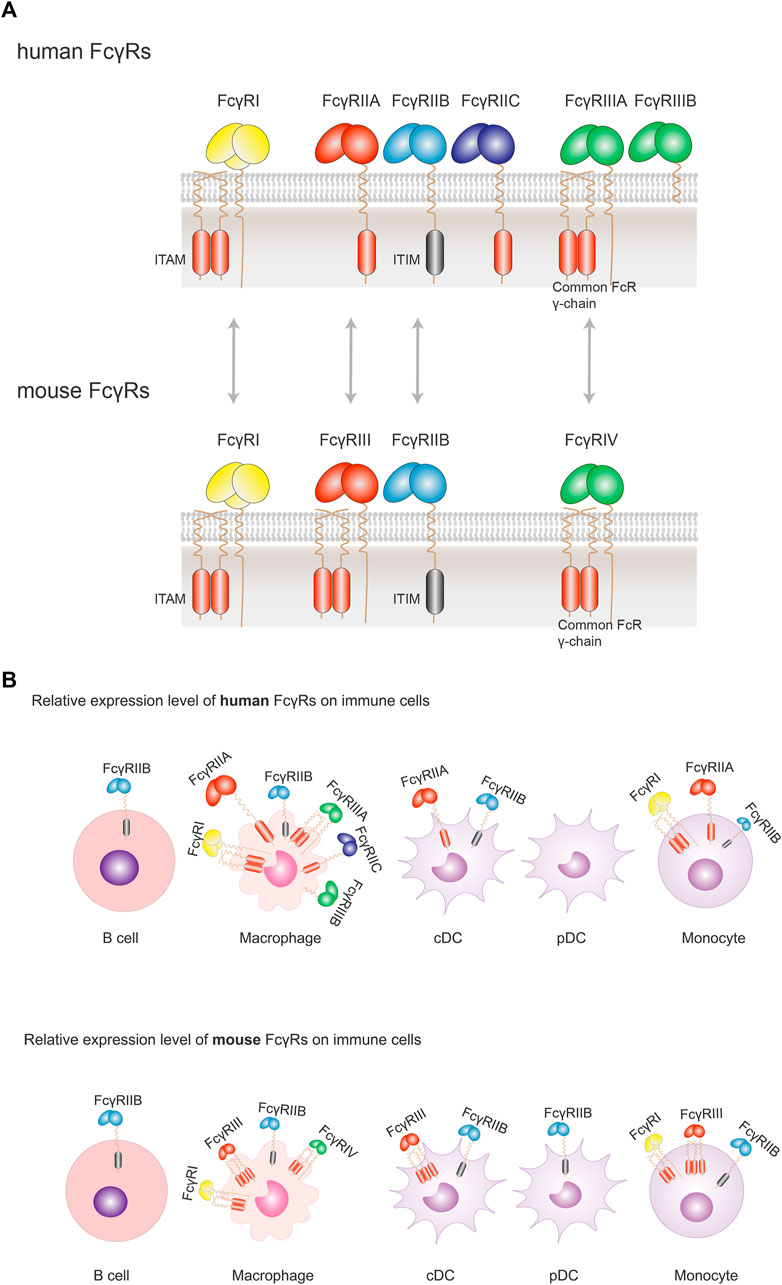
FIGURE 3. Orthologous pairs of FcγRs between human and mouse and their cellular expression. (A) In human, three groups of FcγRs have been described: FcγRI, FcγRIIA/B/C, FcγRIIIA/B. Orthologous pairs identified in mouse include human FcγRI and mouse FcγRI, human FcγRIIA and mouse FcγRIII, human FcγRIIB and Mouse FcγRIIB, and human FcγRIIIA and mouse FcγRIV. All activating FcγRs, except FcγRIIIB, are associated with an immunoreceptor tyrosine-based activation motif (ITAM) either in the intracellular domain (FcγRIIA and FcγRIIC) or associated with the common FcRγ chain (FcγRI and FcγRIIIA). There is only one inhibitory FcγR (FcγRIIB) in human or mouse. FcγRIIB contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) in the intracellular domain. (B) FcγRs are expressed solely or simultaneously at the membrane of the various immune cells. FcγRI, FcγRIIA/B and FcγRIIIA are found on macrophages; FcγRIIA/B on conventional DC (cDC), and FcγRIIB (the sole FcγR expressed) on B cells. When co-expressed, activating FcγRs generally expressed more abundantly with respect to the inhibitory FcγRIIB. Despite a similar expression profile of FcγRs in human and mouse, there are some differences between the two species: FcγRIIB is not expressed on human pDCs, but on mouse pDCs; while co-expressed on the same cells, the inhibitory FcγRIIB is relatively less than activating FcγRs in human, which is not so apparent in mouse. Size of symbols of FcγRs is drawn to reflect their relative expression levels.
Following the binding to antigen-IgG immune complexes (ICs), ITAM of activating FcγRs is phosphorylated by Src kinases and induces downstream signaling, leading to various immune responses. ITAM-mediated functions include phagocytosis, degranulation, antibody-dependent cellular cytotoxicity (ADCC), cytokine, lipid mediator and superoxide production. In contrast, in the case of the inhibitory FcγRIIB engagement, the downstream signal transduction mediated via ITIM results in anti-inflammatory activities. As the only inhibitory FcγR, FcγRIIB plays an important role in preventing exaggerated auto-immunity and maintaining homeostatic immunity to disease prevalence and severity.
4.1.3 FcγR Expression, Affinity and Function Profile
In humans, FcγRs are widely expressed in hematopoietic cells. B cells solely express FcγRIIB. Monocytes express FcγRI and FcγRIIA/B. Both monocyte-derived DCs and DCs from blood, but not plasmacytoid DCs (pDCs), express primarily FcγRIIA/FcγRIIB. Macrophages express all classes of FcγRs. It is worth noting that the activating FcγRs are generally more abundantly expressed than FcγRIIB. FcγRI is the only high-affinity receptor, and is able to bind to monomeric IgG. In contrast, FcγRIIA/B/C and FcγRIIIA/B are low-affinity receptors (Table 3), and therefore binding to FcγRII/IIIs require high-avidity of multivalent antigen-IgGs immune complexes complex (IC). Once antigens are recognized and bound by IgG-Fab, the resulting immune complexes (ICs) enable the Fc portion of the multivalent ICs to bind to low affinity FcγRs, leading to the clustering of multi-homo or hetero FcγRs. The clustering of FcγRs is important for their cytoplasmic signaling activation both for activating FcγRs and inhibitory FcγRIIB (Junker et al., 2020).
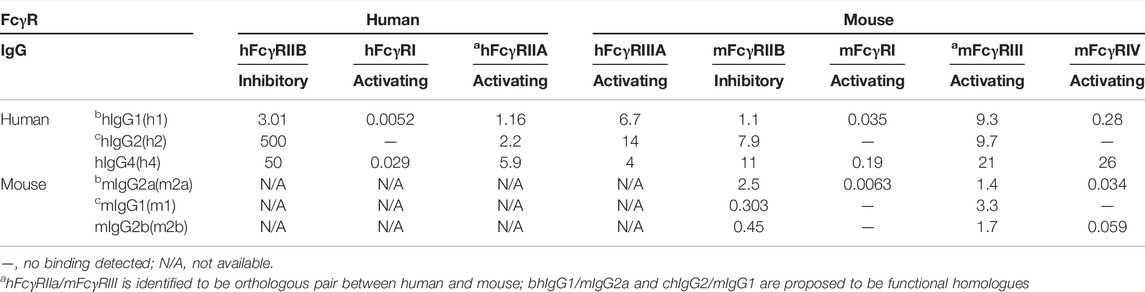
TABLE 3. Affinity in KD (μM) of human and mouse IgG subclasses for FcγRs.
Since activating and inhibitory FcγRs are often co-expressed on the membrane of the same cell, it is the relative abundance of these FcγRs and the strength of the IgG-FcγR interaction that ultimately determines the cellular outcomes. The immune suppressive role of FcγRIIB might explicate its more modest expression profile than that of activating FcγRs. The rarer membrane presence of FcγRIIB suggests a higher binding or activation threshold, for example, requiring a higher valency of multivalent antigens-IgGs-FcγRs complex (Lehmann et al., 2017). Therefore, FcγRIIB won’t be activated too early to disturb the protection functions of the activating FcγRs during infection for example.
Interestingly, besides its immune suppressive function, FcγRIIB was demonstrated to help clustering of immune complexes, as knockout of FcγRIIB resulted in formation of smaller endosomes. In the studies on the phagocytosis of antibody coated nanoparticles, a role for FcγRIIB was identified for the aggregation of multivalent antibody bound nanoparticles. We speculate this clustering ability of FcγRIIB, which facilitates the aggregation of bound antigens-IgGs complexes in the case of phagocytosis, may contribute to the activity of TNFRSF agonistic antibodies via cross-linking, which will be discussed in detail later.
4.1.4 Comparison of IgG/FcγR Interactions in Human and Mouse
The high affinity receptor FcγRI and the sole inhibitory FcγRIIB are highly conserved between human and mouse. Human FcγRIIA and mouse FcγRIII are identified to be orthologous receptors, since they share similarities in terms of genomic localization and sequence in the extracellular portion (Nimmerjahn and Ravetch, 2006, Hirano et al., 2007) (Figure 3A). In mouse, FcγRs show similar expression profile as their human orthologous pairs, except that the expression of FcγRIIB is slightly more abundant and broader among various immune cells than in human (Junker, Gordon et al. 2020) (Figure 3B).
Besides the heterogeneous expression of FcγRs, additional complexity of IgGs- FcγRs system was provided by different IgG subclasses, IgG1, IgG2, IgG3, and IgG4, with distinct binding affinities for different FcγRs (Table 3). Human IgG1 appears to be a functional homolog of mouse IgG2a, and human IgG2 for mouse IgG1. There are some nuanced differences between human and mouse, including the fact that human IgG2 binding affinity of human FcγRIIB is much lower than its orthologous mouse IgG1 to mouse FcγRIIB (Rosales and Uribe-Querol, 2013; Dekkers et al., 2017; Rosales, 2017). Together, that could explain the similarity of those homolog IgGs and FcγRs in terms of their expression profile and binding affinities.
In summary, high similarity is found between human and mouse IgG-FcγR systems system but accompanied with some exceptions. Therefore, cautions should be taken when transferring the data obtained in mouse models into the human system.
Human IgG1 and IgG4 preferably bind to FcγRI, whereas IgG2 fails to bind FcγRI. FcγRI is the only high-affinity IgG receptor. FcγRIIB has a relatively lower affinity for IgG1, IgG2 and IgG4 than FcγRIIA.
Murine IgG2a binds to FcγRI with significantly higher affinity than to other FcγRs, whereas neither IgG1 nor IgG2b binds to FcγRI. Murine IgG1 has a higher binding affinity to FcγRIIB than IgG2a.
Human IgGs bind to mouse FcgγR with similar binding ability to those for human ortholog receptors, with relative affinities IgG1>IgG4>IgG2 and FcγRI>>FcγRIV>FcγRIII>FcγRIIb. However, there are some subtle differences including but not limited to that human IgG2 has a higher binding affinity to mouse FcγRIIB than human FcγRIIB.
4.2 Structural Determinants of Agonistic Antibodies that Mimic TNFSF Ligand to Facilitate the Oligomerization and Activation of TNFRSF Receptors
TNFSF ligands naturally exist on cell membrane as trimers or dimers, which in turn can facilitate the formation of TNFRSF trimer and secondary clustering of oligomers leading to receptor activation. Monoclonal antibodies have shown agonistic activity towards TNFRSF members and been evaluated in the clinic as immune co-stimulation agents for cancer therapy as introduced above (Table 2). One key question is how agonistic antibodies mimic TNFSF ligand to facilitate the oligomerization of the receptors to trigger the downstream signaling cascades. The answer lies within the structural features of the IgG molecule, i.e., its flexible and multiple functional domains consist of bivalent antigen binding Fab arms, conformation determining hinge, and Fc dimer interacting with Fc receptors present on immune cells. It is understood that TNFRSF agonistic antibodies provoke the clustering of TNFRSF in the following ways: 1) bivalent Fab arms with high affinity binding to TNFRSF dimers/trimers; 2) proper hinge conferred by differential disulfide bonds supporting TNFRSF conformational changes for productive receptor clustering and activation; and, 3) Fc binding to membrane bound FcγRs which provide an “immobilized” scaffold to facilitate the aggregation of Fab bound TNFRSF, i.e., cross-linking. Each of these structural determinants individually and collectively attune the agonistic activity of the antibody in vitro and in vivo, with the Fc-FcγR interaction being the dominant factor.
4.2.1 Cross-Linking Mediated by Fc and Fc Gamma Receptors
4.2.1.1 TNFRSF Agonistic Antibodies Showing Dependence on FcγRIIB
The first demonstration of TNFRSF agonism dependency on FcγRIIB was a study focused on the development anti-Fas (TNFRSF6, DR2 or CD95) agonistic antibodies. In vitro studies showed that anti-Fas agonistic mAb Jo2 mediated apoptosis was enhanced by FcγRIIB, but not FcγRI and FcγRIII, on neighboring macrophages (Xu et al. 2003).
Agonistic antibodies targeting DR5, another TNFRSF death receptor, also displayed activities against a variety of tumors. Li et al. reported that the in vivo apoptotic and antitumor activities of these antibodies have an absolute requirement for FcγRIIB. Furthermore, enhancing FcγRIIB engagement via IgG1 S267E variant increased apoptotic and antitumor potency (Li and Ravetch, 2012).
Furthermore, agonist-targeted cells did not need to express FcγRIIB by themselves and FcγRIIB expression on bystander cells was sufficient, as demonstrated by the example that co-culture with CD40−/− (FcγRIIB+/+) B cells allowed anti-CD40 3/23 agonistic mAb to promote proliferation of FcγRIIB−/− (CD40+/+) B cells. This experiment also suggested that FcγRIIB downstream inhibitory signaling pathways were not required for promoting the agonistic antibody activity, as cross-linking mediated by extracellular part of FcγRIIB without intracellular domain is adequate for TNFRSF agonistic co-stimulation activities. More importantly, FcγRIIB has shown to be the only required FcγR for the antitumor activities in vivo (Li and Ravetch, 2013; White et al., 2013; White et al., 2014).
Besides DR2, DR5 and CD40, FcγRIIB-dependent agonism was also demonstrated for antibodies targeting CD137 and OX40 by exploiting FcγR deficient animals and/or antibody variants with defective FcγR binding.
Qi et al. evaluated the antitumor efficacy of two anti-CD137 agonistic antibodies with mouse IgG1 isotype, 3H3 with strong agonistic activity and LOB12.3 with weak agonistic activity in vitro. Both antibodies showed robust antitumor activity in vivo. However, the antitumor activity of LOB12.3-mIgG1 is significantly reduced in Fcgr2b−/− (FcγRIIB deficient) mice, while 3H3-mIgG1 showed a weaker but significant antitumor activity in Fcgr2b−/− mice. In Fcer1g−/− (lacking the common g chain, deficient in FcγRI, FcγRIII, and FcγRIV) and Fcgr3−/− (FcγRIII deficient) mice, both LOB12.3-mIgG1 and 3H3-mIgG1 showed potent anti-tumor activity like in wild type mice. Taken together, these data indicate that FcγRIIB but not the activating FcγRI, FcγRIII, or FcγRIV is required for antitumor activity of agonistic anti-CD137 antibodies in vivo.
Ho et al. identified a new class of CD137 agonist mAbs which showed cross-linking-dependent T-cell co-stimulation activity in vitro (Ho et al., 2020). During further in vivo experiments, when different mouse IgGs subclasses were compared, IgG2a had stronger antitumor activity than the IgG2aDANA variant (D265A-N297A, mutant that eliminates FcγR binding), and both murine IgG1 and IgG2 consistently showed strong efficacy. Because both murine IgG1 and IgG2 engage FcγRIIB and III, Ho et al. evaluated which FcγR was critical for the bioactivity of CD137 mAbs. They demonstrated that antitumor efficacy was maintained in FcγRIII-deficient mice but diminished in FcγRIIB-deficient mice, suggesting the critical role for FcγRIIB to provide cross-linking and to induce antitumor activity in vivo.
The role of FcγRIIB was also demonstrated for anti-OX40 agonist. Campos Carrascosa, van Beek et al. used lymphocytes from resected tumor, tumor-free (TF) tissue and peripheral blood mononuclear cells (PBMC) of 96 patients with hepatocellular and colorectal cancers to test the in vitro T-cell agonistic activity of OX40-targeting antibodies (Campos Carrascosa et al., 2020). They showed that, in contrast to a clinical candidate anti-OX40 IgG1, treatment with an Fc-engineered anti-OX40 IgG1 v12 variant (E223D, G237D, H268D, P271G, Y296D, and A330R, selectively enhanced FcγRIIB binding affinity), stimulated in vitro CD4 and CD8 TIL expansion and shifted their transcriptional landscape toward a pro-survival, inflammatory and chemotactic profile. They also proved that the activity of anti-OX40 IgG1 v12 was dependent on FcγRIIB engagement.
Zhang et al. generated four anti-OX40 agonistic mAbs in mIgG1 and then evaluated their ability to mediate OX40 agonism using reporter cell line in the absence or presence of the mouse B lymphoma cell line A20, which expresses only mouse FcγRIIB (mFcγRIIB). They found that all four mAbs mIgG1 demonstrated the agonistic activity upon mFcγRIIB crosslinking (Zhang et al., 2019). In contrast, all four mAbs mIgG1 were fairly inactive in the absence of A20 cells or in the presence of A20 cells incubated with an anti-mFcγRIIB blocking antibody prior to performing the reporter assays. None of the four mIgG1 D265A variants, which abrogate binding to all mFcγRs, was able to activate OX40.
FcγRIIB was shown to be able to boost the agonism of the engineered anti-OX40 mAb IgG1 with mutations T437R and K248E, that facilitated antibody multimerization upon binding to antigens on cell surface (Zhang et al., 2017). For the note, T437R is a mutation predicted to facilitate the dimerization of CH3 domains, and K248E mutation is predicted to facilitate multimerization by destabilizing the intramolecular CH2:CH3 interface. The combination of the two mutations is predicted to facilitate antibody multimerization upon binding to antigens on cell surface.
These studies demonstrated that there is a positive correlation between the activity of many TNFRSF agonistic antibodies and the capacity of their Fc binding to the inhibitory FcγRIIB.
Conclusive evidence for the essential role of FcγRIIB for certain agonistic antibodies of TNFRSF came from studies using genetic mouse models of Fcγ receptor knockouts. Absolute requirement of FcγRIIB but not activating Fcγ receptors was demonstrated in above mentioned studies for DR2 (Xu et al., 2003), DR5 (Li and Ravetch, 2012), CD40 (Li and Ravetch, 2011; White et al., 2011; Wilson et al., 2011; Dahan et al., 2016), and CD137 (Qi et al., 2019; Ho et al., 2020).
4.2.1.2 Similarities of the TNFRSF Secondary Clustering Mediated by Membrane- Bound Ligands and by Agonistic Antibodies Dependent on FcγRIIB Cross-Linking
In a study to determine if FcγR-dependent agonism is applicable for all the TNFRSF members, a coincidence was identified between the TNFRSF whose activation depended on secondary clustering mediated by membrane- bound ligands and those TNFRSF whose agonistic antibodies required the presence of FcγRIIB (Medler et al., 2019; Kucka and Wajant, 2020). Medler, Nelke et al. analyzed a panel of ∼30 murine antibodies recognizing more than 10 members of the TNFRSF for their capacity to elicit TNFRSF receptor activation. TNFRSF receptor responsive target cells were in co-cultured with empty vector (EV) control or murine FcγRIIB plasmid- transfected HEK293 cells IL8 induction was measured to detect TNFRSF receptor activation. With exception of one antibody against GITR, all other antibodies, recognizing CD27, OX40, 4-1BB, TNFR2, CD40, CD95, TRAILR1, TRAILR2 or Fn14, displayed no or only modest agonistic activity when TNFRSF receptor responsive target cells co-cultured with EV control. But they readily converted to potent receptor agonists in the presence of murine FcγRIIB-expressing cells. The coincidence is that those TNFRSF are only activated by their membrane- bound ligands but not by the soluble ligands. The data suggest that the threshold of activation is more exigent for those TNFRSF whose activation needs the oligomeric clustering mediated by the membrane- bound scaffold. This “immobilized” support is offered either by membrane- bound ligands or IgGs binding to FcγRIIB. It is tempting to postulate that the dependency on FcγRIIB is based on a superior clustering ability of membrane- bound FcγRIIB bridged by IgGs. In a similar model as membrane bound ligands, those FcγRIIB -“immobilized” IgGs facilitate the clustering of their Fab-bound TNFRSF, resulted in the aggregation of FcγRIIBs-IgGs-TNFRSFs complexes, and eventually TNFRSF cytoplasmic activation (Figure 4).
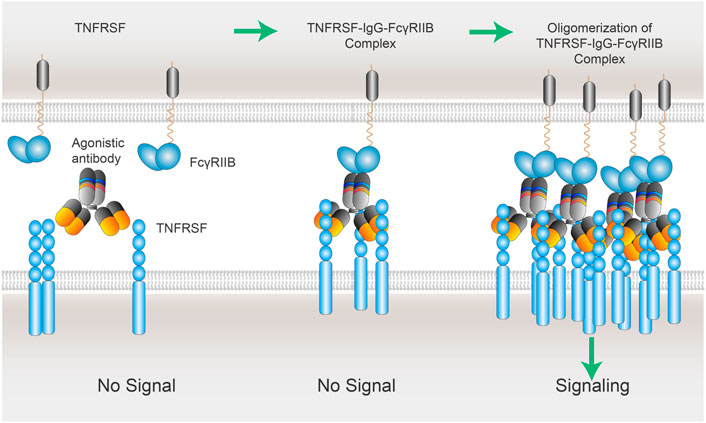
FIGURE 4. xLinkAb working model: the role of FcγRIIB in the oligomerization of TNFSFR-IgG-FcγRIIB complexes. xLinkAb TNFRSF agonistic antibody Fab arms binding to TNFRSF is insufficient to cluster and activate TNFRSF target; xLinkAb IgG-TNFRSF complex engages FcγRIIB with its engineered Fc selectively for FcγRIIB binding, leading to the formation of oligomic TNFRSF-IgG- FcγRIIB complexes and clustering of TNFRSF and signaling activation. The initial step should be IgG-Fab binding to TNFRSF, which is followed by TNFRSF-IgG-Fc binding to FcγRIIB forming TNFRSF-IgG-FcγRIIB complex and secondary clustering of TNFRSF. Activation of TNFRSF signaling depends on the formation of the multivalent TNFRSFs-IgGs-FcγRIIBs complexes and TNFRSF super-clustering.
4.2.1 3 Cross-Linking by Other FcγRs
In addition to FcγRIIB, other Fcγ receptors can cross-link antibodies to enhance their agonistic activity for TNFRSF in vitro. This is consistent with the working model of scaffold effect, which can be achieved by secondary antibodies or by simple physical immobilization on microtiter plate surface. However, FcγRs other than FcγRIIB do not contribute to the antitumor efficacy in vivo in mouse models for the agonistic antibodies whose activity depends on cross-linking. For example, the effect of FcγRIIB on anti-CD40 agonistic activities was investigated both in vitro and in vivo. In vitro studies showed that both mouse FcγRIIB and FcγRIII could promote cross-linking dependent agonistic activity and stimulated proliferation of B cell activity of a rat anti-mouse CD40 mAb, 3/23, in the form of mouse IgG1 (White et al., 2011). In vivo studies, however, demonstrated that the antibody was efficacious in mice with knockout of activating Fcγ receptors. The antibody lost activity only when FcγRIIB was absent (White et al., 2011).
Activating Fcγ receptors are not only dispensable for the antitumor efficacy of agonistic antibodies, but also can be detrimental due to the Fc effector function resulting in elimination of active immune cells by ADCC or ADCP. When tested in hCD40Tg mice, two anti-CD40 human agonistic antibodies ChiLob 7/4 IgG2 and SGN40 IgG2 (very low binding affinity to all FcγRs) stimulated strong expansion of T cells, whereas their corresponding IgG1s (high binding affinity to activating FcγRs) were significantly less active (Yu et al., 2018). White et al. showed that mouse IgG2a antibody (equivalent to human IgG1 capable of ADCC and ADCP) was less effective than mouse IgG1 antibody (Fc effector null). Qi et al. also showed that mouse IgG2a antibody against 4-1BB LOB12.3 had lower antitumor activity than the mouse IgG1 version suggesting the negative impact of effector function.
It is now well accepted that Fc binding to activating Fcγ receptors should be avoided in therapeutic co-stimulation TNFRSF agonistic antibodies.
4.2.2 Fab, Epitope and Ligand Competition
The binding epitope of TNFRSF target are key factors contributing to the intrinsic agonistic activities of the antibody. A correlation was found between the binding epitope and agonistic activity, and the strongest agonists were suggested to be those antibodies binding to the very N-terminus of TNFRSF at CRD-1 which is likely maximally exposed on the surface of the cell, or because of non-competition with natural ligand (Chin et al., 2018). For example, antibodies that bind to CRD1 of CD40 (ChiLob 7/4, SGN40, CP870,893 and Lob 7/2 in same IgG isotype) are stronger agonists, and the CRD2-4 binding mAbs are less agonistic (Yu et al., 2018). However, it is worth noting that ChiLob 7/4 and SGN40, although both anti-CD40 mAbs binding to CRD1 (Vonderheide et al., 2007; Hussein et al. 2010; Johnson et al. 2015), showed much lower agonistic activity than CP870,893 (Yu et al., 2018), suggesting that binding to CRD1 itself doesn’t necessarily guarantee strong agonistic activities.
CD137 agonistic antibody urelumab and utomilumab provided another example of epitope difference (Chin et al., 2018). Urelumab binds CRD1 of CD137 and exhibits strong agonistic activity, while utomilumab binds to CRD3/4 and shows weak activity. The agonistic activity difference was confirmed when these two antibodies were expressed in the same IgG isotype in our lab.
However, the correlation between CRD1 binding and strong agonistic activity is not observed in a study on anti-OX40 agonists. Zhang, Tu et al. generated and characterized a panel of anti-mOX40 mAbs targeting the four mOX40 CRDs and evaluated their in vitro and in vivo agonistic activities along with OX86, a benchmark anti-OX40 agonistic mAb. In striking contrast to agonistic mAbs targeting CD40 and CD137, they found that agonistic mAbs targeting OX40 do not require CRD1 binding and non-ligand blocking for in vivo agonistic function. While all four mAbs enhanced the in vivo immune response, CRD2- and CRD4-binding instead of CRD1-binding anti-mOX40 mAbs showed the highest levels of in vivo agonistic activities in terms of stimulating CD8 Teff cell expansion and anti-tumor activity, highlighting that it remains unknown how the binding epitope affects the intrinsic agonistic potential of mAbs (Zhang et al., 2019).
The possible competition between the agonistic antibodies and endogenous ligands might influence the outcome of TNFRSF signaling activation. Either CD137L or CD40L binds to CRD2-3 of its cognate receptor, and the CRD2-3 binding agonistic antibodies can at least partially compete with the ligand for receptor binding. It is not yet totally understood how this potential competition affects the therapeutic outcome of TNFRSF agonistic antibodies.
The effect of Fab sequence on agonistic activity might be explained by steric acceptability of the membrane-bound receptor. Certain TNFRSF agonistic antibodies can have sufficiently strong agonistic activity without Fc-FcγR cross-linking. This intrinsic agonistic activity could still be affected by Fc-FcγRIIB interaction (vide supra), leading to distinct overall agonistic outcome in the body that will be discussed in more details (vide infra 4.2.4).
4.2.3 Hinge and IgG Flexibility
The flexibility of IgG is also suggested to play a role antibody agonistic activity. IgG2 and more precisely its unique arrangement of disulfide bonds in the hinge region, has been shown to impart superior agonistic activities (White et al. 2015; Yu et al. 2020). Yu et al. dissected the mechanism of an IgG2-mediated, FcγR-independent, agonism. They noted that when the anti-CD40 mAbs were engrafted onto IgG2, the antibodies showed enhanced FcγR-independent agonistic activity. IgG2 is unique among human IgG in its ability to “shuffle” disulfide bonds in its CH1 and hinge regions, resulting in a range of isoforms. The molecule is believed to be synthesized in its “h2A” form having a classical IgG flexible “Y” conformation wherein the heavy chain (HC) Cys127 in CH1 is linked to Cys214 in the light chain (LC). This “h2A” form gradually converted to the “h2B” form in the blood, adopting a more compact shape with the Fab’ arms held in close proximity to the hinge, wherein HC Cys127 form disulfide bonds with the HC hinge Cys232 and LC Cys214 with HC hinge Cys233 (Martinez et al., 2008; Allen et al., 2009). The mutagenesis was used to produce “locked” h2A-like forms (HC C232S or C233S mutation) and h2B-like forms (HC C127S mutant) (Martinez et al., 2008, Allen et al., 2009). White, Chan et al. showed that anti-CD40 agonist ChiLob 7/4 in h2B-like form had increased activity relative to native ChiLob 7/4 h2 to stimulate the proliferation of hCD40 Tg B cells. In contrast, ChiLob 7/4 in h2A-like form totally lost that agonistic activity. The data suggested that the FcγR-independent agonistic activity of h2 is contingent upon the precise conformation of disulfide bonds in its hinge and CH1 domains, and specifically on its ability to adopt the more compact h2B form (White et al., 2015). Some TNFRSF agonistic antibodies were demonstrated to possess autonomous agonistic activity without secondary cross-linking by FcγRIIB (Richman and Vonderheide, 2014). Such antibodies are known as FcγRIIB independent and have sufficient activity in vivo in the absence of FcγRIIB cross-linking. Nevertheless, their activity can still be enhanced when Fc-FcγRIIB cross-linking is available.
Like Fab, the IgG hinge is just one of the structural determinants for antibody overall agonistic activity, which is the integrated sum of these individual components in our body where Fc-FcγR interaction occurs naturally and should be understood as a whole.
4.3 Fc-FcγRIIB Interaction as the Dominant Factor for Therapeutic Agonistic Activities of TNFRSF Antibodies
Physiological TNFRSF receptor activation is induced upon binding to cell-surface TNFSF ligand via cell-cell interaction, resulting in receptor-ligand trimer cross-linking and oligomerization. Clustering of receptors leads to productive docking of intracellular adaptor molecules and assembly of signaling machinery. Antibodies can activate TNFRSF because of their ability to cross-link receptor dimers or trimers on cell surface, but their activity differs widely due to a combination of factors including various Fab, hinge and Fc sequences. Each of the structural features of an antibody contributes to the agonistic activity and can be evaluated in experiments individually. However, the overall agonistic activity is an integration of all interactions in the microenvironment of the tissues. Based on the experimental data, we consider that Fc-FcγRIIB interaction is the dominant factor for therapeutic agonistic activities of TNFRSF antibodies.
4.3.1 FcγRIIB-Dependent TNFRSF Agonistic Antibodies With High Therapeutic Index
Although the importance of FcγRIIB in enhancing agonistic activity of TNFRSF antibodies was recognized as early as 2003 (Xu et al., 2003), it was not until more recently that FcγRIIB-dependent CD137 agonistic antibodies were shown to have superior safety while retained strong antitumor activity (Qi et al., 2019, Ho et al., 2020). Liver toxicity and antitumor efficacy of anti-CD137 antibodies were investigated in preclinical mouse models. Both LOB12.3 and 3H3 antibodies showed similarly strong antitumor efficacy, but they exhibited distinct liver toxicity profiles; 3H3 significantly increased alanine transaminase (ALT) levels, whereas LOB12.3 had minimal impact. Besides elevation of ALT in serum, 3H3 but not LOB12.3 caused immune cell infiltration in the liver. The study revealed that anti-CD137 agonistic antibody-induced antitumor activity and liver toxicity could be separated. Further mechanism studies showed that 3H3 is a strong agonistic antibody in the absence of Fc-FcγRIIB cross-linking while LOB12.3 exhibits agonist activity only when Fc-FcγRIIB cross-linking is available. In vivo, 3H3 induced liver toxicity without Fc-FcγR cross-linking, either as Fc-null mutants or in FcγR knockout mice. In a prior mouse study, the non-FcγR cross-linking dependent 3H3 antibody was shown to induce multifocal mononuclear cell infiltrations in the liver. CD8 T cells, TNF-α, type I IFNs, and IFN-γ were essential for the induction of various aspects of pathology. Additionally, autoimmune mechanism was ruled out, while cytokine-induced inflammation was considered as the primary cause of liver toxicity (Niu et al., 2007).
Ho et al reported a new class of CD137 agonist monoclonal antibody with strong anti-tumor potency without significant transaminitis in vivo, which showed crosslinking-dependent T cell co-stimulation activity in vitro (Ho et al., 2020). Antitumor efficacy was maintained in Fc gamma receptor FcγRIII-deficient mice but diminished in FcγRIIB-deficient mice, confirming the critical role for FcγRIIB to provide cross-linking in vivo.
These landmark preclinical studies provide insights for discovery and clinical development of TNFRSF agonistic antibodies by exploiting the Fc-FcγRIIB cross-linking biology.
4.3.2 The Role of FcγRIIB in vivo
The effect of FcγRIIB on anti-CD40 agonistic activities was investigated both in vitro and in vivo, and anti-CD40 agonism was demonstrated to be enhanced by either mouse FcγRIIB or FcγRIII in vitro while only by FcγRIIB in vivo.
In vivo, FcγRIIB is widely expressed in hematopoietic cells and often co-expressed with other activating FcγRs, more frequently with FcγRIIA. FcγRIIB generally shows a lower expression level than other FcγRs. One exception is on B cells where FcγRIIB is the only FcγR expressed (Junker et al., 2020). Firstly, it is tempting to speculate an important role of B cells in TNFRSF agonistic mAb activities in vivo. Indeed, B cells may concentrate at tumor margins or form tumor-associated immune complex structures such as tumor-associated tertiary lymphoid structures (TLS). In particular, the colocalization of B cells and T cells were reported in breast cancer, malignant melanoma and ovarian cancer (Milne et al. 2009; Ladányi et al. 2011; Nielsen et al. 2012; Iglesia et al. 2014; Sharonov et al., 2020). As mentioned above, FcγRIIB is expressed relatively highly on macrophages and DCs, and low on monocytes. We assume that FcγRIIB-dependent TNFRSF agonist could stimulate immune anti-tumor activities mediated via FcγRIIB expressed on the neighboring macrophages and DCs in tumor microenvironment (TME), with a limited effect on circulating monocytes because of FcγRIIB rareness, which resulted in an optimized efficacy and safety for cancer treatment.
Furthermore, the expression of FcγRIIB is not unchanged but dynamically influenced by cytokine exposure. It is reported that cytokines such as IL-10, IL-6 increase expression of FcγRIIB, while TNF-α and IFN-γ inhibit its expression (Anania et al., 2019).
For the majority of anti-CD40 agonistics in distinct mouse IgG format, mouse IgG1 format was demonstrated to promote an increase in CD8 T cell stimulation and antitumor activities, in contrast, mouse IgG2a was inactive. In light of the requirement for FcγRIIB in anti-CD40 agonistic activity, the difference between mouse isotypes can be explained by the approximately ten-fold higher affinity of IgG1 for FcγRIIB than that of IgG2a (Table 3).
For the TNFRSFs agonistic human IgG, the efficacy of the IgG1 (h1) and IgG2 (h2) isotypes of anti-CD40 are compared in vivo in the context of mouse FcγRs (mFcγR) or human FcγRs (hFcγR). In the context of mFcγR in vivo models, both h1 and h2 isotypes had adjuvant effects on T cell activation, and h2 resulted in a significantly higher T cell response as compared to h1 isotype of the same anti-CD40 clone. Particularly in the case of Lob 7/2, SGN40 and ChiLob 7/4. A remarkable difference between the agonistic activity of h2 and h1 is that the former is FcγR interaction independent and the latter is mFcγRIIB dependent (Richman and Vonderheide, 2014, White et al., 2015).
However, in a delicate study, a panel of humanized TNFRSF and FcγRs mouse models were generated to evaluate human agonistic IgGs in the human FcγR (hFcγR) background. For example, the CD40 humanized mice are crossed to the humanized FcγR mice. These hCD40/hFcγR mice recapitulate hCD40 and hFcγR expression patterns and levels in human, and hFcγR is functionally competent in the mouse background (Dahan et al., 2016). Those mice models were then used to test the agonistic activity of the anti-CD40 antibodies CP-870,893 and ChiLob 7/4 of various IgG subclass.
In the context of these hFcγR in vivo mouse models, h1 was the most active of the human isotypes, particularly in the case of Lob 7/2, SGN40, and ChiLob 7/4, where h1 showed agonism while h2 was less active. CP-870,893 h1 was shown to be unique in evoking very high agonistic activity, which was completely abolished when N297A mutation was introduced in Fc. An even lower activity was observed in deglycosylated form of h2 compared with wild-type h2. As N297A prevents binding to FcγRs and deglycosylated h2 has reduced binding affinity to FcγRIIB compared with h2 wild-type, it was suggested that FcγR-engagement is required for the agonistic activity of the anti-CD40 h1 and FcγRIIB was also involved in agonistic activity of anti-CD40 h2 (Dahan et al., 2016). Higher agonistic activity of h1 than that of h2 could be explained by the higher binding affinity of h1 to FcγRIIB than that of h2 (Table 3).
5 FcγRIIB Dependent TNFRSF Agonistic Antibodies Discovery and Development
5.1 Working Model of TNFRSF Agonistic Antibody Dependent on FcγRIIB Cross-Linking
5.1.1 xLinkAb Model of FcγRIIB-Dependent and IgG-Bridged TNFRSF Clustering
Inspired by the studies on the relationship between the agonistic activities of TNFRSF antibodies and FcγRIIB dependency, we have proposed a cross-link antibody (xLinkAb) working model to optimize therapeutic antibodies by engineering Fc with selective FcγRIIB binding ability to match a desirable Fab to achieve tumor-targeted agonistic activity (Figure 4).
Although the binding to TNFRSF takes place in IgG-Fab region, the potential IgG-Fc binding to FcγRs expressed on surrounding cells has significant and dominant impact on the agonistic activities of IgG. More importantly, FcγRIIB-dependent CD137 agonistic antibodies have been shown to have superior safety while retained strong antitumor activity (Qi et al., 2019; Ho et al., 2020). One possible explanation for the dependency on FcγRIIB for TNFRSF agonistic antibodies is the common clustering ability shared by TNFRSF ligand (TNFSF) and FcγRIIB. Trimer of TNFRSF ligands triggers the formation of TNFSF3-TNFRSF3 complex, which is followed by oligomerization of two and more TNFSF3-TNFRSF3 complexes. Co-stimulation receptors including CD40, CD137, OX40, and GITR require those multi-trimeric structures to fully stimulate their downstream signaling cascades. In the case of TNFRSF agonistic antibodies, the initial step should be IgG-Fab binding to TNFRSF, which is then followed by TNFRSF-IgG-Fc binding to FcγRIIB forming TNFRSF-IgG-FcγRIIB complex and secondary clustering. Activation of TNFRSF signaling depends on the formation of the multivalent TNFRSFs-IgGs-FcγRIIBs complexes and TNFRSF super-clustering.
Analysis of single-cell RNA-seq (scRNA-seq) data and the literature reveals that FcγRIIB shows a unique expression profile on various immune cells enriched in the tumor microenvironment (TME). Firstly, emerging data indicate that intra-tumoral macrophages and dendritic cells, which are critical effectors underlying antibody induced antitumor immunity, express FcγRIIB at highest density (Arce Vargas et al., 2018). Secondly, as discussed earlier, FcγRIIB is the sole FcγR expressed on B cells which are also generally present in TME. In addition, moderate/absent expression of FcγRIIB on monocytes/pDC suggests its less abundance in the blood and normal tissues compared to TME. The cellular expression profile supports the observation that FcγRIIB cross-linking dependent agonistic antibodies could achieve acceptable therapeutic index.
The inhibitory FcγRIIB is low-affinity FcγR, which means it is unable to bind to circulating monomeric IgG. Its binding to IgGs is high-avidity driven thanks to multivalent antigen bound IgG structures. The aggregation of multivalent TNFRSF-IgG-FcγRIIB complexes might be mutually facilitated by Fab-bound TNFRSF clustering and Fc-bound FcγRIIB clustering (Figure 4).
Based on the xLinkAb model, FcγRIIB dependent TNFRSF agonistic antibodies would show optimal agonistic activity in a tumor microenvironment where both co-stimulation target TNFRSF and FcγRIIB are present on infiltrating immune cells and readily available for interaction. Because of the tumor-targeted agonism, FcγRIIB-dependent TNFRSF agonistic antibodies would be better tolerated due to diminished activity in circulation and normal tissues where FcγRIIB and TNFRSF rarely are present in sufficient density.
5.1.2 Consideration and Uncertainty of FcγRIIB-Dependent Agonistic Antibodies
There are several important factors to be considered in engineering and optimizing FcγRIIB-dependent TNFRSF agonistic antibodies for cancer immunotherapy. Firstly, the antigen-binding Fab sequences must be extensively evaluated and only those with low apparent agonistic activity in the absence of cross-linking may be suitable. For example, urelumab (anti-CD137 agonist) and selicrelumab (anti-CD40 agonist) exhibit strong agonistic activity without secondary cross-linking as they recognize the most external CRD domain of either CD137 or CD40, and can efficiently cluster the receptor trimers with the bivalent Fab arms. The strong apparent agonistic activity may be sufficient to provide antitumor efficacy without Fc-FcγRIIB cross-linking. However, their agonistic activity could be further enhanced when the Fc-FcγRs interactions are engaged in vivo. Therefore, they could become super agonists in the body and thus are less tolerated. Additionally, the non-cross-linking dependent TNFRSF agonistic antibodies exhibit widely target agonism without modulation control and may cause on-target and off-tumor side effects. Therefore, the intrinsically strong agonistic antibodies are not desirable candidate for optimization to FcγRIIB dependent agonistic antibodies. On the other hand, not all intrinsically weak agonistic Fab sequences can be used for development of Fc-FcγRIIB cross-linking dependent agonistic antibodies without trial-and-error analysis. It would be ideal that the agonistic activity reaches a desired high level under Fc-FcγRIIB cross-linking conditions while remaining silent or low without cross-linking conditions. Limited by the expression profile of FcγRIIB mainly on B cells, macrophages and DCs accumulated in the tumor microenvironment, FcγRIIB-dependent TNFRSF antibodies are predicted to have a narrower scope of co-stimulating targeted cells and less systemic side effects.
Secondly, selection of hinge may be important. The apparent agonistic activity of a given Fab can change in different hinge contexts as discussed earlier. Thus, hinge can be used as an engineering tool in the optimization process.
Thirdly, Fc-FcγRIIB binding affinity and selectivity should be elaborately evaluated for each TNFRSF target and antibody. Native human Fc does not have a strict selectivity profile for Fc gamma receptors. FcγRIIB is a low affinity receptor for IgGs. There are successful examples in engineering more potent FcγRIIB binding mutants such as S267E (SE) and S267E/L328F (SELF) (Chu et al., 2008; Dahan et al., 2016), but selectivity optimization may be a challenging task. In addition, enhanced binding affinity to FcγRIIB could induce FcγRIIB ITIM activity, which might result in undesired immune effects. Further studies on the effect of FcγRIIB signaling is highly needed.
There is expression of FcγRIIB in normal tissues and especially in liver which may be a liability for liver toxicity. Therefore, Fc variants with high affinity to FcγRIIB may need to be avoided. It is critical to balance the Fab, hinge and Fc activity as whole both in vitro and in vivo settings.
5.2 First Generation Fc-Engineering to Enhance Binding Affinity to FcγRIIB
The initial Fc-engineering for TNFRSF agonistic antibodies focused on enhancing absolute binding affinity to FcγRIIB. The pioneering work was exemplified by APX005M (Filbert et al., 2021), which was engineered to introduce the S267E mutation in IgG1. APX005M strongly bound FcγRIIB (KD = 5.8 × 10−8 M), and required crosslinking for optimal agonistic activity. APX005M has advanced to Phase II clinical development (Table 2).
To improve CD40 agonistic antibodies, IgG1 Fc variants of CP-870,893 carrying the mutations S267E (SE) and S267E/L328F (SELF) were made and they showed 30- and 70-fold increased binding affinity to FcγRIIB, and resulted in increased ability to activate T cells and stronger antitumor activities (Dahan et al., 2016). Due to the high similarity of the extracellular part between FcγRIIA and FcγRIIB, the SE and SELF variations, resulting in increased binding affinity to both the activating FcγRIIA and the inhibitory FcγRIIB, thus a limited increase of the antitumor activity in vivo (Dahan et al., 2016).
Two additional Fc variants, G237D/P238D/P271G/A330R (V9) and G237D/P238D/H268D/P271G/A330R (V11), showed selective enhancement on FcγRIIB binding. CP-870,893 IgG1 with V9 variation (h1-V9) and CP-870,893 IgG1 with V11 variation (h1-V11) resulting in 32- and 97-fold increased binding affinity to hFcγRIIB, respectively, and about 3-fold decreased binding affinity to hFcγRIIA R131. Both h1-V9 and h1-V11 have significantly improved in vivo activity compared to the IgG2 subclass of CP-870,893 (h2). The CP-870,893 h1-V11 results in an increase in T cell activation and anti-tumor activity in vivo compared to h2 and h1-SELF. The increased agonism on T cell activation and anti-tumor activity was also observed in two other anti-CD40 mAbs CD40.1 (CRD2-3 binding) and CD40.2 (CRD1 binding) h1-V11 compared to their h1 or h1-SELF respectively (Dahan et al., 2016), suggesting that selective enhanced binding to FcγRIIB results in stronger agonistic activity and is rather a general rule than specific Fab clones. However, h1-V11 showed absolutely increased binding affinity to FcγRIIB from 3.0 × 10−6 M (KD) to 3.2 × 10−8 M (KD), similar to that for CD40 of 2.7 × 10−8 M (KD) (Dahan et al., 2016). Accompanied with an increased agonistic activity, there might be some bystander undesired effects. It was indeed observed that CP-870,893 h1-V11 caused the most significant decreases in body weight and platelet count after a single injection, compared with h2 or h1-SELF, and a similar phenomenon was observed for another anti-CD40 agonistic antibody CD40.2 (Dahan et al., 2016).
Several newer CD137 agonistic antibodies using IgG4 for FcγRIIB cross-linking have entered the clinic including ADG106 and CTX-471 (Table 2). Given that IgG4 binds to the high affinity FcγRI in addition to FcγRIIB and FcγRIIA, their in vivo activity may not solely depend on FcγRIIB and unwanted CD137 activation may affect their efficacy and safety profile. It is also unknown if these antibodies could activate CD137 as efficiently as the benchmark agonistic antibody urelumab.
5.3 Second Generation Fc-Engineering for FcγRIIb Selectivity on xLinkAb Platform
In our earlier study, we demonstrated for the first time that the antitumor efficacy and liver toxicity characteristics of anti-CD137 agonistic antibodies can be separated based on the agonistic ability and isotype. Proper combination of intrinsic agonistic strength and Fc-FcγRIIB cross-linking collectively determine the antitumor and liver toxicity property of TNFRSFs agonistic antibodies. The xLinkAb model and platform enabled the discovery of the second generation CD137 agonistic antibody LVGN6051 (Qi et al., 2019; Fu et al., 2021). LVGN6051 is a weak agonistic antibody which required FcγRIIB-mediated cross-linking for optimal agonistic activity. In CD137 activation reporter assay, LVGN6051 showed significantly enhanced co-stimulation ability when FcγRIIB was present. Importantly, LVGN6051 showed T cell co-stimulation ability comparable to a urelumab analog and superior to a utomilumab analog when FcγRIIB-expressing cells were present. Furthermore, LVGN6051 showed robust tumor control ability in a wide range of dosage. Most importantly, it did not induce liver toxicity while maintaining potent anti-tumor activity. LVGN6051 has shown an outstanding safety profile in the clinic with promising sign of antitumor activity (Qi et al., 2019; Fu et al., 2021).
LVGN7409 is a recombinant monoclonal antibody against CD40. The modified Fc fragment retains residual binding to FcγRIIB. LVGN7409 activates CD40 signaling in Fc-FcγRIIB cross-link dependent manner and thus operates optimally in a CD40 and FcγRIIB-enriched tumor microenvironment. LVGN7409 demonstrated more robust antitumor efficacy and superior safety profile than published CD40 agonist antibodies in preclinical models. Preliminary clinical safety and activity has been observed (Fu et al., 2022).
6 Conclusion
TNFRSF signaling stimulates the activation, proliferation of various immune cells, including myeloid cells and T cells, providing potential targets for cancer immunotherapy application. However, TNFRSF agonistic antibodies have shown limited success in the clinic, which may be due to limited receptor clustering-mediated signaling or associated dose-limiting toxicities of these early generation products. The studies investigating mechanisms and functions of various TNFRSF agonistic antibodies have identified an important role of FcγRIIB. FcγRIIB has been shown to mediate TNFRSF clustering bridged by TNFRSF antibodies. The agonistic activity of an antibody can be impacted by the Fab, hinge or Fc region, but the Fc-FcγRIIB interaction has been identified as the dominant determinant for the overall agonistic activity outcome (efficacy and toxicity). Strong agonistic antibodies without Fc cross-linking can be identified in vitro, and such antibodies would exhibit agonism upon target binding and become super-agonist when cross-linking of Fc-FcγRs is available. TNFRSF antibodies with weak or no detectable agonistic activity in the absence of Fc cross-linking can exhibit strong agonism when Fc-FcγRIIB is engaged.
Two important discoveries came from the studies of TNFRSF agonistic antibodies in mouse models. Firstly, only Fc-FcγRIIB cross-linking contributes antitumor immunity and efficacy although other FcγRs can cross-link TNFRSR-IgG to enhance agonism. Secondly, TNFRSF agonistic antibodies that rely on Fc-FcγRIIB cross-linking to show activity can induce antitumor efficacy without liver toxicity, indicating feasibility of selective agonism in tumor microenvironment. As the sole inhibitory Fcγ receptor, FcγRIIB has unique structural features and expression profile, which may contribute to these important observations. FcγRIIB is expressed exclusively on B cells that do not express other FcγRs, high on MDSC, widely on macrophages and DCs, and low on monocytes in the blood. Tumor tissues are rich in immune infiltrates including B cells, MDSC, macrophages and DC cells. Thus, FcγRIIB is present in higher density relative to normal tissues. The differential expression profile and level between tumor and normal tissue supports the separation of efficacy and toxicity of TNFRSF agonistic antibodies driven by Fc-FcγRIIB cross-linking.
Therefore, independent research teams from academic, biotech and pharmaceutical companies have been applying Fc engineering approaches to improve tumor-targeted TNFRSF clustering for efficacy and safety. In particular, the conditional TNFRSF agonistic antibodies with engineered Fc for enhanced affinity or selectivity for FcγRIIB, represents a new wave of therapeutic agents entering clinical development. FcγRIIB-dependent crosslinking of agonistic antibodies should be applicable for a majority, if not all, of TNFRSF members including CD40, CD137, OX40, GITR, and CD27. Furthermore, cross-linking dependency could be applied in developing TNFRSF bispecific or multi-specific antibodies, where FcγRIIB could be replaced or supplemented by a tumor antigen or immune target. Consequently, binding to these tumor-selective targets could result in the formation of multivalent tumor-targeted -antibodies-TNFRSFs complexes, which in turn cause the clustering of TNFRSF and activation of downstream signaling selectively in the tumor microenvironment.
Taken together, TNFRSF immune co-stimulation targets hold great potential to expand immunotherapy clinical benefits to more cancer patients. TNFRSF agonistic antibodies with regular IgG structure remain a desirable format to deliver clinical efficacy and safety with a mature manufacturing and commercial platform. It has become evident and important to explore Fc biology and Fc engineering, including our xLinkAb approaches guided by the principle of FcγRIIB cross-linking dependent agonism, to achieve breakthroughs in targeting TNFRSF for cancer immunotherapy.
Author Contributions
LL, YW, KY, MC, GZ, and JW wrote and reviewed the review.
Conflict of Interest
LL, YW and JW are employees of Lyvgen Biopharma, a private biotech company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Janice Marie McCourt for reviewing and editing the manuscript.
References
Adachi, M., Suematsu, S., Suda, T., Watanabe, D., Fukuyama, H., Ogasawara, J., et al. (1996). Enhanced and Accelerated Lymphoproliferation in Fas-Null Mice. Proc. Natl. Acad. Sci. U. S. A. 93 (5), 2131–2136. doi:10.1073/pnas.93.5.2131
Aggarwal, B. B., Gupta, S. C., and Kim, J. H. (2012). Historical Perspectives on Tumor Necrosis Factor and its Superfamily: 25 Years Later, a Golden Journey. Blood 119 (3), 651–665. doi:10.1182/blood-2011-04-325225
Akiba, H., Miyahira, Y., Atsuta, M., Takeda, K., Nohara, C., Futagawa, T., et al. (2000). Critical Contribution of OX40 Ligand to T Helper Cell Type 2 Differentiation in Experimental Leishmaniasis. J. Exp. Med. 191 (2), 375–380. doi:10.1084/jem.191.2.375
Alimzhanov, M. B., Kuprash, D. V., Kosco-Vilbois, M. H., Luz, A., Turetskaya, R. L., Tarakhovsky, A., et al. (1997). Abnormal Development of Secondary Lymphoid Tissues in Lymphotoxin Beta-Deficient Mice. Proc. Natl. Acad. Sci. U. S. A. 94 (17), 9302–9307. doi:10.1073/pnas.94.17.9302
Allen, M. J., Guo, A., Martinez, T., Han, M., Flynn, G. C., Wypych, J., et al. (2009). Interchain Disulfide Bonding in Human IgG2 Antibodies Probed by Site-Directed Mutagenesis. Biochemistry 48 (17), 3755–3766. doi:10.1021/bi8022174
Allen, R. C., Armitage, R. J., Conley, M. E., Rosenblatt, H., Jenkins, N. A., Copeland, N. G., et al. (1993). CD40 Ligand Gene Defects Responsible for X-Linked Hyper-IgM Syndrome. Science 259 (5097), 990–993. doi:10.1126/science.7679801
Anania, J. C., Chenoweth, A. M., Wines, B. D., and Hogarth, P. M. (2019). The Human FcγRII (CD32) Family of Leukocyte FcR in Health and Disease. Front. Immunol. 10. doi:10.3389/fimmu.2019.00464
Ansell, S. M., Flinn, I., Taylor, M. H., Sikic, B. I., Brody, J., Nemunaitis, J., et al. (2020). Safety and Activity of Varlilumab, a Novel and First-In-Class Agonist Anti-CD27 Antibody, for Hematologic Malignancies. Blood Adv. 4 (9), 1917–1926. doi:10.1182/bloodadvances.2019001079
Arce Vargas, F., Furness, A. J. S., Litchfield, K., Joshi, K., Rosenthal, R., Ghorani, E., et al. (2018). Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 33 (4), 649–e4. e644. doi:10.1016/j.ccell.2018.02.010
Assal, A., Kaner, J., Pendurti, G., and Zang, X. (2015). Emerging Targets in Cancer Immunotherapy: beyond CTLA-4 and PD-1. Immunotherapy 7 (11), 1169–1186. doi:10.2217/imt.15.78
Balmanoukian, A. S., Infante, J. R., Aljumaily, R., Naing, A., Chintakuntlawar, A. V., Rizvi, N. A., et al. (2020). Safety and Clinical Activity of MEDI1873, a Novel GITR Agonist, in Advanced Solid Tumors. Clin. Cancer Res. 26 (23), 6196–6203. doi:10.1158/1078-0432.CCR-20-0452
Banks, T. A., Rouse, B. T., Kerley, M. K., Blair, P. J., Godfrey, V. L., Kuklin, N. A., et al. (1995). Lymphotoxin-alpha-deficient Mice. Effects on Secondary Lymphoid Organ Development and Humoral Immune Responsiveness. J. Immunol. 155 (4), 1685–1693.
Banner, D. W., D'Arcy, A., Janes, W., Gentz, R., Schoenfeld, H. J., Broger, C., et al. (1993). Crystal Structure of the Soluble Human 55 Kd TNF Receptor-Human TNF Beta Complex: Implications for TNF Receptor Activation. Cell 73 (3), 431–445. doi:10.1016/0092-8674(93)90132-a
Beatty, G. L., Torigian, D. A., Chiorean, E. G., Saboury, B., Brothers, A., Alavi, A., et al. (2013). A Phase I Study of an Agonist CD40 Monoclonal Antibody (CP-870,893) in Combination with Gemcitabine in Patients with Advanced Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 19 (22), 6286–6295. doi:10.1158/1078-0432.CCR-13-1320
Blazar, B. R., Levy, R. B., Mak, T. W., Panoskaltsis-Mortari, A., Muta, H., Jones, M., et al. (2004). CD30/CD30 Ligand (CD153) Interaction Regulates CD4+ T Cell-Mediated Graft-Versus-Host Disease. J. Immunol. 173 (5), 2933–2941. doi:10.4049/jimmunol.173.5.2933
Bruhns, P., Iannascoli, B., England, P., Mancardi, D. A., Fernandez, N., Jorieux, S., et al. (2009). Specificity and Affinity of Human Fcgamma Receptors and Their Polymorphic Variants for Human IgG Subclasses. Blood 113 (16), 3716–3725. doi:10.1182/blood-2008-09-179754
Bruhns, P., and Jönsson, F. (2015). Mouse and Human FcR Effector Functions. Immunol. Rev. 268 (1), 25–51. doi:10.1111/imr.12350
Bucay, N., Sarosi, I., Dunstan, C. R., Morony, S., Tarpley, J., Capparelli, C., et al. (1998). Osteoprotegerin-Deficient Mice Develop Early Onset Osteoporosis and Arterial Calcification. Genes Dev. 12 (9), 1260–1268. doi:10.1101/gad.12.9.1260
Burugu, S., Dancsok, A. R., and Nielsen, T. O. (2018). Emerging Targets in Cancer Immunotherapy. Semin. Cancer Biol. 52 (Pt 2), 39–52. doi:10.1016/j.semcancer.2017.10.001
Byun, M., Ma, C. S., Akçay, A., Pedergnana, V., Palendira, U., Myoung, J., et al. (2013). Inherited Human OX40 Deficiency Underlying Classic Kaposi Sarcoma of Childhood. J. Exp. Med. 210 (9), 1743–1759. doi:10.1084/jem.20130592
Campos Carrascosa, L., van Beek, A. A., de Ruiter, V., Doukas, M., Wei, J., Fisher, T. S., et al. (2020). FcγRIIB Engagement Drives Agonistic Activity of Fc-Engineered αOX40 Antibody to Stimulate Human Tumor-Infiltrating T Cells. J. Immunother. Cancer 8 (2), e000816. doi:10.1136/jitc-2020-000816
Castigli, E., Scott, S., Dedeoglu, F., Bryce, P., Jabara, H., Bhan, A. K., et al. (2004). Impaired IgA Class Switching in APRIL-Deficient Mice. Proc. Natl. Acad. Sci. U. S. A. 101 (11), 3903–3908. doi:10.1073/pnas.0307348101
Castigli, E., Wilson, S. A., Garibyan, L., Rachid, R., Bonilla, F., Schneider, L., et al. (2005). TACI Is Mutant in Common Variable Immunodeficiency and IgA Deficiency. Nat. Genet. 37 (8), 829–834. doi:10.1038/ng1601
Chin, S. M., Kimberlin, C. R., Roe-Zurz, Z., Zhang, P., Xu, A., Liao-Chan, S., et al. (2018). Structure of the 4-1BB/4-1BBL Complex and Distinct Binding and Functional Properties of Utomilumab and Urelumab. Nat. Commun. 9 (1), 4679. doi:10.1038/s41467-018-07136-7
Chu, S. Y., Vostiar, I., Karki, S., Moore, G. L., Lazar, G. A., Pong, E., et al. (2008). Inhibition of B Cell Receptor-Mediated Activation of Primary Human B Cells by Coengagement of CD19 and FcgammaRIIb with Fc-Engineered Antibodies. Mol. Immunol. 45 (15), 3926–3933. doi:10.1016/j.molimm.2008.06.027
Collette, Y., Gilles, A., Pontarotti, P., and Olive, D. (2003). A Co-evolution Perspective of the TNFSF and TNFRSF Families in the Immune System. Trends Immunol. 24 (7), 387–394. doi:10.1016/s1471-4906(03)00166-2
Compaan, D. M., and Hymowitz, S. G. (2006). The Crystal Structure of the Costimulatory OX40-Ox40l Complex. Structure 14 (8), 1321–1330. doi:10.1016/j.str.2006.06.015
Coward, J., Abed, A., Nagrial, A., and Markman, B. (2021). Phase I Open-Label, Dose Escalation of YH003, an Anti-CD40 Monoclonal Antibody in Combination with Toripalimab (Anti-PD-1 mAb) in Patients with Advanced Solid Tumors. Jco 39 (15_Suppl. l), 2580. doi:10.1200/jco.2021.39.15_suppl.2580
Croft, M., Benedict, C. A., and Ware, C. F. (2013). Clinical Targeting of the TNF and TNFR Superfamilies. Nat. Rev. Drug Discov. 12 (2), 147–168. doi:10.1038/nrd3930
Croft, M., and Siegel, R. M. (2017). Beyond TNF: TNF Superfamily Cytokines as Targets for the Treatment of Rheumatic Diseases. Nat. Rev. Rheumatol. 13 (4), 217–233. doi:10.1038/nrrheum.2017.22
Crow, A. R., Leytin, V., Starkey, A. F., Rand, M. L., and Lazarus, A. H. (2003). CD154 (CD40 Ligand)-Deficient Mice Exhibit Prolonged Bleeding Time and Decreased Shear-Induced Platelet Aggregates. J. Thromb. Haemost. 1 (4), 850–852. doi:10.1046/j.1538-7836.2003.t01-1-00115.x
Cundy, T., Hegde, M., Naot, D., Chong, B., King, A., Wallace, R., et al. (2002). A Mutation in the Gene TNFRSF11B Encoding Osteoprotegerin Causes an Idiopathic Hyperphosphatasia Phenotype. Hum. Mol. Genet. 11 (18), 2119–2127. doi:10.1093/hmg/11.18.2119
Curti, B. D., Kovacsovics-Bankowski, M., Morris, N., Walker, E., Chisholm, L., Floyd, K., et al. (2013). OX40 Is a Potent Immune-Stimulating Target in Late-Stage Cancer Patients. Cancer Res. 73 (24), 7189–7198. doi:10.1158/0008-5472.CAN-12-4174
Dahan, R., Barnhart, B. C., Li, F., Yamniuk, A. P., Korman, A. J., and Ravetch, J. V. (2016). Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcγR Engagement. Cancer Cell 29 (6), 820–831. doi:10.1016/j.ccell.2016.05.001
DeBenedette, M. A., Wen, T., Bachmann, M. F., Ohashi, P. S., Barber, B. H., Stocking, K. L., et al. (1999). Analysis of 4-1BB Ligand (4-1bbl)-Deficient Mice and of Mice Lacking Both 4-1BBL and CD28 Reveals a Role for 4-1BBL in Skin Allograft Rejection and in the Cytotoxic T Cell Response to Influenza Virus. J. Immunol. 163 (9), 4833–4841.
Dekkers, G., Bentlage, A. E. H., Stegmann, T. C., Howie, H. L., Lissenberg-Thunnissen, S., Zimring, J., et al. (2017). Affinity of Human IgG Subclasses to Mouse Fc Gamma Receptors. mAbs 9 (5), 767–773. doi:10.1080/19420862.2017.1323159
Deronic, A., Thagesson, M., Nilsson, A., Ellmark, P., Fält, A., Fält, C., et al. (2021). Mitazalimab, a Potent CD40 Agonist with Potential for Combination with Chemotherapy. Cancer Res. 81, 1593. doi:10.1158/1538-7445.AM2021-1593
Diab, A., Hamid, O., Thompson, J. A., Ros, W., Eskens, F. A. L. M., Doi, T., et al. (2022). A Phase I, Open-Label, Dose-Escalation Study of the OX40 Agonist Ivuxolimab in Patients with Locally Advanced or Metastatic Cancers. Clin. Cancer Res. 28 (1), 71–83. doi:10.1158/1078-0432.CCR-21-0845
Diab, A., El-Khoueiry, A., Eskens, F. A., Ros, W., Thompson, J. A., Konto, C., et al. (2016). A First-In-Human (FIH) Study of PF-04518600 (PF-8600) OX40 Agonist in Adult Patients (Pts) with Select Advanced Malignancies. Ann. Oncol. 27, vi361. doi:10.1093/annonc/mdw378.08
Dostert, C., Grusdat, M., Letellier, E., and Brenner, D. (2019). The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and beyond. Physiol. Rev. 99 (1), 115–160. doi:10.1152/physrev.00045.2017
Dougall, W. C., Glaccum, M., Charrier, K., Rohrbach, K., Brasel, K., De Smedt, T., et al. (1999). RANK Is Essential for Osteoclast and Lymph Node Development. Genes Dev. 13 (18), 2412–2424. doi:10.1101/gad.13.18.2412
Enell Smith, K., Deronic, A., Hägerbrand, K., Norlén, P., and Ellmark, P. (2021). Rationale and Clinical Development of CD40 Agonistic Antibodies for Cancer Immunotherapy. Expert Opin. Biol. Ther. 21 (12), 1635–1646. doi:10.1080/14712598.2021.1934446
Filbert, E. L., Björck, P. K., Srivastava, M. K., Bahjat, F. R., and Yang, X. (2021). APX005M, a CD40 Agonist Antibody with Unique Epitope Specificity and Fc Receptor Binding Profile for Optimal Therapeutic Application. Cancer Immunol. Immunother. 70 (7), 1853–1865. doi:10.1007/s00262-020-02814-2
Finnberg, N., Gruber, J. J., Fei, P., Rudolph, D., Bric, A., Kim, S. H., et al. (2005). DR5 Knockout Mice Are Compromised in Radiation-Induced Apoptosis. Mol. Cell Biol. 25 (5), 2000–2013. doi:10.1128/MCB.25.5.2000-2013.2005
Fu, S., Harb, W. A., Patel, S. P., Lu, C., Halperin, D. M., Hsu, Y. H., et al. (2021). Early Safety and Efficacy from a Phase I Open-Label Clinical Trial of CD137(4-1BB) Agonistic Antibody LVGN6051 as Monotherapy and in Combination with Pembrolizumab. Jco 39 (15_Suppl. l), 2521. doi:10.1200/jco.2021.39.15_suppl.2521
Fu, S., Vandross, A. L., Hsu, Y. H., Shi, N., Jiang, L., Su, F., et al. (2022). Early Safety and Efficacy from a Phase I Open-Label Clinical Study of LVGN7409 (CD40 Agonist Antibody) in Patients with Advanced or Metastatic Malignancies. J. Clin. Oncol. 41 (00_Suppl. l).
Fütterer, A., Mink, K., Luz, A., Kosco-Vilbois, M. H., and Pfeffer, K. (1998). The Lymphotoxin Beta Receptor Controls Organogenesis and Affinity Maturation in Peripheral Lymphoid Tissues. Immunity 9 (1), 59–70.
Gardai, S. J., Neff-LaFord, H., Epp, A., Yang, J., Manley, T., and Law, C.-L. (2016). Abstract 4994: SEA-CD40: from Bench to Bedside. Cancer Res. 76 (14_Suppl. ment), 4994. doi:10.1158/1538-7445.am2016-4994
Gaspal, F. M., Kim, M. Y., McConnell, F. M., Raykundalia, C., Bekiaris, V., and Lane, P. J. (2005). Mice Deficient in OX40 and CD30 Signals Lack Memory Antibody Responses Because of Deficient CD4 T Cell Memory. J. Immunol. 174 (7), 3891–3896. doi:10.4049/jimmunol.174.7.3891
Geva, R., Voskoboynik, M., Dobrenkov, K., Mayawala, K., Gwo, J., Wnek, R., et al. (2020). First-in-human Phase 1 Study of MK-1248, an Anti-glucocorticoid-induced Tumor Necrosis Factor Receptor Agonist Monoclonal Antibody, as Monotherapy or with Pembrolizumab in Patients with Advanced Solid Tumors. Cancer 126 (22), 4926–4935. doi:10.1002/cncr.33133
Girgenrath, M., Weng, S., Kostek, C. A., Browning, B., Wang, M., Brown, S. A., et al. (2006). TWEAK, via its Receptor Fn14, Is a Novel Regulator of Mesenchymal Progenitor Cells and Skeletal Muscle Regeneration. Embo J. 25 (24), 5826–5839. doi:10.1038/sj.emboj.7601441
Glenney, G. W., and Wiens, G. D. (2007). Early Diversification of the TNF Superfamily in Teleosts: Genomic Characterization and Expression Analysis. J. Immunol. 178 (12), 7955–7973. doi:10.4049/jimmunol.178.12.7955
Glisson, B. S., Leidner, R. S., Ferris, R. L., Powderly, J., Rizvi, N. A., Keam, B., et al. (2020). Safety and Clinical Activity of MEDI0562, a Humanized OX40 Agonist Monoclonal Antibody, in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 26 (20), 5358–5367. doi:10.1158/1078-0432.CCR-19-3070
Gopal, A. K., Levy, R., Houot, R., Patel, S. P., Popplewell, L., Jacobson, C., et al. (2020). First-in-Human Study of Utomilumab, a 4-1BB/CD137 Agonist, in Combination with Rituximab in Patients with Follicular and Other CD20+ Non-hodgkin Lymphomas. Clin. Cancer Res. 26 (11), 2524–2534. doi:10.1158/1078-0432.CCR-19-2973
Gourh, P., Arnett, F. C., Tan, F. K., Assassi, S., Divecha, D., Paz, G., et al. (2010). Association of TNFSF4 (OX40L) Polymorphisms with Susceptibility to Systemic Sclerosis. Ann. Rheum. Dis. 69 (3), 550–555. doi:10.1136/ard.2009.116434
Gregory, A. P., Dendrou, C. A., Attfield, K. E., Haghikia, A., Xifara, D. K., Butter, F., et al. (2012). TNF Receptor 1 Genetic Risk Mirrors Outcome of Anti-TNF Therapy in Multiple Sclerosis. Nature 488 (7412), 508–511. doi:10.1038/nature11307
Grilley-Olson, J. E., Curti, B. D., Smith, D. C., Goel, S., Gajewski, T., Markovic, S., et al. (2018). SEA-CD40, a Non-fucosylated CD40 Agonist: Interim Results from a Phase 1 Study in Advanced Solid Tumors. J. Clin. Oncol. 36 (15_Suppl. l), 3093. doi:10.1200/jco.2018.36.15_suppl.3093
Grosse-Wilde, A., Voloshanenko, O., Bailey, S. L., Longton, G. M., Schaefer, U., Csernok, A. I., et al. (2008). TRAIL-R Deficiency in Mice Enhances Lymph Node Metastasis without Affecting Primary Tumor Development. J. Clin. Invest 118 (1), 100–110. doi:10.1172/JCI33061
Guerrini, M. M., Sobacchi, C., Cassani, B., Abinun, M., Kilic, S. S., Pangrazio, A., et al. (2008). Human Osteoclast-Poor Osteopetrosis with Hypogammaglobulinemia Due to TNFRSF11A (RANK) Mutations. Am. J. Hum. Genet. 83 (1), 64–76. doi:10.1016/j.ajhg.2008.06.015
Hansen, A. R., Infante, J. R., McArthur, G., Gordon, M. S., Lesokhin, A. M., Stayner, A.-L., et al. (2016). Abstract CT097: A First-In-Human Phase I Dose Escalation Study of the OX40 Agonist MOXR0916 in Patients with Refractory Solid Tumors. Cancer Res. 76 (14_Suppl. ment), CT097. doi:10.1158/1538-7445.am2016-ct097
Heinhuis, K. M., Carlino, M., Joerger, M., Di Nicola, M., Meniawy, T., Rottey, S., et al. (2020). Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor-Related Protein Agonist Alone or in Combination with Nivolumab for Patients with Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 6 (1), 100–107. doi:10.1001/jamaoncol.2019.3848
Hirano, M., Davis, R. S., Fine, W. D., Nakamura, S., Shimizu, K., Yagi, H., et al. (2007). IgEb Immune Complexes Activate Macrophages through FcgammaRIV Binding. Nat. Immunol. 8 (7), 762–771. doi:10.1038/ni1477
Ho, S. K., Xu, Z., Thakur, A., Fox, M., Tan, S. S., DiGiammarino, E., et al. (2020). Epitope and Fc-Mediated Cross-Linking, but Not High Affinity, Are Critical for Antitumor Activity of CD137 Agonist Antibody with Reduced Liver Toxicity. Mol. Cancer Ther. 19 (4), 1040–1051. doi:10.1158/1535-7163.MCT-19-0608
Hogarth, P. M., and Pietersz, G. A. (2012). Fc Receptor-Targeted Therapies for the Treatment of Inflammation, Cancer and beyond. Nat. Rev. Drug Discov. 11 (4), 311–331. doi:10.1038/nrd2909
Hubert, K. E., Davies, M. H., Stempel, A. J., Griffith, T. S., and Powers, M. R. (2009). TRAIL-deficient Mice Exhibit Delayed Regression of Retinal Neovascularization. Am. J. Pathol. 175 (6), 2697–2708. doi:10.2353/ajpath.2009.090099
Hughes, A. E., Ralston, S. H., Marken, J., Bell, C., MacPherson, H., Wallace, R. G., et al. (2000). Mutations in TNFRSF11A, Affecting the Signal Peptide of RANK, Cause Familial Expansile Osteolysis. Nat. Genet. 24 (1), 45–48. doi:10.1038/71667
Hussein, M., Berenson, J. R., Niesvizky, R., Munshi, N., Matous, J., Sobecks, R., et al. (2010). A Phase I Multidose Study of Dacetuzumab (SGN-40; Humanized Anti-CD40 Monoclonal Antibody) in Patients with Multiple Myeloma. Haematologica 95 (5), 845–848. doi:10.3324/haematol.2009.008003
Ida, H., Kawasaki, E., Miyashita, T., Tanaka, F., Kamachi, M., Izumi, Y., et al. (2004). A Novel Mutation (T61I) in the Gene Encoding Tumour Necrosis Factor Receptor Superfamily 1A (TNFRSF1A) in a Japanese Patient with Tumour Necrosis Factor Receptor-Associated Periodic Syndrome (TRAPS) Associated with Systemic Lupus Erythematosus. Rheumatol. Oxf. 43 (10), 1292–1299. doi:10.1093/rheumatology/keh320
Iglesia, M. D., Vincent, B. G., Parker, J. S., Hoadley, K. A., Carey, L. A., Perou, C. M., et al. (2014). Prognostic B-Cell Signatures Using mRNA-Seq in Patients with Subtype-specific Breast and Ovarian Cancer. Clin. Cancer Res. 20 (14), 3818–3829. doi:10.1158/1078-0432.CCR-13-3368
Infante, J. R., Hansen, A. R., Pishvaian, M. J., Chow, L. Q. M., McArthur, G. A., Bauer, T. M., et al. (2016). A Phase Ib Dose Escalation Study of the OX40 Agonist MOXR0916 and the PD-L1 Inhibitor Atezolizumab in Patients with Advanced Solid Tumors. Jco 34 (15_Suppl. l), 101. doi:10.1200/jco.2016.34.15_suppl.101
Ishii, N., Ndhlovu, L. C., Murata, K., Sato, T., Kamanaka, M., and Sugamura, K. (2003). OX40 (CD134) and OX40 Ligand Interaction Plays an Adjuvant Role during In Vivo Th2 Responses. Eur. J. Immunol. 33 (9), 2372–2381. doi:10.1002/eji.200324031
Ishikawa, M., Vowinkel, T., Stokes, K. Y., Arumugam, T. V., Yilmaz, G., Nanda, A., et al. (2005). CD40/CD40 Ligand Signaling in Mouse Cerebral Microvasculature after Focal Ischemia/reperfusion. Circulation 111 (13), 1690–1696. doi:10.1161/01.CIR.0000160349.42665.0C
Jeon, H. J., Choi, J. H., Jung, I. H., Park, J. G., Lee, M. R., Lee, M. N., et al. (2010). CD137 (4-1BB) Deficiency Reduces Atherosclerosis in Hyperlipidemic Mice. Circulation 121 (9), 1124–1133. doi:10.1161/CIRCULATIONAHA.109.882704
Jeong, S., and Park, S. H. (2020). Co-Stimulatory Receptors in Cancers and Their Implications for Cancer Immunotherapy. Immune Netw. 20 (1), e3–0. doi:10.4110/in.2020.20.e3
Johnson, M., Fakih, M., Bendell, J., Bajor, D., Cristea, M., Tremblay, T., et al. (2017). First in Human Study with the CD40 Agonistic Monoclonal Antibody APX005M in Subjects with Solid Tumors. J. Immunother. Cancer 5 (Suppl. 3), O36.
Johnson, M., Lopez, J., LoRusso, P., Bauman, J., Haggstrom, D., Lagkadinou, E., et al. (2021). 493 First-In-Human Phase 1/2 Trial to Evaluate the Safety and Initial Clinical Activity of DuoBody-Cd40×4-1bb (GEN1042) in Patients with Advanced Solid Tumors. J. Immunother. Cancer 9, A525. doi:10.1136/jitc-2021-sitc2021.493
Johnson, P., Challis, R., Chowdhury, F., Gao, Y., Harvey, M., Geldart, T., et al. (2015). Clinical and Biological Effects of an Agonist Anti-CD40 Antibody: a Cancer Research UK Phase I Study. Clin. Cancer Res. 21 (6), 1321–1328. doi:10.1158/1078-0432.CCR-14-2355
Junker, F., Gordon, J., and Qureshi, O. (2020). Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 11. doi:10.3389/fimmu.2020.01393
Karray, S., Kress, C., Cuvellier, S., Hue-Beauvais, C., Damotte, D., Babinet, C., et al. (2004). Complete Loss of Fas Ligand Gene Causes Massive Lymphoproliferation and Early Death, Indicating a Residual Activity of Gld Allele. J. Immunol. 172 (4), 2118–2125. doi:10.4049/jimmunol.172.4.2118
Khan, W. N., Nilsson, A., Mizoguchi, E., Castigli, E., Forsell, J., Bhan, A. K., et al. (1997). Impaired B Cell Maturation in Mice Lacking Bruton's Tyrosine Kinase (Btk) and CD40. Int. Immunol. 9 (3), 395–405. doi:10.1093/intimm/9.3.395
Kim, N., Odgren, P. R., Kim, D. K., Marks, S. C., and Choi, Y. (2000). Diverse Roles of the Tumor Necrosis Factor Family Member TRANCE in Skeletal Physiology Revealed by TRANCE Deficiency and Partial Rescue by a Lymphocyte-Expressed TRANCE Transgene. Proc. Natl. Acad. Sci. U. S. A. 97 (20), 10905–10910. doi:10.1073/pnas.200294797
Knight, B., Yeoh, G. C., Husk, K. L., Ly, T., Abraham, L. J., Yu, C., et al. (2000). Impaired Preneoplastic Changes and Liver Tumor Formation in Tumor Necrosis Factor Receptor Type 1 Knockout Mice. J. Exp. Med. 192 (12), 1809–1818. doi:10.1084/jem.192.12.1809
Kong, Y. Y., Yoshida, H., Sarosi, I., Tan, H. L., Timms, E., Capparelli, C., et al. (1999). OPGL Is a Key Regulator of Osteoclastogenesis, Lymphocyte Development and Lymph-Node Organogenesis. Nature 397 (6717), 315–323. doi:10.1038/16852
Koon, H. B., Shepard, D. R., Merghoub, T., Schaer, D. A., Sirard, C. A., and Wolchok, J. D. (2016). First-in-human Phase 1 Single-Dose Study of TRX-518, an Anti-human Glucocorticoid-Induced Tumor Necrosis Factor Receptor (GITR) Monoclonal Antibody in Adults with Advanced Solid Tumors. J. Clin. Oncol. 34.
Ku, G., Bendell, J. C., Tolcher, A. W., Hurvitz, S. A., Krishnamurthy, A., El-Khoueiry, A. B., et al. (20202020). 525O - A Phase I Dose Escalation Study of PRS-343, a HER2/4-1BB Bispecific Molecule, in Patients with HER2-Positive Malignancies. Ann. Oncol. 31, S462–S504. doi:10.1016/j.annonc.2020.08.639
Kucka, K., and Wajant, H. (2020). Receptor Oligomerization and its Relevance for Signaling by Receptors of the Tumor Necrosis Factor Receptor Superfamily. Front. Cell Dev. Biol. 8, 615141. doi:10.3389/fcell.2020.615141
Kwon, B. S., Hurtado, J. C., Lee, Z. H., Kwack, K. B., Seo, S. K., Choi, B. K., et al. (2002). Immune Responses in 4-1BB (CD137)-Deficient Mice. J. Immunol. 168 (11), 5483–5490. doi:10.4049/jimmunol.168.11.5483
Ladányi, A., Kiss, J., Mohos, A., Somlai, B., Liszkay, G., Gilde, K., et al. (2011). Prognostic Impact of B-Cell Density in Cutaneous Melanoma. Cancer Immunol. Immunother. 60 (12), 1729–1738.
Lamhamedi-Cherradi, S. E., Zheng, S. J., Maguschak, K. A., Peschon, J., and Chen, Y. H. (2003). Defective Thymocyte Apoptosis and Accelerated Autoimmune Diseases in TRAIL-/- Mice. Nat. Immunol. 4 (3), 255–260. doi:10.1038/ni894
Lee, D. H. (2021). Update of Early Phase Clinical Trials in Cancer Immunotherapy. BMB Rep. 54 (1), 70–88. doi:10.5483/bmbrep.2021.54.1.242
Lee, K. F., Li, E., Huber, L. J., Landis, S. C., Sharpe, A. H., Chao, M. V., et al. (1992). Targeted Mutation of the Gene Encoding the Low Affinity NGF Receptor P75 Leads to Deficits in the Peripheral Sensory Nervous System. Cell 69 (5), 737–749. doi:10.1016/0092-8674(92)90286-l
Lee, S. W., Park, Y., So, T., Kwon, B. S., Cheroutre, H., Mittler, R. S., et al. (2008). Identification of Regulatory Functions for 4-1BB and 4-1BBL in Myelopoiesis and the Development of Dendritic Cells. Nat. Immunol. 9 (8), 917–926. doi:10.1038/ni.1632
Lehmann, C. H. K., Baranska, A., Heidkamp, G. F., Heger, L., Neubert, K., Lühr, J. J., et al. (2017). DC Subset-specific Induction of T Cell Responses upon Antigen Uptake via Fcγ Receptors In Vivo. J. Exp. Med. 214 (5), 1509–1528. doi:10.1084/jem.20160951
Li, F., and Ravetch, J. V. (2013). Antitumor Activities of Agonistic Anti-TNFR Antibodies Require Differential FcγRIIB Coengagement In Vivo. Proc. Natl. Acad. Sci. U. S. A. 110 (48), 19501–19506. doi:10.1073/pnas.1319502110
Li, F., and Ravetch, J. V. (2012). Apoptotic and Antitumor Activity of Death Receptor Antibodies Require Inhibitory Fcγ Receptor Engagement. Proc. Natl. Acad. Sci. U. S. A. 109 (27), 10966–10971. doi:10.1073/pnas.1208698109
Li, F., and Ravetch, J. V. (2011). Inhibitory Fcγ Receptor Engagement Drives Adjuvant and Anti-tumor Activities of Agonistic CD40 Antibodies. Science 333 (6045), 1030–1034. doi:10.1126/science.1206954
Li, J., Sarosi, I., Yan, X. Q., Morony, S., Capparelli, C., Tan, H. L., et al. (2000). RANK Is the Intrinsic Hematopoietic Cell Surface Receptor that Controls Osteoclastogenesis and Regulation of Bone Mass and Calcium Metabolism. Proc. Natl. Acad. Sci. U. S. A. 97 (4), 1566–1571. doi:10.1073/pnas.97.4.1566
Liu, J., Na, S., Glasebrook, A., Fox, N., Solenberg, P. J., Zhang, Q., et al. (2001). Enhanced CD4+ T Cell Proliferation and Th2 Cytokine Production in DR6-Deficient Mice. Immunity 15 (1), 23–34. doi:10.1016/s1074-7613(01)00162-5
Locksley, R. M., Killeen, N., and Lenardo, M. J. (2001). The TNF and TNF Receptor Superfamilies: Integrating Mammalian Biology. Cell 104 (4), 487–501. doi:10.1016/s0092-8674(01)00237-9
Luu, K., Nickles, E., and Schwarz, H. (2020). Destroy, what Destroys You. Oncoimmunology 9 (1), 1685301. doi:10.1080/2162402X.2019.1685301
Maecker, H., Varfolomeev, E., Kischkel, F., Lawrence, D., LeBlanc, H., Lee, W., et al. (2005). TWEAK Attenuates the Transition from Innate to Adaptive Immunity. Cell 123 (5), 931–944. doi:10.1016/j.cell.2005.09.022
Martinez, T., Guo, A., Allen, M. J., Han, M., Pace, D., Jones, J., et al. (2008). Disulfide Connectivity of Human Immunoglobulin G2 Structural Isoforms. Biochemistry 47 (28), 7496–7508. doi:10.1021/bi800576c
Massarelli, E., Segal, N., Ribrag, V., Melero, I., Gangadhar, T., Urba, W., et al. (2016). Clinical Safety and Efficacy Assessment of the CD137 Agonist Urelumab Alone and in Combination with Nivolumab in Patients with Hematologic and Solid Tumor Malignancies. J. Immunother. Cancer 4 (Suppl. 1), O7.
McDermott, M. F., Aksentijevich, I., Galon, J., McDermott, E. M., Ogunkolade, B. W., Centola, M., et al. (1999). Germline Mutations in the Extracellular Domains of the 55 kDa TNF Receptor, TNFR1, Define a Family of Dominantly Inherited Autoinflammatory Syndromes. Cell 97 (1), 133–144. doi:10.1016/s0092-8674(00)80721-7
Medler, J., Nelke, J., Weisenberger, D., Steinfatt, T., Rothaug, M., Berr, S., et al. (2019). TNFRSF Receptor-specific Antibody Fusion Proteins with Targeting Controlled FcγR-independent Agonistic Activity. Cell Death Dis. 10 (3), 224. doi:10.1038/s41419-019-1456-x
Melero, I., Sanmamed, M. F., Calvo, E., Moreno, I., Moreno, V., Guerrero, T. C. H., et al. (2020). 1025MO First-In-Human (FIH) Phase I Study of RO7122290 (RO), a Novel FAP-Targeted 4-1BB Agonist, Administered as Single Agent and in Combination with Atezolizumab (ATZ) to Patients with Advanced Solid Tumours. Ann. Oncol. 31, S707. doi:10.1016/j.annonc.2020.08.1145
Middleton-Hardie, C., Zhu, Q., Cundy, H., Lin, J. M., Callon, K., Tong, P. C., et al. (2006). Deletion of Aspartate 182 in OPG Causes Juvenile Paget's Disease by Impairing Both Protein Secretion and Binding to RANKL. J. Bone Min. Res. 21 (3), 438–445. doi:10.1359/JBMR.051104
Milne, K., Köbel, M., Kalloger, S. E., Barnes, R. O., Gao, D., Gilks, C. B., et al. (2009). Systematic Analysis of Immune Infiltrates in High-Grade Serous Ovarian Cancer Reveals CD20, FoxP3 and TIA-1 as Positive Prognostic Factors. PLoS One 4 (7), e6412. doi:10.1371/journal.pone.0006412
Monreal, A. W., Zonana, J., and Ferguson, B. (1998). Identification of a New Splice Form of the EDA1 Gene Permits Detection of Nearly All X-Linked Hypohidrotic Ectodermal Dysplasia Mutations. Am. J. Hum. Genet. 63 (2), 380–389. doi:10.1086/301984
Morad, G., Helmink, B. A., Sharma, P., and Wargo, J. A. (2021). Hallmarks of Response, Resistance, and Toxicity to Immune Checkpoint Blockade. Cell 184 (21), 5309–5337. doi:10.1016/j.cell.2021.09.020
Muik, A., Garralda, E., Altintas, I., Gieseke, F., Geva, R., Ben-Ami, E., et al. (2022). Preclinical Characterization and Phase I Trial Results of a Bispecific Antibody Targeting PD-L1 and 4-1BB (GEN1046) in Patients with Advanced Refractory Solid Tumors. Cancer Discov. 12, 1248–1265. doi:10.1158/2159-8290.CD-21-1345
Munitic, I., Kuka, M., Allam, A., Scoville, J. P., and Ashwell, J. D. (2013). CD70 Deficiency Impairs Effector CD8 T Cell Generation and Viral Clearance but Is Dispensable for the Recall Response to Lymphocytic Choriomeningitis Virus. J. Immunol. 190 (3), 1169–1179. doi:10.4049/jimmunol.1202353
Newton, K., French, D. M., Yan, M., Frantz, G. D., and Dixit, V. M. (2004). Myodegeneration in EDA-A2 Transgenic Mice Is Prevented by XEDAR Deficiency. Mol. Cell Biol. 24 (4), 1608–1613. doi:10.1128/mcb.24.4.1608-1613.2004
Nguyen, J., Pettmann, J., Kruger, P., and Dushek, O. (2021). Quantitative Contributions of TNF Receptor Superfamily Members to CD8+ T-Cell Responses. Mol. Syst. Biol. 17 (11), e10560. doi:10.15252/msb.202110560
Nielsen, J. S., Sahota, R. A., Milne, K., Kost, S. E., Nesslinger, N. J., Watson, P. H., et al. (2012). CD20+ Tumor-Infiltrating Lymphocytes Have an Atypical CD27- Memory Phenotype and Together with CD8+ T Cells Promote Favorable Prognosis in Ovarian Cancer. Clin. Cancer Res. 18 (12), 3281–3292. doi:10.1158/1078-0432.CCR-12-0234
Nimmerjahn, F., Bruhns, P., Horiuchi, K., and Ravetch, J. V. (2005). FcγRIV: A Novel FcR with Distinct IgG Subclass Specificity. Immunity 23, 41–51.
Nimmerjahn, F., and Ravetch, J. V. (2006). Fcγ Receptors: Old Friends and New Family Members. Immunity 24 (1), 19–28. doi:10.1016/j.immuni.2005.11.010
Niu, L., Strahotin, S., Hewes, B., Zhang, B., Zhang, Y., Archer, D., et al. (2007). Cytokine-mediated Disruption of Lymphocyte Trafficking, Hemopoiesis, and Induction of Lymphopenia, Anemia, and Thrombocytopenia in Anti-CD137-treated Mice. J. Immunol. 178 (7), 4194–4213. doi:10.4049/jimmunol.178.7.4194
Nowak, A. K., Cook, A. M., McDonnell, A. M., Millward, M. J., Creaney, J., Francis, R. J., et al. (2015). A Phase 1b Clinical Trial of the CD40-Activating Antibody CP-870,893 in Combination with Cisplatin and Pemetrexed in Malignant Pleural Mesothelioma. Ann. Oncol. 26 (12), 2483–2490. doi:10.1093/annonc/mdv387
O'Hara, M. H., O'Reilly, E. M., Rosemarie, M., Varadhachary, G., Wainberg, Z. A., Ko, A., et al. (2019). Abstract CT004: A Phase Ib Study of CD40 Agonistic Monoclonal Antibody APX005M Together with Gemcitabine (Gem) and Nab-Paclitaxel (NP) with or without Nivolumab (Nivo) in Untreated Metastatic Ductal Pancreatic Adenocarcinoma (PDAC) Patients. Cancer Res. 79 (13_Suppl. ment), CT004. doi:10.1158/1538-7445.am2019-ct004
Pan, G., Ni, J., Wei, Y. F., Yu, G., Gentz, R., and Dixit, V. M. (1997). An Antagonist Decoy Receptor and a Death Domain-Containing Receptor for TRAIL. Science 277 (5327), 815–818. doi:10.1126/science.277.5327.815
Papadopoulos, K. P., Autio, K. A., Golan, T., Dobrenkov, K., Chartash, E., Li, X. N., et al. (2019). Phase 1 Study of MK-4166, an Anti-human Glucocorticoid-Induced Tumor Necrosis Factor Receptor (GITR) Antibody, as Monotherapy or with Pembrolizumab (Pembro) in Patients (Pts) with Advanced Solid Tumors. J. Clin. Oncol. 37.
Pappu, B. P., Borodovsky, A., Zheng, T. S., Yang, X., Wu, P., Dong, X., et al. (2008). TL1A-DR3 Interaction Regulates Th17 Cell Function and Th17-Mediated Autoimmune Disease. J. Exp. Med. 205 (5), 1049–1062. doi:10.1084/jem.20071364
Pasparakis, M., Alexopoulou, L., Episkopou, V., and Kollias, G. (1996). Immune and Inflammatory Responses in TNF Alpha-Deficient Mice: a Critical Requirement for TNF Alpha in the Formation of Primary B Cell Follicles, Follicular Dendritic Cell Networks and Germinal Centers, and in the Maturation of the Humoral Immune Response. J. Exp. Med. 184 (4), 1397–1411. doi:10.1084/jem.184.4.1397
Pispa, J., Pummila, M., Barker, P. A., Thesleff, I., and Mikkola, M. L. (2008). Edar and Troy Signalling Pathways Act Redundantly to Regulate Initiation of Hair Follicle Development. Hum. Mol. Genet. 17 (21), 3380–3391. doi:10.1093/hmg/ddn232
Postel-Vinay, S., Lam, V. K., Ros, W., Bauer, T. M., Hansen, A. R., Cho, D. C., et al. (2020). Abstract CT150: A First-In-Human Phase I Study of the OX40 Agonist GSK3174998 (GSK998) +/- Pembrolizumab in Patients (Pts) with Selected Advanced Solid Tumors (ENGAGE-1). Cancer Res. 80 (16_Suppl. ment), CT150.
Qi, X., Li, F., Wu, Y., Cheng, C., Han, P., Wang, J., et al. (2019). Optimization of 4-1BB Antibody for Cancer Immunotherapy by Balancing Agonistic Strength with FcγR Affinity. Nat. Commun. 10 (1), 2141. doi:10.1038/s41467-019-10088-1
Richman, L. P., and Vonderheide, R. H. (2014). Role of Crosslinking for Agonistic CD40 Monoclonal Antibodies as Immune Therapy of Cancer. Cancer Immunol. Res. 2 (1), 19–26. doi:10.1158/2326-6066.CIR-13-0152
Rickert, R. C., Jellusova, J., and Miletic, A. V. (2011). Signaling by the Tumor Necrosis Factor Receptor Superfamily in B-Cell Biology and Disease. Immunol. Rev. 244 (1), 115–133. doi:10.1111/j.1600-065X.2011.01067.x
Rieux-Laucat, F., Le Deist, F., Hivroz, C., Roberts, I. A., Debatin, K. M., Fischer, A., et al. (1995). Mutations in Fas Associated with Human Lymphoproliferative Syndrome and Autoimmunity. Science 268 (5215), 1347–1349. doi:10.1126/science.7539157
Rosales, C. (2017). Fcγ Receptor Heterogeneity in Leukocyte Functional Responses. Front. Immunol. 8. doi:10.3389/fimmu.2017.00280
Rosales, C., and Uribe-Querol, E. (2013). Fc Receptors: Cell Activators of Antibody Functions. Adv. Biosci. Biotechnol. 04, 13. doi:10.4236/abb.2013.44a004
Rothe, J., Lesslauer, W., Lötscher, H., Lang, Y., Koebel, P., Köntgen, F., et al. (1993). Mice Lacking the Tumour Necrosis Factor Receptor 1 Are Resistant to TNF-Mediated Toxicity but Highly Susceptible to Infection by Listeria Monocytogenes. Nature 364 (6440), 798–802. doi:10.1038/364798a0
Rüter, J., Antonia, S. J., Burris, H. A., Huhn, R. D., and Vonderheide, R. H. (2010). Immune Modulation with Weekly Dosing of an Agonist CD40 Antibody in a Phase I Study of Patients with Advanced Solid Tumors. Cancer Biol. Ther. 10 (10), 983–993.
Salzer, U., Chapel, H. M., Webster, A. D., Pan-Hammarström, Q., Schmitt-Graeff, A., Schlesier, M., et al. (2005). Mutations in TNFRSF13B Encoding TACI Are Associated with Common Variable Immunodeficiency in Humans. Nat. Genet. 37 (8), 820–828. doi:10.1038/ng1600
Sanborn, R., Hauke, R., Gabrail, N., O’Hara, M., Bhardwaj, N., Bordoni, R., et al. (2020). 405 CDX1140–01, a Phase 1 Dose-Escalation/expansion Study of CDX-1140 Alone (Part 1) and in Combination with CDX-301 (Part 2) or Pembrolizumab (Part 3). J. Immunother. Cancer 8 (Suppl. 3), A246.
Sashio, H., Tamura, K., Ito, R., Yamamoto, Y., Bamba, H., Kosaka, T., et al. (2002). Polymorphisms of the TNF Gene and the TNF Receptor Superfamily Member 1B Gene Are Associated with Susceptibility to Ulcerative Colitis and Crohn's Disease, Respectively. Immunogenetics 53 (12), 1020–1027. doi:10.1007/s00251-001-0423-7
Scheu, S., Alferink, J., Pötzel, T., Barchet, W., Kalinke, U., and Pfeffer, K. (2002). Targeted Disruption of LIGHT Causes Defects in Costimulatory T Cell Activation and Reveals Cooperation with Lymphotoxin Beta in Mesenteric Lymph Node Genesis. J. Exp. Med. 195 (12), 1613–1624. doi:10.1084/jem.20020215
Schiemann, B., Gommerman, J. L., Vora, K., Cachero, T. G., Shulga-Morskaya, S., Dobles, M., et al. (2001). An Essential Role for BAFF in the Normal Development of B Cells through a BCMA-independent Pathway. Science 293 (5537), 2111–2114. doi:10.1126/science.1061964
Schneider, P., Street, S. L., Gaide, O., Hertig, S., Tardivel, A., Tschopp, J., et al. (2001). Mutations Leading to X-Linked Hypohidrotic Ectodermal Dysplasia Affect Three Major Functional Domains in the Tumor Necrosis Factor Family Member Ectodysplasin-A. J. Biol. Chem. 276 (22), 18819–18827. doi:10.1074/jbc.M101280200
Segal, N. H., He, A. R., Doi, T., Levy, R., Bhatia, S., Pishvaian, M. J., et al. (2018). Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 24 (8), 1816–1823. doi:10.1158/1078-0432.CCR-17-1922
Segal, N. H., Logan, T. F., Hodi, F. S., McDermott, D., Melero, I., Hamid, O., et al. (2017). Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin. Cancer Res. 23 (8), 1929–1936. doi:10.1158/1078-0432.CCR-16-1272
Sharonov, G. V., Serebrovskaya, E. O., Yuzhakova, D. V., Britanova, O. V., and Chudakov, D. M. (2020). B Cells, Plasma Cells and Antibody Repertoires in the Tumour Microenvironment. Nat. Rev. Immunol. 20 (5), 294–307. doi:10.1038/s41577-019-0257-x
Shimomura, Y., Sato, N., Miyashita, A., Hashimoto, T., Ito, M., and Kuwano, R. (2004). A Rare Case of Hypohidrotic Ectodermal Dysplasia Caused by Compound Heterozygous Mutations in the EDAR Gene. J. Invest Dermatol 123 (4), 649–655. doi:10.1111/j.0022-202X.2004.23405.x
Singer, G. G., and Abbas, A. K. (1994). The Fas Antigen Is Involved in Peripheral but Not Thymic Deletion of T Lymphocytes in T Cell Receptor Transgenic Mice. Immunity 1 (5), 365–371. doi:10.1016/1074-7613(94)90067-1
Steinhilber, W., and Stegmeyer, H. (1977). Functional Analytic Studies on Mandibular Motility after Fractures of the Mandibular Condyle. Dtsch. Zahnarztl Z 32 (2), 96–98.
Stephens, G. L., McHugh, R. S., Whitters, M. J., Young, D. A., Luxenberg, D., Carreno, B. M., et al. (2004). Engagement of Glucocorticoid-Induced TNFR Family-Related Receptor on Effector T Cells by its Ligand Mediates Resistance to Suppression by CD4+CD25+ T Cells. J. Immunol. 173 (8), 5008–5020. doi:10.4049/jimmunol.173.8.5008
Takahashi, T., Tanaka, M., Brannan, C. I., Jenkins, N. A., Copeland, N. G., Suda, T., et al. (1994). Generalized Lymphoproliferative Disease in Mice, Caused by a Point Mutation in the Fas Ligand. Cell 76 (6), 969–976. doi:10.1016/0092-8674(94)90375-1
Teodorovic, L. S., Riccardi, C., Torres, R. M., and Pelanda, R. (2012). Murine B Cell Development and Antibody Responses to Model Antigens Are Not Impaired in the Absence of the TNF Receptor GITR. PLoS One 7 (2), e31632. doi:10.1371/journal.pone.0031632
Timmerman, J., Herbaux, C., Ribrag, V., Zelenetz, A. D., Houot, R., Neelapu, S. S., et al. (2020). Urelumab Alone or in Combination with Rituximab in Patients with Relapsed or Refractory B-Cell Lymphoma. Am. J. Hematol. 95 (5), 510–520. doi:10.1002/ajh.25757
Tirnitz-Parker, J. E., Viebahn, C. S., Jakubowski, A., Klopcic, B. R., Olynyk, J. K., Yeoh, G. C., et al. (2010). Tumor Necrosis Factor-like Weak Inducer of Apoptosis Is a Mitogen for Liver Progenitor Cells. Hepatology 52 (1), 291–302. doi:10.1002/hep.23663
Tolcher, A. W., Sznol, M., Hu-Lieskovan, S., Papadopoulos, K. P., Patnaik, A., Rasco, D. W., et al. (2017). Phase Ib Study of Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Combination with Pembrolizumab (MK-3475) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 23 (18), 5349–5357. doi:10.1158/1078-0432.CCR-17-1243
Um, J. Y., An, N. H., and Kim, H. M. (2003). TNF-alpha and TNF-Beta Gene Polymorphisms in Cerebral Infarction. J. Mol. Neurosci. 21 (2), 167–171. doi:10.1385/JMN:21:2:167
van Montfrans, J. M., Hoepelman, A. I., Otto, S., van Gijn, M., van de Corput, L., de Weger, R. A., et al. (2012). CD27 Deficiency Is Associated with Combined Immunodeficiency and Persistent Symptomatic EBV Viremia. J. Allergy Clin. Immunol. 129 (3), 787–793.e6. doi:10.1016/j.jaci.2011.11.013
Varfolomeev, E., Kischkel, F., Martin, F., Seshasayee, D., Wang, H., Lawrence, D., et al. (2004). APRIL-deficient Mice Have Normal Immune System Development. Mol. Cell Biol. 24 (3), 997–1006. doi:10.1128/mcb.24.3.997-1006.2004
Vidarsson, G., and van de Winkel, J. G. (1998). Curr. Opin. Infect. Dis. 11, 271. doi:10.1097/00001432-199806000-00002
Vinay, D. S., Choi, B. K., Bae, J. S., Kim, W. Y., Gebhardt, B. M., and Kwon, B. S. (2004). CD137-deficient Mice Have Reduced NK/NKT Cell Numbers and Function, Are Resistant to Lipopolysaccharide-Induced Shock Syndromes, and Have Lower IL-4 Responses. J. Immunol. 173 (6), 4218–4229. doi:10.4049/jimmunol.173.6.4218
Vinay, D. S., Kim, J. D., Asai, T., Choi, B. K., and Kwon, B. S. (2007). Absence of 4 1BB Gene Function Exacerbates Lacrimal Gland Inflammation in Autoimmune-Prone MRL-Faslpr Mice. Invest Ophthalmol. Vis. Sci. 48 (10), 4608–4615. doi:10.1167/iovs.07-0153
Vonderheide, R. H., Burg, J. M., Mick, R., Trosko, J. A., Li, D., Shaik, M. N., et al. (2013). Phase I Study of the CD40 Agonist Antibody CP-870,893 Combined with Carboplatin and Paclitaxel in Patients with Advanced Solid Tumors. Oncoimmunology 2 (1), e23033. doi:10.4161/onci.23033
Vonderheide, R. H., Flaherty, K. T., Khalil, M., Stumacher, M. S., Bajor, D. L., Hutnick, N. A., et al. (2007). Clinical Activity and Immune Modulation in Cancer Patients Treated with CP-870,893, a Novel CD40 Agonist Monoclonal Antibody. J. Clin. Oncol. 25 (7), 876–883. doi:10.1200/JCO.2006.08.3311
Wajant, H. (2015). Principles of Antibody-Mediated TNF Receptor Activation. Cell Death Differ. 22 (11), 1727–1741. doi:10.1038/cdd.2015.109
Wang, E. C., Thern, A., Denzel, A., Kitson, J., Farrow, S. N., and Owen, M. J. (2001). DR3 Regulates Negative Selection during Thymocyte Development. Mol. Cell Biol. 21 (10), 3451–3461. doi:10.1128/MCB.21.10.3451-3461.2001
Wang, H. Y., Ma, C. A., Zhao, Y., Fan, X., Zhou, Q., Edmonds, P., et al. (2013). Antibody Deficiency Associated with an Inherited Autosomal Dominant Mutation in TWEAK. Proc. Natl. Acad. Sci. U. S. A. 110 (13), 5127–5132. doi:10.1073/pnas.1221211110
Ward-Kavanagh, L. K., Lin, W. W., Šedý, J. R., and Ware, C. F. (2016). The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 44 (5), 1005–1019. doi:10.1016/j.immuni.2016.04.019
Warnatz, K., Salzer, U., Rizzi, M., Fischer, B., Gutenberger, S., Böhm, J., et al. (2009). B-cell Activating Factor Receptor Deficiency Is Associated with an Adult-Onset Antibody Deficiency Syndrome in Humans. Proc. Natl. Acad. Sci. U. S. A. 106 (33), 13945–13950. doi:10.1073/pnas.0903543106
Waschke, K. A., Villani, A. C., Vermeire, S., Dufresne, L., Chen, T. C., Bitton, A., et al. (2005). Tumor Necrosis Factor Receptor Gene Polymorphisms in Crohn's Disease: Association with Clinical Phenotypes. Am. J. Gastroenterol. 100 (5), 1126–1133. doi:10.1111/j.1572-0241.2005.40534.x
Watts, T. H. (2005). TNF/TNFR Family Members in Costimulation of T Cell Responses. Annu. Rev. Immunol. 23, 23–68. doi:10.1146/annurev.immunol.23.021704.115839
Wei, S. C., Duffy, C. R., and Allison, J. P. (2018). Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 8 (9), 1069–1086. doi:10.1158/2159-8290.CD-18-0367
White, A. L., Chan, H. T., French, R. R., Beers, S. A., Cragg, M. S., Johnson, P. W., et al. (2013). FcγRΙΙB Controls the Potency of Agonistic Anti-TNFR mAbs. Cancer Immunol. Immunother. 62 (5), 941–948. doi:10.1007/s00262-013-1398-6
White, A. L., Chan, H. T., French, R. R., Willoughby, J., Mockridge, C. I., Roghanian, A., et al. (2015). Conformation of the Human Immunoglobulin G2 Hinge Imparts Superagonistic Properties to Immunostimulatory Anticancer Antibodies. Cancer Cell 27 (1), 138–148. doi:10.1016/j.ccell.2014.11.001
White, A. L., Chan, H. T., Roghanian, A., French, R. R., Mockridge, C. I., Tutt, A. L., et al. (2011). Interaction with FcγRIIB Is Critical for the Agonistic Activity of Anti-CD40 Monoclonal Antibody. J. Immunol. 187 (4), 1754–1763. doi:10.4049/jimmunol.1101135
White, A. L., Dou, L., Chan, H. T., Field, V. L., Mockridge, C. I., Moss, K., et al. (2014). Fcγ Receptor Dependency of Agonistic CD40 Antibody in Lymphoma Therapy Can Be Overcome through Antibody Multimerization. J. Immunol. 193 (4), 1828–1835. doi:10.4049/jimmunol.1303204
Wiens, G. D., and Glenney, G. W. (2011). Origin and Evolution of TNF and TNF Receptor Superfamilies. Dev. Comp. Immunol. 35 (12), 1324–1335. doi:10.1016/j.dci.2011.03.031
Wilson, N. S., Yang, B., Yang, A., Loeser, S., Marsters, S., Lawrence, D., et al. (2011). An Fcγ Receptor-dependent Mechanism Drives Antibody-Mediated Target-Receptor Signaling in Cancer Cells. Cancer Cell 19 (1), 101–113. doi:10.1016/j.ccr.2010.11.012
Xu, J., Foy, T. M., Laman, J. D., Elliott, E. A., Dunn, J. J., Waldschmidt, T. J., et al. (1994). Mice Deficient for the CD40 Ligand. Immunity 1 (5), 423–431. doi:10.1016/1074-7613(94)90073-6
Xu, S., and Lam, K. P. (2001). B-cell Maturation Protein, Which Binds the Tumor Necrosis Factor Family Members BAFF and APRIL, Is Dispensable for Humoral Immune Responses. Mol. Cell Biol. 21 (12), 4067–4074. doi:10.1128/MCB.21.12.4067-4074.2001
Xu, Y., Szalai, A. J., Zhou, T., Zinn, K. R., Chaudhuri, T. R., Li, X., et al. (2003). Fc Gamma Rs Modulate Cytotoxicity of Anti-fas Antibodies: Implications for Agonistic Antibody-Based Therapeutics. J. Immunol. 171 (2), 562–568. doi:10.4049/jimmunol.171.2.562
Yan, M., Brady, J. R., Chan, B., Lee, W. P., Hsu, B., Harless, S., et al. (2001a). Identification of a Novel Receptor for B Lymphocyte Stimulator that Is Mutated in a Mouse Strain with Severe B Cell Deficiency. Curr. Biol. 11 (19), 1547–1552. doi:10.1016/s0960-9822(01)00481-x
Yan, M., Wang, H., Chan, B., Roose-Girma, M., Erickson, S., Baker, T., et al. (2001b). Activation and Accumulation of B Cells in TACI-Deficient Mice. Nat. Immunol. 2 (7), 638–643. doi:10.1038/89790
Yu, X., Chan, H. T. C., Fisher, H., Penfold, C. A., Kim, J., Inzhelevskaya, T., et al. (2020). Isotype Switching Converts Anti-CD40 Antagonism to Agonism to Elicit Potent Antitumor Activity. Cancer Cell 37 (6), 850–866.e7. doi:10.1016/j.ccell.2020.04.013
Yu, X., Chan, H. T. C., Orr, C. M., Dadas, O., Booth, S. G., Dahal, L. N., et al. (2018). Complex Interplay between Epitope Specificity and Isotype Dictates the Biological Activity of Anti-human CD40 Antibodies. Cancer Cell 33 (4), 664–675.e4. doi:10.1016/j.ccell.2018.02.009
Zhang, D., Armstrong, A. A., Tam, S. H., McCarthy, S. G., Luo, J., Gilliland, G. L., et al. (2017). Functional Optimization of Agonistic Antibodies to OX40 Receptor with Novel Fc Mutations to Promote Antibody Multimerization. mAbs 9 (7), 1129–1142. doi:10.1080/19420862.2017.1358838
Zhang, L., Zhao, H., Ma, Y., Zheng, X., Jiang, J., Zhang, Y., et al. (2020). A Phase I, Dose-Escalation Study of ADG106, a Fully Human Anti-CD137 Agonistic Antibody, in Subjects with Advanced Solid Tumors or Relapsed/refractory Non-hodgkin Lymphoma. J. Clin. Oncol. 38 (15_Suppl. l), 3105. doi:10.1200/jco.2020.38.15_suppl.3105
Zhang, P., Tu, G. H., Wei, J., Santiago, P., Larrabee, L. R., Liao-Chan, S., et al. (2019). Ligand-Blocking and Membrane-Proximal Domain Targeting Anti-OX40 Antibodies Mediate Potent T Cell-Stimulatory and Anti-tumor Activity. Cell Rep. 27 (11), 3117–3123.e5. doi:10.1016/j.celrep.2019.05.027
Keywords: FcγRIIB, TNFRSF, CD40, CD137 (4-1BB), agonist, immunotherapy, cancer, cross-linking
Citation: Liu L, Wu Y, Ye K, Cai M, Zhuang G and Wang J (2022) Antibody-Targeted TNFRSF Activation for Cancer Immunotherapy: The Role of FcγRIIB Cross-Linking. Front. Pharmacol. 13:924197. doi: 10.3389/fphar.2022.924197
Received: 20 April 2022; Accepted: 30 May 2022;
Published: 05 July 2022.
Edited by:
Liqiang Pan, Zhejiang University, ChinaReviewed by:
Shahryar Khoshtinat Nikkhoi, Rutgers, The State University of New Jersey, United StatesZhang Baohong, Shanghai Jiao Tong University, China
Copyright © 2022 Liu, Wu, Ye, Cai, Zhuang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jieyi Wang, amlleWkud2FuZ0BseXZnZW4uY29t