 Xue Zhao
Xue Zhao Muna I. Naash
Muna I. Naash Muayyad R. Al-Ubaidi
Muayyad R. Al-Ubaidi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 06 July 2022
Sec. Neuropharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.919667
This article is part of the Research Topic Neuroprotective Mechanisms and Translational Approaches in the Retina View all 9 articles
Dysregulation of retinal metabolism is emerging as one of the major reasons for many inherited retinal diseases (IRDs), a leading cause of blindness worldwide. Thus, the identification of a common regulator that can preserve or revert the metabolic ecosystem to homeostasis is a key step in developing a treatment for different forms of IRDs. Riboflavin (RF) and its derivatives (flavins), flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), are essential cofactors for a wide range of cellular metabolic processes; hence, they are particularly critical in highly metabolically active tissues such as the retina. Patients with RF deficiency (ariboflavinosis) often display poor photosensitivity resulting in impaired low-light vision. We have identified a novel retina-specific RF binding protein called retbindin (Rtbdn), which plays a key role in retaining flavin levels in the neural retina. This role is mediated by its specific localization at the interface between the neural retina and retinal pigment epithelium (RPE), which is essential for metabolite and nutrient exchange. As a consequence of this vital function, Rtbdn’s role in flavin utilization and metabolism in retinal degeneration is discussed. The principal findings are that Rtbdn helps maintain high levels of retinal flavins, and its ablation leads to an early-onset retinal metabolic dysregulation, followed by progressive degeneration of rod and cone photoreceptors. Lack of Rtbdn reduces flavin levels, forcing the neural retina to repurpose glucose to reduce the production of free radicals during ATP production. This leads to metabolic breakdown followed by retinal degeneration. Assessment of the role of Rtbdn in several preclinical retinal disease models revealed upregulation of its levels by several folds prior to and during the degenerative process. Ablation of Rtbdn in these models accelerated the rate of retinal degeneration. In agreement with these in vivo studies, we have also demonstrated that Rtbdn protects immortalized cone photoreceptor cells (661W cells) from light damage in vitro. This indicates that Rtbdn plays a neuroprotective role during retinal degeneration. Herein, we discussed the specific function of Rtbdn and its neuroprotective role in retinal metabolic homeostasis and its role in maintaining retinal health.
Inherited retinal diseases (IRDs) represent a highly variable group of blinding retinal diseases, which are known to be caused by mutations in more than 300 different genes or loci (https://sph.uth.edu/retnet/). The two most commonly mutated genes in IRD patients are the rod visual pigment rhodopsin (RHO) and peripherin 2 (PRPH2, also known as RDS), a retinal-specific tetraspanin protein that is vital for the structure of the photoreceptor outer segment (OS) (Conley and Naash, 2009; Stuck et al., 2016). To investigate how mutations in photoreceptor-specific genes lead to cell death, various animal models expressing mutant versions of these genes have been generated, and the retinal phenotype in many of these models mimicked human retinal diseases (Collin et al., 2020). Although most retinal phenotypes in these models are associated with photoreceptor degeneration, deciphering the mechanism(s) that lead to photoreceptor cell death in most of these models has been elusive. In some cases, patients are born expressing the mutant proteins, yet the symptoms do not develop until the fourth or the fifth decade of life. In addition, in order to treat any of these inherited diseases, the development of specific gene therapy, viral or non-viral, must be designed for the specific mutation. Thus, it became clear that there is an imminent need for a pan-gene-mutation therapy, which galvanized investigations into disease mechanisms beyond the mutant proteins. As a result, in recent years, our research has focused on the metabolic changes caused by the absence of the native protein or the expression of a mutant protein (for example, Rtbdn−/− (Kelley et al., 2017; Sinha et al., 2021), Rd1 (Murenu et al., 2021), mutations in Prph2 (Ding et al., 2004; Conley et al., 2014; Stuck et al., 2014; Stuck et al., 2016), or mutant Prph2/Rtbdn−/− (Genc et al., 2020a; Genc et al., 2020b). After all, metabolism is the source of energy that cells need to achieve homeostasis or to execute cell death and to provide the metabolic intermediates needed for anabolic activities. More and more data are emerging to support the conclusion that metabolic dysregulations are the main common events in initiating IRDs (Punzo et al., 2012; Griciuc et al., 2014; Joyal et al., 2016; Kaplan et al., 2021; Sinha et al., 2021). Thus, identifying common metabolic regulators involved in retinal degeneration that can preserve or revert the metabolic ecosystem to homeostasis is essential for the future development of neuroprotective therapeutic strategies. Due to the complicated and energy-intensive nature of phototransduction, photoreceptors have higher energy demands than any other cell type (Hurley, 2021), as demonstrated by the retina’s high consumption of glucose and oxygen (Chertov et al., 2011; Damsgaard and Country, 2022).
Nutrient availability and metabolic alterations are likely common mechanisms that play roles in retinal degeneration (Du et al., 2015). The diseased retinas may reprogram their metabolism at any time, employing different energy sources or mechanisms before or after the onset of degeneration in order to adapt to stress and changes to homeostasis. As a result, metabolic failure may ensue, eventually leading to degeneration (Sinha et al., 2021). Improper levels of neuroprotective factors can contribute to the degenerative process in the retina. The negative effects of downregulation of these neuroprotective factors may eventually prevail, leading to retinal degeneration (Ly et al., 2016). Harder et al. reported the idea that a decline in retinal pyruvate levels resulting from intraocular pressure changes is linked to dysregulated metabolism of glucose (Harder et al., 2020). This highlights the importance of investigating retinal metabolism to identify essential players involved in maintaining health or in exacerbating disease mechanisms and developing strategies for their use as common neuroprotective therapeutic targets for IRD cases.
In our approach to investigate the metabolic dysregulation in IRDs, we decided to study pan-metabolic effects rather than focusing on a specific metabolite or pathway. The retinal pigment epithelium (RPE) supplies the photoreceptor cells with essential metabolites by transferring nutrients and oxygen from the choroidal blood supply to the neural retina (reviewed by Sparrow et al. (2010) and Hurley (2021)). Among these metabolites is riboflavin (RF, C17H20N4O6), the parent molecule for the cofactors, flavin mononucleotide (FMN, C17H21N4O9P), and flavin adenine dinucleotide (FAD, C27H33N9O15P2) (Figure 1). FMN and FAD, two functionally important cofactors in many redox reactions in metabolism (Powers, 2003), are substantially concentrated in the retina when compared to the blood (Batey and Eckhert, 1990; Batey and Eckhert, 1991; Sinha et al., 2018). RF and its derivatives are involved in various biological processes, including DNA repair, energy metabolism, amino acid synthesis, protein folding, and normal immunological function (Bro-Rasmussen and Horwitt, 1967; Tu et al., 2000; Mazur-Bialy et al., 2013). RF is also an antioxidant, and its absence causes oxidative damage and upregulates stress response in the cell (reviewed by Olfat et al. (2022)). Some of the symptoms seen in individuals lacking RF include growth retardation, hair loss, anemia, nerve impairments, hearing loss, and visual abnormalities [reviewed by Cali et al. (1993) and Mosegaard et al. (2020)]. Ocular abnormalities in humans receiving insufficient RF include corneal lesions, cataract formation, and retinal ganglion cell degeneration, thus, highlighting RF as an essential vitamin in retinal health and survival (Irinoda and Sato, 1954; Venkataswamy, 1967).
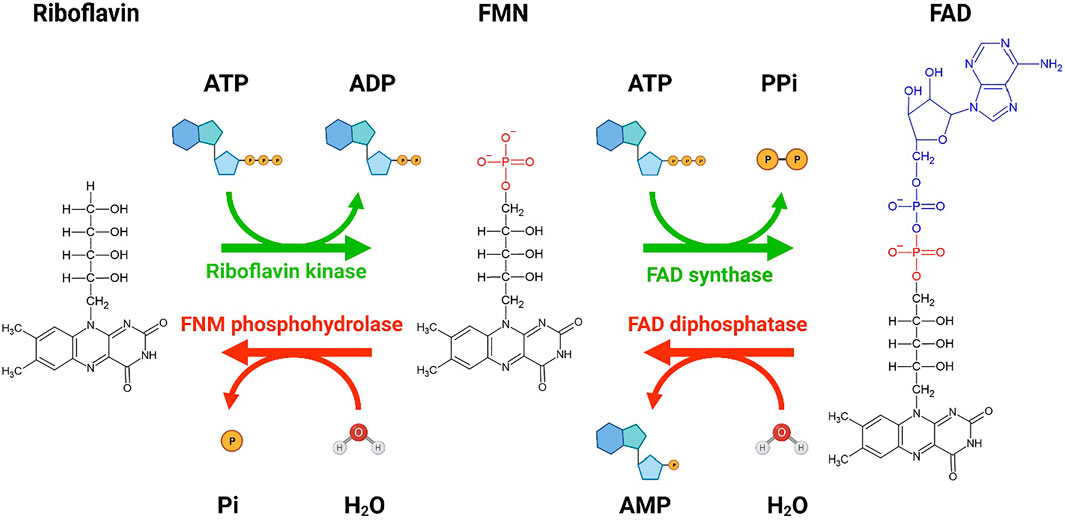
FIGURE 1. A schematic showing the conversion of riboflavin to flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). Bio Render was used to generate the schematic diagram showing the conversion of riboflavin to FMN and FAD by riboflavin kinase and FAD synthase, respectively. Two molecules of ATP are utilized for the conversion of riboflavin to FAD. The reverse conversion of FAD to FMN by FAD diphosphatase and FMN to riboflavin by FMN phosphohydrolase is also shown. The forward conversions are more favorable than the reverse conversion. Re-drawn from Tolomeo et al. (2020). Created with BioRender.
The neural retina’s high flavin content suggests that it harbors a unique mechanism for flavin transport/concentration, but until recently, no such systems have been described. Unbound RF can be reduced by light and produce oxygen radicals which lead to peroxidation of lipids in the photoreceptor OS membranes (Oster et al., 1962; Treadwell et al., 1968; Wiegand et al., 1983; Huvaere et al., 2010). As a result, special proteins (flavoproteins) likely play an important role in the sequestration and utilization of flavins in the retina. During our studies on retinal metabolic homeostasis, we became interested in a novel retina-specific RF-binding protein called retbindin (Rtbdn), which has homology to the chicken RF-binding protein (RBP) (Wistow et al., 2002; Kelley et al., 2015). Rtbdn was identified as an expressed sequence tag in the human retina, which Wistow et al. named retbindin due to its retina specificity and the fact that it shares 27% sequence identity (over 135 residues) with RBP (Wistow et al., 2002). Rtbdn is a ∼30 kDa protein that is only found in the neural retina and not in the RPE (Kelley et al., 2017). Rtbdn is located at the junction between the RPE and the photoreceptor OS tips (Kelley et al., 2015). Due to the high levels of flavin in the retina (Batey and Eckhert, 1990; Batey et al., 1992; Sinha et al., 2018) and the fact that the retina has high metabolic activities [reviewed by Country (2017) and Hurley (2021)], we became interested in studying the role of Rtbdn in the retina. Our assumption was that Rtbdn might be a key player in sustaining the high levels of flavins in the retina and, thus, supporting retinal metabolic activities. Given the significance of flavins in metabolic processes and the lack of knowledge about how the retina obtains and retains high flavin levels, we assessed the role of Rtbdn in regulating retinal flavins (Kelley et al., 2015). We confirmed the extracellular nature of Rtbdn and its ability to electrostatically bind to the plasma membrane (Kelley et al., 2015). We also found that Rtbdn can bind RF in vitro (Kelley et al., 2015) and in vivo (Kelley et al., 2017), confirming the function of Rtbdn as an RF carrier between the retina and the RPE.
To test the neuroprotective abilities of Rtbdn, 661W cells were exposed to 11,000 lux for 1 hour in the presence of membrane preparations isolated from COS-7 cells either untransfected or transiently transfected with a vector to express Rtbdn or RBP (Kelley et al., 2018). The 661W cells were shown to be from a cone-cell lineage (Tan et al., 2004) and are sensitive to exposure to high light intensities (Kelley et al., 2018). Following light exposure in the absence of Rtbdn-containing membrane preparations, ∼70% of the cells died, while 100% of the cells supplied with Rtbdn-containing membrane preparations survived (Kelley et al., 2018). This level of protection was comparable to cells given 100 nM Docosahexaenoic acid (DHA) in the medium (Kelley et al., 2018), which has been shown to have an anti-apoptotic role in the retina (Querques et al., 2011). Furthermore, supplementation with RBP-containing membrane preparations showed an analogous effect as the supplementation with Rtbdn-containing membrane preparations (Kelley et al., 2018).
Rtbdn ablation led to a reduction in rod and cone functions and a time- and dose-dependent retinal degeneration starting as early as P120 and progressing at P240 (Kelley et al., 2017), providing additional evidence for its protective role. This degenerative process was accompanied by a significant reduction in total retinal flavin levels (Kelley et al., 2017). Subsequently, we showed that the absence of Rtbdn in the knockout mice (Rtbdn−/−) and the ensuing reduction in retinal flavins levels led to significant misregulation in the levels of multiple metabolites involved in the citric acid cycle prior to the onset of degeneration (Sinha et al., 2021). Among other metabolic changes, the neural retina of Rtbdn−/− mice displayed a significant reduction in glutathione recycling, accumulation of toxic metabolites, and increased methionine sulfonation, all of which contribute to the degenerative phenotype associated with Rtbdn ablation (Sinha et al., 2021). Sphingolipid metabolism is hampered by the lack of glucose transport through serine production, and toxic hexasylceramide accumulates (Sinha et al., 2021). This causes the neural retina to inhibit glycolysis by inhibiting tetrameric pyruvate kinase M2 (PKM2) formation and instead upregulating the pentose phosphate pathway (PPP) to counteract the oxidative damage. Lipolysis cannot function properly in the presence of diminished flavin cofactors, resulting in acylcarnitine accumulation in the Rtbdn−/− retina. Moreover, as the metabolic load increases, the gap between ATP demand and supply widens, causing the metabolic homeostasis to be disrupted. This is accompanied by a significant decrease in amino acid metabolism in Rtbdn−/− neural retina and RPE (Sinha et al., 2021). The findings from Sinha et al. showed that even small alterations in retinal metabolic homeostasis can shift the balance between a healthy and pathogenic state, ultimately affecting retinal function (Sinha et al., 2021). In fact, we found that ATP levels were reduced in P45 neural retinas in absence of Rtbdn and declined further as the animal aged. Overall, our findings indicate that Rtbdn is an important participant in flavin modulation in the retina and the association between retinal degeneration and the reduction in flavin levels strengthened our hypothesis about the neuroprotective role of Rtbdn.
In a recent publication, Fan et al. (2021) proposed that Rtbdn mediates retinal light damage and has significant retinoid-binding affinity, most notably 11-cis retinal. The authors also claimed that in the absence of Rtbdn, there was no drop in total flavin levels. They based their conclusions on two pieces of evidence. First, they used bio-layer interferometry binding studies to show that Rtbdn has a high binding affinity for retinoids, especially 11-cis retinal. In our studies, we showed that Rtbdn binds RF in vitro, ex vivo, and in vivo systems. The findings of Fan et al. would have been validated should they have performed direct binding assays to confirm the ability of Rtbdn to bind 11-cis retinal or any other retinoids in retinal explants or in vivo. Furthermore, it would have been most informative should Fan et al. measured and demonstrated alterations in retinal retinoids in the Rtbdn knockout retina. As far as the levels of flavin in their Rtbdn−/− mouse line, Fan et al. showed a reduction of 12.5% in RF levels, 3% in FAD, and 4.6% in FMN in aphakic eyeballs (Fan et al., 2021), which contains all eye components except the lens. In fact, it is most likely that the changes in retinal flavin levels in their measurements are masked by the significantly higher flavin levels in the cornea (Batey and Eckhert, 1991), RPE (Sinha et al., 2018), iris, and sclera. Interestingly, we showed the RPE contains ∼20 times more flavin levels than the neural retina (Sinha et al., 2018). It is very clear that they underestimated the reduction in retinal levels of flavin in their Rtbdn−/− line. Regardless, in our studies, we measured flavin levels in isolated neural retinas from our Rtbdn−/− line and showed a 50% reduction when compared to age-matched wild-type neural retinas.
To investigate the neuroprotective effects of Rtbdn, its levels were determined in mouse models of retinal degeneration (RhoP23H/+ and Prph2R172W). Furthermore, Rtbdn was ablated in these models, and the change in the rate of retinal degeneration was determined. Table 1 summarizes the retinal phenotypes observed in these models.

TABLE 1. Inherited retinal degenerative mouse models used in the study of the neuroprotective effects of Rtbdn.
Retinitis pigmentosa (RP) represents inherited retinal dystrophy caused by the progressive loss of photoreceptors, with no effective therapy available thus far. RP affects over 1.5 million patients worldwide, with a prevalence of 1:4,000 (reviewed by Verbakel et al. (2018)). Therefore, identifying the factors that maintain retinal homeostasis and promote photoreceptor survival during the pathogenesis of RP is critical for the treatment of this disease. In most cases of RP, the rod photoreceptors in the peripheral retina begin to degenerate first with a gradual development of night blindness (Hartong et al., 2006). With the increasing loss of rods, the cones gradually die, ultimately resulting in complete blindness, making it more challenging to develop treatment strategies. Although the pathogenesis of RP is still not clear, recent studies have found oxidative stress build-up during rod and cone degeneration in RP (Moreno et al., 2018; Trachsel-Moncho et al., 2018; Newton and Megaw, 2020).
Around 25% of all RP cases are caused by mutations in the rhodopsin gene (RHO) (Hartong et al., 2006), with a proline to histidine change at codon 23 (P23H) in RHO being the first RP mutation identified in human patients (Dryja et al., 1990a; Dryja et al., 1990b). In addition, this mutation also represents the most common cause of Rho-related RP in the US (Sung et al., 1991; Sohocki et al., 2001). Various transgenic and knockin animal models have been used for the investigation of the effects of the P23H mutation (Olsson et al., 1992; Naash et al., 1993; Machida et al., 2000; Ross et al., 2012; LaVail et al., 2018). The most widely used models that mimicked the patient’s phenotype and disease progression are several P23H transgenic rat lines (LaVail et al., 2018), a P23H transgenic mouse line (Naash et al., 1993), and a P23H knock-in mouse line (RhoP23H/+) (Sakami et al., 2011). The mouse models showed severe functional impairments in rods and less functional deficits in cones at an early age, but cone abnormalities become more prominent at about P60 (Naash et al., 1993; Sakami et al., 2011).
To evaluate the role of Rtbdn in retinal degenerative disorders, Rtbdn levels were measured in the retinas of RhoP23H/+ mice at P15 (before the onset of degeneration), P30 (left image in Figure 2B, at the onset of degeneration), and P60 (after the onset of degeneration) (Genc et al., 2020b). In their study, Genc et al. showed significant upregulation of Rtbdn levels prior to and during the degenerative process in the RhoP23H/+ mice (Genc et al., 2020b). Although the pattern of Rtbdn distribution in the RhoP23H/+ retina was still preserved like the wild-type around the inner segment and at the tips of the OS/RPE junction, abnormally large amounts of Rtbdn staining were also seen throughout the OS layer in patches. The identity of these patches is still unknown. Since Rtbdn binds RF and flavins are necessary for a variety of metabolic processes, it is no surprise that Rtbdn is overexpressed before the onset of degeneration and during cell death. This strongly suggests that Rtbdn plays a major role in the cellular stress response, especially knowing that apoptosis requires energy. Notably, the elimination of Rtbdn in the RhoP23H/+ retina at P30 caused a significant reduction in both scotopic a- and photopic b-waves amplitudes (Genc et al., 2020b). Structurally, ablation of Rtbdn in RhoP23H/+ animals (RhoP23H/+/Rtbdn−/−) accelerated the onset of degeneration (see arrowheads in Figure 2B and Figure 3B right images) and increased the progression of the disease phenotype (Genc et al., 2020b), despite the fact that there was no apparent phenotype in the Rtbdn−/− retinas until P120 (Kelley et al., 2017; Genc et al., 2020b). When Rtbdn was eliminated, there were no initial changes in flavin levels at P15 (Genc et al., 2020b), but those levels were reduced in older RhoP23H/+ retinas despite Rtbdn upregulation. This could be the outcome of excessive flavin consumption by the surviving cells or due to reduced delivery by the RPE, which undergoes stress due to increased cellular debris from dying photoreceptor cells and/or changes in its metabolic activity as a result of the metabolic alterations occurring in the neural retina (Sinha et al., 2021) or a decrease in the number of surviving photoreceptor cells (Genc et al., 2020b).
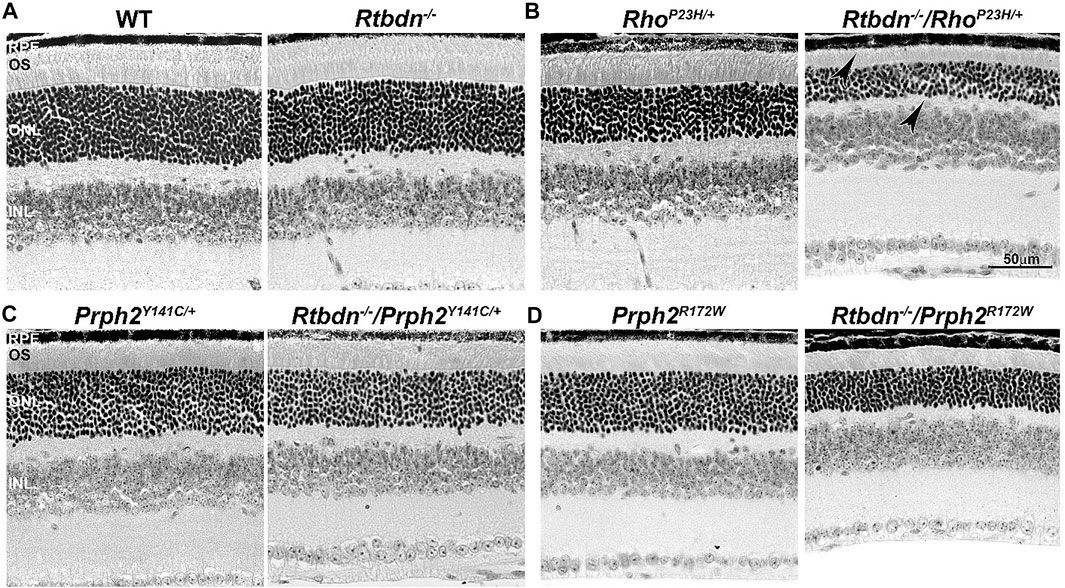
FIGURE 2. Ablation of Rtbdn accelerates retinal phenotype in several models of degeneration. Histologic analysis at the light microscopic level of retinal sections from WT and Rtbdn−/− (A), RhoP23H/+ and Rtbdn−/−/RhoP23H/+ (B), PrphY141C/+ and Rtbdn−/−/Prph2Y141C/+ (C), and PrphR172W and Rtbdn−/−/Prph2R172W (D) mice at P30. Arrowheads point to the degenerated ONL and outer segments shortening in the Rtbdn−/−/RhoP23H/+ model. Scale bar: 50 µm. RPE, retinal pigment epithelium; OS, outer segment; ONL, outer nuclear layer; INL, inner nuclear layer. Images were taken from paraffin-embedded eyes of the relevant genotypes used by Genc et al. (2020a) and Genc et al. (2020b).
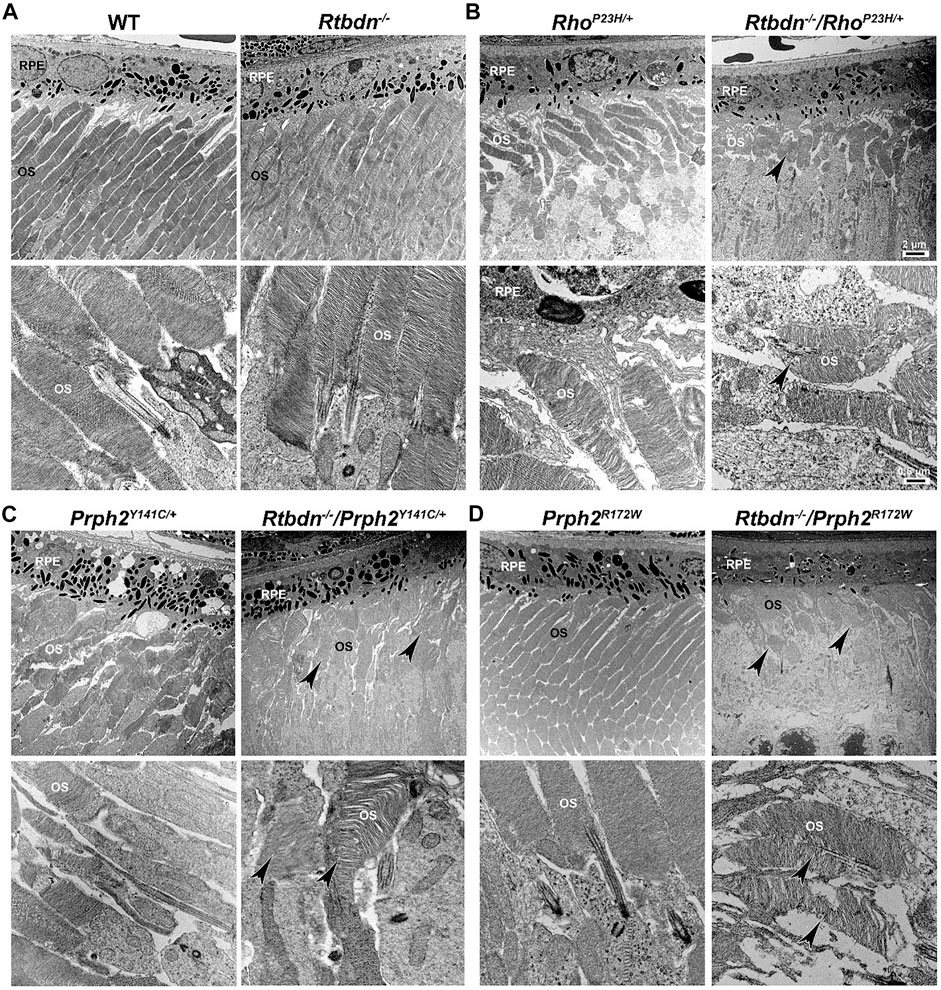
FIGURE 3. Ablation of Rtbdn exacerbated the changes in outer segment ultrastructure in models of retinal degeneration. Effects of Rtbdn ablation at the ultrastructural level in the retinas of disease models. (A–D) Representative TEM images of RPE and OS at two magnifications (5,000× for the upper image and 25,000×for the lower image for each genotype) are presented for the indicated genotypes at P30. Arrowheads point to the shorter and misoriented OSs discs, disorganized and misaligned, or abnormal membrane accumulation when Rtbdn is ablated. Scale bar: 2 µm (A), 0.5 µm (B). RPE, retinal pigment epithelium; OS, outer segment. Images were taken from paraffin-embedded eyes of the relevant genotypes used by Genc et al. (2020a) and Genc et al. (2020b).
The tetraspanin protein Prph2 is a photoreceptor-specific membrane protein that is required for the morphogenesis of both rod and cone OSs rims (Sanyal and Jansen, 1981; Connell et al., 1991; Travis et al., 1991; Stuck et al., 2016). Mutations in the PRPH2 gene are associated with a variety of pathologic conditions, including central areolar choroidal dystrophy, autosomal dominant retinitis pigmentosa, various forms of macular degeneration, and pattern dystrophy mimicking Stargardt/fundus flavimaculatus (Boon et al., 2008; Reeves et al., 2020). Pattern dystrophy (PD) is a dominantly inherited macular disease characterized by lipofuscin buildup in the RPE. Mutations in the PRPH2 gene have been identified to associate with or cause different forms of PD at different times of onset (Francis et al., 2005). It has been hypothesized that mutant PRPH2 leads to PD by disrupting the integrity of the photoreceptor disc membrane, resulting in photoreceptor cell death and accumulation of lipofuscin in the RPE (Francis et al., 2005). One of the mutations in PRPH2 that leads to PD is the Y141C, which is dominantly inherited (Yang et al., 2004). A knockin mouse line for this mutation was generated and showed a retinal phenotype similar to that seen in patients carrying the Y141C mutation (Stuck et al., 2014). Histological assessments of the heterozygous retinas taken from P30 Prph2Y141C/+mice revealed a reduction of about two rows of photoreceptor nuclei (Figure 2C, left image) with significantly shorter OSs (Figure 3C, left images) (Stuck et al., 2014). The Prph2Y141C/+ knock-in mouse model shows impaired rod and cone function, progressive retinal degeneration, and broad diffused yellow flecks in fundus imaging (Stuck et al., 2014).
Similar to RhoP23H/+, Rtbdn levels are significantly increased in Prph2Y141C/+ retinas compared to WT retinas, and ablation of Rtbdn accelerated the degenerative process (Genc et al., 2020b). Prph2Y141C/+/Rtbdn−/− exhibited reduced scotopic a- and photopic b-waves amplitudes and ultrastructural defects (see right images in Figure 2C and Figure 3C) (Genc et al., 2020b). Prph2Y141C/+/Rtbdn−/− OSs were also very short and round and exhibited whorl formation (Figure 3C, right images) (Genc et al., 2020b).
The abovementioned findings indicate that Rtbdn plays a protective role for both rods and cones during photoreceptor degeneration, even though Rtbdn is only expressed by rods (Kelley et al., 2015). Rtbdn is likely to be required for retinal homeostasis because it is involved in the enrichment of retinal flavins by extracellularly binding to RF and potentially increasing the active concentration of RF in the extracellular environment (Genc et al., 2020b).
One of the most common mutations in PRPH2 is at codon 172, where arginine is substituted by tryptophan and is observed in patients diagnosed with either retinitis pigmentosa or macular dystrophy (Wells et al., 1993). This led to the conclusion that the function of certain domains in PRPH2 may be different in cones and rods. A transgenic mouse model for this mutation (Prph2R172W) was generated and showed a retinal phenotype similar to that of the patients (Ding et al., 2004). The R172W animals showed late-onset degeneration that became initially apparent in cones, followed by degeneration of the rods (Ding et al., 2004), consistent with what has been seen in patients affected by this mutation. No significant degeneration is seen in the P30 Prph2R172W retina (Figure 2D, left image) with the normal ultrastructural organization of the discs (Figure 3D, left images).
Genc et al. investigated Rtbdn’s levels in retinal extracts from Prph2R172W mice and reported that Rtbdn levels were significantly upregulated before (at P30) and during (P90) retinal degeneration (Genc et al., 2020a). Again, the absence of Rtbdn (Prph2R172W/Rtbdn−/−) exacerbated functional and structural decline (see right images in Figure 2D and Figure 3D) accompanied by retinal gliosis (Genc et al., 2020a). Overall, the Prph2R172W/Rtbdn−/− mice displayed a significantly more severe phenotype than either Rtbdn−/− or Prph2R172W mice (Genc et al., 2020a).
IRDs are a diverse set of hereditary abnormalities that affect the retina and lead to photoreceptor malfunction and vision loss. Treatments for IRDs are currently unavailable, but new options are rapidly evolving. Advances in vectors (viral and non-viral) and delivery systems to carry therapeutic genes to the retina are very promising. These vectors can deliver the normal gene in cases where gene augmentation is sufficient or neuroprotective genes. Delivery of a specific gene will only provide therapy for those IRDs caused by that mutant gene; however, delivery of a neuroprotective gene allows for a therapy that spans many different IRDs. Therefore, the search for neuroprotective genes is critical for the accomplishment of this goal. We found that the RF-binding protein, Rtbdn, is significantly increased during retinal degeneration, irrespective of the causative mutation or mutant gene. Furthermore, we have shown that ablation of Rtbdn exacerbates the degenerative process. Finally, the addition of Rtbdn-containing membrane preparations to the 661W cells, which are known to be light-sensitive, protected these cells from light-induced cell death. Combined, these data support a protective role for Rtbdn in the retina, making it a promising candidate in the development of a pan-mutant therapy.
Going forward, Rtbdn expressing vectors packaged into non-viral nanoparticles will be used in gene therapy experiments in mouse models of retinal degenerative diseases. Since Rtbdn is extracellular in nature, it will be interesting to investigate whether targeting Rtbdn expression to the RPE is as effective as targeting it to the neural retina. This strategy is motivated by the RPE’s ability to take up therapeutic vectors much more readily than the neural retina. Furthermore, delivery to the RPE can be achieved via suprachoroidal means, which prevents the retinal detachment that occurs with subretinal delivery. We have previously shown that some proteins present in the retina are expressed by the RPE (Kanan et al., 2009). In addition, we believe that Rtbdn is an ideal therapeutic target due to its localization at the OS tips, critical for the interrelated functions of the RPE and retina, and given that many IRDs have secondary RPE and choroidal defects.
To summarize, future investigations on Rtbdn and the mechanism of flavin regulation in the retina are needed to establish how it exerts its neuroprotective effects. However, that should not prevent attempts to determine the protective effects of Rtbdn through gene therapy approaches.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by the National Institute of Health/National Eye Institute (EY010609 and EY018656 MIN and MRA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Larissa Ikelle at the department of Biomedical Engineering at the University of Houston for reading the review and providing helpful comments.
Batey, D. W., and Eckhert, C. D. (1990). Identification of FAD, FMN, and Riboflavin in the Retina by Microextraction and High-Performance Liquid Chromatography. Anal. Biochem. 188, 164–167. doi:10.1016/0003-2697(90)90546-l
Batey, D. W., and Eckhert, C. D. (1991). Analysis of Flavins in Ocular Tissues of the Rabbit. Invest. Ophthalmol. Vis. Sci. 32, 1981–1985.
Batey, D. W., Daneshgar, K. K., and Eckhert, C. D. (1992). Flavin Levels in the Rat Retina. Exp. Eye Res. 54, 605–609. doi:10.1016/0014-4835(92)90139-j
Boon, C. J., den Hollander, A. I., Hoyng, C. B., Cremers, F. P., Klevering, B. J., and Keunen, J. E. (2008). The Spectrum of Retinal Dystrophies Caused by Mutations in the Peripherin/RDS Gene. Prog. Retin Eye Res. 27, 213–235. doi:10.1016/j.preteyeres.2008.01.002
Bro-Rasmussen, F., and Horwitt, M. K. (1967). Riboflavin Requirement of Animal and Man, Related to Protein Requirement or Energy Turnover. Am. J. Clin. Nutr. 20, 507–510. doi:10.1093/ajcn/20.5.507
Cali, E., Dominik, N., Manole, A., and Houlden, H. (1993). “Riboflavin Transporter Deficiency,” in GeneReviews®. Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Beanet al. (Seattle, WA: University of Washington).
Chertov, A. O., Holzhausen, L., Kuok, I. T., Couron, D., Parker, E., Linton, J. D., et al. (2011). Roles of Glucose in Photoreceptor Survival. J. Biol. Chem. 286, 34700–34711. doi:10.1074/jbc.M111.279752
Collin, G. B., Gogna, N., Chang, B., Damkham, N., Pinkney, J., Hyde, L. F., et al. (2020). Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss. Cells 9, 931. doi:10.3390/cells9040931
Conley, S. M., and Naash, M. I. (2009). Focus on Molecules: RDS. Exp. Eye Res. 89, 278–279. doi:10.1016/j.exer.2009.03.023
Conley, S. M., Stuck, M. W., Burnett, J. L., Chakraborty, D., Azadi, S., Fliesler, S. J., et al. (2014). Insights into the Mechanisms of Macular Degeneration Associated with the R172W Mutation in RDS. Hum. Mol. Genet. 23, 3102–3114. doi:10.1093/hmg/ddu014
Connell, G., Bascom, R., Molday, L., Reid, D., McInnes, R. R., and Molday, R. S. (1991). Photoreceptor Peripherin is the Normal Product of the Gene Responsible for Retinal Degeneration in the Rds Mouse. Proc. Natl. Acad. Sci. U. S. A. 88, 723–726. doi:10.1073/pnas.88.3.723
Country, M. W. (2017). Retinal Metabolism: A Comparative Look at Energetics in the Retina. Brain Res. 1672, 50–57. doi:10.1016/j.brainres.2017.07.025
Damsgaard, C., and Country, M. W. (2022). The Opto-Respiratory Compromise: Balancing Oxygen Supply and Light Transmittance in the Retina. Physiol. (Bethesda) 37, 101–113. doi:10.1152/physiol.00027.2021
Ding, X. Q., Nour, M., Ritter, L. M., Goldberg, A. F., Fliesler, S. J., and Naash, M. I. (2004). The R172W Mutation in Peripherin/rds Causes a Cone-Rod Dystrophy in Transgenic Mice. Hum. Mol. Genet. 13, 2075–2087. doi:10.1093/hmg/ddh211
Dryja, T. P., McGee, T. L., Hahn, L. B., Cowley, G. S., Olsson, J. E., Reichel, E., et al. (1990a). Mutations within the Rhodopsin Gene in Patients with Autosomal Dominant Retinitis Pigmentosa. N. Engl. J. Med. 323, 1302–1307. doi:10.1056/NEJM199011083231903
Dryja, T. P., McGee, T. L., Reichel, E., Hahn, L. B., Cowley, G. S., Yandell, D. W., et al. (1990b). A Point Mutation of the Rhodopsin Gene in One Form of Retinitis Pigmentosa. Nature 343, 364–366. doi:10.1038/343364a0
Du, J., Linton, J. D., and Hurley, J. B. (2015). Probing Metabolism in the Intact Retina Using Stable Isotope Tracers. Methods Enzymol. 561, 149–170. doi:10.1016/bs.mie.2015.04.002
Fan, J., Rajapakse, D., Peterson, K., Lerner, J., Parsa, S., Ponduri, A., et al. (2021). Retbindin Mediates Light-Damage in Mouse Retina while its Absence Leads to Premature Retinal Aging. Exp. Eye Res. 209, 108698. doi:10.1016/j.exer.2021.108698
Francis, P. J., Schultz, D. W., Gregory, A. M., Schain, M. B., Barra, R., Majewski, J., et al. (2005). Genetic and Phenotypic Heterogeneity in Pattern Dystrophy. Br. J. Ophthalmol. 89, 1115–1119. doi:10.1136/bjo.2004.062695
Genc, A. M., Makia, M. S., Sinha, T., Conley, S. M., Al-Ubaidi, M. R., and Naash, M. I. (2020a). Elimination of a Retinal Riboflavin Binding Protein Exacerbates Degeneration in a Model of Cone-Rod Dystrophy. Invest. Ophthalmol. Vis. Sci. 61, 17. doi:10.1167/iovs.61.6.17
Genc, A. M., Makia, M. S., Sinha, T., Conley, S. M., Al-Ubaidi, M. R., and Naash, M. I. (2020b). Retbindin: A Riboflavin Binding Protein, is Critical for Photoreceptor Homeostasis and Survival in Models of Retinal Degeneration. Int. J. Mol. Sci. 21, 8083. doi:10.3390/ijms21218083
Griciuc, A., Roux, M. J., Merl, J., Giangrande, A., Hauck, S. M., Aron, L., et al. (2014). Proteomic Survey Reveals Altered Energetic Patterns and Metabolic Failure Prior to Retinal Degeneration. J. Neurosci. 34, 2797–2812. doi:10.1523/JNEUROSCI.2982-13.2014
Harder, J. M., Guymer, C., Wood, J. P. M., Daskalaki, E., Chidlow, G., Zhang, C., et al. (2020). Disturbed Glucose and Pyruvate Metabolism in Glaucoma with Neuroprotection by Pyruvate or Rapamycin. Proc. Natl. Acad. Sci. U. S. A. 117, 33619–33627. doi:10.1073/pnas.2014213117
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis Pigmentosa. Lancet 368, 1795–1809. doi:10.1016/S0140-6736(06)69740-7
Hurley, J. B. (2021). Retina Metabolism and Metabolism in the Pigmented Epithelium: A Busy Intersection. Annu. Rev. Vis. Sci. 7, 665–692. doi:10.1146/annurev-vision-100419-115156
Huvaere, K., Cardoso, D. R., Homem-de-Mello, P., Westermann, S., and Skibsted, L. H. (2010). Light-induced Oxidation of Unsaturated Lipids as Sensitized by Flavins. J. Phys. Chem. B 114, 5583–5593. doi:10.1021/jp9121744
Irinoda, K., and Sato, S. (1954). Contribution to the Ocular Manifestation of Riboflavin Deficiency. Tohoku J. Exp. Med. 61, 93–104. doi:10.1620/tjem.61.93
Joyal, J. S., Sun, Y., Gantner, M. L., Shao, Z., Evans, L. P., Saba, N., et al. (2016). Retinal Lipid and Glucose Metabolism Dictates Angiogenesis through the Lipid Sensor Ffar1. Nat. Med. 22, 439–445. doi:10.1038/nm.4059
Kanan, Y., Hoffhines, A., Rauhauser, A., Murray, A., and Al-Ubaidi, M. R. (2009). Protein Tyrosine-O-Sulfation in the Retina. Exp. Eye Res. 89, 559–567. doi:10.1016/j.exer.2009.05.010
Kaplan, H. J., Wang, W., Piri, N., and Dean, D. C. (2021). Metabolic Rescue of Cone Photoreceptors in Retinitis Pigmentosa. Taiwan J. Ophthalmol. 11, 331–335. doi:10.4103/tjo.tjo_46_21
Kelley, R. A., Al-Ubaidi, M. R., and Naash, M. I. (2015). Retbindin is an Extracellular Riboflavin-Binding Protein Found at the Photoreceptor/retinal Pigment Epithelium Interface. J. Biol. Chem. 290, 5041–5052. doi:10.1074/jbc.M114.624189
Kelley, R. A., Al-Ubaidi, M. R., Sinha, T., Genc, A. M., Makia, M. S., Ikelle, L., et al. (2017). Ablation of the Riboflavin-Binding Protein Retbindin Reduces Flavin Levels and Leads to Progressive and Dose-dependent Degeneration of Rods and Cones. J. Biol. Chem. 292, 21023–21034. doi:10.1074/jbc.M117.785105
Kelley, R. A., Al-Ubaidi, M. R., and Naash, M. I. (2018). Retbindin is Capable of Protecting Photoreceptors from Flavin-Sensitized Light-Mediated Cell Death In Vitro. Adv. Exp. Med. Biol. 1074, 485–490. doi:10.1007/978-3-319-75402-4_60
LaVail, M. M., Nishikawa, S., Steinberg, R. H., Naash, M. I., Duncan, J. L., Trautmann, N., et al. (2018). Phenotypic Characterization of P23H and S334ter Rhodopsin Transgenic Rat Models of Inherited Retinal Degeneration. Exp. Eye Res. 167, 56–90. doi:10.1016/j.exer.2017.10.023
Ly, A., Merl-Pham, J., Priller, M., Gruhn, F., Senninger, N., Ueffing, M., et al. (2016). Proteomic Profiling Suggests Central Role of STAT Signaling during Retinal Degeneration in the Rd10 Mouse Model. J. Proteome Res. 15, 1350–1359. doi:10.1021/acs.jproteome.6b00111
Machida, S., Kondo, M., Jamison, J. A., Khan, N. W., Kononen, L. T., Sugawara, T., et al. (2000). P23H Rhodopsin Transgenic Rat: Correlation of Retinal Function with Histopathology. Invest. Ophthalmol. Vis. Sci. 41, 3200–3209.
Mazur-Bialy, A. I., Buchala, B., and Plytycz, B. (2013). Riboflavin Deprivation Inhibits Macrophage Viability and Activity-A Study on the RAW 264.7 Cell Line. Br. J. Nutr. 110, 509–514. doi:10.1017/S0007114512005351
Moreno, M. L., Mérida, S., Bosch-Morell, F., Miranda, M., and Villar, V. M. (2018). Autophagy Dysfunction and Oxidative Stress, Two Related Mechanisms Implicated in Retinitis Pigmentosa. Front. Physiol. 9, 1008. doi:10.3389/fphys.2018.01008
Mosegaard, S., Dipace, G., Bross, P., Carlsen, J., Gregersen, N., and Olsen, R. K. J. (2020). Riboflavin Deficiency-Implications for General Human Health and Inborn Errors of Metabolism. Int. J. Mol. Sci. 21, 3847. doi:10.3390/ijms21113847
Murenu, E., Kostidis, S., Lahiri, S., Geserich, A. S., Imhof, A., Giera, M., et al. (2021). Metabolic Analysis of Vitreous/Lens and Retina in Wild Type and Retinal Degeneration Mice. Int. J. Mol. Sci. 22, 2345. doi:10.3390/ijms22052345
Naash, M. I., Hollyfield, J. G., al-Ubaidi, M. R., and Baehr, W. (1993). Simulation of Human Autosomal Dominant Retinitis Pigmentosa in Transgenic Mice Expressing a Mutated Murine Opsin Gene. Proc. Natl. Acad. Sci. U. S. A. 90, 5499–5503. doi:10.1073/pnas.90.12.5499
Newton, F., and Megaw, R. (2020). Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes (Basel) 11, 1120. doi:10.3390/genes11101120
Olfat, N., Ashoori, M., and Saedisomeolia, A. (2022). Riboflavin is an Antioxidant: A Review Update. Br. J. Nutr., 1–9. doi:10.1017/S0007114521005031
Olsson, J. E., Gordon, J. W., Pawlyk, B. S., Roof, D., Hayes, A., Molday, R. S., et al. (1992). Transgenic Mice with a Rhodopsin Mutation (Pro23His): A Mouse Model of Autosomal Dominant Retinitis Pigmentosa. Neuron 9, 815–830. doi:10.1016/0896-6273(92)90236-7
Oster, G., Bellin, J. S., and Holmstrom, B. (1962). Photochemistry of Riboflavin. Experientia 18, 249–253. doi:10.1007/BF02148213
Powers, H. J. (2003). Riboflavin (Vitamin B-2) and Health. Am. J. Clin. Nutr. 77, 1352–1360. doi:10.1093/ajcn/77.6.1352
Punzo, C., Xiong, W., and Cepko, C. L. (2012). Loss of Daylight Vision in Retinal Degeneration: Are Oxidative Stress and Metabolic Dysregulation to Blame? J. Biol. Chem. 287, 1642–1648. doi:10.1074/jbc.R111.304428
Querques, G., Forte, R., and Souied, E. H. (2011). Retina and Omega-3. J. Nutr. Metabolism 2011, 748361. doi:10.1155/2011/748361
Reeves, M. J., Goetz, K. E., Guan, B., Ullah, E., Blain, D., Zein, W. M., et al. (2020). Genotype-phenotype Associations in a Large PRPH2-Related Retinopathy Cohort. Hum. Mutat. 41, 1528–1539. doi:10.1002/humu.24065
Ross, J. W., Fernandez de Castro, J. P., Zhao, J., Samuel, M., Walters, E., Rios, C., et al. (2012). Generation of an Inbred Miniature Pig Model of Retinitis Pigmentosa. Invest. Ophthalmol. Vis. Sci. 53, 501–507. doi:10.1167/iovs.11-8784
Sakami, S., Maeda, T., Bereta, G., Okano, K., Golczak, M., Sumaroka, A., et al. (2011). Probing Mechanisms of Photoreceptor Degeneration in a New Mouse Model of the Common Form of Autosomal Dominant Retinitis Pigmentosa Due to P23H Opsin Mutations. J. Biol. Chem. 286, 10551–10567. doi:10.1074/jbc.M110.209759
Sakami, S., Kolesnikov, A. V., Kefalov, V. J., and Palczewski, K. (2014). P23H Opsin Knock-In Mice Reveal a Novel Step in Retinal Rod Disc Morphogenesis. Hum. Mol. Genet. 23, 1723–1741. doi:10.1093/hmg/ddt561
Sanyal, S., and Jansen, H. G. (1981). Absence of Receptor Outer Segments in the Retina of Rds Mutant Mice. Neurosci. Lett. 21, 23–26. doi:10.1016/0304-3940(81)90051-3
Sinha, T., Makia, M., Du, J., Naash, M. I., and Al-Ubaidi, M. R. (2018). Flavin Homeostasis in the Mouse Retina during Aging and Degeneration. J. Nutr. Biochem. 62, 123–133. doi:10.1016/j.jnutbio.2018.09.003
Sinha, T., Du, J., Makia, M. S., Hurley, J. B., Naash, M. I., and Al-Ubaidi, M. R. (2021). Absence of Retbindin Blocks Glycolytic Flux, Disrupts Metabolic Homeostasis, and Leads to Photoreceptor Degeneration. Proc. Natl. Acad. Sci. U. S. A. 118, e2018956118. doi:10.1073/pnas.2018956118
Sohocki, M. M., Daiger, S. P., Bowne, S. J., Rodriquez, J. A., Northrup, H., Heckenlively, J. R., et al. (2001). Prevalence of Mutations Causing Retinitis Pigmentosa and Other Inherited Retinopathies. Hum. Mutat. 17, 42–51. doi:10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K
Sparrow, J. R., Hicks, D., and Hamel, C. P. (2010). The Retinal Pigment Epithelium in Health and Disease. Curr. Mol. Med. 10, 802–823. doi:10.2174/156652410793937813
Stuck, M. W., Conley, S. M., and Naash, M. I. (2014). The Y141C Knockin Mutation in RDS Leads to Complex Phenotypes in the Mouse. Hum. Mol. Genet. 23, 6260–6274. doi:10.1093/hmg/ddu345
Stuck, M. W., Conley, S. M., and Naash, M. I. (2016). PRPH2/RDS and ROM-1: Historical Context, Current Views and Future Considerations. Prog. Retin Eye Res. 52, 47–63. doi:10.1016/j.preteyeres.2015.12.002
Sung, C. H., Davenport, C. M., Hennessey, J. C., Maumenee, I. H., Jacobson, S. G., Heckenlively, J. R., et al. (1991). Rhodopsin Mutations in Autosomal Dominant Retinitis Pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 88, 6481–6485. doi:10.1073/pnas.88.15.6481
Tan, E., Ding, X. Q., Saadi, A., Agarwal, N., Naash, M. I., and Al-Ubaidi, M. R. (2004). Expression of Cone-photoreceptor-specific Antigens in a Cell Line Derived from Retinal Tumors in Transgenic Mice. Invest. Ophthalmol. Vis. Sci. 45, 764–768. doi:10.1167/iovs.03-1114
Tolomeo, M., Nisco, A., Leone, P., and Barile, M. (2020). Development of Novel Experimental Models to Study Flavoproteome Alterations in Human Neuromuscular Diseases: The Effect of Rf Therapy. Int. J. Mol. Sci. 21, 5310. doi:10.3390/ijms21155310
Trachsel-Moncho, L., Benlloch-Navarro, S., Fernández-Carbonell, Á., Ramírez-Lamelas, D. T., Olivar, T., Silvestre, D., et al. (2018). Oxidative Stress and Autophagy-Related Changes during Retinal Degeneration and Development. Cell Death Dis. 9, 812. doi:10.1038/s41419-018-0855-8
Travis, G. H., Sutcliffe, J. G., and Bok, D. (1991). The Retinal Degeneration Slow (Rds) Gene Product is a Photoreceptor Disc Membrane-Associated Glycoprotein. Neuron 6, 61–70. doi:10.1016/0896-6273(91)90122-g
Treadwell, G. E., Cairns, W. L., and Metzler, D. E. (1968). Photochemical Degradation of Flavins. V. Chromatographic Studies of the Products of Photolysis of Riboflavin. J. Chromatogr. 35, 376–388. doi:10.1016/s0021-9673(01)82399-2
Tu, B. P., Ho-Schleyer, S. C., Travers, K. J., and Weissman, J. S. (2000). Biochemical Basis of Oxidative Protein Folding in the Endoplasmic Reticulum. Science 290, 1571–1574. doi:10.1126/science.290.5496.1571
Venkataswamy, G. (1967). Ocular Manifestations of Vitamin B-Complex Deficiency. Br. J. Ophthalmol. 51, 749–754. doi:10.1136/bjo.51.11.749
Verbakel, S. K., van Huet, R. A. C., Boon, C. J. F., den Hollander, A. I., Collin, R. W. J., Klaver, C. C. W., et al. (2018). Non-syndromic Retinitis Pigmentosa. Prog. Retin Eye Res. 66, 157–186. doi:10.1016/j.preteyeres.2018.03.005
Wells, J., Wroblewski, J., Keen, J., Inglehearn, C., Jubb, C., Eckstein, A., et al. (1993). Mutations in the Human Retinal Degeneration Slow (RDS) Gene Can Cause Either Retinitis Pigmentosa or Macular Dystrophy. Nat. Genet. 3, 213–218. doi:10.1038/ng0393-213
Wiegand, R. D., Giusto, N. M., Rapp, L. M., and Anderson, R. E. (1983). Evidence for Rod Outer Segment Lipid Peroxidation Following Constant Illumination of the Rat Retina. Invest. Ophthalmol. Vis. Sci. 24, 1433–1435.
Wistow, G., Bernstein, S. L., Wyatt, M. K., Ray, S., Behal, A., Touchman, J. W., et al. (2002). Expressed Sequence Tag Analysis of Human Retina for the NEIBank Project: Retbindin, an Abundant, Novel Retinal cDNA and Alternative Splicing of Other Retina-Preferred Gene Transcripts. Mol. Vis. 8, 196–204.
Keywords: retbindin, neuroprotection, retinal metabolism, retinal regeneration, riboflavin, flavins
Citation: Zhao X, Tebbe L, Naash MI and Al-Ubaidi MR (2022) The Neuroprotective Role of Retbindin, a Metabolic Regulator in the Neural Retina. Front. Pharmacol. 13:919667. doi: 10.3389/fphar.2022.919667
Received: 22 April 2022; Accepted: 10 June 2022;
Published: 06 July 2022.
Edited by:
Cavit Agca, Sabanci University, TurkeyReviewed by:
Gopalan Gnanaguru, Harvard Medical School, United StatesCopyright © 2022 Zhao, Tebbe, Naash and Al-Ubaidi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muna I. Naash, bW5hYXNoQGNlbnRyYWwudWguZWR1; Muayyad R. Al-Ubaidi, bWFsdWJhaWRAY2VudHJhbC51aC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.