 Yan Ren
Yan Ren Xiuhua Zhong1†
Xiuhua Zhong1† Zhongqi Chen
Zhongqi Chen Xiaoning Zeng
Xiaoning Zeng Yuan Ma
Yuan Ma
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 01 June 2022
Sec. Respiratory Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.916508
This article is part of the Research TopicOxidative Stress Related to Cellular Metabolism in Lung Health and DiseasesView all 10 articles
Switching of airway smooth muscle (ASM) cell phenotype from differentiated-contractile to dedifferentiated-proliferative/synthetic state often occurs in asthmatic subjects with airway dysfunction. Evidence has been provided that chloroquine (an agonist of bitter taste receptors) presented benefits to ASM cell function implicated in asthma. However, the underlying mechanism is unclear. House dust mite (HDM)-sensitized mice were administered with chloroquine or dexamethasone before challenge. BALF and lung tissue were obtained for cell counting, histological analysis or ELISA. Primary cultured ASM cells were stimulated with transforming growth factor (TGF)-β1 or H2O2. Cells and supernatant were collected for the detection of ASM phenotype, ROS level, and proinflammatory cytokine production. In HDM-sensitized mice, chloroquine attenuated airway hyperresponsiveness (AHR), inflammation and remodeling with an inhibition of immunoglobulin E, IL-4/-13, and TGF-β1 in BALF. ASM cell proliferation (PCNA), hypertrophy (α-SMA), and parasecretion (MMP-9 and MMP-13) were strongly suppressed by chloroquine, hinting the rebalance of the heterogeneous ASM populations in asthmatic airway. Our data in vitro indicated that chloroquine markedly restrained maladaptive alteration in ASM phenotype in concert with a remission of ROS. Using H2O2 and PI3K inhibitor (LY294002), we found that the inhibition of oxidative stress level and ROS-AKT signal by chloroquine may serve as a potential mechanism that dedicates to the restoration of the phenotypic imbalance in ASM cells. Overall, the present findings suggested that chloroquine improves asthmatic airway function by controlling ASM cell phenotype shift, sketching a novel profile of chloroquine as a new therapeutic candidate for airway remodeling.
As the major characteristic of asthma (Al-Muhsen et al., 2011), airway remodeling encompasses a range of events such as alterations in airway smooth muscle (ASM), which definitively determine the severity and outcome of the disease (Zhang and Li, 2011). ASM cells retain a remarkable phenotypic plasticity. Insults such as infection, allergens and environmental factors can alter the profile of ASM cells from a differentiated and quiescent “contractile” state to a dedifferentiated and proliferative “synthetic” state (Liu et al., 2015). The “synthetic” ASM cells involve features such as cell hypertrophy, hyperplasia and parasecretion, which contribute to the persistent AHR, irreversible airway obstruction, and exacerbation of inflammation. Hence, a “vicious cycle” occurs. Many strategies have been developed for asthma management, but few proved effective in stopping “the remodeling clock”. Therefore, an urgent need for the development of new therapeutic options targeting airway remodeling has been brought into schedule.
Bitter taste receptors (TAS2Rs) were originally found to be expressed in tongue, responsible for our perception of bitter taste. However, they are also found recently in other tissues including ASM cells which might play a key role in airway remodeling. An upregulation of TAS2Rs was detected in those children with severe asthma. And inhaled bitter tastants helped to decrease the obstruction of asthmatic airway (Deshpande et al., 2010; Pulkkinen et al., 2012). Activation of TAS2Rs by chloroquine and denatonium has been proved to be able to cause the relaxation of ASM (Deshpande et al., 2010; Pulkkinen et al., 2012) and inhibit the secretion of proinflammatory cytokines (Orsmark-Pietras et al., 2013). All these findings suggest TAS2Rs as the potential targets for the management of ASM cell function and open up new avenues for asthma treatment.
Chloroquine (N4-(7-chloroquineolin-4-yl)-1N,N1-diethylpentane-1,4-diamine) (Figure 1A) has been proposed as a classic TAS2Rs agonist in digestive system and respiratory system. It was revealed that chloroquine could lead to relaxation of precontracted airways (Becker et al., 1984; Colacone et al., 1990; Kivity et al., 1990; Grassin-Delyle et al., 2015), as well as inhibition of ASM cell growth and cytokine production (Orsmark-Pietras et al., 2013; Ekoff et al., 2014). Although TAS2Rs have got growing attention in respiratory diseases, the actual effects of chloroquine on ASM cells and the precise mechanism involved in asthma have not been clarified.
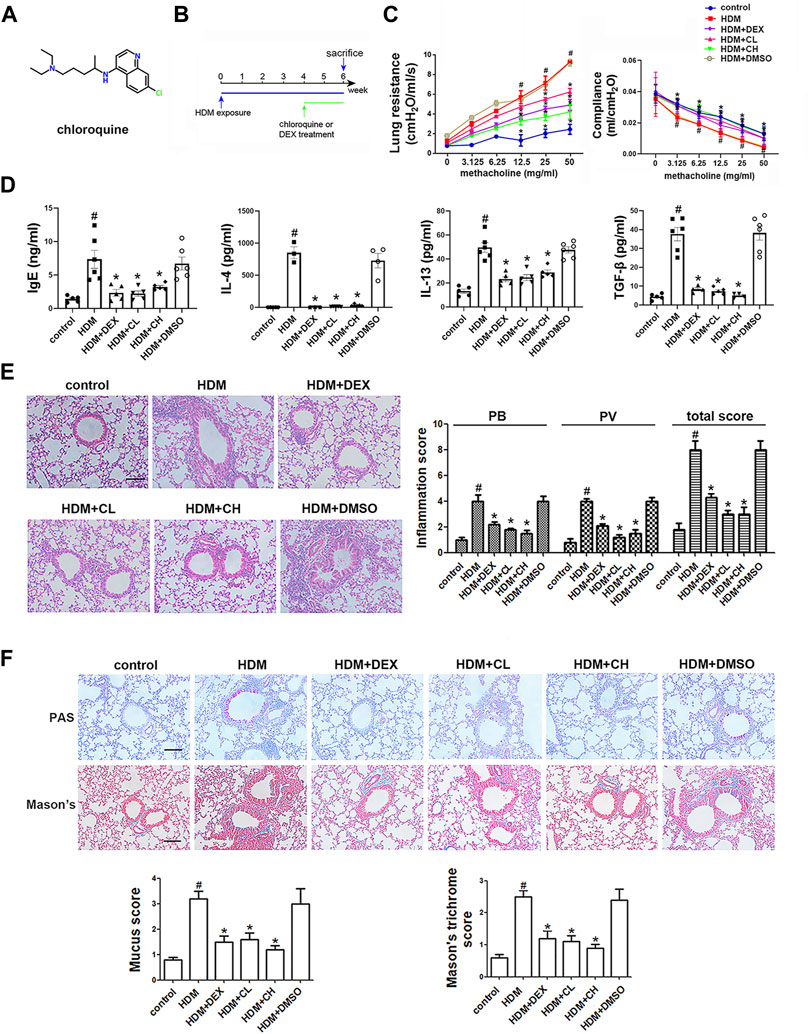
FIGURE 1. Chemical structure of chloroquine and experimental protocol for the chronic asthma model. Treatment with chloroquine alleviated AHR, ameliorated lung dynamic compliance, decreased the levels of IgE, Th2 cytokines, and TGF-β1 in BALF, and inhibited inflammatory cell infiltration, goblet cell hyperplasia, collagen deposition in lung (A) Chemical structure of chloroquine (B) Brief scheme of HDM sensitization and challenge (C) Increasing inhaled doses of methacholine (3.125–50 mg/ml), lung resistance and dynamic compliance in mice (D) The concentrations of HDM-specific IgE, IL-4, IL-13, and TGF-β1 were measured by ELISA (E) Lung sections were stained with H&E to analyze the infiltration of inflammatory cells (magnification, ×400; scale bar, 50 µm). Layers of inflammatory cells were counted, and the total inflammation score was summed with the peribronchial (PB) and perivascular (PV) inflammation scores (F) Lung sections were stained with PAS to assess goblet cell hyperplasia and Masson’s trichrome to evaluate the subepithelial deposition of collagen and fibrosis (magnification, ×400; scale bar, 50 µm). PAS-positive and PAS-negative epithelial cells were counted, and the percentage of PAS-positive cells per bronchiole was calculated. Masson’s trichrome staining analysis of collagen deposition was calculated. Values are the mean ± SEM (n = 6 per group). #p < 0.05 compared with the control group, and *p < 0.05 compared with the HDM group.
In this study, the house dust mite (HDM) model was established to illustrate the impacts of chloroquine during asthma progressing. With an emphasis on the modulation of cellular phenotypes, an in vitro model of human ASM cells stimulated with TGF-β1 was used to elucidate the underlying mechanism involved.
Thirty-six specific pathogen-free female BALB/c mice (18–22 g) aged 6–8 weeks were obtained from the Animal Core Facility of Nanjing Medical University (Nanjing, China). The mice were maintained in a temperature-controlled room under a 12-h dark/12-h light cycle and provided food and water. All experiments involving animal and tissue samples were performed in accordance with the guidelines established by the National Institutes of Health and Nanjing Medical University, and all of the procedures were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (Nanjing, China). The BALB/c mice were randomly divided into the six groups: control group; HDM group, treated with HDM (Greer Laboratories, Lenoir, NC, United States); HDM + CL group, treated with HDM and 1 mg/kg chloroquine (Sigma-Aldrich, St. Louis, MO, United States); HDM + CH group, treated with HDM and 5 mg/kg chloroquine; HDM + dexamethasone group, treated with HDM and 1 mg/kg DEX (Sigma-Aldrich); and HDM + DMSO group, treated with HDM and 0.5% dimethyl sulfoxide (DMSO, Sigma-Aldrich). The mice received intranasal droplets containing the purified HDM extract [25 μg of protein solubilized in 25 μL of phosphate-buffered saline (PBS)] for 5 days/week for up to six consecutive weeks. During the last 2 weeks of the HDM treatment, the mice were intraperitoneally administered chloroquine, DEX or vehicle (DMSO) 1 h prior to the HDM challenge. All treatments were administered under isoflurane anesthesia and ended 24 h before sacrifice. Bronchoalveolar lavage fluid (BALF) and lung tissues were collected for analyses (Figure 1B).
The lung function was evaluated from direct measurements of lung resistance and dynamic compliance in restrained, tracheostomized, mechanically ventilated mice using the FinePointe RC system (Buxco Research Systems, Wilmington, NC, United States) under general anesthesia as described previously (Kerzerho et al., 2013).
The tracheae were exposed, and BALF was collected via lavage with ice-cold PBS (400 μL × 3; 85–90% of the lavage volume was recovered) using a tracheal catheter. The lavage samples from each mouse were centrifuged at 1000 rpm at 4°C for 10 min. The supernatant was collected, divided into four equal portions and frozen at −80°C for enzyme-linked immunosorbent assay (ELISA).
BALF samples were obtained, and the left lung was immersed in 4% paraformaldehyde and then embedded in paraffin. A series of microsections (5 μm) were cut using a microtome and stained with hematoxylin and eosin (H&E) for assessments of inflammatory cell infiltration. An inflammation score was determined as follows: grade 0, no inflammation; grade 1, occasional cuffing with inflammatory cells; and grades 2, 3, and 4, most bronchi or vessels surrounded by a thin layer (1–2 cells), a moderate layer (3–5 cells), or a thick layer (>5 cells) of inflammatory cells, respectively. The total inflammation score was calculated by the addition of the peribronchial (PB) and perivascular (PV) inflammation scores. Periodic acid-Schiff (PAS) staining was used for the quantification of airway goblet cells, and Masson’s trichrome staining was used for the visualization of collagen deposition and fibrosis. Both staining methods were scored as follows: 0, none; 1, <25%; 2, 25–50%; 3, 50–75%; and 4, >75% goblet cells (Ma et al., 2016). The sections were also immunohistochemically stained for matrix metalloproteinase (MMP)-9, MMP-13, proliferating cell nuclear antigen (PCNA) and alpha-smooth muscle actin (α-SMA). For the semiquantitative evaluation of the expression of MMP-9, MMP-13, PCNA and α-SMA, the IOD of the results was analyzed using Image-Pro Plus 6.0 software. The mean percentages of positive epithelial cells in the bronchi were determined in at least five areas at ×400 magnification and assigned to one of the following categories: 0, <5%; 1, 5–25%; 2, 25–50%; 3, 50–75%; and 4, >75% (Ma et al., 2016). The immunostaining intensities of MMP-9, MMP-13, PCNA and α-SMA were scored as 1+ (weak), 2+ (moderate) or 3+ (intense). The percentage of positive epithelial cells and the staining intensities were multiplied to yield a weighted score for each case. Two independent observers who were blinded to the experiment calculated all of the scores, and at least three different fields of each lung section were examined.
The in situ production of ROS in frozen sections was evaluated microscopically using dihydroethidium (DHE) (Beyotime Institute of Biotechnology, Jinan, Shandong, China) (Boldogh et al., 2005). Briefly, snap-frozen lung tissue samples were embedded in Tissue-Tek OCT compound, cryosectioned at 7 mm, allowed to air-dry at room temperature, and stored at -80°C until needed. The slides were placed in PBS for 30 min at room temperature and stained with DHE (10 mm) in PBS for 30 min in a moist chamber in the dark. The slides were rinsed extensively with PBS, covered with a coverslip, and imaged using a fluorescence microscope (Zeiss LSM 5 LIVE, Germany). The DHE fluorescence was quantified by averaging the mean value of the fluorescence intensity using Image-Pro Plus 6.0. Four sections from each mouse were analyzed using this procedure, and the mean value of the fluorescence was calculated.
Normal human ASM cells were purchased from ScienCell Research Laboratories (Carlsbad, CA, United States) and cultured at 37°C in the presence of 5% CO2 in smooth muscle cell medium (ScienCell) supplemented with 20 U/l penicillin, 20 μg/ml streptomycin, 1% smooth muscle cell growth supplement and 2% fetal bovine serum (ScienCell). Cells between passages four and eight were used for the experiments. After serum starvation for 6–8 h (Supplementary Figure S1), the ASM cells were stimulated with 5 ng/ml TGF-β1 (Peprotech) or H2O2 (20 μm, Sigma-Aldrich) alone or in combination with chloroquine (10 μm) or N-acetylcysteine (NAC, 10 mm, Sigma-Aldrich) or LY294002 (10 μm, Sigma-Aldrich). The cells were further cultured for the indicated durations.
LDH assay (Jiancheng, Nanjing, China) was adopted to determine the cytotoxicity of chloroquine in ASM cells. As previously described, ASM cells were planted at a density of 5×103 per well in a 96-well plate and treated with chloroquine for 48 h. Then the supernatant was centrifuged and transferred to be mixed with matrix buffer and coenzyme I application solution. After incubation with 2,4-dinitrophenylhydrazine, 0.4 M NaOH was added into the system to terminate the reaction. The release of LDH from ASM cells was then evaluated by colorimetric assay with a microplate reader (CANY, Shanghai, China) at 450 nm. Each sample was tested in triplicate.
The proliferation of ASM cells was determined using cell counting kits (CCK)-8 and 5-ethynyl-2′-deoxyuridine (EdU) assays. The ASM cells were cultured in a 96-well plate at a density of 5×103 cells per well and treated with TGF-β1 and chloroquine at concentrations from 0.1 to 100 μm for 48 h. The CCK-8 solution (Dojindo Molecular Technologies, Inc. Kumamoto, Japan) was added to the cell culture medium at a dilution of 1:10, and the cultures were incubated for another 1–2 h at 37°C. The absorbance at 450 nm (A450) was measured using a microplate reader (CANY, Shanghai, China). For the EdU assay, the ASM cells were cultured using the aforementioned procedure. After 48 h of treatment, the cells were labelled using an EdU assay kit (Ribobio, Guangzhou, China) according to the manufacturer’s instructions. Images were obtained using a fluorescence microscope (Olympus IX71, Japan). Each sample was measured in triplicate.
ASM cell phenotype was observed using TEM. The ASM cell culture medium was discarded, and the cells were fixed with an electron microscope fixative solution at 4°C for 2 h. The ASM cells were centrifuged at low speed, wrapped with 1% agarose, and rinsed with 0.1 M PBS. The cells were fixed with 1% osmium acid at room temperature for 2 h, rinsed with 0.1 M PBS, and dehydrated using an alcohol and acetone gradient for 15 min. The mixture was subsequently combined with acetone and the embedding agent at a ratio of 1:1, infiltrated for 2–4 h, mixed with acetone and the embedding agent at a ratio of 1:2, infiltrated overnight, treated with the pure embedding agent for 5–8 h, poured into an embedding plate and placed overnight in an oven at 37°C. After penetration, the plate was heated in an oven at 60°C for 48 h for polymerization. Ultrathin slices (60–80 nm) were obtained using a tissue slicer. After double staining with uranium (alcohol solution saturated with 2% uranyl acetate, 15 min) and lead (lead citrate, 15 min), the sections were dried overnight at room temperature and observed and analyzed using TEM.
To examine the effects of chloroquine on airway inflammation and the microenvironment in vitro, the levels of interleukin-8 (IL-8), monocyte chemoattractant protein-1 (MCP-1), soluble intercellular adhesion molecule-1 (ICAM-1), vascular endothelial growth factor (VEGF, R&D Systems, Minneapolis, MN, United States) and isoprostane-8 (IP-8) (Senbeijia Corp. Nanjing, China) were measured. The ASM cells were cultured using the aforementioned procedure. After starvation in serum-free medium, the cells were stimulated with TGF-β1 or H2O2 alone or in combination with chloroquine or NAC for 6–8 h and then subjected to ELISA. The level of HDM-specific immunoglobulin E (IgE, Chondrex, Inc. Redmond, WA, United States) in the serum and the levels of TGF-β1, IL-4, and IL-13 (R&D Systems) in the BALF of the mice were also measured using ELISA according to the manufacturer’s instructions.
The MDA levels in the cell culture medium were determined using the thiobarbituric acid reacting substances (TBARS) assay (Senbeijia Corp. Nanjing, China) as previously described (Ma et al., 2016). MDA reacts with thiobarbituric acid under acidic conditions at 95°C to form a pink-colored complex that can be measured at 532 nm, and 1,3,3-tetra ethoxy propane (TEP) was used as a standard.
The intracellular ROS level was measured using the 2′,7′-dichlorofluorescin diacetate (DCFH-DA, Sigma-Aldrich) assay. Briefly, 1.5×104 ASM cells were seeded into each well of a six-well plate and cultured for 24 h. The cells were then divided into four groups: control group; TGF-β group, treated with 5 ng/ml TGF-β1; TGF-β+C group, treated with 5 ng/ml TGF-β1 and 5 μm chloroquine; and C group, treated with 5 μm chloroquine. The cells were then cultured for an additional 24 h and incubated with 10 μm DCFH-DA for 30 min at 37°C in the dark. The cells were subsequently washed twice with PBS and analyzed within 30 min using a FACScan instrument (Becton Dickinson, San Jose, CA, United States) with an excitation setting of 488 nm. The specific fluorescence signals corresponding to DCFH-DA were determined using a 525-nm bandpass filter.
The cells were homogenized and lysed in RIPA buffer (Sigma-Aldrich) supplemented with a protease inhibitor and a phosphatase inhibitor (Selleck). Equal amounts of proteins were separated using 10% SDS-PAGE. After electrophoresis, the separated proteins were transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, United States) using the wet transfer method. Nonspecific sites were blocked with 5% nonfat milk in TBS Tween 20 [TBST; 25 mm Tris (pH 7.5), 150 mm NaCl, and 0.1% Tween 20] for 2 h, and the blots were incubated with primary antibodies (Cell Signaling Technology, Inc.), including β-actin, anti-phospho-protein kinase B (AKT), anti-AKT, α-SMA, fibronectin and collagen I antibodies overnight at 4°C. Goat anti-rabbit horseradish peroxidase-conjugated IgG (Cell Signaling Technology, Inc.) was used for the detection of antibody binding. The membranes were treated with enhanced chemiluminescence system reagents (Thermo), and the binding of specific antibodies was visualized using a Bio-Rad Gel Doc/ChemiDoc Imaging System and analyzed using Quantity One software.
The data are expressed as the means ± standard errors of the mean (SEM). All of the tests were performed using Prism 6.00 (GraphPad Software, San Diego, CA, United States) and SPSS version 20 (SPSS, Inc. Chicago, IL, United States). To determine the differences between multiple groups, the results were analyzed using one-way analysis of variance for repeated measures followed by Dunnett’s post hoc test. The significance level was set to p < 0.05.
In this study, HDM was used to establish a mouse model of asthma, and chloroquine-mediated alleviation of asthma was examined (Figure 1B; Supplementary Figure S2). Our results showed that chloroquine protected lungs from HDM-induced AHR (as assessed by reduced lung resistance) and preserved lung function (as assessed by increased dynamic compliance) (Figure 1C; Supplementary Figure S3; Supplementary Video S1). A high level of IgE is well established as a feature of asthma. After the sensitization and challenge, HDM-specific IgE level in the BALF was significantly elevated in the HDM and HDM + DMSO groups, and the administration of chloroquine and DEX abolished this increase (Figure 1D). These findings indicated that the asthma model was successful and chloroquine alleviated AHR and HDM-specific IgE level.
Extensive infiltration of inflammatory cells was observed around the respiratory tracts and vessels in the HDM and HDM + DMSO groups (Figure 1E), which indicated that the HDM and HDM + DMSO groups exhibited more severe airway inflammatory responses than the control group. Similarly, airway challenge with HDM notably increased IL-4 and IL-13 levels in the BALF. The administration of chloroquine dose-dependently reduced the IL-4 and IL-13 levels in the BALF compared to the levels in the HDM group (Figure 1D). To evaluate the extent of goblet cell and mucus production, the lung sections were stained with PAS, and the percentage of PAS-positive cells in the airway epithelium was determined. We found that the HDM-challenged mice developed marked goblet cell hyperplasia and mucus hypersecretion in the lumen of the bronchioles (Figure 1F). The mice treated with DEX and high doses of chloroquine had fewer goblet cells in the airway epithelium and reduced mucus scores of 1.5 ± 0.24 and 1.2 ± 0.15 (p < 0.05), respectively. The area of collagen deposition/fibrosis was assessed using Masson’s trichrome staining, and this analysis revealed that the extent of collagen deposition/fibrosis was profoundly enhanced over the interstitia of the airways and vessels of tissues of the HDM-challenged mice compared to the control mice (Figure 1F). The administration of 5 mg/kg chloroquine significantly ameliorated these serious pathophysiological changes, which resulted in a score of 1.1 ± 0.12 (p < 0.05), and modest effects were observed with the administration of DEX and low doses of chloroquine. These findings indicated that chloroquine inhibited airway inflammation, goblet cell proliferation and collagen deposition/fibrosis in the HDM-induced asthma model.
As a phenotypic marker of ASM cells, a-SMA was detected using immunohistochemical techniques. The α-SMA staining densities in the ASM of the HDM-challenged mice were higher than the control mice (p < 0.05). As shown in Figure 2A, the administration of chloroquine markedly decreased the α-SMA-stained smooth muscle layer. Treatment with DEX also decreased the α-SMA staining densities, but these effects were less obvious than the chloroquine effects. The immunohistochemistry assays also showed stronger proliferation-associated protein PCNA, MMP-9 and MMP-13 staining around the bronchioles and in the infiltrated inflammatory cells in the HDM-challenged mice compared to the control mice. The administration of DEX or high doses of chloroquine markedly reversed these increases (Figures 2A,B). To determine whether chloroquine inhibited HDM-induced airway inflammation and remodeling via the scavenging of free radicals, we detected the in situ ROS level in the lung tissue to evaluate the changes resulting from HDM-induced oxidative damage. As shown in Figure 2C, DHE staining was used to assess the in situ ROS level in the lungs. DHE was predominately detected in the airway epithelium in control mice. Challenge with HDM enhanced the fluorescence not only in the inflammatory cells and epithelium, but also ASM cells around the airway, whereas treatment with chloroquine and DEX weakened the fluorescence in those cells of mice. TGF-β1 is a key mediator in asthmatic airway remodeling. After sensitization and challenge, the TGF-β1 level in the BALF was also upregulated in the HDM and HDM + DMSO groups compared to the control group (p < 0.05). Chloroquine and DEX almost reversed this upregulation (Figure 1D). These observations suggested that the protective effect of chloroquine was attributed to the downregulation of TGF-β1.
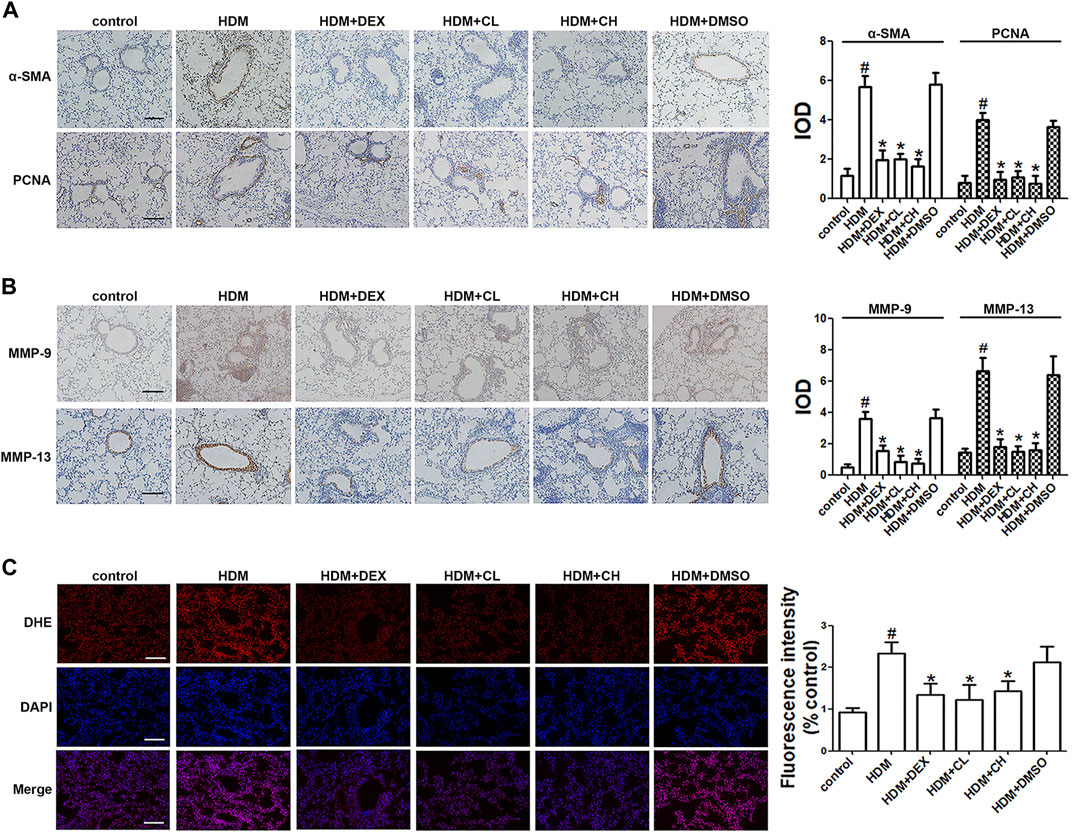
FIGURE 2. Treatment with chloroquine inhibited the expression of α-SMA, PCNA, MMP-9, MMP-13, and in situ ROS levels in lung tissue (A,B) Immunohistochemical staining was performed to assess the distribution of α-SMA, PCNA, MMP-9, and MMP-13 (magnification, ×400; scale bar, 50 µm), and their expression was evaluated, respectively. The staining scores were multiplied by the percentage of positive epithelial cells and staining intensity scores (C) Frozen lung sections were stained with DHE, imaged and analyzed by Image-Pro Plus 6.0. The fluorescence values were expressed as the ratio to the levels in the control cells. Values are the mean ± SEM (n = 6 per group). #p < 0.05 compared with the control group, and *p < 0.05 compared with the HDM group.
The toxicity of chloroquine (1, 2.5, 5, 10, 20, 40, and 80 μm) on human ASM cells as shown in Figure 3A, and the cell viability after 48 h of treatment with 5 μm chloroquine was 97 ± 3% (Figure 3B). Chloroquine decreased the proliferation of ASM cells in vitro in a dose-dependent manner, and the proliferation of human ASM cells was enhanced after TGF-β1 stimulation. The concentration of TGF-β1 (5 ng/ml) induced significant ASM cell proliferation, as determined by the CCK-8 assays, after 48 h of treatment (Figure 3C). Treatment with 5 μm chloroquine markedly decreased TGF-β1-induced ASM cell proliferation from 132 ± 3%–104 ± 2% (p < 0.05) (Figure 3D). Then, we used 5 μm chloroquine for further study and determined whether ROS were involved in TGF-β1-induced proliferation. As shown in Figure 3E, treatment with TGF-β1 activated ROS. A H2DCFDA analysis was performed to evaluate the level of intracellular ROS in ASM cells (Figure 3F). The results suggested that treatment of ASM cells with chloroquine significantly reduced the TGF-β1-induced fluorescence, which reflects the level of oxidative damage. A flow cytometry analysis showed that treatment with chloroquine decreased the intracellular production of ROS from 79 ± 2%–64 ± 4% (p < 0.05) (Figures 3G,H). As shown in Figure 3I, the concentrations of MDA and IP-8 in the TGF-β1-treated group were significantly higher than the control group (p < 0.05), and treatment with chloroquine significantly decreased the increased levels of MDA and IP-8 (p < 0.05).
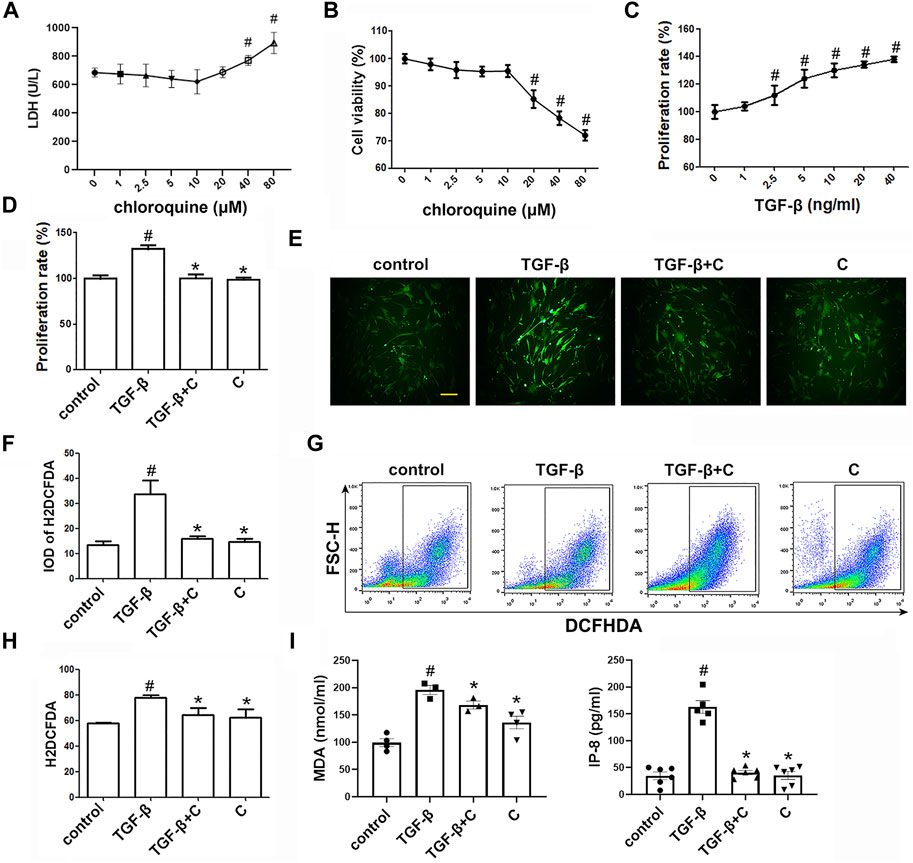
FIGURE 3. Treatment with chloroquine inhibited ASM cell proliferation, ROS generation and MDA/IP-8 production elicited by TGF-β1 (A) Cytotoxicity of chloroquine in human ASM cells (LDH assay) (B) Cell viability with diverse concentrations of chloroquine (CCK-8 assay) (C) Cell proliferation provoked by TGF-β1 (CCK-8 assay) (D) Inhibition of TGF-β1-stimulated cell proliferation by chloroquine (E,F) DCFH-DA fluorescence (green) imaging of ROS with a laser scanning confocal microscope (magnification, ×400; scale bar, 50 µm) (G,H) Fluorescence-activated cell sorting profile of ROS with DCFH-DA by flow cytometry (I) Analysis for MDA and IP-8 content in supernatant. Values are the mean ± SEM of at least four independent experiments performed in triplicate. #p < 0.05 compared with the control, and *p < 0.05 compared with the TGF-β1 group. C, chloroquine (5 μm); TGF-β, TGF-β1 (5 ng/ml).
We used the EdU incorporation assay, which is a more sensitive and specific method (Yu et al., 2009), to evaluate the effects of chloroquine on TGF-β1-induced cell proliferation. As shown in Figure 4A, TGF-β1 stimulation significantly increased the number of cells that incorporated EdU compared to the control, and treatment with chloroquine or NAC (10 mm) clearly limited the TGF-β1-induced proliferation. As previously mentioned, ASM cells tend to proliferative/synthetic phenotype in asthmatic patients, and release chemokines and adhesion molecules to induce inflammatory responses and stimulate eosinophil migration (Zha et al., 2013). To ascertain the anti-inflammatory mechanism of chloroquine, we studied the effects of chloroquine on the TGF-β1-induced upregulation of IL-8, MCP-1, ICAM-1 and VEGF in human ASM cells. The results showed that chloroquine and NAC substantially blocked the TGF-β1-induced upregulation of IL-8, MCP-1, ICAM-1, and VEGF expression in human ASM cells, respectively (Figure 4B). The TGF-β1-mediated phenotype switching of ASM cells was assessed by TEM. The proliferative/synthetic ASM cells appeared flattened with numerous cytoplasmic processes and an elongated oval nucleus containing little or no heterochromatin. The cytoplasm contained mitochondria, highly developed Golgi cisternae and numerous profiles of rough endoplasmic reticulum. The results showed that chloroquine and NAC also reversed TGF-β1-induced proliferative/synthetic ASM cells (Figure 4C). Based on the above results, we conclude that chloroquine rebalances ASM cell phenotype, and the mechanism may be related to the regulation of ROS.
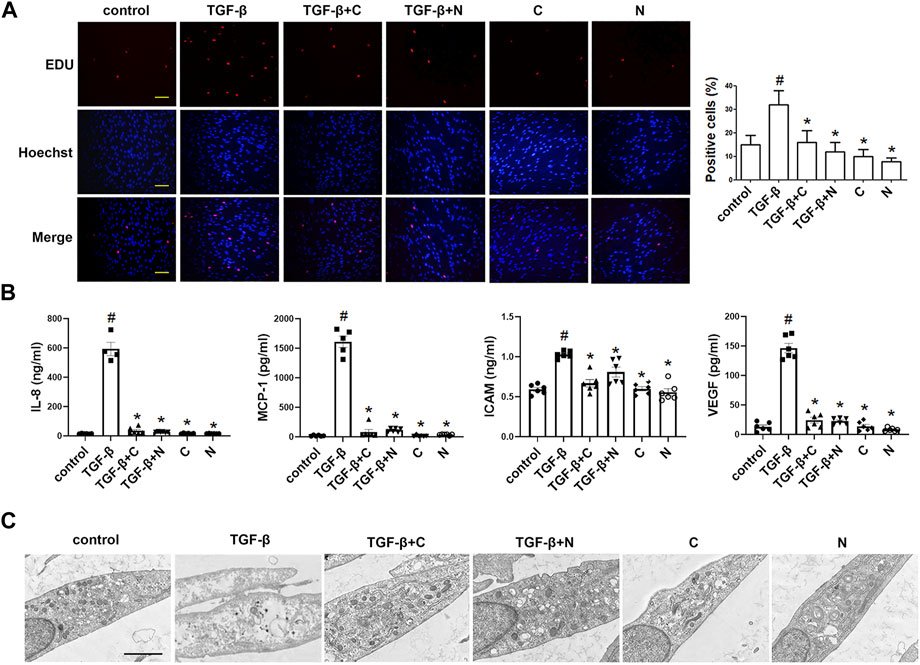
FIGURE 4. Treatment with chloroquine inhibited TGF-β1-induced proliferation, hypersecretion and hypertrophy in ASM cells (A) Effect of chloroquine on TGF-β1-stimulated human ASM cells as assessed by EdU assay (magnification, ×200; scale bar, 100 µm) (B) The IL-8, MCP-1, ICAM-1, and VEGF levels in the supernatant were measured by ELISA (C) Effect of chloroquine on TGF-β1-stimulated human ASM cells as assessed by transmission electron microscope (TEM) (magnification, ×7000; scale bar, 2 µm). Values are the mean ± SEM of at least four independent experiments performed in triplicate. #p < 0.05 compared with the control, and *p < 0.05 compared with the TGF-β1 group. C, chloroquine (5 μm); TGF-β, TGF-β1 (5 ng/ml); N, NAC (10 mm).
Furthermore, we studied whether oxidative stress involves in the ASM cell phenotype switching. As shown in Figures 5A–C, H2O2 (20 μm) stimulation directly increased the number of cells compared to the control, and treatment with chloroquine or NAC clearly limited the H2O2-induced proliferation. Chloroquine and NAC also inhibited H2O2-induced expression of IL-8, MCP-1, ICAM-1, and VEGF in human ASM cells, respectively (Figure 5D). The TEM results showed that the ASM cell phenotype switching induced by H2O2 (20 μm) was almost the same as that induced by TGF-β1 (5 ng/ml), and chloroquine also reversed H2O2-induced proliferative/synthetic ASM cells (Figure 5E). Based on the above results, we conclude that chloroquine rebalances ASM cell phenotype via the ROS pathway.
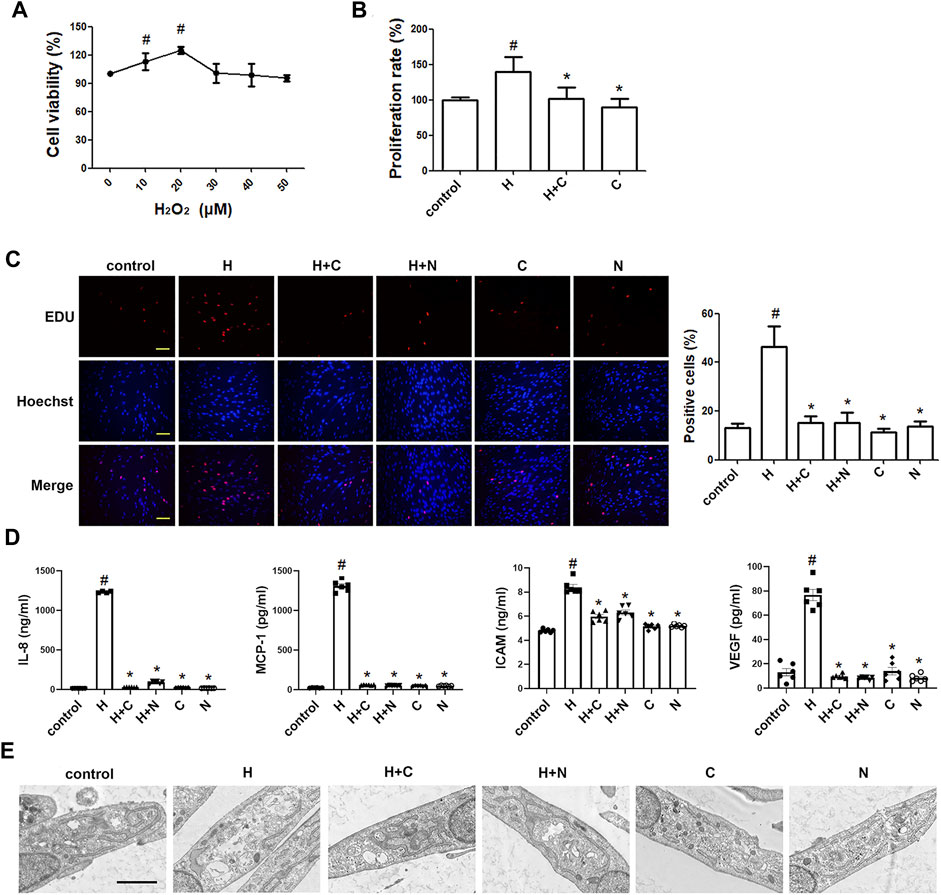
FIGURE 5. Treatment with chloroquine inhibited the H2O2-induced proliferation, hypersecretion and hypertrophy in ASM cells (A) Effect of stimulation with different concentrations of H2O2 on the proliferation of human ASM cells as assessed by CCK-8 assay (B) Effect of chloroquine on H2O2-stimulated human ASM cells as assessed by CCK-8 assay (C) Effect of chloroquine on H2O2-stimulated human ASM cells as assessed by EdU assay (magnification, ×200; scale bar, 100 µm) (D) The IL-8, MCP-1, ICAM-1, and VEGF levels in the supernatant were measured by ELISA (E) Effect of chloroquine on H2O2-stimulated human ASM cells as assessed by TEM (magnification, ×7000; scale bar, 2 µm). Values are the mean ± SEM of at least four independent experiments performed in triplicate. #p < 0.05 compared with the control, and *p < 0.05 compared with the TGF-β1 group. C, chloroquine (5 μm); H, H2O2 (20 μm); N, NAC (10 mm).
To determine the signaling mechanisms underlying the effects of chloroquine on airway remodeling, the phosphorylation status of AKT was investigated. TGF-β1-induced ASM cells were treated with chloroquine and the AKT signal inhibitor LY294002 (Supplementary Figure S4). As shown in Figure 6A, treatment with TGF-β1 for 2 h increased the AKT phosphorylation level, whereas chloroquine and NAC inhibited the TGF-β1-mediated induction of AKT phosphorylation. In addition, chloroquine and LY294002 treatment significantly reversed the effects of TGF-β1 on proliferation- and fibrosis-related indicators (Figure 6B). Based on the above results, we conclude that chloroquine acts as an inhibitor of the AKT signaling pathway, regulates ASM cell hypertrophy, hypersecretion and proliferation.
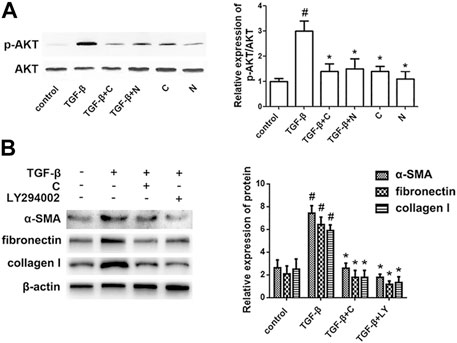
FIGURE 6. Treatment with chloroquine suppressed the activation of the AKT pathway (A) Treatment with chloroquine suppressed the TGF-β1-induced phosphorylation of AKT in ASM cells. The phosphorylation and total protein content of AKT were measured by western blot. The relative density quantification was phosphorylated proteins relative to total proteins (B) The contents of α-SMA, fibronectin and collagen I were measured by western blot. Values are the mean ± SEM of at least four independent experiments performed in triplicate. #p < 0.05 compared with the control, and *p < 0.05 compared with the TGF-β1 group. C, chloroquine (5 μm); TGF-β, TGF-β1 (5 ng/ml); N, NAC (10 mm); LY, LY294002 (10 μm).
Asthmatic airway remodeling entails a wide array of pathophysiologic events such as epithelial damage, mucus gland and goblet cell hyperplasia, subepithelial fibrosis, vascular changes, and increased smooth muscle mass (including hyperplasia, parasecretion and hypertrophy) (Wang J et al., 2018). Several studies demonstrated that patients dying from severe asthma exhibited vigorous increases in the apparent smooth muscle mass within the airway wall (Ebina et al., 1993). ASM cell phenotype switching is deeply implicated in this event and favors persistent AHR and irreversible airway obstruction with a decrease in pulmonary function. ASM cells are found to be skewed towards proliferative/synthetic state in asthma and exhibit characteristics of transcription and division. Organelles associated with the synthetic phenotype predominate in the cytoplasm (Hirst, 1996). ASM cell proliferation and synthesis of cytokines or growth factors make a huge contribution to the imbalance of airway microenvironment and perpetuate the chronicity of asthma (Wang L et al., 2018). Our presented data indicated that TAS2Rs agonist chloroquine exhibited a positive effect on asthmatic mice with a significant improvement in AHR, inflammatory cell infiltration, mucus hypersecretion, collagen deposition and fibrosis. It also reduced IgE and Th2-associated cytokines, which are closely involved in the asthmatic airway microenvironment and remodeling.
As a marker of ASM cells, α-SMA is an essential component of microfilaments which connect to cell membranes (Lin et al., 1989; Colpan et al., 2019). It is pivotal for cell proliferation, hypertrophy, contraction and migration which contribute a lot to the development of airway remodeling and airway hyperresponsiveness (AHR) (Bentley and Hershenson, 2008; Xu et al., 2012; Rao et al., 2014). PCNA is also an indicator of ASM cell proliferation devoted to ASM mass (Kesavan et al., 2013; Cabral-Pacheco et al., 2020). Patients with severe asthma were confirmed to present increased levels of α-SMA and PCNA, associated with higher mortality (Kesavan et al., 2013; Zhao et al., 2013; Cabral-Pacheco et al., 2020). Our results further demonstrated that chloroquine pronouncedly downregulated α-SMA and PCNA expression within asthmatic airway walls, implying a reversion of ASM mass by chloroquine. Further study in vitro even showed that chloroquine suppressed TGF-β1-induced ASM cell proliferation, providing evidence for chloroquine in ASM cell hyperplasia remission in mice.
ASM cell phenotype shifts are often accompanied by a worse parasecretion state. The influx of inflammatory factors within the airways is proposed as a distinct hallmark of asthma (O’Byrne and Postma, 1999). Increased IL-8 level was observed in severe asthma (Tanabe et al., 2014). It promotes ASM cell contraction and migration (Govindaraju et al., 2006), and changes ASM cell features by eliciting the rate of cell proliferation and survival (Halwani et al., 2011). IL-8 can be secreted from ASM cells accompanied by MCP-1, ICAM-1 and VEGF (Lazaar, 2002; Rose et al., 2003; Mukhopadhyay et al., 2014). As a specific endothelial growth factor, VEGF contributes a lot to nonspecific AHR, exerts chemotactic effects on eosinophils, and enhances ASM cell proliferation (Pei et al., 2016). Previous studies showed that Th2-associated cytokines could increase VEGF production which correlated with the severity of asthma (Makinde et al., 2006). All of these suggests deteriorated airway microenvironment as a critical incentive in asthma progressing, facilitating ASM cells to the proliferative/synthetic phenotype, and aggravating a vicious circle of inflammatory response (Lazaar, 2002; Zha et al., 2013; Faiz et al., 2018). In our study, the in vitro findings showed that chloroquine blocked the secretion of these cytokines and adhesion molecules induced by TGF-β1 or H2O2, suggesting a novel option for airway remodeling control by rebalancing ASM cell phenotypes.
Along with this phenotype switching, the microstructure of ASM cells profoundly changed. Proliferative/synthetic ASM cells appear hypertrophy with aberrantly increased cytoplasm, organelles and microfilaments (Jones et al., 2014). Abnormal hypertrophy of ASM cells cause cell dysfunction in contraction, which determines the development and persistence of AHR in asthma (Wang J et al., 2018; Yu et al., 2020). Our TEM results revealed that TGF-β1 or H2O2 induced ASM cells to a proliferative/synthetic state, resulting in numerous cytoplasmic processes with increased profiles of organelles, such as mitochondria, highly developed Golgi cisternae and rough endoplasmic reticulum. Chloroquine markedly reversed the growth of these synthetic organelles, implying an inhibition of the high parasecretion of proliferative/synthetic ASM cells in TGF-β1/H2O2 model with the rebalance of ASM cell phenotypes.
Hyperplasia, parasecretion and hypertrophy are often present in ASM cell phenotype alterations. Other pathophysiologic events such as extracellular matrix (ECM) deposition and fibrosis involved in airway remodeling are also associated with cell phenotype switching. ECM proteins increased in asthmatic airways, particularly collagen I and fibronectin (Roche et al., 1989; Neil et al., 2007), give strong backing to ASM cell survival and proliferation. They have been proved in vitro to enhance the contractile signal (Johnson, 2001; Freyer et al., 2004). In turn, MMPs derived from ASM cells participated actively in ECM deposition and fibrosis (Kesavan et al., 2013; Zhao et al., 2013). Elevated levels of MMP-9 and MMP-13 were found in the serum, sputum and BALF of patients with classic asthma (Greenlee et al., 2007; Mori et al., 2012). MMP-9-deficient animals exhibited reduced airway inflammation, collagen deposition, and peribronchial fibrosis (Halade et al., 2013). Our findings indicated that chloroquine significantly restrained HDM-induced upregulation of MMP-9 and MMP-13, and substantially inhibited the expression of α-SMA, collagen I and fibronectin in ASM cells, sustaining that chloroquine possesses the anti-remodeling potency by controlling ASM cell phenotype. On the other hand, AKT signal inhibitor LY294002 produced the similar effects on the overexpression of α-SMA, collagen I and fibronectin induced by TGF-β1 in vitro, suggesting that chloroquine rebalances ASM cell phenotypes through AKT pathway.
As a serine/threonine kinase, AKT is the key mediator in phosphatidylinositol 3 kinase (PI3K)-initiated signaling closely related to ASM cell proliferation (Xu et al., 2017; Li et al., 2018). Once PI3K is activated, AKT could be phosphorylated (p-AKT) and thought as a flare of PI3K activation (Hu et al., 2019). Evidence from our previous study has suggested that ROS play key roles in AKT signal during TGF-β1 stimulation (Ma et al., 2016). ROS triggered PI3K to amplify the downstream signal, and inactivated phosphatase and tensin homolog (PTEN) which negatively regulates AKT activation (Zhang et al., 2016). It has been reported that ROS upregulated mitochondrial E3 ubiquitin protein ligase 1, induced AKT ubiquitination, and promoted proteasome degradation in head and neck cancer (Bae et al., 2012; Kim et al., 2015; Su et al., 2019). ROS-induced miRNAs modulated AKT phosphorylation to promote cellular senescence in uterine leiomyoma (Xu et al., 2018). Since AKT pathway is critical for the phenotype shifts of ASM cells, the suppression of upstream ROS may have potential implications in asthma therapy.
Our results showed that the level of in situ ROS was increased in the lung tissue obtained from HDM-challenged mice. And chloroquine treatment significantly attenuated the increase and presented a better benefit than DEX. In vitro, our data further demonstrated that chloroquine strongly inhibited the production of intracellular ROS induced by TGF-β1, and reduced the levels of MDA and IP-8. TGF-β1 and increased ROS (H2O2) directly induced ASM cell proliferation involved in airway remodeling. Chloroquine and ROS scavenger NAC notably suppressed AKT phosphorylation during the switching of ASM cell phenotype, highlighting a crucial role of ROS-AKT signaling in the phenotype reversal of ASM cells by chloroquine.
By far, the expression of TAS2Rs has been determined in multiple cell types in airway, including resident (macrophages) and migratory (neutrophils, mast cells, lymphocytes) inflammatory cells, epithelial cells, and ASM cells (Shah et al., 2009; Deshpande et al., 2011; Orsmark-Pietras et al., 2013; Ekoff et al., 2014; Maurer et al., 2015; Tran et al., 2018; Nayak et al., 2019). Activation of TAS2Rs in diverse cells may lead to distinct impacts on cell function (Lee et al., 2012; Sharma et al., 2017). As for ASM cells, a previous study has shown that chloroquine could cause their relaxation via TAS2Rs (Tan and Sanderson, 2014). It also plays a modulatory role in ERK1/2 phosphorylation, which were closely implicated in ASM cell proliferation (Kim et al., 2019). Besides, an antimitogenic potency of TAS2R agonists has been reported in ASM cells by inhibiting the activities of Akt kinase and S6 kinase (Sharma et al., 2016). Then our present work further provided strong evidences that chloroquine prevents ASM from the aberrant proliferation, hypersecretion and hypertrophy by reversing cellular phenotype. These findings may help to offer a potential option for the treatment of asthma, especially for the remission of airway remodeling. By using H2O2 and PI3K inhibitor (LY294002), we further confirmed that the positive effect of chloroquine may rely on the signal of ROS-AKT. The protective properties of chloroquine implicated in asthma should be studied further to determine the value and precise subtype involved in clinical application.
We demonstrated the potential therapeutic action of chloroquine in asthma, revealed its properties and underlying mechanism. Collectively, our findings showed that chloroquine (I) abrogated AHR and attenuated inflammatory cell infiltration, goblet cell hyperplasia and fibrotic expression in the airways (II) decreased the overexpression of α-SMA, PCNA, MMP-9, and MMP-13 in HDM-sensitized mice (III) diminished the elevated levels of IgE, IL-4, IL-13, and TGF-β1 in the BALF (IV) reduced the in situ ROS in the lungs and MDA and IP-8 levels in the BALF (V) reversed TGF-β1 and H2O2-induced proliferative/synthetic ASM cells (proliferation, hypersecretion and hypertrophy); and (VI) suppressed the TGF-β1-induced production of ROS through the inhibition of AKT phosphorylation in human ASM cells. Taken together, our results indicate that chloroquine has the potential to provide beneficial improvements to AHR, airway inflammation and remodeling, and these beneficial effects are most likely due to ROS-AKT pathways. Based on these findings and its recently described bronchodilator and anti-inflammatory properties, chloroquine, as an TAS2R agonist, may be a new therapeutic approach for the treatment of allergic airway diseases.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by the All animal experiments were conducted according to a protocol approved by the Nanjing Medical University (NJMU) Institutional Animal Care and Use Committee (NJMU IACUC-1711002, Nanjing, Jiangsu, China).
YM and XNZ contributed to the design of the study and wrote the manuscript. YR and XHZ were responsible for the cell culture experiments and performed most of the western blot and immunofluorescent staining experiments. HW, YL, and ZC contributed to animal model establishment, sample collection and detection. HW and YL helped to the acquisition and interpretation of the data.
This work was supported by the National Natural Science Foundation of China (NSFC, No. 81700028, No. 81970016, No. 82170031).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Jiangsu Province Hospital Core Facility Center for the vigorous technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.916508/full#supplementary-material
Al-Muhsen, S., Johnson, J. R., and Hamid, Q. (2011). Remodeling in Asthma. J. Allergy Clin. Immunol. 128 (3), 451–454. doi:10.1016/j.jaci.2011.04.047
Bae, S., Kim, S. Y., Jung, J. H., Yoon, Y., Cha, H. J., Lee, H., et al. (2012). Akt Is Negatively Regulated by the MULAN E3 Ligase. Cell Res. 22 (5), 873–885. doi:10.1038/cr.2012.38
Becker, A. B., Simons, K. J., Gillespie, C. A., and Simons, F. E. (1984). The Bronchodilator Effects and Pharmacokinetics of Caffeine in Asthma. N. Engl. J. Med. 310 (12), 743–746. doi:10.1056/NEJM198403223101202
Bentley, J. K., and Hershenson, M. B. (2008). Airway Smooth Muscle Growth in Asthma: Proliferation, Hypertrophy, and Migration. Proc. Am. Thorac. Soc. 5 (1), 89–96. doi:10.1513/pats.200705-063VS
Boldogh, I., Bacsi, A., Choudhury, B. K., Dharajiya, N., Alam, R., Hazra, T. K., et al. (2005). ROS Generated by Pollen NADPH Oxidase Provide a Signal that Augments Antigen-Induced Allergic Airway Inflammation. J. Clin. Invest. 115 (8), 2169–2179. doi:10.1172/JCI24422
Cabral-Pacheco, G. A., Garza-Veloz, I., Castruita-De la Rosa, C., Ramirez-Acuña, J. M., Perez-Romero, B. A., Guerrero-Rodriguez, J. F., et al. (2020). The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Ijms 21 (24), 9739. doi:10.3390/ijms21249739
Colacone, A., Bertolo, L., Wolkove, N., Cohen, C., and Kreisman, H. (1990). Effect of Caffeine on Histamine Bronchoprovocation in Asthma. Thorax 45 (8), 630–632. doi:10.1136/thx.45.8.630
Deshpande, D. A., Robinett, K. S., Wang, W. C., Sham, J. S., An, S. S., and Liggett, S. B. (2011). Bronchodilator Activity of Bitter Tastants in Human Tissue. Nat. Med. 17, 776–778. doi:10.1038/nm0711-776b
Deshpande, D. A., Wang, W. C., McIlmoyle, E. L., Robinett, K. S., Schillinger, R. M., An, S. S., et al. (2010). Bitter Taste Receptors on Airway Smooth Muscle Bronchodilate by Localized Calcium Signaling and Reverse Obstruction. Nat. Med. 16 (11), 1299–1304. doi:10.1038/nm.2237
Ebina, M., Takahashi, T., Chiba, T., and Motomiya, M. (1993). Cellular Hypertrophy and Hyperplasia of Airway Smooth Muscles Underlying Bronchial Asthma. A 3-D Morphometric Study. Am. Rev. Respir. Dis. 148 (3), 720–726. doi:10.1164/ajrccm/148.3.720
Ekoff, M., Choi, J. H., James, A., Dahlén, B., Nilsson, G., and Dahlén, S. E. (2014). Bitter Taste Receptor (TAS2R) Agonists Inhibit IgE-dependent Mast Cell Activation. J. Allergy Clin. Immunol. 134 (2), 475–478. doi:10.1016/j.jaci.2014.02.029
Faiz, A., Weckmann, M., Tasena, H., Vermeulen, C. J., Van den Berge, M., Ten Hacken, N. H. T., et al. (2018). Profiling of Healthy and Asthmatic Airway Smooth Muscle Cells Following Interleukin-1β Treatment: a Novel Role for CCL20 in Chronic Mucus Hypersecretion. Eur. Respir. J. 52 (2), 1800310. doi:10.1183/13993003.00310-2018
Freyer, A. M., Billington, C. K., Penn, R. B., and Hall, I. P. (2004). Extracellular Matrix Modulates Beta2-Adrenergic Receptor Signaling in Human Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 31 (4), 440–445. doi:10.1165/rcmb.2003-0241OC
Govindaraju, V., Michoud, M. C., Al-Chalabi, M., Ferraro, P., Powell, W. S., and Martin, J. G. (2006). Interleukin-8: Novel Roles in Human Airway Smooth Muscle Cell Contraction and Migration. Am. J. Physiol. Cell Physiol. 291 (5), C957–C965. doi:10.1152/ajpcell.00451.2005
Grassin-Delyle, S., Naline, E., and Devillier, P. (2015). Taste Receptors in Asthma. Curr. Opin. Allergy Clin. Immunol. 15 (1), 63–69. doi:10.1097/ACI.0000000000000137
Greenlee, K. J., Werb, Z., and Kheradmand, F. (2007). Matrix Metalloproteinases in Lung: Multiple, Multifarious, and Multifaceted. Physiol. Rev. 87 (1), 69–98. doi:10.1152/physrev.00022.2006
Halade, G. V., Jin, Y. F., and Lindsey, M. L. (2013). Matrix Metalloproteinase (MMP)-9: a Proximal Biomarker for Cardiac Remodeling and a Distal Biomarker for Inflammation. Pharmacol. Ther. 139 (1), 32–40. doi:10.1016/j.pharmthera.2013.03.009
Halwani, R., Al-Abri, J., Beland, M., Al-Jahdali, H., Halayko, A. J., Lee, T. H., et al. (2011). CC and CXC Chemokines Induce Airway Smooth Muscle Proliferation and Survival. J. Immunol. 186 (7), 4156–4163. doi:10.4049/jimmunol.1001210
Henderson, N., Markwick, L. J., Elshaw, S. R., Freyer, A. M., Knox, A. J., and Johnson, S. R. (2007). Collagen I and Thrombin Activate MMP-2 by MMP-14-dependent and -independent Pathways: Implications for Airway Smooth Muscle Migration. Am. J. Physiology-Lung Cell. Mol. Physiology 292 (4), L1030–L1038. doi:10.1152/ajplung.00317.2006
Hirst, S. J. (1996). Airway Smooth Muscle Cell Culture: Application to Studies of Airway Wall Remodelling and Phenotype Plasticity in Asthma. Eur. Respir. J. 9 (4), 808–820. doi:10.1183/09031936.96.09040808
Hu, M., Liu, Y., He, L., Yuan, X., Peng, W., and Wu, C. (2019). Antiepileptic Effects of Protein-Rich Extract from Bombyx Batryticatus on Mice and its Protective Effects against H2O2-Induced Oxidative Damage in PC12 Cells via Regulating PI3K/Akt Signaling Pathways. Oxid. Med. Cell Longev. 2019, 7897584. doi:10.1155/2019/7897584
Johnson, P. R. (2001). Role of Human Airway Smooth Muscle in Altered Extracellular Matrix Production in Asthma. Clin. Exp. Pharmacol. Physiol. 28 (3), 233–236. doi:10.1046/j.1440-1681.2001.03426.x
Jones, R. L., Elliot, J. G., and James, A. L. (2014). Estimating Airway Smooth Muscle Cell Volume and Number in Airway Sections. Sources of Variability. Am. J. Respir. Cell Mol. Biol. 50 (2), 246–252. doi:10.1165/rcmb.2013-0029OC
Kepser, L.-J., Damar, F., De Cicco, T., Chaponnier, C., Prószyński, T. J., Pagenstecher, A., et al. (2019). CAP2 Deficiency Delays Myofibril Actin Cytoskeleton Differentiation and Disturbs Skeletal Muscle Architecture and Function. Proc. Natl. Acad. Sci. U.S.A. 116 (17), 8397–8402. doi:10.1073/pnas.1813351116
Kerzerho, J., Maazi, H., Speak, A. O., Szely, N., Lombardi, V., Khoo, B., et al. (2013). Programmed Cell Death Ligand 2 Regulates TH9 Differentiation and Induction of Chronic Airway Hyperreactivity. J. Allergy Clin. Immunol. 131 (4), 1048–1057. doi:10.1016/j.jaci.2012.09.027
Kesavan, R., Potunuru, U. R., Nastasijević, B., T, A., Joksić, G., and Dixit, M. (2013). Inhibition of Vascular Smooth Muscle Cell Proliferation by gentiana Lutea Root Extracts. PLoS One 8 (4), e61393. doi:10.1371/journal.pone.0061393
Kim, D., Cho, S., Castaño, M. A., Panettieri, R. A., Woo, J. A., and Liggett, S. B. (2019). Biased TAS2R Bronchodilators Inhibit Airway Smooth Muscle Growth by Downregulating Phosphorylated Extracellular Signal-Regulated Kinase 1/2. Am. J. Respir. Cell Mol. Biol. 60 (5), 532–540. doi:10.1165/rcmb.2018-0189OC
Kim, S. Y., Kim, H. J., Kang, S. U., Kim, Y. E., Park, J. K., Shin, Y. S., et al. (2015). Non-thermal Plasma Induces AKT Degradation through Turn-On the MUL1 E3 Ligase in Head and Neck Cancer. Oncotarget 6 (32), 33382–33396. doi:10.18632/oncotarget.5407
Kivity, S., Ben Aharon, Y., Man, A., and Topilsky, M. (1990). The Effect of Caffeine on Exercise-Induced Bronchoconstriction. Chest 97 (5), 1083–1085. doi:10.1378/chest.97.5.1083
Lazaar, A. L. (2002). Airway Smooth Muscle: New Targets for Asthma Pharmacotherapy. Expert. Opin. Ther. Targets 6 (4), 447–459. doi:10.1517/14728222.6.4.447
Lee, R. J., Xiong, G., Kofonow, J. M., Chen, B., Lysenko, A., Jiang, P., et al. (2012). T2R38 Taste Receptor Polymorphisms Underlie Susceptibility to Upper Respiratory Infection. J. Clin. Invest. 122, 4145–4159. doi:10.1172/JCI64240
Li, H., Tang, Z., Chu, P., Song, Y., Yang, Y., Sun, B., et al. (2018). Neuroprotective Effect of Phosphocreatine on Oxidative Stress and Mitochondrial Dysfunction Induced Apoptosis In Vitro and In Vivo: Involvement of Dual PI3K/Akt and Nrf2/HO-1 Pathways. Free Radic. Biol. Med. 120, 228–238. doi:10.1016/j.freeradbiomed.2018.03.014
Lin, Z. X., Eshleman, J., Grund, C., Fischman, D. A., Masaki, T., Franke, W. W., et al. (1989). Differential Response of Myofibrillar and Cytoskeletal Proteins in Cells Treated with Phorbol Myristate Acetate. J. Cell Biol. 108 (3), 1079–1091. doi:10.1083/jcb.108.3.1079
Liu, Y. N., Zha, W. J., Ma, Y., Chen, F. F., Zhu, W., Ge, A., et al. (2015). Galangin Attenuates Airway Remodelling by Inhibiting TGF-Β1-Mediated ROS Generation and MAPK/Akt Phosphorylation in Asthma. Sci. Rep. 5, 11758. doi:10.1038/srep11758
Ma, Y., Zhang, J. X., Liu, Y. N., Ge, A., Gu, H., Zha, W. J., et al. (2016). Corrigendum to 'Caffeic Acid Phenethyl Ester Alleviates Asthma by Regulating the Airway Microenvironment via the ROS-Responsive MAPK/Akt Pathway' [Free Radic. Biol. Med. 101C (2016) 163-175]. Free Radic. Biol. Med. 101, 534–175. doi:10.1016/j.freeradbiomed.2016.09.01210.1016/j.freeradbiomed.2016.10.502
Makinde, T., Murphy, R. F., and Agrawal, D. K. (2006). Immunomodulatory Role of Vascular Endothelial Growth Factor and Angiopoietin-1 in Airway Remodeling. Curr. Mol. Med. 6 (8), 831–841. doi:10.2174/156652406779010795
Maurer, S., Wabnitz, G. H., Kahle, N. A., Stegmaier, S., Prior, B., Giese, T., et al. (2015). Tasting Pseudomonas aeruginosa Biofilms: Human Neutrophils Express the Bitter Receptor T2R38 as Sensor for the Quorum Sensing Molecule N-(3-Oxododecanoyl)-l-homoserine Lactone. Front. Immunol. 6, 369. doi:10.3389/fimmu.2015.00369
Mori, S., Pawankar, R., Ozu, C., Nonaka, M., Yagi, T., and Okubo, K. (2012). Expression and Roles of MMP-2, MMP-9, MMP-13, TIMP-1, and TIMP-2 in Allergic Nasal Mucosa. Allergy Asthma Immunol. Res. 4 (4), 231–239. doi:10.4168/aair.2012.4.4.231
Mukhopadhyay, S., Malik, P., Arora, S. K., and Mukherjee, T. K. (2014). Intercellular Adhesion Molecule-1 as a Drug Target in Asthma and Rhinitis. Respirology 19 (4), 508–513. doi:10.1111/resp.12285
Nayak, A. P., Shah, S. D., Michael, J. V., and Deshpande, D. A. (2019). Bitter Taste Receptors for Asthma Therapeutics. Front. Physiol. 10, 884. doi:10.3389/fphys.2019.00884
O’Byrne, P. M., and Postma, D. S. (1999). The Many Faces of Airway Inflammation: Asthma and Chronic Obstructive Pulmonary Disease. Asthma Research Group. Am. J. Respir. Crit. Care Med. 159 (5 Pt 2), S41–S63.
Orsmark-Pietras, C., James, A., Konradsen, J. R., Nordlund, B., Söderhäll, C., Pulkkinen, V., et al. (2013). Transcriptome Analysis Reveals Upregulation of Bitter Taste Receptors in Severe Asthmatics. Eur. Respir. J. 42 (1), 65–78. doi:10.1183/09031936.00077712
Pei, Q. M., Jiang, P., Yang, M., Qian, X. J., Liu, J. B., and Kim, S. H. (2016). Roxithromycin Inhibits VEGF-Induced Human Airway Smooth Muscle Cell Proliferation: Opportunities for the Treatment of Asthma. Exp. Cell Res. 347 (2), 378–384. doi:10.1016/j.yexcr.2016.08.024
Pulkkinen, V., Manson, M. L., Säfholm, J., Adner, M., and Dahlén, S. E. (2012). The Bitter Taste Receptor (TAS2R) Agonists Denatonium and Chloroquine Display Distinct Patterns of Relaxation of the guinea Pig Trachea. Am. J. Physiol. Lung Cell Mol. Physiol. 303 (11), L956–L966. doi:10.1152/ajplung.00205.2012
Rao K, B., Malathi, N., Narashiman, S., and Rajan, S. T. (2014). Evaluation of Myofibroblasts by Expression of Alpha Smooth Muscle Actin: a Marker in Fibrosis, Dysplasia and Carcinoma. J. Clin. Diagn. Res. 8 (4), ZC14–7. doi:10.7860/JCDR/2014/7820.4231
Roche, W. R., Beasley, R., Williams, J. H., and Holgate, S. T. (1989). Subepithelial Fibrosis in the Bronchi of Asthmatics. Lancet 1 (8637), 520–524. doi:10.1016/s0140-6736(89)90067-6
Rose, C. E., Sung, S. S., and Fu, S. M. (2003). Significant Involvement of CCL2 (MCP-1) in Inflammatory Disorders of the Lung. Microcirculation 10 (3-4), 273–288. doi:10.1038/sj.mn.7800193
Shah, A. S., Ben-Shahar, Y., Moninger, T. O., Kline, J. N., and Welsh, M. J. (2009). Motile Cilia of Human Airway Epithelia Are Chemosensory. Science 325, 1131–1134. doi:10.1126/science.1173869
Sharma, P., Panebra, A., Pera, T., Tiegs, B. C., Hershfeld, A., Kenyon, L. C., et al. (2016). Antimitogenic Effect of Bitter Taste Receptor Agonists on Airway Smooth Muscle Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 310 (4), L365–L376. doi:10.1152/ajplung.00373.2015
Sharma, P., Yi, R., Nayak, A. P., Wang, N., Tang, F., Knight, M. J., et al. (2017). Bitter Taste Receptor Agonists Mitigate Features of Allergic Asthma in Mice. Sci. Rep. 7, 46166. doi:10.1038/srep46166
Su, X., Shen, Z., Yang, Q., Sui, F., Pu, J., Ma, J., et al. (2019). Vitamin C Kills Thyroid Cancer Cells through ROS-dependent Inhibition of MAPK/ERK and PI3K/AKT Pathways via Distinct Mechanisms. Theranostics 9 (15), 4461–4473. doi:10.7150/thno.35219
Tan, X., and Sanderson, M. J. (2014). Bitter Tasting Compounds Dilate Airways by Inhibiting Airway Smooth Muscle Calcium Oscillations and Calcium Sensitivity. Br. J. Pharmacol. 171 (3), 646–662. doi:10.1111/bph.12460
Tanabe, T., Shimokawaji, T., Kanoh, S., and Rubin, B. K. (2014). IL-33 Stimulates CXCL8/IL-8 Secretion in Goblet Cells but Not Normally Differentiated Airway Cells. Clin. Exp. Allergy 44 (4), 540–552. doi:10.1111/cea.12283
Tran, H. T. T., Herz, C., Ruf, P., Stetter, R., and Lamy, E. (2018). Human T2R38 Bitter Taste Receptor Expression in Resting and Activated Lymphocytes. Front. Immunol. 9, 2949. doi:10.3389/fimmu.2018.02949
Wang, J., Shang, Y. X., Cai, X. X., and Liu, L. Y. (2018). Vasoactive Intestinal Peptide Inhibits Airway Smooth Muscle Cell Proliferation in a Mouse Model of Asthma via the ERK1/2 Signaling Pathway. Exp. Cell Res. 364 (2), 168–174. doi:10.1016/j.yexcr.2018.01.042
Wang, L., Feng, X., Hu, B., Xia, Q., Ni, X., and Song, Y. (2018). P2X4R Promotes Airway Remodeling by Acting on the Phenotype Switching of Bronchial Smooth Muscle Cells in Rats. Purinergic Signal 14 (4), 433–442. doi:10.1007/s11302-018-9625-4
Xu, G. N., Yang, K., Xu, Z. P., Zhu, L., Hou, L. N., Qi, H., et al. (2012). Protective Effects of Anisodamine on Cigarette Smoke Extract-Induced Airway Smooth Muscle Cell Proliferation and Tracheal Contractility. Toxicol. Appl. Pharmacol. 262 (1), 70–79. doi:10.1016/j.taap.2012.04.020
Xu, J., Gan, S., Li, J., Wand, D. B., Chen, Y., Hu, X., et al. (2017). Garcinia Xanthochymus Extract Protects PC12 Cells from H2O2-Induced Apoptosis through Modulation of PI3K/AKT and NRF2/HO-1 Pathways. Chin. J. Nat. Med. 15 (11), 825–833. doi:10.1016/S1875-5364(18)30016-5
Xu, X., Kim, J. J., Li, Y., Xie, J., Shao, C., and Wei, J. J. (2018). Oxidative Stress-Induced miRNAs Modulate AKT Signaling and Promote Cellular Senescence in Uterine Leiomyoma. J. Mol. Med. Berl. 96 (10), 1095–1106. doi:10.1007/s00109-018-1682-1
Yu, Q., Yu, X., Zhong, X., Ma, Y., Wu, Y., Bian, T., et al. (2020). Melatonin Modulates Airway Smooth Muscle Cell Phenotype by Targeting the STAT3/Akt/GSK-3β Pathway in Experimental Asthma. Cell Tissue Res. 380 (1), 129–142. doi:10.1007/s00441-019-03148-x
Yu, Y., Arora, A., Min, W., Roifman, C. M., and Grunebaum, E. (2009). EdU Incorporation Is an Alternative Non-radioactive Assay to [(3)H]thymidine Uptake for In Vitro Measurement of Mice T-Cell Proliferations. J. Immunol. Methods 350 (1-2), 29–35. doi:10.1016/j.jim.2009.07.008
Zha, W. J., Qian, Y., Shen, Y., Du, Q., Chen, F. F., Wu, Z. Z., et al. (2013). Galangin Abrogates Ovalbumin-Induced Airway Inflammation via Negative Regulation of NF-Κb. Evid. Based Complement. Altern. Med. 2013, 767689. doi:10.1155/2013/767689
Zhang, J., Wang, X., Vikash, V., Ye, Q., Wu, D., Liu, Y., et al. (2016). ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 4350965. doi:10.1155/2016/4350965
Zhang, W. X., and Li, C. C. (2011). Airway Remodeling: a Potential Therapeutic Target in Asthma. World J. Pediatr. 7 (2), 124–128. doi:10.1007/s12519-011-0264-x
Keywords: chloroquine, airway smooth muscle (ASM) cells, bitter taste receptors (TAS2Rs), TGF-β1, asthma
Citation: Ren Y, Zhong X, Wang H, Chen Z, Liu Y, Zeng X and Ma Y (2022) Chloroquine Attenuates Asthma Development by Restoring Airway Smooth Muscle Cell Phenotype Via the ROS-AKT Pathway. Front. Pharmacol. 13:916508. doi: 10.3389/fphar.2022.916508
Received: 09 April 2022; Accepted: 18 May 2022;
Published: 01 June 2022.
Edited by:
Arunachalam Karuppusamy, Chinese Academy of Sciences (CAS), ChinaCopyright © 2022 Ren, Zhong, Wang, Chen, Liu, Zeng and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoning Zeng, emVuZ194aWFvX25pbmdAaG90bWFpbC5jb20=; Yuan Ma, bWF5dWFuQG5qbXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.