 Hai-Ying Shen1*
Hai-Ying Shen1* Sadie B. Baer1Raey Gesese1
Sadie B. Baer1Raey Gesese1 John M. Cook1Landen Weltha1Shayla Q. Coffman1Jie Wu2
John M. Cook1Landen Weltha1Shayla Q. Coffman1Jie Wu2 Jiang-Fan Chen3
Jiang-Fan Chen3 Ming Gao2Teng Ji4
Ming Gao2Teng Ji4- 1Department of Neuroscience, Legacy Research Institute, Portland, OR, United States
- 2Department of Neurobiology, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, AZ, United States
- 3Molecular Neuropharmacology Laboratory, School of Optometry and Ophthalmology and Eye Hospital, Wenzhou Medical University, Wenzhou, China
- 4Department of Pediatric Neurology, Randall Children’s Hospital, Legacy Emanuel Medical Center, Portland, OR, United States
Adenosinergic activities are suggested to participate in SUDEP pathophysiology; this study aimed to evaluate the adenosine hypothesis of SUDEP and specifically the role of adenosine A2A receptor (A2AR) in the development of a SUDEP mouse model with relevant clinical features. Using a combined paradigm of intrahippocampal and intraperitoneal administration of kainic acid (KA), we developed a boosted-KA model of SUDEP in genetically modified adenosine kinase (ADK) knockdown (Adk+/-) mice, which has reduced ADK in the brain. Seizure activity was monitored using video-EEG methods, and in vivo recording of local field potential (LFP) was used to evaluate neuronal activity within the nucleus tractus solitarius (NTS). Our boosted-KA model of SUDEP was characterized by a delayed, postictal sudden death in epileptic mice. We demonstrated a higher incidence of SUDEP in Adk+/- mice (34.8%) vs. WTs (8.0%), and the ADK inhibitor, 5-Iodotubercidin, further increased SUDEP in Adk+/- mice (46.7%). We revealed that the NTS level of ADK was significantly increased in epileptic WTs, but not in epileptic Adk+/- mutants, while the A2AR level in NTS was increased in epileptic (WT and Adk+/-) mice vs. non-epileptic controls. The A2AR antagonist, SCH58261, significantly reduced SUDEP events in Adk+/- mice. LFP data showed that SCH58261 partially restored KA injection-induced suppression of gamma oscillation in the NTS of epileptic WT mice, whereas SCH58261 increased theta and beta oscillations in Adk+/- mutants after KA injection, albeit with no change in gamma oscillations. These LFP findings suggest that SCH58261 and KA induced changes in local neuronal activities in the NTS of epileptic mice. We revealed a crucial role for NTS A2AR in SUDEP pathophysiology suggesting A2AR as a potential therapeutic target for SUDEP risk prevention.
Introduction
SUDEP is the leading cause of death in individuals with epilepsy, and as yet, no pharmacological intervention is available (Surges et al., 2009; Devinsky et al., 2016; Maguire et al., 2016). Although the mechanisms underlying SUDEP remain elusive, brainstem-related central apnea and cardiac arrest are considered two characteristic hallmarks (Stöllberger and Finsterer, 2004; Hirsch, 2010; Shorvon and Tomson, 2011). Animal models of SUDEP currently remain limited (Devinsky et al., 2016; Pansani et al., 2016; Li and Buchanan, 2019) in representing key features of clinical SUDEP cases, (i.e., the chronic nature of epilepsy and the preceding convulsive seizures), which in turn, impedes efforts in investigating SUDEP mechanisms.
The adenosinergic system has been proposed as one of the potential mechanisms for the pathophysiological development of SUDEP (Shen et al., 2010; Massey et al., 2014; Devinsky et al., 2016; Faingold et al., 2016; Kommajosyula et al., 2016; Ashraf et al., 2021). Findings from animal studies and clinical evidence also suggest a complexity of adenosinergic adaptations in epilepsy and SUDEP, including changes in adenosine metabolism and adenosine receptors (ARs). For instance, adenosine A1 and A2A receptors (A1R and A2AR) are identified to express on synapses in limbic cortical areas (Tetzlaff et al., 1987; Rebola et al., 2003a; Rebola et al., 2005a; Rebola et al., 2005b); an increased A2AR density and a decreased A1R density are shown at excitatory terminals of different limbic areas from animal models of epilepsy and patients with epilepsy (Rebola et al., 2003b; Rebola et al., 2005c; He et al., 2020). The disrupted adenosinergic system, e.g., altered densities of A1R, A2AR, and the adenosine metabolic enzyme, adenosine kinase (ADK) were also seen in different brain areas in patients with temporal lobe epilepsy (TLE) and correlated to SUDEP risk (Patodia et al., 2020). Thus, further characterization of brain area-dependent changes is warranted to reveal the complexity of adenosine and A2AR-mediated regulation actions in SUDEP. During seizure events, adenosine is increased by tremendous consumption of ATP, and sequentially, increased adenosine alters neurotransmission in the brain and acts as a potent endogenous anticonvulsant to terminate seizures (During and Spencer, 1992; Dunwiddie and Masino, 2001). This anticonvulsive effect is mainly due to A1Rs-mediated inhibition of excitatory neurotransmissions (Dunwiddie and Masino, 2001). However, seizure-induced increases of extracellular adenosine can also broadly affect brain regions outside the primary seizure origins to exert a wide spectrum of actions through dominantly distributed A1Rs in the hippocampus, cerebral cortex, and cerebellum and enriched A2ARs in the striatum, nucleus accumbens, and brainstem (Fredholm et al., 2001; Sebastião and Ribeiro, 2009). Indeed, adenosine actions in the brainstem contribute to the central regulation of cardiorespiratory functions (Barraco and Janusz, 1989; Phillis et al., 1997) that are proposed to play a crucial role in SUDEP events (Hirsch, 2010; Massey et al., 2014; Devinsky et al., 2016). Since NTS is a critical hub for cardiorespiratory regulation, manipulation of A2ARs and A1Rs in the NTS affects cardiac and respiratory functions (Minic et al., 2015). Specifically, activation of A2ARs can alter GABAergic neuron activity (Wilson et al., 2004; Minic et al., 2015) and overactivation of A2AR was associated with increased mortality in mice with febrile seizures (Fukuda et al., 2012). These findings indicate an important role of A2ARs and the brainstem in SUDEP pathophysiology.
The metabolic clearance of extracellular adenosine is important for limiting the activation of adenosine receptors and seizure-related death. Clinical studies showed upregulated ADK densities in the brains of TLE patients (Aronica et al., 2011). We previously demonstrated that inhibition of ADK led to increased sudden deaths in mice with acute seizures (Shen et al., 2010), indicating that adenosine removal is essential for the brain to respond to the seizure-induced adenosine surge in the seizing brain to maintain balanced adenosinergic activities, i.e., as an endogenous anticonvulsant affecting seizure risk. Of note, seizure-induced increases of extracellular adenosine were reported in patients with intractable epilepsy (During and Spencer, 1992), and importantly, patients with uncontrolled or refractory seizures are associated with a high risk of SUDEP (Sperling, 2001; Devinsky et al., 2016).
Taken together, we hypothesized that in the brainstem chronic seizure-induced adenosine surges, in combination with abnormal metabolic adenosine removal, may cause fatal overactivation of A2AR and result in cardiorespiratory dysfunctions and increased risk of SUDEP. The present study aimed to evaluate whether targeting A2AR activity can reduce SUDEP risk. To accomplish this objective, we developed a new SUDEP mouse model with relevant clinical features, using a boosted-KA administration paradigm, and we investigated the vulnerability of SUDEP phenotype in mutant Adk+/- mouse (Boison et al., 2002; Palchykova et al., 2010) that was characterized by a ∼50% decrease in ADK and thus impaired adenosine clearance.
Materials and Methods
Animals and Pharmacological Reagents
All animal procedures were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Legacy Research Institute (LRI, No. 120–2018, 114–2020, and 120–2021) and Barrow Neurological Institute (BNI, No. 366) consistent with the principles outlined by the National Institutes of Health. Adk+/- mutants (Boison et al., 2002; Palchykova et al., 2010) and their wild-type (WT) littermates were bred at the LRI (Portland, OR, United States). Adult male mice (bodyweight of 26–30 g) were used for the experiments, which were housed in a temperature- and humidity-controlled room with a 12-h light/dark cycle (lights on at 7:00 a.m.) throughout the experimental period. The reagents used in this study were commercially purchased: Kainic acid (KA, 0222, Tocris), 5-Iodotubercidin (5-ITU, 1745, Tocris), SCH58261 (S4568, Sigma).
Boosted Kainic Acid Model of SUDEP
To develop a SUDEP model with clinically relevant features, we designed a boosted-KA administration paradigm that consists of an intrahippocampal KA (IHKA) injection followed by a single systemic KA (SKA) intraperitoneal (i.p.) injection (Figure 1A). Specifically, 1) for generating chronic epilepsy, adult male Adk+/- and WT mice were subjected to unilateral IHKA (400 ng KA in 200 nl 0.9% saline—a dose used in our previous studies to establish chronic seizures (Shen et al., 2015)) or intrahippocampal injection of 200 nl saline (as sham control) into the right hemisphere, using coordinates (to Bregma): AP = −2.00 mm; ML = ± 1.25 mm; DV = −1.70 mm from procedures published previously (Shen et al., 2015). Ten days after the IHKA or vehicle injection, mice were implanted with bipolar coated stainless steel electrodes (80 μm in diameter; Plastics One) into the right hippocampus using the same coordinates as the IHKA injection (Shen et al., 2015). Animals were maintained in a group in their housing cage throughout the experiment period, except for the v-EEG monitoring period in which the mice were single-housed. 2) For triggering convulsive seizures and possible postictal SUDEP phenotype, one single SKA injection (15 mg/kg, i. p.—a dose that is not expectable to trigger convulsions in naïve mice) was given to the epileptic mice that received IHKA injection 6–8 weeks prior. Then, animals underwent a block of 72-h v-EEG recording for evaluation of possible SUDEP phenotype, and EEG seizure activities were also determined and analyzed (see next section).
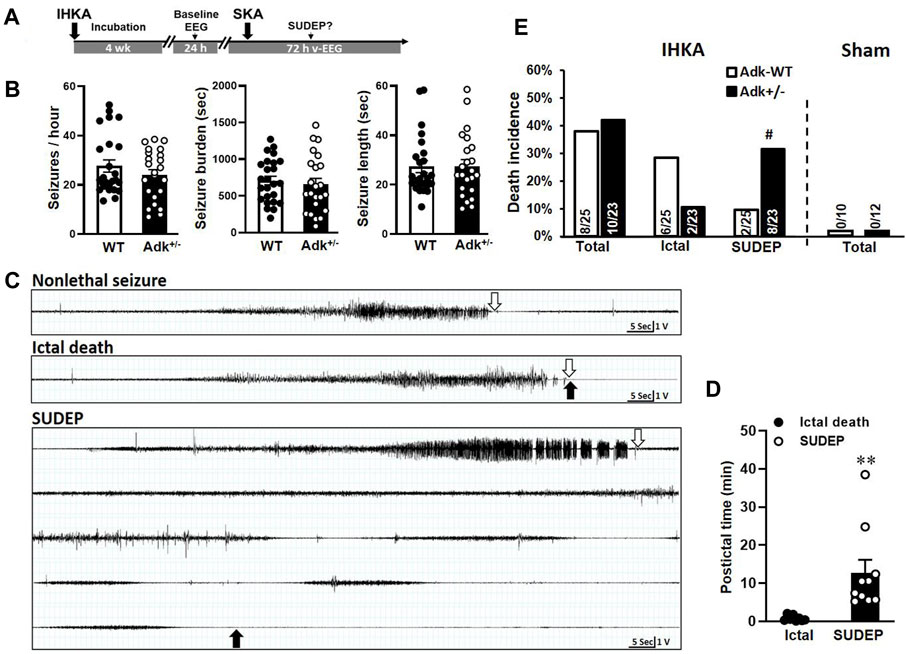
FIGURE 1. Boosted kainic acid (KA) model of SUDEP. (A) Boosted KA administration paradigm that consists of an intrahippocampal KA (IHKA) injection followed by a single systemic KA (SKA) intraperitoneal (i.p.) injection. (B) Baseline EEG evaluation of Adk+/- mice and their WT littermates at 4 weeks after IHKA injection. (C) Representative EEG traces of non-lethal seizure (upper panel), ictal death (middle panel), and SUDEP event (lower panel). Open arrows indicate the stop points of seizures; solid arrows indicate the point of death occurrence. (D) The postictal period from the end of the seizure until the death occurred. (E) Rates of ictal death, SUDEP, and total mortality in mice received IHKA injection (IHKA) or vehicle control (Sham). Data are mean ± SEM. #p < 0.05 vs. SUDEP in Adk-WT, Fisher’s exact test, two-sided; **p < 0.01 vs. ictal death, unpaired t-test.
Video-Electroencephalogram Recording and Analysis
The video-electroencephalogram (v-EEG) was performed according to previously published methods (Shen et al., 2015). Mice were singly housed while tethered for the acquisition of the EEG recordings. Four weeks after IHKA injection, each mouse was subjected to a baseline EEG evaluation of an epileptic phenotype with a block of 24-h EEG monitoring and recording (P511/P122 Grass Instruments, Astro-Med, West Warwick, RI). Electrical brain activity was digitized (ML880 PowerLab 16/30; AD Instruments, Colorado Springs, CO) and quantification of EEG seizure activity was determined as in our previous work (Shen et al., 2015). For evaluation of possible SUDEP phenotype and pharmacological pretreatment on SUDEP risk, animals underwent a block of 72-h v-EEG recording, starting prior to the pretreatment of A2AR antagonist, SCH58261 (3 mg/kg), ADK inhibitor, 5-ITU (2 mg/kg), or vehicle (1.5% DMSO in saline) i. p. and lasting for 72 h. EEG seizure activities were determined and analyzed as aforementioned.
Immunohistochemistry
For immunohistochemistry assessment of chronic epilepsy-induced biochemistry changes, a set of mice (n = 22) was sacrificed 6 weeks post-IHKA or vehicle injection after completion of EEG evaluation but without receiving systemic KA injection. Mice were transcardially perfused with 4% formaldehyde; the dissected brains were postfixed in 4% formaldehyde and cryoprotected in 30% sucrose PBS solution before sectioning into 30 μm sagittal sections using a cryostat (VT 1000 S, Leica, Bannockburn) (Shen et al., 2015). For staining, tissue sections were pretreated with citrate buffer at 80°C for 30 min; then blocked in goat blocking buffer (GBB, containing 2% goat serum, 0.05% Triton X-100 and 1% BSA) for 1 h and incubated at 1% sodium tetraborate for 30 min at room temperature. Pretreated sections were then incubated at 4°C for 48 h in GBB containing corresponding primary antibodies with the indicated dilution: ADK (A304-280A, Bethy Labs, 1:1,000), A2AR (A2A-GP-Af1000, Frontier Institute, 1:100), or cAMP (MAB2146, R&D Systems, 1:200); followed by incubation of corresponding secondary antibodies: Goat anti-guinea pig IgG H&L, Alexa Fluor 488 (A11073, Thermo, 1:500), Donkey anti-rabbit IgG H&L, Alexa Fluor 555 (A-31572, Thermo, 1:1,000), Donkey anti-mouse IgG H&L, Alexa Fluor 647 (ab150107, Abcam, 1:500), or NeuroTrace 435/455 Blue Fluorescent Nissl Stain (N21479, Thermo, 1: 1,000), for 90 min at room temperature. Sections were then washed and mounted on slides. Once dried, sections were cover-slipped with Vectashield Antifade Mounting Medium (H-1000) for fluorescence microscopy observation on a Leica TCS SPE confocal laser-scanning microscope (LAS X 3.1.2.16221). Three independent sections were stained for each method. All sections were processed in parallel using identical solutions and incubation times, while stain slices without either primary or secondary antibodies were used for controls.
Image Quantification of Densitometry
High-resolution digital images were acquired under identical conditions using the LasX software system (Leica, Buffalo Grove, IL, United States). Fluorescence intensity analysis was performed using Leica Application Suite analysis software (Leica, Buffalo Grove, IL) or ImageJ software (ImageJ, US. National Institutes of Health, Bethesda, MD; ImageJ.nih.gov/ij/). All image processing was applied identically across experimental groups. The NTS region was selected as shown in Figure 2A, and immunoreactive material was measured in the same designated area of NTS for each sample and expressed as relative optical density (ROD) by area. Three levels were measured for each mouse, and data analysis is expressed as the mean ± SEM of ROD. The average levels in treatment groups were normalized according to that in the corresponding control group (as baseline).
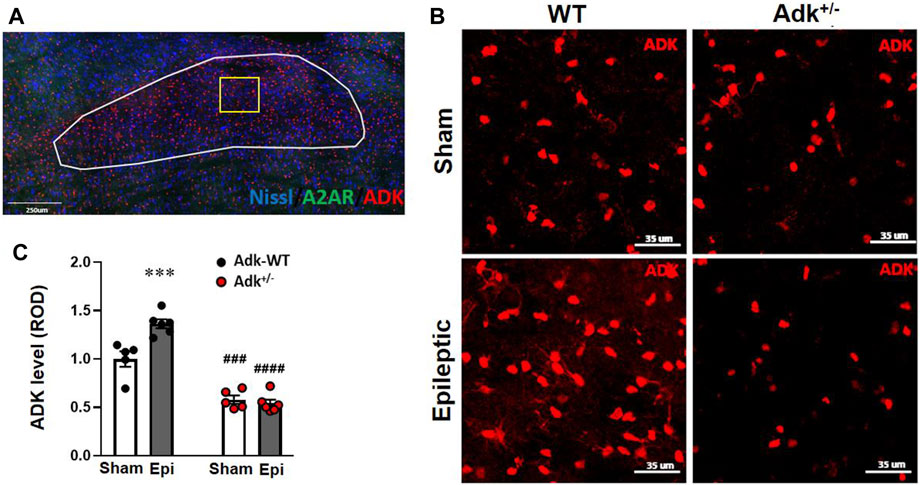
FIGURE 2. ADK changes in the NTS of IHKA modeled epileptic mice. (A) Representative immunofluorescence (IF) image and an indication of selection of NTS region. (B) Representative images of IF staining for ADK in the NTS of IHKA modeled epileptic mice (Epileptic) vs. sham controls (Sham). (C) Quantitative analysis of the NTS expression levels of ADK (presented as relative optical density, ROD) in IHKA modeled epileptic (Epi) mice vs. sham controls (Sham). Data are mean ± SEM. ***p < 0.001 vs. sham controls of same genotype; ###p < 0.001 vs. WT sham controls; ####p < 0.0001 vs. WT epileptics; unpaired t-test. Scale bar = 35 μm.
Electrophysiology Recording of Local Field Potential in the NTS
In vivo LFP in the NTS was recorded with similar procedures as our previous work (Gao et al., 2007) using the below coordinates (to Bregma): AP = −7.35 mm; ML = 0.20 mm; DV = 4.50 mm, with animals under anesthesia (isoflurane; induction 3.0%, maintenance 1.5%). For LFP recordings, the signals were collected by tetrodes and amplified by a 16-channel amplifier (Plexon DigiAmp; bandpass filtered at 0.1–300 Hz, 2,000× gain, sampled at 2 k Hz). First, a baseline LFP was recorded for 30 min, then mice received pretreatment of SCH58261 (3 mg/kg, i. p.) or 0.9% saline (0.3 ml, i. p.) as control with continued recording; 30 min later mice were given a single KA dose (15 mg/kg, i. p.) and recording was continued for another 30 min. For LFP analysis, the raw data of the 2 min prior to the onset of drug administration were selected as representing ongoing LFP activity. A time-frequency transformation was performed (Hanning window; FFT size, 256) with NeuroExplorer, and the spectral power was calculated for each frequency resolution. The spectral power from all frequencies included within the bandwidth was averaged. LFP signals were divided into different frequency bands: theta (2–12 Hz), beta (15–35 Hz), and gamma (36–95 Hz).
Statistical Analyses
All data were analyzed using GraphPad Prism software. The quantitative data are presented as mean ± SEM and were analyzed using one-way ANOVA, two-way ANOVA, or t-tests, as appropriate. The categorical data were analyzed using Fisher’s exact test or Chi-square test, as appropriate. A p-value < 0.05 was considered significant.
Results
Establishment of a Boosted KA Mouse Model of SUDEP
We established a new SUDEP model using a boosted-KA paradigm (Figure 1A) and tested the vulnerability of SUDEP risk in Adk+/- mutants that have approximately 50% reduction in ADK protein level in the brain and compromised ability to metabolize adenosine (Boison et al., 2002; Palchykova et al., 2010). This model consists of two phases - chronic epilepsy modeled by IHKA injection (400 ng) and a potential phenotypic SUDEP (i.e., delayed postictal sudden death) event triggered by a single SKA (15 mg/kg) challenge (Figure 1A). We first assessed the epileptic features between Adk+/- and WT mice at 4 weeks post-IHKA (or vehicle) injections as a baseline EEG evaluation. No EEG seizures were observed in sham control animals with the intrahippocampal injection of saline (WT n = 10 and Adk+/- n = 12), whereas spontaneous recurrent electrographic seizures were developed in IHKA-injected WTs and Adk+/- mutants. There was no significant difference in seizure-onset frequencies (p = 0.2778), seizure burden (i.e., total duration of seizure activity, p = 0.5995), and average length of seizures (p = 0.9687), between IHKA-injected Adk+/- vs. WT mice (n = 23–25 per genotype, unpaired t-test, two-tailed) (Figure 1B).
After baseline EEG evaluations, mice were subjected to SKA (15 mg/kg, i. p.) to trigger possible SUDEP events. The v-EEG monitoring data showed SKA injection-induced convulsive seizures in all the epileptic animals, which eventually resulted in two outcomes: non-lethal seizures or lethal seizures (Figure 1C). Importantly, v-EEG monitoring demonstrated two distinctive phenotypes of lethal seizures: 1) ictal death - which occurred immediately after the end of SKA-induced seizures (Figure 1C, middle panel), or 2) delayed postictal death (aka, SUDEP event (Ryvlin et al., 2013)) - defined as a sudden death that occurred without coexisting behavioral and/or electrographic EEG seizures (based on v-EEG monitoring) for more than 5 min (Figure 1C, lower panel). In contrast, SKA (15 mg/kg, i. p.) did not trigger any death in non-epileptic sham control mice without prior IHKA (but intrahippocampal saline) injection, regardless of their genotypes (n = 10–12 per genotype) (Figure 1E, right panel). The v-EEG analysis showed that the SUDEP events occurred in a period of 12.73 ± 3.40 min after the last v-EEG-recorded seizure (Figure 1D), with the longest seizure-free period prior to a SUDEP event being 38.5 min. The periods from last seizure to death occurrence were significantly different between defined ictal deaths vs. SUDEP events (p = 0.0067, unpaired t-test, n = 8–10/phenotype) (Figure 1D). Remarkably, the occurrences of SUDEP events in epileptic Adk+/- mutants (34.8%, 8/23) was significantly higher than epileptic WTs (8.0%, 2/25, p = 0.0335, Fisher’s exact test, two-sided), whereas the ictal seizure death rate was not significantly different between Adk+/- mutants (8.7%, 2/23) and WTs (24.0%, 6/25) (p = 0.2487, Fisher’s exact test, two-sided), and the seizure-related total mortality was not significantly different between Adk+/- mutants (43.5%, 10/23) and WTs (32.0%, 8/25) (p = 0.5524, Fisher’s exact test, two-sided) (Figure 1E, left panel). This suggests that Adk+/- mice, with impaired adenosine removal, are more vulnerable to SUDEP risk.
Chronic Epilepsy-Induced Adenosinergic Changes in the Brainstem of Mice
To understand the underlying mechanisms of chronic epilepsy-associated SUDEP, we evaluated the molecular changes related to adenosinergic activity with immunofluorescence staining of ADK, A2AR, and cAMP in the NTS. The NTS level of ADK (Figures 2B,C) was altered by Adk mutation and IHKA-modeled epilepsy (genotype factor, p < 0.0001 F(1,18) = 189.2; modeling factor, p = 0.0012, F(1,18) = 14.69; interaction, p = 0.002, F(1,18) = 22.21; two-way ANOVA, n = 5–6 per group). Specifically, the basal NTS level of ADK in sham Adk+/- mutants was lower than sham WTs (p < 0.0002), at 57.9% of the level of sham WTs (Figures 2B,C). The epileptic WTs had significantly (36.5%) increased NTS ADK vs. sham WTs (p = 0.0006, unpaired t-test). Of note, epileptic Adk+/- mutants showed no increase in NTS ADK level vs. sham mutants (p = 0.9514, unpaired t-test), whereas NTS ADK level in epileptic Adk+/- mutants was significantly lower vs. epileptic WTs (p < 0.0001, unpaired t-test) (Figures 2B,C). This suggests an epilepsy-induced compensatory increase of ADK in the NTS of WTs whereas Adk+/-mice were devoid of this change. Furthermore, NTS A2AR level was altered by IHKA epilepsy vs. sham controls (p < 0.0001, F(1,18) = 166.5, modeling factor, two-way ANOVA, n = 5-6 per group), in both epileptic Adk+/- and WT mice vs. their corresponding sham controls (p < 0.0001 and p < 0.0001, unpaired t-test) (Figures 3A,B). These indicate an epilepsy-induced adaptive A2AR increase in the NTS of both genotypes, regardless of ADK levels.
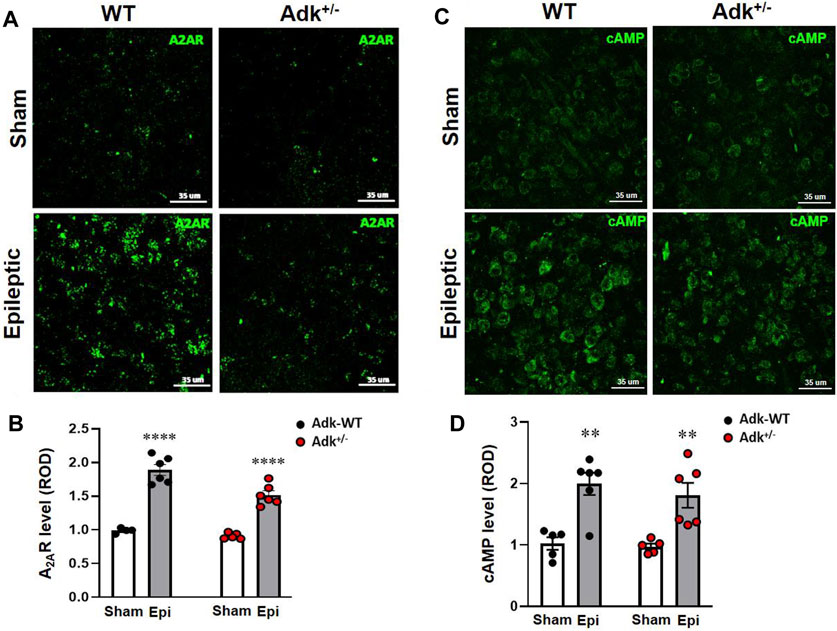
FIGURE 3. Changes of A2AR and cAMP in the NTS of IHKA modeled epileptic mice. Representative images of IF staining for A2AR (A) and cAMP (C) in the NTS of IHKA modeled epileptic mice (Epileptic) vs. sham controls (Sham). Quantitative analysis of the NTS densities of A2AR (B) and cAMP (D) (presented as relative optical density, ROD) in IHKA modeled epileptic (Epi) mice vs. sham controls (Sham). Data are mean ± SEM. **p < 0.01 and ****p < 0.0001, vs. sham controls within same genotype, unpaired t-test. Scale bar = 35 μm.
To further explore A2AR activation-related changes, we evaluated cAMP in the NTS (Figure 3C). IHKA-modeled epilepsy significantly increased NTS cAMP levels (p < 0.0001, F(1,18) = 32.17, treatment effect, two-way ANOVA, n = 5-6 per group) without a difference between genotypes (p = 0.4676, F(1,18) = 0.5506, two-way ANOVA) (Figure 3D). Specifically, NTS cAMP levels were increased in both epileptic WTs and Adk+/- mutant vs. their corresponding non-epileptic sham controls (p = 0.0022, p = 0.0080, t-test, n = 5–6 per group) (Figure 3D), which was in line with the changing pattern of A2AR in the NTS (Figure 3B). Together, these biochemistry findings revealed that chronic epilepsy resulted in increased ADK levels for the removal of excessive adenosine, and increased A2AR activities were in line with increased cAMP in the NTS.
Blockade of A2AR Activation Reduced the SUDEP Risk in Epileptic Mice
Having demonstrated perturbations in the adenosinergic pathway in the NTS induced by epilepsy, we further explored whether the increased A2AR activity contributes to the risk of SUDEP events. We tested if suppressing A2AR activity, with A2AR antagonist SCH58261, can reduce the risk of SUDEP and whether ADK inhibition can exacerbate SUDEP risk. A cohort of epileptic Adk+/- mutants and WTs was generated by IHKA-injection and confirmed via EEG evaluation for their baseline epileptic features (Figure 4A). They then were randomly assigned to three groups to receive pretreatment of SCH58261 (3 mg/kg, i. p, n = 11–12), 5-ITU (2 mg/kg, i. p, n = 15), or vehicle (n = 17); and 30 min later each mouse was given a single injection of SKA (15 mg/kg, i. p.) followed by v-EEG monitoring for 72 h (Figure 4A). Figure 4B demonstrates the epileptic features of each group of mice at 4 weeks post-IHKA injections as their baseline EEG evaluation. There was no significant difference in seizure-onset frequencies between groups (n = 11–17 per group) (p = 0.7769, F(2, 81) = 0.2532, two-way ANOVA) or between genotypes (p = 0.9867 F(1, 81) = 0.0002, genotype effect, two-way ANOVA) (Figure 4B, upper panel); no significant difference was observed in seizure burden between groups (p = 0.8933, F(2, 81) = 0.1130, two-way ANOVA) or between genotypes (p = 0.2321, F(1, 81) = 1.450, genotype effect, two-way ANOVA) (Figure 4B, middle panel); also no difference was shown in average seizure length across groups (p = 0.5270, F(2, 81) = 0.6456) and genotypes (p = 0.1338, F(1, 81) = 2.294) (Figure 4B, lower panel).
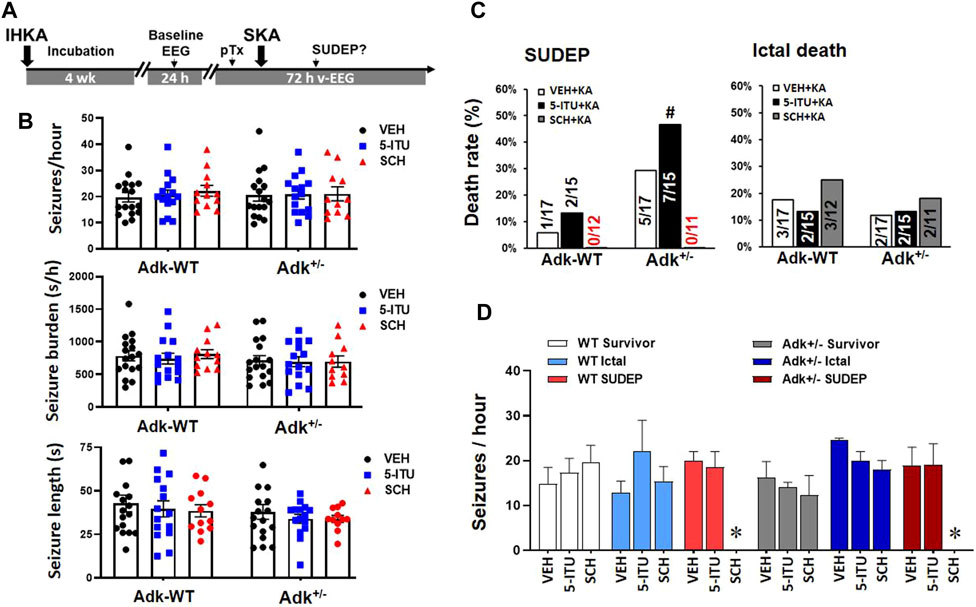
FIGURE 4. A2AR antagonist SCH58261 reduced SUDEP occurrence in epileptic mice. (A) The paradigm of pharmacological pretreatment (pTx) in the boosted KA model of SUDEP. (B) EEG validation of epileptic phenotypes in each group of Adk+/- mice and their littermates (Adk-WT) at 4 weeks after IHKA injection, prior to the experimental SKA (15 mg/kg, i. p.) injections. (C) Rates of ictal death, SUDEP, and total mortality among groups with pretreatment of i. p. injection of vehicle (VEH), 5-Iodotubercidin (5-ITU, 2 mg/kg), and SCH58261 (SCH, 3 mg/kg). (D) Baseline EEG evaluation of seizure onset frequencies (seizure/hour) in mice grouped as survivors, ictal death, and SUDEP, with different pretreatments of a VEH, 5-ITU, or SCH. * Indicating zero animals with SUDEP existing in the marked (SCH) group. #p < 0.05 vs. same treatment group in Adk-WT mice, Chi-square test.
The v-EEG analysis showed that the SUDEP occurrences increased in 5-ITU pretreatment (46.7%, 7/15), whereas SCH58261-pretreatment drastically reduced SUDEP events (0%, 0/11) in Adk+/-mutants vs. vehicle-pretreated controls (29.4%, 5/17) (X2(2, N=43) = 6.901, p = 0.0317). The SCH58261-pretreatment also reduced SUDEP events in WTs (0%, 0/11), but 5-ITU pretreatment did not significantly increase SUDEP onset vs. vehicle pretreated WTs (X2(2, N=44) = 1.904, p = 0.3860) (Figure 4C). These findings indicate that A2AR overactivation could contribute to increased SUDEP risk, and A2AR blockade efficiently reduced the SUDEP risk. Retrospective analysis of their baseline EEG showed that no significant differences were found in seizure onset frequencies among mice of survival, ictal death, and SUDEP groups that received different treatments (F(5,10) = 1.799, p = 0.2007, one-way ANOVA) (Figure 4D). This suggests that the baseline epileptic phenotype was not linked to their seizure-related death phenotypes.
A2AR Blockade Disinhibits Seizure-suppressed Coordination of Activity in the NTS
To mechanistically dissect the A2AR effects on the coordination of activity in the NTS, SCH58261 or saline (as control) was administrated 30 min prior to KA injection in a separated cohort of IHKA-modeled Adk+/- and WT mice (n = 4-6 per group), and LFP of NTS was recorded to reflect the neural activity of the assembly of cells surrounding the recording site (Bantikyan et al., 2009). We compared the LFP signals in epileptic Adk+/- and WT mice following pretreatment with saline or SCH58261, and afterward with KA injections (Figure 5). The raw LFP signals were divided into three frequency bands, i.e., theta, 2–12 Hz; beta, 15–35 Hz; gamma, 36–95 Hz (Figure 5A). The heat map of the power spectrum of LFP signals shows an overt continuous reduced gamma response after KA in WT mice with saline pretreatment (Figure 5B). Analysis of averaged power spectrums between the genotypes demonstrated that the baseline oscillation power of gamma was significantly lower in Adk+/- mice vs. WTs (p = 0.0037, unpaired t-test) while their baseline theta or beta powers were not different (p = 0.8656 and p = 0.9081, unpaired t-test, n = 5-6 per group) (Figure 5C).
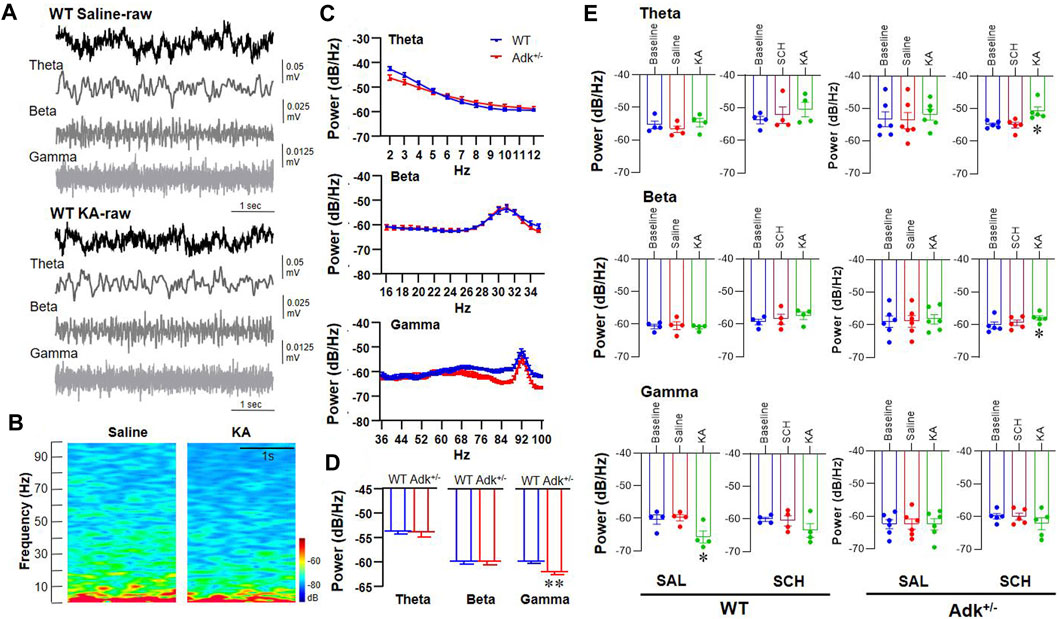
FIGURE 5. Electrophysiological LFP recording in the NTS of Adk+/- and WT epileptic mice. (A) Typical LFP signals from one WT mouse after saline treatment (top) and subsequent KA injection (bottom). The first row of both upper (WT Saline-raw) and lower (WT KA-raw) panels showing a 5 s of the raw trace; the second to fourth rows in both panels showing a 5 s of filtered signals (Theta, Beta, and Gamma, respectively). (B) The power spectrum of the above LFP signals showing a reduced gamma response after KA injection. (C) The averaged power spectrums of LFP from WT and Adk+/- mice, indicating a lower baseline gamma response in Adk+/- mice. (D) The histogram showing a significant decrease in baseline gamma response in Adk+/- mice. (E) Histograms of the averaged power spectrum of LFP signals showing quantitative changes in theta (top panel), beta (middle panel) and gamma (bottom panel) bands from WT and Adk+/- mice after injections of saline followed KA or SCH followed KA. Data are mean ± SEM. *p < 0.05 vs. corresponding baseline, one-way ANOVA, in (E); **p < 0.01 vs. WT baseline, paired t-test, in (D).
Further analysis showed that the saline- or SCH58261-pretreatment per se did not cause LFP power changes in theta (F(2,9) = 1.801, p = 0.2189), beta (F(2,9) = 1.017, p = 0.3997), or gamma (F(2, 9) = 0.0742, p = 0.9290, one-way ANOVA) band vs. baselines (Figure 5E). KA injection following saline pretreatment decreased the gamma oscillation in WTs (F(2, 6) = 9.504, p = 0.0138, one-way ANOVA; p = 0.0077 vs. saline, p = 0.0115 vs. baseline, paired t-test), but the reduction of gamma oscillation was not seen when following SCH58261 pretreatment (F(2, 6) = 3.057, p = 0.1215, one-way ANOVA; p = 0.0967 vs. SCH58261, p = 0.0328 vs. baseline, paired t-test), suggesting that SCH58231 partially blocked the KA suppression on gamma. The inferred role of A2AR is similar to that previously reported (Pietersen et al., 2009), although in this previous study kainate enhanced cortical gamma power. Also, while the baseline power of gamma oscillation in Adk+/- mutants was significantly lower than WTs (Figure 5D), no gamma power change resulted after KA injection following saline pretreatment (F(2, 10) = 0.0021, p = 0.9979, one-way ANOVA; p = 0.9509 vs. saline, p = 0.9649 vs. baseline, paired t-test) (Figure 5E, lower panel). Similarly, no KA-induced changes in theta (F(2, 10) = 0.6913, p = 0.5234) or beta (F(2, 10) = 0.3417, p = 0.7185, one-way ANOVA) were found in Adk+/- mice when following saline pretreatment. Remarkably, the SCH58261 pretreatment significantly increased the theta (F(2, 8) = 8.959, p = 0.0091, one-way ANOVA; p = 0.0056, paired t-test) and beta powers (F(2, 8) = 5.452, p = 0.0321, one-way ANOVA; p = 0.0119, paired t-test) in Adk+/- mice post-KA injection (Figure 5E, upper and middle panels). Together, we demonstrated the differential effects of KA-induced LFP power changes and SCH58261 manipulation on KA effects between WT and Adk+/- mice.
Discussion
While the mechanisms of SUDEP remain elusive (Stöllberger and Finsterer, 2004; Hirsch, 2010; Shorvon and Tomson, 2011), studies suggested that abnormalities in the adenosinergic system may play an important role in SUDEP events (Shen et al., 2010; Boison, 2012; Devinsky et al., 2016). We hypothesized that repeated seizure-induced adenosine increases in the brainstem can result in potentially fatal overactivation of A2AR while decreased ADK can exacerbate fatality. We developed a new SUDEP model characterized by a delayed postictal death phenotype in mice with chronic epilepsy. Our findings suggest an enhanced A2AR activity in the NTS of epileptic mice - while LFP alteration can result from a local modulation by A2AR and/or potentially by a long-distance network effect triggered by A2AR in different brain regions - and provide experimental evidence supporting A2AR as a therapeutic target for SUDEP prevention. To better understand the role of A2AR in SUDEP and its therapeutic potential, the following aspects warrant further discussion.
SUDEP Animal Models and Clinically Relevant Phenotypes
The unmet need in developing pharmacological preventative therapies against SUDEP was not only due to inadequate understanding of SUDEP mechanisms (Hirsch, 2010; Shorvon and Tomson, 2011; Stöllberger and Finsterer, 2004) but also was compromised by limitations of available SUDEP animal models (Surges et al., 2009; Devinsky et al., 2016; Pansani et al., 2016). Of the previous animal models used for SUDEP research, most carry genetic mutations that mimic several major clinical conditions related to sudden death (e.g., cardiac arrhythmia, arrest, and coincidence of seizures) (Massey et al., 2014; Scheffer and Nabbout, 2019; Tu et al., 2011). Also, there is a lack of data in systematically characterizing the types of death (such as ictal death vs. delayed postictal death). These include rodents with mutations in the SCN1A gene (mimics Dravet syndrome) (Scheffer and Nabbout, 2019), SCN5A gene (mimics Brugada syndrome), KCNH2, KCNQ1 genes (mimics Long QT syndrome), etc. (Tu et al., 2011). These genetic mutations could be potential causes of SUDEP; however, while they can result in pathophysiological changes, e.g., prolonged cardiac action potential, ventricular tachycardia, syncope, and sudden death, these genetic deficits were rarely reported in clinical SUDEP cases or patients with TLE—the most common form of epilepsy involved in SUDEP cases (Massey et al., 2014; Patodia et al., 2020; Devinsky, 2011). Facing these limitations, we developed a new SUDEP mouse model using a boosted-KA paradigm that mimics major, clinically relevant features of SUDEP cases, including: 1) a chronic nature of spontaneous seizures (Figure 1B) (Shen et al., 2015); and 2) a delayed postictal death phenotype - i.e., SUDEP event - one of the central revelations of the MORTEMUS study (Ryvlin et al., 2013), dissociated from prior convulsive and/or electrographic seizures (Figure 1C). Notably, the average latency to the SUDEP event is substantially longer than the delay seen in most (but not all) epilepsy patients reported in the MORTEMUS study (Ryvlin et al., 2013). A limitation of this study is the need for cardiopulmonary monitoring, which should be undertaken in future studies to fully characterize this model. It may be of interest to note that the adenosine modulation system also has direct cardiorespiratory effects which are affected in the global genetic and pharmacological manipulation attempted in this study. Additionally, future work could optimize our paradigm. For example, we utilized 15 mg/kg KA to trigger behavior seizures in epileptic mice; a lower KA dose may achieve a less severe behavioral seizure phenotype, while still triggering a SUDEP event in epileptic animals, resulting in a longer postictal period before SUDEP occurrence. These models have the potential for better characterization of adenosinergic changes in other brain regions in SUDEP, e.g., hippocampus, cortex, and other brainstem nuclei. Nevertheless, this adenosinergic SUDEP mouse model provides a novel tool for the SUDEP research field while it warrants continued optimization. The biological sex variable should also be investigated and addressed in future studies.
Disturbances in the Adenosinergic Signaling Pathway Resulted in SUDEP
Adenosinergic activities are tightly linked to the etiological and pathophysiological outcomes of seizures and epilepsy (Ashraf et al., 2021; Faingold et al., 2016; Kommajosyula et al., 2016; Masino et al., 2014; O'Brien, 1988; Shen et al., 2014). Acute and chronic seizures can trigger repeated adenosine surges and increase adenosine tone in the brain, which can act as an endogenous anticonvulsant and also reset neuron network stability by affecting neurotransmission at the synapse (Masino et al., 2014; O'Brien, 1988). It is increasingly established that two main systems are contributing to the extracellular adenosine that engages the adenosine modulation system in the brain: the activity of equilibrative nucleoside transporters (ENTs) and of ADK mostly associated with a global A1R function and CD73-mediated formation of ATP-derived adenosine tightly associated with A2AR activation (Cunha, 2016). Seizure-induced adenosine surges can result in changes in ADK (Aronica et al., 2011), which together with adaptive alterations of the density and activity of adenosine receptors (He et al., 2020; Rebola et al., 2005c) maintain balanced adenosine activities in epileptic sites. Despite Adk+/− mutants having reduced endogenous brain ADK, intriguingly, no overt genotype effect of Adk+/− mutants was observed in IHKA-induced chronic seizures (Figure 1B). This could be attributed to potential adaptations of mutants to genetic knockdown of ADK during their development, or it could be masked by undetermined compensatory changes in the adenosinergic pathway: changes in additional adenosine metabolic pathways, e.g., adenosine deaminase (to process adenosine into inosine and then into hypoxanthine), and/or changes in adenosine receptors in the hippocampus of Adk+/− mutants. These potential changes may separately and/or synergistically affect the seizure phenotypes in Adk+/− mice. The complexity and seeming discrepancy of the relationship between the ADK/adenosine and seizure phenotypes were also shown in the Sandau et al. study with similar ADK deletion in the hippocampus (Sandau et al., 2016), in which the AdkΔbrain mice (i.e., Nestin-Cre+/−:Adkfl/fl mice) were used with the characterized conditional Adk gene deletion, which caused brain-wide ADK deficiency and increased synaptic adenosine levels. However, AdkΔbrain mice surprisingly showed an increase in spontaneous seizures and susceptibility to seizure induction compared to their WT littermates (Sandau et al., 2016). The findings from AdkΔbrain mice and our Adk+/− mutants indicated a complexity between ADK/adenosine and seizure (onset) phenotypes, which is also a suggestive indication supporting the contention that studying only ADK as a controller of the adenosine levels may well be inadequate. The work of Sandau et al. (2016) did not consider the possibility that changes in ATP release and ect-nucleotidase activity might have occurred in the tested Adk transgenic mice. Nevertheless, whether genetic ADK knockdown can yield possible preconditioning effects against the development of epilepsy warrants additional evaluation.
In the brainstem, we demonstrated that chronic seizures led to a compensatory increase in NTS ADK (of WT mice), an area outside the hippocampal seizure origin; epileptic Adk+/- mice did not experience any ADK level increases, which may have contributed to their increased death rates. The NTS level of A2ARs was enhanced in epileptic mice along with increased cAMP, suggesting an increased output of A2AR activation. Notably, in epileptic Adk+/- mice, the combination of impaired adenosine removal potential with increased A2ARs in the NTS could result in lethal suppression on cardiorespiratory reflexes of chemo- and baroreceptors inputs (Wilson et al., 2004; Zoccal et al., 2014; Minic et al., 2015). Mechanistically, A2ARs can alter GABAergic activities in several aspects: 1) A2AR controls the depolarization-evoked GABA release in synaptosomes in the hippocampus (Cunha and Ribeiro, 2000) and striatum (Kirk and Richardson, 1994); 2) A2AR controls the activity of GABAergic interneurons, increasing synchronization in hippocampal networks (Rombo et al., 2015) and most evidently controlling adaptive plastic changes in GABAergic synapses in the prefrontal cortex (Kerkhofs et al., 2018); 3) previous studies in different animal models of epilepsy suggested that the neuroprotection afforded by A2AR blockade might involve a rebalance of GABAergic transmission (Seo et al., 2020); 4) A2AR are critical mediators of the stability of GABAergic synapses (Gomez-Castro et al., 2021). Thus, overactivation of A2ARs in the NTS can attenuate depolarization-evoked GABA release (Saransaari and Oja, 2005) and affect oscillations of GABAergic interneurons (Buzsáki and Wang, 2012). Meanwhile, it has been shown that A2AR is located in glutamatergic synapses (Rebola et al., 2005b) acting as a controller of plasticity processes at glutamatergic synapses, either in the hippocampus (Costenla et al., 2011), dorsal or ventral (Reis et al., 2019) hippocampus, the striatum (Shen et al., 2008; Li et al., 2015), the amygdala (Simões et al., 2016), or the prefrontal cortex (Kerkhofs et al., 2018). Overactivated A2AR may also lead to altered synaptic glutamatergic activities in NTS neurons.
Remarkably, A2AR antagonist SCH58261 disinhibited KA-induced continuous suppression in gamma oscillation and enhanced theta and beta oscillations in anesthesia preparation (Figure 5E), likely preventing the lethal suppression of NTS neuronal activity during and after seizures (Kuo et al., 2016). Theta, beta, and gamma oscillations not only represent activity changes of local neurons in the recorded area but also activities from specific brain circuits of information flow (Massimini et al., 2004). Our study demonstrated that under chronic epileptic condition, KA and/or acute seizure tends to decrease the slow-wave synchrony, suggesting decreased communications between brain regions. SCH58261 pretreatment can restore KA-induced inhibition on slow waves, indicating the disinhibition is mainly mediated by A2AR activation. In the mammalian cortex, neural communication is organized by 30–100 Hz gamma oscillation, with gamma frequency related to processing speed in neural networks (Insel et al., 2012). Furthermore, this fast band typically requires interplay between excitatory and inhibitory transmission (Buzsáki et al., 2012). However, we need to bear in mind that though LFP recorded from the anesthetized animal has been accepted in epilepsy studies (Williams et al., 2016; Wenzel et al., 2017) to reflect synchronous activities of neuronal assemblies, the anesthetized states might mask changes in seizure activity, respiration, and potential neurobehavioral outcomes. The LFP findings aimed to mechanistically demonstrate the local neuronal activities in the NTS, linked to IHKA-induced chronic epilepsy and/or affected by adenosine-A2AR activities.
In the NTS, neurons are segregated into neuronal clusters, which receive distinct cardiorespiratory afferents and regulate their function by regulating NMDAR- and GABAAR-mediated excitatory/inhibitory synaptic plasticity (Bantikyan et al., 2009). We propose that, in ADK+/− mice, the inhibitory transmission may predominate in the NTS because of its impaired capacity to remove the endogenous adenosine, which may lead to a potential tonic suppression of cardiorespiratory function, and a higher vulnerability to SUDEP risk. As such, the additional KA injection can cause a higher sudden death rate in ADK−/+ mice. While the cell type- and/or neurotransmitter-dependent actions of A2AR in the NTS warrant further characterization to reveal the complexity of A2AR-mediated regulation in NTS circuits, a report from Derera et al (2017) supports the role of A2AR in the NTS in an increased risk of cardiorespiratory dysfunction and sudden death in TLE patients (Derera et al., 2017). Enhanced A2AR signaling may not be limited to NTS; it may affect other autonomic brainstem structures that potentially contribute to SUDEP. Our findings indicate a crucial role of A2AR in the pathophysiology of SUDEP and suggest that antagonism of A2AR may be a therapeutic strategy for SUDEP prevention.
Adenosinergic Intervention—do the Timing and Subtype of Receptor Matter?
With a broader consideration, seizures also trigger a release of ATP (Wieraszko et al., 1989; Augusto et al., 2021) and an increased extracellular catabolism of ATP into adenosine (Bonan et al., 2000; Bruno et al., 2003; Nicolaidis et al., 2005), which sustains A2AR activation (Augusto et al., 2013; Carmo et al., 2019; Gonçalves et al., 2019; Alçada-Morais et al., 2021; Augusto et al., 2021) that is critically associated with seizure-induced neurodegeneration (Cognato et al., 2010; Canas et al., 2018; Augusto et al., 2021). This evidence is of key importance to understand the role of A2AR and the limitations in studying only the relation between ADK and adenosine neuromodulation without considering the whole limb of ATP release and ectonucleotidase activity selectively associated with A2AR activation (Augusto et al., 2013; Carmo et al., 2019; Gonçalves et al., 2019; Alçada-Morais et al., 2021; Augusto et al., 2021). Strangely, it has never been tested if decreased ADK results in aberrantly increased ATP release upon neuronal activation.
Further, due to the complexity of the adenosinergic actions in the CNS and diverse and wide distributions of A1R and A2AR across brain regions (O'Brien, 1988; Chen et al., 2014; Fredholm et al., 2005; Chen et al., 2013), the application of adenosinergic interventions to prevent SUDEP deserves deeper discussion. In the hippocampus, A1R suppresses the ictal firing of excitatory neurons; conversely, hippocampal A2AR activation is proconvulsant and proseizure (Zeraati et al., 2006; El Yacoubi et al., 2009). Clinical findings revealed upregulated hippocampal A2ARs in patients with TLE, which supports the notion of applying A2AR antagonists without exacerbating epileptic seizures. In the brainstem, a working hypothesis is that A2AR activation leads to suppression of GABAergic inhibitory neurons and their mediated cardiorespiratory functions, whereas A1R activation promotes opposite effects (Phillis et al., 1997). These diverse effects of A2AR vs. A1R limit the potential application of the widely used, non-selective adenosine receptor antagonist, caffeine. Indeed, the cause-and-effect relationship between caffeine and epileptic seizures has long been debated (Kaufman and Sachdeo, 2003; Samsonsen et al., 2013) and was recently reviewed (Tescarollo et al., 2020) with due care to distinguish the effects of acutely administered caffeine compared to the ‘chronic’ consumption of caffeine, the latter attenuating epileptic-like phenotypes. Rather, specific antagonism of A2AR can reduce the adenosine surge-related brainstem suppression, while avoiding interference with A1R-mediated anticonvulsive actions. Last but not least, antiepileptic drug-resistant/refractory patients are linked to a high risk of SUDEP, while having tonic-clonic seizures is considered the greatest risk factor (Annegers and Coan, 1999). With the recent FDA approval of the A2AR antagonist istradefylline for PD treatment (Chen and Cunha, 2020), our results indicate istradefylline should be examined for repurposing for epileptic patients at risk of SUDEP. Together, A2AR antagonists may be a potential add-on anti-SUDEP approach. This approach may provide an important reduction of SUDEP with a remaining question as to the suppression of seizures.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Legacy Research Institute and Barrow Neurological Institute.
Author Contributions
SB, RG, and JC performed research and analyzed data; LW and SC performed research, MG performed research, analyzed data, and wrote the paper; JW and J-FC contributed unpublished reagents/analytic tools and edited the paper; TJ designed research and analyzed data; H-YS designed and performed research, analyzed data, and wrote the paper.
Funding
The research was supported by grants from the Good Samaritan Foundation of Legacy Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors wish to thank Danielle Osborne and Joseph Frascella for their discussion and intellectual contribution.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.910535/full#supplementary-material
References
Alçada-Morais, S., Gonçalves, N., Moreno-Juan, V., Andres, B., Ferreira, S., Marques, J. M., et al. (2021). Adenosine A2A Receptors Contribute to the Radial Migration of Cortical Projection Neurons through the Regulation of Neuronal Polarization and Axon Formation. Cereb. Cortex 31, 5652–5663. doi:10.1093/cercor/bhab188
Annegers, J. F., and Coan, S. P. (1999). SUDEP: Overview of Definitions and Review of Incidence Data. Seizure 8, 347–352. doi:10.1053/seiz.1999.0306
Aronica, E., Zurolo, E., Iyer, A., de Groot, M., Anink, J., Carbonell, C., et al. (2011). Upregulation of Adenosine Kinase in Astrocytes in Experimental and Human Temporal Lobe Epilepsy. Epilepsia 52, 1645–1655. doi:10.1111/j.1528-1167.2011.03115.x
Ashraf, O., Huynh, T., Purnell, B. S., Murugan, M., Fedele, D. E., Chitravanshi, V., et al. (2021). Suppression of Phrenic Nerve Activity as a Potential Predictor of Imminent Sudden Unexpected Death in Epilepsy (SUDEP). Neuropharmacology 184, 108405. doi:10.1016/j.neuropharm.2020.108405
Augusto, E., Matos, M., Sévigny, J., El-Tayeb, A., Bynoe, M. S., Müller, C. E., et al. (2013). Ecto-5'-nucleotidase (CD73)-Mediated Formation of Adenosine Is Critical for the Striatal Adenosine A2A Receptor Functions. J. Neurosci. 33, 11390–11399. doi:10.1523/JNEUROSCI.5817-12.2013
Augusto, E., Gonçalves, F. Q., Real, J. E., Silva, H. B., Pochmann, D., Silva, T. S., et al. (2021). Increased ATP Release and CD73-Mediated Adenosine A2A Receptor Activation Mediate Convulsion-Associated Neuronal Damage and Hippocampal Dysfunction. Neurobiol. Dis. 157, 105441. doi:10.1016/j.nbd.2021.105441
Bantikyan, A., Song, G., Feinberg-Zadek, P., and Poon, C. S. (2009). Intrinsic and Synaptic Long-Term Depression of NTS Relay of Nociceptin- and Capsaicin-Sensitive Cardiopulmonary Afferents Hyperactivity. Pflugers Arch. 457, 1147–1159. doi:10.1007/s00424-008-0571-9
Barraco, R. A., and Janusz, C. A. (1989). Respiratory Effects of 5'-ethylcarboxamidoadenosine, an Analog of Adenosine, Following Microinjections into the Nucleus Tractus Solitarius of Rats. Brain Res. 480, 360–364. doi:10.1016/0006-8993(89)90208-4
Boison, D., Scheurer, L., Zumsteg, V., Rülicke, T., Litynski, P., Fowler, B., et al. (2002). Neonatal Hepatic Steatosis by Disruption of the Adenosine Kinase Gene. Proc. Natl. Acad. Sci. U. S. A. 99, 6985–6990. doi:10.1073/pnas.092642899
Bonan, C. D., Walz, R., Pereira, G. S., Worm, P. V., Battastini, A. M., Cavalheiro, E. A., et al. (2000). Changes in Synaptosomal Ectonucleotidase Activities in Two Rat Models of Temporal Lobe Epilepsy. Epilepsy Res. 39, 229–238. doi:10.1016/s0920-1211(00)00095-4
Bruno, A. N., Oses, J. P., Amaral, O., Coitinho, A., Bonan, C. D., Battastini, A. M., et al. (2003). Changes in Nucleotide Hydrolysis in Rat Blood Serum Induced by Pentylenetetrazol-Kindling. Brain Res. Mol. Brain Res. 114, 140–145. doi:10.1016/s0169-328x(03)00168-2
Buzsáki, G., and Wang, X. J. (2012). Mechanisms of Gamma Oscillations. Annu. Rev. Neurosci. 35, 203–225. doi:10.1146/annurev-neuro-062111-150444
Buzsáki, G., Anastassiou, C. A., and Koch, C. (2012). The Origin of Extracellular Fields and Currents--EEG, ECoG, LFP and Spikes. Nat. Rev. Neurosci. 13, 407–420. doi:10.1038/nrn3241
Canas, P. M., Porciúncula, L. O., Simões, A. P., Augusto, E., Silva, H. B., Machado, N. J., et al. (2018). Neuronal Adenosine A2A Receptors Are Critical Mediators of Neurodegeneration Triggered by Convulsions. eNeuro 5, ENEURO.0385-18.2018. doi:10.1523/ENEURO.0385-18.2018
Carmo, M., Gonçalves, F. Q., Canas, P. M., Oses, J. P., Fernandes, F. D., Duarte, F. V., et al. (2019). Enhanced ATP Release and CD73-Mediated Adenosine Formation Sustain Adenosine A2A Receptor Over-activation in a Rat Model of Parkinson's Disease. Br. J. Pharmacol. 176, 3666–3680. doi:10.1111/bph.14771
Chen, J. F., and Cunha, R. A. (2020). The Belated US FDA Approval of the Adenosine A2A Receptor Antagonist Istradefylline for Treatment of Parkinson's Disease. Purinergic Signal 16, 167–174. doi:10.1007/s11302-020-09694-2
Chen, J. F., Eltzschig, H. K., and Fredholm, B. B. (2013). Adenosine Receptors as Drug Targets-Wwhat Are the Challenges? Nat. Rev. Drug Discov. 12, 265–286. doi:10.1038/nrd3955
Chen, J. F., Lee, C. F., and Chern, Y. (2014). Adenosine Receptor Neurobiology: Overview. Int. Rev. Neurobiol. 119, 1–49. doi:10.1016/B978-0-12-801022-8.00001-5
Cognato, G. P., Agostinho, P. M., Hockemeyer, J., Müller, C. E., Souza, D. O., and Cunha, R. A. (2010). Caffeine and an Adenosine A(2A) Receptor Antagonist Prevent Memory Impairment and Synaptotoxicity in Adult Rats Triggered by a Convulsive Episode in Early Life. J. Neurochem. 112, 453–462. doi:10.1111/j.1471-4159.2009.06465.x
Costenla, A. R., Diógenes, M. J., Canas, P. M., Rodrigues, R. J., Nogueira, C., Maroco, J., et al. (2011). Enhanced Role of Adenosine A(2A) Receptors in the Modulation of LTP in the Rat hippocampus upon Ageing. Eur. J. Neurosci. 34, 12–21. doi:10.1111/j.1460-9568.2011.07719.x
Cunha, R. A., and Ribeiro, J. A. (2000). Purinergic Modulation of [(3)H]GABA Release from Rat Hippocampal Nerve Terminals. Neuropharmacology 39, 1156–1167. doi:10.1016/s0028-3908(99)00237-3
Cunha, R. A. (2016). How Does Adenosine Control Neuronal Dysfunction and Neurodegeneration? J. Neurochem. 139, 1019–1055. doi:10.1111/jnc.13724
Derera, I. D., Delisle, B. P., and Smith, B. N. (2017). Functional Neuroplasticity in the Nucleus Tractus Solitarius and Increased Risk of Sudden Death in Mice with Acquired Temporal Lobe Epilepsy. eNeuro 4, ENEURO.0319-17.2017. doi:10.1523/ENEURO.0319-17.2017
Devinsky, O., Hesdorffer, D. C., Thurman, D. J., Lhatoo, S., and Richerson, G. (2016). Sudden Unexpected Death in Epilepsy: Epidemiology, Mechanisms, and Prevention. Lancet Neurol. 15, 1075–1088. doi:10.1016/S1474-4422(16)30158-2
Devinsky, O. (2011). Sudden, Unexpected Death in Epilepsy. N. Engl. J. Med. 365, 1801–1811. doi:10.1056/NEJMra1010481
Dunwiddie, T. V., and Masino, S. A. (2001). The Role and Regulation of Adenosine in the Central Nervous System. Annu. Rev. Neurosci. 24, 31–55. doi:10.1146/annurev.neuro.24.1.31
During, M. J., and Spencer, D. D. (1992). Adenosine: a Potential Mediator of Seizure Arrest and Postictal Refractoriness. Ann. Neurol. 32, 618–624. doi:10.1002/ana.410320504
El Yacoubi, M., Ledent, C., Parmentier, M., Costentin, J., and Vaugeois, J. M. (2009). Adenosine A2A Receptor Deficient Mice Are Partially Resistant to Limbic Seizures. Naunyn Schmiedeb. Arch. Pharmacol. 380, 223–232. doi:10.1007/s00210-009-0426-8
Faingold, C. L., Randall, M., and Kommajosyula, S. P. (2016). Susceptibility to Seizure-Induced Sudden Death in DBA/2 Mice Is Altered by Adenosine. Epilepsy Res. 124, 49–54. doi:10.1016/j.eplepsyres.2016.05.007
Fredholm, B. B., IJzerman, A. P., Jacobson, K. A., Klotz, K. N., and Linden, J. (2001). International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol. Rev. 53, 527–552.
Fredholm, B. B., Chen, J. F., Masino, S. A., and Vaugeois, J. M. (2005). Actions of Adenosine at its Receptors in the CNS: Insights from Knockouts and Drugs. Annu. Rev. Pharmacol. Toxicol. 45, 385–412. doi:10.1146/annurev.pharmtox.45.120403.095731
Fukuda, M., Suzuki, Y., Hino, H., and Ishii, E. (2012). Over-activation of Adenosine A(2A) Receptors and Sudden Unexpected Death in Epilepsy. Epilepsy Behav. 23, 387–388. doi:10.1016/j.yebeh.2011.12.022
Gao, M., Liu, C. L., Yang, S., Jin, G. Z., Bunney, B. S., and Shi, W. X. (2007). Functional Coupling between the Prefrontal Cortex and Dopamine Neurons in the Ventral Tegmental Area. J. Neurosci. 27, 5414–5421. doi:10.1523/JNEUROSCI.5347-06.2007
Gomez-Castro, F., Zappettini, S., Pressey, J. C., Silva, C. G., Russeau, M., Gervasi, N., et al. (2021). Convergence of Adenosine and GABA Signaling for Synapse Stabilization during Development. Science 374, eabk2055. doi:10.1126/science.abk2055
Gonçalves, F. Q., Lopes, J. P., Silva, H. B., Lemos, C., Silva, A. C., Gonçalves, N., et al. (2019). Synaptic and Memory Dysfunction in a β-amyloid Model of Early Alzheimer's Disease Depends on Increased Formation of ATP-Derived Extracellular Adenosine. Neurobiol. Dis. 132, 104570. doi:10.1016/j.nbd.2019.104570
He, X., Chen, F., Zhang, Y., Gao, Q., Guan, Y., Wang, J., et al. (2020). Upregulation of Adenosine A2A Receptor and Downregulation of GLT1 Is Associated with Neuronal Cell Death in Rasmussen's Encephalitis. Brain Pathol. 30, 246–260. doi:10.1111/bpa.12770
Hirsch, L. J. (2010). Is Sudden Unexpected Death in Epilepsy Due to Postictal Brain Shutdown? Ann. Neurol. 68, 773–775. doi:10.1002/ana.22242
Insel, N., Patron, L. A., Hoang, L. T., Nematollahi, S., Schimanski, L. A., Lipa, P., et al. (2012). Reduced Gamma Frequency in the Medial Frontal Cortex of Aged Rats during Behavior and Rest: Implications for Age-Related Behavioral Slowing. J. Neurosci. 32, 16331–16344. doi:10.1523/JNEUROSCI.1577-12.2012
Kaufman, K. R., and Sachdeo, R. C. (2003). Caffeinated Beverages and Decreased Seizure Control. Seizure 12, 519–521. doi:10.1016/s1059-1311(03)00048-7
Kerkhofs, A., Canas, P. M., Timmerman, A. J., Heistek, T. S., Real, J. I., Xavier, C., et al. (2018). Adenosine A2A Receptors Control Glutamatergic Synaptic Plasticity in Fast Spiking Interneurons of the Prefrontal Cortex. Front. Pharmacol. 9, 133. doi:10.3389/fphar.2018.00133
Kirk, I. P., and Richardson, P. J. (1994). Adenosine A2a Receptor-Mediated Modulation of Striatal [3H]GABA and [3H]acetylcholine Release. J. Neurochem. 62, 960–966. doi:10.1046/j.1471-4159.1994.62030960.x
Kommajosyula, S. P., Randall, M. E., and Faingold, C. L. (2016). Inhibition of Adenosine Metabolism Induces Changes in Post-ictal Depression, Respiration, and Mortality in Genetically Epilepsy Prone Rats. Epilepsy Res. 119, 13–19. doi:10.1016/j.eplepsyres.2015.11.001
Kuo, J., Zhao, W., Li, C. S., Kennedy, J. D., and Seyal, M. (2016). Postictal Immobility and Generalized EEG Suppression Are Associated with the Severity of Respiratory Dysfunction. Epilepsia 57, 412–417. doi:10.1111/epi.13312
Li, R., and Buchanan, G. F. (2019). Scurrying to Understand Sudden Expected Death in Epilepsy: Insights From Animal Models. Epilepsy Curr. 19, 390–396. doi:10.1177/1535759719874787
Li, W., Silva, H. B., Real, J., Wang, Y. M., Rial, D., Li, P., et al. (2015). Inactivation of Adenosine A2A Receptors Reverses Working Memory Deficits at Early Stages of Huntington's Disease Models. Neurobiol. Dis. 79, 70–80. doi:10.1016/j.nbd.2015.03.030
Maguire, M. J., Jackson, C. F., Marson, A. G., and Nolan, S. J. (2016). Treatments for the Prevention of Sudden Unexpected Death in Epilepsy (SUDEP). Cochrane Database Syst. Rev. 7, CD011792. doi:10.1002/14651858.CD011792.pub2
Masino, S. A., Kawamura, M., and Ruskin, D. N. (2014). Adenosine Receptors and Epilepsy: Current Evidence and Future Potential. Int. Rev. Neurobiol. 119, 233–255. doi:10.1016/B978-0-12-801022-8.00011-8
Massey, C. A., Sowers, L. P., Dlouhy, B. J., and Richerson, G. B. (2014). Mechanisms of Sudden Unexpected Death in Epilepsy: the Pathway to Prevention. Nat. Rev. Neurol. 10, 271–282. doi:10.1038/nrneurol.2014.64
Massimini, M., Huber, R., Ferrarelli, F., Hill, S., and Tononi, G. (2004). The Sleep Slow Oscillation as a Traveling Wave. J. Neurosci. 24, 6862–6870. doi:10.1523/JNEUROSCI.1318-04.2004
Minic, Z., O'Leary, D. S., and Scislo, T. J. (2015). NTS Adenosine A2a Receptors Inhibit the Cardiopulmonary Chemoreflex Control of Regional Sympathetic Outputs via a GABAergic Mechanism. Am. J. Physiol. Heart Circ. Physiol. 309, H185–H197. doi:10.1152/ajpheart.00838.2014
Nicolaidis, R., Bruno, A. N., Sarkis, J. J., and Souza, D. O. (2005). Increase of Adenine Nucleotide Hydrolysis in Rat Hippocampal Slices after Seizures Induced by Quinolinic Acid. Neurochem. Res. 30, 385–390. doi:10.1007/s11064-005-2613-4
O'Brien, D. R. (1988). The Adenosine Hypothesis of Epilepsy. Med. Hypotheses 27, 281–284. doi:10.1016/0306-9877(88)90007-2
Palchykova, S., Winsky-Sommerer, R., Shen, H. Y., Boison, D., Gerling, A., and Tobler, I. (2010). Manipulation of Adenosine Kinase Affects Sleep Regulation in Mice. J. Neurosci. 30, 13157–13165. doi:10.1523/JNEUROSCI.1359-10.2010
Pansani, A. P., Colugnati, D. B., Scorza, C. A., de Almeida, A. C., Cavalheiro, E. A., and Scorza, F. A. (2016). Furthering Our Understanding of SUDEP: the Role of Animal Models. Expert Rev. Neurother. 16, 561–572. doi:10.1586/14737175.2016.1169925
Patodia, S., Paradiso, B., Garcia, M., Ellis, M., Diehl, B., Thom, M., et al. (2020). Adenosine Kinase and Adenosine Receptors A1 R and A2A R in Temporal Lobe Epilepsy and Hippocampal Sclerosis and Association with Risk Factors for SUDEP. Epilepsia 61, 787–797. doi:10.1111/epi.16487
Phillis, J. W., Scislo, T. J., and O'Leary, D. S. (1997). Purines and the Nucleus Tractus Solitarius: Effects on Cardiovascular and Respiratory Function. Clin. Exp. Pharmacol. Physiol. 24, 738–742. doi:10.1111/j.1440-1681.1997.tb02124.x
Pietersen, A. N., Lancaster, D. M., Patel, N., Hamilton, J. B., and Vreugdenhil, M. (2009). Modulation of Gamma Oscillations by Endogenous Adenosine through A1 and A2A Receptors in the Mouse hippocampus. Neuropharmacology 56, 481–492. doi:10.1016/j.neuropharm.2008.10.001
Rebola, N., Pinheiro, P. C., Oliveira, C. R., Malva, J. O., and Cunha, R. A. (2003). Subcellular Localization of Adenosine A(1) Receptors in Nerve Terminals and Synapses of the Rat hippocampus. Brain Res. 987, 49–58. doi:10.1016/s0006-8993(03)03247-5
Rebola, N., Coelho, J. E., Costenla, A. R., Lopes, L. V., Parada, A., Oliveira, C. R., et al. (2003). Decrease of Adenosine A1 Receptor Density and of Adenosine Neuromodulation in the hippocampus of Kindled Rats. Eur. J. Neurosci. 18, 820–828. doi:10.1046/j.1460-9568.2003.02815.x
Rebola, N., Canas, P. M., Oliveira, C. R., and Cunha, R. A. (2005). Different Synaptic and Subsynaptic Localization of Adenosine A2A Receptors in the hippocampus and Striatum of the Rat. Neuroscience 132, 893–903. doi:10.1016/j.neuroscience.2005.01.014
Rebola, N., Rodrigues, R. J., Lopes, L. V., Richardson, P. J., Oliveira, C. R., and Cunha, R. A. (2005). Adenosine A1 and A2A Receptors Are Co-expressed in Pyramidal Neurons and Co-localized in Glutamatergic Nerve Terminals of the Rat hippocampus. Neuroscience 133, 79–83. doi:10.1016/j.neuroscience.2005.01.054
Rebola, N., Porciúncula, L. O., Lopes, L. V., Oliveira, C. R., Soares-da-Silva, P., and Cunha, R. A. (2005). Long-term Effect of Convulsive Behavior on the Density of Adenosine A1 and A 2A Receptors in the Rat Cerebral Cortex. Epilepsia 46 Suppl 5, 159–165. doi:10.1111/j.1528-1167.2005.01026.x
Reis, S. L., Silva, H. B., Almeida, M., Cunha, R. A., Simões, A. P., and Canas, P. M. (2019). Adenosine A1 and A2A Receptors Differently Control Synaptic Plasticity in the Mouse Dorsal and Ventral hippocampus. J. Neurochem. 151, 227–237. doi:10.1111/jnc.14816
Rombo, D. M., Newton, K., Nissen, W., Badurek, S., Horn, J. M., Minichiello, L., et al. (2015). Synaptic Mechanisms of Adenosine A2A Receptor-Mediated Hyperexcitability in the hippocampus. Hippocampus 25, 566–580. doi:10.1002/hipo.22392
Ryvlin, P., Nashef, L., Lhatoo, S. D., Bateman, L. M., Bird, J., Bleasel, A., et al. (2013). Incidence and Mechanisms of Cardiorespiratory Arrests in Epilepsy Monitoring Units (MORTEMUS): a Retrospective Study. Lancet Neurol. 12, 966–977. doi:10.1016/S1474-4422(13)70214-X
Samsonsen, C., Bråthen, G., Reimers, A., Helde, G., and Brodtkorb, E. (2013). Is Dietary Caffeine Involved in Seizure Precipitation? Epilepsy Behav. 28, 147–150. doi:10.1016/j.yebeh.2013.05.003
Sandau, U. S., Colino-Oliveira, M., Jones, A., Saleumvong, B., Coffman, S. Q., Liu, L., et al. (2016). Adenosine Kinase Deficiency in the Brain Results in Maladaptive Synaptic Plasticity. J. Neurosci. 36, 12117–12128. doi:10.1523/JNEUROSCI.2146-16.2016
Saransaari, P., and Oja, S. S. (2005). GABA Release Modified by Adenosine Receptors in Mouse Hippocampal Slices under Normal and Ischemic Conditions. Neurochem. Res. 30, 467–473. doi:10.1007/s11064-005-2682-4
Scheffer, I. E., and Nabbout, R. (2019). SCN1A-related Phenotypes: Epilepsy and beyond. Epilepsia 60 Suppl 3, S17–S24. doi:10.1111/epi.16386
Sebastião, A. M., and Ribeiro, J. A. (2009). Adenosine Receptors and the Central Nervous System. Handb. Exp. Pharmacol. 193, 471–534. doi:10.1007/978-3-540-89615-9_16
Seo, S., Song, Y., Gu, S. M., Min, H. K., Hong, J. T., Cha, H. J., et al. (2020). D-limonene Inhibits Pentylenetetrazole-Induced Seizure via Adenosine A2A Receptor Modulation on GABAergic Neuronal Activity. Int. J. Mol. Sci. 21. doi:10.3390/ijms21239277
Shen, H. Y., Coelho, J. E., Ohtsuka, N., Canas, P. M., Day, Y. J., Huang, Q. Y., et al. (2008). A Critical Role of the Adenosine A2A Receptor in Extrastriatal Neurons in Modulating Psychomotor Activity as Revealed by Opposite Phenotypes of Striatum and Forebrain A2A Receptor Knock-Outs. J. Neurosci. 28, 2970–2975. doi:10.1523/JNEUROSCI.5255-07.2008
Shen, H. Y., Li, T., and Boison, D. (2010). A Novel Mouse Model for Sudden Unexpected Death in Epilepsy (SUDEP): Role of Impaired Adenosine Clearance. Epilepsia 51, 465–468. doi:10.1111/j.1528-1167.2009.02248.x
Shen, H. Y., Sun, H., Hanthorn, M. M., Zhi, Z., Lan, J. Q., Poulsen, D. J., et al. (2014). Overexpression of Adenosine Kinase in Cortical Astrocytes and Focal Neocortical Epilepsy in Mice. J. Neurosurg. 120, 628–638. doi:10.3171/2013.10.JNS13918
Shen, H. Y., van Vliet, E. A., Bright, K. A., Hanthorn, M., Lytle, N. K., Gorter, J., et al. (2015). Glycine Transporter 1 Is a Target for the Treatment of Epilepsy. Neuropharmacology 99, 554–565. doi:10.1016/j.neuropharm.2015.08.031
Shorvon, S., and Tomson, T. (2011). Sudden Unexpected Death in Epilepsy. Lancet 378, 2028–2038. doi:10.1016/S0140-6736(11)60176-1
Simões, A. P., Machado, N. J., Gonçalves, N., Kaster, M. P., Simões, A. T., Nunes, A., et al. (2016). Adenosine A2A Receptors in the Amygdala Control Synaptic Plasticity and Contextual Fear Memory. Neuropsychopharmacology 41, 2862–2871. doi:10.1038/npp.2016.98
Sperling, M. R. (2001). Sudden Unexplained Death in Epilepsy. Epilepsy Curr. 1, 21–23. doi:10.1046/j.1535-7597.2001.00012.x
Stöllberger, C., and Finsterer, J. (2004). Cardiorespiratory Findings in Sudden Unexplained/unexpected Death in Epilepsy (SUDEP). Epilepsy Res. 59, 51–60. doi:10.1016/j.eplepsyres.2004.03.008
Surges, R., Thijs, R. D., Tan, H. L., and Sander, J. W. (2009). Sudden Unexpected Death in Epilepsy: Risk Factors and Potential Pathomechanisms. Nat. Rev. Neurol. 5, 492–504. doi:10.1038/nrneurol.2009.118
Tescarollo, F. C., Rombo, D. M., DeLiberto, L. K., Fedele, D. E., Alharfoush, E., Tomé, Â. R., et al. (2020). Role of Adenosine in Epilepsy and Seizures. J. Caffeine Adenosine Res. 10, 45–60. doi:10.1089/caff.2019.0022
Tetzlaff, W., Schubert, P., and Kreutzberg, G. W. (1987). Synaptic and Extrasynaptic Localization of Adenosine Binding Sites in the Rat hippocampus. Neuroscience 21, 869–875. doi:10.1016/0306-4522(87)90043-1
Tu, E., Bagnall, R. D., Duflou, J., and Semsarian, C. (2011). Post-mortem Review and Genetic Analysis of Sudden Unexpected Death in Epilepsy (SUDEP) Cases. Brain Pathol. 21, 201–208. doi:10.1111/j.1750-3639.2010.00438.x
Wenzel, M., Hamm, J. P., Peterka, D. S., and Yuste, R. (2017). Reliable and Elastic Propagation of Cortical Seizures In Vivo. Cell Rep. 19, 2681–2693. doi:10.1016/j.celrep.2017.05.090
Wieraszko, A., Goldsmith, G., and Seyfried, T. N. (1989). Stimulation-dependent Release of Adenosine Triphosphate from Hippocampal Slices. Brain Res. 485, 244–250. doi:10.1016/0006-8993(89)90567-2
Williams, A. J., Zhou, C., and Sun, Q. Q. (2016). Enhanced Burst-Suppression and Disruption of Local Field Potential Synchrony in a Mouse Model of Focal Cortical Dysplasia Exhibiting Spike-Wave Seizures. Front. Neural Circuits 10, 93. doi:10.3389/fncir.2016.00093
Wilson, C. G., Martin, R. J., Jaber, M., Abu-Shaweesh, J., Jafri, A., Haxhiu, M. A., et al. (2004). Adenosine A2A Receptors Interact with GABAergic Pathways to Modulate Respiration in Neonatal Piglets. Respir. Physiol. Neurobiol. 141, 201–211. doi:10.1016/j.resp.2004.04.012
Zeraati, M., Mirnajafi-Zadeh, J., Fathollahi, Y., Namvar, S., and Rezvani, M. E. (2006). Adenosine A1 and A2A Receptors of Hippocampal CA1 Region Have Opposite Effects on Piriform Cortex Kindled Seizures in Rats. Seizure 15, 41–48. doi:10.1016/j.seizure.2005.10.006
Keywords: adenosine A2A receptor, NTS, nucleus tractus solitarius, brainstem, SUDEP (sudden unexplained death in epilepsy), local field potential, adenosine kinase
Citation: Shen H-Y, Baer SB, Gesese R, Cook JM, Weltha L, Coffman SQ, Wu J, Chen J-F, Gao M and Ji T (2022) Adenosine-A2A Receptor Signaling Plays a Crucial Role in Sudden Unexpected Death in Epilepsy. Front. Pharmacol. 13:910535. doi: 10.3389/fphar.2022.910535
Received: 01 April 2022; Accepted: 16 May 2022;
Published: 09 June 2022.
Edited by:
Francisco Ciruela, University of Barcelona, SpainReviewed by:
Rodrigo A. Cunha, University of Coimbra, PortugalAnna Maria Pugliese, University of Florence, Italy
Copyright © 2022 Shen, Baer, Gesese, Cook, Weltha, Coffman, Wu, Chen, Gao and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai-Ying Shen, aHNoZW5AZG93bmV1cm9iaW9sb2d5Lm9yZw==