 Daohua Jiang
Daohua Jiang Jiangtao Zhang
Jiangtao Zhang Zhanyi Xia
Zhanyi Xia- 1Laboratory of Soft Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Key Laboratory of Molecular Biophysics of MOE, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, China
Voltage-gated sodium (NaV) channels are responsible for the rapid rising-phase of action potentials in excitable cells. Over 1,000 mutations in NaV channels are associated with human diseases including epilepsy, periodic paralysis, arrhythmias and pain disorders. Natural toxins and clinically-used small-molecule drugs bind to NaV channels and modulate their functions. Recent advances from cryo-electron microscopy (cryo-EM) structures of NaV channels reveal invaluable insights into the architecture, activation, fast inactivation, electromechanical coupling, ligand modulation and pharmacology of eukaryotic NaV channels. These structural analyses not only demonstrate molecular mechanisms for NaV channel structure and function, but also provide atomic level templates for rational development of potential subtype-selective therapeutics. In this review, we summarize recent structural advances of eukaryotic NaV channels, highlighting the structural features of eukaryotic NaV channels as well as distinct modulation mechanisms by a wide range of modulators from natural toxins to synthetic small-molecules.
Introduction
Voltage-gated sodium (NaV) channels are a family of integrated membrane proteins, which selectively conduct sodium ions across cell membrane in response to depolarizing stimuli (Catterall, 2000; Hille, 2001). The primary function of NaV channels was related to the generation of action potentials (Hodgkin And Huxley, 1952a; Hodgkin And Huxley, 1952b). Subsequently, the voltage-dependent activation, sodium selectivity, fast inactivation and components of NaV channels were characterized by extensive biophysical and biochemical studies (Armstrong and Bezanilla, 1973; Tamkun and Catterall, 1981; Weigele and Barchi, 1982; Hartshorne and Catterall, 1984; Stühmer et al., 1987). Since the first NaV channel gene was cloned by Noda in 1984 (Noda et al., 1984), nine highly conserved NaV channel subtypes (NaV1.1- NaV1.9) in humans have been identified (Catterall et al., 2005). These channels have specific tissue-expression patterns. NaV1.1, NaV1.2 and NaV1.3 are mainly expressed in the central nervous system (CNS), which are crucial for nerve excitability. Hundreds of mutations in these channels cause inherited epilepsy and other form diseases of hyperexcitability (Meisler and Kearney, 2005; Catterall et al., 2008; Catterall, 2014; Huang et al., 2017). Phenytoin, lamotrigine, and carbamazepine are clinical drugs for treatment of epilepsy that act as NaV channel blockers (Zuliani et al., 2012; Catterall, 2014). NaV1.4 and NaV1.5 are predominantly expressed in skeletal muscle and cardiomyocytes, respectively. Malfunction of these two NaV channels are associated with periodic paralysis, myotonia and cardiac arrhythmias (Sokolov et al., 2007; Catterall et al., 2020a). Class I anti-arrhythmic drugs, which block NaV1.5 to remove the abnormal component, are broadly used for treating arrhythmias. Based on the rate of binding and unbinding, the Class I anti-arrhythmic drugs are divided into three subclasses: IA (e.g., quinidine), IB (e.g., lidocaine) and IC (e.g., flecainide) (Kowey, 1998; Huang et al., 2020). NaV1.6 is widely distributed in both CNS and the peripheral nervous system (PNS). The unique features of NaV1.6 such as generating persistent current and resurgent current, contribute to neuronal excitability and repetitive neuronal firing (Raman and Bean, 1997; Raman et al., 1997). Mutations in NaV1.6 are related to epilepsy, ataxia and dystonia (O'Brien and Meisler, 2013). NaV1.7, NaV1.8 and NaV1.9 are highly expressed in PNS, which are closely related to pain perception (Bennett et al., 2019; Dib-Hajj and Waxman, 2019). NaV1.7 was also reported to be essential for odour sensation (Weiss et al., 2011). Many programs have been established in searching for NaV1.7 and NaV1.8 selective inhibitors as potential analgesics (Kingwell, 2019; Alsaloum et al., 2020). In addition, a wide range of natural toxins from animal or plant venoms target the NaV channels and modulate channel functions by binding in at least six distinct receptor sites (Catterall et al., 2007).
Further studies revealed that NaV channels are widely distributed in eukaryotes, as well as in prokaryotes (Koishi et al., 2004; Ren et al., 2001) and marine unicellular phytoplankton (Helliwell et al., 2020; Helliwell et al., 2019), highlighting the evolutionary conservation of NaV channels. The metazoan NaV channels are composed of a large pore-forming α-subunit and one or two auxiliary β-subunits (β1-β4) (Figure 1A) (Catterall, 2000; Hartshorne and Catterall, 1984; Isom et al., 1994; O'Malley and Isom, 2015). The α-subunit consists of 24-transmembrane helices, which are divided into four homologous domains (DI-DIV). The four domains are connected by intracellular loops. The loop between DIII and DIV contains a highly conserved fast inactivation gate, which mediates the fast inactivation of NaV channels (West et al., 1992; Goldin, 2003). The first four transmembrane segments (S1-S4) form the voltage-sensing domain (VSD), the S5 and S6 form the pore module (PM). Distinct from the metazoan NaV channels, NaV channels from bacteria and the unicellular phytoplankton are formed by four identical protomers in a homotetrameric organization (Ren et al., 2001; Helliwell et al., 2020). In 2001, Sato pioneered the structural study of NaV channels reporting a ∼19 Å cryo-electron microscopy (cryo-EM) map of NaV channel from the electrical eel (Sato et al., 2001). However, the low-resolution map did not permit model building of the NaV channel. The crystal structure of voltage-gated potassium (KV) channel also provided useful homologous template for NaV channels (Long et al., 2005). Since 2011, Catterall lab and other groups reported high-resolution crystal structures of prokaryotic NaV (BacNaV) channels (Payandeh et al., 2011; Payandeh et al., 2012; Zhang et al., 2012; Lenaeus et al., 2017; Sula et al., 2017; Wisedchaisri et al., 2019), which revealed structural basis for bacterial NaV channel architecture, activation and inactivation, gating, ion conductance and selectivity at atomic level. These bacterial NaV channels had also been used as templates to investigate the structural pharmacology of human NaV channels (Bagnéris et al., 2014; Boiteux et al., 2014; Ahuja et al., 2015; Tang et al., 2016; Gamal El-Din et al., 2018; Wisedchaisri et al., 2021). Recent advances from cryo-EM structures of the eukaryotic NaV channel NaVPas (Shen et al., 2017; Shen et al., 2018; Clairfeuille et al., 2019), NaV1.4 (Yan et al., 2017; Pan et al., 2018), NaV1.7 (Shen et al., 2019), NaV1.2 (Pan et al., 2019), NaV1.5 (Jiang et al., 2020; Jiang et al., 2021a; Li et al., 2021a; Li et al., 2021b; Jiang et al., 2021c), NaV1.1 (Pan et al., 2021) and NaV1.3 (Li et al., 2022) reveal detailed structural basis for the eukaryotic NaV channel subunits assembly, fast inactivation, modulation by natural animal toxins, and pore blockade by anti-arrhythmic drugs (Figures 1B,C). In this review, we highlight the structural features of the eukaryotic NaV channels and the distinct molecular mechanisms for NaV channel modulation by natural toxins and clinical drugs.
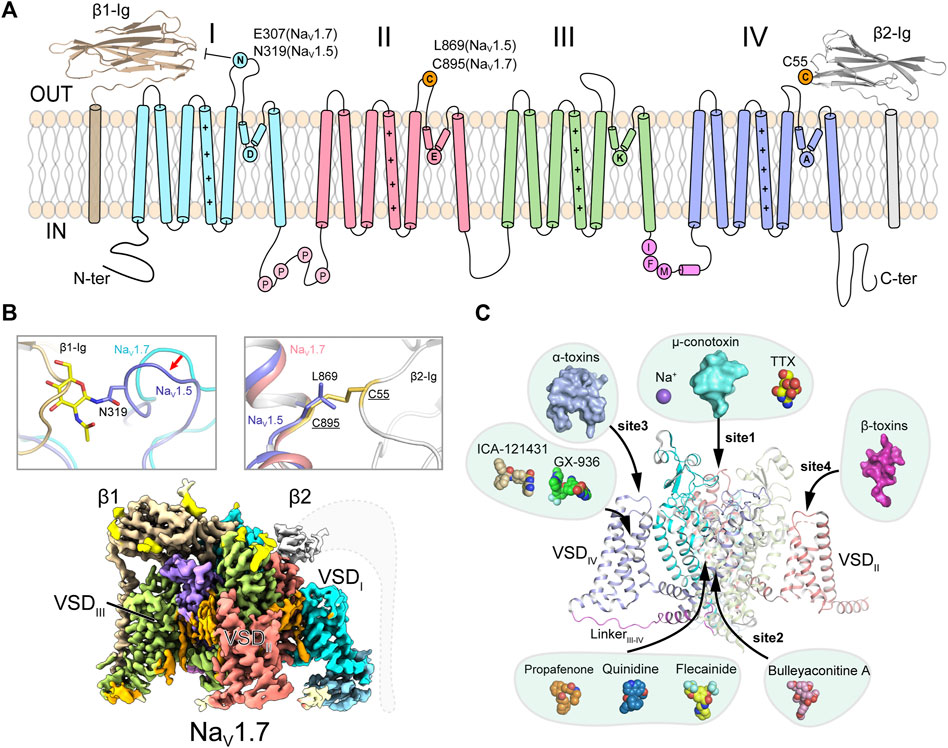
FIGURE 1. Topology and architecture of mammalian NaV channels. (A) Topology of mammalian NaV channels. DI, DII, DIII, IFM-motif and DIV of α-subunit, β1 and β2 subunit are colored in cyan, red, green, purple, blue, brown and gray, respectively. A unique N-glycosylation site in NaV1.5 and the cysteine residue for linking β2 are highlighted. P represents phosphorylation sites. (B) Subunits assembly of mammalian NaV channels. A sugar moiety of N319 glycosylation and L869 block the binding of β1 and β2 to NaV1.5 (PDB code: 6UZ3), respectively (Upper panels). Cryo-EM map of human NaV1.7/β1/β2 (lower panel, EMDB code: EMD-33292). Dashed lines indicate the invisible region of β2 subunit. (C) Multiple ligand binding sites in mammalian NaV channel α-subunit. Sodium ion and small-molecule modulators shown in spheres, polypeptide toxins shown in surface.
Assembly of Voltage-Gated Sodium Channels
The homotetrameric bacterial NaVAb had been an ideal model for investigating molecular mechanisms of NaV channel activation, inactivation, gating, electromechanical coupling and mutation induced channelopathies (Payandeh et al., 2011; Payandeh et al., 2012; Lenaeus et al., 2017; Jiang et al., 2018; Wisedchaisri et al., 2019) (Reviewed in Ref (Catterall et al., 2020b)). However, there are significant structural and functional differences between the homotetrameric BacNaV channels and the asymmetric eukaryotic NaV channels. The eukaryotic NaV channels withheld their detailed structural features owing to their extremely low yield of recombinant expression and sample heterogeneity. In 2017, Nieng Yan lab reported the ground breaking high-resolution cryo-EM structure of a eukaryotic NaV channel NaVPas from the American cockroach (Shen et al., 2017). Although NaVPas was found to be non-functional in heterologous expression system and lack of the conserved fast inactivation gate, the NaVPas structure does provide important insights into the architecture of NaV channel α-subunit, four VSDs in distinct conformations and the asymmetric selectivity filter (SF). In addition, the structure proved that human NaV channels could possibly be resolved to high-resolution by the advanced cryo-EM technique. After extensive efforts had been conducted to improve the sample quality (Jiang et al., 2021b; Shen et al., 2021), cryo-EM structures of the electrical eel and human NaV1.4-β1, human NaV1.7-β1-β2, human NaV1.2-β2, rat and human NaV1.5, human NaV1.1-β4 and human NaV1.3-β1-β2 were subsequently reported at 3–4 Å resolution (Yan et al., 2017; Pan et al., 2018; Pan et al., 2019; Shen et al., 2019; Jiang et al., 2020; Li et al., 2021b; Pan et al., 2021; Li et al., 2022). These structures reveal detailed insights into the assembly of NaV channel α- and β-subunits, allosteric inhibition mechanism of the fast inactivation and multiple ligand binding sites (Figures 1B,C).
These high-resolution structures suggest that the overall structures of the NaV channels are structurally conserved. The transmembrane core-regions closely resemble the bacterial NaV channels, which are formed similarly in a domain-swapped manner, e.g., VSD and PM of one domain are separated by a S4S5 linker helix, thus VSD of one domain closely engages PM from the other domain. Distinct from the BacNaV channels, the metazoan NaV channels possess large extracellular loops (ECLs) between S5 and S6 helices of each domain. Multiple glycosylation sites on the ECLs were visualized from these structures, which are important for the channel maturation and modulation. On the cytoplasmic side, the N-terminal domain (NTD), long loops of DI-DII and DII-DIII, and the C-terminal domain (CTD) were invisible to cryo-EM, which are probably due to mobility of these regions. These intracellular regions were reported to be essential for NaV channel function and modulation (West et al., 1991; Mantegazza et al., 2001; Scheuer, 2011; Clatot et al., 2012). However, it’s challenging to capture a snapshot of the transient modulation state such as phosphorylation. The relative short loop between DIII-DIV of the mammalian NaV channels, which contains the fast inactivation particle Ile-Phe-Met (IFM) motif, were all clearly resolved in a nearly identical conformation (Pan et al., 2018; Pan et al., 2019; Shen et al., 2019; Jiang et al., 2020; Pan et al., 2021). The IFM-motif binds to a hydrophobic pocket adjacent to the intracellular activation gate, rather than directly blocking the activation gate, elucidating a unique local allosteric inhibition mechanism for the fast inactivation.
The β-subunits (β1-β4) bind to the α-subunit and regulate the NaV channel properties such as cell surface expression, voltage-dependent activation and gating kinetics (Hartshorne and Catterall, 1984; Isom et al., 1994; O'Malley and Isom, 2015; Vijayaragavan et al., 2001). The four β-subunits share conserved topology of an extracellular immunoglobulin-like domain and a single transmembrane helix (Das et al., 2016; Gilchrist et al., 2013; Namadurai et al., 2014). The NaV1.4-β1, NaV1.7-β1-β2, NaV1.2-β2, NaV1.1-β4 and NaV1.3-β1-β2 complex structures confirmed that the β1 noncovalently binds to the α-subunit, whereas β2 or β4 is linked to the α-subunit via a disulfide bond (Figures 1A,B). Surprisingly, the rat and human NaV1.5 structures showed that the β1 binding is blocked by a sugar moiety of a unique glycosylation modification in the ECLI, and the cysteine for linking β2 or β4 is substituted by a Leu869 in NaV1.5 or an Ala822 in NaV1.8 (Figure 1B) (Jiang et al., 2020; Li et al., 2021a). These structural observations demonstrate that the β subunits bind weakly to NaV1.5, which is in agreement with previous biophysical reports that the β subunits have negligible effect on the kinetics or voltage-dependence of NaV1.5 (Makita et al., 1994; Qu et al., 1995). However, pathogenic mutations in the NaV β-subunits were reported to be associated with cardiac arrhythmias (Ruan et al., 2009; Watanabe et al., 2009; Olesen et al., 2011). These results indicate that the β-subunits might be important for NaV1.5 folding or trafficking to the plasma membrane, rather than modifying NaV1.5 functional properties. It has also been reported that the β-subunits play other physiological functions unrelated to NaV1.5, such as cell adhesion in the cardiomyocytes (Patino et al., 2011; Namadurai et al., 2015).
Ion Path of NaV Channels
Hille tested the sodium selectivity of NaV channels and estimated a ∼3.0 Å by ∼5.0 Å constriction site at the extracellular side (Hille, 1971a; b; 1972; 1975). Point-mutagenesis studies revealed that an Asp in DI, Glu in DII, Lys in DIII and Ala in DIV, often termed the DEKA-locus, are responsible for the sodium selectivity (Favre et al., 1996; Sun et al., 1997). The crystal structure of the bacterial NaVAb provides the first atomic model for investigating the molecular mechanism of sodium selectivity (Payandeh et al., 2011). At the center of NaV channels, four pore-loops (P-loop) and S6 helices form an ion conductance path across the membrane. The ion path consists of a narrow extracellular constriction site termed selectivity filter (SF), a large central cavity (CC) and a size-variable intracellular activation gate (AG) (Figure 2A). Unlike the asymmetric DEKA-locus of the metazoan NaV channels, four identical P-loops (TLESWSM) of NaVAb constitute the SF with diameter of ∼4.6 Å by ∼4.6 Å, close to Hille’s estimation (Hille, 1975; Payandeh et al., 2011). The SF of NaVAb is significantly wider than that of potassium channels, suggesting that sodium may pass through the SF in partially hydrated form (Doyle et al., 1998; Hille, 1971a; Payandeh et al., 2011; Yellen, 2002). The four Glu at +3 position of the SF loop determine sodium selectivity, which form a high-field strength site to coordinate sodium ions (Figure 2B). Substitution of the TLESWSM with TLDSWDD converts the sodium channel to a highly calcium selective channel (Yue et al., 2002; Tang et al., 2014), suggesting that the ion path of sodium and calcium channels are closely related. Crystal structure of the chimeric calcium channel CaVAb revealed high affinity binding sites for Ca2+ supporting the stepwise “knock-off” permeation mechanism, which are not observed in the BacNaV channel structures (Payandeh et al., 2011; Zhang et al., 2012; Tang et al., 2014). These observations suggested that the NaV and CaV channel are evolutionarily highly-related, the subtle compositional differences in the SFs discriminate Na+ and Ca2+ ions. Based on the BacNaV structures and molecular dynamics simulation studies, potential models had been proposed to explain the sodium selectivity (Corry and Thomas, 2012; Chakrabarti et al., 2013; Naylor et al., 2016). However, these possible mechanisms may not be fully applicable to the metazoan NaV channels, because the asymmetric DEKA-locus is absent in the BacNaV channels.
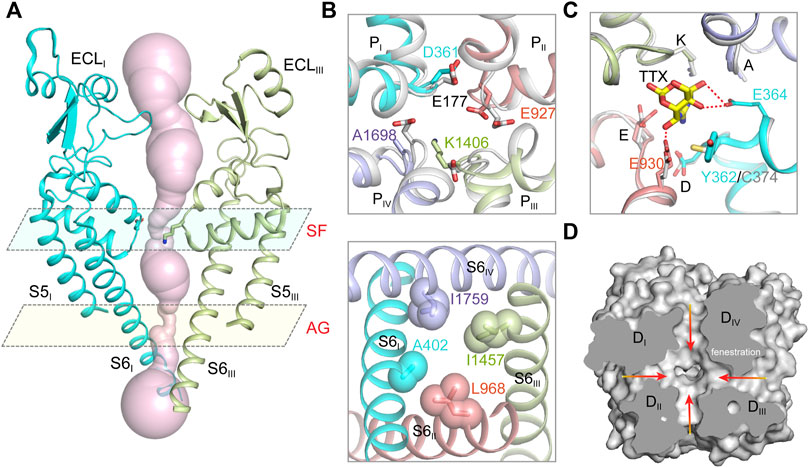
FIGURE 2. The ion path of mammalian NaV channels. (A) The pore radii of human NaV1.7 (PDB code: 7XM9) calculated by HOLE. Only two opposing subunits shown for clarity. The constriction sites of selectivity filter (SF) and intracellular activation gate (AG) are highlighted. (B) A close-up view of the SF (top panel) and AG (bottom panel) of human NaV1.7 (PDB code: 7XM9). The SF of NaV1.7 was aligned with the SF of NaVAb (gray, PDB code: 3RVY). The side-chains of key residues in the SF and the AG are depicted in sticks. (C) The TTX binding site in NaV1.7 (PDB code: 6J8I). The TTX-insensitive channel NaV1.5 (PDB code: 6UZ3) shown in gray. (D) Fenestrations of human NaV1.7 (PDB code: 7XM9). Red arrows indicate potential access for lipids or pore-blockers.
The cryo-EM structures of metazoan NaV channels revealed that the SF is comparable to that of the BacNaV structures (Jiang et al., 2020; Pan et al., 2019; Pan et al., 2018; Shen et al., 2019; Shen et al., 2017). The Asp in DI and Glu in DII of the DEKA-locus provide the high-field strength site similar to the BacNaV channels (Figure 2B), which coordinates and facilitates partially dehydration of sodium ions (Payandeh et al., 2011; Chakrabarti et al., 2013). The Lys in DIII of the DEKA-locus was consistently found pointing its sidechain deep inside the SF and narrowing the SF in one dimension, which meets Hille’s estimation (Hille, 1971b, 1975; Jiang et al., 2020). A positively-charged Lys embedded in the SF would likely block the ion permeation rather than conduct. Considering pKa value of Lys-NH2 can be dramatically affected by its interacting chemical environments (Isom et al., 2011), one possibility is that the pKa of the Lys is significantly decreased and its positive charge is delocalized by nearby carbonyl groups (Jiang et al., 2020). In this case, the neutralized Lys may serve as an energetically favorable coordinating ligand for Na+ or Li+, whereas the larger alkali cations like K+, Rb+ and Cs+ are energetically unfavorable.
The tetrodotoxin (TTX) from puffer fish is a highly NaV channel specific and highly lethal neurotoxin, which was initially used to discriminate Na+ and K+ currents in the squid giant axons and to map the location of the SF (Kao, 1966; Noda et al., 1989). Based on the binding affinity, NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.6 and NaV1.7 are classified as TTX-sensitive channels with TTX affinity at nanomolar level; NaV1.5, NaV1.8 and NaV1.9 are TTX-insensitive channels with micromolar affinity. A Tyr or Phe residue located in the DI P-loop was found as the key determinant for the TTX sensitivity, this residue is substituted by a Cys or Ser in the TTX-insensitive NaV channels (Sivilotti et al., 1997; Sunami et al., 2000). The detailed binding site for TTX and another guanidinium neurotoxin Saxitoxin (STX), termed site-1, was depicted by the high-resolution cryo-EM complex structures of NaVPas and NaV1.7 (Shen et al., 2018; Shen et al., 2019). The positively-charged TTX or STX forms extensive electrostatic interactions with the negatively-charged vestibule of the NaV channels, that physically blocks ions entering the SF. The Tyr on DI P-loop of NaV1.7 forms an additional π-cation interaction with the TTX or STX, which is absent in NaV1.5 (Figure 2C). Consequently, the affinity of TTX for NaV1.7 is over 500-fold higher than that for NaV1.5. These structures provide an excellent example showing that a single residue at the receptor site could confer isoform selectivity. Recently, guanidinium neurotoxin analogues ST-2262 and ST-2530 were discovered as potent and selective NaV1.7 inhibitors (Pajouhesh et al., 2020), suggesting that the TTX receptor site is a potential therapeutic site. Because the blockade of the guanidinium neurotoxin is state-independent, high affinity and isoform selectivity is critical to minimize potential side effects. The structures of human NaV channel complexed with the guanidinium neurotoxins should facilitate rational development of isoform selective candidate drugs.
The Central Cavity of NaV Channels
The bulky central cavity of NaV channels connects the extracellular SF and the intracellular activation gate, but also opens to four fenestrations formed by adjacent PMs (Figure 2D). Lipids or small-molecule drugs access the central cavity through these connecting tunnels to modulate channel properties. The central cavity accommodates multiple receptor sites for site-2 neurotoxins (Deuis et al., 2017), local anesthetic, anti-epilepsy and antiarrhythmic drugs (Hille, 1977; Hondeghem and Katzung, 1984). Batrachotoxin, veratridine and aconitine are natural alkaloids, which modulate the voltage-dependent activation, inactivation and ion selectivity of NaV channels (Catterall et al., 2007; Quandt and Narahashi, 1982; Ulbricht, 1969). The site-2 neurotoxins usually activate NaV channels at hyperpolarized membrane potential or increase the open probability, thus they are considered as NaV channel activators (Deuis et al., 2017). Bulleyaconitine A (BLA), the active substance of the traditional herb Aconitum bulleyanum plant (Tang et al., 1986; Wang et al., 2007), exhibits strong inhibition of NaV channels in a use-dependent manner. The complex structure of human NaV1.3-β1-β2 with BLA reveals a binding site for the site-2 neurotoxin (Li et al., 2022). BLA binds tightly to the central cavity of NaV1.3 via multiple polar and non-polar interactions with P-loops and S6 helices of DI and DII (Figure 3A). The BLA engages V416 and L420 on DI-S6 helix, N972 and L976 on DII-S6 helix (Figure 3B), which are in line with the biophysical studies that these residues are important for batrachotoxin, grayanotoxin or veratridine binding (Wang and Wang, 1998; Ishii et al., 1999; Wang et al., 2000; Wang et al., 2001). However, DIII-S6 and DIV-S6 helices, which were reported to be involved in batrachotoxin binding (Wang and Wang, 1999; Deuis et al., 2017), have no direct contact with BLA in the NaV1.3-BLA complex structure. These results suggest that the site-2 neurotoxins may bind to overlapping but not identical receptor sites in the central cavity. The binding of BLA partially blocks the ion path, which causes the strong inhibitory effect on NaV1.3. The weak activation potency of BLA probably because BLA sticks into the DI-DII fenestration, which prevents the closure of the pore-lining S6 helices of DI and DII during state transition (Figure 3C). Owing to the shape and chemistry differences, site-2 neurotoxins with strong activation potency such as batrachotoxin and veratridine likely bind in the central cavity partially overlapping with the BLA site, which stabilize the pore in open conformation and cause less pore blockade.
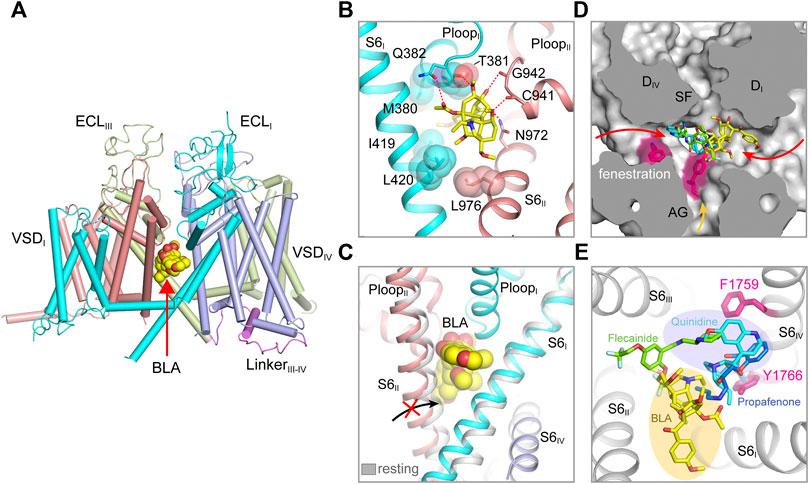
FIGURE 3. Pore modulators of mammalian NaV channels. (A) The binding site of site-2 neurotoxin BLA in NaV1.3 (PDB code: 7W77). BLA is depicted in spheres. (B) A close-up view of the BLA binding site. Key interacting residues are shown side chains in sticks. BLA is shown in sticks. (C) BLA binding prevents S6II helix shifting to the resting state. (D) Superposition of NaV structure bound BLA (PDB code: 7W77), Flecainide (PDB code: 6UZ0), Propafenone (PDB code: 7FBS) and Quinidine (PDB code: 6LQA) in the pore. Arrows indicate possible access path for the drugs. (E) Top-down view of the BLA binding site and the antiarrhythmic drug binding sites from panel (D).
The structures of NaV1.5 in complex with antiarrhythmic drugs flecainide, propafenone and quinidine reveal antiarrhythmic drugs binding sites in the central cavity (Jiang et al., 2021a; Jiang et al., 2020; Li et al., 2021a), which are distinct from the BLA binding site (Figures 3D,E). All three drugs block NaV1.5 in use-dependent manner. The Class IC drug flecainide binds in the central cavity close to DII-DIII fenestration. Its positively-charged piperidine nitrogen lies beneath the SF, which prevents the exit of Na+ from the SF. Meanwhile, the hydrophobic side of the piperidine ring faces the sidechain of Phe1759 (rat NaV1.5), the key residue for antiarrhythmic and local anesthetic drugs binding (Ragsdale et al., 1994). Mutation of this Phe1759 to Ala significantly drops the affinity of flecainide (Liu et al., 2003; Ragsdale et al., 1996; Wang et al., 2003). One of the two hydrophobic trifluoromethoxy tails of flecainide is inserted into the DII-DIII fenestration, which suggests that flecainide may access the binding sites through the fenestrations (Gamal El-Din et al., 2018; Nguyen et al., 2019). Compared to class IA (e.g., quinidine) and IB (e.g., lidocaine) drugs, flecainide is larger in size and more hydrophobic, the higher affinity and slower binding kinetics of flecainide and other class IC drugs likely reflect their stronger interaction with NaV1.5. The structure of human NaV1.5-quinidine reveals a quinidine binding site which is also located in the central cavity (Li et al., 2021a). Quinidine lies under the SF and physically blocks the ion path. The quinolone ring of quinidine overlaps with the piperidine ring of flecainide (Figure 3D). Unlike the flecainide binding without causing obvious local conformational changes, the quinidine binding was reported to induce the side chain rotation of Tyr1767 (human NaV1.5) and a slightly smaller activation gate compared to the human NaV1.5 structure with a Glu1784Lys mutation (Li et al., 2021a; Pan et al., 2021). However, similar “up” and “down” conformations of the Tyr1755 at the equivalent position were also observed in the NaV1.7 structures (Shen et al., 2019). In addition, the side chain rotation of Tyr1767 could also be affected by the pathogenic Glu1784Lys mutation, which alters the NaV1.5 gating by significantly shifting the voltage-dependent activation toward depolarized membrane potential and the voltage-dependent inactivation toward hyperpolarized membrane potential (Makita et al., 2008).
Class IC drug propafenone preferentially blocks open NaV1.5 (Kohlhardt and Fichtner, 1988). The open state structure of NaV1.5 was achieved by importing the IFM/QQQ mutations (termed NaV1.5/QQQ) to remove the fast inactivation gate, also with the help of propafenone blocking the constant opening activity of the NaV1.5/QQQ during protein expression (Jiang et al., 2021a). The NaV1.5/QQQ structure revealed a high affinity binding site for propafenone in the central cavity. Propafenone engages both the Phe1762 and Tyr1769 (rat NaV1.5) on DIV S6 helix by forming π-π stacking interaction and on-edge van der Waals interactions, respectively (Figure 3E). The positively-charged amino group blocks the exit of Na+ from the SF, which is similar to the piperidine of flecainide and the quinolone ring of quinidine (Jiang et al., 2020; Li et al., 2021a). The open activation gate of NaV1.5/QQQ is wide enough for propafenone passing through, which suggests that propafenone probably access its high affinity binding site through the open activation gate. In the resting state, the intracellular activation gate is closed, the antiarrhythmic and local anesthetic drugs may access their receptor sites through the fenestrations in a less efficient manner (Figure 3D) (Gamal El-Din et al., 2018; Catterall et al., 2020a), thus exhibiting low affinity blocking. Structural superposition of the NaV isoforms show that the wall of the central cavity is nearly identical, suggesting it’s very difficult to achieve selective receptor site inside the central cavity. Thus, the state- and use-dependent block of the antiarrhythmic and local anesthetic drugs relies on the membrane potential and firing frequency (Hille, 1977; Hondeghem and Katzung, 1984), which allows the drugs to access their active sites in the favorable states without affecting the channels in unfavorable states (Catterall et al., 2020a).
Fast Inactivation of Sodium Channel
Fast inactivation is one of the hallmark features of eukaryotic NaV channels (Goldin, 2003). The fast inactivation of NaV channels is crucial for preventing hyperexcitability and priming firing frequency (Catterall, 2000; Catterall et al., 2017). In 1973, Armstrong found that the fast inactivation was dramatically destructed by intracellularly applying proteolytic enzyme in the squid giant axons (Armstrong et al., 1973), indicating that the fast inactivation gate is located in the cytosol. Site-direct antibody studies identified that the fast inactivation gate is located in the loop between DIII and DIV (Vassilev et al., 1988; Stühmer et al., 1989). More detailed point mutagenesis studies confirmed a triple-hydrophobic Ile-Phe-Met (IFM) motif in the DIII-DIV loop is responsible for the fast inactivation (West et al., 1992; Eaholtz et al., 1994). The cysteine substitution accessibility results indicated that the IFM-motif is buried in a receptor site inaccessible to solvent during inactivation process (Kellenberger et al., 1996). Further extensive site-directed mutagenesis screening studies mapped the IFM-motif receptor site which is constituted by the S4-S5 linkers of DIII and DIV, and also the cytoplasmic end of DIV-S6 helix (McPhee et al., 1994, 1998; Lerche et al., 1997; Smith and Goldin, 1997). Bases on these results, a hinged-lid mechanism for the fast inactivation was proposed, that is, the IFM-motif acts as a hydrophobic latch that binds to its receptor site to close the activation gate. Furthermore, the fast inactivation was found to be electromechanically coupled to the activation of the VSDIV (Chanda and Bezanilla, 2002; Capes et al., 2013).
The solution structure of the DIII and DIV linker showed that the flexible IFM-motif is tethered to an α-helix (Rohl et al., 1999), suggesting that the IFM-motif is readily available to bind to its receptor site once close to it. The detailed binding mode of the IFM-motif was consistently revealed by the cryo-EM structures of the eukaryotic NaV channels (Jiang et al., 2020; Pan et al., 2019; Pan et al., 2018; Shen et al., 2019; Yan et al., 2017). In agreement with the mutagenesis studies and the hinged-lid model, the IFM-motif is embedded in a hydrophobic receptor site formed by the DIV-S6 helix and the S4-S5 linkers of DIII and DIV, adjacent to the intracellular activation gate (Figure 4A). The binding pose of the IFM-motif is further stabilized by extensive hydrophobic interactions and hydrogen-bond network (Jiang et al., 2020; Pan et al., 2018). In particular, the activated conformation of VSDIII and VSDIV are required for the formation of the IFM-motif receptor site (Jiang et al., 2020). These structures also showed that the activation gate is in the non-conductive state with a diameter of <5 Å, elucidating an allosteric inhibition mechanism for the fast inactivation (Figures 2A, 4A). Meanwhile, the electromechanical coupling was observed to be disrupted by α-Scorpion toxins, which specifically bind to the VSDIV and trap it in a deactivated state (Clairfeuille et al., 2019; Jiang et al., 2021c). There structures provide a structural framework for the fast inactivation of sodium channels. Pathogenic mutations target the fast inactivation gate and cause life-threatening diseases. For example, mapping the location of the arrhythmia associated mutations in NaV1.5 revealed that the gain-of-function mutations are dense around the fast inactivation gate (Jiang et al., 2020). These gain-of-function mutations impair the fast inactivation, thus cause the channel to generate large abnormal persistent currents or repetitive firing that lead to arrhythmias such as Long-QT syndrome type-3 (Clancy and Kass, 2005).
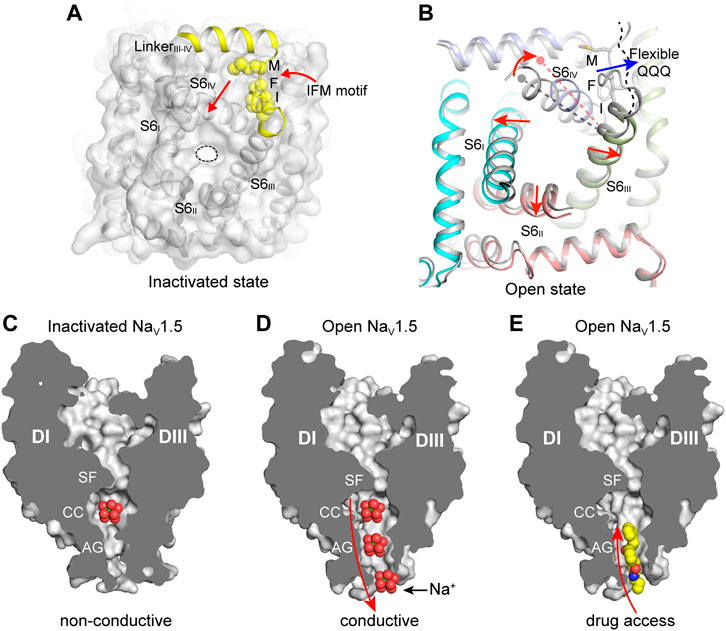
FIGURE 4. The IFM-motif mediated fast inactivation and the open activation gate. (A) Allosteric inhibition of mammalian NaV channels by the IFM-motif (PDB code: 6J8I). The pore domain viewed from cytosol is shown in surface. The IFM-motif shown in yellow spheres. (B) Releasing of the IFM-motif leads to channel opening. Activation gate comparison between rat NaV1.5 (PDB code: 6UZ3) and NaV1.5/QQQ (PDB code: 7FBS). Red arrows indicate the S6 helix shift. (C) Non-conductive ion path of the inactivated state NaV channel (PDB code: 6UZ3). (D) Na+ conductive ion path of the open-state NaV channel (PDB code: 7FBS). (E) The open activation gate of NaV channel provides potential accessing path for the open-state blockers. Propafenone is shown in spheres.
Sodium ions across the ion path are believed to undergo de-hydration by the SF and re-hydration in the central cavity, then pass the activation gate in hydrated form. The open state structures of BacNaV channels have shown that the activation gate is wide enough for conducting hydrated Na+ (Lenaeus et al., 2017; Sula et al., 2017). The overall structure of the open state NaV1.5/QQQ is similar to the inactivated state NaV1.5 structures (Jiang et al., 2021a; Jiang et al., 2020). However, releasing of the IFM-motif from its receptor site caused marked local conformational changes in the activation gate (Figure 4B). Especially, The DIV-S6 helix shifts toward the receptor site by ∼6 Å at the cytoplasmic end. The resulting dilated activation gate of the NaV1.5/QQQ is sufficient for free passing of hydrated Na+, also provides an access for pore-blocking drugs such as propafenone (Figures 4C–E) (Jiang et al., 2021a).
VSD and Gating Modifier Toxins
The VSDs control both the activation and inactivation of the eukaryotic NaV channels in response to membrane potential changes. The molecular gating mechanisms of NaV channels have been reviewed by Ahern (Ahern et al., 2016) and Catterall (Catterall et al., 2017; 2020b). The NaV channel amino acid sequence revealed that the fourth helix (S4) contains at least four positively-charged Arg or Lys (gating-charges) repeated in a pattern of every three-residues (Noda et al., 1984). In light of the sequence and the biophysical results, the “sliding-helix” and “helical-screw” models were soon proposed to explain the voltage sensing, both models suggest that the positive gating-charges on the S4 helix serve as voltage sensors which move up and down in the membrane in response to membrane potential changes (Catterall, 1986; Guy and Seetharamulu, 1986). The BacNaV structures captured in the resting and activated states show that the gating-charges underwent a two helical-turns shift, supporting the “sliding-helix” or “helical-screw” models (Payandeh et al., 2011; Sula et al., 2017; Wisedchaisri et al., 2019; Zhang et al., 2012). Because there is no membrane potential in purification conditions (0 mV), most NaV structures were determined in the activated state (Figures 5A,B). The S4 helix is wrapped by the S1-S3 helices, forming a V-shaped aqueous cleft toward the extracellular side. Three of the four gating-charges in the activated NaVAb structure adopt the activated “up” conformation, which are neutralized by extracellular negatively-charged clusters (ENC). Wisedchaisri obtained the resting state structure of NaVAb by a combination of importing positive voltage-shifting mutations and cysteine disulfide-bond lock (Wisedchaisri et al., 2019). Compared to the activated VSD, the VSD in the resting state shifts the S4 helix two helical-turns downward to the intracellular side, while the conformation of the S1-S3 helices remains unchanged (Figures 5A,B). The inward movement of the S4 helix further bends the S4-S5 linker helix via an elbow-like turn. The twisted S4-S5 linker directly causes the bending and rotation of the S6 helix to close the activation gate. These structures define a possible general mechanism for electromechanical coupling of voltage-gated ion channels.
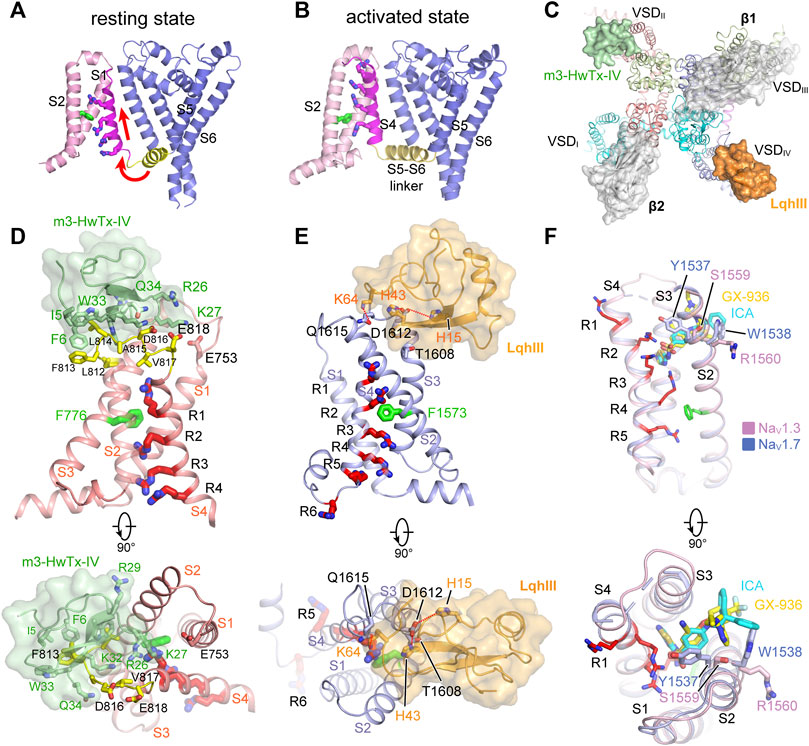
FIGURE 5. GMTs bind to VSDs of NaV channels. (A,B) Resting (PDB code: 6P6W) and activated (PDB code: 3RVY) state structure of NaVAb. Red arrows indicate conformational shifts between resting and activated state NaVAb. The gating charges on the S4 helix shown side chains in sticks. (C) Site-3 toxin LqhIII (PDB code: 7K18) and site-4 toxin HwTxIV (PDB code: 7K48) bind to VSDIV and VSDII of NaV channel, respectively. The Ig-like domain of β1 and β2 subunits project on the VSDIII and VSDI, respectively, which block the accessibility for potential GMTs binding to VSDIII and VSDI. (D) High affinity binding site for site-4 toxin HwTxIV in the deactivated VSDII (PDB code: 7K48). (E) Detailed binding site for α-Scorpion toxin LqhIII in the deactivated VSDIV (PDB code: 7K18). (F) Detailed binding site for aryl sulfonamide derivatives ICA121431 and GX-936 in the activated VSDIV of NaV1.3 (PDB code: 7W7F) and NaV1.7 (PDB code: 5EK0), respectively.
Distinct from the homotetrameric BacNaV channels, metazoan NaV channels possess four non-identical VSDs (Noda et al., 1984; Catterall et al., 2017). The VSDI and VSDII have four gating-charges, VSDIII has five gating-charges, and VSDIV has six to eight gating-charges. The four asymmetric VSDs response asynchronously to membrane depolarization (Chanda and Bezanilla, 2002; Ahern et al., 2016). This asynchronous activation is closely related to the voltage-dependent activation and fast inactivation of NaV channels. According to the asynchronous gating model (Ahern et al., 2016), activation of VSDI-VSDIII precedes VSDIV leading to channel opening, and VSDIV activates subsequently to induce the fast inactivation (Chanda and Bezanilla, 2002; Chanda et al., 2004; Capes et al., 2013). The mammalian NaV channel structures confirmed the asymmetric activation of the four VSDs (Pan et al., 2018; Pan et al., 2019; Shen et al., 2019; Jiang et al., 2020). The VSDI and VSDII closely resemble the activated VSD of NaVAb, R1-R3 adopt the “up” conformation above the hydrophobic constriction stie (HCS). The VSDIII is more activated, K1-R4 point up and R5 is stuck in the HCS. In contrast, the VSDIV is less activated, R1-R4 are located above the HCS, whereas R5-R6 point downward interacting with the intracellular negatively-charged clusters (INC). The VSDIII and VSDIV also recover slower than the VSDI and VSDII, which is probably because the VSDIII and VSDIV carry more gating charges that limit their moving rate across the HCS (Capes et al., 2013; Goldschen-Ohm et al., 2013; Jiang et al., 2020).
Modulation of NaV Channels by Site-4 Toxins
Many natural gating modifier toxins (GMTs) target the VSDs and alter NaV channel properties (Catterall et al., 2007). These GMTs are important tools to study the NaV channel properties because of their high-affinity and specific binding mode (Lazdunski et al., 1986). Among them, a group of polypeptide toxins from spiders or scorpions specifically bind to the VSDII of NaV channels, classified as site-4 neurotoxins, which are used as weapons to paralyze prey (Catterall et al., 2007; Dufton and Rochat, 1984; Rochat et al., 1979). The site-4 toxins were found to inhibit the activation of NaV channels (Sokolov et al., 2008; Xiao et al., 2008), or cause abnormal activation (Cestèle et al., 1998). The binding site for the site-4 toxins is located in the VSDII, especially the extracellular loops linking S1-S2 and S3-S4 (Cestèle et al., 1998; Marcotte et al., 1997). Two site-4 toxins, Protoxin-II (β/ω-theraphotoxin-Tp2a; ProTx-II) from the Peruvian green velvet tarantula Thrixopelma pruriens (Middleton et al., 2002) and Huwentoxin-IV (μ-theraphotoxin-Hs2a; HwTx-IV) from the Chinese bird tarantula Haplopelma schmidti (Peng et al., 2002), show higher potency in inhibiting NaV1.7 than other NaV isoforms. Shen reported the cryo-EM structures of human NaV1.7 in complex with ProTx-II and HwTx-IV, showing that ProTx-II binds to both the VSDII and VSDIV, whereas HwTx-IV only binds to the VSDII (Figure 5C) (Shen et al., 2019). However, the EM density for ProTx-II and HwTx-IV are insufficient to define a detailed binding site. The binding of the two toxins appear only to induce subtle local conformational changes in the activated VSDs (Shen et al., 2019). It has been shown that ProTx-II has higher affinity to the resting state of NaV1.7 than the activated state (Sokolov et al., 2008; Xu et al., 2019). These observations suggest that the binding poses of ProTx-II and HwTx-IV in the NaV1.7 structures may reflect a low-affinity binding state. Meanwhile, Xu reported the ProTx-II bound structures of a chimeric bacterial NaVAb with VSDII of NaV1.7 (designated as NaVAb-NaV1.7VSDII) in different activation states (Xu et al., 2019). By sorting cryo-EM images of the NaVAb-NaV1.7VSDII-ProTxII yielded two maps in distinct conformations. The VSDs of the major class are in the activated conformation, while the structure of the minor class clearly showed that ProTx-II binding shifted the S4 helix ∼10 Å downward into a deactivated state. The deactivated NaVAb-NaV1.7VSDII-ProTxII may represent the high-affinity binding state for ProTx-II. However, the limited resolution prevented revealing a clearer picture for the high-affinity binding site (Xu et al., 2019). Subsequently, Wisedchaisri refined the chimeric NaVAb-NaV1.7VS2A construct by importing a voltage-shifting mutation to stabilize the channel in the resting state even under positive membrane potential (Wisedchaisri et al., 2021), and reported the structure of the NaVAb-NaV1.7VS2A complexed with a modified HwTx-IV (m3-HwTx-IV) (Rahnama et al., 2017; Revell et al., 2013). The NaVAb-NaV1.7VS2A:m3-HwTx-IV structure unveiled the high affinity binding site for m3-HwTx-IV and demonstrated that the m3-HwTx-IV inhibits the channel by locking the VSDII in the resting state (Wisedchaisri et al., 2021). The m3-HwTx-IV forms extensive interactions with the S3-S4 loop (811ELFLADVE818) of the VSDII, which is consistent with the deactivated NaVAb-NaV1.7VSDII-ProTxII structure (Xu et al., 2019) and the site-direct mutagenesis studies (Xiao et al., 2008; Xiao et al., 2010). Among the S3-S4 loop (811ELFLADVE818) of the VSDII, the F813G mutation drops the affinity of ProTx-II by ∼9-fold, but does not affect the binding of HwTx-IV (Xiao et al., 2010); by contrast, the E818C mutation significantly increases the affinity of HwTx-IV, whereas it has little effect on ProTx-II (Xiao et al., 2010; Xu et al., 2019). These opposite effects suggest that the detailed binding poses for the HwTx-IV and ProTx-II are different, despite both of the toxins bind to the same region of the VSDII (Figure 5D). The NaVAb-NaV1.7VSDII-ProTxII and NaVAb-NaV1.7VS2A:m3-HwTx-IV structures elucidate a common inhibition mechanism for the site-4 toxins (Figures 6A,B), that is, the positively-charged residues in the C-terminus of the toxins insert their side-chains into the cleft of the VSDII and trap the VSDII in the resting state to prevent the activation (Xu et al., 2019; Wisedchaisri et al., 2021).
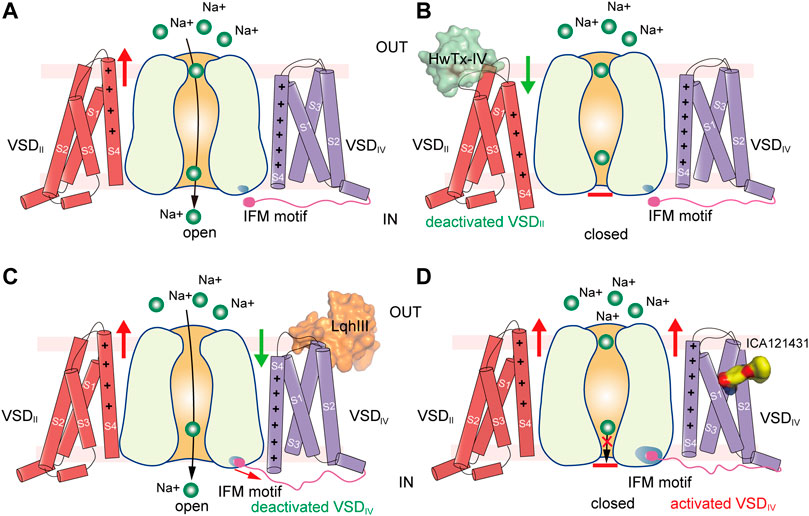
FIGURE 6. Putative models for modulation of VSDs of NaV channels. (A) Activation of VSDI and VSDII cause channel opening. Green balls represent sodium ions. Red arrow indicates activation of the VSD. (B) Site-4 toxin HwTx-IV traps VSDII in a deactivated state and inhibits channel opening. Green arrow indicates deactivation of the VSD. (C) α-Scorpion toxin LqhIII traps VSDIV in a deactivated state and inhibits fast inactivation. (D) Aryl sulfonamide analogue ICA121431 binds to activated VSDIV and inhibits channel opening.
The site-4 toxins ProTxII and HwTx-IV are more potent in inhibiting NaV1.7 than other NaV subtypes (Schmalhofer et al., 2008). NaV1.7 plays crucial roles in pain sensation (Bennett et al., 2019; Dib-Hajj and Waxman, 2019), thus, selective inhibition of NaV1.7 may bring potentially non-addictive medical benefits in treating chronic pain. Given the fact that NaV1.7 are in the resting state for most of the time in the nociceptive afferents (Wisedchaisri et al., 2021), the resting-state trapping mechanism revealed by the above structures suggests that optimization of these gating-modifier toxins could potentially generate effective and selective analgesics. These structures also provide structural templates for future development of new analgesics targeting the resting-state NaV channels.
Modulation of NaV Channel by α-Scorpion Toxins
α-Scorpion toxins belong to a family of peptide neurotoxins which inhibit fast inactivation of NaV channels, causing prolonged and/or repetitive action potentials (Catterall et al., 2007; Jiang et al., 2021c; Kopeyan et al., 1974). Site-directed mutagenesis studies have localized the binding site of α-Scorpion toxins in the S3-S4 loop of VSDIV, termed site-3 (Rogers et al., 1996; Thomsen and Catterall, 1989). In addition, a negatively-charged Asp or Glu residue on S3 of VSDIV was identified as the key determinant for the α-Scorpion toxins binding (Benzinger et al., 1998; Rogers et al., 1996). These findings also support the electromechanical coupling between the fast inactivation gate and the VSDIV. The α-Scorpion toxin LqhIII and AaHII are lethal toxins extracted from venoms of the “deathstalker scorpion” Leiurus quinquestriatus hebraeus and the Androctonus australis Hector “man killer” scorpion (Chen and Heinemann, 2001; Martin et al., 1987), both of which dramatically inhibit the fast inactivation of the NaV channels. In particular, LqhIII exhibits higher affinity to NaV1.5 and extremely slow disassociation rate (Chen and Heinemann, 2001). The cryo-EM structures of rat NaV1.5-LqhIII and the chimeric NaVPas-NaV1.7VSDIV-AaHII reveal detailed binding site for the α-Scorpion toxins (Clairfeuille et al., 2019; Jiang et al., 2021c). Consistent with previous mutagenesis studies, LqhIII and AaHII engage the S3-S4 loop of VSDIV by forming broad interaction interface (Figure 5C). The D1612 in DIV-S3 of NaV1.5 and D1586 in DIV-S3 of NaV1.7 form critical polar interactions with LqhIII and AaHII, respectively. The toxins binding shifts the S4 helix about two helical-turns inward and traps the VSDIV in an intermediate activated state (Figure 5E). The relatively flexible positively-charged C-terminal tails of LqhIII and AaHII dock into the extracellular aqueous cleft of VSDIV, displacing the gating-charges and preventing them moving upward. As expected, the two structures show very similar overall binding sites for the α-Scorpion toxins, and also similar conformational shifts in the VSDIV induced the toxins binding (Clairfeuille et al., 2019; Jiang et al., 2021c). Furthermore, the NaV1.5-LqhIII structure reveals a common gating mechanism for the α-Scorpion toxins (Figure 6C). That is, the α-Scorpion toxins specifically recognize the high-affinity binding site in the VSDIV and trap the VSDIV in the intermediate activated state, then deactivation of the VSDIV destabilizes the fast inactivation gate thus favors the channel opening (Jiang et al., 2021c).
Although the site-3 toxins and site-4 toxins bind to distinct receptor sites and cause different effects on NaV channels, these two types of neurotoxins use similar VSD trapping mechanism to modulation channel gating (Figures 5D,E). The different modulation effects are because that the activation and fast inactivation of NaV channels rely on the activation of VSDI-VSDII and VSDIV, respectively (Chanda and Bezanilla, 2002). From the NaV1.4-β1, NaV1.7-β1-β2, NaV1.2-β2, NaV1.1-β4 and NaV1.3-β1-β2 complex structures, we have known that β1 subunit projects its Ig-like domain on the VSDIII, β2 and β4 project their Ig-like domain on the VSDI, respectively (Figures 1B, 5C). Consequently, the accessibility of VSDIII and VSDI for potential neurotoxins are blocked by the β subunits, which may explain that only a few neurotoxins were reported to bind to VSDIII or VSDI. For instance, Hm-3, from the crab spider Heriaeus melloteei, was reported to inhibit NaV1.4 by binding to the VSDI with micromolar affinity (Männikkö et al., 2018). Recently, a spider toxin Gr4b from Grammostola rosea appears to selectively impair fast inactivation of NaV1.9 by binding to its VSDIII (Peng et al., 2021). However, the detailed binding sites and the underlying mechanisms for those toxins need further investigation. Meanwhile, because the β subunits bind loosely to NaV1.5 and NaV1.8 (Jiang et al., 2020), this raises a possibility that candidate modulators such as engineered neurotoxins or nanobodies can target NaV1.5 or NaV1.8 by binding to the VSDIII or VSDI without affecting other β subunit-bound NaV isoforms.
Aryl Sulfonamides Selectively Binds to VSDIV
The apo-form NaV channel structures show that activation of the VSDIV tightens the fast inactivation gate to close the channel (Jiang et al., 2020; Pan et al., 2018; Yan et al., 2017). Thus, trapping the VSDIV in the activated conformation inhibits the opening of NaV channels. In agreement with this concept, a family of synthetic aryl sulfonamide derivatives exhibit potent and selective inhibition of NaV channel isoforms via binding to the VSDIV (McCormack et al., 2013). For instance, PF-04856264 and GX-936 selectively and potently inhibit NaV1.7 (Ahuja et al., 2015; McCormack et al., 2013), whereas ICA121431 exhibits potent inhibition of NaV1.3/NaV1.1 with a factor of >1000-fold over other isoforms (McCormack et al., 2013). The promising selective inhibition is of great interest for developing potentially non-addictive analgesics (Alsaloum et al., 2020). The crystal structure of the chimeric NaVAb-NaV1.7VSDIV in complex with GX-936 revealed the first binding site for the aryl sulfonamide analogues (Ahuja et al., 2015). The GX-936 sticks deep into the extracellular aqueous cleft of the VSDIV, and its negatively-charged warhead engages the fourth gating-charge (R4), locking the VSDIV in the activated conformation (Figure 5F). Subsequently, the complex structure of human NaV1.3/β1/β2-ICA121431 confirmed the conserved binding site for the aryl sulfonamide analogues (Li et al., 2022). The conserved warhead of the aryl sulfonamide derivatives determines the potency via strong electrostatic interactions with the positively-charges R4, which is supported by the R4A mutation dramatically decreasing the affinity of GX-936 by >2000-fold (Ahuja et al., 2015). In addition, the S1559/R1560 on S2 helix of the NaV1.3-VSDIV are responsible for recognizing the tail of ICA121431, and Y1537/W1538 at the equivalent position on NaV1.7 are more favorable for GX-936 binding (Ahuja et al., 2015; Li et al., 2022; McCormack et al., 2013). Superposition of the ICA121431 bound NaV1.3-VSDIV with the deactivated LqhIII bound NaV1.5-VSDIV shows that the gating charges in the deactivated VSDIV cannot form proper interactions with the anionic warhead, elucidating why the aryl sulfonamide derivatives are in favor of binding to the activated VSDIV. In the NaV1.3/β1/β2-ICA121431 structure, the fast inactivation gate binds tightly to its receptor site resulting in a non-conductive activation gate (Figure 6D), which provides a full-picture for understanding the allosteric inhibition mechanism of the aryl sulfonamide antagonists (Li et al., 2022).
Several aryl sulfonamide derivatives, such as PF-05089771, GDC-0276 and RG6029, which selectively inhibit NaV1.7 (Alexandrou et al., 2016; McDonnell et al., 2018; Rothenberg et al., 2019; Alsaloum et al., 2020), have failed in Phase I or Phase II clinical trials because of low efficacy (Kingwell, 2019; Alsaloum et al., 2020). These discouraging results suggest that isoform selectivity is not the only challenge for developing candidate analgesics targeting pain related NaV channels.
Summary and Prospects
NaV channels play fundamental roles in electrical signaling. Extensive studies on the biophysical characterization, gene sequence, physiological functions, ligands modulation and pharmacology of NaV channels have greatly enriched the knowledge of NaV channels. During the last a few years, cryo-EM structures of NaV channels from nerve, cardiomyocytes and skeletal muscle were resolved in different functional states or with the binding of distinct modulators. These structures provide in-depth mechanistic insights into the architecture, activation, inactivation, ion selectivity, electromechanical coupling, ligand modulation, and structural pharmacology of NaV channels. The structure-based drug design will be accelerated by those structural studies, which could potentially generate more efficient, safer and selective drugs for the treatment of NaV channel associated diseases. The cryo-EM technique will have broader application prospects in validating the structural pharmacology of NaV channels, also in expanding the fundamental understanding of NaV channel structure and function. We anticipate that more mammalian NaV channel structures in different functional states including the resting-state will be achieved, and high-resolution structures will also be very important to unveil more detailed information such as ion selectivity and lipid-channel interactions.
Author Contributions
DJ wrote the manuscript, ZX and JZ prepared the figures, and all authors reviewed and revised the paper.
Funding
This work is supported by funding from the Institute of Physics, Chinese Academy of Sciences (E0VK101 to DJ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahern, C. A., Payandeh, J., Bosmans, F., and Chanda, B. (2016). The Hitchhiker's Guide to the Voltage-Gated Sodium Channel Galaxy. J. Gen. Physiol. 147, 1–24. doi:10.1085/jgp.201511492
Ahuja, S., Mukund, S., Deng, L., Khakh, K., Chang, E., Ho, H., et al. (2015). Structural Basis of Nav1.7 Inhibition by an Isoform-Selective Small-Molecule Antagonist. Science 350, aac5464. doi:10.1126/science.aac5464
Alexandrou, A. J., Brown, A. R., Chapman, M. L., Estacion, M., Turner, J., Mis, M. A., et al. (2016). Subtype-Selective Small Molecule Inhibitors Reveal a Fundamental Role for Nav1.7 in Nociceptor Electrogenesis, Axonal Conduction and Presynaptic Release. PLoS One 11, e0152405. doi:10.1371/journal.pone.0152405
Alsaloum, M., Higerd, G. P., Effraim, P. R., and Waxman, S. G. (2020). Status of Peripheral Sodium Channel Blockers for Non-addictive Pain Treatment. Nat. Rev. Neurol. 16, 689–705. doi:10.1038/s41582-020-00415-2
Armstrong, C. M., and Bezanilla, F. (1973). Currents Related to Movement of the Gating Particles of the Sodium Channels. Nature 242, 459–461. doi:10.1038/242459a0
Armstrong, C. M., Bezanilla, F., and Rojas, E. (1973). Destruction of Sodium Conductance Inactivation in Squid Axons Perfused with Pronase. J. Gen. Physiol. 62, 375–391. doi:10.1085/jgp.62.4.375
Bagnéris, C., DeCaen, P. G., Naylor, C. E., Pryde, D. C., Nobeli, I., Clapham, D. E., et al. (2014). Prokaryotic NavMs Channel as a Structural and Functional Model for Eukaryotic Sodium Channel Antagonism. Proc. Natl. Acad. Sci. U. S. A. 111, 8428–8433. doi:10.1073/pnas.1406855111
Bennett, D. L., Clark, A. J., Huang, J., Waxman, S. G., and Dib-Hajj, S. D. (2019). The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 99, 1079–1151. doi:10.1152/physrev.00052.2017
Benzinger, G. R., Kyle, J. W., Blumenthal, K. M., and Hanck, D. A. (1998). A Specific Interaction between the Cardiac Sodium Channel and Site-3 Toxin Anthopleurin B. J. Biol. Chem. 273, 80–84. doi:10.1074/jbc.273.1.80
Boiteux, C., Vorobyov, I., French, R. J., French, C., Yarov-Yarovoy, V., and Allen, T. W. (2014). Local Anesthetic and Antiepileptic Drug Access and Binding to a Bacterial Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 111, 13057–13062. doi:10.1073/pnas.1408710111
Capes, D. L., Goldschen-Ohm, M. P., Arcisio-Miranda, M., Bezanilla, F., and Chanda, B. (2013). Domain IV Voltage-Sensor Movement Is Both Sufficient and Rate Limiting for Fast Inactivation in Sodium Channels. J. Gen. Physiol. 142, 101–112. doi:10.1085/jgp.201310998
Catterall, W. A., Cestèle, S., Yarov-Yarovoy, V., Yu, F. H., Konoki, K., and Scheuer, T. (2007). Voltage-gated Ion Channels and Gating Modifier Toxins. Toxicon 49, 124–141. doi:10.1016/j.toxicon.2006.09.022
Catterall, W. A., Dib-Hajj, S., Meisler, M. H., and Pietrobon, D. (2008). Inherited Neuronal Ion Channelopathies: New Windows on Complex Neurological Diseases. J. Neurosci. 28, 11768–11777. doi:10.1523/JNEUROSCI.3901-08.2008
Catterall, W. A. (2000). From Ionic Currents to Molecular Mechanisms: the Structure and Function of Voltage-Gated Sodium Channels. Neuron 26, 13–25. doi:10.1016/s0896-6273(00)81133-2
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 57, 397–409. doi:10.1124/pr.57.4.4
Catterall, W. A., Lenaeus, M. J., and Gamal El-Din, T. M. (2020a). Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 60, 133–154. doi:10.1146/annurev-pharmtox-010818-021757
Catterall, W. A. (1986). Molecular Properties of Voltage-Sensitive Sodium Channels. Annu. Rev. Biochem. 55, 953–985. doi:10.1146/annurev.bi.55.070186.004513
Catterall, W. A. (2014). Sodium Channels, Inherited Epilepsy, and Antiepileptic Drugs. Annu. Rev. Pharmacol. Toxicol. 54, 317–338. doi:10.1146/annurev-pharmtox-011112-140232
Catterall, W. A., Wisedchaisri, G., and Zheng, N. (2017). The Chemical Basis for Electrical Signaling. Nat. Chem. Biol. 13, 455–463. doi:10.1038/nchembio.2353
Catterall, W. A., Wisedchaisri, G., and Zheng, N. (2020b). The Conformational Cycle of a Prototypical Voltage-Gated Sodium Channel. Nat. Chem. Biol. 16, 1314–1320. doi:10.1038/s41589-020-0644-4
Cestèle, S., Qu, Y., Rogers, J. C., Rochat, H., Scheuer, T., and Catterall, W. A. (1998). Voltage Sensor-Trapping: Enhanced Activation of Sodium Channels by Beta-Scorpion Toxin Bound to the S3-S4 Loop in Domain II. Neuron 21, 919–931. doi:10.1016/s0896-6273(00)80606-6
Chakrabarti, N., Ing, C., Payandeh, J., Zheng, N., Catterall, W. A., and Pomès, R. (2013). Catalysis of Na+ Permeation in the Bacterial Sodium Channel Na(V)Ab. Proc. Natl. Acad. Sci. U. S. A. 110, 11331–11336. doi:10.1073/pnas.1309452110
Chanda, B., Asamoah, O. K., and Bezanilla, F. (2004). Coupling Interactions between Voltage Sensors of the Sodium Channel as Revealed by Site-specific Measurements. J. Gen. Physiol. 123, 217–230. doi:10.1085/jgp.200308971
Chanda, B., and Bezanilla, F. (2002). Tracking Voltage-dependent Conformational Changes in Skeletal Muscle Sodium Channel during Activation. J. Gen. Physiol. 120, 629–645. doi:10.1085/jgp.20028679
Chen, H., and Heinemann, S. H. (2001). Interaction of Scorpion Alpha-Toxins with Cardiac Sodium Channels: Binding Properties and Enhancement of Slow Inactivation. J. Gen. Physiol. 117, 505–518. doi:10.1085/jgp.117.6.505
Clairfeuille, T., Cloake, A., Infield, D. T., Llongueras, J. P., Arthur, C. P., Li, Z. R., et al. (2019). Structural Basis of α-scorpion Toxin Action on Nav Channels. Science 363. doi:10.1126/science.aav8573
Clancy, C. E., and Kass, R. S. (2005). Inherited and Acquired Vulnerability to Ventricular Arrhythmias: Cardiac Na+ and K+ Channels. Physiol. Rev. 85, 33–47. doi:10.1152/physrev.00005.2004
Clatot, J., Ziyadeh-Isleem, A., Maugenre, S., Denjoy, I., Liu, H., Dilanian, G., et al. (2012). Dominant-negative Effect of SCN5A N-Terminal Mutations through the Interaction of Na(v)1.5 α-subunits. Cardiovasc Res. 96, 53–63. doi:10.1093/cvr/cvs211
Corry, B., and Thomas, M. (2012). Mechanism of Ion Permeation and Selectivity in a Voltage Gated Sodium Channel. J. Am. Chem. Soc. 134, 1840–1846. doi:10.1021/ja210020h
Das, S., Gilchrist, J., Bosmans, F., and Van Petegem, F. (2016). Binary Architecture of the Nav1.2-β2 Signaling Complex. Elife 5. doi:10.7554/eLife.10960
Deuis, J. R., Mueller, A., Israel, M. R., and Vetter, I. (2017). The Pharmacology of Voltage-Gated Sodium Channel Activators. Neuropharmacology 127, 87–108. doi:10.1016/j.neuropharm.2017.04.014
Dib-Hajj, S. D., and Waxman, S. G. (2019). Sodium Channels in Human Pain Disorders: Genetics and Pharmacogenomics. Annu. Rev. Neurosci. 42, 87–106. doi:10.1146/annurev-neuro-070918-050144
Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., et al. (1998). The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 280, 69–77. doi:10.1126/science.280.5360.69
Dufton, M. J., and Rochat, H. (1984). Classification of Scorpion Toxins According to Amino Acid Composition and Sequence. J. Mol. Evol. 20, 120–127. doi:10.1007/BF02257372
Eaholtz, G., Scheuer, T., and Catterall, W. A. (1994). Restoration of Inactivation and Block of Open Sodium Channels by an Inactivation Gate Peptide. Neuron 12, 1041–1048. doi:10.1016/0896-6273(94)90312-3
Favre, I., Moczydlowski, E., and Schild, L. (1996). On the Structural Basis for Ionic Selectivity Among Na+, K+, and Ca2+ in the Voltage-Gated Sodium Channel. Biophysical J. 71, 3110–3125. doi:10.1016/S0006-3495(96)79505-X
Gamal El-Din, T. M., Lenaeus, M. J., Zheng, N., and Catterall, W. A. (2018). Fenestrations Control Resting-State Block of a Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 115, 13111–13116. doi:10.1073/pnas.1814928115
Gilchrist, J., Das, S., Van Petegem, F., and Bosmans, F. (2013). Crystallographic Insights into Sodium-Channel Modulation by the β4 Subunit. Proc. Natl. Acad. Sci. U. S. A. 110, E5016–E5024. doi:10.1073/pnas.1314557110
Goldin, A. L. (2003). Mechanisms of Sodium Channel Inactivation. Curr. Opin. Neurobiol. 13, 284–290. doi:10.1016/s0959-4388(03)00065-5
Goldschen-Ohm, M. P., Capes, D. L., Oelstrom, K. M., and Chanda, B. (2013). Multiple Pore Conformations Driven by Asynchronous Movements of Voltage Sensors in a Eukaryotic Sodium Channel. Nat. Commun. 4, 1350. doi:10.1038/ncomms2356
Guy, H. R., and Seetharamulu, P. (1986). Molecular Model of the Action Potential Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 83, 508–512. doi:10.1073/pnas.83.2.508
Hartshorne, R. P., and Catterall, W. A. (1984). The Sodium Channel from Rat Brain. Purification and Subunit Composition. J. Biol. Chem. 259, 1667–1675. doi:10.1016/s0021-9258(17)43460-0
Helliwell, K. E., Chrachri, A., Koester, J. A., Wharam, S., Taylor, A. R., Wheeler, G. L., et al. (2020). A Novel Single-Domain Na+-Selective Voltage-Gated Channel in Photosynthetic Eukaryotes. Plant Physiol. 184, 1674–1683. doi:10.1104/pp.20.00889
Helliwell, K. E., Chrachri, A., Koester, J. A., Wharam, S., Verret, F., Taylor, A. R., et al. (2019). Alternative Mechanisms for Fast Na+/Ca2+ Signaling in Eukaryotes via a Novel Class of Single-Domain Voltage-Gated Channels. Curr. Biol. 29, 1503–e6.e1506. doi:10.1016/j.cub.2019.03.041
Hille, B. (1975). Ionic Selectivity, Saturation, and Block in Sodium Channels. A Four-Barrier Model. J. Gen. Physiol. 66, 535–560. doi:10.1085/jgp.66.5.535
Hille, B. (1977). Local Anesthetics: Hydrophilic and Hydrophobic Pathways for the Drug-Receptor Reaction. J. Gen. Physiol. 69, 497–515. doi:10.1085/jgp.69.4.497
Hille, B. (1971a). The Hydration of Sodium Ions Crossing the Nerve Membrane. Proc. Natl. Acad. Sci. U. S. A. 68, 280–282. doi:10.1073/pnas.68.2.280
Hille, B. (1972). The Permeability of the Sodium Channel to Metal Cations in Myelinated Nerve. J. Gen. Physiol. 59, 637–658. doi:10.1085/jgp.59.6.637
Hille, B. (1971b). The Permeability of the Sodium Channel to Organic Cations in Myelinated Nerve. J. Gen. Physiol. 58, 599–619. doi:10.1085/jgp.58.6.599
Hodgkin, A. L., and Huxley, A. F. (1952a). Currents Carried by Sodium and Potassium Ions through the Membrane of the Giant Axon of Loligo. J. Physiol. 116, 449–472. doi:10.1113/jphysiol.1952.sp004717
Hodgkin, A. L., and Huxley, A. F. (1952b). The Components of Membrane Conductance in the Giant Axon of Loligo. J. Physiol. 116, 473–496. doi:10.1113/jphysiol.1952.sp004718
Hondeghem, L. M., and Katzung, B. G. (1984). Antiarrhythmic Agents: the Modulated Receptor Mechanism of Action of Sodium and Calcium Channel-Blocking Drugs. Annu. Rev. Pharmacol. Toxicol. 24, 387–423. doi:10.1146/annurev.pa.24.040184.002131
Huang, C. L., Wu, L., Jeevaratnam, K., and Lei, M. (2020). Update on Antiarrhythmic Drug Pharmacology. J. Cardiovasc Electrophysiol. 31, 579–592. doi:10.1111/jce.14347
Huang, W., Liu, M., Yan, S. F., and Yan, N. (2017). Structure-based Assessment of Disease-Related Mutations in Human Voltage-Gated Sodium Channels. Protein Cell. 8, 401–438. doi:10.1007/s13238-017-0372-z
Ishii, H., Kinoshita, E., Kimura, T., Yakehiro, M., Yamaoka, K., Imoto, K., et al. (1999). Point-mutations Related to the Loss of Batrachotoxin Binding Abolish the Grayanotoxin Effect in Na(+) Channel Isoforms. Jpn. J. Physiol. 49, 457–461. doi:10.2170/jjphysiol.49.457
Isom, D. G., Castañeda, C. A., Cannon, B. R., and García-Moreno, B. (2011). Large Shifts in pKa Values of Lysine Residues Buried inside a Protein. Proc. Natl. Acad. Sci. U. S. A. 108, 5260–5265. doi:10.1073/pnas.1010750108
Isom, L. L., De Jongh, K. S., and Catterall, W. A. (1994). Auxiliary Subunits of Voltage-Gated Ion Channels. Neuron 12, 1183–1194. doi:10.1016/0896-6273(94)90436-7
Jiang, D., Banh, R., Gamal El-Din, T. M., Tonggu, L., Lenaeus, M. J., Pomès, R., et al. (2021a). Open-state Structure and Pore Gating Mechanism of the Cardiac Sodium Channel. Cell. 184, 5151–e11.e5111. doi:10.1016/j.cell.2021.08.021
Jiang, D., Gamal El-Din, T., Zheng, N., and Catterall, W. A. (2021b). Expression and Purification of the Cardiac Sodium Channel NaV1.5 for Cryo-EM Structure Determination. Methods Enzymol. 653, 89–101. doi:10.1016/bs.mie.2021.01.030
Jiang, D., Gamal El-Din, T. M., Ing, C., Lu, P., Pomès, R., Zheng, N., et al. (2018). Structural Basis for Gating Pore Current in Periodic Paralysis. Nature 557, 590–594. doi:10.1038/s41586-018-0120-4
Jiang, D., Shi, H., Tonggu, L., Gamal El-Din, T. M., Lenaeus, M. J., Zhao, Y., et al. (2020). Structure of the Cardiac Sodium Channel. Cell. 180, 122–e10. doi:10.1016/j.cell.2019.11.041
Jiang, D., Tonggu, L., Gamal El-Din, T. M., Banh, R., Pomès, R., Zheng, N., et al. (2021c). Structural Basis for Voltage-Sensor Trapping of the Cardiac Sodium Channel by a Deathstalker Scorpion Toxin. Nat. Commun. 12, 128. doi:10.1038/s41467-020-20078-3
Kao, C. Y. (1966). Tetrodotoxin, Saxitoxin and Their Significance in the Study of Excitation Phenomena. Pharmacol. Rev. 18, 997–1049.
Kellenberger, S., Scheuer, T., and Catterall, W. A. (1996). Movement of the Na+ Channel Inactivation Gate during Inactivation. J. Biol. Chem. 271, 30971–30979. doi:10.1074/jbc.271.48.30971
Kingwell, K. (2019). Nav1.7 Withholds its Pain Potential. Nat. Rev. Drug Discov. doi:10.1038/d41573-019-00065-0
Kohlhardt, M., and Fichtner, H. (1988). Block of Single Cardiac Na+ Channels by Antiarrhythmic Drugs: the Effect of Amiodarone, Propafenone and Diprafenone. J. Membr. Biol. 102, 105–119. doi:10.1007/BF01870449
Koishi, R., Xu, H., Ren, D., Navarro, B., Spiller, B. W., Shi, Q., et al. (2004). A Superfamily of Voltage-Gated Sodium Channels in Bacteria. J. Biol. Chem. 279, 9532–9538. doi:10.1074/jbc.M313100200
Kopeyan, C., Martinez, G., Lissitzky, S., Miranda, F., and Rochat, H. (1974). Disulfide Bonds of Toxin II of the Scorpion Androctonus Australis Hector. Eur. J. Biochem. 47, 483–489. doi:10.1111/j.1432-1033.1974.tb03716.x
Kowey, P. R. (1998). Pharmacological Effects of Antiarrhythmic Drugs. Review and Update. Arch. Intern Med. 158, 325–332. doi:10.1001/archinte.158.4.325
Lazdunski, M., Frelin, C., Barhanin, J., Lombet, A., Meiri, H., Pauron, D., et al. (1986). Polypeptide Toxins as Tools to Study Voltage-Sensitive Na+ Channels. Ann. N. Y. Acad. Sci. 479, 204–220. doi:10.1111/j.1749-6632.1986.tb15571.x
Lenaeus, M. J., Gamal El-Din, T. M., Ing, C., Ramanadane, K., Pomès, R., Zheng, N., et al. (2017). Structures of Closed and Open States of a Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 114, E3051–E3060. doi:10.1073/pnas.1700761114
Lerche, H., Peter, W., Fleischhauer, R., Pika-Hartlaub, U., Malina, T., Mitrovic, N., et al. (1997). Role in Fast Inactivation of the IV/S4-S5 Loop of the Human Muscle Na+ Channel Probed by Cysteine Mutagenesis. J. Physiol. 505 ( Pt 2) (Pt 2), 345–352. doi:10.1111/j.1469-7793.1997.345bb.x
Li, X., Xu, F., Xu, H., Zhang, S., Gao, Y., Zhang, H., et al. (2022). Structural Basis for Modulation of Human NaV1.3 by Clinical Drug and Selective Antagonist. Nat. Commun. 13, 1286. doi:10.1038/s41467-022-28808-5
Li, Z., Jin, X., Wu, T., Huang, G., Wu, K., Lei, J., et al. (2021a). Structural Basis for Pore Blockade of the Human Cardiac Sodium Channel Nav 1.5 by the Antiarrhythmic Drug Quinidine*. Angew. Chem. Int. Ed. Engl. 60, 11474–11480. doi:10.1002/anie.202102196
Li, Z., Jin, X., Wu, T., Zhao, X., Wang, W., Lei, J., et al. (2021b). Structure of Human Na V 1.5 Reveals the Fast Inactivation-Related Segments as a Mutational Hotspot for the Long QT Syndrome. Proc. Natl. Acad. Sci. U.S.A. 118. doi:10.1073/pnas.2100069118
Liu, H., Atkins, J., and Kass, R. S. (2003). Common Molecular Determinants of Flecainide and Lidocaine Block of Heart Na+ Channels: Evidence from Experiments with Neutral and Quaternary Flecainide Analogues. J. Gen. Physiol. 121, 199–214. doi:10.1085/jgp.20028723
Long, S. B., Campbell, E. B., and Mackinnon, R. (2005). Crystal Structure of a Mammalian Voltage-dependent Shaker Family K+ Channel. Science 309, 897–903. doi:10.1126/science.1116269
Makita, N., Behr, E., Shimizu, W., Horie, M., Sunami, A., Crotti, L., et al. (2008). The E1784K Mutation in SCN5A Is Associated with Mixed Clinical Phenotype of Type 3 Long QT Syndrome. J. Clin. Invest. 118, 2219–2229. doi:10.1172/JCI34057
Makita, N., Bennett, P. B., and George, A. L. (1994). Voltage-gated Na+ Channel Beta 1 Subunit mRNA Expressed in Adult Human Skeletal Muscle, Heart, and Brain Is Encoded by a Single Gene. J. Biol. Chem. 269, 7571–7578. doi:10.1016/s0021-9258(17)37325-8
Männikkö, R., Shenkarev, Z. O., Thor, M. G., Berkut, A. A., Myshkin, M. Y., Paramonov, A. S., et al. (2018). Spider Toxin Inhibits Gating Pore Currents Underlying Periodic Paralysis. Proc. Natl. Acad. Sci. U. S. A. 115, 4495–4500. doi:10.1073/pnas.1720185115
Mantegazza, M., Yu, F. H., Catterall, W. A., and Scheuer, T. (2001). Role of the C-Terminal Domain in Inactivation of Brain and Cardiac Sodium Channels. Proc. Natl. Acad. Sci. U. S. A. 98, 15348–15353. doi:10.1073/pnas.211563298
Marcotte, P., Chen, L. Q., Kallen, R. G., and Chahine, M. (1997). Effects of Tityus Serrulatus Scorpion Toxin Gamma on Voltage-Gated Na+ Channels. Circ. Res. 80, 363–369. doi:10.1161/01.res.80.3.363
Martin, M. F., Rochat, H., Marchot, P., and Bougis, P. E. (1987). Use of High Performance Liquid Chromatography to Demonstrate Quantitative Variation in Components of Venom from the Scorpion Androctonus Australis Hector. Toxicon 25, 569–573. doi:10.1016/0041-0101(87)90293-5
McCormack, K., Santos, S., Chapman, M. L., Krafte, D. S., Marron, B. E., West, C. W., et al. (2013). Voltage Sensor Interaction Site for Selective Small Molecule Inhibitors of Voltage-Gated Sodium Channels. Proc. Natl. Acad. Sci. U. S. A. 110, E2724–E2732. doi:10.1073/pnas.1220844110
McDonnell, A., Collins, S., Ali, Z., Iavarone, L., Surujbally, R., Kirby, S., et al. (2018). Efficacy of the Nav1.7 Blocker PF-05089771 in a Randomised, Placebo-Controlled, Double-Blind Clinical Study in Subjects with Painful Diabetic Peripheral Neuropathy. Pain 159, 1465–1476. doi:10.1097/j.pain.0000000000001227
McPhee, J. C., Ragsdale, D. S., Scheuer, T., and Catterall, W. A. (1998). A Critical Role for the S4-S5 Intracellular Loop in Domain IV of the Sodium Channel Alpha-Subunit in Fast Inactivation. J. Biol. Chem. 273, 1121–1129. doi:10.1074/jbc.273.2.1121
McPhee, J. C., Ragsdale, D. S., Scheuer, T., and Catterall, W. A. (1994). A Mutation in Segment IVS6 Disrupts Fast Inactivation of Sodium Channels. Proc. Natl. Acad. Sci. U. S. A. 91, 12346–12350. doi:10.1073/pnas.91.25.12346
Meisler, M. H., and Kearney, J. A. (2005). Sodium Channel Mutations in Epilepsy and Other Neurological Disorders. J. Clin. Invest. 115, 2010–2017. doi:10.1172/JCI25466
Middleton, R. E., Warren, V. A., Kraus, R. L., Hwang, J. C., Liu, C. J., Dai, G., et al. (2002). Two Tarantula Peptides Inhibit Activation of Multiple Sodium Channels. Biochemistry 41, 14734–14747. doi:10.1021/bi026546a
Namadurai, S., Balasuriya, D., Rajappa, R., Wiemhöfer, M., Stott, K., Klingauf, J., et al. (2014). Crystal Structure and Molecular Imaging of the Nav Channel β3 Subunit Indicates a Trimeric Assembly. J. Biol. Chem. 289, 10797–10811. doi:10.1074/jbc.M113.527994
Namadurai, S., Yereddi, N. R., Cusdin, F. S., Huang, C. L., Chirgadze, D. Y., and Jackson, A. P. (2015). A New Look at Sodium Channel β Subunits. Open Biol. 5, 140192. doi:10.1098/rsob.140192
Naylor, C. E., Bagnéris, C., DeCaen, P. G., Sula, A., Scaglione, A., Clapham, D. E., et al. (2016). Molecular Basis of Ion Permeability in a Voltage-Gated Sodium Channel. EMBO J. 35, 820–830. doi:10.15252/embj.201593285
Nguyen, P. T., DeMarco, K. R., Vorobyov, I., Clancy, C. E., and Yarov-Yarovoy, V. (2019). Structural Basis for Antiarrhythmic Drug Interactions with the Human Cardiac Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 116, 2945–2954. doi:10.1073/pnas.1817446116
Noda, M., Shimizu, S., Tanabe, T., Takai, T., Kayano, T., Ikeda, T., et al. (1984). Primary Structure of Electrophorus Electricus Sodium Channel Deduced from cDNA Sequence. Nature 312, 121–127. doi:10.1038/312121a0
Noda, M., Suzuki, H., Numa, S., and Stühmer, W. (1989). A Single Point Mutation Confers Tetrodotoxin and Saxitoxin Insensitivity on the Sodium Channel II. FEBS Lett. 259, 213–216. doi:10.1016/0014-5793(89)81531-5
O'Brien, J. E., and Meisler, M. H. (2013). Sodium Channel SCN8A (Nav1.6): Properties and De Novo Mutations in Epileptic Encephalopathy and Intellectual Disability. Front. Genet. 4, 213. doi:10.3389/fgene.2013.00213
O'Malley, H. A., and Isom, L. L. (2015). Sodium Channel β Subunits: Emerging Targets in Channelopathies. Annu. Rev. Physiol. 77, 481–504. doi:10.1146/annurev-physiol-021014-071846
Olesen, M. S., Jespersen, T., Nielsen, J. B., Liang, B., Møller, D. V., Hedley, P., et al. (2011). Mutations in Sodium Channel β-subunit SCN3B Are Associated with Early-Onset Lone Atrial Fibrillation. Cardiovasc Res. 89, 786–793. doi:10.1093/cvr/cvq348
Pajouhesh, H., Beckley, J. T., Delwig, A., Hajare, H. S., Luu, G., Monteleone, D., et al. (2020). Discovery of a Selective, State-independent Inhibitor of NaV1.7 by Modification of Guanidinium Toxins. Sci. Rep. 10, 14791. doi:10.1038/s41598-020-71135-2
Pan, X., Li, Z., Huang, X., Huang, G., Gao, S., Shen, H., et al. (2019). Molecular Basis for Pore Blockade of Human Na+ Channel Nav1.2 by the μ-conotoxin KIIIA. Science 363, 1309–1313. doi:10.1126/science.aaw2999
Pan, X., Li, Z., Zhou, Q., Shen, H., Wu, K., Huang, X., et al. (2018). Structure of the Human Voltage-Gated Sodium Channel Nav1.4 in Complex with β1. Science 362. doi:10.1126/science.aau2486
Pan, X., Li, Z., Jin, X., Zhao, Y., Huang, G., Huang, X., et al. (2021). Comparative Structural Analysis of Human Na V 1.1 and Na V 1.5 Reveals Mutational Hotspots for Sodium Channelopathies. Proc. Natl. Acad. Sci. U.S.A. 118. doi:10.1073/pnas.2100066118
Patino, G. A., Brackenbury, W. J., Bao, Y., Lopez-Santiago, L. F., O'Malley, H. A., Chen, C., et al. (2011). Voltage-gated Na+ Channel β1B: a Secreted Cell Adhesion Molecule Involved in Human Epilepsy. J. Neurosci. 31, 14577–14591. doi:10.1523/JNEUROSCI.0361-11.2011
Payandeh, J., Gamal El-Din, T. M., Scheuer, T., Zheng, N., and Catterall, W. A. (2012). Crystal Structure of a Voltage-Gated Sodium Channel in Two Potentially Inactivated States. Nature 486, 135–139. doi:10.1038/nature11077
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The Crystal Structure of a Voltage-Gated Sodium Channel. Nature 475, 353–358. doi:10.1038/nature10238
Peng, K., Shu, Q., Liu, Z., and Liang, S. (2002). Function and Solution Structure of Huwentoxin-IV, a Potent Neuronal Tetrodotoxin (TTX)-sensitive Sodium Channel Antagonist from Chinese Bird Spider Selenocosmia Huwena. J. Biol. Chem. 277, 47564–47571. doi:10.1074/jbc.M204063200
Peng, S., Chen, M., Xiao, Z., Xiao, X., Luo, S., Liang, S., et al. (2021). A Novel Spider Toxin Inhibits Fast Inactivation of the Nav1.9 Channel by Binding to Domain III and Domain IV Voltage Sensors. Front. Pharmacol. 12, 778534. doi:10.3389/fphar.2021.778534
Qu, Y., Isom, L. L., Westenbroek, R. E., Rogers, J. C., Tanada, T. N., McCormick, K. A., et al. (1995). Modulation of Cardiac Na+ Channel Expression in Xenopus Oocytes by Beta 1 Subunits. J. Biol. Chem. 270, 25696–25701. doi:10.1074/jbc.270.43.25696
Quandt, F. N., and Narahashi, T. (1982). Modification of Single Na+ Channels by Batrachotoxin. Proc. Natl. Acad. Sci. U. S. A. 79, 6732–6736. doi:10.1073/pnas.79.21.6732
Ragsdale, D. S., McPhee, J. C., Scheuer, T., and Catterall, W. A. (1996). Common Molecular Determinants of Local Anesthetic, Antiarrhythmic, and Anticonvulsant Block of Voltage-Gated Na+ Channels. Proc. Natl. Acad. Sci. U. S. A. 93, 9270–9275. doi:10.1073/pnas.93.17.9270
Ragsdale, D. S., McPhee, J. C., Scheuer, T., and Catterall, W. A. (1994). Molecular Determinants of State-dependent Block of Na+ Channels by Local Anesthetics. Science 265, 1724–1728. doi:10.1126/science.8085162
Rahnama, S., Deuis, J. R., Cardoso, F. C., Ramanujam, V., Lewis, R. J., Rash, L. D., et al. (2017). The Structure, Dynamics and Selectivity Profile of a NaV1.7 Potency-Optimised Huwentoxin-IV Variant. PLoS One 12, e0173551. doi:10.1371/journal.pone.0173551
Raman, I. M., and Bean, B. P. (1997). Resurgent Sodium Current and Action Potential Formation in Dissociated Cerebellar Purkinje Neurons. J. Neurosci. 17, 4517–4526. doi:10.1523/jneurosci.17-12-04517.1997
Raman, I. M., Sprunger, L. K., Meisler, M. H., and Bean, B. P. (1997). Altered Subthreshold Sodium Currents and Disrupted Firing Patterns in Purkinje Neurons of Scn8a Mutant Mice. Neuron 19, 881–891. doi:10.1016/s0896-6273(00)80969-1
Ren, D., Navarro, B., Xu, H., Yue, L., Shi, Q., and Clapham, D. E. (2001). A Prokaryotic Voltage-Gated Sodium Channel. Science 294, 2372–2375. doi:10.1126/science.1065635
Revell, J. D., Lund, P. E., Linley, J. E., Metcalfe, J., Burmeister, N., Sridharan, S., et al. (2013). Potency Optimization of Huwentoxin-IV on hNav1.7: a Neurotoxin TTX-S Sodium-Channel Antagonist from the Venom of the Chinese Bird-Eating Spider Selenocosmia Huwena. Peptides 44, 40–46. doi:10.1016/j.peptides.2013.03.011
Rochat, H., Bernard, P., and Couraud, F. (1979). Scorpion Toxins: Chemistry and Mode of Action. Adv. Cytopharmacol. 3, 325–334.
Rogers, J. C., Qu, Y., Tanada, T. N., Scheuer, T., and Catterall, W. A. (1996). Molecular Determinants of High Affinity Binding of Alpha-Scorpion Toxin and Sea Anemone Toxin in the S3-S4 Extracellular Loop in Domain IV of the Na+ Channel Alpha Subunit. J. Biol. Chem. 271, 15950–15962. doi:10.1074/jbc.271.27.15950
Rohl, C. A., Boeckman, F. A., Baker, C., Scheuer, T., Catterall, W. A., and Klevit, R. E. (1999). Solution Structure of the Sodium Channel Inactivation Gate. Biochemistry 38, 855–861. doi:10.1021/bi9823380
Rothenberg, M. E., Tagen, M., Chang, J. H., Boyce-Rustay, J., Friesenhahn, M., Hackos, D. H., et al. (2019). Safety, Tolerability, and Pharmacokinetics of GDC-0276, a Novel NaV1.7 Inhibitor, in a First-In-Human, Single- and Multiple-Dose Study in Healthy Volunteers. Clin. Drug Investig. 39, 873–887. doi:10.1007/s40261-019-00807-3
Ruan, Y., Liu, N., and Priori, S. G. (2009). Sodium Channel Mutations and Arrhythmias. Nat. Rev. Cardiol. 6, 337–348. doi:10.1038/nrcardio.2009.44
Sato, C., Ueno, Y., Asai, K., Takahashi, K., Sato, M., Engel, A., et al. (2001). The Voltage-Sensitive Sodium Channel Is a Bell-Shaped Molecule with Several Cavities. Nature 409, 1047–1051. doi:10.1038/35059098
Scheuer, T. (2011). Regulation of Sodium Channel Activity by Phosphorylation. Semin. Cell. Dev. Biol. 22, 160–165. doi:10.1016/j.semcdb.2010.10.002
Schmalhofer, W. A., Calhoun, J., Burrows, R., Bailey, T., Kohler, M. G., Weinglass, A. B., et al. (2008). ProTx-II, a Selective Inhibitor of NaV1.7 Sodium Channels, Blocks Action Potential Propagation in Nociceptors. Mol. Pharmacol. 74, 1476–1484. doi:10.1124/mol.108.047670
Shen, H., Li, Z., Jiang, Y., Pan, X., Wu, J., Cristofori-Armstrong, B., et al. (2018). Structural Basis for the Modulation of Voltage-Gated Sodium Channels by Animal Toxins. Science 362. doi:10.1126/science.aau2596
Shen, H., Liu, D., Wu, K., Lei, J., and Yan, N. (2019). Structures of Human Nav1.7 Channel in Complex with Auxiliary Subunits and Animal Toxins. Science 363, 1303–1308. doi:10.1126/science.aaw2493
Shen, H., Yan, N., and Pan, X. (2021). Structural Determination of Human Nav1.4 and Nav1.7 Using Single Particle Cryo-Electron Microscopy. Methods Enzymol. 653, 103–120. doi:10.1016/bs.mie.2021.03.010
Shen, H., Zhou, Q., Pan, X., Li, Z., Wu, J., and Yan, N. (2017). Structure of a Eukaryotic Voltage-Gated Sodium Channel at Near-Atomic Resolution. Science 355. doi:10.1126/science.aal4326
Sivilotti, L., Okuse, K., Akopian, A. N., Moss, S., and Wood, J. N. (1997). A Single Serine Residue Confers Tetrodotoxin Insensitivity on the Rat Sensory-neuron-specific Sodium Channel SNS. FEBS Lett. 409, 49–52. doi:10.1016/s0014-5793(97)00479-1
Smith, M. R., and Goldin, A. L. (1997). Interaction between the Sodium Channel Inactivation Linker and Domain III S4-S5. Biophys. J. 73, 1885–1895. doi:10.1016/S0006-3495(97)78219-5
Sokolov, S., Kraus, R. L., Scheuer, T., and Catterall, W. A. (2008). Inhibition of Sodium Channel Gating by Trapping the Domain II Voltage Sensor with Protoxin II. Mol. Pharmacol. 73, 1020–1028. doi:10.1124/mol.107.041046
Sokolov, S., Scheuer, T., and Catterall, W. A. (2007). Gating Pore Current in an Inherited Ion Channelopathy. Nature 446, 76–78. doi:10.1038/nature05598
Stühmer, W., Conti, F., Suzuki, H., Wang, X. D., Noda, M., Yahagi, N., et al. (1989). Structural Parts Involved in Activation and Inactivation of the Sodium Channel. Nature 339, 597–603. doi:10.1038/339597a0
Stühmer, W., Methfessel, C., Sakmann, B., Noda, M., and Numa, S. (1987). Patch Clamp Characterization of Sodium Channels Expressed from Rat Brain cDNA. Eur. Biophys. J. 14, 131–138. doi:10.1007/BF00253837
Sula, A., Booker, J., Ng, L. C., Naylor, C. E., DeCaen, P. G., and Wallace, B. A. (2017). The Complete Structure of an Activated Open Sodium Channel. Nat. Commun. 8, 14205. doi:10.1038/ncomms14205
Sun, Y. M., Favre, I., Schild, L., and Moczydlowski, E. (1997). On the Structural Basis for Size-Selective Permeation of Organic Cations through the Voltage-Gated Sodium Channel. Effect of Alanine Mutations at the DEKA Locus on Selectivity, Inhibition by Ca2+ and H+, and Molecular Sieving. J. Gen. Physiol. 110, 693–715. doi:10.1085/jgp.110.6.693
Sunami, A., Glaaser, I. W., and Fozzard, H. A. (2000). A Critical Residue for Isoform Difference in Tetrodotoxin Affinity Is a Molecular Determinant of the External Access Path for Local Anesthetics in the Cardiac Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 97, 2326–2331. doi:10.1073/pnas.030438797
Tamkun, M. M., and Catterall, W. A. (1981). Reconstitution of the Voltage-Sensitive Sodium Channel of Rat Brain from Solubilized Components. J. Biol. Chem. 256, 11457–11463. doi:10.1016/s0021-9258(19)68422-x
Tang, L., Gamal El-Din, T. M., Payandeh, J., Martinez, G. Q., Heard, T. M., Scheuer, T., et al. (2014). Structural Basis for Ca2+ Selectivity of a Voltage-Gated Calcium Channel. Nature 505, 56–61. doi:10.1038/nature12775
Tang, L., Gamal El-Din, T. M., Swanson, T. M., Pryde, D. C., Scheuer, T., Zheng, N., et al. (2016). Structural Basis for Inhibition of a Voltage-Gated Ca2+ Channel by Ca2+ Antagonist Drugs. Nature 537, 117–121. doi:10.1038/nature19102
Tang, X. C., Liu, X. J., Lu, W. H., Wang, M. D., and Li, A. L. (1986). Studies on the Analgesic Action and Physical Dependence of Bulleyaconitine A. Yao Xue Xue Bao 21, 886–891.
Thomsen, W. J., and Catterall, W. A. (1989). Localization of the Receptor Site for Alpha-Scorpion Toxins by Antibody Mapping: Implications for Sodium Channel Topology. Proc. Natl. Acad. Sci. U. S. A. 86, 10161–10165. doi:10.1073/pnas.86.24.10161
Ulbricht, W. (1969). The Effect of Veratridine on Excitable Membranes of Nerve and Muscle. Ergeb Physiol. 61, 18–71. doi:10.1007/BFb0111446
Vassilev, P. M., Scheuer, T., and Catterall, W. A. (1988). Identification of an Intracellular Peptide Segment Involved in Sodium Channel Inactivation. Science 241, 1658–1661. doi:10.1126/science.241.4873.1658
Vijayaragavan, K., O'Leary, M. E., and Chahine, M. (2001). Gating Properties of Na(v)1.7 and Na(v)1.8 Peripheral Nerve Sodium Channels. J. Neurosci. 21, 7909–7918. doi:10.1523/jneurosci.21-20-07909.2001
Wang, C. F., Gerner, P., Wang, S. Y., and Wang, G. K. (2007). Bulleyaconitine A Isolated from Aconitum Plant Displays Long-Acting Local Anesthetic Properties In Vitro and In Vivo. Anesthesiology 107, 82–90. doi:10.1097/01.anes.0000267502.18605.ad
Wang, G. K., Quan, C., Seaver, M., and Wang, S. Y. (2000). Modification of Wild-type and Batrachotoxin-Resistant Muscle Mu1 Na+ Channels by Veratridine. Pflugers Arch. 439, 705–713. doi:10.1007/s004249900229
Wang, G. K., Russell, C., and Wang, S. Y. (2003). State-dependent Block of Wild-type and Inactivation-Deficient Na+ Channels by Flecainide. J. Gen. Physiol. 122, 365–374. doi:10.1085/jgp.200308857
Wang, S. Y., Barile, M., and Wang, G. K. (2001). Disparate Role of Na(+) Channel D2-S6 Residues in Batrachotoxin and Local Anesthetic Action. Mol. Pharmacol. 59, 1100–1107. doi:10.1124/mol.59.5.1100
Wang, S. Y., and Wang, G. K. (1999). Batrachotoxin-resistant Na+ Channels Derived from Point Mutations in Transmembrane Segment D4-S6. Biophys. J. 76, 3141–3149. doi:10.1016/S0006-3495(99)77465-5
Wang, S. Y., and Wang, G. K. (1998). Point Mutations in Segment I-S6 Render Voltage-Gated Na+ Channels Resistant to Batrachotoxin. Proc. Natl. Acad. Sci. U. S. A. 95, 2653–2658. doi:10.1073/pnas.95.5.2653
Watanabe, H., Darbar, D., Kaiser, D. W., Jiramongkolchai, K., Chopra, S., Donahue, B. S., et al. (2009). Mutations in Sodium Channel β1- and β2-subunits Associated with Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2, 268–275. doi:10.1161/CIRCEP.108.779181
Weigele, J. B., and Barchi, R. L. (1982). Functional Reconstitution of the Purified Sodium Channel Protein from Rat Sarcolemma. Proc. Natl. Acad. Sci. U. S. A. 79, 3651–3655. doi:10.1073/pnas.79.11.3651
Weiss, J., Pyrski, M., Jacobi, E., Bufe, B., Willnecker, V., Schick, B., et al. (2011). Loss-of-function Mutations in Sodium Channel Nav1.7 Cause Anosmia. Nature 472, 186–190. doi:10.1038/nature09975
West, J. W., Numann, R., Murphy, B. J., Scheuer, T., and Catterall, W. A. (1991). A Phosphorylation Site in the Na+ Channel Required for Modulation by Protein Kinase C. Science 254, 866–868. doi:10.1126/science.1658937
West, J. W., Patton, D. E., Scheuer, T., Wang, Y., Goldin, A. L., and Catterall, W. A. (1992). A Cluster of Hydrophobic Amino Acid Residues Required for Fast Na(+)-Channel Inactivation. Proc. Natl. Acad. Sci. U. S. A. 89, 10910–10914. doi:10.1073/pnas.89.22.10910
Wisedchaisri, G., Tonggu, L., Gamal El-Din, T. M., McCord, E., Zheng, N., and Catterall, W. A. (2021). Structural Basis for High-Affinity Trapping of the NaV1.7 Channel in its Resting State by Tarantula Toxin. Mol. Cell. 81, 38–e4. doi:10.1016/j.molcel.2020.10.039
Wisedchaisri, G., Tonggu, L., McCord, E., Gamal El-Din, T. M., Wang, L., Zheng, N., et al. (2019). Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Cell. 178, 993–e12.e1012. doi:10.1016/j.cell.2019.06.031
Xiao, Y., Bingham, J. P., Zhu, W., Moczydlowski, E., Liang, S., and Cummins, T. R. (2008). Tarantula Huwentoxin-IV Inhibits Neuronal Sodium Channels by Binding to Receptor Site 4 and Trapping the Domain Ii Voltage Sensor in the Closed Configuration. J. Biol. Chem. 283, 27300–27313. doi:10.1074/jbc.M708447200
Xiao, Y., Blumenthal, K., Jackson, J. O., Liang, S., and Cummins, T. R. (2010). The Tarantula Toxins ProTx-II and Huwentoxin-IV Differentially Interact with Human Nav1.7 Voltage Sensors to Inhibit Channel Activation and Inactivation. Mol. Pharmacol. 78, 1124–1134. doi:10.1124/mol.110.066332
Xu, H., Li, T., Rohou, A., Arthur, C. P., Tzakoniati, F., Wong, E., et al. (2019). Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell. 176, 702–e14. doi:10.1016/j.cell.2019.01.047
Yan, Z., Zhou, Q., Wang, L., Wu, J., Zhao, Y., Huang, G., et al. (2017). Structure of the Nav1.4-β1 Complex from Electric Eel. Cell. 170, 470–e11.e411. doi:10.1016/j.cell.2017.06.039
Yellen, G. (2002). The Voltage-Gated Potassium Channels and Their Relatives. Nature 419, 35–42. doi:10.1038/nature00978
Yue, L., Navarro, B., Ren, D., Ramos, A., and Clapham, D. E. (2002). The Cation Selectivity Filter of the Bacterial Sodium Channel, NaChBac. J. Gen. Physiol. 120, 845–853. doi:10.1085/jgp.20028699
Zhang, X., Ren, W., DeCaen, P., Yan, C., Tao, X., Tang, L., et al. (2012). Crystal Structure of an Orthologue of the NaChBac Voltage-Gated Sodium Channel. Nature 486, 130–134. doi:10.1038/nature11054
Keywords: voltage-gated sodium channel, cryo-EM, pharmocology, gating mechanism, drug modulation mechanism
Citation: Jiang D, Zhang J and Xia Z (2022) Structural Advances in Voltage-Gated Sodium Channels. Front. Pharmacol. 13:908867. doi: 10.3389/fphar.2022.908867
Received: 31 March 2022; Accepted: 23 May 2022;
Published: 03 June 2022.
Edited by:
Chia-Hsueh Lee, St. Jude Children’s Research Hospital, United StatesReviewed by:
Rong Shen, The University of Chicago, United StatesHeike Wulff, University of California, Davis, United States
Copyright © 2022 Jiang, Zhang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daohua Jiang, jiangdh@iphy.ac.cn