
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 06 July 2022
Sec. Pharmacology of Infectious Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.903438
This article is part of the Research Topic Phytochemicals Targeting Autophagy in Treatment of Bacterial Infection and Malignancies View all 5 articles
 Priyanka Basak1Priyanka Maitra1Uzma Khan1Kalyani Saha1Satya Sundar Bhattacharya2
Priyanka Basak1Priyanka Maitra1Uzma Khan1Kalyani Saha1Satya Sundar Bhattacharya2 Moumita Dutta3
Moumita Dutta3 Sushmita Bhattacharya1*
Sushmita Bhattacharya1*Antibiotic treatment plays an essential role in preventing Shigella infection. However, incidences of global rise in antibiotic resistance create a major challenge to treat bacterial infection. In this context, there is an urgent need for newer approaches to reduce S. flexneri burden. This study largely focuses on the role of the herbal compound capsaicin (Caps) in inhibiting S. flexneri growth and evaluating the molecular mechanism behind bacterial clearance. Here, we show for the first time that Caps inhibits intracellular S. flexneri growth by inducing autophagy. Activation of autophagy by Caps is mediated through transcription factor TFEB, a master regulator of autophagosome biogenesis. Caps induced the nuclear localization of TFEB. Activation of TFEB further induces the gene transcription of autophagosomal genes. Our findings revealed that the inhibition of autophagy by silencing TFEB and Atg5 induces bacterial growth. Hence, Caps-induced autophagy is one of the key factors responsible for bacterial clearance. Moreover, Caps restricted the intracellular proliferation of S. flexneri-resistant strain. The efficacy of Caps in reducing S. flexneri growth was confirmed by an animal model. This study showed for the first time that S. flexneri infection can be inhibited by inducing autophagy. Overall observations suggest that Caps activates TFEB to induce autophagy and thereby combat S. flexneri infection.
Shigella flexneri, a common gastrointestinal pathogen, causes bacillary dysentery that affects millions every year worldwide (Nandy et al., 2011; Taneja and Mewara, 2016; Williams and Berkley, 2018; McQuade et al., 2020). According to the available information, S. flexneri infection is highly predominant in developing nations. Children below 5 years of age are affected with severe morbidity and mortality (Chang et al., 2016; Baker and The, 2018; Khalil et al., 2018; Chanin et al., 2019; Chen et al., 2020). The pathogen invades the intestinal epithelial and immune cells, causing ulcerative lesions in the gut under severe conditions (Killackey et al., 2016). To date, the only treatment option is antibiotics as no licensed vaccine has been developed (Hosangadi et al., 2019). Recently, the reverse vaccinology approach has been considered to improve vaccine research against Shigella (Hajialibeigi et al., 2021). Excessive use of antibiotics has led to a significant rise in antibiotic-resistant bacteria (Puzari et al., 2018; Ranjbar and Farahani, 2019). Hence, in the current scenario, there is an imperative need to search for alternative approaches in treating S. flexneri infection.
Presently, host-directed therapy is considered as an alternative therapeutic approach for the eradication of invasive pathogens (Rubinsztein et al., 2012; Kaufmann et al., 2018). Host-directed therapeutics can target the disease-causing signaling pathways and help eradicate microbes (Noad et al., 2017; Young et al., 2020). Autophagy plays a significant role among different host response mechanisms in combating infectious pathogens. Autophagy is a self-degradation mechanism used to eliminate unwanted materials like pathogens from the cell. Exploitation of autophagy has emerged as a new approach toward intracellular pathogens like Mycobacterium tuberculosis and Salmonella typhimurium (Kim et al., 2012; Ammanathan et al., 2020). In the context of infectious diseases, autophagy inducers such as vitamin D and antibiotics isoniazid and bedaquiline are reported to inhibit the proliferation of Mycobacterium tuberculosis and H. pylori (Yuk et al., 2009; Hu et al., 2019; Kim et al., 2019; Giraud-Gatineau et al., 2020). Similar strategies have also been used against viruses like HIV (Jo et al., 2013; Sharma et al., 2018). However, there are no such interventions reported against S. flexneri.
As autophagy is an important host defense mechanism occurring within the cell to clear pathogens, S. flexneri has evolved multiple ways to prevent autophagic recognition (Ogawa et al., 2005; Ra et al., 2016). S. flexneri circumvents autophagic degradation by releasing effector proteins (icsB and virA) and establishes itself in the intracellular niche of the colonic environment. Moreover, it has been reported earlier that Atg4b mutant cells inhibited S. flexneri associated maturation of autophagy (Ogawa et al., 2005; Weddle and Agaisse, 2018). Additionally, septin cages are known to promote autophagy; however, Shigella can polymerize actin and avoid septin caging (Krokowski et al., 2018; Lobato-Márquez et al., 2019). Hence, S. flexneri has developed an intricate machinery to avoid autophagy and establish infection within the host. As drug resistance is a major problem to tackle S. flexneri, thus boosting autophagy can curb intracellular S. flexneri infection.
Autophagy induction occurs by inducing several signaling molecules like mTOR, Akt, and AMPK (Floto et al., 2007; Rubinsztein et al., 2012; Young et al., 2020). Recent studies have shown the importance of a transcription factor known as TFEB (Ammanathan et al., 2020). TFEB activation leads to upregulation of autophagy genes. Boosting gene transcription induces host defense machinery and helps in the eradication of pathogens. Therefore, we searched for potent inducers of autophagy.
Capsaicin, a dietary compound from chilli plants, is reported to have anticancer properties (Lin S. R. et al., 2017). Emerging evidences have pointed out that capsaicin also possess antimicrobial activities against gastrointestinal pathogens (Füchtbauer et al., 2021). However, to date, there are no reports of its antibacterial action against Shigella flexneri.
Herein, we exploited the mechanism of autophagy induced by capsaicin to inhibit bacterial growth. This study showed for the first time that Caps induced an autophagy-dependent clearance of intracellular S. flexneri. Our findings indicate that Caps is a potential autophagy inducer that helps reduce S. flexneri infection.
Shigella flexneri (sf2457T) serotype 2a, plasmid-cured Shigella flexneri (sf2457T)2a, and Shigella flexneri-resistant strain (NA/CIP/NOR/OFX/TET/S/C/AM/E/ST) (BCH12702) were obtained from Dr Asish Mukherjee and Dr. Hemanta Koley (NICED, Kolkata, India). Bacteria were routinely cultured in Mueller Hinton Broth (Himedia) at 37°C incubator with a shaker.
HT-29 (ATCC HTB-38) and murine macrophage RAW264.7 (ATCC TIB-71) cells were used during this study. HT-29 cells were cultured with a McCoy’s 5A medium (Sigma-Aldrich) containing 10% heat inactivated FBS (Sigma-Aldrich, United States), 1% non-essential amino acids, and 1% penicillin–streptomycin (Himedia, India). Graphs represent CFU/ml of intracellular S. flexneri. RAW264.7 cells were grown in RPMI1640 with 10% FBS and 1% penstrep. Experiments were conducted in cells with maximum three to five passages. Cells were maintained in a 37 °C humidified incubator with 5% CO2.
The eGFP-LC3 plasmid was a gift from Prof. Parimal Karmakar (Department of Biotechnology, Jadavpur University, India). pEGFP-N1-TFEB plasmid was a gift from Dr. Ravi Manjithaya, Autophagy lab, JNCASR, Bangalore. Control EGFP plasmid was a kind gift from Dr Santa Sabuj Das, ICMR, NICED, Kolkata. The plasmids were transformed in DH5α and purified by the mini-prep plasmid isolation kit (Promega). The plasmid DNA was suspended in nuclease free water and stored at −20°C.
Cells were seeded in 96-well plates (1×104 cells/ml). At this seeding density, cells were treated with varying concentrations of Caps (16 μM, 32 µM) keeping an uninfected DMSO control and incubated for 24 h. Cell viability was checked using a Colorimetric Cell Viability Kit IV (MTT) (Promokine) followed by the addition of 20 μl of MTT reagent and incubated for 4 h at 37°C in a CO2 incubator. Thereafter, 300 μl of DMSO was added to solubilize the purple crystal formazan, OD was measured at 570 nm, and % viability was calculated and graphically represented.
GFP-LC3 (2 ug/well) plasmid was transfected in HT 29 and Raw264.7 cells using lipofectamine 2000 reagent (Invitrogen. United States). At 85% confluency, cells were transfected with [plasmid (ug): lipofectamine (ul) = 1:3] following the manufacturer’s protocol. After transfection for 48 h, cells were pre-treated with caps (16 µM) followed by S. flexneri infection. For Shigella immunofluorescence, GFP-LC3B-transfected cells were infected with S. flexneri. 400 µl of S. flexneri from an overnight culture of O.D (0.5–0.6) was added in intestinal cells and macrophages. Cells were further subjected to immunofluorescence. Briefly, fixed cells were incubated with anti-Shigella antibody (1:250) (ab65282) dilution. Subsequently, TRITC-conjugated anti rabbit secondary antibody (1:2000) (Cat# AP132R) was used. Finally, coverslips were mounted on a ProLong™ Gold Antifade reagent containing DAPI (ThermoFisher) and observed under an inverted confocal microscope (Carl Zeiss LSM 710). GFP-positive cells per sample were selected for observation.
The following siRNAs were purchased from IDT and used for the knockdown of those particular genes: ATG5 siRNA (Design ID hs. Ri. ATG5.13.1) and TFEB siRNA (Design ID hs. Ri. TFEB.13.1). HT-29 cells at 60%–70% confluency were transfected with 12 pmol of siRNA/Scrambled siRNA, and 2 ul of lipofectamine was added per well in a 6-well plate. Forty-eight hours after transfection, the infection assay was performed in cells infected with S. flexneri with and without Caps pre-treatment. Cells were lysed for CFU/ml counting.
For fluorescence microscopy, cells were grown on coverslips and Caps treatment was done for 2 h. After fixation with 4% formaldehyde, cells were further blocked in PBS containing 3% BSA and 0.1% Triton X100 for 1 h at room temperature. Overnight incubation was performed with anti-TFEB antibody (1:250 dilution). Following that, fixed cells were kept in a blocking buffer containing TRITC-conjugated anti-rabbit secondary antibody (1:2000) (Cat# AP132R). Finally, the coverslips mounting upon glass slides were done by adding a drop of ProLong™ Gold Antifade reagent containing DAPI (ThermoFisher). Images were scanned on a Zeiss LSM 710 confocal system (Carl Zeiss).
Intestinal and macrophage cells (1X105) were cultured in 6-well plates for 24 h at 37 °C and 5% CO2 prior to infection. Cells were further incubated overnight in antibiotic free media. S. flexneri 2457t colonies (red) were picked from Congo red plates as Congo red indicates the retention of virulence plasmid. S. flexneri was grown overnight in MHB at 37°C with shaking and sub-cultured to an OD600 of 0.5–0.6. Bacteria pellets were collected by centrifuging the culture at 12,000 g for 5 min at 4°C and further washed with PBS. Pellets were then re-suspended in incomplete tissue culture media. Intestinal cells were pre-treated with Caps (1.6, 3.2, 4.8, 6.4, 8, 16, 32 µM, or vehicle control (DMSO), for 2, 4, 6, 8, 12, and 24 h in incomplete media. Then, infection with S. flexneri was performed at an MOI of 200:1. The infected plates were centrifuged for 15 min at 700 g for synchronization and incubated for 2 h to allow bacteria to enter the cells. Cells were thoroughly washed and treated with 50 μg/ml of gentamicin for 2 h, killing the extracellular bacteria. Further washing with PBS was done, and then cells were lysed using 0.1% triton X. The cell lysates were plated in MHA at 37°C for 18 h to count the number of colonies (CFU/ml). For post-treatment analysis, cells were infected with bacteria and then exposed to Caps for different time points. After incubation and infection, gentamicin was used to kill the extracellular bacteria, and further lysis was done and plated. This assay was also performed using S. flexneri highly resistant strain (BCH12702) with or without Caps 12 h post-treatment to compare invasion. Results were compared to 3-methyladenine (#SAE0107, sigma)-treated cells. For infection in macrophages, cells were preincubated with Caps for 2 h followed by 30 min of S. flexneri infection (MOI100). After infection, gentamicin was used for killing the extracellular bacteria and further lysis was done and plated. For post-infection analysis, cells were infected with S flexneri (MOI100) for 30 min followed by Caps treatment for 4 h. After that, gentamicin was used to kill the extracellular bacteria, and further lysis was performed and plated.
siATG5, siTFEB-transfected cells were pre-treated with Caps and infected for gentamicin survival assay to count CFU/ml compared to scrambled siRNA. TFEB overexpression was also performed by lipofectamine 2000 using pEGFP-N1-TFEB plasmid and empty vector (pcDNA3-EGFP).
RNA isolation was performed using the RNA isolation kit (zymo research) from treated and untreated cells. TRIzol reagent was used for colon samples collected from mice. RNA samples were converted to c-DNAs using the Thermo Scientific c-DNAs synthesis kit. Real-Time PCR was done using SYBR green kit of Applied Biosystems. The expression patterns were analyzed using the ΔΔCt method and normalized using the internal control GAPDH. ΔΔCt were calculated by the following formula:
Human primer sequences used were listed as follows:

Differential expression of genes involved in autophagy pathway was analyzed using a human autophagy PCR array. This array contained 84 key genes in a 96-well plate format. Fold change was plotted in a column graph to observe the differences among uninfected, infected, and infected with Caps. PCR array (#PAHS-084Z) (RT2 Profiler™ PCR Array Human Autophagy) was performed to understand the complete autophagy profile of the control and treated samples following the manufacturer's instructions (Qiagen). RNA isolation was performed from two groups of cells. cDNA was obtained using cDNA synthesis reagents and mixed with SYBR Green Mastermix (provided inside the kit). Quantitative real-time PCR was done in Applied biosystems StepOnePlus system. Obtained melt curves and ΔΔCt values were analyzed in free web-based RT2 Profiler™ PCR array data analysis software.
Untreated, treated, and infected cell lysates were prepared in RIPA (radioimmunoprecipitation assay buffer) lysis buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 1 mM PMSF) (protease and phosphatase inhibitor added). Lysates were centrifuged at 7,000 rpm for 20 min at 4°C. Nuclear and cytosolic extracts were prepared by using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents of Thermo Scientific (Cat#78833) following the manufacturer’s instructions.
After protein estimation, lysates were boiled in SDS-PAGE sample buffer and run on 10% or 12.5% SDS- PAGE gel at 120 V. Gels were transferred to PVDF membrane. Blocking was performed with 5% skimmed milk dissolved in TBST (20 mM Tris–HCl, 150 mM NaCl, 0.1% Tween20) buffer for 1 h at room temperature, and the membranes were incubated overnight with primary antibodies at 4 °C. The primary antibodies utilized are rabbit monoclonal anti-GAPDH antibody (Cat#D16H11) (1:2,000), rabbit polyclonal Anti-SQSTM1/P62 antibody (Cat# ab91526) (1:1,000), rabbit monoclonal anti-ATG5 (Cat #ab228668) (1:1,000), rabbit monoclonal anti-beclin1 antibody (Cat #ab207612) (1:1,000), rabbit polyclonal anti-LC3B antibody (Cat #ab51520) (1:1,000), and rabbit monoclonal anti-TFEB antibody (Cat #D2070). Following primary antibody treatment, membranes were kept in a shaker at room temperature for 2 h with horseradish peroxidase (HRP) conjugated goat anti-rabbit secondary antibody (dilution 1:10,000) or goat anti-mouse secondary antibody (dilution 1:10,000). Membranes were finally washed with TBST for 30 min. Bands were observed in the ChemiDoc MP Imaging System (Biorad) using Millipore immobilon western chemiluminescent HRP substrate (luminol and hydrogen peroxide) as a substrate.
A broth dilution assay was performed in the presence of DMSO as a control, 16, 32, 64, and 80 µM Caps, and nalidixic acid (10 μg/ml) as a positive control in separate flasks containing MHB. A total of 105 S. flexneri from overnight culture was added to the flasks. O.D.600nm was measured at regular intervals and graphically represented.
Shigella flexneri (BCH12702) was treated with antibiotics (NA/CIP/NOR/OFX/TET/S/C/AM/E/ST) at different concentrations. The minimum inhibitory concentrations of the antibiotics were determined by the CLSI broth microdilution method. The test was performed by serially diluting the antibiotics in LB media and incubating for 24 h in a 37°C incubator. The minimum inhibitory concentration (MIC) represents the lowest concentration of the antibiotics that resulted in a lack of visible bacterial growth. Tabular representation shows the MIC (µg/ml) and cutoff values for resistance according to CLSI guidelines.
Uninfected and infected cells (HT-29 and RAW264.7) treated with or without Caps were fixed in 3% glutaraldehyde in 0.1 M sodium cacodylate buffer. Further fixation with 1% osmium tetroxide continued, and then dehydrated with increasing grades of acetone, eventually embedded in Agar 100 resin, and polymerized at 60 °C. Ultrathin sections (around 40–50 nm thickness) were made using a Leica Ultracut UCT ultramicrotome (Leica Microsystems, Germany), picked up on nickel grids, and dual-stained with 2% aqueous uranyl acetate and 0.2% lead citrate. We examined cell sections using an FEI Tecnai 12 Biotwin transmission electron microscope (FEI, Hillsboro, OR, United States) at an accelerating voltage of 100 kV. Percentage autophagosome formation was calculated: Cell number containing autophagosome/total cell number x 100%Autophagic bodies were outlined with the help of Adobe Photoshop.
HT-29 cells were treated with Caps for 4 h. After incubation, crosslinking was done in 1% formaldehyde and processed according to the protocol of the CHIP assay kit (#7-295; Merck Millipore). Briefly, cells were lysed in SDS lysis buffer and pelleted. After processing, chromatin was immunoprecipitated with anti-TFEB antibodies (5 µg/sample). The chromatin fraction, which lacks a primary antibody, was taken as “input.” Real-time PCR was performed using the following chip primer assemblies. Primers were designed for amplifying CLEAR elements in the promoter region of TFEB using primer: MAP1LC3B-forward; 5′-GAAGGCTCGGGACAAAAGCAG-3′, reverse; 5′-GTGGGTGGCTTCCGGGGAG-3’. The PCR cycle was conducted in accordance with the manufacturer’s instructions. The data were graphically represented as the % of input.
The TFEB transcription factor assay was performed using the RayBio® Human TFEB TF-Activity Assay Kit Protocol. Nuclear extract collected from control and Caps-treated cells were used as samples. Samples were added to wells coated with oligonucleotides containing CLEAR sequence. After the addition of primary antibody, HRP-conjugated secondary antibody was added. This step was followed by the addition of the TMB substrate reagent, the reaction was stopped with stop solution, and O.D. was collected at 450 nm by a spectrophotometer. The fold change was calculated and graphically represented.
Male BALB/c (8 weeks) mice weighing 20–22 g mice were used for S. flexneri infection. Animals were adjusted under standard laboratory conditions. Mice were provided with a standard diet and water ad libitum. IAEC (Institutional Animal Ethical Committee), NICED, Kolkata (PRO/157/-July 2022) guidelines were followed during all the experiments. Animals were maintained in the animal house with 75% humidity, and specific pathogen-free healthy individuals were selected.
After fasting for 6 h, mice were injected with pathogenic Shigella flexneri (sf2457T) intraperitoneally (Yang et al., 2014). After 2 h, Caps treatment was performed. In the experimental design, four groups (n = 4) were created accordingly: Group 1. Control: Only DMSO was used as the vehicle. Group 2. Infected: Received S. flexneri suspension (0.5 × 109 CFU/ml) in PBS. Group 3. Infected + Caps: Received S. flexneri suspension followed by Caps (20 mg kg−1 body weight) treatment for 2 h. Group 4. Caps: Received Caps (20 mg kg−1 body weight). Each group was kept in an individual cage. Experiments were repeated in triplicate (n = 3). The total number of mice in each group was 12.
After 2 h of Caps treatment, all the animals were sacrificed. Colon from drug-treated, untreated, infected, and control mice was aseptically removed. The crushed colon was plated for bacterial colony count. The tissues were kept at −80 °C until further experiments were performed.
Colon samples were collected following dissection, washed in PBS buffer, and further homogenized in an ice-cold RIPA lysis buffer for western blot and in TRIzol reagent (RNAiso Plus, TaKaRa) for RNA isolation. The homogenates were centrifuged at 12,000 rpm at 4 °C for 10 min. The supernatants collected were further utilized for experiments. RNA was isolated, and RNA to cDNA conversion was performed using a Verso cDNA Synthesis Kit (Thermo Scientific) followed by qRT PCR.
Test information is presented as mean ± S.E.M. Statistical analyses were performed, and bar graphs were processed in GraphPad Prism 5 after the data processing in Microsoft Excel. For comparison between two groups, an unpaired t-test was performed, and for multiple comparison, one-way ANOVA is performed. For multiple variants, two-way ANOVA was performed. Significance level has been marked as * for p < 0.05, which implies significant, ** for p < 0.01, which implies very significant, and *** for p < 0.001, which implies highly significant.
The antibacterial effects of capsaicin (Caps) are reported against Vibrio cholerae, H. pylori and other bacteria (Füchtbauer et al., 2021). However, there are no such reports of capsaicin-induced inhibition of intracellular S. flexneri growth. Here, we tested the effect of Caps on the intracellular clearance of S. flexneri. For this approach, we used an intracellular invasion assay in an S. flexneri infection model. Briefly, Caps was added in HT-29 cells for 2 h followed by S. flexneri infection to check intracellular bacterial clearance. Caps at different doses (16 and 32 μM) significantly reduced S. flexneri replication (Figure 1A). In addition, Caps significantly (16 μM) reduced S. flexneri multiplication after pre-treatment at different time points (2, 4, 6, 12, and 24 h) in HT-29 cells (Figure 1B). Moreover, we checked the effect of Caps by posttreatment analysis. S. flexneri growth was inhibited significantly at 12 h by Caps. However, at 24 h, cell death occurred in infected cells; hence, data are not significant (Figure 1C).
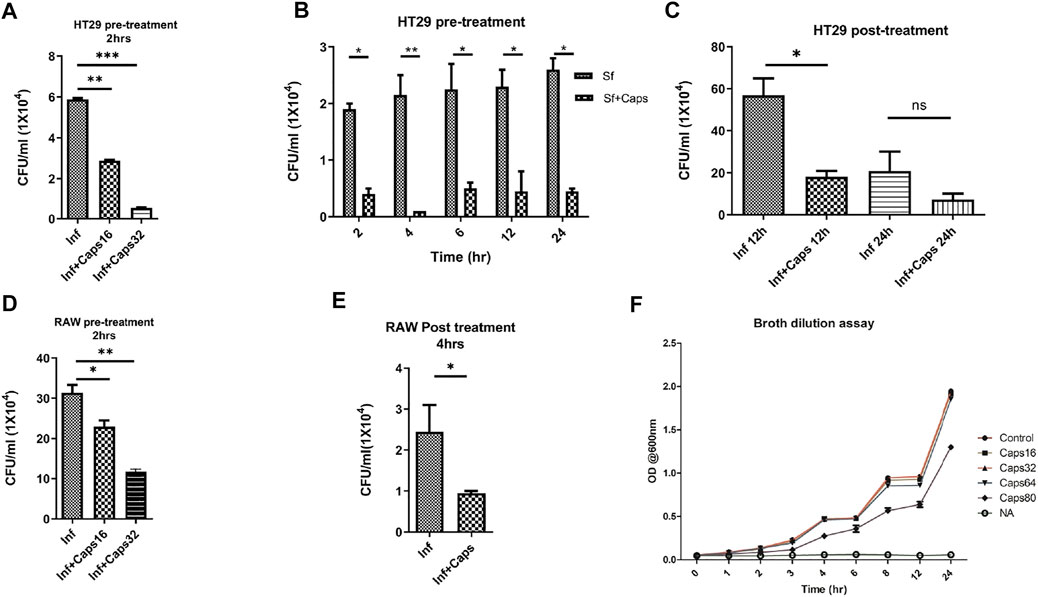
FIGURE 1. Intracellular S. flexneri growth was inhibited by capsaicin (Caps) in both intestinal and macrophage cells. (A) Infection assay was performed in HT29 cells with different concentrations of capsaicin (Caps). Briefly, cells were pre-treated (16 µM, 32 µM) with Caps for 2 h followed by S. flexneri infection (MOI 200) for 2 h. Finally, gentamicin treatment was done to kill extracellular bacteria. Cells were lysed and plated overnight for counting the number of colonies (CFU/ml). Graphs represent CFU/ml of intracellular S. flexneri. One-way ANOVA was performed. (B) Caps pre-treatment (16 µM) for 2 h followed by S. flexneri (MOI 200) infection at different time points (2, 4, 6, 8, 12, 24 h) was done. Two-way ANOVA was performed. (C) HT29 cells were infected with MOI200 followed by post-treatment with Caps (16 µM) for 12 and 24 h. One-way ANOVA was performed. Cells were lysed and plated to count the number of colonies. (D) RAW 264.7 cells were infected with S. flexneri MOI 100 for 30 min after 2 h Caps pre-treatment. Cells were lysed and plated to count the number of colonies One-way ANOVA was performed. (E) RAW264.7 cells were infected with S. flexneri MOI 100, followed by post-treatment with Caps (16 µM) for 4 h. Graph represents CFU/ml of intracellular S. flexneri. One-way ANOVA was performed. (F) A broth dilution assay was performed in the presence of DMSO control, 16, 32, 64, and 80 µM Caps and nalidixic acid (10 μg/ml) (NA) followed by the addition of 105 S. flexneri overnight culture. The OD at 600 nm was measured at regular intervals. Graphs were represented using GraphPad Prism 5 as mean ± SEM (n = 3); Significance was calculated; *p < 0.05, **p < 0.01, ***p < 0.001.
Similarly, we validated the results in macrophages, as macrophages are the immune cells and site of infection for S. flexneri during pathogenesis. Caps pre-treatment for 2 h significantly reduced bacterial growth in a dose-dependent manner (Figure 1D). Moreover, posttreatment analysis in macrophages showed that Caps is effective at 4 h in inhibiting S. flexneri growth (Figure 1E). Upon compound addition, bacterial growth was significantly reduced. However, the inhibition of bacterial burden could be due to a probable antibacterial property of Caps. Hence, we checked the effect of Caps on S. flexneri by the broth dilution method for extracellular bactericidal properties. There is no apparent change in bacterial growth for treated (Caps 16, 32, 64, and 80 μM) and untreated samples (Figure 1F). Collectively, the data revealed that exposure to Caps inhibits intracellular S. flexneri proliferation.
As capsaicin has no direct antibacterial action against S. flexneri, we questioned the probable mechanism involved in reducing intracellular bacterial growth. Previously, it was identified that Caps overexpressed autophagy proteins in nasopharyngeal cancer cells (Lin S. R. et al., 2017). However, there are no such reports on intestinal cells. Here, we examined the expression of autophagy proteins in intestinal epithelial cells by western blots. Caps overexpressed the autophagy proteins Atg5 and beclin1 in a dose-dependent manner in HT29 cells after treatment for 2 h (Figure 2A). Beclin1 is an autophagy regulator involved in autophagosome formation, while Atg5 helps in autophagosome maturation and LC3B lipidation (Brier et al., 2019). P62 degradation, another marker of autophagy initiation, takes place due to Caps treatment. Next, we examined the effect of Caps on autophagy genes at different concentrations. Caps significantly augmented the expression of MAP1LC3B and Atg5 genes in a dose-dependent manner (Figure 2B). To further assess the effect of Caps on autophagic gene expression, a PCR array was performed. Differential gene expression was observed due to Caps treatment in intestinal cells for 4 h. More than 78% of the genes are augmented by Caps alone at a dose of 16 µM (Figure 2C). As autophagy induction is often linked with cellular toxicity, we checked the toxicity of Caps in both intestinal and macrophage cells. Cytotoxicity assay of Caps in intestinal and macrophage cells showed insignificant effects due to Caps treatment for 24 h (Supplementary Figure S1). Together, these results indicate that Caps induces autophagic gene and protein expression in intestinal cells.
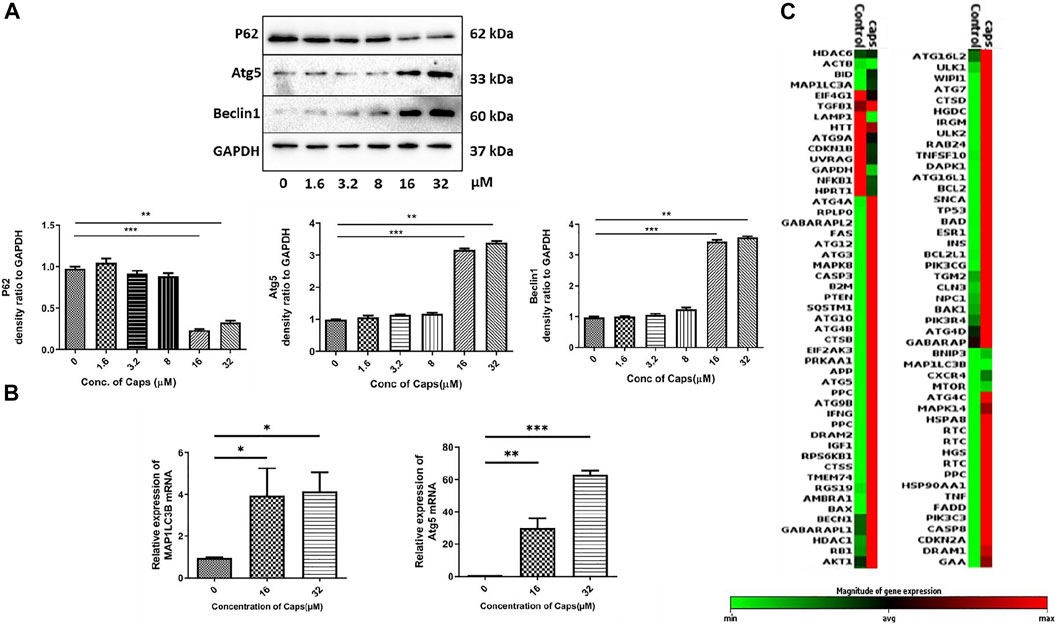
FIGURE 2. Capsaicin (Caps) induces autophagy in intestinal cells. (A) HT29 cells were treated with Caps (1.6, 3.2, 8, 16, 32 µM) or DMSO for 2 h. Expression of major autophagy-associated marker proteins P62, Atg5, and Beclin1 was observed in western blot. GAPDH was used as a loading control. Densitometric analyses are graphically represented. (B) HT29 cells were treated with Caps (16, 32 µM) or DMSO for 2 h followed by qRT-PCR, Expression of the autophagy-associated marker genes (Map1LC3B, Atg5) was quantified using GAPDH as a housekeeping gene. One-way ANOVA was performed. (C) PCR array was performed with control and only Caps-treated samples. Caps treatment was done for 4 h. Heat map generated represents differential gene expression. All experiments were performed in triplicate. Graphs were generated using GraphPad Prism 5 were represented as mean ± SEM (n = 3); significance was calculated; *p < 0.05, **p < 0.01, ***p < 0.001.
In addition to the stimulation of autophagic gene expression by Caps, we examined the potential effect of Caps on autophagosome formation by transmission electron microscopy (TEM) and confocal microscopy. The images showed autophagosome formation in cells treated with Caps (Figure 3A). Caps treatment alone showed 50% autophagosome formation in intestinal cells. Similar observations were validated by confocal microscopy. It is known that autophagosome formation is associated with membrane-bound LC3B. GFP-LC3B-transfected cells were exposed to Caps. Caps (16 µM) treatment for 2 h resulted in LC3B puncta formation in intestinal cells (Figure 3B). Together, these results confirm that Caps induces autophagosome formation in intestinal cells.
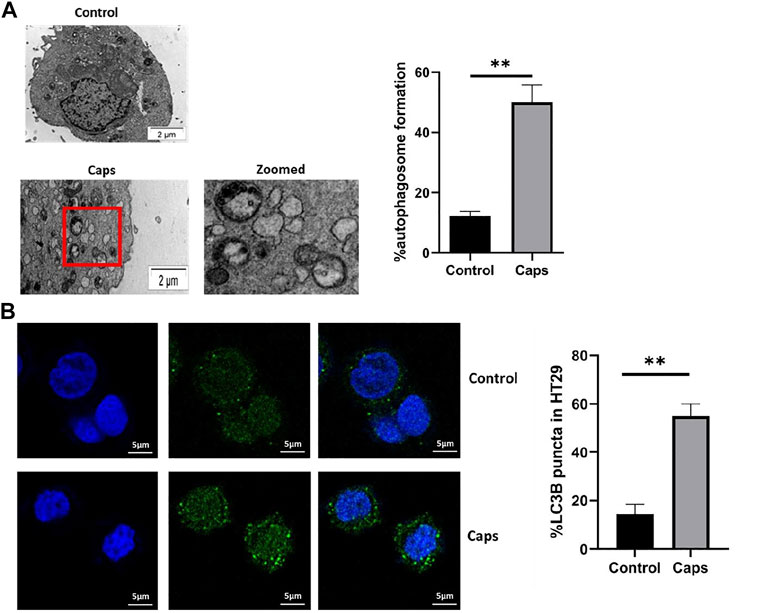
FIGURE 3. Treatment with Caps enhances autophagosome formation. (A) Electron microscopic (TEM) pictures showed the presence of autophagosome formation in control and Caps-treated HT-29 cells for 4 h. Zoomed images of the marked area. Scale bar: 2 μm. Graphical representation of % autophagosome formation as observed in TEM. Unpaired t-test was performed. (B) Confocal microscopic images of GFP-LC3B transfected intestinal cells (HT-29) showed LC3B puncta (green) formation. Briefly, cells were transfected with GFP-LC3B plasmid, then treated with Caps for 4 h, and visualized under a confocal microscope. Graph representing the percentage of LC3B puncta formation. All experiments were performed in triplicate. Unpaired t-test was performed. Graphs generated using GraphPad Prism 5 were represented as mean ± SEM (n = 3); significance was calculated; *p < 0.05, **p < 0.01, ***p < 0.001.
Host-induced capture of pathogens by autophagosomes occurs during autophagy-mediated degradation. As S. flexneri is known to escape from autophagy, we checked the capture of bacteria by autophagosomes due to Caps treatment in infected cells. Caps treatment showed the presence of autophagosome-like structures containing bacteria. We calculated the percentage of autophagosomes and observed that approximately 40% autophagosome formation takes place in S. flexneri-infected cells pre-treated with Caps (Figure 4A). Similar observations were observed in macrophage cells. Capture of bacteria by autophagosome was observed in drug-treated cells (Figure 4B). All these findings direct that Caps treatment induced autophagosome formation in S. flexneri-infected cells.
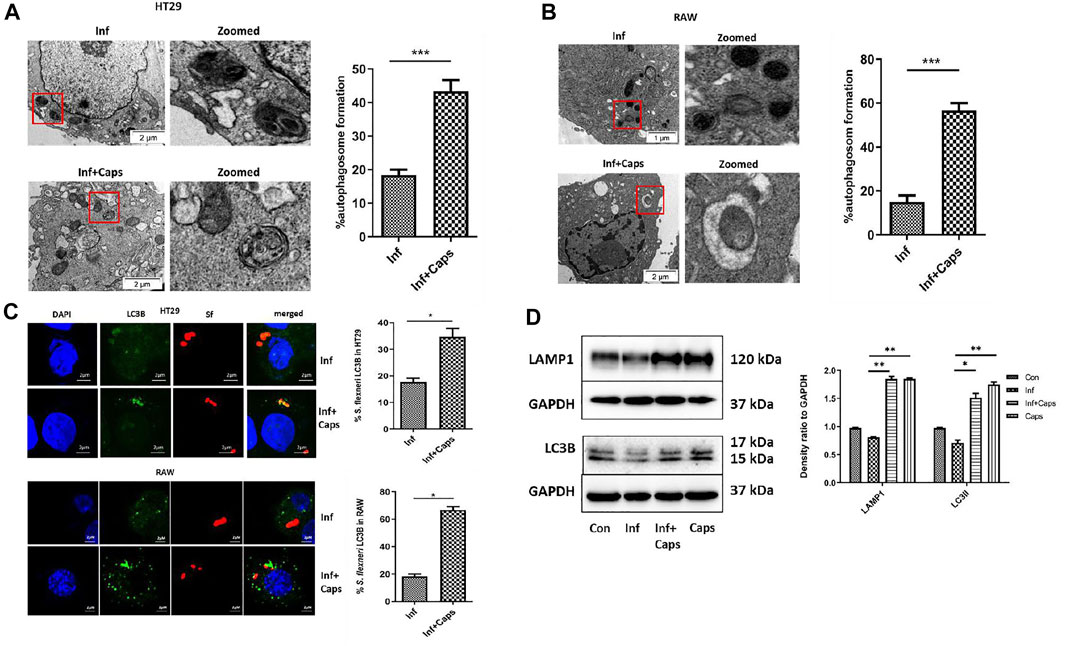
FIGURE 4. Caps treatment results in the engulfment of bacteria by autophagosome. (A) Transmission electron microscopy (TEM) pictures showed the presence of autophagosome formation in infected intestinal cells with and without Caps treatment. Zoomed images of the marked area. Scale bar: 2 μm. Graphical representation of % autophagosome formation as observed in TEM. Unpaired t-test was performed. (B) Transmission electron microscopy (TEM) pictures of autophagosome formation in infected and infected + Caps RAW267.4 cells. Zoomed images of the marked area. Scale bar: 2 μm. (C) Microscopic images of GFP-LC3B transfected intestinal cells (HT-29) showed the recruitment of S. flexneri (red) to LC3B (green). Briefly, cells were transfected with GFP-LC3B plasmid, then treated with Caps for 2 h followed by S. flexneri infection for 2 h. Graph representing the percentage of bacteria localizing with LC3B. Microscopy images of GFP-LC3B transfected macrophages (RAW 267.4) infected with bacteria and treated with and without Caps. Graph representing the number of bacteria recruited to LC3B. Unpaired t-test was performed. Scale bar: 2 μM. (D) HT29 cells were pre-treated with Caps for 2 h followed by infection. After infection, cells were lysed. Cell lysates were subjected to immunoblotting. Increase in LC3B and LAMP1 expression was shown by western blotting, and densitometric analyses are graphically represented [Caps = Only Caps treatment at a dose of 16 µM]. Graphs were represented using GraphPad Prism 5 as mean ± SEM (n = 3); one-way ANOVA was performed. Significance was calculated; *p < 0.05, **p < 0.01, ***p < 0.001.
To confirm the capture of S. flexneri by autophagosomes, confocal microscopy was performed. Therefore, both macrophages and intestinal cells were transfected with GFP-LC3B, treated with Caps
Following our investigations, we tried to find out the impact of autophagy inhibition on S. flexneri growth. We used autophagy inhibitors like 3-methyl adenine (3MA). As expected, 3MA increased intracellular S. flexneri proliferation. In the presence of 3MA, the anti-shigella activity of Caps was impaired, suggesting that the killing of bacteria is dependent on autophagy (Figure 5A). To rule out the nonspecific effect of inhibitors (3MA) a major autophagy player Atg5 was knocked down using siRNA to inhibit autophagy. Successful Atg5 knockdown was confirmed by western blot (Figure 5B). In Atg5 knockdown cells, Caps treatment failed to inhibit S. flexneri intracellular growth (Figure 5C).
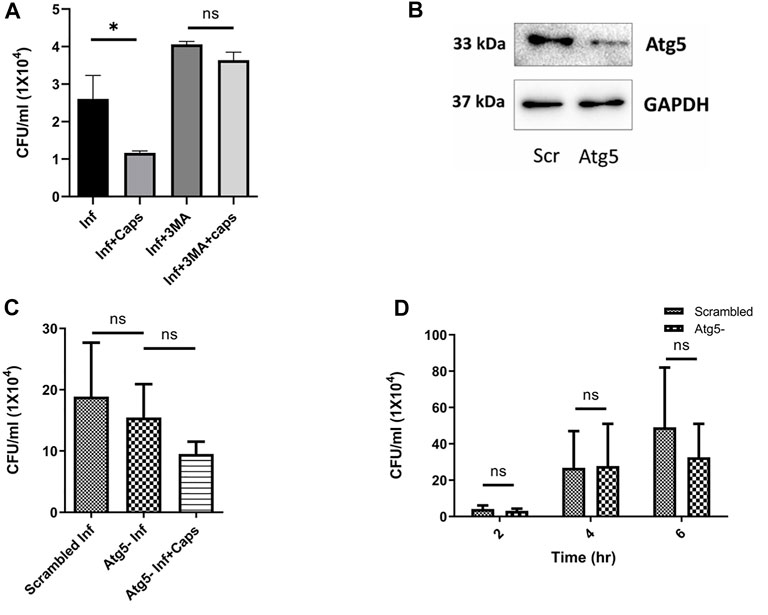
FIGURE 5. Autophagy inhibition induced bacterial growth. (A) Intestinal cells were treated with 3MA (5 mM), Caps (16µM) for 2 h, further infected with S. flexneri for 2 h followed by gentamicin treatment. Infection assay was performed. Graph represents intracellular bacterial count. One-way ANOVA was performed. (B) Cells were transfected with scrambled and Atg5 siRNA. Western blot shows the Atg5 expression in scrambled and knockdown cells. GAPDH was kept as a housekeeping control. (C) Infection assay was performed in scrambled and Atg5 knockdown cells treated with Caps for 2 h followed by infection. CFU/ml was graphically represented. One-way ANOVA was performed. (D) Infection assay was performed in scrambled and Atg5 knockdown cells for different time points (2, 4, 6 h). Briefly, after transfection with scrambled and Atg5 siRNA, cells were infected for different time points. Cells were lysed and plated for CFU count. Graphs were represented using GraphPad Prism 5 as mean ± SEM (n = 3); significance was calculated by two-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001.
Furthermore, to observe the effect of Atg5 knockdown in detail, we infected Atg5 knockdown cells for different time intervals (Figure 5D). Bacterial growth was unaffected due to the blockade of autophagy. These observations indicate that Caps-induced autophagy is responsible for S. flexneri death.
As Caps induced the transcription of autophagy genes, we searched for a potential transcription factor responsible for autophagosomal and lysosomal biogenesis. TFEB, a transcription factor, is reported to enhance autophagosome biogenesis. It binds to the promoter of CLEAR network (coordinated, lysosomal expression, and regulation) and induces autophagy genes (Polito et al., 2014; Sardiello, 2016; Vega-Rubin-de-Celis et al., 2017; Fan et al., 2018; Puertollano et al., 2018). HT29 cells were treated with Caps for 2 h followed by immunofluorescence. Herein, we observed an 80% increase in the nuclear expression of TFEB in Caps-treated cells as compared to that of untreated cells by the immunofluorescence assay (Figure 6A). To confirm TFEB nuclear localization and autophagic gene transcription, we performed promoter assays. Consistently, Caps augmented TFEB promoter activity significantly in treated cells as compared to that of untreated cells by binding to CLEAR sequence (Figure 6B). ChIP assay after chromatin immunoprecipitation also confirmed higher TFEB binding to MAP1LC3B promoter in cells treated with Caps (Figure 6C). Therefore, Caps triggered autophagic gene transcription via TFEB. Moreover, we examined the effect of Caps by immunoblotting. Caps treatment enhanced the nuclear expression of TFEB in both S. flexneri-infected and uninfected cells, whereas Caps reduced the cytosolic expression of TFEB (Figure 6D). These results suggest that Caps induced autophagic gene transcription by stimulating the nuclear translocation of TFEB.
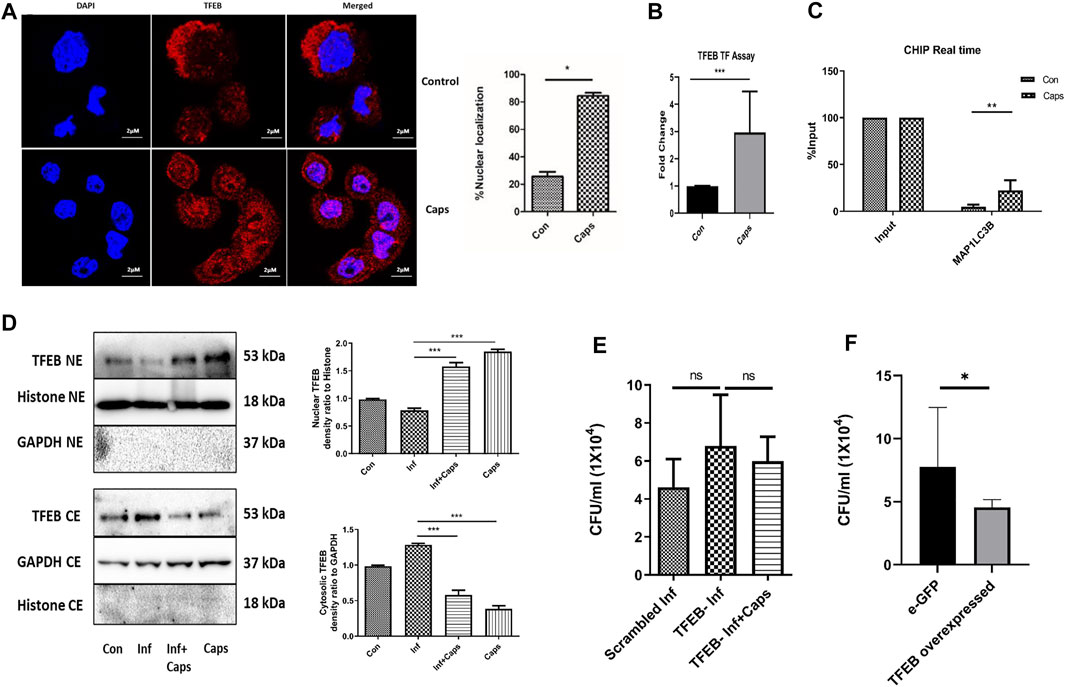
FIGURE 6. Caps treatment induced TFEB nuclear localization. (A) Confocal microscopic data showing nuclear localization of TFEB upon Caps treatment. Briefly, HT29 cells were treated with Caps (16 μM) for 2 h followed by immunofluorescence microscopy. Scale bar: 2 μm. (B) Caps treated and untreated cells were subjected to TFEB transcription assay. Transcription factor assay was performed to show the activation of TFEB binding to CLEAR sequence by Caps. Unpaired t-test was performed. (C) CHIP assay was performed with or without anti-TFEB antibody (input). Expression of MAP1LC3B was checked and compared to input in real-time PCR assay for untreated and Caps-treated intestinal cells. The data were represented graphically and compared to input; two-way ANOVA was performed. (D) Western blot analysis was performed for determining the protein level of TFEB in both cytosolic (CE) and nuclear extracts (NE) after Caps treatment in S. flexneri infected and uninfected cells. GAPDH was kept as a housekeeping control for cytoplasmic cell extracts. Histone was used as a control for nuclear extracts. Densitometric analyses are shown and graphically represented. One-way ANOVA was performed. (E) Cells were transfected with scrambled siRNA or TFEB siRNA. After transfection, cells were treated with Caps for 2 h and infected with S. flexneri for 2 h followed by gentamicin treatment. Cell lysis was performed and plated. One-way ANOVA was performed. (F) Cells were transfected with an empty vector eGFP or eGFP-TFEB plasmid followed by S. flexneri infection. Cells were lysed and plated for colony count. Unpaired t-test was performed. Graphs were represented using GraphPad Prism 5 as mean ± SEM (n = 3); Significance was calculated; *p < 0.05, **p < 0.01, ***p < 0.001.
To evaluate the relevance of TFEB activation in S. flexneri proliferation, we silenced TFEB by siRNA. TFEB knockdown itself has no effect on S. flexneri growth; however, Caps treatment in TFEB-silenced cells fails to clear intracellular bacterial burden (Figure 6E). Similarly, the overexpression of eGFP-TFEB plasmid in HT 29 cells resulted in the reduction of S. flexneri growth significantly (Figure 6F). Silencing of TFEB by siRNA and overexpression in intestinal cells were confirmed by immunoblot analysis (Supplementary Figure S3). Taken together, these findings indicate that TFEB is a major player in reducing bacterial growth by Caps.
To assess the efficacy of Caps in controlling S. flexneri infection, we checked the effect of Caps against a resistant strain of S. flexneri. We observed that Caps is effective in inhibiting the resistant S. flexneri strain growth significantly at a higher dose of 32 µM after 12 h post treatment. Supplementary Table S1 shows the MIC values of the resistant strain against different antibiotics (Figure 7A). Finally, we validated the effect of Caps in mouse model. Mice were infected by intraperitoneal challenge with S. flexneri 2457T strain (Figure 7B). The dose of Caps was selected in the infection model based on the available data of capsaicin (Rollyson et al., 2014). After 2 h of infection, mice were treated with Caps (20 mg/kg body weight) for 2 h and then sacrificed for further studies. We took the colon from infected and Caps-treated mice samples. The bacterial count showed a decrease in S. flexneri-infected mice that received Caps (Figure 7C). Additionally, we examined the effect of Caps on MAP1LC3B gene expression in mice colon tissues. Caps significantly enhanced the MAP1LC3B gene expression in infected mice as compared to that in untreated mice. Caps alone also increased the MAP1LC3B gene expression significantly (Figure 7D). We assessed the effect of Caps on p62 degradation, another marker of autophagy induction. Caps treatment induced p62 degradation in both infected and uninfected mice tissues (Figure 7E). These data confirm that Caps has the potential to be used as a therapeutic against S. flexneri.
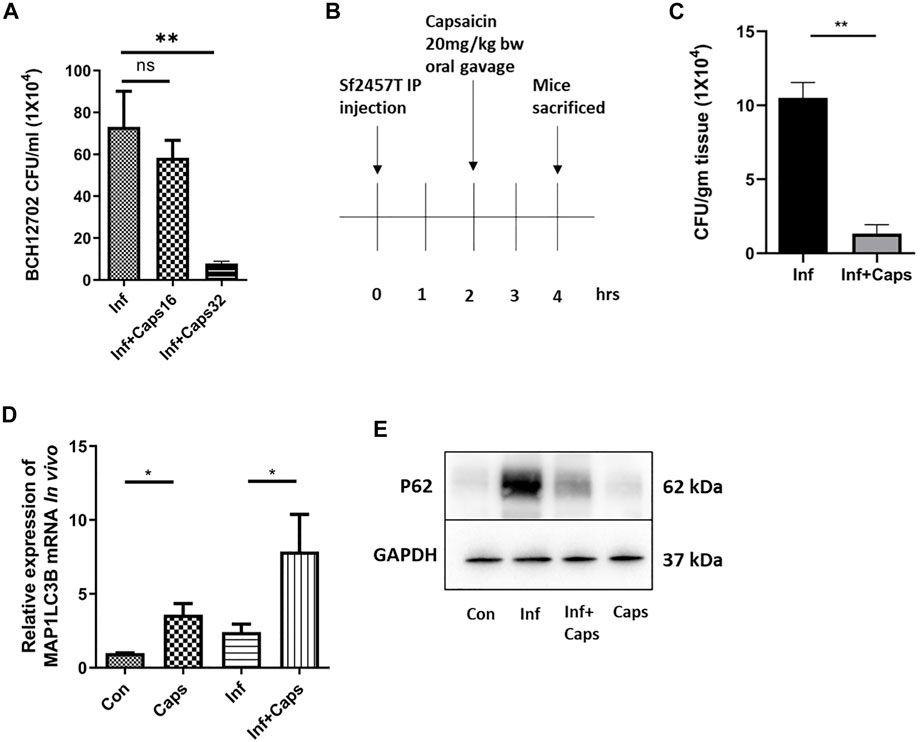
FIGURE 7. Caps reduced bacterial growth in both cellular and in vivo conditions. (A) Cells were infected with resistant S. flexneri 2a (BCH12702) strain followed by 16 μM, 32 µM Caps post-treatment for 12 h and CFU/ml was graphically represented. One-way ANOVA was performed. (B) Caps treatment in Shigella flexneri 2457T-infected BALB/c mice. Briefly, mice were infected for 2 h followed by Caps (20 mg/kg b. w.) treatment for 2 h. Four groups (N = 4) were created accordingly: Group 1. Control: only DMSO was used as the vehicle. Group 2. Infected: Received S. flexneri suspension (0.5 × 109 CFU/ml) in PBS. Group 3. Infected + Caps: Received S. flexneri suspension followed by Caps (20 mg kg−1 body weight) treatment for 2 h. Group 4. Caps: Received Caps (20 mg kg−1 body weight). Each group was kept in an individual cage. Experiments were repeated in triplicate (n = 3). Total number of mice in each group were 12. (C) CFU was counted and plotted for S. flexneri sf2457T infected and infected + Caps-treated crushed colon samples. Unpaired t-test was performed. (D) qRT-PCR was performed for the expression of the autophagy-associated marker gene MAP1LC3B in control, infected, infected with caps treatment, and only Caps-treated mice tissues using GAPDH as a housekeeping gene control. One-way ANOVA was performed. (E) Expression of major autophagy-associated marker protein P62 was observed in western blot. GAPDH was used as a loading control. Graphs were represented using GraphPad Prism 5 as mean ± SEM (n = 3); significance was calculated: *p < 0.05, **p < 0.01, ***p < 0.001.
There is a constant interplay between the host and pathogen to maintain cellular homeostasis. Both play diverse strategies to overcome each other. One of the powerful strategies to boost host defense is autophagy. However, intracellular pathogens evade autophagy to establish successful infection. In contrast, boosting autophagy results in the clearance of intracellular bacteria and thereby inhibits infection. Small-molecule enhancers of autophagy are reported to control bacterial infections like Mycobacterium and Salmonella (Kim et al., 2012; Noad et al., 2017; Ammanathan et al., 2020). Recent studies have also focused on major autophagy players, such as mTOR, akt, and TFEB, a transcription factor controlling autophagy to reduce bacterial burden (Floto et al., 2007; Lin Y.-T et al., 2017). Hence, in this era of antimicrobial resistance, there is an urgent need to identify new antimicrobial compounds to overcome the problem of resistance mechanisms by targeting host factors like autophagy.
This study is the first evidence to show that capsaicin (Caps) induces autophagy for the intracellular killing of S. flexneri. Caps, a known anticancer herbal compound, is able to stimulate autophagy in intestinal cells and macrophages. Autophagy, in turn, inhibited the intracellular infiltration of S. flexneri. This is a novel approach in inhibiting S. flexneri infection. Previous reports indicate that Caps induces autophagy in tumor cells by overexpressing the autophagy proteins beclin1, Atg5, and LC3B (Jin et al., 2014). Consistent with these studies in cancer cells, we showed that Caps also augmented autophagy at a lower dose in intestinal epithelial cells at the protein level. This dose is nontoxic to cells, as reported earlier (Campbell-Valois et al., 2015). Further, we confirmed insignificant toxicity in HT29 cells and macrophages. Our findings showed for the first time that Caps treatment resulted in the overexpression of autophagy genes in intestinal cells. Unlike our knowledge of the overexpression of autophagy proteins by Caps, the transcriptional upregulation of autophagy by Caps has not been reported earlier. Furthermore, Caps induced autophagosome formation in intestinal and macrophage cells. Transcriptional activation of autophagy resulted in autophagosome formation.
In addition to autophagic stimulation, Caps attributed to the killing of S. flexneri in intestinal epithelial cells and macrophages. Along with bactericidal effects, our microscopic observations indicated autophagosome formation in Caps-treated cells. Electron and confocal microscopic studies revealed that bacteria are associated with autophagosomes in drug-treated cells. Consistently, we observed that Caps robustly increased LC3B puncta formation and targeted bacteria to autophagosome. In plasmid-cured avirulent strain of S. flexneri (lacking T3SS), Caps is unable to target bacteria, as most of the bacteria are outside the cells. Published reports have shown that the IcsB, VirA, and other proteins released by T3SS are required for the intracellular infiltration of S. flexneri (Ogawa et al., 2005; Vakifahmetoglu-Norberg et al., 2015). Hence, Caps targeted only intracellular bacteria. However, we checked whether Caps can directly kill the bacteria, but there is no direct effect of Caps on S. flexneri. Thus, it is confirmed that the anti-shigella effect of Caps is autophagy-dependent.
Previous reports of S. flexneri showed that Atg5, an essential autophagy protein, is needed for LC3 recruitment to Shigella flexneri. Here, we observed that the intracellular growth of S. flexneri remains unaffected due to the knockdown of Atg5 in intestinal cells. Caps treatment in Atg5 knockdown cells resulted in the loss of pathogen restriction, suggesting that autophagy plays an essential role in the inhibition of bacterial growth.
Having established that autophagy is the major player in Caps-mediated S. flexneri clearance, we explored the mechanistic details of autophagy induction by Caps. We observed that autophagic gene transcription is enhanced due to nuclear localization of TFEB (Sardiello, 2016). Drug treatment enhanced the nuclear translocation of TFEB in both infected and uninfected cells.
It has been reported that TFEB is a transcriptional regulator of autophagosomal biogenesis (Fan et al., 2018). It induces the transcription of several autophagosomal and lysosomal genes. Moreover, TFEB enhancers are currently reported for the development of therapeutics against intracellular pathogens (Ammanathan et al., 2020). Hence, it is a targeted transcription factor. Here, we observed for the first time that Caps induces TFEB binding to promoter elements to augment autophagic gene transcription.
Previous studies have shown that the depletion of TFEB induces the multiplication of pathogens by downregulating autophagy during infection (Gray et al., 2016). Similarly, in this study, TFEB knockdown in intestinal cells retained S. flexneri multiplication, and Caps treatment was unable to restrict bacterial growth, suggesting the importance of TFEB in controlling S. flexneri infection. In contrast, the overexpression of TFEB reduced bacterial growth. Therefore, TFEB is involved in Caps-mediated transcription of autophagy genes, resulting in the enhanced intracellular killing of S. flexneri. Moreover, capsaicin is known to selectively bind to the TRPV1 receptor and induce calcium influx (Liang et al., 2018; Allais et al., 2020). TRPV1 is expressed in intestinal cells and is known to induce autophagy (Chen et al., 2021; Wang et al., 2021). Hence, Caps-induced autophagy might be due to TRPV1 activation.
In the current study, we proved that the antimicrobial effect of Caps is mediated through autophagy. Moreover, we observed that Caps inhibited S. flexneri resistant strain growth at a higher dose. Thus, it is evident that probably Caps can address the problem of antimicrobial resistance in near future. Recent studies of Salmonella showed the effect of autophagy inducing compounds in mice (Ammanathan et al., 2020). Hence, we explored Caps’s effectivity in an in vivo mouse model. We observed that Caps increased the clearance of S. flexneri in intestinal tissues of infected mice. Here, we also showed that Caps treatment in in vivo mouse model induced autophagy. Thus, Caps is effective in preclinical models to boost autophagy and simultaneously decrease S. flexneri burden.
In conclusion, the emergence of antibiotic resistance requires a constant effort for the development of new therapies, which can boost host mechanisms to enhance bactericidal activity. Our data represent an important novel finding that drugs such as Caps manipulate host cellular defense mechanisms such as autophagy to achieve bactericidal effects against S. flexneri. The mechanistic details of Caps-activated autophagy uncovered a novel role of TFEB to be a potential target in treating S. flexneri infection. This is the first evidence of TFEB activation by Caps. Caps induced TFEB nuclear translocation by dephosphorylation of TFEB. TFEB in nucleus, in turn, binds to the promoter of autophagy genes to boost autophagy. Autophagy induction helps reduce S. flexneri growth and thereby inhibits shigellosis (Figure 8) Hence, a new alternative approach has been unraveled for the treatment of S. flexneri infection.
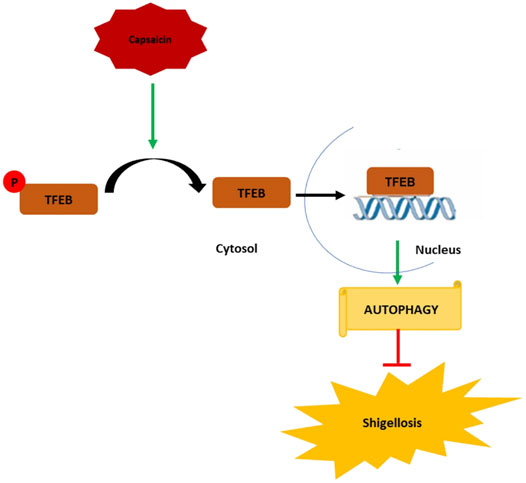
FIGURE 8. Graphical representation of the mechanism of action of capsaicin (Caps). Caps inhibits TFEB phosphorylation. Dephosphorylated TFEB moves from cytosol to nucleus and binds to the promoter site of autophagy genes resulting in the upregulation of autophagy-associated marker genes. Induction of autophagy inhibits shigellosis by inhibiting S. flexneri growth in both in vitro and in vivo models.
This study will open new avenues in treating shigellosis in the near future. More inducers of TFEB nuclear translocation, such as Caps, may be used to strengthen the host response against pathogens. However, Caps may not act as the only drug for the treatment of shigellosis, as most autophagy inducers are used in combination with other antibiotics for the treatment of bacterial pathogenesis. Host-directed therapy could be a new approach in adjunct to the use of antibiotics for infectious diseases. Moreover, Caps, as an autophagy inducer, might be effective against other intracellular pathogens.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by IAEC (Institutional Animal Ethical Committee), NICED, Kolkata (PRO/157/-July 2022).
PB: investigation and writing—review and editing. PM: investigation and data curation. UK: investigation and data curation. KS: methodology and validation. SB: writing—review and editing and data curation. MD: validation. SB: supervision and writing—review and editing, writing—original draft, funding acquisition, and investigation.
This work was funded by ICMR Extramural Project 59/01/2019/Online/BMS/TRM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Dr. Shanta Dutta, Director, ICMR-National Institute of Cholera and Enteric Diseases (NICED), for her support in providing funding and infrastructure, Dr. Santasabuj Das, ICMR-NICED, for helping with confocal microscopy, real-time PCR, providing antibodies, EGFP plasmid, and RAW Cell line, Dr. Nabendu Sekhar Chatterjee ICMR-NICED for chemicals and reagents, Dr. Asish K Mukhopadhyay ICMR-NICED for bacterial strains, Dr. Mamta Chawla Sarkar ICMR-NICED for antibodies, Dr. Avinash Bajaj, Regional Centre for Biotechnology, Faridabad, for the kind gift of HT29 cell line, Prof. Parimal Karmakar, Jadavpur University, for eGFP-LC3 plasmid, and Dr. Ravi Manjithaya, JNCASR, Bangalore, for pEGFP-N1-TFEB plasmid. The authors also thank the Council for Scientific and Industrial Research (CSIR) and Department of Biotechnology (DBT) for providing fellowship to the authors.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.903438/full#supplementary-material.
Allais, L., Verschuere, S., Maes, T., De Smet, R., Devriese, S., Gonzales, G. B., et al. (2020). Translational Research into the Effects of Cigarette Smoke on Inflammatory Mediators and Epithelial TRPV1 in Crohn's Disease. PloS one 15 (8), e0236657. doi:10.1371/journal.pone.0236657
Ammanathan, V., Mishra, P., Chavalmane, A. K., Muthusamy, S., Jadhav, V., Siddamadappa, C., et al. (2020). Restriction of Intracellular Salmonella Replication by Restoring TFEB-Mediated Xenophagy. Autophagy 16 (9), 1584–1597. doi:10.1080/15548627.2019.1689770
Baker, S., and The, H. C. (2018). Recent Insights into Shigella: A Major Contributor to the Global Diarrhoeal Disease Burden. Curr. Opin. Infect. Dis. 31 (5), 449–454. doi:10.1097/QCO.0000000000000475
Brier, L. W., Ge, L., Stjepanovic, G., Thelen, A. M., Hurley, J. H., and Schekman, R. (2019). Regulation of LC3 Lipidation by the Autophagy-specific Class III Phosphatidylinositol-3 Kinase Complex. Mol. Biol. Cell 30 (9), 1098–1107. doi:10.1091/mbc.E18-11-0743
Campbell-Valois, F. X., Sachse, M., Sansonetti, P. J., and Parsot, C. (2015). Escape of Actively Secreting Shigella Flexneri from ATG8/LC3-Positive Vacuoles Formed during Cell-To-Cell Spread Is Facilitated by IcsB and VirA. MBio 6 (3), e02567–14. doi:10.1128/mBio.02567-14
Chang, Z., Zhang, J., Ran, L., Sun, J., Liu, F., Luo, L., et al. (2016). The Changing Epidemiology of Bacillary Dysentery and Characteristics of Antimicrobial Resistance of Shigella Isolated in China from 2004-2014. BMC Infect. Dis. 16 (1), 685. doi:10.1186/s12879-016-1977-1
Chanin, R. B., Nickerson, K. P., Llanos-Chea, A., Sistrunk, J. R., Rasko, D. A., Kumar, D. K. V., et al. (2019). Shigella Flexneri Adherence Factor Expression in In Vivo-like Conditions. Msphere 4 (6), e00751–19. doi:10.1128/mSphere.00751-19
Chen, Q., Rui, J., Hu, Q., Peng, Y., Zhang, H., Zhao, Z., et al. (2020). Epidemiological Characteristics and Transmissibility of Shigellosis in Hubei Province, China, 2005 - 2017. BMC Infect. Dis. 20 (1), 272. doi:10.1186/s12879-020-04976-x
Chen, M., Dong, X., Deng, H., Ye, F., Zhao, Y., Cheng, J., et al. (2021). Targeting TRPV1-Mediated Autophagy Attenuates Nitrogen Mustard-Induced Dermal Toxicity. Signal Transduct. Target. Ther. 6 (1), 1–13. doi:10.1038/s41392-020-00389-z
Fan, Y., Lu, H., Liang, W., Garcia-Barrio, M. T., Guo, Y., Zhang, J., et al. (2018). Endothelial TFEB (Transcription Factor EB) Positively Regulates Postischemic Angiogenesis. Circ. Res. 122 (7), 945–957. doi:10.1161/CIRCRESAHA.118.312672
Floto, R. A., Sarkar, S., Perlstein, E. O., Kampmann, B., Schreiber, S. L., and Rubinsztein, D. C. (2007). Small Molecule Enhancers of Rapamycin-Induced TOR Inhibition Promote Autophagy, Reduce Toxicity in Huntington's Disease Models and Enhance Killing of Mycobacteria by Macrophages. Autophagy 3 (6), 620–622. doi:10.4161/auto.4898
Füchtbauer, S., Mousavi, S., Bereswill, S., and Heimesaat, M. M. (2021). Antibacterial Properties of Capsaicin and its Derivatives and Their Potential to Fight Antibiotic Resistance - A Literature Survey. Eur. J. Microbiol. Immunol. (Bp) 11 (1), 10–17. doi:10.1556/1886.2021.00003
Giraud-Gatineau, A., Coya, J. M., Maure, A., Biton, A., Thomson, M., Bernard, E. M., et al. (2020). The Antibiotic Bedaquiline Activates Host Macrophage Innate Immune Resistance to Bacterial Infection. Elife 9, e55692. doi:10.7554/eLife.55692
Gray, M. A., Choy, C. H., Dayam, R. M., Ospina-Escobar, E., Somerville, A., Xiao, X., et al. (2016). Phagocytosis Enhances Lysosomal and Bactericidal Properties by Activating the Transcription Factor TFEB. Curr. Biol. 26 (15), 1955–1964. doi:10.1016/j.cub.2016.05.070
Hajialibeigi, A., Amani, J., and Gargari, S. L. M. (2021). Identification and Evaluation of Novel Vaccine Candidates against Shigella Flexneri through Reverse Vaccinology Approach. Appl. Microbiol. Biotechnol. 105 (3), 1159–1173. doi:10.1007/s00253-020-11054-4
Hosangadi, D., Smith, P. G., and Giersing, B. K. (2019). Considerations for Using ETEC and Shigella Disease Burden Estimates to Guide Vaccine Development Strategy. Vaccine 37 (50), 7372–7380. doi:10.1016/j.vaccine.2017.09.083
Hu, W., Zhang, L., Li, M. X., Shen, J., Liu, X. D., Xiao, Z. G., et al. (2019). Vitamin D3 Activates the Autolysosomal Degradation Function against Helicobacter pylori through the PDIA3 Receptor in Gastric Epithelial Cells. Autophagy 15 (4), 707–725. doi:10.1080/15548627.2018.1557835
Jin, J., Lin, G., Huang, H., Xu, D., Yu, H., Ma, X., et al. (2014). Capsaicin Mediates Cell Cycle Arrest and Apoptosis in Human Colon Cancer Cells via Stabilizing and Activating P53. Int. J. Biol. Sci. 10 (3), 285–295. doi:10.7150/ijbs.7730
Jo, E. K., Yuk, J. M., Shin, D. M., and Sasakawa, C. (2013). Roles of Autophagy in Elimination of Intracellular Bacterial Pathogens. Front. Immunol. 4, 97. doi:10.3389/fimmu.2013.00097
Kaufmann, S. H. E., Dorhoi, A., Hotchkiss, R. S., and Bartenschlager, R. (2018). Host-directed Therapies for Bacterial and Viral Infections. Nat. Rev. Drug Discov. 17 (1), 35–56. doi:10.1038/nrd.2017.162
Khalil, I. A., Troeger, C., Blacker, B. F., Rao, P. C., Brown, A., Atherly, D. E., et al. (2018). Morbidity and Mortality Due to shigella and Enterotoxigenic Escherichia coli Diarrhoea: The Global Burden of Disease Study 1990-2016. Lancet Infect. Dis. 18 (11), 1229–1240. doi:10.1016/S1473-3099(18)30475-4
Killackey, S. A., Sorbara, M. T., and Girardin, S. E. (2016). Cellular Aspects of Shigella Pathogenesis: Focus on the Manipulation of Host Cell Processes. Front. Cell Infect. Microbiol. 6, 38. doi:10.3389/fcimb.2016.00038
Kim, J. J., Lee, H. M., Shin, D. M., Kim, W., Yuk, J. M., Jin, H. S., et al. (2012). Host Cell Autophagy Activated by Antibiotics Is Required for Their Effective Antimycobacterial Drug Action. Cell Host Microbe 11 (5), 457–468. doi:10.1016/j.chom.2012.03.008
Kim, Y. S., Silwal, P., Kim, S. Y., Yoshimori, T., and Jo, E. K. (2019). Autophagy-activating Strategies to Promote Innate Defense against Mycobacteria. Exp. Mol. Med. 51 (12), 1–10. doi:10.1038/s12276-019-0290-7
Krokowski, S., Lobato-Márquez, D., Chastanet, A., Pereira, P. M., Angelis, D., Galea, D., et al. (2018). Septins Recognize and Entrap Dividing Bacterial Cells for Delivery to Lysosomes. Cell Host Microbe 24 (6), 866–e4. doi:10.1016/j.chom.2018.11.005
Liang, Q., Lv, X., Cai, Q., Cai, Y., Zhao, B., and Li, G. (2018). Novobiocin, a Newly Found TRPV1 Inhibitor, Attenuates the Expression of TRPV1 in Rat Intestine and Intestinal Epithelial Cell Line IEC-6. Front. Pharmacol. 9, 1171. doi:10.3389/fphar.2018.01171
Lin, S. R., Fu, Y. S., Tsai, M. J., Cheng, H., and Weng, C. F. (2017). Natural Compounds from Herbs that Can Potentially Execute as Autophagy Inducers for Cancer Therapy. Int. J. Mol. Sci. 18 (7), 1412. doi:10.3390/ijms18071412
Lin, Y.-T., Wang, H.-C., Hsu, Y.-C., Cho, C.-L., Yang, M.-Y., and Chien, C.-Y. (2017). Capsaicin Induces Autophagy and Apoptosis in Human Nasopharyngeal Carcinoma Cells by Downregulating the PI3K/AKT/mTOR Pathway. Ijms 18 (7), 1343. doi:10.3390/ijms18071343
Lobato‐Márquez, D., Krokowski, S., Sirianni, A., Larrouy‐Maumus, G., and Mostowy, S. (2019). A Requirement for Septins and the Autophagy Receptor P62 in the Proliferation of Intracellular Shigella. Cytoskeleton 76 (1), 163–172. doi:10.1002/cm.21453
McQuade, E. T. R., Shaheen, F., Kabir, F., Rizvi, A., Platts-Mills, J. A., Aziz, F., et al. (2020). Epidemiology of Shigella Infections and Diarrhea in the First Two Years of Life Using Culture-independent Diagnostics in 8 Low-Resource Settings. PLoS Negl. Trop. Dis. 14 (8), e0008536. doi:10.1371/journal.pntd.0008536
Nandy, S., Dutta, S., Ghosh, S., Ganai, A., Rajahamsan, J., Theodore, R. B., et al. (2011). Foodborne-associated Shigella Sonnei, India, 2009 and 2010. Emerg. Infect. Dis. 17 (11), 2072–2074. doi:10.3201/eid1711.110403
Noad, J., Von Der Malsburg, A., Pathe, C., Michel, M. A., Komander, D., and Randow, F. (2017). LUBAC-synthesized Linear Ubiquitin Chains Restrict Cytosol-Invading Bacteria by Activating Autophagy and NF-Κb. Nat. Microbiol. 2 (7), 17063. doi:10.1038/nmicrobiol.2017.63
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of Intracellular Shigella from Autophagy. Science 307 (5710), 727–731. doi:10.1126/science.1106036
Polito, V. A., Li, H., Martini-Stoica, H., Wang, B., Yang, L., Xu, Y., et al. (2014). Selective Clearance of Aberrant Tau Proteins and Rescue of Neurotoxicity by Transcription Factor EB. EMBO Mol. Med. 6 (9), 1142–1160. doi:10.15252/emmm.201303671
Puertollano, R., Ferguson, S. M., Brugarolas, J., and Ballabio, A. (2018). The Complex Relationship between TFEB Transcription Factor Phosphorylation and Subcellular Localization. EMBO J. 37 (11), e98804. doi:10.15252/embj.201798804
Puzari, M., Sharma, M., and Chetia, P. (2018). Emergence of Antibiotic Resistant Shigella Species: A Matter of Concern. J. Infect. Public Health 11 (4), 451–454. doi:10.1016/j.jiph.2017.09.025
Ra, E. A., Lee, T. A., Won Kim, S., Park, A., Choi, H. J., Jang, I., et al. (2016). TRIM31 Promotes Atg5/Atg7-independent Autophagy in Intestinal Cells. Nat. Commun. 7 (1), 11726. doi:10.1038/ncomms11726
Ranjbar, R., and Farahani, A. (2019). Shigella: Antibiotic-Resistance Mechanisms and New Horizons for Treatment. Infect. Drug Resist 12, 3137–3167. doi:10.2147/IDR.S219755
Rollyson, W. D., Stover, C. A., Brown, K. C., Perry, H. E., Stevenson, C. D., McNees, C. A., et al. (2014). Bioavailability of Capsaicin and its Implications for Drug Delivery. J. Control Release 196, 96–105. doi:10.1016/j.jconrel.2014.09.027
Rubinsztein, D. C., Codogno, P., and Levine, B. (2012). Autophagy Modulation as a Potential Therapeutic Target for Diverse Diseases. Nat. Rev. Drug Discov. 11 (9), 709–730. doi:10.1038/nrd3802
Sardiello, M. (2016). Transcription Factor EB: From Master Coordinator of Lysosomal Pathways to Candidate Therapeutic Target in Degenerative Storage Diseases. Ann. N. Y. Acad. Sci. 1371 (1), 3–14. doi:10.1111/nyas.13131
Sharma, V., Verma, S., Seranova, E., Sarkar, S., and Kumar, D. (2018). Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 6, 147. doi:10.3389/fcell.2018.00147
Taneja, N., and Mewara, A. (2016). Shigellosis: Epidemiology in India. Indian J. Med. Res. 143 (5), 565–576. doi:10.4103/0971-5916.187104
Vakifahmetoglu-Norberg, H., Xia, H. G., and Yuan, J. (2015). Pharmacologic Agents Targeting Autophagy. J. Clin. Invest. 125 (1), 5–13. doi:10.1172/JCI73937
Vega-Rubin-de-Celis, S., Peña-Llopis, S., Konda, M., and Brugarolas, J. (2017). Multistep Regulation of TFEB by MTORC1. Autophagy 13 (3), 464–472. doi:10.1080/15548627.2016.1271514
Wang, C., Huang, W., Lu, J., Chen, H., and Yu, Z. (2021). TRPV1-Mediated Microglial Autophagy Attenuates Alzheimer’s Disease-Associated Pathology and Cognitive Decline. Front. Pharmacol. 12, 763866. doi:10.3389/fphar.2021.763866
Weddle, E., and Agaisse, H. (2018). Spatial, Temporal, and Functional Assessment of LC3-dependent Autophagy in Shigella Flexneri Dissemination. Infect. Immun. 86 (8), e00134–18. doi:10.1128/IAI.00134-18
Williams, P. C. M., and Berkley, J. A. (2018). Guidelines for the Treatment of Dysentery (Shigellosis): A Systematic Review of the Evidence. Paediatr. Int. Child. Health 38 (Suppl. 1), S50–S65. doi:10.1080/20469047.2017.1409454
Yang, J. Y., Lee, S. N., Chang, S. Y., Ko, H. J., Ryu, S., and Kweon, M. N. (2014). A Mouse Model of Shigellosis by Intraperitoneal Infection. J. Infect. Dis. 209 (2), 203–215. doi:10.1093/infdis/jit399
Young, C., Walzl, G., and Du Plessis, N. (2020). Therapeutic Host-Directed Strategies to Improve Outcome in Tuberculosis. Mucosal Immunol. 13 (2), 190–204. doi:10.1038/s41385-019-0226-5
Keywords: Shigella flexneri, autophagy, TFEB, capsaicin, gene transcription
Citation: Basak P, Maitra P, Khan U, Saha K, Bhattacharya SS, Dutta M and Bhattacharya S (2022) Capsaicin Inhibits Shigella flexneri Intracellular Growth by Inducing Autophagy. Front. Pharmacol. 13:903438. doi: 10.3389/fphar.2022.903438
Received: 24 March 2022; Accepted: 16 May 2022;
Published: 06 July 2022.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Sue Twine, National Research Council Canada (NRC-CNRC), CanadaCopyright © 2022 Basak, Maitra, Khan, Saha, Bhattacharya, Dutta and Bhattacharya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sushmita Bhattacharya, ZHVyZ2FzdXNobWl0YUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.