 Li Chen1†
Li Chen1† Shizhong Zheng
Shizhong Zheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 17 May 2022
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.891069
This article is part of the Research TopicMolecular Mechanisms and Drug Discovery for Liver FibrosisView all 5 articles
Liver fibrosis is a repair process of chronic liver injuries induced by toxic substances, pathogens, and inflammation, which exhibits a feature such as deposition of the extracellular matrix. The initiation and progression of liver fibrosis heavily relies on excessive activation of hepatic stellate cells (HSCs). The activated HSCs express different kinds of chemokine receptors to further promote matrix remodulation. The long-term progression of liver fibrosis will contribute to dysfunction of the liver and ultimately cause hepatocellular carcinoma. The liver also has abundant innate immune cells, including DCs, NK cells, NKT cells, neutrophils, and Kupffer cells, which conduct complicated functions to activation and expansion of HSCs and liver fibrosis. Autophagy is one specific type of cell death, by which the aberrantly expressed protein and damaged organelles are transferred to lysosomes for further degradation, playing a crucial role in cellular homeostasis. Autophagy is also important to innate immune cells in various aspects. The previous studies have shown that dysfunction of autophagy in hepatic immune cells can result in the initiation and progression of inflammation in the liver, directly or indirectly causing activation of HSCs, which ultimately accelerate liver fibrosis. Given the crosstalk between innate immune cells, autophagy, and fibrosis progression is complicated, and the therapeutic options for liver fibrosis are quite limited, the exploration is essential. Herein, we review the previous studies about the influence of autophagy and innate immunity on liver fibrosis and the molecular mechanism to provide novel insight into the prevention and treatment of liver fibrosis.
Liver fibrosis is a chronic disease caused by liver injuries that are stimulated by several factors, such as excessive alcohol consumption, virus infection (including hepatitis B and hepatitis C), non-alcohol steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), and autoimmune hepatitis (Affo et al., 2017; Kakino et al., 2018; Parola and Pinzani 2019). As a result of multiple kinds of liver damage and diseases, liver fibrosis has a large prevalence and high mortality in China and also worldwide (Moon et al., 2020). Liver fibrosis will gradually result in hepatocellular carcinoma without timely treatment (Affo et al., 2017). The features of liver fibrosis are excessive accumulation of extracellular matrix (ECM), which consists of glycoproteins, collagen, and proteoglycans (Karsdal et al., 2017). Hepatic stellate cells (HSCs) are recognized as the main cause for initiation and progression of liver fibrosis (Li X. et al., 2020). The hepatic damage causes the activation of HSCs, which are changed from the quiescent status to myofibroblasts, and leads to production and accumulation of the ECM, ultimately causing liver fibrosis (Zhang et al., 2016; Chen et al., 2019; Dewidar et al., 2019). The progression of liver fibrosis will lead to mild-to-moderate liver fibrosis, and nearly 30% of the patients will develop cirrhosis, in which more than 80% of patients will finally progress to hepatocellular carcinoma (Pang et al., 2018). Notably, liver fibrosis is a reversible pathological condition, lacking effective treatment in addition to surgical resection or transplantation (Baglieri et al., 2019). However, if the disease developed to terminal stage, the surgical operation will be invalid. As a result, it is significant to prevent the initiation and progression of liver fibrosis.
There are different factors involved in the activation of HSCs, including toxins, hepatitis, autoimmune disorders, and steatohepatitis. HSC activation contains two major stages, which are named as initiation and perpetuation, to stimulate the HSCs to acquire a myofibroblast-like phenotype (Higashi et al., 2017). Early activation of HSCs is driven by paracrine and autocrine growth factors, mainly including platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), and transforming growth factor (TGF-β) (Borkham-Kamphorst and Weiskirchen 2016; Xu et al., 2016; Feng et al., 2019; Schumacher et al., 2020). The release of these growth factors is regulated at the transcriptional level. Moreover, the continuous stimuli for HSC activation will result in alteration of cell behaviors, such as proliferation, fibrogenesis, matrix degradation, and cytokine release, finally causing continuous activation (Zhou et al., 2019). Overall, these cellular changes will contribute to ECM accumulation. The ECM mainly comprises α smooth muscle actin (α-SMA) and collagen, and the excessive accumulation of the ECM will cause damage to normal liver structure and function (Schnieder et al., 2020).
The role of autophagy in the process of liver fibrosis is complex and bidirectional, which has received particular attention in recent years. From the current research, the upregulation of autophagy can aggravate liver fibrosis mainly by promoting HSC activation in HSC autophagy, especially lipophagy can provide energy for its activation (Hou et al., 2022). The inhibitory effect of upregulated autophagy on liver fibrosis is mainly reflected in the protective effect of hepatocyte and LSEC autophagy and the anti-inflammatory effect of immune cell autophagy (Hammoutene et al., 2020). In addition, various signaling pathway studies suggest that the upregulation of HSC autophagy aggravates liver fibrosis (Liu et al., 2020). Autophagy of different kinds of cells and different levels of autophagy of different substrates may have biased or different mechanisms on liver fibrosis (Li Y. et al., 2020). In addition, its complexity may be related to the type of disease and period of fibrosis.
In the traditional conception, the liver is a metabolic organ conducting the function of substance circulation and energy regeneration. However, the unique anatomical characteristics of the liver, including abundant blood supply and rich microarchitecture of vessels, have empowered the liver with a profound feature of immune function (Jenne and Kubes 2013). The liver is abundant in multiple immune cells, which contribute to immune regulation of hepatic function. Compared with adaptive immunity, the innate immune response is activated much faster, especially in acute liver injury, where the host has little time to trigger an effective adaptive immune response. From this perspective, the innate immune system may provide a more profound contribution than the adaptive immune system (Wu et al., 2010). A coordinated network of innate immune cells, inducing Kupffer cells (KCs), natural killer cells (NKs), innate lymphoid cells (ILCs), mucosal-associated invariant T cells (MAITs), dendritic cells (DCs), and invariant NKT cells (iNKTs), are involved in the inflammation of the liver to encounter the invading microorganisms, toxic damage, and pathological challenges (Hossain and Kubes 2019). Typically, KCs are responsible for first detecting the occurrence of exogenous stimulation, releasing the proinflammatory cytokines (such as IL-6, TNF-α, and IL-6), and chemokines (e.g., CCL-2, CCL-4, and CXCL-1) (Abdullah and Knolle 2017). The soluble factors contribute to the recruitment of monocytes and neutrophils to the liver. Neutrophils play an important role in innate immune response by capturing and destroying the pathogens, performing its function by producing reactive oxygen species (ROS), stretching neutrophil extracellular traps (NETs), and conducting the function of phagocytosis (Yang et al., 2019). DCs conduct the functions by bridging the gap between innate immune reaction and adaptive immune response. DCs maintain quiescent conditions under a healthy environment, but they move to the lymph node after stimulation and present antigens to T cells to further activate adaptive immune reaction (Soysa et al., 2017). In addition, the abovementioned cell types and the innate-like lymphocytes, such as MAITs, ILCs, NKs, and iNKTs, are also significant in shaping the hepatic immune microenvironment by secreting different kinds of cytokines (Chen and Tian 2020; Niehaus et al., 2020; Gan et al., 2021). The cytokines and chemokines further facilitate the expression of adhesive molecules such as VCAM1 and ICAM1 to stimulate the secretion of hepatic sinusoidal endothelial cells and ECM formation (Roh and Seki 2018).
When the liver encounters sustained damage, the inflammatory cells will generate a huge amount of chemokines and cytokines to exert its defensive reaction (Figure 1). Meanwhile, the HSCs also express different kinds of chemokine receptors. The chemokine receptors dysregulated in HSCs contain CXC chemokine receptor 3 (CXCR3); C–C chemokine receptor (CCR) family, including CCR5 and CCR7(Seki et al., 2009). The chemokine family chain in HSCs contains the chemokine ligand (CCL) family, which includes CCL2, CCL3, and CCL5; in addition, the CXC chemokine ligand (CXCL) family contains CXCL1 and CXCL8–CXCL10 (Weiskirchen and Tacke 2014; Yan et al., 2020). Generally, the chemokines bind to their receptors to promote HSC migration and ECM generation. However, the function of chemokines and their receptors in liver fibrosis is a double-edged sword. For instance, the overexpression of CCR1, CCR5, and CXCL4 can contribute to progression of liver fibrosis, whereas the stimulation of CXCR3 and CXCL9 exert the protective function of liver fibrosis (Bruns et al., 2014; Arsent’eva et al., 2015; Ambade et al., 2019).
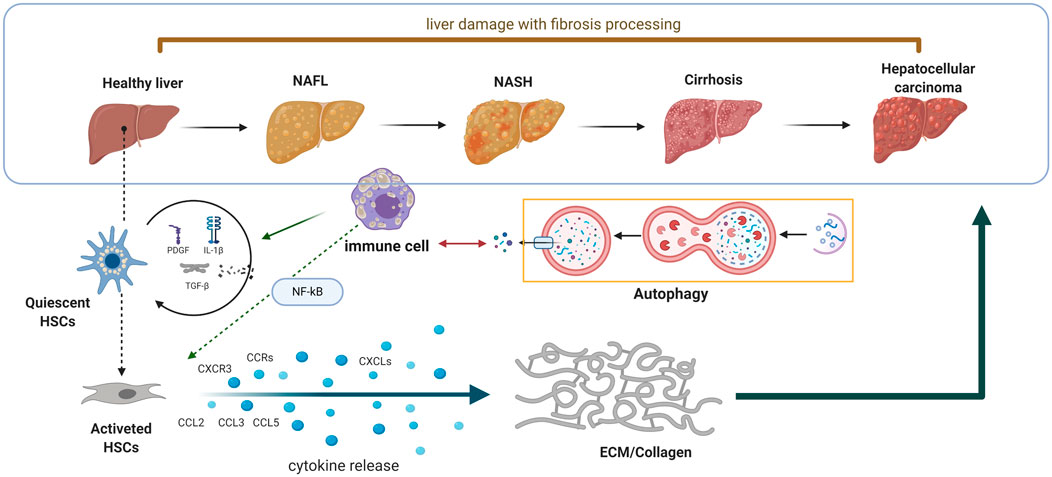
FIGURE 1. Schematic diagram of the interaction between autophagy and innate immunity in the process of liver fibrosis. When the liver encounters sustained damage, the immune cells will generate huge amount of chemokines and cytokines to exert its defensive reaction. Meanwhile, the HSCs also express different kinds of chemokine receptors to further promote matrix remodulation.
In addition, to the chemokines and their receptors in the initiation and progression of liver fibrosis, the secreted cytokines also enhance the inflammatory effect. The immune cells and parenchymal cells secrete interleukin-1β, transforming growth factor (TGF-β), tumor necrosis factor-α (TNF-α), and other kinds of cytokines to enhance the inflammatory effect of hepatocytes and activated HSCs (Mridha et al., 2017; Fabre et al., 2018; Kakino et al., 2018). The downstream pathways of these cytokines were also partially elucidated in the previous studies. For instance, the TGF-β/Smad signaling is deeply involved in HSC activation. After activation by TGF-β/Smad signaling, the HSCs will, in turn, generate TGF-β in a manner of autocrine function, which consequently enhances the activation of the TGF-β/Smad pathway. In addition, the NF-κB pathway is also associated with liver fibrosis. The stimuli of the NF-κB pathway include the proinflammatory factors including interleukins (IL-1, IL-2, and IL-6) and TNF-α (Luedde and Schwabe 2011). The signaling of the NF-κB pathway in fibrosis also conducts its function in a manner of autocrine function, for the activation of the NF-κB signal will contribute to the translocation of IκB in the nucleus and upregulate the expression of proinflammatory cytokines, finally leading to cascade amplification of the proinflammatory effect and HSC activation (Bai et al., 2018).
Generally, considering the crucial role of hepatic innate immune regulation, the liver is not only a metabolic organ but also an important immune organ that wipes out invading pathogens and endogenous harmful stimuli.
Autophagy is one specific type of cell death, by which the aberrantly expressed protein and damaged organelles are transferred to lysosomes for further degradation. The initiation and process of autophagy involve the formation of multiple biofilm structures. The autophagosomes are formed, fusing with lysosomes to form autolysosomes, which consequently conduct the function of degradation (Li Y. et al., 2020; Lorincz and Juhasz 2020).
The autophagy flux is regulated by a series of evolutionarily conserved proteins. For instance, the translation products of ATG genes are responsible for the formation of autophagosomes, which could be divided into two major stages: nucleation and elongation (Li and Zhang 2019). The proteins of Atg1/ULK kinase and phosphatidylinositol 3 (PI3K) kinase and the downstream PI3P effectors are critical to the nucleation stage, and the Atg8 and Atg12 are significantly involved in the elongation stage (Mizushima 2010). In addition, there still exists a homolog of yeast Atg8, microtubule-associated protein 1 light chain 3 (LC3), localizing to the autophagosome membrane after posttranslational modification (Lorincz and Juhasz 2020). The clipping product of the C-terminal fragment of LC3 is called LC3-I, which could be activated by Atg7 and transferred to Atg3, and it combines with phosphatidylethanolamine in a manner of covalent bonding, ultimately converting to LC3-II, whose number is closely related to the number of autophagosomes. Under the condition of an adverse situation or pathological factors, the LC3-I will rapidly transform to LC3-II to enhance the autophagy flux, and the expression of LC3-I and LC3-II is an effective parameter to monitor autophagic activity (Huang and Liu 2015). Moreover, the complex of p62/SQSTM1 directly binds to LC3 and is incorporated into the autophagosome to facilitate the process of autophagy, which will be degraded by autophagy ultimately (Katsuragi et al., 2015).
Autophagy commonly occurs in nearly all cell types and is crucial for sustaining cell homeostasis, cell survival, differentiation, and growth (Hu et al., 2019; Yan et al., 2019). Due to its wide distribution and function in nucleated cells, the maintenance of regular autophagy is essential to cellular function (Xiang et al., 2020). It is widely recognized that dysregulation of autophagy is closely related to the development of various diseases, such as malignant tumors (Mowers et al., 2018; Jiang et al., 2019), auto-immune disease (Cosin-Roger et al., 2017), metabolic disease (Yan et al., 2017; Madhavi et al., 2019), and neurodegeneration (Plaza-Zabala et al., 2017; Liu et al., 2019), including liver fibrosis (Qu et al., 2017; Meng et al., 2018; Liu et al., 2019). Similar to the function of autophagy in other types of diseases, autophagy also exerts complicated roles in liver fibrosis. The role of autophagy in liver fibrosis depends on the cell type and stage of the disease. For example, in hepatocytes, autophagy has been demonstrated to have a protective role (Allaire et al., 2019; Hammoutene et al., 2020). In Atg5 liver-specific KO mice, the number of fibrosis-related genes, such as Col-1, α-SMA, and TGF-β, was increased in hepatocytes. While in the wild-type mice, saracatinib markedly suppressed the expression of α-SMA in HSCs and decreased the TGF-β–induced CTGF expression by increasing autophagy flux in hepatocytes (Li Y. et al., 2020). In HSCs, the function of autophagy becomes paradoxical. On the one hand, it has been proposed that autophagy in HSCs might induce their activation through lipophagy, a selective type of lipid droplet degradation (Hammoutene et al., 2020). On the other hand, converse findings suggest that increased autophagy in HSCs attenuates liver fibrosis. In a recent study, researchers demonstrated that mammalian target of rapamycin (mTOR), an inhibitor of autophagy, actually promotes HSC activation and liver fibrosis through extracellular vesicle (EV) release, while restoring autophagy in HSCs attenuates liver fibrosis by inhibiting the release of fibrogenic EVs(Gao et al., 2020).
As is known, the innate immune cells of the liver are crucial to the influence of liver fibrosis; the stability of liver immune function is an important factor to maintain normal liver function (Wu et al., 2020; Sepulveda-Crespo et al., 2021). Autophagy balances inflammation in innate immunity, and when liver injury occurs, the autophagy function of liver innate immune cells will also change. A failure in autophagy functions is often manifested as dysregulated inflammation in animal models and human diseases (Deretic and Levine 2018). In liver diseases, the functions of immune cells are more complex, and the interaction between autophagy and immune cells may be the underlying molecular mechanism for the complex regulatory functions of immune cells. The formation and progression of fibrosis is a complicated process, which involves the crosstalk between autophagy and innate immune cells (Figure 2). Therefore, a deep understanding of the cellular function of the molecular mechanism of autophagy in liver fibrosis is essential to its therapy.
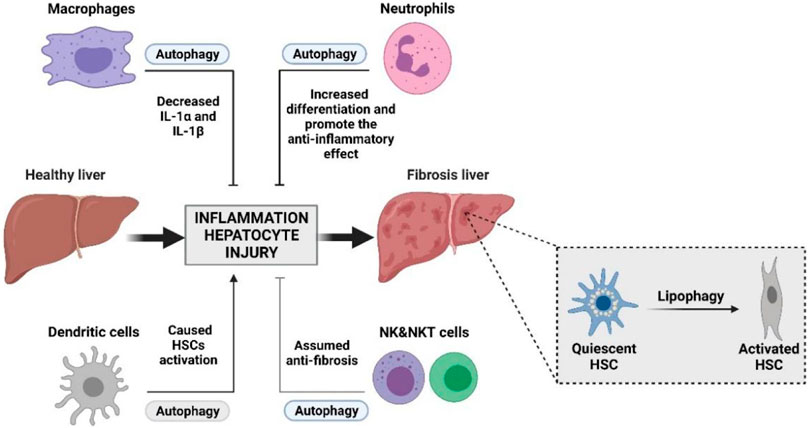
FIGURE 2. Interplay between autophagy and innate immunity during liver damage with fibrosis processing. Innate immunity plays an important role in liver fibrosis. Activation of HSCs and the remodeling of the hepatic microenvironment by accumulation of the ECM is essential to the initiation and progression of liver fibrosis. The interplay between autophagy and innate immunity may become an important part of intervention in the treatment of liver fibrosis.
The formation and progression of fibrosis is a complicated process, which involves the crosstalk between autophagy and innate immune cells and molecules. Many studies have reported the autophagy function of some major immune cells in liver disease. Here, we summarized the relationship between these major immune cells and autophagy in the process of liver fibrosis as below.
Kupffer cells (KCs) are hepatic macrophages that are a heterogenous population of non-parenchymal cells, comprising 90% of all macrophages, and can be divided into resident and infiltrating macrophages. Kupffer cells are essential for sensing tissue damage and initiating inflammatory response, while infiltrating monocytes/macrophages are associated with chronic inflammatory fiber formation (Ji et al., 2020), (Lodder et al., 2015). Recent studies have shown that during the regression of fibrosis, infiltrating monocytes/macrophages differentiate into “repair” macrophages, which have the function of promoting the regression of fibrosis (Zhu et al., 2018), (Zhou et al., 2018). It is now recognized that KCs play an important role in the pathogenesis of hepatic fibrosis and may be a potential therapeutic target for hepatic fibrosis (Cheng et al., 2021). KCs are involved in the initiation and progression in different stages. In the initiation stage, KCs interact with damage-associated molecular patterns (DAMPs) released by injured or dying hepatocytes, thereby promoting the activation, polarization, and recruitment of KCs. Correspondingly, the activation of KCs induces the secretion of proinflammatory and fibrotic cytokines that act as proinflammatory drivers and promote the activation and fibrotic response of HSCs (Pradere et al., 2013). Current studies have confirmed that HSCs can interact with KCs to promote the generation of the immune microenvironment around HSCs and remodeling of the ECM. The interaction between KCs and HSCs is a complex process involving various cytokines and chemokines (e.g., transforming growth factor (TGF)-β, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and CCL2) involved in the activation of HSCs. In liver tissue and KCs of animals with CCl4-induced fibrosis, the expression of TNF-like ligand 1A was significantly increased, which in turn stimulated the secretion of PDGF-BB, TNF-α, and IL-1β, thereby driving the activation and proliferation of HSCs (Matsuda et al., 2018). In addition, in the period of progression, KCs also release matrix metalloproteinases (MMPs) to remodel the extracellular matrix which further propels liver fibrosis (Cai et al., 2018; Ge et al., 2020). However, some studies have shown that stimulating KCs can also play an antifibrotic role under certain pathological conditions. For example, one study showed that in CCl4-induced liver fibrosis, IL-22 polarized M2 KCs through the STAT3 pathway, exerting an anti-inflammatory effect (Su et al., 2021). Another study showed that inhibition of dipeptidyl peptidase 4 (DPP4) skews KCs toward the anti-inflammatory M2 phenotype, thereby alleviating insulin resistance, steatohepatitis, and fibrosis in cholesterol high-fat diet models (Sakai et al., 2020).
In the initiation and progression of liver fibrosis, autophagy in KCs is a potent protective factor for the liver. Autophagy of KCs protects hepatocytes upon liver injury and inhibits hepatic inflammation and fibrogenesis by suppression of IL-1α/β secretion induced by ROS in the mice with CCl4 treatment and N-diethylnitrosamine (DEN)–fed rats (Sun et al., 2017). It also limits IL1A and IL1B secretion, thus alleviating the recruitment of other inflammatory cells. While knockout of Atg5, a key player in autophagy, has been proven to aggravate liver fibrosis (Lodder et al., 2015), IL-7 has been confirmed to promote liver injury and inflammation by suppressing the autophagy of KCs (Zhu et al., 2018). IL-7/IL-7R signaling inhibits Schistosoma japonicum egg antigen–triggered macrophage autophagy through amp-activated protein kinase (AMPK) signaling, thereby aggravating liver fibrosis in Schistosoma japonicum infection (Zhu et al., 2018). Furthermore, spermine can alleviate liver injury by hindering the proinflammatory response of KCs by activating autophagy, which also contributes to the polarization from M2 to M1 (Zhou et al., 2018). Considering the benefits of autophagy, modulating the autophagic function of KCs is an encouraging antifibrotic strategy that deserves further study.
Neutrophils account for nearly 60% of circulating leukocytes in human bodies, defending the microbial infections in the first line. Neutrophils exert the function of killing pathogens by directly swallowing, producing reactive oxygen species (ROS), and stretching neutrophil extracellular traps (NETs) (Papayannopoulos 2018). In addition to the traditional recognition of the function carried out by neutrophils, emerging evidence also illustrates that neutrophils can interact with other immune cells, including NKs, DCs, and lymphocytes. Neutrophils interact with other immune cells via secreting cytokines (such as CCL3 and CCL20) or expressing molecules on the cell membrane, such as peptides.
A systematic review has analyzed the correlation between the neutrophil-to-lymphocyte ratio (NLR) and liver fibrosis, which found that a high NLR was related to mild progression of liver fibrosis in patients infected by HBV, but the patients with chronic HCV infection showed no significant relationships. Generally, the relative number of neutrophils in blood could be associated with the liver fibrosis stage and used as a biomarker in cirrhotic patients (Peng et al., 2018; Pokora Rodak et al., 2018). For mechanism, the previous study found that neutrophils in the liver could induce the proinflammatory effect of macrophages by upregulating miR-223 and downregulating its targeted gene, NLRP3, finally contributing to the spontaneous resolution of liver fibrosis (Calvente et al., 2019).
Accumulative studies have shed light on the role of autophagy in the function of neutrophils, which could further explain the effect of crosstalk between autophagy and neutrophils. During sepsis, the neutrophil autophagy facilitates NET formation and enhances the ability of antibiosis (Park et al., 2017). In a model of ATG16L1-deficient mice, the amount of ROS has increased and the capability of Salmonella typhimurium clearance is impaired, which illustrated that autophagy is beneficial to neutrophils in the function of antibiosis. Moreover, autophagy is also important to neutrophil differentiation. Neutrophils are short-lived mediators of innate immunity, which require timely refreshment and constant replenishment. The impairment of autophagy by knocking out ATG7 was proven to enhance glycolytic activity but decreased mitochondrial respiration, reduced ATP production, and enhanced lipid droplets, finally destructing neutrophil differentiation (Riffelmacher et al., 2017). Taken together, we can assume that properly enhanced autophagy in neutrophils will increase their differentiation and promote the anti-inflammatory effect in the liver, which contribute to the remission of liver fibrosis.
Dendritic cells (DCs) are heterogenous innate immune cells located in the liver, performing the function of presenting antigens to other immune cells, which are the crucial linkage between innate and adaptive immunity (Théry and Amigorena 2001). The absolute amount of DCs in the liver is not large; thus, hepatic DCs tend to be tolerogenic apart from being immunogenic. After treatment with thioacetamide and leptin, the DCs showed great capability to remodel the proinflammatory environment of liver fibrosis (Izawa et al., 2014). In the condition of liver fibrosis, the number of DCs dramatically increases and obtains an immunogenic phenotype. The previous study has shown that DCs extracted from tissues of liver fibrosis could activate HSCs directly in vitro by upregulating the expression of ICAM-1 (Blois et al., 2014). In addition, the overexpression of DCs can activate inflammatory signaling in HSCs and enhance their activation. Thus, the aberrant dynamics of DCs is responsible for the initiation and progression of liver fibrosis, whereas the deterioration of fibrosis increases the population of DCs reciprocally. After being activated by damaged hepatocytes, innate immune cells, especially DCs, recruit and activate CD8+T effector cells, which in turn aggravate hepatocyte injury by releasing cytotoxic cytokines (Parola and Pinzani 2019). DCs are critical for the regulation of liver immunity, and irregular DC activity can induce the activation of T cells and HSCs to contribute to the pathological inflammation–rich environment and fibrogenesis (Miao et al., 2015). A recent study on the mechanism of kinsenoside’s antihepatic fibrosis showed that CD8+ T cell activation could be blocked by specifically upregulating PD-L1 expression on DCs, while reducing the glycolysis of DCs and keeping DCs in the immature state, improved the liver fibrosis index (Xiang et al., 2022).
As a mediator to deliver signals to adaptive immune cells and contribute to their activation, one important function of DCs is to present antigens in the form of major histocompatibility complex (MHC) class II. Lysosomal degradation is essential to pathogen degradation and antigen presentation. Intriguingly, the autophagy flux contains the formation of lysosomes, and in some cases, the autophagosomes fused with lysosomes are conducted by autophagy and are also used to degrade pathogens. Thus, autophagy in DCs can serve an effective role to trigger the adaptive immune system (Germic et al., 2019). Upregulation of ATG5 promotes the expression of CD36 and enhances MHC-II antigen presentation in DCs, which demonstrates that autophagy positively results in DC activity (Oh and Lee 2019). In the LPS-induced injury model, the researchers observed enhanced autophagy in DCs. The mechanism of this autophagy may be caused by local hypoxia to upregulate the phosphorylation levels of AKT, ERK, P38, and NF-κB, thereby activating DCs with inflammatory factors such as IL-1β, IL-18, and TNF-α release (Monaci et al., 2020). These inflammatory factors are bound to aggravate liver fibrosis. From previous studies, we find that at least three distinct types of canonical autophagy coexist in dendritic cells, including microautophagy, which involves lysosomal capturing of cytoplasmic components through various modifications on its membrane; chaperone-mediated induced autophagy; and macroautophagy (Ghislat and Lawrence 2018). Even though limited evidence has clarified the function of DC autophagy in liver fibrosis, it can be speculated that excessive autophagy in DCs promotes their activation to remodel the hepatic immune environment and ultimately cause HSC activation. Therefore, proper inhibition of autophagy in hepatic DCs will be beneficial to the prevention and treatment of liver fibrosis.
Compared with DCs, the proportion of NKs in hepatocytes is much higher than that in peripheral blood, with NKs accounting for nearly 50% of lymphocytes (Abel et al., 2018). Similar to the literal description, the biological function of NKs is in attacking pathogens to maintain homeostasis. NKs are cytotoxic and have effector functions with cytokines, and the ability to be cytotoxic makes it important for these cells to be able to distinguish between target cells and healthy “self” cells. NK cells have a variety of activated and inhibited cell surface receptors that regulate NK cell activity (Vivier et al., 2008). NKs exert potent function in antifibrosis in two different ways. On the one hand, NKs attack and kill HSCs directly in the early stage of HSC activation, but if the HSCs were in the quiescent condition or fully activated, the NKs do not function. On the other hand, NKs release IFN-γ to induce the apoptosis of HSCs (Luci et al., 2019). Meanwhile, the secreted IFN-γ can upregulate the expression of NKG2D and TRAIL on NKs to enhance the NK activities of killing HSCs in a manner of autocrine function (Glassner et al., 2012). Moreover, the fully activated HSCs can release transforming growth factor-beta (TGF-beta) to impair the killing effect of NKs, which explains the dysfunction of NKs in killing fully activated HSCs (Shi et al., 2017). Hence, the utilization of TGF-beta inhibitors is a possible method to strengthen the capability of NKs in the treatment of liver fibrosis.
Recent experimental evidence has preliminarily illustrated the function of autophagy in NKs. NKs can induce mitophagy in the injury caused by a virus infection and maintain the survival of NKs by clearing the damaged mitochondria inside the cell. In addition, autophagy is essential in the process of NK transition from effector cells to long-life memory cells. Drug induction of autophagy enhances memory formation of NKs through an ATG3-dependent mechanism so that NKs can maintain the ability to eliminate the virus and other pathogenic factors (O'Sullivan et al., 2015). Deletion of ATG5, a vital protein involved in autophagy formation, was proved to cause progressive mitochondrial damage and excessive generation of ROS to interrupt the development of NKs and even death (O'Sullivan et al., 2016). In addition, the interaction between ATG7 (another player essential to autophagy) and FOXO1 (forkhead box O1) contributes to the formation of autophagy flux in the cytosol of immature NKs to accelerate the maturation (Huang et al., 2019). These findings demonstrate that proper activation of autophagy is critical to the maturation of NKs, which subsequently enhance the capability of antifibrosis in the liver.
Natural killer T cells (NKTs) are innate-like T cell subsets that are abundant in the human liver. NKTs are quite different from the conventional T cells, for they recognize glycolipid antigens presented by the MHC-I molecule, CD1d, which is usually expressed on the cellular membrane of T cells and NKs (Exley et al., 2017). According to the expression type of T cell receptor (TCR), response to glycolipid antigen, and CD1d dependence during development, NKTs can be divided into three types: type I, type II, and type III NKTs. Type I NKTs, also known as invariant NKT cells (iNKTs), express the constant TCRα chain, co-expressed the Vβ chain, and are activated by the α galactose amide delivered by the CD1d molecule. iNKTs account for 95% of the total number of lymphocytes in the liver. The narrow definition of NKTs refers to iNKT cells (Wang and Yin 2015), which are also the cell types that we summarize here. After activation, the NKTs release proinflammatory cytokines and interact with other types of innate immune cells (Nilsson et al., 2020). NKTs can not only kill target cells directly but also produce a variety of cytokines, thus playing an important role in liver injury, liver fibrosis, liver regeneration, and the occurrence and development of liver cancer (Gao et al., 2009). Similar to NKs, the activation of NKTs also exerts an antifibrosis role by directly killing partially activated HSCs and rapidly secreting IFN-γ to induce apoptosis of HSCs (Ravichandran et al., 2019). Activated HSCs usually have high expression of NKG2D (natural killer group 2 member D) ligand Rae1(retinoic acid early inducible 1), IL-30 stimulates the high expression of NKG2D in NKTs, and NKTs eventually kill the active HSCs and improve liver fibrosis by combining nKG2D-RAE1 with highly specific targeted activated HSCs (Mitra et al., 2014).
Previous studies have suggested that autophagy is deeply involved in the development and maturation of NKTs. By deleting the expression of ATG7, an important gene in autophagy flux formation, the thymic NKT development is blocked at the very early stage, which is different from conventional T cells. Meanwhile, during the NKT differentiation, the phenomena of autophagy significantly increase (Salio et al., 2014). If autophagy was blocked by deleting ATG5 or ATG7, the number of NKTs is significantly reduced, and the function of NKTs is disrupted (Pei et al., 2015).
The function of autophagy in NKTs, which is also involved in the progression of liver fibrosis, is lacking investigation. However, we can assume that the proper existence of autophagy in NKTs will promote their maturation and facilitate the function of killing HSCs.
To date, emerging evidence has proven that innate immunity plays an important role in liver fibrosis. Activation of HSCs and the remodeling of the hepatic microenvironment by the accumulation of the ECM are essential to the initiation and progression of liver fibrosis. The innate immune cells, such as Kupffer cells, NK cells, NKT cells, and neutrophils, are essential to prevent fibrosis by either directly attacking HSCs or releasing cytokines to facilitate the apoptosis of HSCs, consequently maintaining homeostasis in the liver (Table 1). However, the M2 type of Kupffer cells and DCs generally contribute to the progression of liver fibrosis. For these innate immune cells, the proper activation of autophagy is beneficial to their maturation and biological function, such as the production of cytokines.

TABLE 1. Relationship between major innate immune cells and autophagy in liver fibrosis.
Due to the complication of innate immune cell types and their functions in fibrosis, the function of autophagy depends on innate immune cells. The functions of autophagy are various in cells. For most of the immune cells, the proper extent of autophagy can promote their development and maturation, while excessive autophagy will cause a sharp decrease. Thus, for some innate cell types, such as M1 type of Kupffer cells, NK cells, NKT cells, and neutrophils, the appropriate degree of autophagy can promote their development and maturation, which finally exert the function of antifibrosis; however, the pathological autophagy of these cells can enhance the progression of liver fibrosis. Nevertheless, the agents that trigger autophagy in the M2 type of Kupffer cells and DCs are beneficial to patients suffering from liver fibrosis.
Given the complicated conditions of innate immune cells and autophagy, the therapeutic strategy which targets autophagy in innate immune cells is difficult. In the future, accurate determination of innate cell types in the liver is important before using drugs to stimulate or inhibit autophagy. Meanwhile, immunomodulators in clinical operations should be taken into consideration in the therapy of liver fibrosis.
In summary, the observations in the review strongly support the conclusion that autophagy is crucial for the morphological and biological function of the innate immune system in the liver to prevent the initiation and progression of hepatic fibrosis.
FZ and SZ provided the writing ideas and guided the revision of manuscript content. LC and SX summarized the literature and wrote the manuscript. DK, FW, and ZL provided ideas for figures and revised the manuscript.
This work was supported by the National Natural Science Foundation of China (82073914 and 81870423), the Jiangsu Province Traditional Chinese Medicine Science and Technology Development Plan Project (QN202112), and the Joint Project of Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica and Yangtze River Pharmaceutical (JKLPSE202005).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdullah, Z., and Knolle, P. A. (2017). Liver Macrophages in Healthy and Diseased Liver. Pflugers Arch. 469, 553–560. doi:10.1007/s00424-017-1954-6
Abel, A. M., Yang, C., Thakar, M. S., and Malarkannan, S. (2018). Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 9, 1869. doi:10.3389/fimmu.2018.01869
Affo, S., Yu, L. X., and Schwabe, R. F. (2017). The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 12, 153–186. doi:10.1146/annurev-pathol-052016-100322
Allaire, M., Rautou, P. E., Codogno, P., and Lotersztajn, S. (2019). Autophagy in Liver Diseases: Time for Translation? J. Hepatol. 70, 985–998. doi:10.1016/j.jhep.2019.01.026
Ambade, A., Lowe, P., Kodys, K., Catalano, D., Gyongyosi, B., Cho, Y., et al. (2019). Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol-Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology 69, 1105–1121. doi:10.1002/hep.30249
Arsent'eva, N. A., Semenov, A. V., Lyubimova, N. E., Ostankov, Y. V., Elezo, D. S., Kudryavtsev, I. V., et al. (2015). Chemokine Receptors CXCR3 and CCR6 and Their Ligands in the Liver and Blood of Patients with Chronic Hepatitis C. Bull. Exp. Biol. Med. 160, 252–255. doi:10.1007/s10517-015-3142-z
Baglieri, J., Brenner, D. A., and Kisseleva, T. (2019). The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 20, 1723. doi:10.3390/ijms20071723
Bai, F., Huang, Q., Wei, J., Lv, S., Chen, Y., Liang, C., et al. (2018). Gypsophila elegans Isoorientin-2″-O-α-L-Arabinopyranosyl Ameliorates Porcine Serum-Induced Immune Liver Fibrosis by Inhibiting NF-Κb Signaling Pathway and Suppressing HSC Activation. Int. Immunopharmacology 54, 60–67. doi:10.1016/j.intimp.2017.10.028
Blois, S. M., Piccioni, F., Freitag, N., Tirado-González, I., Moschansky, P., Lloyd, R., et al. (2014). Dendritic Cells Regulate Angiogenesis Associated with Liver Fibrogenesis. Angiogenesis 17, 119–128. doi:10.1007/s10456-013-9382-5
Borkham-Kamphorst, E., and Weiskirchen, R. (2016). The PDGF System and its Antagonists in Liver Fibrosis. Cytokine Growth Factor. Rev. 28, 53–61. doi:10.1016/j.cytogfr.2015.10.002
Bruns, I., Lucas, D., Pinho, S., Ahmed, J., Lambert, M. P., Kunisaki, Y., et al. (2014). Megakaryocytes Regulate Hematopoietic Stem Cell Quiescence through CXCL4 Secretion. Nat. Med. 20, 1315–1320. doi:10.1038/nm.3707
Cai, X., Li, Z., Zhang, Q., Qu, Y., Xu, M., Wan, X., et al. (2018). CXCL6-EGFR-induced Kupffer Cells Secrete TGF-Β1 Promoting Hepatic Stellate Cell Activation via the SMAD2/BRD4/C-MYC/EZH2 Pathway in Liver Fibrosis. J. Cel Mol Med 22, 5050–5061. doi:10.1111/jcmm.13787
Calvente, C. J., Tameda, M., Johnson, C. D., Del Pilar, H., Lin, Y. C., Adronikou, N., et al. (2019). Neutrophils Contribute to Spontaneous Resolution of Liver Inflammation and Fibrosis via microRNA-223. J. Clin. Invest. 129, 4091–4109. doi:10.1172/JCI122258
Chen, Y., and Tian, Z. (2020). Roles of Hepatic Innate and Innate-like Lymphocytes in Nonalcoholic Steatohepatitis. Front. Immunol. 11, 1500. doi:10.3389/fimmu.2020.01500
Chen, Z., Jain, A., Liu, H., Zhao, Z., and Cheng, K. (2019). Targeted Drug Delivery to Hepatic Stellate Cells for the Treatment of Liver Fibrosis. J. Pharmacol. Exp. Ther. 370, 695–702. doi:10.1124/jpet.118.256156
Cheng, D., Chai, J., Wang, H., Fu, L., Peng, S., and Ni, X. (2021). Hepatic Macrophages: Key Players in the Development and Progression of Liver Fibrosis. Liver Int. 41, 2279–2294. doi:10.1111/liv.14940
Cosin-Roger, J., Simmen, S., Melhem, H., Atrott, K., Frey-Wagner, I., Hausmann, M., et al. (2017). Hypoxia Ameliorates Intestinal Inflammation through NLRP3/mTOR Downregulation and Autophagy Activation. Nat. Commun. 8, 98. doi:10.1038/s41467-017-00213-3
Deretic, V., and Levine, B. (2018). Autophagy Balances Inflammation in Innate Immunity. Autophagy 14, 243–251. doi:10.1080/15548627.2017.1402992
Dewidar, B., Meyer, C., Dooley, S., and Meindl-Beinker, A. N. (2019). TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated. Cells, 8, 1419. doi:10.3390/cells8111419
Exley, M. A., Wilson, S. B., and Balk, S. P. (2017). Isolation and Functional Use of Human NKT Cells. Curr. Protoc. Immunol. 119, 14–20. doi:10.1002/cpim.33
Fabre, T., Molina, M. F., Soucy, G., Goulet, J. P., Willems, B., Villeneuve, J. P., et al. (2018). Type 3 Cytokines IL-17A and IL-22 Drive TGF-β-dependent Liver Fibrosis. Sci. Immunol. 3, eaar7754. doi:10.1126/sciimmunol.aar7754
Feng, J., Wang, C., Liu, T., Li, J., Wu, L., Yu, Q., et al. (2019). Procyanidin B2 Inhibits the Activation of Hepatic Stellate Cells and Angiogenesis via the Hedgehog Pathway during Liver Fibrosis. J. Cel Mol Med 23, 6479–6493. doi:10.1111/jcmm.14543
Gan, J., Zheng, S. J., Mao, X. R., and Li, J. F. (2021). Invariant Natural Killer T Cells: Not to Be Ignored in Liver Disease. J. Dig. Dis. 22, 136–142. doi:10.1111/1751-2980.12968
Gao, B., Radaeva, S., and Park, O. (2009). Liver Natural Killer and Natural Killer T Cells: Immunobiology and Emerging Roles in Liver Diseases. J. Leukoc. Biol. 86, 513–528. doi:10.1189/JLB.0309135
Gao, J., Wei, B., De Assuncao, T. M., Liu, Z., Hu, X., Ibrahim, S., et al. (2020). Hepatic Stellate Cell Autophagy Inhibits Extracellular Vesicle Release to Attenuate Liver Fibrosis. J. Hepatol. 73, 1144–1154. doi:10.1016/j.jhep.2020.04.044
Ge, J., Yan, Q., Wang, Y., Cheng, X., Song, D., Wu, C., et al. (2020). IL-10 Delays the Degeneration of Intervertebral Discs by Suppressing the P38 MAPK Signaling Pathway. Free Radic. Biol. Med. 147, 262–270. doi:10.1016/j.freeradbiomed.2019.12.040
Germic, N., Frangez, Z., Yousefi, S., and Simon, H. U. (2019). Regulation of the Innate Immune System by Autophagy: Monocytes, Macrophages, Dendritic Cells and Antigen Presentation. Cell Death Differ 26, 715–727. doi:10.1038/s41418-019-0297-6
Ghislat, G., and Lawrence, T. (2018). Autophagy in Dendritic Cells. Cell Mol Immunol 15, 944–952. doi:10.1038/cmi.2018.2
Glässner, A., Eisenhardt, M., Krämer, B., Körner, C., Coenen, M., Sauerbruch, T., et al. (2012). NK Cells from HCV-Infected Patients Effectively Induce Apoptosis of Activated Primary Human Hepatic Stellate Cells in a TRAIL-, FasL- and NKG2D-dependent Manner. Lab. Invest. 92, 967–977. doi:10.1038/labinvest.2012.54
Hammoutene, A., Biquard, L., Lasselin, J., Kheloufi, M., Tanguy, M., Vion, A. C., et al. (2020). A Defect in Endothelial Autophagy Occurs in Patients with Non-alcoholic Steatohepatitis and Promotes Inflammation and Fibrosis. J. Hepatol. 72, 528–538. doi:10.1016/j.jhep.2019.10.028
Higashi, T., Friedman, S. L., and Hoshida, Y. (2017). Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv. Drug Deliv. Rev. 121, 27–42. doi:10.1016/j.addr.2017.05.007
Hossain, M., and Kubes, P. (2019). Innate Immune Cells Orchestrate the Repair of Sterile Injury in the Liver and beyond. Eur. J. Immunol. 49, 831–841. doi:10.1002/eji.201847485
Hou, L. S., Zhang, Y. W., Li, H., Wang, W., Huan, M. L., Zhou, S. Y., et al. (2022). The Regulatory Role and Mechanism of Autophagy in Energy Metabolism-Related Hepatic Fibrosis. Pharmacol. Ther. 234, 108117. doi:10.1016/j.pharmthera.2022.108117
Hu, Y. X., Han, X. S., and Jing, Q. (2019). Autophagy in Development and Differentiation. Adv. Exp. Med. Biol. 1206, 469–487. doi:10.1007/978-981-15-0602-4_22
Huang, P., Wang, F., Yang, Y., Lai, W., Meng, M., Wu, S., et al. (2019). Hematopoietic-Specific Deletion of Foxo1 Promotes NK Cell Specification and Proliferation. Front. Immunol. 10, 1016. doi:10.3389/fimmu.2019.01016
Huang, R., and Liu, W. (2015). Identifying an Essential Role of Nuclear LC3 for Autophagy. Autophagy 11, 852–853. doi:10.1080/15548627.2015.1038016
Izawa, T., Murakami, H., Wijesundera, K. K., Golbar, H. M., Kuwamura, M., and Yamate, J. (2014). Inflammatory Regulation of Iron Metabolism during Thioacetamide-Induced Acute Liver Injury in Rats. Exp. Toxicol. Pathol. 66, 155–162. doi:10.1016/j.etp.2013.12.002
Jenne, C. N., and Kubes, P. (2013). Immune Surveillance by the Liver. Nat. Immunol. 14, 996–1006. doi:10.1038/ni.2691
Ji, G., Ma, L., Yao, H., Ma, S., Si, X., Wang, Y., et al. (2020). Precise Delivery of Obeticholic Acid via Nanoapproach for Triggering Natural Killer T Cell-Mediated Liver Cancer Immunotherapy. Acta Pharm. Sin B 10, 2171–2182. doi:10.1016/j.apsb.2020.09.004
Jiang, G. M., Tan, Y., Wang, H., Peng, L., Chen, H. T., Meng, X. J., et al. (2019). The Relationship between Autophagy and the Immune System and its Applications for Tumor Immunotherapy. Mol. Cancer 18, 17. doi:10.1186/s12943-019-0944-z
Kakino, S., Ohki, T., Nakayama, H., Yuan, X., Otabe, S., Hashinaga, T., et al. (2018). Pivotal Role of TNF-α in the Development and Progression of Nonalcoholic Fatty Liver Disease in a Murine Model. Horm. Metab. Res. 50, 80–87. doi:10.1055/s-0043-118666
Karsdal, M. A., Nielsen, S. H., Leeming, D. J., Langholm, L. L., Nielsen, M. J., Manon-Jensen, T., et al. (2017). The Good and the Bad Collagens of Fibrosis - Their Role in Signaling and Organ Function. Adv. Drug Deliv. Rev. 121, 43–56. doi:10.1016/j.addr.2017.07.014
Katsuragi, Y., Ichimura, Y., and Komatsu, M. (2015). p62/SQSTM1 Functions as a Signaling Hub and an Autophagy Adaptor. FEBS J. 282, 4672–4678. doi:10.1111/febs.13540
Li, W., and Zhang, L. (2019). Regulation of ATG and Autophagy Initiation. Adv. Exp. Med. Biol. 1206, 41–65. doi:10.1007/978-981-15-0602-4_2
Li, X., Liu, R., Wang, Y., Zhu, W., Zhao, D., Wang, X., et al. (2020a). Cholangiocyte-Derived Exosomal lncRNA H19 Promotes Macrophage Activation and Hepatic Inflammation under Cholestatic Conditions. Cells 9, 190. doi:10.3390/cells9010190
Li, Y., Liu, R., Wu, J., and Li, X. (2020b). Self-eating: Friend or Foe? the Emerging Role of Autophagy in Fibrotic Diseases. Theranostics 10, 7993–8017. doi:10.7150/thno.47826
Liu, N., Feng, J., Lu, X., Yao, Z., Liu, Q., Lv, Y., et al. (2019). Isorhamnetin Inhibits Liver Fibrosis by Reducing Autophagy and Inhibiting Extracellular Matrix Formation via the TGF-β1/Smad3 and TGF-Β1/p38 MAPK Pathways. Mediators Inflamm. 2019, 6175091. doi:10.1155/2019/6175091
Liu, Y. M., Cong, S., Cheng, Z., Hu, Y. X., Lei, Y., Zhu, L. L., et al. (2020). Platycodin D Alleviates Liver Fibrosis and Activation of Hepatic Stellate Cells by Regulating JNK/c-JUN Signal Pathway. Eur. J. Pharmacol. 876, 172946. doi:10.1016/j.ejphar.2020.172946
Lodder, J., Denaës, T., Chobert, M. N., Wan, J., El-Benna, J., Pawlotsky, J. M., et al. (2015). Macrophage Autophagy Protects against Liver Fibrosis in Mice. Autophagy 11, 1280–1292. doi:10.1080/15548627.2015.1058473
Lorincz, P., and Juhasz, G. (2020). Autophagosome-Lysosome Fusion. J. Mol. Biol. 432, 2462–2482. doi:10.1016/j.jmb.2019.10.028
Luci, C., Vieira, E., Perchet, T., Gual, P., and Golub, R. (2019). Natural Killer Cells and Type 1 Innate Lymphoid Cells Are New Actors in Non-alcoholic Fatty Liver Disease. Front. Immunol. 10, 1192. doi:10.3389/fimmu.2019.01192
Luedde, T., and Schwabe, R. F. (2011). NF-κB in the Liver-Llinking Injury, Fibrosis and Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 8, 108–118. doi:10.1038/nrgastro.2010.213
Madhavi, Y. V., Gaikwad, N., Yerra, V. G., Kalvala, A. K., Nanduri, S., and Kumar, A. (2019). Targeting AMPK in Diabetes and Diabetic Complications: Energy Homeostasis, Autophagy and Mitochondrial Health. Curr. Med. Chem. 26, 5207–5229. doi:10.2174/0929867325666180406120051
Matsuda, M., Tsurusaki, S., Miyata, N., Saijou, E., Okochi, H., Miyajima, A., et al. (2018). Oncostatin M Causes Liver Fibrosis by Regulating Cooperation between Hepatic Stellate Cells and Macrophages in Mice. Hepatology 67, 296–312. doi:10.1002/hep.29421
Meng, D., Li, Z., Wang, G., Ling, L., Wu, Y., and Zhang, C. (2018). Carvedilol Attenuates Liver Fibrosis by Suppressing Autophagy and Promoting Apoptosis in Hepatic Stellate Cells. Biomed. Pharmacother. 108, 1617–1627. doi:10.1016/j.biopha.2018.10.005
Miao, Q., Bian, Z., Tang, R., Zhang, H., Wang, Q., Huang, S., et al. (2015). Emperipolesis Mediated by CD8 T Cells Is a Characteristic Histopathologic Feature of Autoimmune Hepatitis. Clin. Rev. Allergy Immunol. 48, 226–235. doi:10.1007/s12016-014-8432-0
Mitra, A., Satelli, A., Yan, J., Xueqing, X., Gagea, M., Hunter, C. A., et al. (2014). IL-30 (IL27p28) Attenuates Liver Fibrosis through Inducing NKG2D-Rae1 Interaction between NKT and Activated Hepatic Stellate Cells in Mice. Hepatology 60, 2027–2039. doi:10.1002/hep.27392
Mizushima, N. (2010). The Role of the Atg1/ULK1 Complex in Autophagy Regulation. Curr. Opin. Cel Biol 22, 132–139. doi:10.1016/j.ceb.2009.12.004
Monaci, S., Aldinucci, C., Rossi, D., Giuntini, G., Filippi, I., Ulivieri, C., et al. (2020). Hypoxia Shapes Autophagy in LPS-Activated Dendritic Cells. Front. Immunol. 11, 573646. doi:10.3389/fimmu.2020.573646
Moon, A. M., Singal, A. G., and Tapper, E. B. (2020). Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 18, 2650–2666. doi:10.1016/j.cgh.2019.07.060
Mowers, E. E., Sharifi, M. N., and Macleod, K. F. (2018). Functions of Autophagy in the Tumor Microenvironment and Cancer Metastasis. FEBS J. 285, 1751–1766. doi:10.1111/febs.14388
Mridha, A. R., Wree, A., Robertson, A. A. B., Yeh, M. M., Johnson, C. D., Van Rooyen, D. M., et al. (2017). NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J. Hepatol. 66, 1037–1046. doi:10.1016/j.jhep.2017.01.022
Niehaus, C. E., Strunz, B., Cornillet, M., Falk, C. S., Schnieders, A., Maasoumy, B., et al. (2020). MAIT Cells Are Enriched and Highly Functional in Ascites of Patients with Decompensated Liver Cirrhosis. Hepatology 72, 1378–1393. doi:10.1002/hep.31153
Nilsson, J., Hörnberg, M., Schmidt-Christensen, A., Linde, K., Nilsson, M., Carlus, M., et al. (2020). NKT Cells Promote Both Type 1 and Type 2 Inflammatory Responses in a Mouse Model of Liver Fibrosis. Sci. Rep. 10, 21778. doi:10.1038/s41598-020-78688-2
O'sullivan, T. E., Geary, C. D., Weizman, O. E., Geiger, T. L., Rapp, M., Dorn, G. W., et al. (2016). Atg5 Is Essential for the Development and Survival of Innate Lymphocytes. Cell Rep 15, 1910–1919. doi:10.1016/j.celrep.2016.04.082
O'sullivan, T. E., Johnson, L. R., Kang, H. H., and Sun, J. C. (2015). BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 43, 331–342. doi:10.1016/j.immuni.2015.07.012
Oh, D. S., and Lee, H. K. (2019). Autophagy Protein ATG5 Regulates CD36 Expression and Anti-tumor MHC Class II Antigen Presentation in Dendritic Cells. Autophagy 15, 2091–2106. doi:10.1080/15548627.2019.1596493
Pang, Y., Kartsonaki, C., Turnbull, I., Guo, Y., Clarke, R., Chen, Y., et al. (2018). Diabetes, Plasma Glucose, and Incidence of Fatty Liver, Cirrhosis, and Liver Cancer: A Prospective Study of 0.5 Million People. Hepatology 68, 1308–1318. doi:10.1002/hep.30083
Papayannopoulos, V. (2018). Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol. 18, 134–147. doi:10.1038/nri.2017.105
Park, S. Y., Shrestha, S., Youn, Y. J., Kim, J. K., Kim, S. Y., Kim, H. J., et al. (2017). Autophagy Primes Neutrophils for Neutrophil Extracellular Trap Formation during Sepsis. Am. J. Respir. Crit. Care Med. 196, 577–589. doi:10.1164/rccm.201603-0596OC
Parola, M., and Pinzani, M. (2019). Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol. Aspects Med. 65, 37–55. doi:10.1016/j.mam.2018.09.002
Pei, B., Zhao, M., Miller, B. C., Véla, J. L., Bruinsma, M. W., Virgin, H. W., et al. (2015). Invariant NKT Cells Require Autophagy to Coordinate Proliferation and Survival Signals during Differentiation. J. Immunol. 194, 5872–5884. doi:10.4049/jimmunol.1402154
Peng, Y., Li, Y., He, Y., Wei, Q., Xie, Q., Zhang, L., et al. (2018). The Role of Neutrophil to Lymphocyte Ratio for the Assessment of Liver Fibrosis and Cirrhosis: a Systematic Review. Expert Rev. Gastroenterol. Hepatol. 12, 503–513. doi:10.1080/17474124.2018.1463158
Plaza-Zabala, A., Sierra-Torre, V., and Sierra, A. (2017). Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 18, 598. doi:10.3390/ijms18030598
Pokora Rodak, A., Kiciak, S., and Tomasiewicz, K. (2018). Neutrophil-lymphocyte Ratio and Mean Platelet Volume as Predictive Factors for Liver Fibrosis and Steatosis in Patients with Chronic Hepatitis B. Ann. Agric. Environ. Med. 25, 690–692. doi:10.26444/aaem/99583
Pradere, J. P., Kluwe, J., De Minicis, S., Jiao, J. J., Gwak, G. Y., Dapito, D. H., et al. (2013). Hepatic Macrophages but Not Dendritic Cells Contribute to Liver Fibrosis by Promoting the Survival of Activated Hepatic Stellate Cells in Mice. Hepatology 58, 1461–1473. doi:10.1002/hep.26429
Qu, Y., Zhang, Q., Cai, X., Li, F., Ma, Z., Xu, M., et al. (2017). Exosomes Derived from miR-181-5p-Modified Adipose-Derived Mesenchymal Stem Cells Prevent Liver Fibrosis via Autophagy Activation. J. Cel Mol Med 21, 2491–2502. doi:10.1111/jcmm.13170
Ravichandran, G., Neumann, K., Berkhout, L. K., Weidemann, S., Langeneckert, A. E., Schwinge, D., et al. (2019). Interferon-γ-dependent Immune Responses Contribute to the Pathogenesis of Sclerosing Cholangitis in Mice. J. Hepatol. 71, 773–782. doi:10.1016/j.jhep.2019.05.023
Riffelmacher, T., Clarke, A., Richter, F. C., Stranks, A., Pandey, S., Danielli, S., et al. (2017). Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 47, 466–e5. doi:10.1016/j.immuni.2017.08.005
Roh, Y. S., and Seki, E. (2018). Chemokines and Chemokine Receptors in the Development of NAFLD. Adv. Exp. Med. Biol. 1061, 45–53. doi:10.1007/978-981-10-8684-7_4
Sakai, Y., Chen, G., Ni, Y., Zhuge, F., Xu, L., Nagata, N., et al. (2020). DPP-4 Inhibition with Anagliptin Reduces Lipotoxicity-Induced Insulin Resistance and Steatohepatitis in Male Mice. Endocrinology 161, bqaa139. doi:10.1210/endocr/bqaa139
Salio, M., Puleston, D. J., Mathan, T. S., Shepherd, D., Stranks, A. J., Adamopoulou, E., et al. (2014). Essential Role for Autophagy during Invariant NKT Cell Development. Proc. Natl. Acad. Sci. U S A. 111, E5678–E5687. doi:10.1073/pnas.1413935112
Schnieder, J., Mamazhakypov, A., Birnhuber, A., Wilhelm, J., Kwapiszewska, G., Ruppert, C., et al. (2020). Loss of LRP1 Promotes Acquisition of Contractile-Myofibroblast Phenotype and Release of Active TGF-Β1 from ECM Stores. Matrix Biol. 88, 69–88. doi:10.1016/j.matbio.2019.12.001
Schumacher, J. D., Kong, B., Wu, J., Rizzolo, D., Armstrong, L. E., Chow, M. D., et al. (2020). Direct and Indirect Effects of Fibroblast Growth Factor (FGF) 15 and FGF19 on Liver Fibrosis Development. Hepatology 71, 670–685. doi:10.1002/hep.30810
Seki, E., De Minicis, S., Gwak, G. Y., Kluwe, J., Inokuchi, S., Bursill, C. A., et al. (2009). CCR1 and CCR5 Promote Hepatic Fibrosis in Mice. J. Clin. Invest. 119, 1858–1870. doi:10.1172/jci37444
Sepulveda-Crespo, D., Resino, S., and Martinez, I. (2021). Strategies Targeting the Innate Immune Response for the Treatment of Hepatitis C Virus-Associated Liver Fibrosis. Drugs 81, 419–443. doi:10.1007/s40265-020-01458-x
Shi, J., Zhao, J., Zhang, X., Cheng, Y., Hu, J., Li, Y., et al. (2017). Activated Hepatic Stellate Cells Impair NK Cell Anti-fibrosis Capacity through a TGF-β-dependent Emperipolesis in HBV Cirrhotic Patients. Sci. Rep. 7, 44544. doi:10.1038/srep44544
Soysa, R., Wu, X., and Crispe, I. N. (2017). Dendritic Cells in Hepatitis and Liver Transplantation. Liver Transpl. 23, 1433–1439. doi:10.1002/lt.24833
Su, S. B., Qin, S. Y., Xian, X. L., Huang, F. F., Huang, Q. L., Zhangdi, H. J., et al. (2021). Interleukin-22 Regulating Kupffer Cell Polarization through STAT3/Erk/Akt Crosstalk Pathways to Extenuate Liver Fibrosis. Life Sci. 264, 118677. doi:10.1016/j.lfs.2020.118677
Sun, K., Xu, L., Jing, Y., Han, Z., Chen, X., Cai, C., et al. (2017). Autophagy-deficient Kupffer Cells Promote Tumorigenesis by Enhancing mtROS-NF-κB-IL1α/β-dependent Inflammation and Fibrosis during the Preneoplastic Stage of Hepatocarcinogenesis. Cancer Lett. 388, 198–207. doi:10.1016/j.canlet.2016.12.004
Théry, C., and Amigorena, S. (2001). The Cell Biology of Antigen Presentation in Dendritic Cells. Curr. Opin. Immunol. 13, 45–51. doi:10.1016/s0952-7915(00)00180-1
Vivier, E., Tomasello, E., Baratin, M., Walzer, T., and Ugolini, S. (2008). Functions of Natural Killer Cells. Nat. Immunol. 9, 503–510. doi:10.1038/ni1582
Wang, H., and Yin, S. (2015). Natural Killer T Cells in Liver Injury, Inflammation and Cancer. Expert Rev. Gastroenterol. Hepatol. 9, 1077–1085. doi:10.1586/17474124.2015.1056738
Weiskirchen, R., and Tacke, F. (2014). Cellular and Molecular Functions of Hepatic Stellate Cells in Inflammatory Responses and Liver Immunology. Hepatobiliary Surg. Nutr. 3, 344–363. doi:10.3978/j.issn.2304-3881.2014.11.03
Wu, Y., Huang, M., Sun, H., Zhou, X., Zhou, R., Gu, G., et al. (2020). Role of Innate Immunity in Pediatric Post-transplant Idiopathic Liver Fibrosis. Front. Immunol. 11, 2111. doi:10.3389/fimmu.2020.02111
Wu, Z., Han, M., Chen, T., Yan, W., and Ning, Q. (2010). Acute Liver Failure: Mechanisms of Immune-Mediated Liver Injury. Liver Int. 30, 782–794. doi:10.1111/j.1478-3231.2010.02262.x
Xiang, H., Zhang, J., Lin, C., Zhang, L., Liu, B., and Ouyang, L. (2020). Targeting Autophagy-Related Protein Kinases for Potential Therapeutic Purpose. Acta Pharm. Sin B 10, 569–581. doi:10.1016/j.apsb.2019.10.003
Xiang, M., Liu, T., Tian, C., Ma, K., Gou, J., Huang, R., et al. (2022). Kinsenoside Attenuates Liver Fibro-Inflammation by Suppressing Dendritic Cells via the PI3K-AKT-FoxO1 Pathway. Pharmacol. Res. 177, 106092. doi:10.1016/j.phrs.2022.106092
Xu, F., Liu, C., Zhou, D., and Zhang, L. (2016). TGF-β/SMAD Pathway and its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 64, 157–167. doi:10.1369/0022155415627681
Yan, S., Huda, N., Khambu, B., and Yin, X. M. (2017). Relevance of Autophagy to Fatty Liver Diseases and Potential Therapeutic Applications. Amino Acids 49, 1965–1979. doi:10.1007/s00726-017-2429-y
Yan, X., Zhou, R., and Ma, Z. (2019). Autophagy-Cell Survival and Death. Adv. Exp. Med. Biol. 1206, 667–696. doi:10.1007/978-981-15-0602-4_29
Yan, Z., Qin, C., Zhao, C., and Fang, Z. (2020). Research Progress of Nanomaterial-Mediated Photodynamic Therapy in Tumor Treatment. J. Nanoparticle Res. 22, 12405. doi:10.1007/s11051-020-05030-2
Yang, W., Tao, Y., Wu, Y., Zhao, X., Ye, W., Zhao, D., et al. (2019). Neutrophils Promote the Development of Reparative Macrophages Mediated by ROS to Orchestrate Liver Repair. Nat. Commun. 10, 1076. doi:10.1038/s41467-019-09046-8
Zhang, C. Y., Yuan, W. G., He, P., Lei, J. H., and Wang, C. X. (2016). Liver Fibrosis and Hepatic Stellate Cells: Etiology, Pathological Hallmarks and Therapeutic Targets. World J. Gastroenterol. 22, 10512–10522. doi:10.3748/wjg.v22.i48.10512
Zhou, J., Huang, N., Guo, Y., Cui, S., Ge, C., He, Q., et al. (2019). Combined Obeticholic Acid and Apoptosis Inhibitor Treatment Alleviates Liver Fibrosis. Acta Pharm. Sin B 9, 526–536. doi:10.1016/j.apsb.2018.11.004
Zhou, S., Gu, J., Liu, R., Wei, S., Wang, Q., Shen, H., et al. (2018). Spermine Alleviates Acute Liver Injury by Inhibiting Liver-Resident Macrophage Pro-inflammatory Response through ATG5-dependent Autophagy. Front. Immunol. 9, 948. doi:10.3389/fimmu.2018.00948
Keywords: liver fibrosis, autophagy, innate immunity, HSCs, immune cells
Citation: Chen L, Kong D, Xia S, Wang F, Li Z, Zhang F and Zheng S (2022) Crosstalk Between Autophagy and Innate Immunity: A Pivotal Role in Hepatic Fibrosis. Front. Pharmacol. 13:891069. doi: 10.3389/fphar.2022.891069
Received: 07 March 2022; Accepted: 29 March 2022;
Published: 17 May 2022.
Edited by:
Xiaojiaoyang Li, Beijing University of Chinese Medicine, ChinaReviewed by:
Jin Liu, Hong Kong Baptist University, Hong Kong SAR, ChinaCopyright © 2022 Chen, Kong, Xia, Wang, Li, Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Zhang, emhhbmdmZW5nMjAxM0BuanVjbS5lZHUuY24=; Shizhong Zheng, bnl0d3NAbmp1Y20uZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.