
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 19 May 2022
Sec. Respiratory Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.880878
This article is part of the Research TopicMechanisms of Oxidative Cellular Injury: Redox Imbalance, Signaling Pathways and Implications for Human Lung DiseasesView all 5 articles
 Anantha Harijith1
Anantha Harijith1 Prathima Basa1Alison Ha1,2Jaya Thomas1
Prathima Basa1Alison Ha1,2Jaya Thomas1 Anjum Jafri3
Anjum Jafri3 Panfeng Fu4
Panfeng Fu4 Peter M. MacFarlane1
Peter M. MacFarlane1 Thomas M. Raffay1
Thomas M. Raffay1 Viswanathan Natarajan4,5Tara Sudhadevi1*
Viswanathan Natarajan4,5Tara Sudhadevi1*Management of acute respiratory distress involves O2 supplementation, which is lifesaving, but causes severe hyperoxic acute lung injury (HALI). NADPH oxidase (NOX) could be a major source of reactive oxygen species (ROS) in hyperoxia (HO). Epithelial cell death is a crucial step in the development of many lung diseases. Alveolar type II (AT2) cells are the metabolically active epithelial cells of alveoli that serve as a source of AT1 cells following lung injury. The aim of this study was to determine the possible role of AT2 epithelial cell NOX4 in epithelial cell death from HALI. Wild type (WT), Nox4fl/fl (control), and Nox4−/−Spc-Cre mice were exposed to room air (NO) or 95% O2 (HO) to investigate the structural and functional changes in the lung. C57BL/6J WT animals subjected to HO showed increased expression of lung NOX4 compared to NO. Significant HALI, increased bronchoalveolar lavage cell counts, increased protein levels, elevated proinflammatory cytokines and increased AT2 cell death seen in hyperoxic Nox4fl/fl control mice were attenuated in HO-exposed Nox4−/−Spc-Cre mice. HO-induced expression of NOX4 in MLE cells resulted in increased mitochondrial (mt) superoxide production and cell apoptosis, which was reduced in NOX4 siRNA silenced cells. This study demonstrates a novel role for epithelial cell NOX4 in accelerating lung epithelial cell apoptosis from HALI. Deletion of the Nox4 gene in AT2 cells or silencing NOX4 in lung epithelial cells protected the lungs from severe HALI with reduced apoptosis and decreased mt ROS production in HO. These results suggest NOX4 as a potential target for the treatment of HALI.
Supplemental oxygen therapy is often inevitable in the treatment of serious lung diseases like COVID-19 pneumonia (Dias-Freitas et al., 2016). However, prolonged exposure of patients with acute respiratory distress syndrome (ARDS) to a high fraction of inspired oxygen (FiO2) i.e., hyperoxia (HO) causes severe hyperoxic acute lung injury (HALI). HO overwhelms the antioxidant defense systems of the body leading to oxidative stress, triggering several pathological processes in the lung. This results in severe lung damage with subsequent deterioration of respiratory function (Kallet and Matthay, 2013). Various experimental strategies to reduce increased ROS showed some beneficial roles in animal models; however, were ineffective clinically (Welty-Wolf et al., 1997; Otterbein et al., 1999). Hence, there is an immediate need to identify new targets and understand mechanism(s) of HO-induced ROS generation in different cell types in the lung for the development of potential therapies.
Cellular respiration is the prime source for generation of ROS (Auten and Davis, 2009). NADPH oxidases (NOXs) are also major sources of cellular ROS and have been a focus of extensive research due to their critical function in producing ROS under normal and patho-physiological conditions (Mittal et al., 2014; Panday et al., 2015). Over-activity or increased expression of NOX proteins can lead to oxidative stress causing cellular damage (Tarafdar and Pula, 2018). While NOX1, 2, 3, and 5 generate superoxide, NOX4 produces hydrogen peroxide (H2O2) (Takac et al., 2011; Nisimoto et al., 2014). Interestingly, NOX4 is localized both in the mitochondrial and cytosolic compartment of mammalian cells (Carnesecchi et al., 2009; Brown and Griendling, 2009; Pendyala et al., 2009) as well as in the nucleus (Anilkumar et al., 2013). NOX2/NOX4 activation by HO has been shown to promote oxidative stress (Parinandi et al., 2003; Chowdhury et al., 2005; Usatyuk et al., 2007; Pendyala et al., 2011) in endothelial cells. Dual Oxidase 2 in mouse lung epithelia was found to be essential for HO-induced acute lung injury (Kim et al., 2014; Dias-Freitas et al., 2016). NOX2 has been extensively studied in lungs and generates high levels of ROS in neutrophils as a central mechanism of host defense against microbial infection (Paiva and Bozza, 2014). NOX2, for its activity, needs assembly of other cytosolic components such as p47phox, p67phox and Rac1 in the plasma membrane (Bernard et al., 2014). In contrast, NOX4 is constitutively active, seems not to require any other component for its activity and generates low levels of H2O2 under basal conditions; however, not much is known about its role in HALI. NOX4 has recently gained attention as it is now known to regulate several pathological processes including cellular differentiation (Lee et al., 2014), host defense (Kim et al., 2017), and tissue fibrosis (Hecker et al., 2009).
The pathophysiology of HALI suggests AT2 to be one of the most vulnerable lung cell types. AT2 cells are essential for lung regeneration after injury (Olajuyin et al., 2019). It was demonstrated that NOX4 deficiency or acute treatment with a NOX4 inhibitor blunted TGFβ1-induced alveolar epithelial cell death and prevented subsequent pulmonary fibrosis in a murine model of lung fibrosis (Carnesecchi et al., 2011). NOX4 also mediates myofibroblast activation and fibrogenic responses to lung injury (Hecker et al., 2009). There is growing evidence that suggests NOX4 as a potential target for the treatment of ARDS/ALI patients during both the acute and the proliferative stage (Manoury et al., 2005; Bernard et al., 2017). Our data have shown elevated NOX4 expression in whole lung tissue as well as lung epithelial cells upon HO treatment which led to our hypothesis that AT2 NOX4 mediates HALI and AT2 cell specific deletion of Nox4 will attenuate HO injury.
All experimental procedures were approved by the Institutional Animal Care and Use Committee (protocol #2020-0052) of the Case Western Reserve University (CWRU). All animals were treated in accordance with the NIH guidelines for the care and use of laboratory animals. Nox4fl/fl mice used in this study were described previously (Matsushima et al., 2013; Fu et al., 2021). To determine the role of AT2 epithelial cell NOX4 in HO-mediated lung inflammatory injury, mice harboring a deletion of Nox4 in AT2 cells (Nox4−/−Spc-Cre) were generated. The Nox4fl/fl and the Nox+/-Spc-Cre mice were gifted by Dr. V Natarajan, Dept. of Pharmacology and Regenerative Medicine, University of Illinois at Chicago. Both male and female mice, 8 weeks old, were used for the experiments. Temporal deletion of Nox4 in Nox+/-Spc-Cre mice was achieved by intraperitoneal injection of tamoxifen (200 mg/kg dissolved in corn oil) for five consecutive days. The animals were used for experiments 2 weeks later. Nox4fl/fl mice that served as controls also received the same dosage and time course of tamoxifen injection and experimentation. The mice were exposed to room air/ normoxia (NO) or 95% hyperoxia (HO) for 66 h. A 66 h timepoint was chosen based on the preliminary data collected at either 48 or 72 h. At 48 h, there was no significant injury observed (data not shown) and at 72 h, the animals appeared seriously morbid. Following exposure to NO or HO, the mice were humanely euthanized to collect lungs for histology, bronchoalveolar lavage (BAL) and other analyses. Euthanasia was by anesthetic overdose (intraperitoneal injection of a 100 mg/kg ketamine and 10 mg/kg xylazine mix) followed by exsanguination. Treatment groups of 6–10 mice (both male and female) were used for each of the experiments and representative data are shown.
The animals were euthanized, the trachea intubated, and the left lung lobe was inflated with neutral buffered formalin at 20 cm of H2O pressure. Following fixation for another 24 h, the lungs were processed for paraffin embedding and sectioning. Lung tissue was cut into 5 μm sections for subsequent alveolar analyses after hematoxylin and eosin (H&E) staining. We have used two serial sections (six fields/section) from each animal and a total of six animals per group. Assessment of acute lung injury was done for infiltration of neutrophils, alveolar wall thickening and deposition of proteinaceous debris (Matute-Bello et al., 2011). Briefly, a scoring system with scores of 0, 1 and 2 was used for each of the three criteria mentioned. An average score of 0 indicated absence of injury, 1–2 indicated mild to moderate injury and two indicated severe injury. Scoring was done for all animals and averaged for a group.
To assess the evaluation of Nox4 deletion in AT2 cells, the lung tissue sections from Nox4fl/fl and Nox4−/−Spc-Cre mice (Nox4 deleted) were pretreated with antigen retrieval buffer (10 mM citrate), followed by blocking nonspecific binding with appropriate serum. Sections were then subjected to immunofluorescence co-staining for NOX4 (1:300; 14347-1-AP; Proteintech, Rosemont, IL, United States) and surfactant protein-C/ SPC (1:50; sc-7705; Santacruz, Dallas, TX, United States) to co-localize NOX4 deletion in AT2 cells. This was followed by incubation with respective secondary antibodies (Alexa Fluor 488 and Alexa Fluor 594 from Invitrogen at 1:500) followed by mounting using Vectashield mounting medium containing 4’,6-diamidino-2-phenylindole/DAPI (Vector Laboratories, Burlingame, CA, United States). Appropriate negative controls were run by omitting the primary antibody to confirm nonspecific staining. Images of the immunostained sections were captured using a fluorescence microscope (Leica Microsystems, Wetzlar, Germany) equipped with a Rolera XR CCD camera (Q-Imaging, Surrey, BC, Canada) mounted on a microscope. ImageJ software was used to analyze the stained lung sections and the intensity of staining was measured per area.
BAL collection was based on our previous protocol (Ha et al., 2020; Sudhadevi et al., 2021). Briefly, the mice were sacrificed, the trachea were exposed, the tubing were inserted and secured, and the whole lung was lavaged with 1 ml of PBS. Cells in the BAL fluid was counted using a cell counter (Bio-Rad). The total protein concentration in BAL fluid was measured using the BCA protein assay according to the manufacturer’s instructions (#23225; Pierce). For cytokine analysis in the BAL fluid, Luminex technology and reagents (R&D Systems, Minneapolis, MN, United States) were used. Murine TNF-α, IL-6, macrophage inflammatory protein 2 (MIP-2), keratinocyte-derived chemokine (KC), and MIP-1
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was used per manufacturer’s instructions (The ApopTag Peroxidase In Situ Apoptosis Detection Kit, S7100; Millipore Sigma, Billerica, United States). Sections were counterstained with methyl green. The number of TUNEL positive cells were counted in all areas of the section using a light microscope (×20 magnification) and the average number of TUNEL-positive cells per field per animal were calculated.
Mice after exposure to NO or HO were anesthetized intraperitoneally with ketamine (100 mg/kg) and xylazine (10 mg/kg), tracheostomized with a metal 18 G cannula and connected to a flexiVent ventilator (SCIREQ, Montréal, QC, Canada). Baseline measurements were performed using forced-oscillation maneuvers and calculated using the ventilator software (flexiWare 5.1, Version 7.2; SCIREQ). Following a recruitment breath to 30 cm H2O, three measurements of respiratory system resistance (Rrs), respiratory system elastance (Ers), and dynamic compliance (Crs) were performed (Snapshot 150), as well as lung tissue impedance parameters of tissue resistance (G) and tissue elastance (H) (Quick Prime-3). Mean values of Rrs, Ers, Crs, G and H for each animal are reported.
Mouse lung epithelial cells (MLE-12) cultured in DMEM-F12 medium containing 10% FBS and antibiotic (Sigma) were used for the in vitro experiments. The cells (∼80% confluence) were exposed to HO (95% O2) as described before (Harijith et al., 2016) using a humidity-controlled airtight Billups- Rothenberg modulator incubator chamber (Billups-Rothenberg, CA, United States), flushed with 95% O2:5% CO2 gas. The concentration of O2 inside was continuously monitored using a digital oxygen monitor. The buffering capacity of the cell culture medium was maintained at a pH ∼7.4 during the period of HO exposure.
Protein expression was detected in the mouse lungs and MLE-12 cells by immunoblotting as described earlier (Harijith et al., 2013; Bandela et al., 2021). Lung tissues were pulverized (Bessman tissue pulverizer, # 189475; Spectrum Laboratories Inc.) using a pulverizer, sonicated in RIPA buffer (Sigma, in the ratio 10 volumes of lysis buffer to 1 part by weight of lung tissue) and centrifuged to collect the supernatant. Similarly, cells exposed to HO were lysed in RIPA buffer, sonicated and the supernatants were subjected to protein quantification using the BCA protein assay (Pierce Chemical, Rockford, IL, United States). Samples (20 µg) were subjected to SDS-gel electrophoresis (ThermoFisher, DE, United States) and immunoblotting (#03500216; Hoefer, Fisher Scientific). The following antibodies were used: NOX4 antibody (1:300; 14347-1-AP, Proteintech, IL, United States),
MLE-12 cells at ∼50% confluence were transfected with scRNA (scrambled) or siRNA specific for NOX4 (sc-41587; Santacruz, CA, United States) using Gene Silencer transfection reagent (T500750; Gelantis, San Diego, CA, United States) in serum-free DMEM medium (Sigma) according to manufacturer’s recommendations. After 3 h of transfection, the serum-free medium was replaced with complete MLE-12 medium containing 10% FBS. The cells were cultured for an additional 48h/72 h prior to experiments.
MLE-12 cells were pretreated with 10 µM
Mitochondrial superoxide generation in MLE1-12 cells upon HO exposure and NOX4 silencing was determined using the MitoSOX Red mitochondrial superoxide indicator (Invitrogen, Greenville, NC, United States), as per the manufacturer’s protocol. The cells were loaded with 5 µM MitoSOX reagent for 15 min. The washed cells were subjected to live cell imaging using a confocal microscope (Leica HyVolution SP8) at ×63 magnification.
MLE-12 cells were cultured in phenol red free medium and then exposed to specific treatments. The cell culture supernatants were collected and H2O2 concentration was measured using Amplex Red Hydrogen Peroxide/Peroxidase kit (Invitrogen, United States), according to the manufacturer’s instruction.
Flow cytometry was used to assess apoptosis. Mouse lung epithelial cells subjected to various treatments were stained with Annexin V and 7-AAD (PE Annexin V apoptosis kit, #559763; BD, United States) according to the manufacturer’s instructions. Data was analyzed using a BD LSR II (BD Biosciences, United States) flow cytometer. Gating was set based on unstained and single-stained populations.
All data are expressed as mean ± SEM from a minimum of three independent experiments. GraphPad Prism 9 software was used for statistical analysis. Student’s t-test or two-way ANOVA (Tukey’s post hoc tests) were used for comparison of two or more groups respectively. The level of significance was set to ****p < 0.0001, ***p < 0.001, **p <0.01, and *p < 0.05.
Exposure of wild-type (WT) mice to HO (95% O2) for 66 h stimulated NOX4 protein expression in the lungs (∼1.5-fold increase compared to normoxia, NO) (Figures 1A,B).
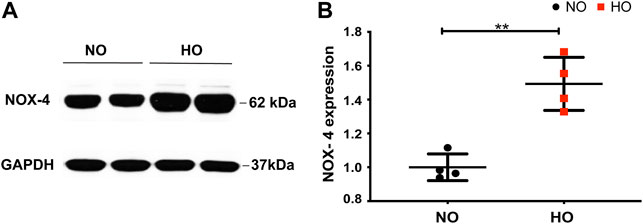
FIGURE 1. HO induces NOX4 expression in lung tissue. To determine the role of NOX4 in HALI, wild type (WT) mice (C57BL/6J male, ∼25 g body weight) were exposed to either NO or HO (95% oxygen) for 66 h. Total lung tissue protein was then subjected to SDS-PAGE followed by immunoblotting with anti-NOX4 antibody. (A) HO significantly enhanced the expression of NOX4 as compared to NO (B) Densitometric quantification of NOX4. The data were normalized to GAPDH. n = 4. **p < 0.01.
Nox4−/−Spc-Cre animals were generated and validated. Immunofluorescence staining of lung tissue sections demonstrated co-localization of NOX4 (green) with SPC (red) in the lung alveolar epithelial cells of Nox4fl/fl mice but not in the tamoxifen induced Nox4−/−Spc-Cre mice (Figure 2A), indicating a significant knockdown of Nox4 as quantified by ImageJ analysis (2722 AU for Nox4fl/fl lung tissue as compared to 932 AU for Nox4−/−Spc-Cre) (Figure 2B). Western blot of lysates with AT2 cells isolated from Nox4−/−Spc-Cre animals showed significant NOX4 knockdown compared to Nox4fl/fl (Figure 2C).

FIGURE 2. NOX4 expression is downregulated in the lungs and isolated AT2 cells from Nox4−/−Spc-Cre mice. Nox+/-Spc-Cre animals were injected with tamoxifen to generate Nox4−/−Spc-Cre animals and IHC was performed on the harvested lungs. (A) Representative immunofluorescent micrographs of lung tissue showing individual staining as well as co-staining of NOX4 and AT2 pneumocyte marker SPC (merge of green and red). Co-localization of NOX4 (green) and SPC (red) could be seen in lungs of Nox4fl/fl mice, but not in Nox4−/−Spc-Cre mice as indicated with white arrows (B) Quantified data of NOX4 fluorescence intensity from NOX4-SPC co-stained lung tissue (C) Expression of NOX4 in primary AT2 cells isolated from Nox4fl/fl and Nox4−/−Spc-Cre mice lung. n = 3 ****p < 0.0001.
The role of NOX4 in HALI was investigated by exposing Nox4fl/fl and Nox4−/−Spc-Cre mice to NO or HO. As shown in Figure 3A, H & E staining of lung tissues harvested from Nox4fl/fl exposed to HO demonstrated significant HALI as determined by increased infiltration of neutrophils, alveolar wall thickening and presence of proteinaceous debris in the alveolar space. ATS scoring was used for quantification of lung injury and showed significantly less injury in the hyperoxic Nox4−/−Spc-Cre mice (0.38 which is 3.5 fold compared to HO) (Figure 3B) compared to HO (1.16 which is ∼10 fold compared to NO). Increased cell count, as well as protein levels in BAL fluid, were also observed with HO exposure (Figures 3C,D). These responses were noted to be significantly lower in the Nox4−/−Spc-Cre mice. A significant elevation of the inflammatory cytokines (Figure 4), (A) KC (∼91 pg/ml in HO against ∼5 pg/ml in NO), (B) TNF-
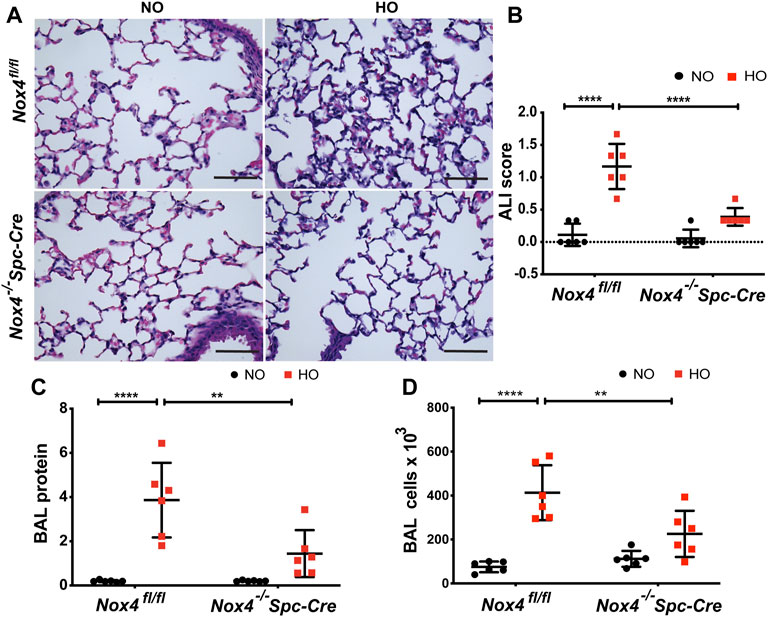
FIGURE 3. Genetic Deletion of Nox4 in AT2 cells of mice reduced HALI. Nox4fl/fl control mice and Nox4−/−Spc-Cre mice induced with tamoxifen and were subjected to NO or HO for 66 h. (A) Representative H&E photomicrographs of lung tissue sections from Nox4fl/fl and Nox4−/−Spc-Cre mice exposed to NO or HO. Quantitative data of (B) ALI scores based on the infiltrating neutrophils, alveolar wall thickening and proteinaceous debris deposition (C) BAL protein and (D) cell counts were elevated in HO exposed control mice and were attenuated by epithelial deletion of Nox4. Scale bar = 100 µm. n = 6. ****p < 0.0001, ***p < 0.001, **p < 0.01 and *p < 0.05.
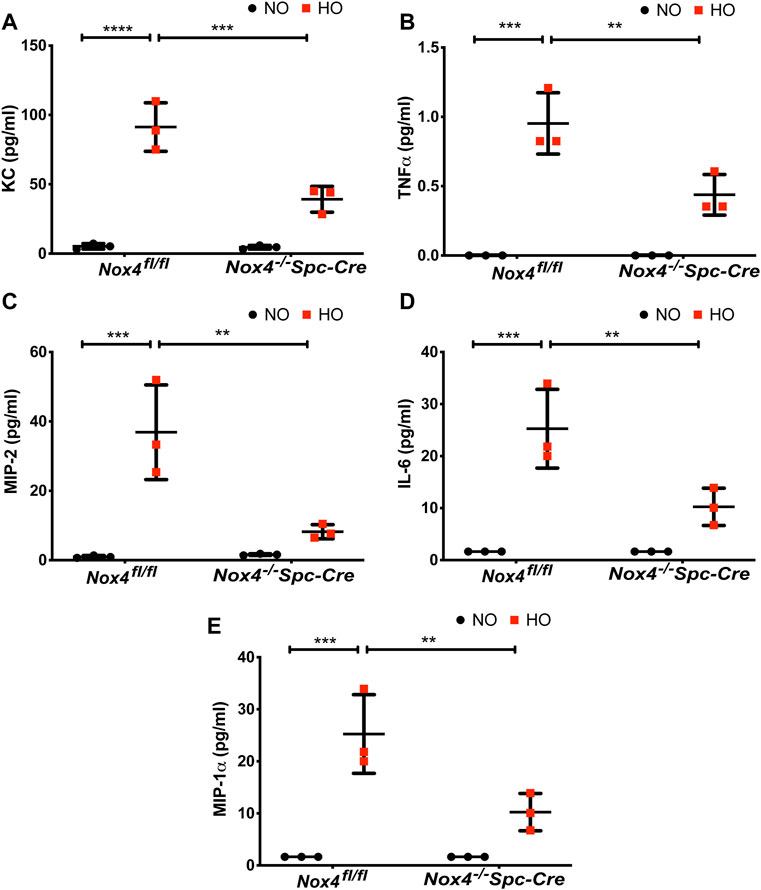
FIGURE 4. Nox4 deletion in AT2 cells of mice reduced inflammatory cytokine profile. Nox4fl/fl control mice and Nox4−/−Spc-Cre mice were subjected to NO or HO for 66 h.The BAL fluid collected were subjected to multiplex assays to determine the cytokine profiles. (A) KC, (B) TNF-
Next, we investigated epithelial cell apoptosis in HALI. TUNEL staining analysis in Nox4fl/fl HO group showed a massive increase in the percentage of TUNEL-positive cells (∼10% in HO as compared to ∼1% in NO) indicating increased AT2 cell death. In contrast, no significant increase in TUNEL-positive cells (∼4% as compared to HO) was observed in Nox4−/−Spc-Cre HO mice (Figure 5A). The differences between the HO control and the HO Nox4−/−Spc-Cre mice were statistically significant (Figure 5B).
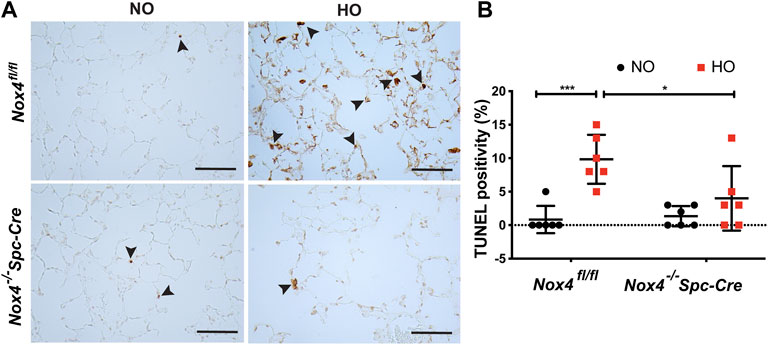
FIGURE 5. Deletion of Nox4 reduced AT2 cell death induced by HALI. (A) Representative immunohistochemical TUNEL assay micrographs of lung tissues from Nox4fl/fl and Nox4−/−Spc-Cre mice exposed to NO or HO (B) The quantified data indicated a lower percentage of TUNEL-positive apoptotic cells in the hyperoxic Nox4−/−Spc-Cre group. n = 6. ***p < 0.001, **p < 0.01 and *p < 0.05.
Hyperoxia resulted in higher baseline respiratory system resistance (Rrs: 0.75 cmH2O.s/ml) and respiratory system elastance (Ers: 35.93 cmH2O/ml) when compared to NO controls. These were reduced significantly to 0.44 cmH2O.s/ml and 25.38 cmH2O/ml in hyperoxic animals by genetic deletion of Nox4 (Figures 6A,B) as compared to control HO group. We also observed significant changes in lung tissue impedance parameters, such as tissue resistance/tissue dampening (G) and tissue elastance (H). The G increased from 3.27 to 6.4 cmH2O/ml which is reduced to 3.36 cmH2O/ml in the Nox4−/−Spc-Cre group. H increased from 25 to 34 cmH2O/ml but remained low in the Nox4 deleted group (∼24 cmH2O/ml) (Figures 6C,D). Compliance is the inverse of elastance, and we noted a decrease in Crs (∼0.03 cmH2O/ml) in the HO group which was significantly increased to ∼0.04 cmH2O/ml in the Nox4−/−Spc-Cre group (Figure 6E).
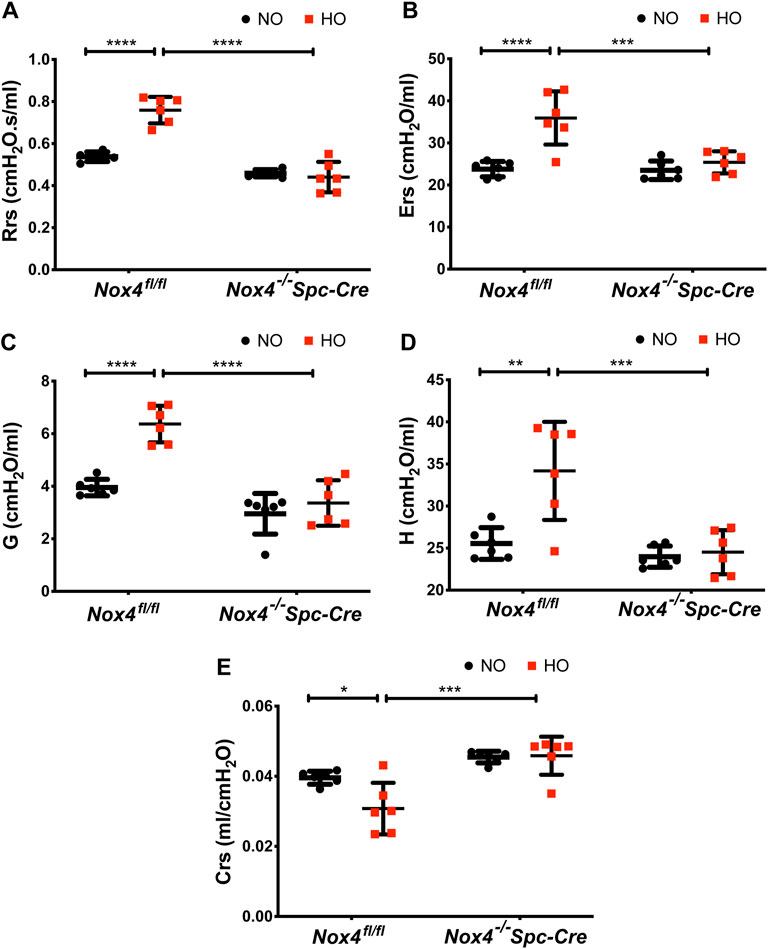
FIGURE 6. Deletion of Nox4 in AT2 cells prevented the sustained lung injury induced by HO. HO caused an increase in baseline (A) respiratory system resistance (Rrs), (B) respiratory system elastance (Ers), (C) tissue dampening/tissue resistance (G), (D) tissue elasticity (H) and a decrease in (E) dynamic compliance (Crs) following HO in control Nox4fl/fl mice as measured by forced oscillatory maneuvers on a flexiVent commercial ventilator. These responses were reduced upon knocking out Nox4. n = 6. ****p < 0.0001, ***p < 0.001, **p < 0.01 and *p < 0.05.
Proteins extracted from primary MLE-12 cells exposed to HO showed increased expression of NOX4. Quantitative analysis by ImageJ showed significant increase in NOX4 expression (∼1.5 fold) (Figures 7A,B).
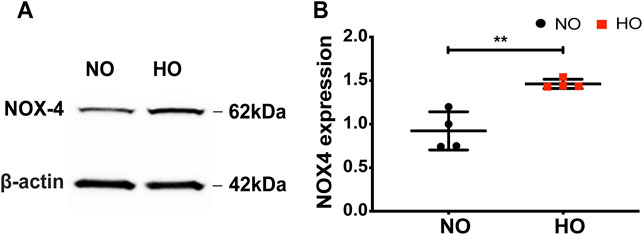
FIGURE 7. HO induces NOX4 expression in lung epithelial cells. (A) MLE-12 cells were exposed to NO or HO and total cell lysates were subjected to Western blotting with anti-NOX4 antibody. Increased expression of NOX4 was observed in the HO group compared to NO (B) Densitometric quantification of NOX4. The data were normalized to GAPDH. n = 4. **p < 0.01 and *p < 0.05.
The role of NOX4 in HO-induced mt ROS was investigated. HO considerably increased MitoSOX fluorescence intensity (∼2 fold more as compared to NO) as well as H2O2 release (∼2.7 µM as compared to 1.2 µM for NO). Silencing NOX4 in MLE-12 cells using NOX4 siRNA for 24 h significantly attenuated HO-induced mitochondrial superoxide production (∼1.2 fold as compared to HO) as determined by MitoSOX (Figures 8A,B) as well as H2O2 release (∼1.6 µM as compared to HO) determined by the Amplex red assay (Figure 8C). Western blot of MLE-12 cells confirmed NOX4 knockdown in the siRNA treated group compared to scRNA (Figure 8D). These results show a role for NOX4 in HO-mediated mt ROS production in lung epithelial cells.
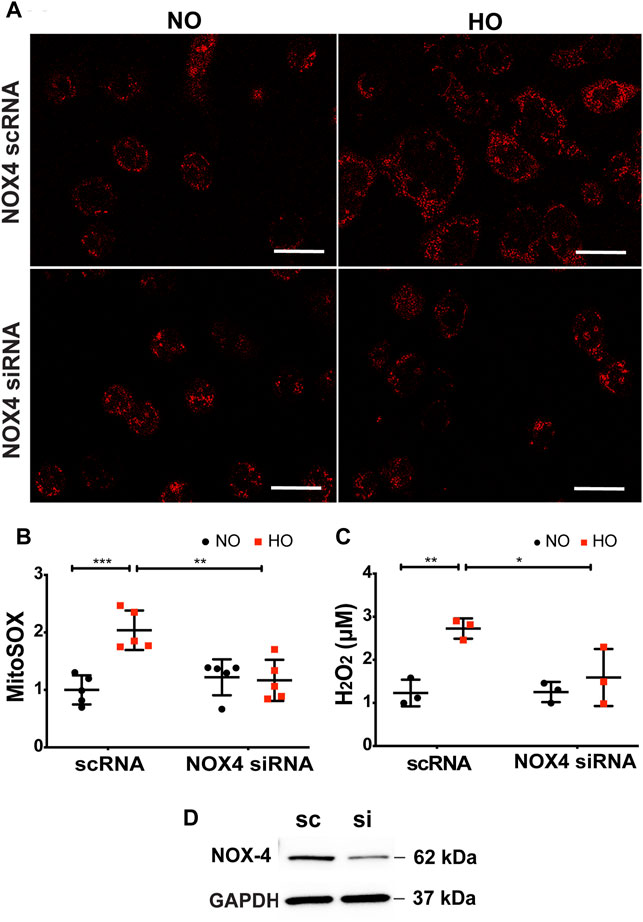
FIGURE 8. Silencing NOX4 reduces HO-induced increase in mt ROS production and apoptosis. (A) Representative confocal images of scRNA and NOX4 siRNA treated MLE-12 cells exposed to NO or HO and stained with MitoSOX (B) Quantitative data of mt ROS intensity measured as arbitrary units showed a decrease in HO-induced mt ROS with inhibition of NOX4 activity (C) Amplex red H2O2 assay of the cell culture supernatant showed increased release of H2O2 from MLE-12 cells exposed to HO compared to NOX4 siRNA treated cell supernatant (D) Immunoblots for NOX4 protein showed a significant reduction in the siRNA treated cells relative to control scRNA cells confirming efficient transfection. n = 3. ***p < 0.001, **p < 0.01, *p < 0.05.
Exposure of MLE-12 cells to HO for 48 h significantly increased the percentage of Annexin V positive early apoptotic cells and this increase was attenuated with NOX4 silencing (Figures 9A,B) suggesting a pathological role for NOX4 in promoting apoptosis. The average percentage fold change was ∼4.5 for HO as compared to NO and this was reduced to ∼2.5 for NOX4 silenced cells.
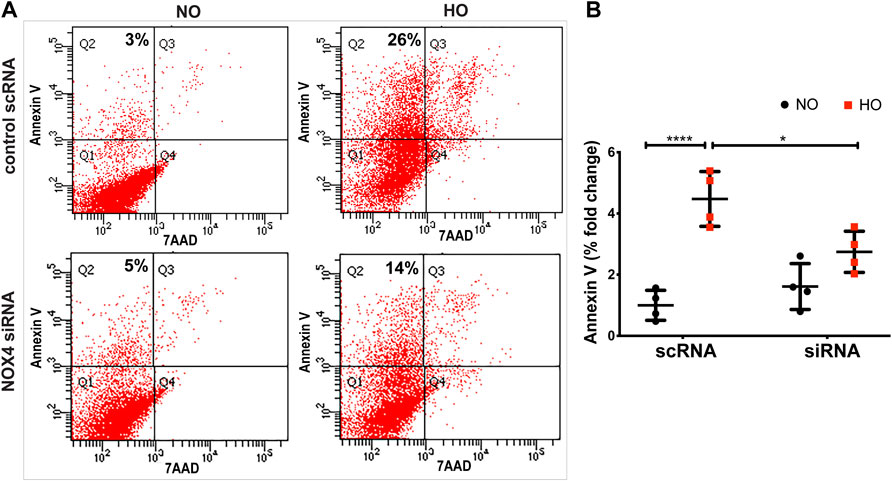
FIGURE 9. NOX4 expression regulates HO-induced apoptosis in lung epithelial cells. MLE-12 cells were transfected with NOX4 siRNA or control scrambled RNA for 24 h before HO exposure and apoptosis was assessed by flow cytometry. (A) Representative dot plots of cells stained with Annexin and 7-AAD (B) Graphs representing the percent fold-change of Annexin V- positive cells showing an increase in percentage fold change of apoptosis in HO group which was reduced in the siRNA treated cells. n = 3. ***p < 0.001, *p < 0.05.
HO induced an increase in mt ROS production as determined by MitoSOX (∼10 AU in HO as compared to ∼5 AU for NO). Scavenging mitochondrial superoxide with MitoTEMPO reduced this HO-induced mitochondrial superoxide production to ∼4.5 AU (Figure 10). Also, the HO-induced increase in the percentage of epithelial cell apoptosis (∼5% fold change compared to NO) was attenuated (∼2.5 fold as compared to HO) by pretreatment with MitoTEMPO (Figure 11).
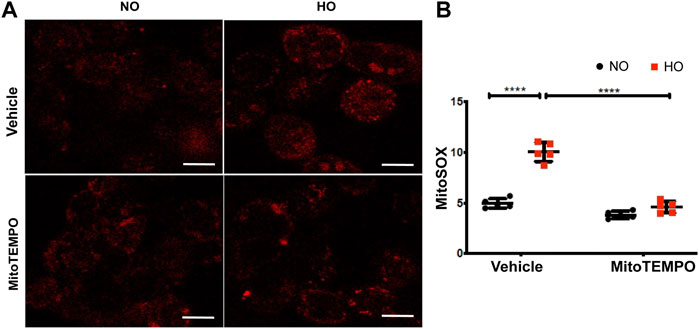
FIGURE 10. HO-induced ROS generation in MLE-12 cells is attenuated by MitoTEMPO. MLE-12 cells were pretreated with vehicle or MitoTEMPO (10 µm) for 1 h and challenged with HO for 48 h. Mitochondrial superoxide production was assayed by MitoSOX red reagent (5 µm) (A) Representative images of MLE-12 cells stained with MitoSOX and (B) Quantified fluorescence intensity measured as arbitrary units showed an increase in MitoSOX staining with HO and decrease with MitoTEMPO treatment. n = 3. ****p < 0.0001.
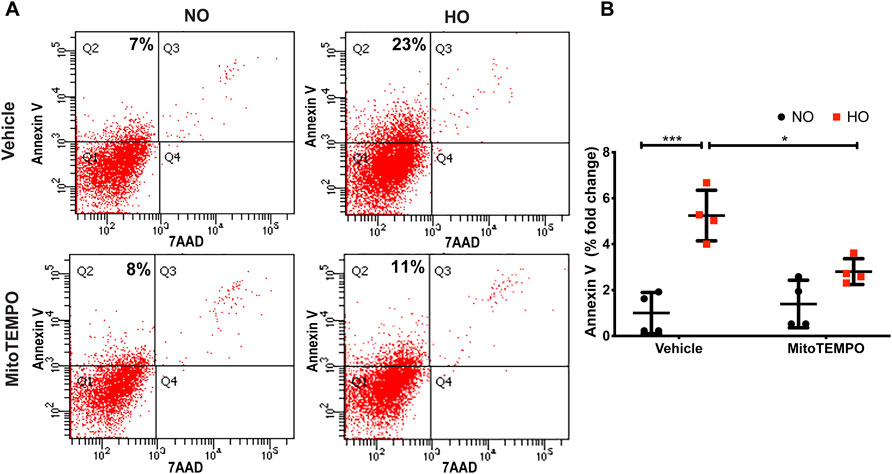
FIGURE 11. MitoTEMPO attenuates HO-induced apoptosis in MLE-12 cells. MLE-12 cells were pretreated with 10uM MitoTEMPO for 1 h followed by HO for 48 h. The cells were stained with Annexin V and 7-AAD and were subjected to flow cytometry. (A) Representative dot plots of cells after staining with Annexin V and 7-AAD (B) Quantification of the percentage of Annexin V- positive apoptotic cells which is expressed as fold-change showing increased apoptotic cells in HO which is significantly reduced with MitoTEMPO treatment. n = 4. ***p < 0.001, *p < 0.05.
Prolonged exposure to hyperoxia (HO) can result in lung tissue damage, which can lead to the development of both acute and chronic lung injury (Northway et al., 1967; Wispe and Roberts, 1987). Despite recent advances, the precise molecular mechanisms by which HO induces lung injury remain unresolved and was the subject of this study. We observed an increase in the expression of NOX4 in WT mice exposed to HO and genetic deletion of Nox4 in lung AT2 epithelial cells (Nox4−/−Spc-Cre) conferred protection against HO-induced HALI.
Reactive oxygen species (ROS) play a significant role in HALI and induces death of different cell types (Buccellato et al., 2004; Bhandari, 2008; Attaye et al., 2017). The pathophysiology of HALI begins with oxidative stress induced destruction of alveolar-capillary barriers followed by an extensive inflammatory response augmented by chemotactic factors (Slutsky, 1999; Bhandari and Elias, 2006). Further, endothelial barrier disruption is followed by death of the Type I pneumocytes. The Type II pneumocytes are more tolerant to HO, though evidence of DNA damage to these cells has been reported (Roper et al., 2004). As several lung cell types are involved in the process of ROS generation, it is imperative to understand the involvement of specific type of cells as well as subcellular organelles in this process. In this study, using cell-specific knockdown of Nox4 in the AT2 cells of the mouse lung, we have defined a novel role for mitochondrial NOX4 in lung AT2 epithelial cell in ROS generation leading to lung epithelial cell apoptosis.
The NOX family, comprising of NOX1–5, are differentially expressed in mammalian cells (Bedard and Krause, 2007; Pendyala and Natarajan, 2010). ROS generation by NADPH oxidases has a context-dependent effect. It has a remarkable role in host defense against invading pathogens in addition to established roles in cellular signaling (Bernard et al., 2017), cell growth (Reddy et al., 2011), cell differentiation and survival (Cucoranu et al., 2005; Hecker et al., 2009). It also contributes to cell death and or cellular senescence (Geiszt et al., 2000). NOX enzymes are involved in the development of various lung diseases. Lung injury induces oxidative stress via NOX proteins (Sadikot et al., 2006; Fu et al., 2013). Lung epithelial cells express NOX2 and NOX4, of which NOX4 is involved in epithelial apoptosis (Carnesecchi et al., 2011). It has been demonstrated in NOX4-deficient mice that ROS generation by NOX4 is crucial for the induction of alveolar epithelial cell death and subsequent development of lung fibrosis (Carnesecchi et al., 2011). It is well known that in viral infections, NOX4 upregulation might be part of the response to the infection leading to protective apoptosis of the infected cell (Grandvaux et al., 2007). However, the role of AT2 NOX4 in regulating HALI is unclear. In this study, we have demonstrated a critical role for AT2 specific NOX4 in promoting oxidative stress and lung injury induced by HO, as AT2 specific knockdown of Nox4 showed protection against pulmonary injury. We found elevated levels of NOX4 in the lungs of HO exposed mice as well as the lung epithelial cells exposed to HO. He et al. showed that NOX4 was elevated in lung macrophages from subjects with asbestosis, and mice harboring a deletion of NOX4 in lung macrophages were protected from asbestos-induced fibrosis (He et al., 2019).
HALI is characterized by an extensive inflammatory response and impaired gas exchange leading to pulmonary edema, respiratory failure and death (Klekamp et al., 1999; Slutsky, 1999). In our study, we noted HALI evidenced by a significant increase in infiltrating neutrophils, alveolar wall thickening and proteinaceous debris deposition in the HO group which was reduced with Nox4 deletion. Elevated BAL proteins, cell counts, and cytokines were observed in HO control mice which was reduced in mice with AT2 specific genetic deletion of Nox4. A similar protection from Pseudomonas aeruginosa induced lung injury was observed upon deletion of Nox4 in lung epithelium. Nox4 deletion reduced ROS generation, histone acetylation, and inflammation induced by the bacteria (Fu et al., 2021). Exposure to HO in Nox4fl/fl mice resulted in significantly higher Rrs, Ers, G, and H with a decreased Crs when compared to NO controls (Figure 6). Conversely, lung mechanics measured in Nox4−/−Spc-Cre mice exposure to HO did not significantly differ from NO Nox4−/−Spc-Cre mice. We also observed significant changes in lung mechanic measurements between the HO exposed Nox4fl/fl mice and HO exposed Nox4−/−Spc-Cre mice, indicating protection from HALI lung function changes when Nox4 was selectively deleted from the AT2 cells.
Studies indicate that multimodal cell death occurs in lung epithelial cells upon exposure to prolonged HO (Kazzaz et al., 1996; Jyonouchi et al., 1998; Mantell et al., 2002). There are several methods to assess epithelial cell apoptosis (Waxman et al., 1998, 11; Buckley et al., 1998; Mantell et al., 2002; Wang et al., 2003; Zhang et al., 2003; Zhang and Foda, 2004). Our study, based on TUNEL assay reports, showed increased apoptosis of AT2 cells in the HO exposed mouse lungs which was reduced with Nox4 deletion. It has been noted that AT2 epithelial cells isolated from HO-exposed rat lungs and cultured cells exhibited apoptotic characteristics (Buckley et al., 1998). Complementary to our results, NOX4 deficiency was demonstrated to decrease bleomycin-induced death of alveolar epithelial cells (Carnesecchi et al., 2011). In another study, ROS released from fibroblasts led to epithelial cell death in pulmonary fibrosis (Waghray et al., 2005). Our results suggest that AT2 NOX4 induced mt ROS and caused epithelial cell death.
We noted that NOX4, under HO, augmented mt ROS production leading to epithelial cell apoptosis. ROS outlines diverse oxidant molecules possessing distinctive biological functions and properties and biological functions (Sies et al., 2022). We focused on H2O2 and O2.-. MitoSOX was used to measure mitochondrial superoxide and amplex red assay the H2O2 levels. The HO-induced surge in mt ROS (superoxide and H2O2) was reduced upon down-regulation of Nox4 with Nox4 siRNA treatment and scavenging of mt ROS by MitoTEMPO (Figures 8, 9). Eventhough these methods are widely used, there are concerns regarding the accuracy of these methods in measuring intracellular ROS (Kalyanaraman, 2020). Novel genetically encoded fluorescent H2O2 probes and superoxide sensors are available to study intracellular ROS (Kalyanaraman, 2020; Mailloux, 2021). NOX4 has been shown to induce mt ROS and modulate mitochondrial biogenesis in cultured macrophages exposed to asbestos and targeting redox signaling prevented the profibrotic polarization of lung macrophages by reducing the production of profibrotic molecules (He et al., 2019). The mechanism by which NOX4-induced mt ROS lead to epithelial cell death is yet to be investigated. NOX-induced mt ROS leading to mitochondrial biogenesis and profibrotic phenotype in macrophages has been demonstrated (He et al., 2019). Suppression of mitochondrial biogenesis and bioenergetics in lung fibroblasts by NOX4 via Nrf2 pathway is also reported (Bernard et al., 2017). NOX4-derived ROS is demonstrated to be a crucial down-stream mediator of TGF-β1-induced alveolar epithelial cell death. NOX4 deficiency and acute inhibition of NOX4 prevented TGF-β1-induced cell death in primary alveolar epithelial cells (Carnesecchi et al., 2011). It is likely that TGF-β1 is not the only factor contributing to NOX4 expression in the bleomycin model. Indeed, NOX4 upregulation may be a part of cellular stress responses, such as endoplasmic reticulum stress (Pedruzzi et al., 2004; Koli et al., 2008). As NOX4 is downstream of TGF-β, inhibition of NOX4 would be expected to be more effective than TGF-β1 inhibition in preventing the development of epithelial cell death and the subsequent injury in the lungs.
Our data demonstrates increased expression of NOX4 in hyperoxic mouse lung tissues as well as mouse lung epithelial (MLE-12) cells suggesting a key role for NOX4 in HALI. Genetic deletion of Nox4 in lung epithelium AT2 cells protected mice from HALI. A mechanistic link between NOX4 and mt ROS leading to epithelial cell death in lungs is established (Figure 12). These results suggest NOX4/ mt ROS as potential therapeutic targets for the treatment of HALI.
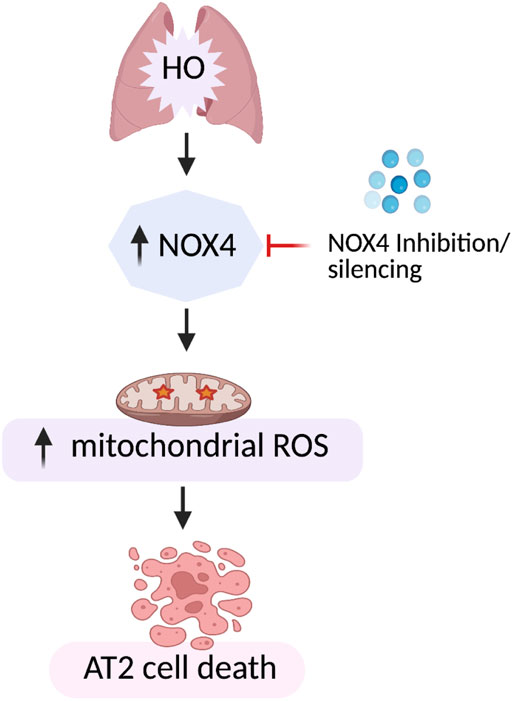
FIGURE 12. Model of HO-induced apoptosis of lung epithelial cells. HO causes an increase in NOX4 expression leading to increased mt ROS production resulting in lung epithelial cell apoptosis. This HO-induced mt ROS production and apoptosis was reduced by silencing NOX4 or mt ROS scavenging by MitoTEMPO.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee (protocol #2020-0052) of the Case Western Reserve University.
Conception and design: AnH, VN, and TS, Investigation: TS, PB, AlH, JT, AJ, and PF, Formal analysis and interpretation of data: AnH, VN, TS, PM, and TR, Writing, review and editing: AnH, VN, TS, PM, and TR, Resources: AnH and VN, Supervision: AnH, VN, and TS. All the authors have read the manuscript and approved its submission.
This work was supported in part by Transitional Grant # 18TPA34230095 from American Heart Association and by R01HD090887-01A1 from Eunice Kennedy Shriver National Institute of Child Health and Human Development to AnH. The funding institutions played no role in the design of the study, conduct of experiments, data collection, analysis, and interpretation, or in writing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The pictorial schema was created using Biorender.
Anilkumar, N., San Jose, G., Sawyer, I., Santos, C. X., Sand, C., Brewer, A. C., et al. (2013). A 28-kDa Splice Variant of NADPH Oxidase-4 Is Nuclear-Localized and Involved in Redox Signaling in Vascular Cells. Arterioscler Thromb. Vasc. Biol. 33, e104. doi:10.1161/ATVBAHA.112.300956
Attaye, I., Smulders, Y. M., de Waard, M. C., Oudemans-van Straaten, H. M., Smit, B., Van Wijhe, M. H., et al. (2017). The Effects of Hyperoxia on Microvascular Endothelial Cell Proliferation and Production of Vaso-Active Substances. Intensive Care Med. Exp. 5, 22. doi:10.1186/s40635-017-0135-4
Auten, R. L., and Davis, J. M. (2009). Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatr. Res. 66, 121–127. doi:10.1203/PDR.0b013e3181a9eafb
Bandela, M., Suryadevara, V., Fu, P., Reddy, S. P., Bikkavilli, K., Huang, L. S., et al. (2021). Role of Lysocardiolipin Acyltransferase in Cigarette Smoke-Induced Lung Epithelial Cell Mitochondrial ROS, Mitochondrial Dynamics, and Apoptosis. Cell Biochem Biophys 80, 203–216. doi:10.1007/s12013-021-01043-3
Bedard, K., and Krause, K. H. (2007). The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 87, 245–313. doi:10.1152/physrev.00044.2005
Bernard, K., Hecker, L., Luckhardt, T. R., Cheng, G., and Thannickal, V. J. (2014). NADPH Oxidases in Lung Health and Disease. Antioxid. Redox Signal. 20, 2838–2853. doi:10.1089/ars.2013.5608
Bernard, K., Logsdon, N. J., Miguel, V., Benavides, G. A., Zhang, J., Carter, A. B., et al. (2017). NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-Derived 2-like 2 (Nrf2)-dependent Pathway. J. Biol. Chem. 292, 3029–3038. doi:10.1074/jbc.M116.752261
Bhandari, V., and Elias, J. A. (2006). Cytokines in Tolerance to Hyperoxia-Induced Injury in the Developing and Adult Lung. Free Radic. Biol. Med. 41, 4–18. doi:10.1016/j.freeradbiomed.2006.01.027
Bhandari, V. (2008). Molecular Mechanisms of Hyperoxia-Induced Acute Lung Injury. Front. Biosci. 13, 6653–6661. doi:10.2741/3179
Brown, D. I., and Griendling, K. K. (2009). Nox Proteins in Signal Transduction. Free Radic. Biol. Med. 47, 1239–1253. doi:10.1016/j.freeradbiomed.2009.07.023
Buccellato, L. J., Tso, M., Akinci, O. I., Chandel, N. S., and Budinger, G. R. (2004). Reactive Oxygen Species Are Required for Hyperoxia-Induced Bax Activation and Cell Death in Alveolar Epithelial Cells. J. Biol. Chem. 279, 6753–6760. doi:10.1074/jbc.M310145200
Buckley, S., Barsky, L., Driscoll, B., Weinberg, K., Anderson, K. D., and Warburton, D. (1998). Apoptosis and DNA Damage in Type 2 Alveolar Epithelial Cells Cultured from Hyperoxic Rats. Am. J. Physiol. 274, L714–L720. doi:10.1152/ajplung.1998.274.5.L714
Carnesecchi, S., Deffert, C., Donati, Y., Basset, O., Hinz, B., Preynat-Seauve, O., et al. (2011). A Key Role for NOX4 in Epithelial Cell Death during Development of Lung Fibrosis. Antioxid. Redox Signal. 15, 607–619. doi:10.1089/ars.2010.3829
Carnesecchi, S., Deffert, C., Pagano, A., Garrido-Urbani, S., Métrailler-Ruchonnet, I., Schäppi, M., et al. (2009). NADPH Oxidase-1 Plays a Crucial Role in Hyperoxia-Induced Acute Lung Injury in Mice. Am. J. Respir. Crit. Care Med. 180, 972–981. doi:10.1164/rccm.200902-0296OC
Chowdhury, A. K., Watkins, T., Parinandi, N. L., Saatian, B., Kleinberg, M. E., Usatyuk, P. V., et al. (2005). Src-mediated Tyrosine Phosphorylation of P47phox in Hyperoxia-Induced Activation of NADPH Oxidase and Generation of Reactive Oxygen Species in Lung Endothelial Cells. J. Biol. Chem. 280, 20700–20711. doi:10.1074/jbc.M411722200
Cucoranu, I., Clempus, R., Dikalova, A., Phelan, P. J., Ariyan, S., Dikalov, S., et al. (2005). NAD(P)H Oxidase 4 Mediates Transforming Growth Factor-Beta1-Induced Differentiation of Cardiac Fibroblasts into Myofibroblasts. Circ. Res. 97, 900–907. doi:10.1161/01.RES.0000187457.24338.3D
Dias-Freitas, F., Metelo-Coimbra, C., and Roncon-Albuquerque, R. (2016). Molecular Mechanisms Underlying Hyperoxia Acute Lung Injury. Respir. Med. 119, 23–28. doi:10.1016/j.rmed.2016.08.010
Fu, P., Mohan, V., Mansoor, S., Tiruppathi, C., Sadikot, R. T., and Natarajan, V. (2013). Role of Nicotinamide Adenine Dinucleotide Phosphate-Reduced Oxidase Proteins in Pseudomonas Aeruginosa-Induced Lung Inflammation and Permeability. Am. J. Respir. Cel Mol Biol 48, 477–488. doi:10.1165/rcmb.2012-0242OC
Fu, P., Ramchandran, R., Sudhadevi, T., Kumar, P. P. K., Krishnan, Y., Liu, Y., et al. (2021). NOX4 Mediates Pseudomonas Aeruginosa-Induced Nuclear Reactive Oxygen Species Generation and Chromatin Remodeling in Lung Epithelium. Antioxidants (Basel) 10, 477. doi:10.3390/antiox10030477
Geiszt, M., Kopp, J. B., Várnai, P., and Leto, T. L. (2000). Identification of Renox, an NAD(P)H Oxidase in Kidney. Proc. Natl. Acad. Sci. U S A. 97, 8010–8014. doi:10.1073/pnas.130135897
Grandvaux, N., Soucy-Faulkner, A., and Fink, K. (2007). Innate Host Defense: Nox and Duox on Phox's Tail. Biochimie 89, 1113–1122. doi:10.1016/j.biochi.2007.04.008
Ha, A. W., Sudhadevi, T., Ebenezer, D. L., Fu, P., Berdyshev, E. V., Ackerman, S. J., et al. (2020). Neonatal Therapy with PF543, a Sphingosine Kinase 1 Inhibitor, Ameliorates Hyperoxia-Induced Airway Remodeling in a Murine Model of Bronchopulmonary Dysplasia. Am. J. Physiol. Lung Cel Mol Physiol 319, L497–L512. doi:10.1152/ajplung.00169.2020
Harijith, A., Pendyala, S., Ebenezer, D. L., Ha, A. W., Fu, P., Wang, Y. T., et al. (2016). Hyperoxia-induced P47phox Activation and ROS Generation Is Mediated through S1P Transporter Spns2, and S1P/S1P1&2 Signaling axis in Lung Endothelium. Am. J. Physiol. Lung Cel Mol Physiol 311, L337–L351. doi:10.1152/ajplung.00447.2015
Harijith, A., Pendyala, S., Reddy, N. M., Bai, T., Usatyuk, P. V., Berdyshev, E., et al. (2013). Sphingosine Kinase 1 Deficiency Confers protection against Hyperoxia-Induced Bronchopulmonary Dysplasia in a Murine Model: Role of S1P Signaling and Nox Proteins. Am. J. Pathol. 183, 1169–1182. doi:10.1016/j.ajpath.2013.06.018
He, C., Larson-Casey, J. L., Davis, D., Hanumanthu, V. S., Longhini, A. L. F., Thannickal, V. J., et al. (2019). NOX4 Modulates Macrophage Phenotype and Mitochondrial Biogenesis in Asbestosis. JCI Insight 4, e126551. doi:10.1172/jci.insight.126551
Hecker, L., Vittal, R., Jones, T., Jagirdar, R., Luckhardt, T. R., Horowitz, J. C., et al. (2009). NADPH Oxidase-4 Mediates Myofibroblast Activation and Fibrogenic Responses to Lung Injury. Nat. Med. 15, 1077–1081. doi:10.1038/nm.2005
Jyonouchi, H., Sun, S., Abiru, T., Chareancholvanich, S., and Ingbar, D. H. (1998). The Effects of Hyperoxic Injury and Antioxidant Vitamins on Death and Proliferation of Human Small Airway Epithelial Cells. Am. J. Respir. Cel Mol Biol 19, 426–436. doi:10.1165/ajrcmb.19.3.2862m
Kallet, R. H., and Matthay, M. A. (2013). Hyperoxic Acute Lung Injury. Respir. Care 58, 123–141. doi:10.4187/respcare.01963
Kalyanaraman, B. (2020). “Pitfalls of Reactive Oxygen Species (ROS) Measurements by Fluorescent Probes and Mitochondrial Superoxide Determination Using MitoSOX,” in Measuring Oxidants and Oxidative Stress in Biological Systems. Editors L. J. Berliner,, and N. L. Parinandi (Cham (CH): Springer). Available at: http://www.ncbi.nlm.nih.gov/books/NBK566438/ (Accessed April 7, 2022). doi:10.1007/978-3-030-47318-1_2
Kazzaz, J. A., Xu, J., Palaia, T. A., Mantell, L., Fein, A. M., and Horowitz, S. (1996). Cellular Oxygen Toxicity. Oxidant Injury without Apoptosis. J. Biol. Chem. 271, 15182–15186. doi:10.1074/jbc.271.25.15182
Kim, J. H., Lee, J., Bae, S. J., Kim, Y., Park, B. J., Choi, J. W., et al. (2017). NADPH Oxidase 4 Is Required for the Generation of Macrophage Migration Inhibitory Factor and Host Defense against Toxoplasma Gondii Infection. Sci. Rep. 7, 6361. doi:10.1038/s41598-017-06610-4
Kim, M. J., Ryu, J. C., Kwon, Y., Lee, S., Bae, Y. S., Yoon, J. H., et al. (2014). Dual Oxidase 2 in Lung Epithelia Is Essential for Hyperoxia-Induced Acute Lung Injury in Mice. Antioxid. Redox Signal. 21, 1803–1818. doi:10.1089/ars.2013.5677
Klekamp, J. G., Jarzecka, K., and Perkett, E. A. (1999). Exposure to Hyperoxia Decreases the Expression of Vascular Endothelial Growth Factor and its Receptors in Adult Rat Lungs. Am. J. Pathol. 154, 823–831. doi:10.1016/S0002-9440(10)65329-1
Koli, K., Myllärniemi, M., Keski-Oja, J., and Kinnula, V. L. (2008). Transforming Growth Factor-Beta Activation in the Lung: Focus on Fibrosis and Reactive Oxygen Species. Antioxid. Redox Signal. 10, 333–342. doi:10.1089/ars.2007.1914
Lee, J. E., Cho, K. E., Lee, K. E., Kim, J., and Bae, Y. S. (2014). Nox4-mediated Cell Signaling Regulates Differentiation and Survival of Neural Crest Stem Cells. Mol. Cell 37, 907–911. doi:10.14348/molcells.2014.0244
Mailloux, R. J. (2021). An Update on Methods and Approaches for Interrogating Mitochondrial Reactive Oxygen Species Production. Redox Biol. 45, 102044. doi:10.1016/j.redox.2021.102044
Manoury, B., Nenan, S., Leclerc, O., Guenon, I., Boichot, E., Planquois, J. M., et al. (2005). The Absence of Reactive Oxygen Species Production Protects Mice against Bleomycin-Induced Pulmonary Fibrosis. Respir. Res. 6, 11. doi:10.1186/1465-9921-6-11
Mantell, L. L., Shaffer, T. H., Horowitz, S., Foust, R., Wolfson, M. R., Cox, C., et al. (2002). Distinct Patterns of Apoptosis in the Lung during Liquid Ventilation Compared with Gas Ventilation. Am. J. Physiol. Lung Cel Mol Physiol 283, L31–L41. doi:10.1152/ajplung.00037.2001
Matsushima, S., Kuroda, J., Ago, T., Zhai, P., Park, J. Y., Xie, L. H., et al. (2013). Increased Oxidative Stress in the Nucleus Caused by Nox4 Mediates Oxidation of HDAC4 and Cardiac Hypertrophy. Circ. Res. 112, 651–663. doi:10.1161/CIRCRESAHA.112.279760
Matute-Bello, G., Downey, G., Moore, B. B., Groshong, S. D., Matthay, M. A., Slutsky, A. S., et al. (2011). An Official American Thoracic Society Workshop Report: Features and Measurements of Experimental Acute Lung Injury in Animals. Am. J. Respir. Cel Mol Biol 44, 725–738. doi:10.1165/rcmb.2009-0210ST
Mittal, M., Siddiqui, M. R., Tran, K., Reddy, S. P., and Malik, A. B. (2014). Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 20, 1126–1167. doi:10.1089/ars.2012.5149
Nisimoto, Y., Diebold, B. A., Cosentino-Gomes, D., Constentino-Gomes, D., and Lambeth, J. D. (2014). Nox4: A Hydrogen Peroxide-Generating Oxygen Sensor. Biochemistry 53, 5111–5120. doi:10.1021/bi500331y
Northway, W. H., Rosan, R. C., and Porter, D. Y. (1967). Pulmonary Disease Following Respirator Therapy of Hyaline-Membrane Disease. Bronchopulmonary Dysplasia. N. Engl. J. Med. 276, 357–368. doi:10.1056/NEJM196702162760701
Olajuyin, A. M., Zhang, X., and Ji, H. L. (2019). Alveolar Type 2 Progenitor Cells for Lung Injury Repair. Cell Death Discov 5, 63–11. doi:10.1038/s41420-019-0147-9
Otterbein, L. E., Kolls, J. K., Mantell, L. L., Cook, J. L., Alam, J., and Choi, A. M. (1999). Exogenous Administration of Heme Oxygenase-1 by Gene Transfer Provides protection against Hyperoxia-Induced Lung Injury. J. Clin. Invest. 103, 1047–1054. doi:10.1172/JCI5342
Paiva, C. N., and Bozza, M. T. (2014). Are Reactive Oxygen Species Always Detrimental to Pathogens? Antioxid. Redox Signal. 20, 1000–1037. doi:10.1089/ars.2013.5447
Panday, A., Sahoo, M. K., Osorio, D., and Batra, S. (2015). NADPH Oxidases: an Overview from Structure to Innate Immunity-Associated Pathologies. Cell Mol Immunol 12, 5–23. doi:10.1038/cmi.2014.89
Parinandi, N. L., Kleinberg, M. A., Usatyuk, P. V., Cummings, R. J., Pennathur, A., Cardounel, A. J., et al. (2003). Hyperoxia-induced NAD(P)H Oxidase Activation and Regulation by MAP Kinases in Human Lung Endothelial Cells. Am. J. Physiol. Lung Cel Mol Physiol 284, L26–L38. doi:10.1152/ajplung.00123.2002
Pedruzzi, E., Guichard, C., Ollivier, V., Driss, F., Fay, M., Prunet, C., et al. (2004). NAD(P)H Oxidase Nox-4 Mediates 7-Ketocholesterol-Induced Endoplasmic Reticulum Stress and Apoptosis in Human Aortic Smooth Muscle Cells. Mol. Cel Biol 24, 10703–10717. doi:10.1128/MCB.24.24.10703-10717.2004
Pendyala, S., Gorshkova, I. A., Usatyuk, P. V., He, D., Pennathur, A., Lambeth, J. D., et al. (2009). Role of Nox4 and Nox2 in Hyperoxia-Induced Reactive Oxygen Species Generation and Migration of Human Lung Endothelial Cells. Antioxid. Redox Signal. 11, 747–764. doi:10.1089/ars.2008.2203
Pendyala, S., Moitra, J., Kalari, S., Kleeberger, S. R., Zhao, Y., Reddy, S. P., et al. (2011). Nrf2 Regulates Hyperoxia-Induced Nox4 Expression in Human Lung Endothelium: Identification of Functional Antioxidant Response Elements on the Nox4 Promoter. Free Radic. Biol. Med. 50, 1749–1759. doi:10.1016/j.freeradbiomed.2011.03.022
Pendyala, S., and Natarajan, V. (2010). Redox Regulation of Nox Proteins. Respir. Physiol. Neurobiol. 174, 265–271. doi:10.1016/j.resp.2010.09.016
Reddy, M. M., Fernandes, M. S., Salgia, R., Levine, R. L., Griffin, J. D., and Sattler, M. (2011). NADPH Oxidases Regulate Cell Growth and Migration in Myeloid Cells Transformed by Oncogenic Tyrosine Kinases. Leukemia 25, 281–289. doi:10.1038/leu.2010.263
Roper, J. M., Mazzatti, D. J., Watkins, R. H., Maniscalco, W. M., Keng, P. C., and O'Reilly, M. A. (2004). In Vivo exposure to Hyperoxia Induces DNA Damage in a Population of Alveolar Type II Epithelial Cells. Am. J. Physiol. Lung Cel Mol Physiol 286, L1045–L1054. doi:10.1152/ajplung.00376.2003
Sadikot, R. T., Zeng, H., Joo, M., Everhart, M. B., Sherrill, T. P., Li, B., et al. (2006). Targeted Immunomodulation of the NF-kappaB Pathway in Airway Epithelium Impacts Host Defense against Pseudomonas aeruginosa. J. Immunol. 176, 4923–4930. doi:10.4049/jimmunol.176.8.4923
Sies, H., Belousov, V. V., Chandel, N. S., Davies, M. J., Jones, D. P., Mann, G. E., et al. (2022). Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cel Biol, 1–17. doi:10.1038/s41580-022-00456-z Epub ahead of print.
Slutsky, A. S. (1999). Lung Injury Caused by Mechanical Ventilation. Chest 116, 9S–15S. doi:10.1378/chest.116.suppl_1.9s-a
Sudhadevi, T., Jafri, A., Ha, A. W., Basa, P., Thomas, J. M., Fu, P., et al. (2021). Hyperoxia-induced S1P1 Signaling Reduced Angiogenesis by Suppression of TIE-2 Leading to Experimental Bronchopulmonary Dysplasia. Cel Biochem Biophys 79, 561–573. doi:10.1007/s12013-021-01014-8
Takac, I., Schröder, K., Zhang, L., Lardy, B., Anilkumar, N., Lambeth, J. D., et al. (2011). The E-Loop Is Involved in Hydrogen Peroxide Formation by the NADPH Oxidase Nox4. J. Biol. Chem. 286, 13304–13313. doi:10.1074/jbc.M110.192138
Tarafdar, A., and Pula, G. (2018). The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 19, 3824. doi:10.3390/ijms19123824
Usatyuk, P. V., Romer, L. H., He, D., Parinandi, N. L., Kleinberg, M. E., Zhan, S., et al. (2007). Regulation of Hyperoxia-Induced NADPH Oxidase Activation in Human Lung Endothelial Cells by the Actin Cytoskeleton and Cortactin. J. Biol. Chem. 282, 23284–23295. doi:10.1074/jbc.M700535200
Waghray, M., Cui, Z., Horowitz, J. C., Subramanian, I. M., Martinez, F. J., Toews, G. B., et al. (2005). Hydrogen Peroxide Is a Diffusible Paracrine Signal for the Induction of Epithelial Cell Death by Activated Myofibroblasts. FASEB J. 19, 854–856. doi:10.1096/fj.04-2882fje
Wang, X., Ryter, S. W., Dai, C., Tang, Z. L., Watkins, S. C., Yin, X. M., et al. (2003). Necrotic Cell Death in Response to Oxidant Stress Involves the Activation of the Apoptogenic Caspase-8/bid Pathway. J. Biol. Chem. 278, 29184–29191. doi:10.1074/jbc.M301624200
Waxman, A. B., Einarsson, O., Seres, T., Knickelbein, R. G., Warshaw, J. B., Johnston, R., et al. (1998). Targeted Lung Expression of Interleukin-11 Enhances Murine Tolerance of 100% Oxygen and Diminishes Hyperoxia-Induced DNA Fragmentation. J. Clin. Invest. 101, 1970–1982. doi:10.1172/JCI1337
Welty-Wolf, K. E., Simonson, S. G., Huang, Y. C., Kantrow, S. P., Carraway, M. S., Chang, L. Y., et al. (1997). Aerosolized Manganese SOD Decreases Hyperoxic Pulmonary Injury in Primates. II. Morphometric Analysis. J. Appl. Physiol. (1985) 83, 559–568. doi:10.1152/jappl.1997.83.2.559
Wispe, J. R., and Roberts, R. J. (1987). Molecular Basis of Pulmonary Oxygen Toxicity. Clin. Perinatol 14, 651–666. doi:10.1016/s0095-5108(18)30755-3
Zhang, X., Shan, P., Sasidhar, M., Chupp, G. L., Flavell, R. A., Choi, A. M., et al. (2003). Reactive Oxygen Species and Extracellular Signal-Regulated Kinase 1/2 Mitogen-Activated Protein Kinase Mediate Hyperoxia-Induced Cell Death in Lung Epithelium. Am. J. Respir. Cel Mol Biol 28, 305–315. doi:10.1165/rcmb.2002-0156OC
Keywords: NADPH oxidase 4, mitochondrial ROS, alveolar type 2 epithelial cell, apoptosis, hyperoxic acute lung injury
Citation: Harijith A, Basa P, Ha A, Thomas J, Jafri A, Fu P, MacFarlane PM, Raffay TM, Natarajan V and Sudhadevi T (2022) NOX4 Mediates Epithelial Cell Death in Hyperoxic Acute Lung Injury Through Mitochondrial Reactive Oxygen Species. Front. Pharmacol. 13:880878. doi: 10.3389/fphar.2022.880878
Received: 22 February 2022; Accepted: 18 April 2022;
Published: 19 May 2022.
Edited by:
Bhagavatula Moorthy, Baylor College of Medicine, United StatesReviewed by:
Stephanie Moore, University of Alabama at Birmingham, United StatesCopyright © 2022 Harijith, Basa, Ha, Thomas, Jafri, Fu, MacFarlane, Raffay, Natarajan and Sudhadevi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tara Sudhadevi, dHhzNjcxQGNhc2UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.