 Xiangning Cui
Xiangning Cui Yang Zhang
Yang Zhang Yingdong Lu
Yingdong Lu Mi Xiang
Mi Xiang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 26 April 2022
Sec. Respiratory Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.879204
This article is part of the Research Topic Oxidative Stress Related to Cellular Metabolism in Lung Health and Diseases View all 10 articles
Pulmonary diseases are main causes of morbidity and mortality worldwide. Current studies show that though specific pulmonary diseases and correlative lung-metabolic deviance own unique pathophysiology and clinical manifestations, they always tend to exhibit common characteristics including reactive oxygen species (ROS) signaling and disruptions of proteostasis bringing about accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER). ER is generated by the unfolded protein response. When the adaptive unfolded protein response (UPR) fails to preserve ER homeostasis, a maladaptive or terminal UPR is engaged, leading to the disruption of ER integrity and to apoptosis, which is called ER stress. The ER stress mainly includes the accumulation of misfolded and unfolded proteins in lumen and the disorder of Ca2+ balance. ROS mediates several critical aspects of the ER stress response. We summarize the latest advances in of the UPR and ER stress in the pathogenesis of pulmonary disease and discuss potential therapeutic strategies aimed at restoring ER proteostasis in pulmonary disease.
The endoplasmic reticulum (ER) is a membrane organelle widely found. In eukaryotic cells with multiple functions. The ER coordinates secretory and transmembrane proteins folding and translocation, serves as a place to regulate the cellular stress response (Chen and Cubillos-Ruiz, 2021). The ER controls cellular Ca2+ uptake, storage and signaling, keeping the intracellular Ca2+ homeostasis. Meanwhile, the ER takes part in the generation of phospholipids including cholesterol (Sozen and Ozer, 2017). The ER lumen is the main site where proteins synthesize in cells. The accumulation of some misfold or unfolded proteins within the ER or the disruption of Ca2+ homeostasis can lead to the disequilibrium of ER homeostasis, which is called ER stress (Di Conza and Ho, 2020). The stability of the environment within the ER is the guarantee of regulating its normal function. In the early stage of ER stress, the induction of unfolded protein response (UPR) can reduce protein synthesis and enhance the degradation function of ER, thus lowering the burden of ER and maintaining intracellular homeostasis, which is a pro-survival response. With the aggravation of ER, the activation of UPR is inhibited, resulting in Ca2+ uptake/release barrier of ER or protein processing/transporting obstacle (Song et al., 2018). The signaling is accordingly transformed from survival to apoptosis, and the expression of ER stress-specific pro-apoptotic transcription factor CHOP is activated, further inhibiting the activity of AKT, and promoting cell apoptosis (Sen, 2019). The normal physiological function of ER is closely related to the redox state, and increasing studies suggest the production of ROS has become a primary part of ER stress response, possibly acting as an upstream signaling (Zhang et al., 2019a). The lung is in direct contact with the outside world, and external stimuli can easily lead to mis-synthetic proteins and cause the occurrence of pulmonary diseases including pulmonary fibrosis, pulmonary infections, asthma, COPD (chronic obstructive pulmonary disease) and lung cancer (Liu et al., 2017; Katzen and Beers, 2020). Several pathophysiological factors such as hypoxia, inflammation, and metabolic disorders can trigger ER stress. Conversely, ER stress induces inflammation and oxidative stress, maladaptive UPR induces apoptosis, and disruption of interaction between the ER and other organelles in epithelial cells can negatively affect pulmonary disease. In this context, we introduced the relationship between ROS and ER stress, summarized current research on the molecular mechanisms of ER stress in pulmonary disease, and raised several promising therapeutic strategies targeting ER stress that might prevent or treat pulmonary disease.
ROS are oxygen radicals in organisms, including oxygen and oxygen-containing highly reactive molecules (such as superoxide anions, tissue peroxides, and free radicals), serving as an intracellular and inter-cellular messenger which regulates many signaling molecules (Incalza et al., 2018). ROS are generated as a byproduct in cells by mitochondria and other cellular organelles (Boengler et al., 2017). ROS modulate different cell signaling pathways, which are mostly mediated by these transcription factors NF-κB and STAT3, hypoxia-inducible factor-1α, kinases, cytokines, and other proteins; these signaling pathways have been demonstrated to be related with inflammation, proliferation, cellular transformation, angiogenesis, as well as cell metabolism in cancer cells (Prasad et al., 2017). Continuous studies have confirmed ROS production in hypoxia and ischemia models, and found that ER proteins were oxidized and modified, suggesting that ER functions can be destroyed by ROS (Xu et al., 2017; Wang et al., 2018a; Singh-Mallah et al., 2019). ROS production on ER is generated by delivering electrons to O2 by NADH-cytochrome P450 reductase to form O2−, with electrons delivered to O2 by the electron transport chain on the nuclear membrane, assisted by NADH (Delaunay-Moisan and Appenzeller-Herzog, 2015; Zeeshan et al., 2016). Changes of intracellular redox state promotes the production of oxygen radicals and apoptosis inducer activation, resulting in apoptosis and aggravate intracellular redox state variation, then affect cell physiological state, initiate oxidative stress, and trigger the activation of apoptotic signal activation. Furtherly, calcium imbalance, oxidative stress are also important factors to induce ER stress (Kaminskyy and Zhivotovsky, 2014; Prasad et al., 2017).
The ER is a multifunctional organelle that forms the access into the secretory pathway which is made up of smooth and rough endoplasmic reticulum. The ER is crucial in maintaining cellular calcium homeostasis, lipid biosynthesis and secretory, and transmembrane protein folding and translocation (Rowland and Voeltz, 2012; Schwarz and Blower, 2016).The oxidative environment of ER lumen promotes disulfide bond formation, allowing complex secretion and transmembrane protein folding. The process of protein folding is regulated by a variety of chaperones, folding enzymes and cofactors (Kim et al., 2013a; Saibil, 2013). The ER takes part in lipids production in cells such as cholesterol and glycerophospholipids regulated by multiple ER-localized lipid biosynthetic enzymes including GTPase SAR1B and SURF4 (Wang et al., 2021). When cholesterol levels in the ER turn too high, SREBP undergo conformational changes that hinder it from leaving the ER. Correspondingly, homeostasis of cholesterol is restored (Brown et al., 2018; Lim et al., 2019). The ER also participates in mediating calcium homeostasis in cells. In quiescent cells, the level of free Ca2+ in cytoplasm was lower than that in extracellular space and ER lumen. Disruption of intracellular Ca2+ homeostasis is related with inflammatory responses. Research suggest that increase in IP3R (inositol 1,4,5-trisphosphate receptor) mediated Ca2+ release can cause inflammatory pathophysiology of ventilator-induced lung injury in mice models via ER stress (Ye et al., 2021). During chaperone-assisted disulfide bond formation between polypeptide chain substrates, two electrons are provided to the cysteine residue within the PDI active site. This transfer of electrons results in the reduction of the PDI active site and oxidation of the substrate, suggesting main ER-originating ROS production process. Studies have confirmed that the ER membrane-associated protein ERO1 (Endoplasmic reticulum oxidoreductin-1) oxidizes PDI, indicating that ERO1 is closely associated with protein load in the ER and can trigger ROS generation and contribute to ER stress. In addition, experimental analysis revealed that UP to 25% of ROS is produced by disulfide bonds in the endoplasmic reticulum during mass oxidation. Therefore, the close relationship between ROS and endoplasmic reticulum homeostasis is further suggested.
As is illustrated above, accumulation of some misfolded or unfolded proteins within the ER or disruption of Ca2+ homeostasis can cause ER stress. Under normal circumstance, the stability of the environment within ER is the guarantee for the normal function of ER. In the early stage, ER stress reduces protein biosynthesis and enhances ER degradation function by inducing unfolded protein response (UPR), thus reducing ER burden, and maintaining intracellular homeostasis, which in essence is a protective means. Changes of intracellular redox state initiate oxidative stress and trigger the activation of apoptosis signals. Meanwhile, Ca2+ imbalance and oxidative stress are also important factors to induce ER stress.
ER is an important site for secretory protein folding and various post-translational modifications such as glycosylation and disulfide bond formation. The homeostasis regulation of ER ensures that correctly folded proteins proceed to the next step of modification, while slowly folded or unfolded proteins are retained in the ER for proteasome degradation through ER related protein degradation, which is the unfolded protein response (UPR).
Under physiological conditions, three independent branches regulate the UPR, mediated by ER stress receptor proteins, PERK (RNA-dependent protein kinase (PKR)-like ER kinase), ATF6 (activating transcription factor 6), and IRE1α (Inositol-requiring enzyme-1α). These three individual proteins respectively bind to ER chaperone GRP78 (glucose-regulated protein 78) and remain inactive and serve as sensors and effectors for enhanced ER stress (Oakes and Papa, 2015; Kopp et al., 2019). When the nucleotide-binding domain of GRP78 coordinates with luminal domains of IRE1α and PERK, GRP78 is converted from a molecular chaperone to an ER stress sensor, accelerating further UPR activation (Kopp et al., 2019). These mechanisms can monitor the deviant conditions on the ER lumen, deliver signals to the cytosol, then transfer them the nucleus and activate corresponding downstream responses (Figure 1) (Schwarz and Blower, 2016).
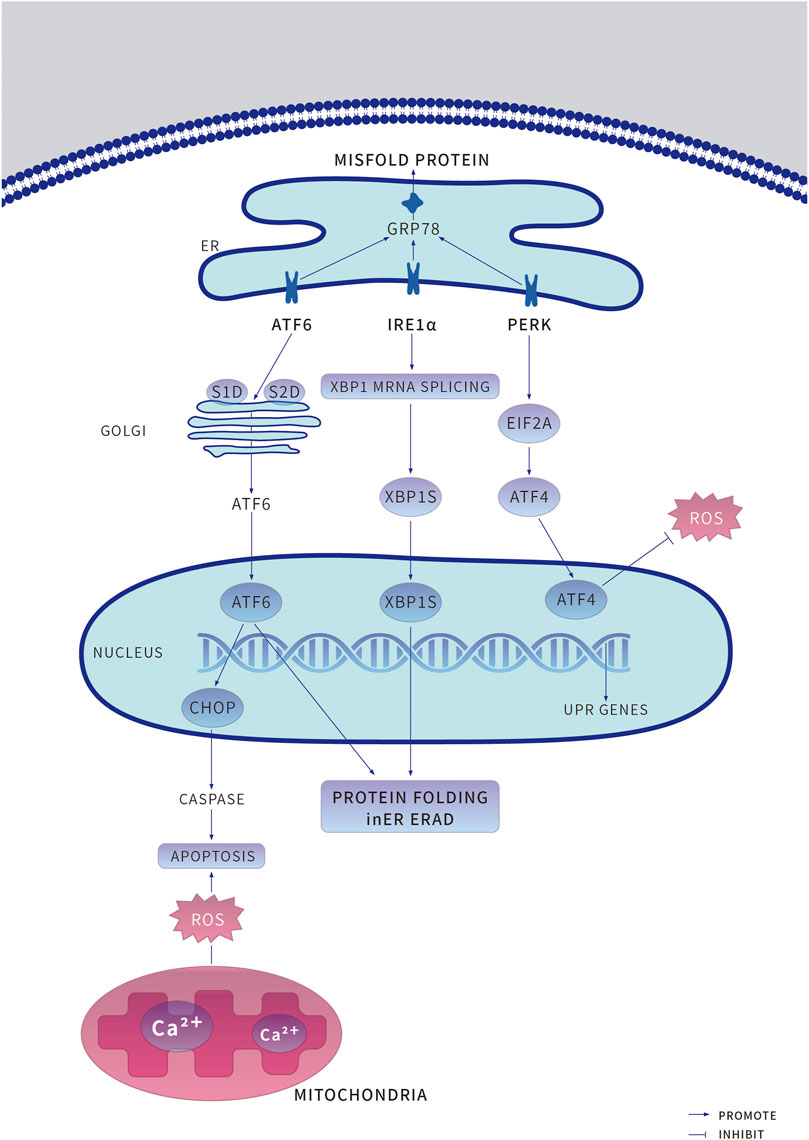
FIGURE 1. Under endoplasmic reticulum (ER) stress, the adaptive unfolded protein response (UPR) is induced to preserve protein homeostasis and to promote cell survival. An increased accumulation of unfolded or misfolded proteins causes the ER chaperone binding immunoglobulin protein (GRP78) to bind to misfolded proteins accumulated in the ER, resulting in the release of the UPR sensors protein kinase RNA-like ER kinase (PERK), Transcriptional transcription factor 6 (ATF6) and inositol-requiring protein 1α (IRE1α) and initiation of the respective proteins dimerization and phosphorylation of PERK and IRE1α activate their downstream pathways, PERK -- phosphorylated Eukaryotic Initiation factor 2α. Eukaryotic initiation factor 2A (EIF2A) -- ATF4 and IRE1α -- Spliced X-box-binding protein 1 (XBP1s), to promote cell survival. ATF6 is recruited to the Golgi apparatus for processing by the enzymes site-1 proteases (S1P) and site-1 proteases (S2P) to release cleaved ATF6, which enters the nucleus to induce the expression of target genes to promote cell survival. ERAD, ER-associated degradation; ROS, reactive oxygen species.
PERK, a crucial ER stress sensor of the UPR, is particularly enriched at the MAMs (mitochondria-associated ER membranes), and capable of preventing mRNA translation under ER stress, thus inhibiting protein synthesis, and folding (Verfaillie et al., 2012). ATF4 (activating transcription factor 4) is a stress-induced transcription factor which is at always upregulated in cells, controls the expression of several adaptive genes that protect cells from stress, such as hypoxia or amino acid limitation. But under sustained stress conditions, ATF4 can initiate the transcription of CHOP (C/EBP homologous protein) to induce cell apoptosis (Wortel et al., 2017). PERK phosphorylates eIF2α (eukaryotic initiation factor 2) to decrease the GTP-bound (guanosine triphosphate, GTP) form and allow translation of ATF4 (Han et al., 2013; Luhr et al., 2019; Vanhoutte et al., 2021). Meanwhile, CHOP can bind to BOK (B-cell lymphoma 2 varian killer) and Caspase to act as an antiapoptotic agent (Carpio et al., 2015).
ATF6 is a type II transmembrane protein that induces ER stress response. As an up-regulating chaperone, it is an important element of the ERAD (endoplasmic reticulum—associated degradation) pathway (Jin et al., 2017). The unfolded proteins in the ER lumen separate ATF6 from GRP78, then ATF6 is transferred from the ER membrane to the Golgi apparatus as vesicles; the lumen domain of ATF6 is cleaved by S1P (serine protease site-1), the n-terminal is cleaved by metalloproteinase S2P (site-2 protease), forming the ATF6f with n-terminal cytoplasmic domain. As a result, several ER stress response element genes such as GRP78, GRP94, CHOP can be activated, and promote the ER associated protein degradation gene XBP1 (X-box binding protein 1) transcription. The ATF6f can bind XBP1 and control the expression of some specific genes (Zhang et al., 2019b; Pachikov et al., 2021).
IRE1α is a transmembrane protein, which is composed of n-terminal ER lumen sensor domain, a single transmembrane domain, and c-terminal cytoplasmic protein kinase effector, responsible for protein kinase and endonuclease activity. When ER stress happens, IRE1α cavity structure domain is dissociative with GRP78, causing cavity structure domain two polymerization to further activate the activity of enzymes, catalyze the transcription of XBP1 to upregulate multiple folding enzymes, including glycosylation, redox enzymes degradation to rectify ER homeostasis. IRE1α also degrades specific mRNA in a tissue-specific manner through RIDD (regulated IRE1-dependent mRNA decay) and activates related kinases through binding of adaptor proteins including JNK (C-Jun amino-terminal kinase) and ASK1 (apoptotic signal-regulated kinase 1) pathways (Lerner et al., 2012; Hwang and Qi, 2018; Huang et al., 2019).
Adaptive UPR is the initial defense against ER stress to keep the balance of ER homeostasis. However, persistent UPR activation can transform into maladaptive UPR, leading to drastic cell injuries even cell death. Incorrectly or terminally misfolded proteins are retained and degraded within the ER. As ER stress cells evolve further with the integrated stress response, cells actively mobilize stress response proteins to resist the deleterious effects caused by stress triggers. Meanwhile, cells adjust ER function to adapt to new internal environmental change requirements. On the other hand, some stress protein regulatory genes that may cause cell death, such as CHOP, will decide according to the situation to finally clear those ER stress cells that are simply unable to return to a normal functional state (Shirakawa et al., 2013; Hotamisligil and Davis, 2016; Urra et al., 2016; Hetz and Papa, 2018). In ER stress response, the activation of IRE-1, PERK, and ATF6 induces CHOP, promotes the activation of CHOP, and significantly increases its expression, leading to apoptosis. IRE-1-mediated splicing of XBP1 induced UPR can promote cell survival, but its overexpression can also promote cell apoptosis. Activated IRE-1 can combine with TRAF2 (TNF-receptor-associated factor 2) and ASK1 (apoptotic signal-regulated kinase 1) to form into the IRE-1-TRAF2-ASK1 complex on ER outer membrane, which activates JNK and P38 MAP (mitogen-activated protein kinase) (Matsuzawa et al., 2002). CHOP expression is induced by phosphorylation of ser78/81 in the transcriptional activation domain of P38 MAP kinase. After ER stress induced GRP78 dissociation from ATF6, the cytoplasmic ATF6 n-terminal domain also dissociated from ER membrane and formed a 50 ku active fragment. It was found that the activated ATF6 entered the nucleus and bound to the NF-Y (nuclear transcription factor-Y) and bound to the promoter ER stress element with homologous or heterologous dimer structure. The complex then induces the transcriptional expression of ER stress genes including CHOP (Feng et al., 2019). PERK is also a transmembrane protein. After dissociation from GRP78, PERK is activated by self-dimerization and phosphorylation of the intracellular domain. Activated PERK phosphorylates serine of the eukaryotic translation initiation factor eIF2α. Phosphorylated eIF2α cannot accept the exchange of GTP-GDP by eIFβ, thus slowing or suspending protein synthesis, thereby promoting the activation of CHOP, and thus inducing cell metabolism and apoptosis (Pillai, 2005; Kopp et al., 2019).
ROS can directly attack the free thionyl group, necessary to maintain the protein foldase activity, leading to oxidative modification of proteins in the ER lumen, and then induce abnormal function of ER molecular chaperones, resulting in the accumulation of unfold proteins and retention in the ER lumen, ultimately triggering ER stress. At the same time, the expression of the chaperone protein GRP78 significantly increased and dissociated from the three transmembrane proteins to bind unfolded proteins to enhance ER protein folding ability and reduce protein accumulation in ER. After dissociation of GRP78, ER stress receptor proteins are activated to trigger UPR generation, reduce the load of ER by limiting the synthesis of unfold proteins, and trigger their respective induced pro-survival response. The PERK-activated-pro-survival pathway inhibits protein translation and synthesis by limiting the entry of abnormally folded proteins into ER and reducing ER’s pressure for new protein folding (Borges and Lake, 2014; Hetz et al., 2015; Khanna et al., 2021). PDI (phosphorylation of protein disulfide isomerases), fundamental enzymes involved in ER protein folding, allows these proteins to combine with luminal IRE1α, then moderating excessive IRE1α activity to protect against cell injuries induced by maladaptive UPR (Zhang et al., 2019a). PDI is a kind of oxidoreductase which can catalyze disulfide bonds and it is the correct sequence of cysteine residue pairs with different redox potentials and substrate specificity (Ellgaard and Ruddock, 2005). Cysteine residues within the active site of PDI provide two electrons to assist disulfide bond formation, which results in reduction of the active site of PDI and oxidation of the substrate (Fu et al., 2020). The formation of disulfide bond in the ER is catalyzed by ERO1 (endoplasmic reticulum oxidoreductin 1) family of sulfhydryl oxidases. ERO1 oxidizes PDI and introduces disulfides into ER client proteins. ERO1 can couple disulfide transfer to PDI with reduction of molecular oxygen, forming hydrogen peroxide. Hence, ERO1 activity is a underlying source of ER-derived oxidative stress (Tavender and Bulleid, 2010). Research has found that ROS inhibitor MnTMPyP can inhibit the increase of ROS in ER stress and delay the activation of UPR, suggesting that ROS is the upstream signal molecule that triggers ERS-mediated apoptosis pathway and initiates ER stress-mediated apoptosis (Xu et al., 2018).
UPR as a protective means at the cellular level reduces the accumulation of unfolded or misfolded proteins on the ER. In the early stage of ER stress, ER mainly restore the normal function of ER for survival through activation of UPR to protect cellular damage caused by ER stress, which is a pro-survival response (Senft and Ronai, 2015; Frakes and Dillin, 2017).
The endoplasmic reticulum is an important calcium reservoir, which regulates ER function, membrane transport and internal homeostasis through maintaining the balance of Ca2+ concentration inside and outside the cell. As a messenger molecule, Ca2+ participates in cell proliferation, differentiation, movement, muscle contraction, hormone secretion, glycogen metabolism, neuronal excitability, and other physiological activities, and then regulates gene and protein expression, secretion, metabolism, and apoptosis (Frakes and Dillin, 2017). Current studies have shown that disruption of calcium homeostasis in the ER can initiate early apoptosis signals (Rizzuto et al., 2012; Orrenius et al., 2015; Marchi et al., 2018). A study revealed that sodium nitrite (anti-tumor drug) can induce ER stress-mediated apoptosis in human NB4 cells in acute leukemia, certified selenite-induced production of ROS is the upstream of the JNK/ATF2 axis, application of ROS inhibitors partially inhibited the intracellular Ca2+ increase caused by sodium nitrite, suggesting that ROS is involved in the sodium nitrite-induced ERS-mediated apoptosis (An et al., 2013). Aoyama elucidated that rotenone, a mitochondrial complex I inhibitor, can cause ER hyperplasia, ER membrane lipid oxidative damage, increase the permeability of ER, cause the increase of cytosol-free Ca2+ and then induce ER stress and apoptosis. After using the ER stress inhibitor SUN N8075, mitochondrial ROS production obviously reduced and Ca2+ elevation induced by rotenone was significantly weakened, indicating the increase of intracellular free Ca2+ induced by rotenone is stimulated by ROS (Aoyama et al., 2021). Although some specific mechanisms are still being studied, ROS production and the increase of ER Ca2+ level are both crucial events in ER stress-mediated apoptosis.
At resting state, the concentration of Ca2+ in the ER is higher than that in the cytoplasm. There are three channels related with Ca2+ uptake and release for maintaining Ca2+ homeostasis on the ER: 1. Ca2+-release channels: RyRs (ryanodine receptors) and IP3Rs; 2. Ca2+ -ATPase or SERCA (sarcoplasmic-endoplasmic-reticulum Ca2+-ATPase) in transporting Ca2+ from cytoplasm to ER; 3. Ca2+-binding protein in ER cavity (such as calnexin and calreticulin) (Ong and Ambudkar, 2020). Studies have confirmed that the IP3R and RyR distributed on the sarcoplasmic reticulum can be regulated by the redox response (Ong and Ambudkar, 2020). ER includes the NOX subtypes of NADPH oxidase and co-exists with calnexin and calreticulin. The NOX subtype contains the main catalytic subunit GP91Phox (NOX2), which produces ROS mainly involved in cell signaling transduction. Recent studies suggest that NOX1 and NOX4 (NADPH Oxidase 4) can act on PDI, that is, ROS outside the mitochondria may also regulate ER function. Moreover, research has demonstrated NOX4 can act as a proximal signaling intermediate to transduce ER stress-related conditions to the UPR, and sulfonate and modify it through interaction with IRE1 to cause ER stress even cell injuries (Kim et al., 2016; Ochoa et al., 2018; Lee et al., 2020).
In the phosphatidylinositol signaling pathway, extracellular signaling molecules bind to the G-protein-coupled receptor on the cell surface to activate phospholipase C on the plasma membrane and hydrolyze PIP2 (4, 5-diphosphate phosphatidylinositol) into 2 s messengers, IP3 (1, 4, 5-triphosphate inositol) and DG (diacylglycerol). Extracellular signals are converted into intracellular signals, and this signaling system is also known as the double messenger system (Kankanamge et al., 2021). IP3R is a widely expressed channel for Ca2+ stores. IP3R can control the release of Ca2+ from stores into cytoplasm once being activated by IP3 and Ca2+ signaling at a lower concentration, and finally trigger downstream events. The closure of the IP3R channel caused by a rise in intracellular Ca2+ signals and the activation of the Ca2+ pump corporately restores the calcium store to a normal level (Zhang et al., 2020). Activation of NADPH oxidase in ER can improve the sensitivity of ER to IP3. NADPH oxidase-originated ROS can directly act on IP3R distributed on ER, causing its conformational changes, result in increased cytosolic free Ca2+ concentration, initiate intracellular Ca2+ signaling system, and regulate biological effects through Ca2+ dependent protein kinase II activity (Hu et al., 2000; Sakurada et al., 2019).
RyR is one of the intracellular Ca2+ release channels in the sarcoplasmic reticulum of muscle cells and endoplasmic reticulum of other cells and is the largest membrane protein molecule ever known. RyR in mammals can be divided into three subtypes: RyR1 (skeletal muscle type), RyR2 (myocardial type) and RyR3 (brain type). The expression level of RyR1 was highest in skeletal muscle, and RyR2 was mainly expressed in cardiomyocytes (Petrou et al., 2017) and some brain tissues. In cardiomyocytes, Ca2+ release of sarcoplasmic reticulum RyR2 is activated by influx of Ca2+ release through L-shaped calcium channels in the cell membrane, known as calcium-induced calcium release. RyR3 is widely distributed and expressed in many tissues. It is also activated by calcium-induced calcium release (Berridge, 2016). The increase of intracellular partial redox potential is the trigger point for important cellular functions. Studies found that RyR in the sarcoplasmic reticulum is sensitive to local redox potential, and mild oxidative stress can slightly increase intracellular redox potential, significantly enhance RyR activity and sensitize Ca2+ release mechanism, suggesting that ROS bidirectional regulation of ER Ca2+ may be concentration-dependent (Santulli et al., 2017; Kushnir et al., 2018). ROS promotes the synthesis of cADPR (cyclic adenosine diphosphate ribose) at low concentrations and opens RyR together with calmodulin. On the contrary, ROS leads to the oxidation of sulfhydryl groups of RyR and sarcoplasmic reticulum-related proteins into disulfide bonds, damaging Ca2+ release mechanisms (Avila et al., 2019; Kobayashi et al., 2021).
In resting conditions, Ca2+ is absorbed by the cytoplasm into the ER cavity through SERCA pump and released into the cytoplasm by RyR and IP3R channels. Most of the Ca2+ released into the cytoplasm will be recaptured by the Ca2+ pump to maintain the Ca2+ concentration in the ER. Therefore, Ca2+ accumulation in ER is mainly accomplished through the activity of SERCA pump (Ivanova et al., 2020). Ca2+-ATPase, as a known promoter of ER stress, since oxidative damage suppresses Ca2+-ATPase activity, ROS can affect Ca2+ storage within ER by inhibiting Ca2+-ATPase, and hence affecting intracellular Ca2+ homeostasis and causing ER stress (Krylatov et al., 2018). Studies have revealed ROS production and ER Ca2+ pump were sensitive to oxidative damage in ischemia and hypoxia cell models. ROS can make proteins in ER cavity be oxidized and retained, thus inhibiting protein synthesis, and leading to cell apoptosis. Puteney et al. found that Ca2+ releasing from the ER via IP3R causes decreased ER calcium reserves, Ca2+ can enter the cell from the extracellular, a process known as SOCE (store-operated Ca2+ entry) (Putney, 1986) Ivan demonstrated that exposure to environmental or cell-intrinsic ROS can affect Ca2+ homeostasis, modify multiple pathways and cause cell injuries (Bogeski et al., 2012). ROS production can induce Ca2+ release within the ER to cause ER stress, whereas Ca2+ release within the ER further stimulates Ca2+ aggregation on the mitochondria, causing mitochondrial damage and subsequently inducing apoptosis. As illustrated above, calreticulin is the main Ca2+ binding chaperone in the ER lumen, which binds to Ca2+ through the C-domain in a high capacity and increases the Ca2+ storage capacity of the ER lumen (Venkatesan et al., 2021). Leta found that calreticulin acts directly with SERCA2b glycosylated C-terminal tails to regulate Ca2+ storage in ER. When ER Ca2+ is sufficient, the activity of ATP-ase can be inhibited and intracellular Ca2+ transport to ER stops (Johnson et al., 2014).
Elevated ROS levels send feedback signals that stimulate ER stress. ER stress promotes the correct folding of the wrong proteins and degrades the proteins that cannot be folded. Transient ER stress helps to restore cell homeostasis, but long-term and high-intensity ERS can lead to multiple pulmonary diseases.
As ER stress aggravates, persistent UPR triggers programmed cell death. The occurrence and development of a variety of lung diseases are closely related to intracellular ROS, ER stress and UPR. (Figure 2)
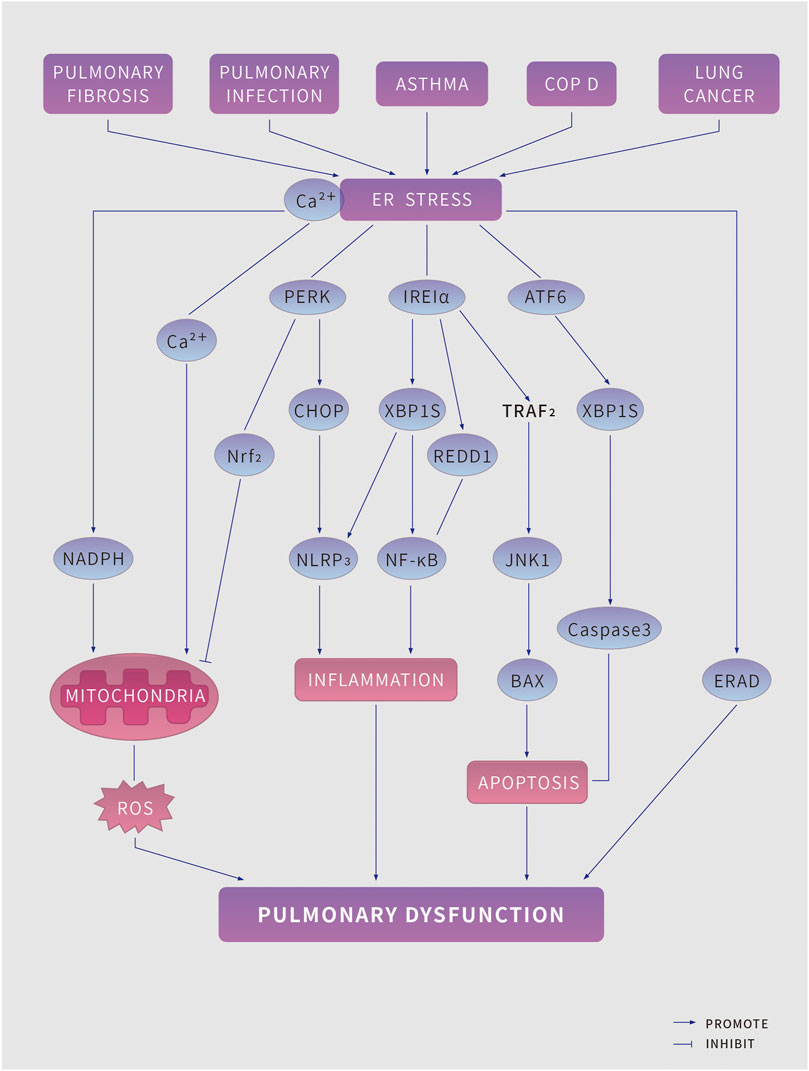
FIGURE 2. ER stress in pulmonary disease. Pulmonary pathologies, including pulmonary fibrosis, pulmonary infection, asthma, Chronic obstructive pulmonary disease and lung cancer lead to endoplasmic reticulum (ER) stress and induce the unfolded protein response (UPR) (which has three branches, mediated by protein kinase RNA-like ER kinase (PERK), Inositol-requiring protein 1α (IRE1α) and activating factor 6 (ATF6) to tackle ER stress. However, if the UPR fails to restore ER homeostasis, it might induce risk factors for pulmonary disease, including increased reactive oxygen species (ROS) production, inflammation, and apoptosis, which further aggravate pulmonary disease. BAX, BCL-2-associated X protein; CHOP, C/EBP homologous protein; ERAD, ER-associated degradation; JNK1, JUN N-terminal kinase 1; The NK-kappa B, nuclear factor kappa B predominate; NLRP3, NACHT, LRR and PYD domains-containing protein 3; NADPH, nicotinamide adenine dinucleotide phosphate; NRF2, nuclear factor erythroid 2-related factor 2; REDD1, protein regulated in development and DNA damage response 1; TRAF2, TNF receptor associated factor 2; XBP1s, spliced X-box-binding protein 1.
Pulmonary fibrosis is a kind of lung diseases characterized by inflammatory damage and tissue structure destruction. Pulmonary fibrosis is usually caused by excessive normal tissue repair resulting in vast proliferation of fibroblasts and abnormal accumulation of extracellular matrix. Cells activate UPR signaling to maintain ER homeostasis. Although the short-time UPR is beneficial to maintaining the cellular protein balance, it can cause lung cell dysfunction or even apoptosis when the UPR is stimulation is overwhelmed (Noble et al., 2012; Hetz et al., 2020).
IPF (idiopathic fibrosis of the lung) is a serious interstitial lung disease that can cause progressive loss of lung function, and its pathological mechanism is constantly being updated. Many cells in the lung parenchyma, including alveolar epithelial cells, macrophages, and fibroblasts, are affected by UPR during the pathological process of IPF. CE (Alveolar epithelial cells) are mainly divided into AEC I (alveolar epithelial cells I), and AEC II (alveolar epithelial cells II). AEC II cells are the progenitor cells required to maintain alveolar integrity. Loss of AEC II function and apoptosis are considered as important processes in the initial stage and development of IPF (Hetz et al., 2020). Researchers took samples of peripheral transplanted lung tissues, analyzed through protein detection, and found that AEC II in IPF can produce much UPR related proteins such as ATF6, ATF4 and CHOP. On the contrary, it does not exist in COPD and normal organ donors, indicating that in IPF patients, UPR activation can lead to the apoptosis of AEC II cells in the inner surface of the fibrotic site and be replaced by fibrous tissue (Korfei et al., 2008; Lawson et al., 2011). Timothy and his colleagues used tunicamycin to induce UPR activation, which enhanced AEC death and pulmonary fibrosis in IPF mice models, demonstrating UPR activation may increase AEC vulnerability, lead to enhanced lung injuries and IPF occurrence. Although UPR cannot directly aggravate the occurrence of IPF, it can improve the incidence probability of IPF in patients (Kropski et al., 2012). In many IPF patients, herpesviruses have been found to be closely associated with the occurrence of IPF. Herpesviruses are frequently found in the IPF lungs and can induce the UPR. Modification and maturation of viral glycosylated envelope proteins during virus replication may induce ER stress, while ER stress play double-acting roles in virus infection. Herpesvirus infection can directly promote UPR activation, causing decreased immune function induced malignant pulmonary fibrosis and IPF (Lee et al., 2009; Tanjore et al., 2012; Liu et al., 2021).
Pulmonary cystic fibrosis (CPF or CF) is a congenital disease with familial autosomal recessive inheritance. It is most common among white North Americans and rarely seen in other races. As an exocrine gland lesion, the gastrointestinal tract and respiratory tract are often affected (Elborn, 2016). The airway of patients with CF presents a persistent and intense inflammatory response, and AMs (alveolar macrophage) play key roles in the pathogenesis of CF (Hey et al., 2021). Some AMs can secrete many inflammatory mediators such as TNF-α and IL-6 induction (Lambertsen et al., 2012). XBP1 cut by IRE1 pathway mRNA in UPR affects peripheral macrophages and produces inflammatory effects (Lubamba et al., 2015). It was found that the UPR activated the expression of XBP1 in AMs in CF patients, and lipopolysaccharide was associated with the increased level of transcription factor XBP-1 in the UPR. Knockdown of XBP-1 in cultured dTHP-1 (differentiated human acute monocytic leukemia-1) cells reduced the inflammatory response of CF patients. Inflammatory factors in CF patients can be regulated by the expression of XBP-1 in UPR, and AMs in CF patients can promote the occurrence of airway inflammation in CF patients by upregulating xBP-1-mediated cytokines (Zhu et al., 2012; Lubamba et al., 2015). CFTR (cystic fibrosis transmembrane conductance regulator) is a central regulator of chlorine channel and epithelial function. Loss of CFTR function in airway can lead to mucus thickening, reduced mucosal ciliary clearance, chronic infection, and respiratory failure. UPR can induce the binding of ATF6 to the minimum promoter of CFTR, leading to the inhibition of CFTR transcription and reduce expression of CFTR (Trouvé et al., 2021).
Pulmonary infection refers to the infectious disease of pulmonary parenchyma and interstitial tissues, mainly caused by virus, bacteria, or other microbial infection, mainly in the form of pneumonia in clinical. UPR signaling play important roles in the secretion of inflammatory factors. Since inflammatory cells tend to have large secretory requirements and are therefore particularly dependent on a well-developed and abundant ER, the presence of infection-induced stimuli may induce an intracellular UPR response. Inflammatory factors such as TNF- (tumor necrosis factor), IL-1 (interleukin-1), and IL-6 can also cause the activation of three UPR pathways in cells, thus causing the acute phase response of lung inflammation (Grootjans et al., 2016). UPR activation and infection are mutually reinforcing and antagonizing each other (Bettigole and Glimcher, 2015).
There are many causes of the acute pneumonia response, where acute lung injury is the most important cause. In acute pneumonia caused by acute lung injury, the UPR activation plays an important role in cellular autophagy, as well as in apoptosis (Bettigole and Glimcher, 2015). In lipopolysaccharide-induced acute lung injury mice model and human alveolar epithelial cell model, the UPR inhibitor 4-PBA (4-phenylbutyric acid) blocks the activation of NF-κB pathway and reduces the release of pro-inflammatory mediators including TNF-α, IL-1β and IL-6. 4-PBA reduces the activation of inflammation by inhibiting UPR pathway and the mechanism is likely to be related to PERK-eIF2α-mediated translation reduction promoting NF-κB activation, suggesting that UPR is a crucial promoter of lipopolysaccharide-induced inflammation. 4-PBA leads to decreased autophagy, which plays a protective role in acute pneumonia through the classical AKT/mTOR signaling pathway and exacerbates lipopolysaccharide-induced A549 cytotoxicity through the 3-MA (autophagy inhibitor 3-methyladenine). 4-PBA inhibits UPR activation and has protective effects on autophagy in acute pneumonia models. UPR can stimulate the activation of NF-κB pathway, leading to the translocation of free NF-κB into the nucleus, possibly through conformational changes in cytoplasmic regions induced by IRE1α autophosphorylation. The IREα -TRAF2 complex binds to the adaptor protein TNF-α receptor-associated factor 2 to recruit IκB kinase and phosphorylates IκB, resulting in degradation of IκB and nuclear NF-κB translocation (Urano et al., 2000). Jiang et al. investigated whether ER stress during simulated microgravity induced endothelial inflammation and apoptosis in HUVECs (human umbilical vein endothelial cells), and they found that the increase of apoptosis in HUVECs during clinorotation was significantly suppressed by inhibiting ER stress, iNOS activity, NF-κB/IκB, and the NLRP3 inflammasome signaling pathway, indicating ER stress-dependent activation of iNOS/NO-NF-κB signaling and NLRP3 inflammasome contributes to chronic injury endothelial inflammation and apoptosis associated with microgravity (Jiang et al., 2020).
Aspergillus fumigatus is an opportunistic pathogen that is responsible for a life-threatening fungal infection called as invasive aspergillosis. The sensitization of AF (aspergillus fumigatus) was associated with severe allergic pulmonary inflammation, and allergic bronchopulmonary aspergillosis (ABPA) was the most common disease in allergic mycosis (Krishnan and Askew, 2014). Studies found that in clinical studies, glucoregulatory protein GRP78 was increased in lung tissues of patients with allergic bronchopulmonary aspergillus. Lee found that utilizing of the ER stress inhibitor or a ROS scavenger improved AF-induced allergic inflammation. The PI3K-δ inhibitor reduced AF-induced mitochondrial ROS generation and ER stress accordingly ameliorated. They revealed that PI3K-δ regulates AF-induced steroid-resistant eosinophilic allergic lung inflammation through ER stress (Lee et al., 2016). Wang revealed the effects and mechanisms of resveratrol on fungus-induced allergic airway inflammation, and they demonstrated resveratrol alleviated the AF-exposed allergic inflammation and apoptosis through inhibiting ER stress via Akt/mTOR pathway, exerting therapeutic effects on the fungus-induced allergic lung disorder (Wang et al., 2022). These studies revealed the UPR is a critical step in ABPA production.
The bronchial epithelium contains a variety of cell types with high protein synthesis and secretion, which are prone to ER stress (Schögler et al., 2019). In bronchial asthma, the existence of airway inflammation state and other factors are considered to induce UPR, and they are mutually causal and interactive with airway inflammation. Current studies suggest that ER stress is closely related with the dysregulation of Ca2+ homeostasis in airway epithelial cells, abnormal secretion of hyaluronic acid and mucin, recruitment of inflammatory cells by cytokines, and abnormal immunomodulatory state in the pathogenesis of bronchial asthma (Russell and Brightling, 2017). The expression of UPR marker protein GRP78 was significantly increased in peripheral blood monocytes and bronchoalveolar lavage fluid of asthmatic mice models, suggesting that the UPR pathway may cause the development of asthma by activating NF-κB, indicating UPR can also stimulate the activity of immune, and get involved in immune response (Kim et al., 2013b; Kim and Lee, 2015). The CHOP protein (also known as GADD153 or DDIT-3) is an important signal molecule in ER stress, and its specific mechanism in asthma has been gradually studied. It has been found that T cells treated with curcumin can induce activation of the PERK and IRE1 pathways in UPR and induce upregulation of stimulating transcription factors XBP-1 and CHOP in CD4+ lymphocytes and T lymphocytes, ultimately leading to apoptosis of T cells (Zhang et al., 2012; Lin et al., 2019). ORMDL3 (Orosomucoid like 3) has been considered as significant regulator of asthma in genetic association studies, David and his colleagues found transfection of ORMDL3s in human bronchial epithelial cells in vitro induced expression of metalloproteases (MMP-9), CC chemokines (CCL-20), CXC chemokines (IL-8, CXC-10, CXCL-11) and selectively activated ATF6 and SERCA2B. They furtherly revealed ORMDL3 may be associated with ER-UPR pathway in asthma. It was reported that the expression of ORMDL3 increased UPR response, and related genes signaling pathways of UPR were significantly upregulated. Ca2+ released by ER calcium influx decreased in ORMDL3 knockout models, and UPR was correspondingly weakened. The increased expression of ORMDL3 gene in asthmatic patients is probably caused by regulating UPR pathway affecting Ca2+ concentration (Cantero-Recasens et al., 2010).
COPD (chronic obstructive pulmonary disease) is a kind of pulmonary disease characterized by persistent respiratory symptoms and airflow restriction, which is not completely reversible and develops progressively, mainly affecting the lungs (Labaki and Rosenberg, 2020). COPD is often associated with smoking, and smoking exposure damages the proteasome itself, resulting in massive accumulation of insoluble proteins in airway epithelial cells, alveolar epithelial cells, and macrophages, ultimately causing UPR. COPD patients have more mucus cells in their respiratory tract, often secreting a lot of mucus. IRE1β (inositol-requiring enzyme 1beta) of airway goblet cells plays an important role in mucin cell phenotype and secretion of mucin5AC and mucin5B (Cloots et al., 2021). Michael found that in HEK293 cells models with purified protein, IRE1α is closely related with IRE1β diminishing expression and inhibiting signaling. IRE1β can assemble with and inhibit IRE1α to suppress stress induced XBP1 splicing to affect the UPR (Grey et al., 2020). Research have shown that hypoxic stimulation can activate the dissociation of molecular chaperone GRP78 and activate UPR to repair protease and autophagy functions of cells. Studies also found that the expression of UPR signaling molecules including GRP78, XBP1, CHOP and other proteins were not found in every COPD patient, suggesting that the UPR pathway was activated only in a part of COPD population (Min et al., 2011; van 't Wout et al., 2015). Mitochondrial oxidative stress is specifically significant in COPD, and Nrf2 (nuclear factor-E2-related factor 2) is a major regulator of antioxidant response element-driven cytoprotective protein expression. ER stress-mediated UPR and insufficiency of antioxidant Nrf2 has been implicated in cigarette smoking-induced COPD, suggesting the potential of Nrf2 in the treatment of COPD (Fratta Pasini et al., 2016; Liu et al., 2019).
Lung cancer is the largest tumor threatening human beings (Nasim et al., 2019). Lung cancer cells often tackle ischemia, hypoxia, metabolism, and other changes. To cope with these extreme factors, the cells themselves also produce corresponding changes, resulting in the activation of UPR pathway. The role of ER stress and the UPR is well demonstrated in a lung cancer (Dastghaib et al., 2021).
The most common type of lung cancer is adenocarcinoma, comprising around 40% of all lung cancer cases, and lung adenocarcinoma is still one of the most aggressive and rapidly fatal tumor types with overall survival less than 5 years (Denisenko et al., 2018). Many studies have shown that adenocarcinoma is often accompanied by activation of the UPR pathway. Lu found that ER stress can induce the high expression of the transcription factor XBP1 in 2D and 3D culture lung adenocarcinoma cells and promote the proliferation of lung adenocarcinoma in 3D culture in vitro. Knockdown the expression of XBP1 can block cell growth induced by Tm/Tg treatment. Meanwhile, LOX gene, as a key downstream sensor of XBP1, can block XBP1-induced cell proliferation by knocking down the expression of LOX, suggesting that XBP1 can regulate the proliferation of lung adenocarcinoma cells through LOX (Yang et al., 2017). It was found that asterosaponin 1 increased ER dilation and cytosolic Ca2+ concentration, enhanced the expression of GRP78 and GRP94 in a dose-and-time-dependent manner and increased the expression of CHOP, Caspase-4 and JNK, the three UPR related apoptotic molecules. Finally, the proliferation of A549 cells was inhibited and apoptosis was promoted (Zhao et al., 2011).
Large cell carcinoma is a barely descriptive term indicating a subtype of lung cancer with no specific features of small-cell lung cancer, adenocarcinoma or squamous cell carcinoma (Weissferdt, 2014). Azam et al. have found that GRP78 is closely related to the regulation of large cell carcinoma. The expression of GRP78 in tumor of patients with large cell carcinoma is more than three times that of the negative control. Both miR-495 and miR-199a-5p can promote the malignant transformation of tumor cells. Increased GRP78 in cells can downregulate Mir-495 and Mir-199a-5p in NSCLC, and overexpression or inhibition of miR-495 and miR-199a-5p can also significantly change the expression of GRP78 and XBP1 level. UPR and microRNA interact to promote the occurrence of cancer (Ahmadi et al., 2017). Pau and his colleagues found that the anti-cancer molecule ABTL0812 increases cellular long-chain dihydroceramides by impairing DEGS1 (delta 4-desaturase, sphingolipid 1) activity, which resulted in persistent ER stress and activated UPR via ATF4-DDIT3-TRIB3 that ultimately promotes cytotoxic autophagy in cancer cells (Muñoz-Guardiola et al., 2021). Studies have proved that in large-cell lung carcinoma H460 treated with cisplatin, the cell viability is distinctly reduced after adding ERS inhibitor 4-PBA (4-phenylbutyric acid). Intracellular caspase-3 gene and cytochrome C were significantly upregulated and promoted cell apoptosis, confirming that chemotherapy drugs can activate UPR and enhance drug resistance of tumor cells (Shi et al., 2016). Blocking the adaptive pathway of ER stress or promoting the apoptotic pathway could be a promising anti-cancer strategy.
Given the significant characteristic in the pathogenesis of pulmonary disease, strategies to target ER proteostasis, UPR signaling, and ROS are crucial for disease intervention. We discuss strategies as following.
4-PBA. 4-phenyl butyric acid, a low-molecular-weight aromatic fatty acid, affect the activation of LPS-induced inflammation human alveolar epithelial cell in acute lung injury models. It further prevents the activation of the NF-κB pathway, decreases the release of the inflammatory mediators through regulating ER stress (Zeng et al., 2017).
TUDCA. TUDCA (Tauroursodeoxycholic) is a hydrophilic bile acid with chaperone properties that is produced endogenously in humans at very low levels. Tong found that the TUDCA can moderate pulmonary ER stress and epithelial-mesenchymal transition in bleomycin-induced lung fibrosis (Tong et al., 2021). TUDCA can inhibit CHOP expression and the bleomycin -induced pulmonary fibrosis and inflammation, suggesting TUDCA may be a promising strategy for preventing pulmonary fibrosis (Tanaka et al., 2015).
EIF2A phosphorylation levels are controlled by two phosphatase complexes, hydrophobic the protein Phosphatase 1 (PP1) catalytic subunit complexed with either constitutively expressed PPP1R15B (PP1 regulatory subunit 15B) or the ER Stress-inducible isoform PPP1R15A, which is an ATF4 -- CHOP Target (PP1). Salubrinal is a cell-permeable, small-molecule inhibitor of the PP1 complex (Liu et al., 2014). Wang found that Salubrinal attenuated A549 cells from Paraquat-induced damages through upregulating the PERK-eIF2α signaling (Wang et al., 2018b). Salubrinal may be a promising therapeutic strategy to suppress ER stress in pulmonary dysfunctions.
Metformin is an antidiabetic drug that has been proved to be beneficial to other diseases independent of its glycaemia effects. Francesca found that metformin may ameliorate cigarette smoke-induced pathologies of emphysematous COPD through the AMPK (AMP-activated protein kinase) pathway (Polverino et al., 2021).
Qin demonstrated ER stress-induced cell death was mediated by autophagy which was partly attributed to the inactivation of the mTOR (mammalian target of rapamycin) (Qin et al., 2010). Tian found that ginkgo biloba leaf extract attenuates atherosclerosis in streptozotocin-induced diabetic ApoE−/− mice by Inhibiting ER stress via restoration of autophagy through the mTOR Signaling Pathway (Tian et al., 2019), which inspired us that the mTOR pathway is expected to become a promising target of ER stress-induced lung diseases.
AβM acid can resolve the inflammatory and UPR in inflammatory diseases (Vander Zanden et al., 2019). Emily proved that AβM acid-based therapy can inhibit the allergen-induced UPR and allergic airway disease in mice via preferential binding of ATF6α, suggesting a novel avenue to treat allergic asthma using AβM (Nakada et al., 2019).
Pharmacologic arm selective UPR signaling pathway activation is emerging as a promising strategy to improve imbalances in ER proteostasis implicated in diverse diseases. The compound 147 was considered to activate ATF6-regulated signaling associated with localized metabolic activation and selective covalent modification of ER stress (Paxman et al., 2018). Therefore, the compound 147 is a promising strategy in preventing ER stress-induced pulmonary disease.
Melatonin is an anti-inflammatory molecule, which has been effective in acute lung injury induced by many conditions (Tordjman et al., 2017). Zhao revealed that melatonin obviously inhibited the activation of NLRP3 inflammation, ER stress and epithelial-mesenchymal transition during bleomycin-mediated pulmonary fibrosis in mice models (Paxman et al., 2018).
Li found that ursolic acid derivative has preventive effects on particulate matter 2.5-induced COPD, causes obvious suppression in PM2.5-induced increase in oxidative stress markers and inflammatory cytokines, suggesting ursolic acid derivative can be a promising therapeutic treatment for PM2.5-induced COPD (Li et al., 2020).
Resveratrol, a natural polyphenol, is beneficial in supporting its numerous biological properties including antioxidant, anti-inflammatory, ant obesity, antidiabetic, and ant-ischemic activities. The compound also has beneficial effects on cognitive function and lung health (Ahmadi et al., 2021). Wang demonstrated that resveratrol attenuates inflammation and apoptosis through alleviating ER stress via Akt/mTOR pathway in fungus-induced allergic airways inflammation, providing therapeutic strategies for fungus-induced allergic lung disease (Wang et al., 2022).
Diets poor in antioxidants are related with lung dysfunction and a risk factor for the development of COPD. Dietaries include Vitamin C (ascorbic acid), Vitamin E (α-tocopherol), resveratrol, and flavonoids has always been studied in improve lung function or clinical features of COPD (Rana, 2020). Andras implied ascorbate may be involved in oxidative protein folding and creates a link between the disulfide bond formation (oxidative protein folding) and hydroxylation, providing promising strategies in preventing and treating pulmonary disease (Szarka and Lőrincz, 2014).
NRF2-mediated heme oxygenase was upregulated by UPR activation in PC12 cells. and GCLC (glutamate cysteine ligase catalytic) subunit expression increases. Heme oxygenase and GCLC can lead to COPD disease aggravation, indicating increasing Nrf2 activity as a treatment strategy in COPD (Digaleh et al., 2013; Niforou et al., 2014; Wu et al., 2015).
Lifestyle adjustments including physical exercise, smoking cessation and diet are a novel approach to modulate ER stress to protect against pulmonary disease (Itoh et al., 2013; Pedersen and Saltin, 2015; Schuller, 2019).
ROS-ER stress-associated therapy is becoming recognized as a crucial treatment strategy for pulmonary disease. Efficiently targeting ER stress in an organelle-specific or organ-specific manner holds promise for preventing and treating pulmonary disease.
Massive attention has been paid to the role of ROS and ER stress in the pathogenesis of pulmonary disease over the past years. The ER coordinates secretory and transmembrane proteins folding and translocation, controls cellular calcium uptake, storage and signaling.
The ER stress promotes the correct folding of faulty proteins and the degradation of misfolding proteins. Transient ER stress contributes to the homeostasis of cells, while sustained and overwhelmed ER stress can lead to several lung diseases. Unfold protein response is the most important reaction in ER stress. UPR and calcium initial signaling are key mechanisms of ER stress-mediated apoptosis. We summarized the mechanisms of ER stress, key regulators, related pathways, and associated lung diseases, including pulmonary fibrosis, pulmonary infections, asthma, COPD, and lung cancers. We also provided latest interventions target ER stress and ROS in pulmonary disease.
ER homeostasis and ER stress, especially the biological entities of ER stress, have been extensively described in many reports. However, there are still many unanswered questions. Although some protein-regulatory promoter compounds targeting ER stress or UPR components have shown potential in preventing and treating lung disease, clinical trials evaluating these agents targeting ER stress or UPR are still lacking. ROS serves as an upstream signal of ER stress, and the study of their internal relationship and mechanism of action need us further understand and provide new ideas for the development of drugs for pulmonary diseases, and further research is needed to provide a new approach for tumor treatment.
YZ researched data for the paper, contributed to discussion of the content, wrote the article, and reviewed the article before the submission. XC had great contribution in subject planning and modified the manuscript. MX and YL retrieved and organized documents.
This work was funded by grants from the National Natural Science Foundation of China (No. 81973842).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahmadi, A., Hayes, A. W., and Karimi, G. (2021). Resveratrol and Endoplasmic Reticulum Stress: A Review of the Potential Protective Mechanisms of the Polyphenol. Phytother Res. 35 (10), 5564–5583. doi:10.1002/ptr.7192
Ahmadi, A., Khansarinejad, B., Hosseinkhani, S., Ghanei, M., and Mowla, S. J. (2017). miR-199a-5p and miR-495 Target GRP78 within UPR Pathway of Lung Cancer. Gene 620, 15–22. doi:10.1016/j.gene.2017.03.032
An, J. J., Shi, K. J., Wei, W., Hua, F. Y., Ci, Y. L., Jiang, Q., et al. (2013). The ROS/JNK/ATF2 Pathway Mediates Selenite-Induced Leukemia NB4 Cell Cycle Arrest and Apoptosis In Vitro and In Vivo. Cell Death Dis 4, e973. doi:10.1038/cddis.2013.475
Aoyama, Y., Inagaki, S., Aoshima, K., Iwata, Y., Nakamura, S., Hara, H., et al. (2021). Involvement of Endoplasmic Reticulum Stress in Rotenone-Induced Leber Hereditary Optic Neuropathy Model and the Discovery of New Therapeutic Agents. J. Pharmacol. Sci. 147 (2), 200–207. doi:10.1016/j.jphs.2021.07.003
Avila, G., de la Rosa, J. A., Monsalvo-Villegas, A., and Montiel-Jaen, M. G. (2019). Ca2+ Channels Mediate Bidirectional Signaling between Sarcolemma and Sarcoplasmic Reticulum in Muscle Cells. Cells 9 (1). doi:10.3390/cells9010055
Berridge, M. J. (2016). The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 96 (4), 1261–1296. doi:10.1152/physrev.00006.2016
Bettigole, S. E., and Glimcher, L. H. (2015). Endoplasmic Reticulum Stress in Immunity. Annu. Rev. Immunol. 33, 107–138. doi:10.1146/annurev-immunol-032414-112116
Boengler, K., Kosiol, M., Mayr, M., Schulz, R., and Rohrbach, S. (2017). Mitochondria and Ageing: Role in Heart, Skeletal Muscle and Adipose Tissue. J. Cachexia Sarcopenia Muscle 8 (3), 349–369. doi:10.1002/jcsm.12178
Bogeski, I., Kilch, T., and Niemeyer, B. A. (2012). ROS and SOCE: Recent Advances and Controversies in the Regulation of STIM and Orai. J. Physiol. 590 (17), 4193–4200. doi:10.1113/jphysiol.2012.230565
Borges, C. R., and Lake, D. F. (2014). Oxidative Protein Folding: Nature's Knotty challenge. Antioxid. Redox Signal. 21 (3), 392–395. doi:10.1089/ars.2014.5946
Brown, M. S., Radhakrishnan, A., and Goldstein, J. L. (2018). Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 87, 783–807. doi:10.1146/annurev-biochem-062917-011852
Cantero-Recasens, G., Fandos, C., Rubio-Moscardo, F., Valverde, M. A., and Vicente, R. (2010). The Asthma-Associated ORMDL3 Gene Product Regulates Endoplasmic Reticulum-Mediated Calcium Signaling and Cellular Stress. Hum. Mol. Genet. 19 (1), 111–121. doi:10.1093/hmg/ddp471
Carpio, M. A., Michaud, M., Zhou, W., Fisher, J. K., Walensky, L. D., and Katz, S. G. (2015). BCL-2 Family Member BOK Promotes Apoptosis in Response to Endoplasmic Reticulum Stress. Proc. Natl. Acad. Sci. U S A. 112 (23), 7201–7206. doi:10.1073/pnas.1421063112
Chen, X., and Cubillos-Ruiz, J. R. (2021). Endoplasmic Reticulum Stress Signals in the Tumour and its Microenvironment. Nat. Rev. Cancer 21 (2), 71–88. doi:10.1038/s41568-020-00312-2
Cloots, E., Simpson, M. S., De Nolf, C., Lencer, W. I., Janssens, S., and Grey, M. J. (2021). Evolution and Function of the Epithelial Cell-specific ER Stress Sensor IRE1β. Mucosal Immunol. 14 (6), 1235–1246. doi:10.1038/s41385-021-00412-8
Dastghaib, S., Kumar, P. S., Aftabi, S., Damera, G., Dalvand, A., Sepanjnia, A., et al. (2021). Mechanisms Targeting the Unfolded Protein Response in Asthma. Am. J. Respir. Cel Mol Biol 64 (1), 29–38. doi:10.1165/rcmb.2019-0235TR
Delaunay-Moisan, A., and Appenzeller-Herzog, C. (2015). The Antioxidant Machinery of the Endoplasmic Reticulum: Protection and Signaling. Free Radic. Biol. Med. 83, 341–351. doi:10.1016/j.freeradbiomed.2015.02.019
Denisenko, T. V., Budkevich, I. N., and Zhivotovsky, B. (2018). Cell Death-Based Treatment of Lung Adenocarcinoma. Cel Death Dis 9 (2), 117. doi:10.1038/s41419-017-0063-y
Di Conza, G., and Ho, P. C. (2020). ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 9 (3). doi:10.3390/cells9030695
Digaleh, H., Kiaei, M., and Khodagholi, F. (2013). Nrf2 and Nrf1 Signaling and ER Stress Crosstalk: Implication for Proteasomal Degradation and Autophagy. Cell Mol Life Sci 70 (24), 4681–4694. doi:10.1007/s00018-013-1409-y
Elborn, J. S. (2016). Cystic Fibrosis. Lancet 388 (10059), 2519–2531. doi:10.1016/S0140-6736(16)00576-6
Ellgaard, L., and Ruddock, L. W. (2005). The Human Protein Disulphide Isomerase Family: Substrate Interactions and Functional Properties. EMBO Rep. 6 (1), 28–32. doi:10.1038/sj.embor.7400311
Feng, K., Ge, Y., Chen, Z., Li, X., Liu, Z., Li, X., et al. (2019). Curcumin Inhibits the PERK-eIF2α-CHOP Pathway through Promoting SIRT1 Expression in Oxidative Stress-Induced Rat Chondrocytes and Ameliorates Osteoarthritis Progression in a Rat Model. Oxid Med. Cel Longev 2019, 8574386. doi:10.1155/2019/8574386
Frakes, A. E., and Dillin, A. (2017). The UPRER: Sensor and Coordinator of Organismal Homeostasis. Mol. Cel 66 (6), 761–771. doi:10.1016/j.molcel.2017.05.031
Fratta Pasini, A. M., Ferrari, M., Stranieri, C., Vallerio, P., Mozzini, C., Garbin, U., et al. (2016). Nrf2 Expression Is Increased in Peripheral Blood Mononuclear Cells Derived from Mild-Moderate Ex-Smoker COPD Patients with Persistent Oxidative Stress. Int. J. Chron. Obstruct Pulmon Dis. 11, 1733–1743. doi:10.2147/COPD.S102218
Fu, J., Gao, J., Liang, Z., and Yang, D. (2020). PDI-regulated Disulfide Bond Formation in Protein Folding and Biomolecular Assembly. Molecules 26 (1). doi:10.3390/molecules26010171
Grey, M. J., Cloots, E., Simpson, M. S., LeDuc, N., Serebrenik, Y. V., De Luca, H., et al. (2020). IRE1β Negatively Regulates IRE1α Signaling in Response to Endoplasmic Reticulum Stress. J. Cel Biol 219 (2). doi:10.1083/jcb.201904048
Grootjans, J., Kaser, A., Kaufman, R. J., and Blumberg, R. S. (2016). The Unfolded Protein Response in Immunity and Inflammation. Nat. Rev. Immunol. 16 (8), 469–484. doi:10.1038/nri.2016.62
Han, J., Back, S. H., Hur, J., Lin, Y. H., Gildersleeve, R., Shan, J., et al. (2013). ER-stress-induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat. Cel Biol 15 (5), 481–490. doi:10.1038/ncb2738
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Erratum: Proteostasis Control by the Unfolded Protein Response. Nat. Cel Biol 17 (7), 1088–1138. doi:10.1038/ncb3221
Hetz, C., and Papa, F. R. (2018). The Unfolded Protein Response and Cell Fate Control. Mol. Cel 69 (2), 169–181. doi:10.1016/j.molcel.2017.06.017
Hetz, C., Zhang, K., and Kaufman, R. J. (2020). Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cel Biol 21 (8), 421–438. doi:10.1038/s41580-020-0250-z
Hey, J., Paulsen, M., Toth, R., Weichenhan, D., Butz, S., Schatterny, J., et al. (2021). Epigenetic Reprogramming of Airway Macrophages Promotes Polarization and Inflammation in Muco-Obstructive Lung Disease. Nat. Commun. 12 (1), 6520. doi:10.1038/s41467-021-26777-9
Hotamisligil, G. S., and Davis, R. J. (2016). Cell Signaling and Stress Responses. Cold Spring Harb Perspect. Biol. 8 (10). doi:10.1101/cshperspect.a006072
Hu, Q., Zheng, G., Zweier, J. L., Deshpande, S., Irani, K., and Ziegelstein, R. C. (2000). NADPH Oxidase Activation Increases the Sensitivity of Intracellular Ca2+ Stores to Inositol 1,4,5-trisphosphate in Human Endothelial Cells. J. Biol. Chem. 275 (21), 15749–15757. doi:10.1074/jbc.M000381200
Huang, S., Xing, Y., and Liu, Y. (2019). Emerging Roles for the ER Stress Sensor IRE1α in Metabolic Regulation and Disease. J. Biol. Chem. 294 (49), 18726–18741. doi:10.1074/jbc.REV119.007036
Hwang, J., and Qi, L. (2018). Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR Pathways. Trends Biochem. Sci. 43 (8), 593–605. doi:10.1016/j.tibs.2018.06.005
Incalza, M. A., D'Oria, R., Natalicchio, A., Perrini, S., Laviola, L., and Giorgino, F. (2018). Oxidative Stress and Reactive Oxygen Species in Endothelial Dysfunction Associated with Cardiovascular and Metabolic Diseases. Vascul Pharmacol. 100, 1–19. doi:10.1016/j.vph.2017.05.005
Itoh, M., Tsuji, T., Nemoto, K., Nakamura, H., and Aoshiba, K. (2013). Undernutrition in Patients with COPD and its Treatment. Nutrients 5 (4), 1316–1335. doi:10.3390/nu5041316
Ivanova, H., Vervliet, T., Monaco, G., Terry, L. E., Rosa, N., Baker, M. R., et al. (2020). Bcl-2-Protein Family as Modulators of IP3 Receptors and Other Organellar Ca2+ Channels. Cold Spring Harb Perspect. Biol. 12 (4). doi:10.1101/cshperspect.a035089
Jiang, M., Wang, H., Liu, Z., Lin, L., Wang, L., Xie, M., et al. (2020). Endoplasmic Reticulum Stress-dependent Activation of iNOS/NO-NF-Κb Signaling and NLRP3 Inflammasome Contributes to Endothelial Inflammation and Apoptosis Associated with Microgravity. FASEB J. 34 (8), 10835–10849. doi:10.1096/fj.202000734R
Jin, J. K., Blackwood, E. A., Azizi, K., Thuerauf, D. J., Fahem, A. G., Hofmann, C., et al. (2017). ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 120 (5), 862–875. doi:10.1161/CIRCRESAHA.116.310266
Johnson, J. S., Kono, T., Tong, X., Yamamoto, W. R., Zarain-Herzberg, A., Merrins, M. J., et al. (2014). Pancreatic and Duodenal Homeobox Protein 1 (Pdx-1) Maintains Endoplasmic Reticulum Calcium Levels through Transcriptional Regulation of Sarco-Endoplasmic Reticulum Calcium ATPase 2b (SERCA2b) in the Islet β Cell. J. Biol. Chem. 289 (47), 32798–32810. doi:10.1074/jbc.M114.575191
Kaminskyy, V. O., and Zhivotovsky, B. (2014). Free Radicals in Cross Talk between Autophagy and Apoptosis. Antioxid. Redox Signal. 21 (1), 86–102. doi:10.1089/ars.2013.5746
Kankanamge, D., Ubeysinghe, S., Tennakoon, M., Pantula, P. D., Mitra, K., Giri, L., et al. (2021). Dissociation of the G Protein βγ from the Gq-Plcβ Complex Partially Attenuates PIP2 Hydrolysis. J. Biol. Chem. 296, 100702. Jan-Jun. doi:10.1016/j.jbc.2021.100702
Katzen, J., and Beers, M. F. (2020). Contributions of Alveolar Epithelial Cell Quality Control to Pulmonary Fibrosis. J. Clin. Invest. 130 (10), 5088–5099. doi:10.1172/JCI139519
Khanna, M., Agrawal, N., Chandra, R., and Dhawan, G. (2021). Targeting Unfolded Protein Response: a New Horizon for Disease Control. Expert Rev. Mol. Med. 23, e1. doi:10.1017/erm.2021.2
Kim, S. H., Kim, K. Y., Yu, S. N., Seo, Y. K., Chun, S. S., Yu, H. S., et al. (2016). Silibinin Induces Mitochondrial NOX4-Mediated Endoplasmic Reticulum Stress Response and its Subsequent Apoptosis. BMC Cancer 16, 452. doi:10.1186/s12885-016-2516-6
Kim, S. R., Kim, D. I., Kang, M. R., Lee, K. S., Park, S. Y., Jeong, J. S., et al. (2013). Endoplasmic Reticulum Stress Influences Bronchial Asthma Pathogenesis by Modulating Nuclear Factor κB Activation. J. Allergy Clin. Immunol. 132 (6), 1397–1408. doi:10.1016/j.jaci.2013.08.041
Kim, S. R., and Lee, Y. C. (2015). Endoplasmic Reticulum Stress and the Related Signaling Networks in Severe Asthma. Allergy Asthma Immunol. Res. 7 (2), 106–117. doi:10.4168/aair.2015.7.2.106
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M., and Hartl, F. U. (2013). Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 82, 323–355. doi:10.1146/annurev-biochem-060208-092442
Kobayashi, T., Kurebayashi, N., and Murayama, T. (2021). The Ryanodine Receptor as a Sensor for Intracellular Environments in Muscles. Int. J. Mol. Sci. 22 (19). doi:10.3390/ijms221910795
Kopp, M. C., Larburu, N., Durairaj, V., Adams, C. J., and Ali, M. M. U. (2019). UPR Proteins IRE1 and PERK Switch BiP from Chaperone to ER Stress Sensor. Nat. Struct. Mol. Biol. 26 (11), 1053–1062. doi:10.1038/s41594-019-0324-9
Korfei, M., Ruppert, C., Mahavadi, P., Henneke, I., Markart, P., Koch, M., et al. (2008). Epithelial Endoplasmic Reticulum Stress and Apoptosis in Sporadic Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 178 (8), 838–846. doi:10.1164/rccm.200802-313OC
Krishnan, K., and Askew, D. S. (2014). The Fungal UPR: a Regulatory Hub for Virulence Traits in the Mold Pathogen Aspergillus fumigatus. Virulence 5 (2), 334–340. doi:10.4161/viru.26571
Kropski, J. A., Lawson, W. E., and Blackwell, T. S. (2012). Right Place, Right Time: the Evolving Role of Herpesvirus Infection as a "second Hit" in Idiopathic Pulmonary Fibrosis. Am. J. Physiol. Lung Cel Mol Physiol 302 (5), L441–L444. doi:10.1152/ajplung.00335.2011
Krylatov, A. V., Maslov, L. N., Voronkov, N. S., Boshchenko, A. A., Popov, S. V., Gomez, L., et al. (2018). Reactive Oxygen Species as Intracellular Signaling Molecules in the Cardiovascular System. Curr. Cardiol. Rev. 14 (4), 290–300. doi:10.2174/1573403X14666180702152436
Kushnir, A., Wajsberg, B., and Marks, A. R. (2018). Ryanodine Receptor Dysfunction in Human Disorders. Biochim. Biophys. Acta Mol. Cel Res 1865 (11 Pt B), 1687–1697. doi:10.1016/j.bbamcr.2018.07.011
Labaki, W. W., and Rosenberg, S. R. (2020). Chronic Obstructive Pulmonary Disease. Ann. Intern. Med. 173 (3), ITC17–ITC32. doi:10.7326/AITC202008040
Lambertsen, K. L., Biber, K., and Finsen, B. (2012). Inflammatory Cytokines in Experimental and Human Stroke. J. Cereb. Blood Flow Metab. 32 (9), 1677–1698. doi:10.1038/jcbfm.2012.88
Lawson, W. E., Cheng, D. S., Degryse, A. L., Tanjore, H., Polosukhin, V. V., Xu, X. C., et al. (2011). Endoplasmic Reticulum Stress Enhances Fibrotic Remodeling in the Lungs. Proc. Natl. Acad. Sci. U S A. 108 (26), 10562–10567. doi:10.1073/pnas.1107559108
Lee, D. Y., Lee, J., and Sugden, B. (2009). The Unfolded Protein Response and Autophagy: Herpesviruses Rule!. J. Virol. 83 (3), 1168–1172. doi:10.1128/JVI.01358-08
Lee, H. Y., Kim, H. K., Hoang, T. H., Yang, S., Kim, H. R., and Chae, H. J. (2020). The Correlation of IRE1α Oxidation with Nox4 Activation in Aging-Associated Vascular Dysfunction. Redox Biol. 37, 101727. doi:10.1016/j.redox.2020.101727
Lee, K. S., Jeong, J. S., Kim, S. R., Cho, S. H., Kolliputi, N., Ko, Y. H., et al. (2016). Phosphoinositide 3-Kinase-δ Regulates Fungus-Induced Allergic Lung Inflammation through Endoplasmic Reticulum Stress. Thorax 71 (1), 52–63. doi:10.1136/thoraxjnl-2015-207096
Lerner, A. G., Upton, J. P., Praveen, P. V., Ghosh, R., Nakagawa, Y., Igbaria, A., et al. (2012). IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab 16 (2), 250–264. doi:10.1016/j.cmet.2012.07.007
Li, C., Chen, J., Yuan, W., Zhang, W., Chen, H., and Tan, H. (2020). Preventive Effect of Ursolic Acid Derivative on Particulate Matter 2.5-induced Chronic Obstructive Pulmonary Disease Involves Suppression of Lung Inflammation. IUBMB Life 72 (4), 632–640. doi:10.1002/iub.2201
Lim, C. Y., Davis, O. B., Shin, H. R., Zhang, J., Berdan, C. A., Jiang, X., et al. (2019). ER-lysosome Contacts Enable Cholesterol Sensing by mTORC1 and Drive Aberrant Growth Signalling in Niemann-Pick Type C. Nat. Cel Biol 21 (10), 1206–1218. doi:10.1038/s41556-019-0391-5
Lin, R., Sun, Y., Ye, W., Zheng, T., Wen, J., and Deng, Y. (2019). T-2 Toxin Inhibits the Production of Mucin via Activating the IRE1/XBP1 Pathway. Toxicology 424, 152230. doi:10.1016/j.tox.2019.06.001
Liu, G., Pei, F., Yang, F., Li, L., Amin, A. D., Liu, S., et al. (2017). Role of Autophagy and Apoptosis in Non-small-cell Lung Cancer. Int. J. Mol. Sci. 18 (2). doi:10.3390/ijms18020367
Liu, Q., Gao, Y., and Ci, X. (2019). Role of Nrf2 and its Activators in Respiratory Diseases. Oxid Med. Cel Longev 2019, 7090534. doi:10.1155/2019/7090534
Liu, Y., Li, G., and Wang, B. (2021). Herpesvirus and Endoplasmic Reticulum Stress. Sheng Wu Gong Cheng Xue Bao 37 (1), 67–77. doi:10.13345/j.cjb.200226
Liu, Y., Wang, J., Qi, S. Y., Ru, L. S., Ding, C., Wang, H. J., et al. (2014). Reduced Endoplasmic Reticulum Stress Might Alter the Course of Heart Failure via Caspase-12 and JNK Pathways. Can. J. Cardiol. 30 (3), 368–375. doi:10.1016/j.cjca.2013.11.001
Lubamba, B. A., Jones, L. C., O'Neal, W. K., Boucher, R. C., and Ribeiro, C. M. (2015). X-Box-Binding Protein 1 and Innate Immune Responses of Human Cystic Fibrosis Alveolar Macrophages. Am. J. Respir. Crit. Care Med. 192 (12), 1449–1461. doi:10.1164/rccm.201504-0657OC
Luhr, M., Torgersen, M. L., Szalai, P., Hashim, A., Brech, A., Staerk, J., et al. (2019). The Kinase PERK and the Transcription Factor ATF4 Play Distinct and Essential Roles in Autophagy Resulting from Tunicamycin-Induced ER Stress. J. Biol. Chem. 294 (20), 8197–8217. doi:10.1074/jbc.RA118.002829
Marchi, S., Patergnani, S., Missiroli, S., Morciano, G., Rimessi, A., Wieckowski, M. R., et al. (2018). Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 69, 62–72. doi:10.1016/j.ceca.2017.05.003
Matsuzawa, A., Nishitoh, H., Tobiume, K., Takeda, K., and Ichijo, H. (2002). Physiological Roles of ASK1-Mediated Signal Transduction in Oxidative Stress- and Endoplasmic Reticulum Stress-Induced Apoptosis: Advanced Findings from ASK1 Knockout Mice. Antioxid. Redox Signal. 4 (3), 415–425. doi:10.1089/15230860260196218
Min, T., Bodas, M., Mazur, S., and Vij, N. (2011). Critical Role of Proteostasis-Imbalance in Pathogenesis of COPD and Severe Emphysema. J. Mol. Med. (Berl) 89 (6), 577–593. doi:10.1007/s00109-011-0732-8
Muñoz-Guardiola, P., Casas, J., Megías-Roda, E., Solé, S., Perez-Montoyo, H., Yeste-Velasco, M., et al. (2021). The Anti-cancer Drug ABTL0812 Induces ER Stress-Mediated Cytotoxic Autophagy by Increasing Dihydroceramide Levels in Cancer Cells. Autophagy 17 (6), 1349–1366. doi:10.1080/15548627.2020.1761651
Nakada, E. M., Bhakta, N. R., Korwin-Mihavics, B. R., Kumar, A., Chamberlain, N., Bruno, S. R., et al. (2019). Conjugated Bile Acids Attenuate Allergen-Induced Airway Inflammation and Hyperresponsiveness by Inhibiting UPR Transducers. JCI Insight 4 (9). doi:10.1172/jci.insight.98101
Nasim, F., Sabath, B. F., and Eapen, G. A. (2019). Lung Cancer. Med. Clin. North. Am. 103 (3), 463–473. doi:10.1016/j.mcna.2018.12.006
Niforou, K., Cheimonidou, C., and Trougakos, I. P. (2014). Molecular Chaperones and Proteostasis Regulation during Redox Imbalance. Redox Biol. 2, 323–332. doi:10.1016/j.redox.2014.01.017
Noble, P. W., Barkauskas, C. E., and Jiang, D. (2012). Pulmonary Fibrosis: Patterns and Perpetrators. J. Clin. Invest. 122 (8), 2756–2762. doi:10.1172/JCI60323
Oakes, S. A., and Papa, F. R. (2015). The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. 10, 173–194. doi:10.1146/annurev-pathol-012513-104649
Ochoa, C. D., Wu, R. F., and Terada, L. S. (2018). ROS Signaling and ER Stress in Cardiovascular Disease. Mol. Aspects Med. 63, 18–29. doi:10.1016/j.mam.2018.03.002
Ong, H. L., and Ambudkar, I. S. (2020). The Endoplasmic Reticulum-Plasma Membrane Junction: A Hub for Agonist Regulation of Ca2+ Entry. Cold Spring Harb Perspect. Biol. 12 (2). doi:10.1101/cshperspect.a035253
Orrenius, S., Gogvadze, V., and Zhivotovsky, B. (2015). Calcium and Mitochondria in the Regulation of Cell Death. Biochem. Biophys. Res. Commun. 460 (1), 72–81. doi:10.1016/j.bbrc.2015.01.137
Pachikov, A. N., Gough, R. R., Christy, C. E., Morris, M. E., Casey, C. A., LaGrange, C. A., et al. (2021). The Non-canonical Mechanism of ER Stress-Mediated Progression of Prostate Cancer. J. Exp. Clin. Cancer Res. 40 (1), 289. doi:10.1186/s13046-021-02066-7
Paxman, R., Plate, L., Blackwood, E. A., Glembotski, C., Powers, E. T., Wiseman, R. L., et al. (2018). Pharmacologic ATF6 Activating Compounds Are Metabolically Activated to Selectively Modify Endoplasmic Reticulum Proteins. Elife 7. doi:10.7554/eLife.37168
Pedersen, B. K., and Saltin, B. (2015). Exercise as Medicine - Evidence for Prescribing Exercise as Therapy in 26 Different Chronic Diseases. Scand. J. Med. Sci. Sports 25 Suppl 3 (Suppl. 3), 1–72. doi:10.1111/sms.12581
Petrou, T., Olsen, H. L., Thrasivoulou, C., Masters, J. R., Ashmore, J. F., and Ahmed, A. (2017). Intracellular Calcium Mobilization in Response to Ion Channel Regulators via a Calcium-Induced Calcium Release Mechanism. J. Pharmacol. Exp. Ther. 360 (2), 378–387. doi:10.1124/jpet.116.236695
Pillai, S. (2005). Birth Pangs: the Stressful Origins of Lymphocytes. J. Clin. Invest. 115 (2), 224–227. doi:10.1172/JCI24238
Polverino, F., Wu, T. D., Rojas-Quintero, J., Wang, X., Mayo, J., Tomchaney, M., et al. (2021). Metformin: Experimental and Clinical Evidence for a Potential Role in Emphysema Treatment. Am. J. Respir. Crit. Care Med. 204 (6), 651–666. doi:10.1164/rccm.202012-4510OC
Prasad, S., Gupta, S. C., and Tyagi, A. K. (2017). Reactive Oxygen Species (ROS) and Cancer: Role of Antioxidative Nutraceuticals. Cancer Lett. 387, 95–105. doi:10.1016/j.canlet.2016.03.042
Putney, J. W. (1986). A Model for Receptor-Regulated Calcium Entry. Cell Calcium 7 (1), 1–12. doi:10.1016/0143-4160(86)90026-6
Qin, L., Wang, Z., Tao, L., and Wang, Y. (2010). ER Stress Negatively Regulates AKT/TSC/mTOR Pathway to Enhance Autophagy. Autophagy 6 (2), 239–247. doi:10.4161/auto.6.2.11062
Rana, S. V. S. (2020). Endoplasmic Reticulum Stress Induced by Toxic Elements-A Review of Recent Developments. Biol. Trace Elem. Res. 196 (1), 10–19. doi:10.1007/s12011-019-01903-3
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as Sensors and Regulators of Calcium Signalling. Nat. Rev. Mol. Cel Biol 13 (9), 566–578. doi:10.1038/nrm3412
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic Reticulum-Mitochondria Contacts: Function of the junction. Nat. Rev. Mol. Cel Biol 13 (10), 607–625. doi:10.1038/nrm3440
Russell, R. J., and Brightling, C. (2017). Pathogenesis of Asthma: Implications for Precision Medicine. Clin. Sci. (Lond) 131 (14), 1723–1735. doi:10.1042/CS20160253
Saibil, H. (2013). Chaperone Machines for Protein Folding, Unfolding and Disaggregation. Nat. Rev. Mol. Cel Biol 14 (10), 630–642. doi:10.1038/nrm3658
Sakurada, R., Odagiri, K., Hakamata, A., Kamiya, C., Wei, J., and Watanabe, H. (2019). Calcium Release from Endoplasmic Reticulum Involves Calmodulin-Mediated NADPH Oxidase-Derived Reactive Oxygen Species Production in Endothelial Cells. Int. J. Mol. Sci. 20 (7). doi:10.3390/ijms20071644
Santulli, G., Nakashima, R., Yuan, Q., and Marks, A. R. (2017). Intracellular Calcium Release Channels: an Update. J. Physiol. 595 (10), 3041–3051. doi:10.1113/JP272781
Schögler, A., Caliaro, O., Brügger, M., Oliveira Esteves, B. I., Nita, I., Gazdhar, A., et al. (2019). Modulation of the Unfolded Protein Response Pathway as an Antiviral Approach in Airway Epithelial Cells. Antivir. Res 162, 44–50. doi:10.1016/j.antiviral.2018.12.007
Schuller, H. M. (2019). The Impact of Smoking and the Influence of Other Factors on Lung Cancer. Expert Rev. Respir. Med. 13 (8), 761–769. doi:10.1080/17476348.2019.1645010
Schwarz, D. S., and Blower, M. D. (2016). The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cel Mol Life Sci 73 (1), 79–94. doi:10.1007/s00018-015-2052-6
Sen, N. (2019). ER Stress, CREB, and Memory: A Tangled Emerging Link in Disease. Neuroscientist 25 (5), 420–433. doi:10.1177/1073858418816611
Senft, D., and Ronai, Z. A. (2015). UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends Biochem. Sci. 40 (3), 141–148. doi:10.1016/j.tibs.2015.01.002
Shi, S., Tan, P., Yan, B., Gao, R., Zhao, J., Wang, J., et al. (2016). ER Stress and Autophagy Are Involved in the Apoptosis Induced by Cisplatin in Human Lung Cancer Cells. Oncol. Rep. 35 (5), 2606–2614. doi:10.3892/or.2016.4680
Shirakawa, J., Togashi, Y., Sakamoto, E., Kaji, M., Tajima, K., Orime, K., et al. (2013). Glucokinase Activation Ameliorates ER Stress-Induced Apoptosis in Pancreatic β-cells. Diabetes 62 (10), 3448–3458. doi:10.2337/db13-0052
Singh-Mallah, G., Nair, S., Sandberg, M., Mallard, C., and Hagberg, H. (2019). The Role of Mitochondrial and Endoplasmic Reticulum Reactive Oxygen Species Production in Models of Perinatal Brain Injury. Antioxid. Redox Signal. 31 (9), 643–663. doi:10.1089/ars.2019.7779
Song, S., Tan, J., Miao, Y., and Zhang, Q. (2018). Crosstalk of ER Stress-Mediated Autophagy and ER-Phagy: Involvement of UPR and the Core Autophagy Machinery. J. Cel Physiol 233 (5), 3867–3874. doi:10.1002/jcp.26137
Sozen, E., and Ozer, N. K. (2017). Impact of High Cholesterol and Endoplasmic Reticulum Stress on Metabolic Diseases: An Updated Mini-Review. Redox Biol. 12, 456–461. doi:10.1016/j.redox.2017.02.025
Szarka, A., and Lőrincz, T. (2014). The Role of Ascorbate in Protein Folding. Protoplasma 251 (3), 489–497. doi:10.1007/s00709-013-0560-5
Tanaka, Y., Ishitsuka, Y., Hayasaka, M., Yamada, Y., Miyata, K., Endo, M., et al. (2015). The Exacerbating Roles of CCAAT/enhancer-binding Protein Homologous Protein (CHOP) in the Development of Bleomycin-Induced Pulmonary Fibrosis and the Preventive Effects of Tauroursodeoxycholic Acid (TUDCA) against Pulmonary Fibrosis in Mice. Pharmacol. Res. 99, 52–62. doi:10.1016/j.phrs.2015.05.004
Tanjore, H., Blackwell, T. S., and Lawson, W. E. (2012). Emerging Evidence for Endoplasmic Reticulum Stress in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Physiol. Lung Cel Mol Physiol 302 (8), L721–L729. doi:10.1152/ajplung.00410.2011
Tavender, T. J., and Bulleid, N. J. (2010). Molecular Mechanisms Regulating Oxidative Activity of the Ero1 Family in the Endoplasmic Reticulum. Antioxid. Redox Signal. 13 (8), 1177–1187. doi:10.1089/ars.2010.3230
Tian, J., Popal, M. S., Liu, Y., Gao, R., Lyu, S., Chen, K., et al. (2019). Erratum to "Ginkgo Biloba Leaf Extract Attenuates Atherosclerosis in Streptozotocin-Induced Diabetic ApoE-/- Mice by Inhibiting Endoplasmic Reticulum Stress via Restoration of Autophagy through the mTOR Signaling Pathway". Oxid Med. Cel Longev 2019, 3084083. doi:10.1155/2019/3084083
Tong, B., Fu, L., Hu, B., Zhang, Z. C., Tan, Z. X., Li, S. R., et al. (2021). Tauroursodeoxycholic Acid Alleviates Pulmonary Endoplasmic Reticulum Stress and Epithelial-Mesenchymal Transition in Bleomycin-Induced Lung Fibrosis. BMC Pulm. Med. 21 (1), 149. doi:10.1186/s12890-021-01514-6
Tordjman, S., Chokron, S., Delorme, R., Charrier, A., Bellissant, E., Jaafari, N., et al. (2017). Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol 15 (3), 434–443. doi:10.2174/1570159X14666161228122115
Trouvé, P., Férec, C., and Génin, E. (2021). The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells 10 (11). doi:10.3390/cells10112980
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 287 (5453), 664–666. doi:10.1126/science.287.5453.664
Urra, H., Dufey, E., Avril, T., Chevet, E., and Hetz, C. (2016). Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2 (5), 252–262. doi:10.1016/j.trecan.2016.03.007
van 't Wout, E. F., van Schadewijk, A., van Boxtel, R., Dalton, L. E., Clarke, H. J., Tommassen, J., et al. (2015). Virulence Factors of Pseudomonas aeruginosa Induce Both the Unfolded Protein and Integrated Stress Responses in Airway Epithelial Cells. Plos Pathog. 11 (6), e1004946. doi:10.1371/journal.ppat.1004946
Vander Zanden, C. M., Wampler, L., Bowers, I., Watkins, E. B., Majewski, J., and Chi, E. Y. (2019). Fibrillar and Nonfibrillar Amyloid Beta Structures Drive Two Modes of Membrane-Mediated Toxicity. Langmuir 35 (48), 16024–16036. doi:10.1021/acs.langmuir.9b02484
Vanhoutte, D., Schips, T. G., Vo, A., Grimes, K. M., Baldwin, T. A., Brody, M. J., et al. (2021). Thbs1 Induces Lethal Cardiac Atrophy through PERK-ATF4 Regulated Autophagy. Nat. Commun. 12 (1), 3928. doi:10.1038/s41467-021-24215-4
Venkatesan, A., Satin, L. S., and Raghavan, M. (2021). Roles of Calreticulin in Protein Folding, Immunity, Calcium Signaling and Cell Transformation. Prog. Mol. Subcell Biol. 59, 145–162. doi:10.1007/978-3-030-67696-4_7
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK Is Required at the ER-Mitochondrial Contact Sites to Convey Apoptosis after ROS-Based ER Stress. Cell Death Differ 19 (11), 1880–1891. doi:10.1038/cdd.2012.74
Wang, H. F., Wang, Z. Q., Ding, Y., Piao, M. H., Feng, C. S., Chi, G. F., et al. (2018). Endoplasmic Reticulum Stress Regulates Oxygen-Glucose Deprivation-Induced Parthanatos in Human SH-Sy5y Cells via Improvement of Intracellular ROS. CNS Neurosci. Ther. 24 (1), 29–38. doi:10.1111/cns.12771
Wang, R., Sun, D.-Z., Song, C.-Q., Xu, Y.-M., Liu, W., Liu, Z., et al. (2018). Eukaryotic Translation Initiation Factor 2 Subunit α (eIF2α) Inhibitor Salubrinal Attenuates Paraquat-Induced Human Lung Epithelial-like A549 Cell Apoptosis by Regulating the PERK-eIF2α Signaling Pathway. Toxicol. Vitro 46, 58–65. doi:10.1016/j.tiv.2017.10.006
Wang, S., Gong, L., Mo, Y., Zhang, J., Jiang, Z., Tian, Z., et al. (2022). Resveratrol Attenuates Inflammation and Apoptosis through Alleviating Endoplasmic Reticulum Stress via Akt/mTOR Pathway in Fungus-Induced Allergic Airways Inflammation. Int. Immunopharmacol 103, 108489. doi:10.1016/j.intimp.2021.108489
Wang, X., Wang, H., Xu, B., Huang, D., Nie, C., Pu, L., et al. (2021). Receptor-Mediated ER Export of Lipoproteins Controls Lipid Homeostasis in Mice and Humans. Cel Metab 33 (2), 350–e7. doi:10.1016/j.cmet.2020.10.020
Weissferdt, A. (2014). Large Cell Carcinoma of Lung: On the Verge of Extinction? Semin. Diagn. Pathol. 31 (4), 278–288. doi:10.1053/j.semdp.2014.06.005
Wortel, I. M. N., van der Meer, L. T., Kilberg, M. S., and van Leeuwen, F. N. (2017). Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 28 (11), 794–806. doi:10.1016/j.tem.2017.07.003
Wu, P. S., Yen, J. H., Kou, M. C., and Wu, M. J. (2015). Luteolin and Apigenin Attenuate 4-Hydroxy-2-Nonenal-Mediated Cell Death through Modulation of UPR, Nrf2-ARE and MAPK Pathways in PC12 Cells. PLoS One 10 (6), e0130599. doi:10.1371/journal.pone.0130599
Xu, Y., Sun, D., Song, C., Wang, R., and Dong, X. (2018). MnTMPyP Inhibits Paraquat-Induced Pulmonary Epithelial-like Cell Injury by Inhibiting Oxidative Stress. J. Toxicol. Sci. 43 (9), 545–555. doi:10.2131/jts.43.545
Xu, Y., Wang, W., Jin, K., Zhu, Q., Lin, H., Xie, M., et al. (2017). Perillyl Alcohol Protects Human Renal Tubular Epithelial Cells from Hypoxia/reoxygenation Injury via Inhibition of ROS, Endoplasmic Reticulum Stress and Activation of PI3K/Akt/eNOS Pathway. Biomed. Pharmacother. 95, 662–669. doi:10.1016/j.biopha.2017.08.129
Yang, Y., Cheng, B. J., Jian, H., Chen, Z. W., Zhao, Y., Yu, Y. F., et al. (2017). XBP1-LOX Axis Is Critical in ER Stress-Induced Growth of Lung Adenocarcinoma in 3D Culture. Am. J. Transl Res. 9 (2), 700–707.
Ye, L., Zeng, Q., Ling, M., Ma, R., Chen, H., Lin, F., et al. (2021). Inhibition of IP3R/Ca2+ Dysregulation Protects Mice from Ventilator-Induced Lung Injury via Endoplasmic Reticulum and Mitochondrial Pathways. Front. Immunol. 12, 729094. doi:10.3389/fimmu.2021.729094
Zeeshan, H. M., Lee, G. H., Kim, H. R., and Chae, H. J. (2016). Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 17 (3), 327. doi:10.3390/ijms17030327
Zeng, M., Sang, W., Chen, S., Chen, R., Zhang, H., Xue, F., et al. (2017). 4-PBA Inhibits LPS-Induced Inflammation through Regulating ER Stress and Autophagy in Acute Lung Injury Models. Toxicol. Lett. 271, 26–37. doi:10.1016/j.toxlet.2017.02.023
Zhang, X., Huang, R., Zhou, Y., Zhou, W., and Zeng, X. (2020). IP3R Channels in Male Reproduction. Int. J. Mol. Sci. 21 (23). doi:10.3390/ijms21239179
Zhang, X., Zhang, H. Q., Zhu, G. H., Wang, Y. H., Yu, X. C., Zhu, X. B., et al. (2012). A Novel Mono-Carbonyl Analogue of Curcumin Induces Apoptosis in Ovarian Carcinoma Cells via Endoplasmic Reticulum Stress and Reactive Oxygen Species Production. Mol. Med. Rep. 5 (3), 739–744. doi:10.3892/mmr.2011.700
Zhang, Y., Cen, J., Jia, Z., Hsiao, C. D., Xia, Q., Wang, X., et al. (2019). Hepatotoxicity Induced by Isoniazid-Lipopolysaccharide through Endoplasmic Reticulum Stress, Autophagy, and Apoptosis Pathways in Zebrafish. Antimicrob. Agents Chemother. 63 (5). doi:10.1128/AAC.01639-18
Zhang, Z., Zhang, L., Zhou, L., Lei, Y., Zhang, Y., and Huang, C. (2019). Redox Signaling and Unfolded Protein Response Coordinate Cell Fate Decisions under ER Stress. Redox Biol. 25, 101047. doi:10.1016/j.redox.2018.11.005
Zhao, Y., Zhu, C., Li, X., Zhang, Z., Yuan, Y., Ni, Y., et al. (2011). Asterosaponin 1 Induces Endoplasmic Reticulum Stress-Associated Apoptosis in A549 Human Lung Cancer Cells. Oncol. Rep. 26 (4), 919–924. doi:10.3892/or.2011.1358
Zhu, X. M., Yao, F. H., Yao, Y. M., Dong, N., Yu, Y., and Sheng, Z. Y. (2012). Endoplasmic Reticulum Stress and its Regulator XBP-1 Contributes to Dendritic Cell Maturation and Activation Induced by High Mobility Group Box-1 Protein. Int. J. Biochem. Cel Biol 44 (7), 1097–1105. doi:10.1016/j.biocel.2012.03.018
Keywords: er stress, unfolded protein response, pulmonary disease, reactive oxygen spieces, oxidative stress
Citation: Cui X, Zhang Y, Lu Y and Xiang M (2022) ROS and Endoplasmic Reticulum Stress in Pulmonary Disease. Front. Pharmacol. 13:879204. doi: 10.3389/fphar.2022.879204
Received: 19 February 2022; Accepted: 11 March 2022;
Published: 26 April 2022.
Edited by:
Arunachalam Karuppusamy, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Lingrui Zhang, Purdue University, United StatesCopyright © 2022 Cui, Zhang, Lu and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Zhang, eWFuZ2hlZG9uZ2hhaUAxNjMuY29t
†These authors share first authorship and share co-first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.