
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
TECHNOLOGY AND CODE article
Front. Pharmacol., 26 April 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.874746
This article is part of the Research TopicChemoinformatics Approaches to Structure- and Ligand-Based Drug Design, Volume IIView all 20 articles
 Vishwesh Venkatraman1*†
Vishwesh Venkatraman1*† Thomas H. Colligan2
Thomas H. Colligan2 George T. Lesica2
George T. Lesica2 Daniel R. Olson2
Daniel R. Olson2 Jeremiah Gaiser2
Jeremiah Gaiser2 Conner J. Copeland2
Conner J. Copeland2 Travis J. Wheeler2*†
Travis J. Wheeler2*† Amitava Roy2,3†
Amitava Roy2,3†The SARS-CoV2 pandemic has highlighted the importance of efficient and effective methods for identification of therapeutic drugs, and in particular has laid bare the need for methods that allow exploration of the full diversity of synthesizable small molecules. While classical high-throughput screening methods may consider up to millions of molecules, virtual screening methods hold the promise of enabling appraisal of billions of candidate molecules, thus expanding the search space while concurrently reducing costs and speeding discovery. Here, we describe a new screening pipeline, called drugsniffer, that is capable of rapidly exploring drug candidates from a library of billions of molecules, and is designed to support distributed computation on cluster and cloud resources. As an example of performance, our pipeline required ∼40,000 total compute hours to screen for potential drugs targeting three SARS-CoV2 proteins among a library of ∼3.7 billion candidate molecules.
The war against viruses is largely fought using vaccines and therapeutic drugs. As of December 2021, there are 55 FDA-approved vaccines against 19 human viruses (FDA, 2021), while only three viruses are targeted by approved antiviral drugs (FDA, 2020b). This disparity is particularly visible in the context of the ongoing SARS-CoV2 pandemic, in which vaccines were produced at a remarkable speed and with excellent effectiveness (FDA, 2020a; Wouters et al., 2021), while effective antiviral agents (Mahase, 2021; Jayk Bernal et al., 2022) only arrived 2 years into the pandemic, and with very limited availability. Despite vaccine success, there remains a vital need for development of effective antiviral drugs due to a combination of vaccine hesitancy, incomplete vaccine availability, breakthrough infection risk, and the continued emergence of viral variants (Kaplan and Milstein, 2021). Beyond SARS-CoV2, the cost and limited exploratory scope of current drug discovery pipelines will hamper efforts to quickly respond to future pandemic needs, and are an obstacle to development of antiviral drugs for viruses primarily afflicting relatively poor populations (Adamson et al., 2021).
Modern drug development efforts rely on high-throughput screening (HTS) analysis, which involves automated physical evaluation of activity across a library of thousands to millions of candidate small-molecule drugs (Berdigaliyev and Aljofan, 2020). HTS can be complemented by computer-aided drug design (CADD) and virtual screening (VS), in which interactions between small-molecules and a targets are estimated using computational models. In particular, computational analysis holds the promise of enabling expansion of the number of considered molecules from millions to billions.
VS strategies are traditionally divided into two categories: ligand-based (LBVS) and structure-based (SBVS) methods. In LBVS methods, a known active ligand is used as the basis for a search for chemically and structurally similar molecules (Ripphausen et al., 2011), with no consideration of the target protein. In SBVS approaches, small molecules are computationally docked into target binding sites to estimate their activities (Maia et al., 2020); this approach depends on availability of structural information, and is computationally intensive. The two methods can be integrated either by combining results (Wilson and Lill, 2011; Wang et al., 2020), or by using LBVS methods to quickly establish a set of candidates subjected to subsequent SBVS docking analysis (Drwal and Griffith, 2013).
Table 1 provides a list of various open access VS tools. For large scale virtual screening of compound libraries, software pipelines such as VSpipe Álvarez-Carretero et al. (2018), VirtualFlow Gorgulla et al. (2020, 2021), AMIDEDarme et al. (2021) have been used. Many of these approaches make use of SBVS and facilitate the use of a variety of docking Bender et al. (2021) programs with significant emphasis on scaling the calculations. Recent GPU acceleration of docking (Santos-Martins et al., 2021) has improved throughput, but resource requirements are still exceedingly high. For example, an effort to performing one billion docking assays was reported to require 664K GPU hours and 4.64M core hours for a single VS analysis (Acharya et al., 2020). With the aim of automating hit-selection protocols and minimizing human intervention, artificial intelligence-driven VS. pipeline have also been introduced Gentile et al. (2020), Gentile et al. (2021); Yaacoub et al. (2021).
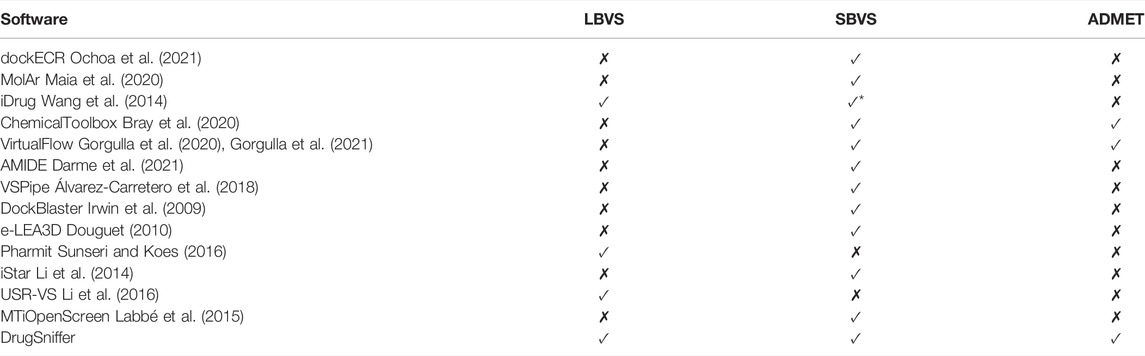
TABLE 1. Several open access software tools for virtual screening. In a number of the tools, such as dockECR and VirtualFlow, multiple docking programs are used to predict scores between a single target or multiple targets (merging and shrinking approach) and a library of compounds. The AMIDE software carries out large-scale chemical ligand docking over a large dataset of proteins with the aim of identifying potential side effects of new drugs. iDrug, Pharmit (for structure-based pharmacophore modeling), iStar, e-LEA3D, USR-VS (3D shape-based similarity), MTiOpenScreen and ChemicalToolbox are web-based platforms for computer-aided drug design. ChemicalToolbox allows for integration with other tools and workflows (molecular dynamics) that are part of the Galaxy software framework (https://galaxyproject.org/). e-LEA3D uses a de novo drug design strategy in which fragments or combination of fragments that fit a QSAR model or the binding site of a protein are identified. * iDrug uses a pocket structure to define the pharmacophore descriptors needed for LBVS. However, they do not explicitly calculate the interaction between a ligand and the pocket, such as docking. In our opinion, they are marginally SBVS.
Herein, we describe our development and release of an open source, massively-scalable LBVS-filtered SBVS pipeline, called drugsniffer, that is designed to achieve the goal of virtually screening bioactive drugs from datasets of billions of probably-synthesizable small molecules in a much-reduced time budget. Drugsniffer is easy to install and manages the distribution of computation across cluster or cloud resources. It reduces the computational burden to 10s of thousands of compute hours for search across a library of billions of candidate molecules, and provides a framework in which future methodological advances can be incorporated and evaluated. Using an early iteration of drugsniffer, we assessed ∼3.7B molecules for binding potential against 3 SARS-CoV2 proteins (22 binding pockets), with total computational investment of ∼40 K compute hours. The results of our analysis were accepted as a finalist in Joint European Disruptive Initiative (JEDI) “billion molecules against COVID19” challenge (Le et al., 2021).
Drugsniffer consists of the following phases (see Figure 1): 1) select the protein target and determine its structure, 2) identify binding pockets, 3) design de novo ligands for each pocket, 4) use these as seeds to identify similar molecules in a large composite database of synthesizable small molecules, 5) perform in silico docking assays on these candidates, 6) apply a new neural network model to predict and rank binding affinity based on features of the docked poses, 7) identify potential toxicity of compounds using a custom ADMET filter. In this section, we describe these stages in detail, then discuss our application of an early implementation of the pipeline to the JEDI COVID19 Grand Challenge.
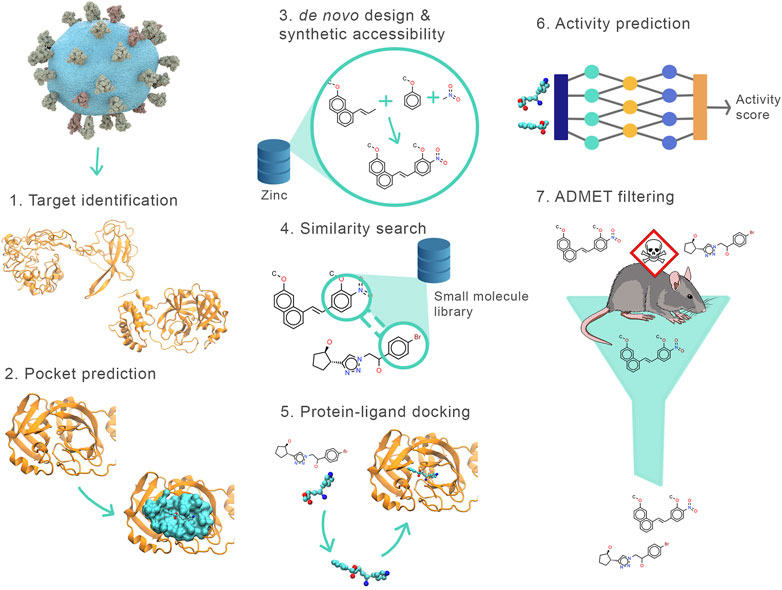
FIGURE 1. Outline of the drugsniffer virtual screening pipeline. The stages include (1) model the targets (e.g., using AlphaFold or crystal structure where available), (2) identify possible binding sites/pockets (e.g., using FPocket), (3) design multiple de novo ligands for the target pockets using AutoGrow, (4) use the designed molecules as seeds to identify similar compounds from small-molecule libraries (using ECFP4 fingerprints as found in RDKit), (5) dock the molecules (using AutoDock Vina) identified by the similarity search and calculate the interaction energy between the target and the docked poses, (6) re-score the best-docked poses of all the molecules using our new scoring function (terms for the function are provided by SMINA and DLIGAND2), (7) identify potentially toxic compounds using our fast ADMET analyzer (using FP-ADMET).
The first step in the drug screening process is the selection of the target protein–the user must provide a structural model for the selected protein. Drugsniffer is agnostic about the source of the structural model, and will work with experimentally-validated or computationally-predicted structures. Though protein structures may be retrieved from a variety of sources, we have had good experiences with ChimeraX (Pettersen et al., 2021), which, for example, supports retrieval of structures from the Protein Data Bank (Berman et al., 2000)) or prediction using AlphaFold2 (Jumper et al., 2021). AlphaFold2 achieved remarkable accuracy in the CASP14 competition; for example, in 92.5% of predictions, all side chain atoms are predicted with error ≤ 5 Å (Pereira et al., 2021). This accuracy is unprecedented for computational models, and these models may provide insight into the diversity of conformations that extend beyond the single conformer of a crystal-based structure. Even so, a substantial fraction of the predicted atoms, primarily from the flexible parts of the proteins, may not be modeled correctly by AlphaFold2. We encourage users to evaluate the overall (IDDT) and residue-specific (pLDDT) scores to evaluate the predicted accuracy of the overall and pocket regions of an AlphaFold2 model.
In addition to a target protein structure, drugsniffer must be provided with at least one pocket descriptor, as well as a preferred pocket box size. The most reliable way of detecting a ligand-binding pocket is a user’s prior knowledge about the binding pocket from experience, experimental evidence, and literature search. Computational identification of a pocket-like region is challenging and an active area of research (Zhao et al., 2020). The drugsniffer pipeline includes a copy of the cavity detection software Fpocket (Le Guilloux et al., 2009) only because it is a stand-alone free program. We encourage users to use multiple pocket search algorithms, such as FTMAP Kozakov et al. (2015), POCASA Yu et al. (2010), and molecular dynamics simulations, and use their judgment to define a pocket-like region in the protein. The current implementation of the drugsniffer pipeline produces an FPOCKET output that includes all predicted pockets; the user is tasked with manually reviewing these and identifying the subset for which the downstream drug discovery stages should be performed, e.g., using ChimeraX or PyMol (Oliveira et al., 2014). Pocket descriptors identified outside of the drugsniffer pipeline may be provided as an alternative or supplementary source of predicted pockets. Box size must be determined for each pocket; we recommend basing this on the scheme proposed by (Feinstein and Brylinski, 2015).
Following manual pocket identification, drugsniffer accepts as input the set of targeted pockets, and proceeds in an automatic fashion through the remaining stages. In the first stage, a large number of candidate ligand molecules are designed from scratch using the software AutoGrow4 (Spiegel and Durrant, 2020), which employs a genetic algorithm to evolve ligands from building blocks obtained from the ZINC library (Sterling and Irwin, 2015). AutoGrow4 utilizes a diversity score that acts as a secondary fitness metric and is used to select seed compounds that are structurally unique from previous generations. The molecules are subsequently docked into the pockets of the specified target protein using QuickVina (Alhossary et al., 2015) which is a faster version of Autodock Vina. Docked results are ranked based on the Vina docking score of the top ranking pose. A Lipinski RO5 filter is used to exclude candidate structures that do not satisfy drug-like criteria. The NIH filter (Jadhav et al., 2010) is also included to screen against compounds containing undesirable functional groups. AutoGrow4 performs in silico chemical reactions (Durrant and McCammon, 2012) derived from a set of robust organic reactions (Hartenfeller et al., 2011) to generate new child compounds from a parent molecule. These reaction-based structural transformations are used to increase the likelihood of the designed molecules being synthetically accessible. However, a drawback of using pre-defined reaction schemes is that they may match reaction handles and fail to consider the presence of competing functionalities that can compromise the reaction outcome (Ghiandoni et al., 2020; Meyers et al., 2021). By default, the pipeline runs AutoGrow4 for 10 generations, and captures 150 de novo molecules from each of the final three generations. Drugsniffer can optionally forgo this AutoGrow4 step, and instead accept a collection of ligands provided by the user–these may be sourced from some prior de novo computation, or from a collection of co-crystallized protein-ligand complexes.
The motivation for employing de novo ligand design is to produce drug-like compounds that can mimic known inhibitors or potentially active ligands with a diversity of chemical structures. While the molecules produced by AutoGrow4 are predicted to be synthesizable, factors such as establishing synthetic routes, material procurement, costs and time involved are difficult to predict. We therefore sought to build on the value of these designed molecules through an LBVS search strategy in which the de novo molecules serve as seeds in a search for similar compounds within a massive library of molecules.
We compiled a collection of molecules from various small-molecule libraries, with the aim of capturing a large diversity of molecules that either already exist, or are likely-synthesizable and can be made to order (see Table 2). The Enamine collection includes more than 1 billion compounds that comply with Lipinski’s rule of five (RO5) criteria and are expected to be realized in 1–3 synthesis steps. The Synthetically Accessible Virtual Inventory (SAVI) (Patel et al., 2020) contains over 1 billion reliably-synthesizable compounds generated through expert-system rules. GDB-13 (Blum and Reymond, 2009) also contains over 1 billion compounds (containing up to 13 atoms of C, N, O, S, and Cl = , generated according to chemical stability and synthetic feasibility rules. PubChem (Kim et al., 2020), ZINC (Sterling and Irwin, 2015), and Molport are curated collections of commercially-available molecules. SweetLead (Novick et al., 2013) and DrugBank (Wishart et al., 2017) contain drugs that are in use or in clinical trials, and may therefore facilitate repurposing of established drugs. We removed molecules containing salts, because downstream docking methods fail in the face the apparent disjoint molecules. The full de-duplicated collection contains ∼3.7 billion unique molecules.
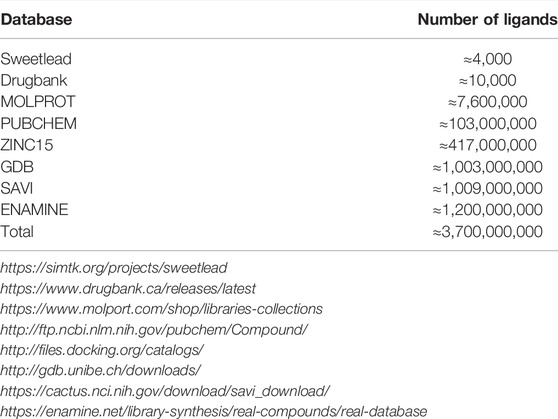
TABLE 2. The small molecule databases searched as part of the VS protocol.
To identify library-sourced compounds similar to the de novo seeds produced by AutoGrow4, 1024-bit ECFP4 fingerprints (O’Boyle and Sayle, 2016) are computed for all ∼3.7 billion library compounds. The ECFP4 fingerpint is a 1024-element binary vector that encodes structural and chemical features. Though a multitude of fingerprint strategies exist, ECFP4 has been reported to effectively rank diverse structures by similarity (O’Boyle and Sayle, 2016). Future releases of drugsniffer will enable selection of other fingerprints, or related similarity measures. ECFP4 fingerprints are computed using RDKIT (https://www.rdkit.org), then stored as a sequence of 1,024 bit vectors, so that a library of 3.7 billion molecules is represented by a ∼475 Gbyte fingerprint database. Fingerprints are similarly computed for all seeds. A measure of similarity between two molecules is computed by comparing the 1024-bit fingerprints of each molecule, using the Tanimoto coefficient (aka Jacaard index): the ratio of the intersecting set (number of bits set to one in both fingerprints) to the union set (number of bits set to one in at least one of the two fingerprints) (Bajusz et al., 2015). Similar (“neighbor”) molecules are identified by computing the Tanimoto coefficient for each seed against each molecule in the fingerprint database using SIMD vectorized bit-level comparison over 1,024 representative bits per molecule. By default, neighbors with Tanimoto similarity
For the seed-neighbor molecules identified by the similarity search, initial 3D coordinates are generated from the SMILES representations using OpenBabel (O’Boyle et al., 2011a). Diverse low-energy conformers for the molecules are generated using the Confab (O’Boyle et al., 2011b), then the lowest energy conformation is retained. These optimized structures of neighbors are docked into their respective targets using AutoDock Vina (Trott and Olson, 2010). The number of docking poses produced and the exhaustiveness parameter for the search for each ligand are parameterized by the user; the default values are 9 and 4, respectively.
AutoDock Vina reports a set of molecular poses within the pocket, along with a value representing a prediction of the quality of each docked pose. Because this prediction is only a loose estimate of actual binding affinity, a variety of post hoc re-scoring methods have been devised [e.g., see (Koes et al., 2013a; Chen et al., 2019; McNutt et al., 2021)]. Drugsniffer can report either the Autodock Vina score, the SMINA (Koes et al., 2013b) rescoring value, or the result of a new neural network re-scoring strategy that we have produced for this workflow (dock2bind, which is the default). Drugsniffer supports retraining of this model with domain-specific binding affinity data, and also will accept an alternate re-scoring function that is injected by the user into the drugsniffer wokflow by providing a Docker container meeting a simple documented API.
For each docked pose, our dock2bind receives 16 pose descriptors calculated by SMINA, along with the DFIRE estimate of protein–ligand potential (Chen et al., 2019), and computes a new affinity estimate for the pose. This estimate is a value between 0 and 1 and can be thought of as the model’s confidence that the molecule binds to the pocket, constrained by the specific pose. See Figure 2 for model details. Ligand-protein pairs were taken from the DUD-E benchmark (Mysinger et al., 2012) and LIT-PCBA (Tran-Nguyen et al., 2020). To train the model, docked poses were generated for ∼14,000 ligand-protein pairs from the DUD-E dataset, along with ∼800,000 decoy ZINC-sourced compounds docked to the same protein partners. These were supplemented with an additional ∼4,000 ligand-protein complexes from LIT-PCBA, and ∼121,000 decoys docked to the same proteins. The active:decoy ratio is intended to reflect the large actual classification imbalance (most molecules are inactive for any specific target). For each target, 9 docked poses were produced, and the pose with the best SMINA score was provided to the dock2bind model for training.
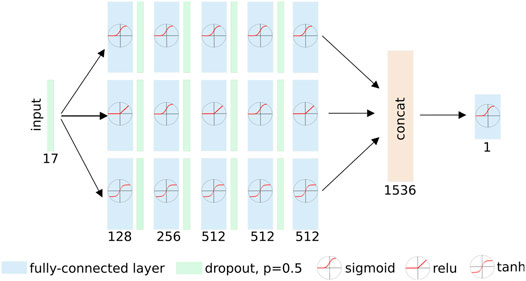
FIGURE 2. Affinity prediction model. The model consists of three separate paths from input to output, each composed of five sequential fully-connected layers. Each path uses a separate set of activation functions, allowing the network to learn diverse representations of the input. The outputs of the three paths are concatenated and passed through a final fully-connected (FC) layer that emits a prediction of binding or non-binding. Fully-connected (FC) layers are represented with blue blocks. The number of nodes in each FC layer is indicated below the block. Activation functions applied to the output of the FC layers are shown in circles. The model was trained for 2000 epochs and batch size 8,192 with the Adam (Diederik and Ba, 2014) optimizer using default β1,2 parameters and a learning rate of 0.001. Dropout (Srivastava et al., 2014) with p = 0.5 was applied after each fully connected layer during training, and also during validation.
Drugsniffer includes a suite of models to predict properties tied to bioavailability and safety. Owing to their ease of computation, molecular fingerprints have been frequently used to predict these properties (Kim and Nam, 2017; Ai et al., 2018; Yang et al., 2019). Fingerprint-based classification models were trained on experimental data available [see (Venkatraman, 2021)] for solubility in dimethyl sulfoxide (DMSO), blood brain barrier permeability, human intestinal absorption (HIA), AMES mutagenicity, HERG cardiotoxicity, drug induced liver injury (DILI), Cytochrome p450 interaction (CYP3A4 and CYP2C9 isoforms), metabolic stability and acute LD50 toxicity based on the criteria defined by the Environmetal Protection Agency (EPA). For each property, various fingerprints (Hinselmann et al., 2011) (substructure and extended/functional connectivity fingerprints) were evaluated for their discriminant ability and the fingerprint model [using random forests (Breiman, 2001)] yielding the best balanced accuracy (Brodersen et al., 2010; Venkatraman, 2021) was retained. The drugsniffer pipeline applies these models to the list of candidates produced by previous stages, and appends the resultant vector of properties to the affinity prediction results. The models can be accessed at https://gitlab.com/vishsoft/fpadmet.
Drugsniffer is implemented as a Nextflow workflow (Di Tommaso et al., 2017) that orchestrates the activity of a curated set of open source tools, and supports analysis in cluster (SLURM) and cloud (AWS) environments. Table 3 lists the different software tools that are used in the workflow. The workflow depends on a collection of Docker containers and runner scripts wrapping each of our own tools as well as the external open source tools included in the analysis pipeline. This organizing principle makes it possible for the user to configure and run drugsniffer without concern for dependencies. Docker container files, NextFlow scripts, and tool code are all available via GitHub (https://github.com/TravisWheelerLab/drug-sniffer). Versioned Docker container images are published in the GitHub containiner registry, and the full library of ∼3.7 billion molecules (with pre-computed fingerprints) is housed in a persistent OSF repository (Soderberg, 2018) and. Instructions for download and use are found at http://drugsniffer.org.

TABLE 3. Software used in the VS pipeline.
In May 2020, the Joint European Disruptive Initiative (JEDI) launched a “Grand Challenge” competition intended to motivate development of methods capable of searching a library of billions of molecules for those with potentially good binding affinity for target SARS CoV2 proteins. We developed drugsniffer to meet these goals, and submitted candidate molecules identified with an early version of the piepeline. Our submissions have reached the finalist stage, and are currently under experimental review. Here, we describe how our pipeline was used to prepare our submission, and document the differences between the version of the pipeline used for our JEDI submission and its current released form.
To begin, we selected three target proteins: RNA dependent RNA polymerase (Non-structural Protein 12, aka NSP12), 3C like protease (3CLPro), and Nucleocapsid protein (N). At the time of the analysis, no whole-protein experimental structure was available for any of the targets and AlphaFold2 was not yet released. We therefore downloaded models created by I-TASSER (Yang et al., 2015), and added hydrogen atoms with CHARMM (Brooks et al., 2009).
Candidate binding pockets for the three selected targets were identified using a combination of literature search and results from the tools FTMAP (Kozakov et al., 2015) and POCASA (Yu et al., 2010) (drugsniffer incorporates Fpocket in lieu of these, because its license allows redistribution). Seven pocket-like regions were identified: 2 each for N and 3CLpro, and 3 for NSP12. Some of the pocket-like regions were too large to be occupied by a typical-sized ligand. Consequently, the larger pocket-like regions were subdivided into smaller pockets. A total of 22 pockets were finalized as targets: 8 each for N and NSP12 and 6 for 3CLPro. We searched the literature to identify any glycosylation sites for the three selected targets and did not find any. We also used N-GlyDe (Pitti et al., 2019) to identify any potential sites for N-linked glycans. Our predicted glycosylation sites are residue 269 of N and residues 767 and 911 of NSP12. As none of the glycosylation sites were near any of the predicted binding pockets, we did not consider glycosylation for our later docking exercises.
The next several pipeline stages were run as in the current release of the pipeline, including de novo ligand design, molecular similarity search, and protein-ligand docking. AutoGrow4 was run for 25 generations, over five independant runs. In total, 31,962 seed molecules were identified by AutoGrow4 (12,227 for nsp12 pockets, 14,334 for N pockets, and 5,401 for 3CLPro pockets). Molecular similarity search identified ∼97,000 library compounds with Tanimoto similarity
Here, we have described the stages and availability of a new pipeline for exploring a pre-built library of billions of likely-synthesizable molecules for a small set of candidate molecules that are expected to show good binding affinity to a user-provided protein structure and pocket descriptor. As a proof of principle, we used a variant of this pipeline to identify drug candidates from our library of ∼3.7 billion molecules, targeting 22 pockets in 3 proteins associated with SARS-CoV2, resulting in a list of ∼30,000 candidate compounds. This collection was submitted for analysis to the JEDI “Grand Challenge,” and were advanced to “finalist” status; experimental review of a subset of these molecules is underway. Compute time for the total search for candidate molecules for all 22 pockets was ∼40,000 CPU hours. By distributing workload across a cluster, the analysis required only a few days. In addition to these run time results, we explored the efficacy of our custom docking re-scoring model, as well as the outcomes of ADMET and synthesizability analysis.
To quantitatively evaluate our model, a test set was developed from DUD-E and LIT-PCBA, consisting of complexes involving proteins not found in the training set. A total of ∼3000 DUD-E ligand-protein pairs, ∼186,000 decoys for DUD-E proteins, ∼900 LIT-PCBA ligand-protein pairs, and ∼27,000 decoys for LIT-PCBA. No hyperparameter tuning was performed on any of the models so a validation set was unnecessary. To test the efficacy of our method of ranking potential binders, we compared our method to a variety of open-source implementations of affinity-predicting methods, including Vina’s default method, the SMINA default score, and the NNScore and RF-score (version 3) from the Open Drug Discovery Toolkit (Wójcikowski et al., 2015) (ODDT). Figure 3 shows the performance of the model architecture trained on different subsets of the data.
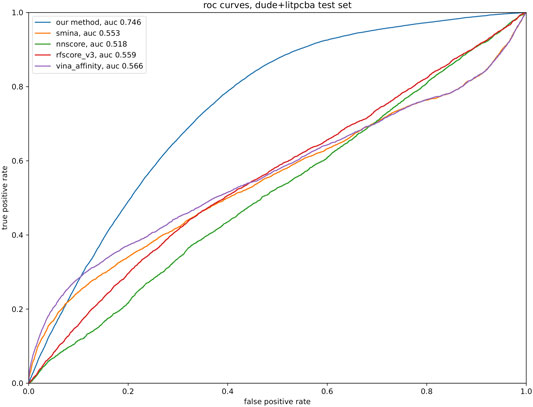
FIGURE 3. Test data consisting of 3,900 ligand-protein pairs and 213,000 decoy-protein pairs was analyzed with the tools listed in the legend, with the relevant tool producing a binding affinity estimate for each pair. Default parameters were used for all tools; our model was trained as described in the text. A ROC curve was produced for each tool, based on the sorted list of predicted affinity.
Figure 4 shows the distribution of the ADMET properties for the ∼30,000 compounds that were submitted to the JEDI competition. For the most part, the shortlisted compounds were predicted to have favourable ADMET properties. Our ML model for DILI (Venkatraman, 2021) predicts a majority (∼85%) of the compounds to be hepatotoxic. The DILI model however only provides a binary (yes/no) prediction and does not indicate the level of the underlying DILI severity. A strict application of the models (i.e., selecting only those compounds that are deemed to be favourable across all calculated properties) yielded a set of 1,635 compounds. Many ADMET properties are affected by the dosage, route and frequency. For better assessment of ADMET, knowledge of the underlying mechanisms is required. Given that it is far from trivial to prioritize one property over the other (leading to varying application of the filter), we have used the model predictions as a guide rather than a filter. With respect to synthesizability, ∼79% of molecules identified by the pipeline were predicted to require three or fewer predicted reaction steps.
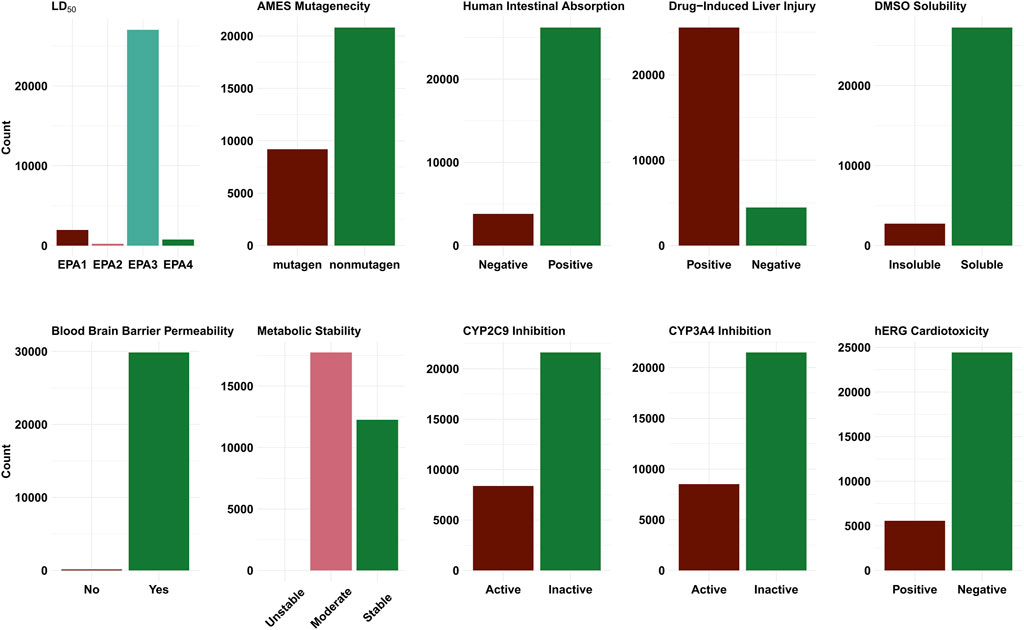
FIGURE 4. Bar plots showing the distribution of the predicted classes for the different ADMET endpoints. EPA1 corresponds to the (LD50 ≤ 50 mg/kg) the highest toxicity category. EPA2 (moderately toxic) includes chemicals with 50
Virtual screening has seen a recent rise in prominence, supported by improved computational methods across the range of analyses represented in the drugsniffer pipeline. The ongoing pandemic has highlighted the need for improved speed and increased exploratory scope of virtual screening methods. Relatedly, the development of low-cost virtual screening methods holds the promise of improving opportunities for development of drugs targeting diseases prevalent in low-income regions, for which economic incentives discourage expensive high-throughput screening assays. We developed drugsniffer as a preliminary tool to meet this need, exploring billions of candidate molecules for a target protein pocket in a few thousand compute hours–relatively modest resources available to most HPC infrastructures. Even with its development, each of the stages of the drugsniffer pipeline will be well-served by methodological advances. We highlight a few such areas of opportunity here, and observe that drugsniffer can easily adapt to incorporate advances along these lines, due to its modular nature.
With the development and release of AlphaFold2 and similar structure prediction methods, structure prediction is perhaps no longer a general bottleneck in the drug discovery problem, though some protein types still suffer from relatively uncertain predictions. Pocket identification remains a challenge, and most current techniques can detect pockets only with ∼60% accuracy (Zhao et al., 2020). Advances in this field will reduce the dependency on expert manual analysis of structures and pockets.
Drugsniffer will also be improved by development of advances in de novo molecule production (where limitations include wall clock run time and molecule synthesizability and utility), molecular similarity search (where current molecule-centric approaches fail to account for pocket-specific interaction profiles), and docking-based affinity prediction (where re-scoring methods produce only modestly enrichment for actives vs. decoys (see Figure 3) and may not generalize well to structures that are not represented in the training set). Drugsniffer will be expanded by including molecular dynamics simulations to consider multiple conformations of a pocket region and refining binding energy estimation of shortlisted ligands. It should be emphasized that the scope of the drugsniffer pipeline is to identify possible ligands with high enrichment factors. Users should carry out such MD or QM studies on the possible ligands predicted by the drugsniffer for a more accurate prediction of binding affinity or to investigate the effect of protonation states in binding. Due to their approximate nature, docking forcefields are insensitive to such details.
Publicly available datasets were analyzed in this study. This data can be found here: http://drugsniffer.org.
VV, AR, and TW designed the pipeline, along with the study of application to SARS-CoV2 proteins; they also supervised efforts of others and collectively wrote the first draft of the manuscript. AR and VV developed approaches for identifying proteins, structures, pockets, and de novo seeds; they also collected the molecule library. TW, VV, and JG developed methods for molecule fingerprinting and rapid neighbour identification, and applied to SARS-Cov2 data. CC and GL incorporated docking into the pipeline. TC and DO developed the machine learning model for docking re-scoring. GL developed the NextFlow workflow, and all associated Docker images. All authors contributed to the manuscript edits.
VV acknowledges financial support from the Research Council of Norway (Grant No. 262152). AR acknowledges funding from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Department of Health and Human Services under BCBB Support Services Contract HHSN316201300006W/HHSN27200002 to MSC, Inc. The remainder of co-authors acknowledge support from the National Institute of General Medical Sciences (NIH NIGMS, R01GM132600) and the Genomic Science program (GSP) of the Office of Biological and Environmental Research in the Department of Energy (DE-SC0021216).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This work would not have been possible without the computational resources of the NIH HPC Biowulf cluster, and the University of Montana’s Griz Shared Computing Cluster (GSCC). We thank Rose Perry-Gottschalk, NIAID, RTB, NIH for help with the visual arts.
Acharya, A., Agarwal, R., Baker, M. B., Baudry, J., Bhowmik, D., Boehm, S., et al. (2020). Supercomputer-based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19. J. Chem. Inf. Model. 60, 5832–5852. doi:10.1021/acs.jcim.0c0101010.26434/chemrxiv.12725465
Adamson, C. S., Chibale, K., Goss, R. J. M., Jaspars, M., Newman, D. J., and Dorrington, R. A. (2021). Antiviral Drug Discovery: Preparing for the Next Pandemic. Chem. Soc. Rev. 50, 3647–3655. doi:10.1039/d0cs01118e
Ai, H., Chen, W., Zhang, L., Huang, L., Yin, Z., Hu, H., et al. (2018). Predicting Drug-Induced Liver Injury Using Ensemble Learning Methods and Molecular Fingerprints. Toxicol. Sci. 165, 100–107. doi:10.1093/toxsci/kfy121
Alhossary, A., Handoko, S. D., Mu, Y., and Kwoh, C.-K. (2015). Fast, Accurate, and Reliable Molecular Docking with QuickVina 2. Bioinformatics 31, 2214–2216. doi:10.1093/bioinformatics/btv082
Álvarez-Carretero, S., Pavlopoulou, N., Adams, J., Gilsenan, J., and Tabernero, L. (2018). VSpipe, an Integrated Resource for Virtual Screening and Hit Selection: Applications to Protein Tyrosine Phospahatase Inhibition. Molecules 23, 353. doi:10.3390/molecules23020353
Bajusz, D., Rácz, A., and Héberger, K. (2015). Why Is Tanimoto index an Appropriate Choice for Fingerprint-Based Similarity Calculations? J. Cheminf. 7, 1–13. doi:10.1186/s13321-015-0069-3
Bender, B. J., Gahbauer, S., Luttens, A., Lyu, J., Webb, C. M., Stein, R. M., et al. (2021). A Practical Guide to Large-Scale Docking. Nat. Protoc. 16, 4799–4832. doi:10.1038/s41596-021-00597-z
Berdigaliyev, N., and Aljofan, M. (2020). An Overview of Drug Discovery and Development. Future Med. Chem. 12, 939–947. doi:10.4155/fmc-2019-0307
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. doi:10.1093/nar/28.1.235
Blum, L. C., and Reymond, J.-L. (2009). 970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13. J. Am. Chem. Soc. 131, 8732–8733. doi:10.1021/ja902302h
Bray, S. A., Lucas, X., Kumar, A., and Grüning, B. A. (2020). The ChemicalToolbox: Reproducible, User-Friendly Cheminformatics Analysis on the Galaxy Platform. J. Cheminform. 12, 40. doi:10.1186/s13321-020-00442-7
Brodersen, K. H., Ong, C. S., Stephan, K. E., and Buhmann, J. M. (2010). “The Balanced Accuracy and its Posterior Distribution,” in 2010 20th International Conference on Pattern Recognition, 3121–3124. doi:10.1109/icpr.2010.764
Brooks, B. R., Brooks, C. L., Mackerell, A. D., Nilsson, L., Petrella, R. J., Roux, B., et al. (2009). Charmm: the Biomolecular Simulation Program. J. Comp. Chem. 30, 1545–1614. doi:10.1002/jcc.21287
Chen, P., Ke, Y., Lu, Y., Du, Y., Li, J., Yan, H., et al. (2019). DLIGAND2: an Improved Knowledge-Based Energy Function for Protein–Ligand Interactions Using the Distance-Scaled, Finite, Ideal-Gas Reference State. J. Cheminf. 11. doi:10.1186/s13321-019-0373-4
Coley, C. W., Rogers, L., Green, W. H., and Jensen, K. F. (2018). SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 58, 252–261. doi:10.1021/acs.jcim.7b00622
Darme, P., Dauchez, M., Renard, A., Voutquenne-Nazabadioko, L., Aubert, D., Escotte-Binet, S., et al. (2021). AMIDE V2: High-Throughput Screening Based on AutoDock-GPU and Improved Workflow Leading to Better Performance and Reliability. Int. J. Mol. Sci. 22, 7489. doi:10.3390/ijms22147489
Di Tommaso, P., Chatzou, M., Floden, E. W., Barja, P. P., Palumbo, E., and Notredame, C. (2017). Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 35, 316–319. doi:10.1038/nbt.3820
Diederik, K., and Ba, J. L. (2014). ADAM: A Method for Stochastic Optimization. AIP Conf. Proc. 1631, 58–62. doi:10.1063/1.4902458
Douguet, D. (2010). e-LEA3D: a Computational-Aided Drug Design Web Server. Nucleic Acids Res. 38, W615–W621. doi:10.1093/nar/gkq322
Drwal, M. N., and Griffith, R. (2013). Combination of Ligand-And Structure-Based Methods in Virtual Screening. Drug Discov. Today Technol. 10, e395–e401. doi:10.1016/j.ddtec.2013.02.002
Durrant, J. D., and McCammon, J. A. (2012). Autoclickchem: Click Chemistry In Silico. Plos Comput. Biol. 8, 1–7. doi:10.1371/journal.pcbi.1002397
Feinstein, W. P., and Brylinski, M. (2015). Calculating an Optimal Box Size for Ligand Docking and Virtual Screening against Experimental and Predicted Binding Pockets. J. Cheminf. 7. doi:10.1186/s13321-015-0067-5
Gentile, F., Agrawal, V., Hsing, M., Ton, A.-T., Ban, F., Norinder, U., et al. (2020). Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 6, 939–949. doi:10.1021/acscentsci.0c00229
Gentile, F., Fernandez, M., Ban, F., Ton, A.-T., Mslati, H., Perez, C. F., et al. (2021). Automated Discovery of Noncovalent Inhibitors of SARS-CoV-2 Main Protease by Consensus Deep Docking of 40 Billion Small Molecules. Chem. Sci. 12, 15960–15974. doi:10.1039/d1sc05579h
Ghiandoni, G. M., Bodkin, M. J., Chen, B., Hristozov, D., Wallace, J. E. A., Webster, J., et al. (2020). Enhancing Reaction-Based De Novo Design Using a Multi-Label Reaction Class Recommender. J. Comput. Aided Mol. Des. 34, 783–803. doi:10.1007/s10822-020-00300-6
Gorgulla, C., Boeszoermenyi, A., Wang, Z.-F., Fischer, P. D., Coote, P. W., Das, K. M. P., et al. (2020). An Open-Source Drug Discovery Platform Enables Ultra-large Virtual Screens. Nature 580, 663–668. doi:10.1038/s41586-020-2117-z
Gorgulla, C., Çınaroğlu, S. S., Fischer, P. D., Fackeldey, K., Wagner, G., and Arthanari, H. (2021). VirtualFlow Ants-Ultra-Large Virtual Screenings with Artificial Intelligence Driven Docking Algorithm Based on Ant colony Optimization. Int. J. Mol. Sci. 22, 5807. doi:10.3390/ijms22115807
Hartenfeller, M., Eberle, M., Meier, P., Nieto-Oberhuber, C., Altmann, K.-H., Schneider, G., et al. (2011). A Collection of Robust Organic Synthesis Reactions for In Silico Molecule Design. J. Chem. Inf. Model. 51, 3093–3098. doi:10.1021/ci200379p
Hinselmann, G., Rosenbaum, L., Jahn, A., Fechner, N., and Zell, A. (2011). jCompoundMapper: An Open Source Java Library and Command-Line Tool for Chemical Fingerprints. J. Cheminf 3. doi:10.1186/1758-2946-3-3
Irwin, J. J., Shoichet, B. K., Mysinger, M. M., Huang, N., Colizzi, F., Wassam, P., et al. (2009). Automated Docking Screens: a Feasibility Study. J. Med. Chem. 52, 5712–5720. doi:10.1021/jm9006966
Jadhav, A., Ferreira, R. S., Klumpp, C., Mott, B. T., Austin, C. P., Inglese, J., et al. (2010). Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease. J. Med. Chem. 53, 37–51. doi:10.1021/jm901070c
Jayk Bernal, A., Gomes da Silva, M. M., Musungaie, D. B., Kovalchuk, E., Gonzalez, A., Delos Reyes, V., et al. (2022). Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N. Engl. J. Med. 386, 509–520. doi:10.1056/NEJMoa2116044
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly Accurate Protein Structure Prediction with Alphafold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kaplan, R. M., and Milstein, A. (2021). Influence of a COVID-19 Vaccine’s Effectiveness and Safety Profile on Vaccination Acceptance. Proc. Natl. Acad. Sci. U.S.A. 118, e2021726118. doi:10.1073/pnas.2021726118
Kim, E., and Nam, H. (2017). Prediction Models for Drug-Induced Hepatotoxicity by Using Weighted Molecular Fingerprints. BMC Bioinform 18. doi:10.1186/s12859-017-1638-4
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., et al. (2020). PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 49, D1388–D1395. doi:10.1093/nar/gkaa971
Koes, D. R., Baumgartner, M. P., and Camacho, C. J. (2013a). Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 53, 1893–1904. doi:10.1021/ci300604z
Koes, D. R., Baumgartner, M. P., and Camacho, C. J. (2013b). Lessons Learned in Empirical Scoring with Smina from the Csar 2011 Benchmarking Exercise. J. Chem. Inf. Model. 53, 1893–1904. doi:10.1021/ci300604z
Kozakov, D., Grove, L. E., Hall, D. R., Bohnuud, T., Mottarella, S. E., Luo, L., et al. (2015). The FTMap Family of Web Servers for Determining and Characterizing Ligand-Binding Hot Spots of Proteins. Nat. Protoc. 10, 733–755. doi:10.1038/nprot.2015.043
Labbé, C. M., Rey, J., Lagorce, D., Vavruša, M., Becot, J., Sperandio, O., et al. (2015). MTiOpenScreen: a Web Server for Structure-Based Virtual Screening. Nucleic Acids Res. 43, W448–W454. doi:10.1093/nar/gkv306
Le Guilloux, V., Schmidtke, P., and Tuffery, P. (2009). Fpocket: an Open Source Platform for Ligand Pocket Detection. BMC Bioinform 10, 1–11. doi:10.1186/1471-2105-10-168
Le, T., Hempel, T., Winter, R., Olsson, S., Raich, L., Elez, K., et al. (2021). JEDI Billion Molecules against Covid-19: Compounds Synthesized. doi:10.6084/m9.figshare.14458896
Li, H., Leung, K.-S., Ballester, P. J., and Wong, M.-H. (2014). Istar: A Web Platform for Large-Scale Protein-Ligand Docking. PLoS One 9, e85678. doi:10.1371/journal.pone.0085678
Li, H., Leung, K.-S., Wong, M.-H., and Ballester, P. J. (2016). USR-VS: a Web Server for Large-Scale Prospective Virtual Screening Using Ultrafast Shape Recognition Techniques. Nucleic Acids Res. 44, W436–W441. doi:10.1093/nar/gkw320
Mahase, E. (2021). Covid-19: Pfizer’s Paxlovid Is 89% Effective in Patients at Risk of Serious Illness, Company Reports. Br. Med. J. 375, n2713. doi:10.1136/bmj.n2713
Maia, E. H. B., Assis, L. C., de Oliveira, T. A., da Silva, A. M., and Taranto, A. G. (2020). Structure-based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 8. doi:10.3389/fchem.2020.00343
McNutt, A. T., Francoeur, P., Aggarwal, R., Masuda, T., Meli, R., Ragoza, M., et al. (2021). Gnina 1.0: Molecular Docking with Deep Learning. J. Cheminf. 13, 1–20. doi:10.1186/s13321-021-00522-2
Meyers, J., Fabian, B., and Brown, N. (2021). De Novo molecular Design and Generative Models. Drug Discov. Today 26, 2707–2715. doi:10.1016/j.drudis.2021.05.019
Mysinger, M. M., Carchia, M., Irwin, J. J., and Shoichet, B. K. (2012). Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 55, 6582–6594. doi:10.1021/jm300687e
Novick, P. A., Ortiz, O. F., Poelman, J., Abdulhay, A. Y., and Pande, V. S. (2013). SWEETLEAD: an In Silico Database of Approved Drugs, Regulated Chemicals, and Herbal Isolates for Computer-Aided Drug Discovery. PLoS ONE 8, e79568. doi:10.1371/journal.pone.0079568
O’Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011a). Open Babel: An Open Chemical Toolbox. J. Cheminf. 3, 1–14. doi:10.1186/1758-2946-3-33
O’Boyle, N. M., and Sayle, R. A. (2016). Comparing Structural Fingerprints Using a Literature-Based Similarity Benchmark. J. Cheminf. 8. doi:10.1186/s13321-016-0148-0
O’Boyle, N. M., Vandermeersch, T., Flynn, C. J., Maguire, A. R., and Hutchison, G. R. (2011b). Confab - Systematic Generation of Diverse Low-Energy Conformers. J. Cheminf. 3. doi:10.1186/1758-2946-3-8
Ochoa, R., Palacio-Rodriguez, K., Clemente, C. M., and Adler, N. S. (2021). dockECR: Open Consensus Docking and Ranking Protocol for Virtual Screening of Small Molecules. J. Mol. Graph. Model. 109, 108023. doi:10.1016/j.jmgm.2021.108023
Oliveira, S. H., Ferraz, F. A., Honorato, R. V., Xavier-Neto, J., Sobreira, T. J., and de Oliveira, P. S. (2014). Kvfinder: Steered Identification of Protein Cavities as a Pymol Plugin. BMC Bioinform 15, 1–8. doi:10.1186/1471-2105-15-197
Patel, H., Ihlenfeldt, W.-D., Judson, P. N., Moroz, Y. S., Pevzner, Y., Peach, M. L., et al. (2020). SAVI, In Silico Generation of Billions of Easily Synthesizable Compounds through Expert-System Type Rules. Sci. Data 7. doi:10.1038/s41597-020-00727-4
Pereira, J., Simpkin, A. J., Hartmann, M. D., Rigden, D. J., Keegan, R. M., and Lupas, A. N. (2021). High-accuracy Protein Structure Prediction in Casp14. Proteins: Struct. Funct. Bioinformatics 89, 1687–1699. doi:10.1002/prot.26171
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). Ucsf Chimerax: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 30, 70–82. doi:10.1002/pro.3943
Pitti, T., Chen, C.-T., Lin, H.-N., Choong, W.-K., Hsu, W.-L., and Sung, T.-Y. (2019). N-glyde: a Two-Stage N-Linked Glycosylation Site Prediction Incorporating Gapped Dipeptides and Pattern-Based Encoding. Sci. Rep. 9, 1–11. doi:10.1038/s41598-019-52341-z
Ripphausen, P., Nisius, B., and Bajorath, J. (2011). State-of-the-art in Ligand-Based Virtual Screening. Drug Discov. Today 16, 372–376. doi:10.1016/j.drudis.2011.02.011
Santos-Martins, D., Solis-Vasquez, L., Tillack, A. F., Sanner, M. F., Koch, A., and Forli, S. (2021). Accelerating AutoDock4 with GPUs and Gradient-Based Local Search. J. Chem. Theor. Comput. 17, 1060–1073. doi:10.1021/acs.jctc.0c01006
Soderberg, C. K. (2018). Using Osf to Share Data: A Step-by-step Guide. Adv. Methods Practices Psychol. Sci. 1, 115–120. doi:10.1177/2515245918757689
Spiegel, J. O., and Durrant, J. D. (2020). AutoGrow4: an Open-Source Genetic Algorithm for De Novo Drug Design and lead Optimization. J. Cheminf. 12. doi:10.1186/s13321-020-00429-4
Srivastava, N., Hinton, G., Krizhevsky, A., Sutskever, I., and Salakhutdinov, R. (2014). Dropout: a Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 15, 1929–1958.
Sterling, T., and Irwin, J. J. (2015). ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 55, 2324–2337. doi:10.1021/acs.jcim.5b00559
Sunseri, J., and Koes, D. R. (2016). Pharmit: Interactive Exploration of Chemical Space. Nucleic Acids Res. 44, W442–W448. doi:10.1093/nar/gkw287
Tran-Nguyen, V.-K., Jacquemard, C., and Rognan, D. (2020). LIT-PCBA: An Unbiased Data Set for Machine Learning and Virtual Screening. J. Chem. Inf. Model. 60, 4263–4273. doi:10.1021/acs.jcim.0c00155
Trott, O., and Olson, A. J. (2010). Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comp. Chem. 31, 455–461. doi:10.1002/jcc.21334
Venkatraman, V. (2021). FP-ADMET: a Compendium of Fingerprint-Based ADMET Prediction Models. J. Cheminf. 13. doi:10.1186/s13321-021-00557-5
Wang, X., Chen, H., Yang, F., Gong, J., Li, S., Pei, J., et al. (2014). Idrug: a Web-Accessible and Interactive Drug Discovery and Design Platform. J. Cheminform. 6, 28. doi:10.1186/1758-2946-6-28
Wang, Z., Sun, H., Shen, C., Hu, X., Gao, J., Li, D., et al. (2020). Combined Strategies in Structure-Based Virtual Screening. Phys. Chem. Chem. Phys. 22, 3149–3159. doi:10.1039/c9cp06303j
Wilson, G. L., and Lill, M. A. (2011). Integrating Structure-Based and Ligand-Based Approaches for Computational Drug Design. Future Med. Chem. 3, 735–750. doi:10.4155/fmc.11.18
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2017). DrugBank 5.0: a Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 46, D1074–D1082. doi:10.1093/nar/gkx1037
Wójcikowski, M., Zielenkiewicz, P., and Siedlecki, P. (2015). Open Drug Discovery Toolkit (ODDT): a New Open-Source Player in the Drug Discovery Field. J. Cheminform. 7, 26. doi:10.1186/s13321-015-0078-2
Wouters, O. J., Shadlen, K. C., Salcher-Konrad, M., Pollard, A. J., Larson, H. J., Teerawattananon, Y., et al. (2021). Challenges in Ensuring Global Access to COVID-19 Vaccines: Production, Affordability, Allocation, and Deployment. The Lancet 397, 1023–1034. doi:10.1016/s0140-6736(21)00306-8
Yaacoub, J. C., Gleave, J., Gentile, F., Stern, A., and Cherkasov, A. (2021). DD-GUI: A Graphical User Interface for Deep Learning-Accelerated Virtual Screening of Large Chemical Libraries (Deep Docking). Bioinformatics 38, 1146–1148. doi:10.1093/bioinformatics/btab771
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-Tasser Suite: Protein Structure and Function Prediction. Nat. Methods 12, 7–8. doi:10.1038/nmeth.3213
Yang, M., Tao, B., Chen, C., Jia, W., Sun, S., Zhang, T., et al. (2019). Machine Learning Models Based on Molecular Fingerprints and an Extreme Gradient Boosting Method lead to the Discovery of JAK2 Inhibitors. J. Chem. Inf. Model. 59, 5002–5012. doi:10.1021/acs.jcim.9b00798
Yu, J., Zhou, Y., Tanaka, I., and Yao, M. (2010). Roll: a New Algorithm for the Detection of Protein Pockets and Cavities with a Rolling Probe Sphere. Bioinformatics 26, 46–52. doi:10.1093/bioinformatics/btp599
Keywords: virtual screeening, machine learning, computer aided drug design, de novo design, SARS-C0V-2, protein-ligand docking
Citation: Venkatraman V, Colligan TH, Lesica GT, Olson DR, Gaiser J, Copeland CJ, Wheeler TJ and Roy A (2022) Drugsniffer: An Open Source Workflow for Virtually Screening Billions of Molecules for Binding Affinity to Protein Targets. Front. Pharmacol. 13:874746. doi: 10.3389/fphar.2022.874746
Received: 12 February 2022; Accepted: 04 April 2022;
Published: 26 April 2022.
Edited by:
Adriano D. Andricopulo, University of Sao Paulo, BrazilReviewed by:
Bruno Villoutreix, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2022 Venkatraman, Colligan, Lesica, Olson, Gaiser, Copeland, Wheeler and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vishwesh Venkatraman, dmlzaHdlc2gudmVua2F0cmFtYW5AbnRudS5ubw==; Travis J. Wheeler, dHJhdmlzLndoZWVsZXJAdW1vbnRhbmEuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.