
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 13 June 2022
Sec. Pharmacology of Ion Channels and Channelopathies
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.872645
This article is part of the Research TopicIon Channels in Health and DiseaseView all 7 articles
 Ilaria Mosca1†
Ilaria Mosca1† Ilaria Rivolta2†Audrey Labalme3†Paolo Ambrosino4Barbara Castellotti5Cinzia Gellera5
Ilaria Rivolta2†Audrey Labalme3†Paolo Ambrosino4Barbara Castellotti5Cinzia Gellera5 Tiziana Granata6
Tiziana Granata6 Elena Freri6Anna Binda2
Elena Freri6Anna Binda2 Gaetan Lesca3
Gaetan Lesca3 Jacopo C. DiFrancesco6,7
Jacopo C. DiFrancesco6,7 Maria Virginia Soldovieri1*
Maria Virginia Soldovieri1* Maurizio Taglialatela8*
Maurizio Taglialatela8*Pathogenic variants in KCNQ2 encoding for Kv7.2 potassium channel subunits have been found in patients affected by widely diverging epileptic phenotypes, ranging from Self-Limiting Familial Neonatal Epilepsy (SLFNE) to severe Developmental and Epileptic Encephalopathy (DEE). Thus, understanding the pathogenic molecular mechanisms of KCNQ2 variants and their correlation with clinical phenotypes has a relevant impact on the clinical management of these patients. In the present study, the genetic, biochemical, and functional effects prompted by two variants, each found in a non-familial SLNE or a DEE patient but both affecting nucleotides at the KCNQ2 intron 6-exon 7 boundary, have been investigated to test whether and how they affected the splicing process and to clarify whether such mechanism might play a pathogenetic role in these patients. Analysis of KCNQ2 mRNA splicing in patient-derived lymphoblasts revealed that the SLNE-causing intronic variant (c.928-1G > C) impeded the use of the natural splice site, but lead to a 10-aa Kv7.2 in frame deletion (Kv7.2 p.G310Δ10); by contrast, the DEE-causing exonic variant (c.928G > A) only had subtle effects on the splicing process at this site, thus leading to the synthesis of a full-length subunit carrying the G310S missense variant (Kv7.2 p.G310S). Patch-clamp recordings in transiently-transfected CHO cells and primary neurons revealed that both variants fully impeded Kv7.2 channel function, and exerted strong dominant-negative effects when co-expressed with Kv7.2 and/or Kv7.3 subunits. Notably, Kv7.2 p.G310S, but not Kv7.2 p.G310Δ10, currents were recovered upon overexpression of the PIP2-synthesizing enzyme PIP5K, and/or CaM; moreover, currents from heteromeric Kv7.2/Kv7.3 channels incorporating either Kv7.2 mutant subunits were differentially regulated by changes in PIP2 availability, with Kv7.2/Kv7.2 G310S/Kv7.3 currents showing a greater sensitivity to PIP2 depletion when compared to those from Kv7.2/Kv7.2 G310Δ10/Kv7.3 channels. Altogether, these results suggest that the two variants investigated differentially affected the splicing process at the intron 6-exon 7 boundary, and led to the synthesis of Kv7.2 subunits showing a differential sensitivity to PIP2 and CaM regulation; more studies are needed to clarify how such different functional properties contribute to the widely-divergent clinical phenotypes.
KCNQ2 and KCNQ3 genes encode for Kv7.2 and Kv7.3 voltage-gated K+ channel subunits each showing six transmembrane segments (S1-S6) and cytoplasmic N- and C-termini of variable length; the pore domain is encompassed by the S5-S6 segments and the intervening linker of each subunit, whereas the transmembrane segments between S1 and S4 form the voltage-sensing domain (VSD). The C-terminal region of Kv7 subunits contains domains (called from A to D) required for homo- or heteromeric subunit assembly and for a complex network of mutually interacting molecules, such as phosphatidylinositol 4,5-bisphosphate (PIP2), calmodulin, syntaxyn, A-kinase-anchoring proteins, which allow Kv7 channel regulation by pKC, and ankyrin-G (Soldovieri et al., 2011). Kv7.2/Kv7.3 channels are mainly expressed in neuronal cells, where they contribute to a slowly activating and deactivating K+ current called IKM (or M-current) as it is inhibited by the activation of M1 receptors (Selyanko et al., 2000). The molecular pathways leading to M-current inhibition include pKC phosphorylation of the Kv7.2 subunits, dissociation of CaM from the channel and a consequent reduced affinity towards PIP2 (Kosenko et al., 2012).
IKM controls neuronal excitability in the sub-threshold range for action potential generation (Marrion, 1997; Wang et al., 1998); thus, it is not surprising that pathogenic variants in KCNQ2 and KCNQ3 genes have been identified in epileptic disorders, ranging from Self-Limiting Familial Neonatal Epilepsy (SLFNE), characterized by seizures occurring in the first days of life and disappearing after few months, without significant effects on the subsequent neuropsychomotor development, to developmental and epileptic encephalopathies (DEEs), most often characterized by severe epileptic conditions, associated to variable degrees of social, cognitive, motor, language, and behavioral impairments, and pharmacoresistance (Weckhuysen et al., 2012; Weckhuysen et al., 2013). Although the causes for this differential developmental trajectories are still largely unknown, some genotype-phenotype correlations have emerged; in fact, while most variants causing SLFNE are nonsense, splice, or frameshifts randomly distributed throughout the gene, those causing DEE are almost invariably missense and cluster in four hot-spots channel domains exerting critical functional roles (Millichap et al., 2016).
Characterization of the specific effects prompted by each KCNQ2 variant on channel function could allow to identify distinct pathogenetic mechanisms, thus corroborating genotype-phenotype correlations between novel KCNQ2 variants and disease severity, with a potentially-relevant impact on the clinical management of these patients. Functional studies, mainly performed with missense variants, have revealed that, when compared with SLFNE-causing variants, associated with haploinsufficiency, most of the DEE-causing substitutions prompt strong loss-of-function (LoF) (Miceli et al., 2013), dominant-negative (Orhan et al., 2014) or gain-of-function (GoF) (Miceli et al., 2015; Millichap et al., 2017) effects. No study has yet been performed in KCNQ2 splice-site variants. Therefore, the genetic, biochemical, and functional effects prompted by two variants, each found in a SLNE (Soldovieri et al., 2014) or a DEE patient (Soldovieri et al., 2020) but both affecting nucleotides at the KCNQ2 intron 6-exon 7 boundary, have been investigated in the present study to test whether and how they affected the splicing process and to clarify whether such mechanism might play a pathogenetic role in the probands carrying these two variants.
Parents of each patient signed an informed consent for the participation, according to the French bioethics law. The clinical features of the two patients herein investigated have been previously described (Soldovieri et al., 2014; Soldovieri et al., 2020).
Briefly, the girl with early-onset DEE caused by the heterozygous NM_172107.4:c.928 G > A mutation described in Soldovieri et al. (2020) showed burst-suppression pattern at the EEG, recurrent seizures with multifocal onset occurring during the first days of life, and was refractory to different therapeutic trials with phenobarbital, phenytoin, levetiracetam, vigabatrin and pyridoxine. The neurologic evolution was characterized by severe psychomotor delay, absent speech, apostural tetraparesis, and strabismus. A significant decrease of the epileptiform activity was observed following introduction of gabapentin, a newly-identified Kv7 activator (Manville and Abbott, 2018) able to counteract in vitro mutation-induced LoF effects (Soldovieri et al., 2020).
The male patient carrying a heterozygous NM_172107.4:c.928-1G > C mutation described in Soldovieri et al. (2014; Individual II-I in family 9) had seizures in the neonatal period but no recurrence thereafter, normal EEG and normal neurodevelopment; the variant was not detected in either parent, and occurred de novo, thus confirming the diagnosis of non-familial Self-Limiting Neonatal Epilepsy (SLNE; Soldovieri et al., 2014).
For the c.928-1G > C variant, the validation of the effects on Kv7.2 mRNAs splicing was performed as previously described (Soldovieri et al., 2014). Briefly, mRNA was extracted from Epstein-Barr Virus-immortalized lymphoblastoid cells with the RNeasy minikit (Qiagen, Courtaboeuf, France). Retrotranscription was performed using the Expand RT kit (Invitrogen, Saint Aubin, France). PCR was performed with primers in exon 5 (forward: 5′-TACGCGGATGCACTCTGG-3′) and exon 8 (reverse: 5′-GTGGAGTGCAGGTCTGTGC-3′); PCR products were subsequently sequenced.
For the c.928G > A variant, RNA was extracted from Blood samples collected on PAXgene Blood Tube using the PAXgene Blood RNA Kit (Qiagen). cDNA was synthesized from RNA using the SuperScriptTM II RT (ThermoFisher Scientific) according to the manufacturer instructions with primers in exon 6 (forward: 5′-CCTTGCGGCAACCTTCAC-3′) and exon 8 (reverse: 5′- GCTCGTAGTACTGCCACG-3′). cDNA amplicons were separated on a gel electrophoresis using a Labchip GX (Perkin Elmer). Both variants were submitted to ClinVar database (SCV001423824.1 and SCV000484597.1).
As reported (Soldovieri et al., 2020), c.928G > A/p.G310S variant was engineered in a pcDNA3.1-Kv7.2 plasmid for electrophysiological and western-blot experiments, as well as in a dual-tagged Enhanced Green Fluorescent Protein-Kv7.2-hemagglutinin (EGFP-Kv7.2-HA) plasmid used for experiments in neurons, by Quick-change mutagenesis (Agilent Technologies), accordingly to previously described procedure (Miceli et al., 2012). In the EGFP-Kv7.2-HA chimeric construct, in addition to an EGFP at the cytoplasmic N-terminus, an HA epitope was inserted in the extracellular loop that connects transmembrane domains S1 and S2 of Kv7.2 subunits (Schwake et al., 2000; Soldovieri et al., 2006). By contrast, the cDNA for the Kv7.2 p.G310Δ10 variant was engineered by Eurofins service (Eurofins Genomics, Germany); then, a BspeEI/BstXI restriction cassette encompassing variant position was extracted and inserted into the dual-tagged EGFP-Kv7.2-HA plasmid, previously reacted with the same enzymes. Successful cloning was checked by direct sequencing.
Methods for CHO cell growth and primary cortical neurons isolation, as well as protocols for wild-type and mutant cDNAs expression by transient transfection have been previously described (Soldovieri et al., 2016; Soldovieri et al., 2020). Total cDNA in the transfection mixture was kept constant at 4 μg (for electrophysiological recordings in CHO cells), 2 μg (for electrophysiological recordings in neurons), or 6 μg (for Western-blotting experiments).
Expression of wild-type or variant Kv7.2 subunits in CHO cells in total lysate or plasma membrane-enriched fractions was investigated by surface biotinylation and western-blotting analysis, as described (Soldovieri et al., 2014). Channel subunits were identified using mouse monoclonal anti-Kv7.2 primary antibodies (clone N26A/23, dilution 1:1,000; Antibodies Inc., Davis, CA), followed by horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibodies (clone NA931V; dilution 1:5,000; GE Healthcare, Little Chalfont, United Kingdom).
Macroscopic current recordings were performed in CHO cells (24 h after transfection) or in neurons (48 h after transfection), as previously reported (Miceli et al., 2013; Binda et al., 2018). Current densities were obtained dividing the current amplitude measured at each test voltage by the cell capacitance (pA/pF). The conductance was calculated by normalizing the currents measured at the end of each pulse to the driving force. Experimental data were fitted to a Boltzmann equation of the following form: y=max/[1+exp(V1/2−V)/k] to obtain the voltage of half activation (V½) and the slope factor (k).
Experiments on primary cortical neurons were performed either in voltage-clamp mode, using protocols similar to those applied on CHO cells, or in current-clamp mode, to record membrane resting potential. Data were acquired with a Multiclamp 700B amplifier and Digidata 1440A (Axon Instruments, Molecular Device) and pClamp 10.3 software (Molecular Devices) and analyzed with Clampfit 10.3 software (Molecular Devices).
Experiments with the Danio rerio voltage-sensing phosphatase (DrVSP) were performed as previously reported (Ambrosino et al., 2018). Briefly, a 0 mV step lasting 1.5 s was used to activate Kv7 currents, followed by a +100 mV step of variable length (from 0.1 to 1 s, with increments of 0.1 s) to activate DrVSP, and by a second 0 mV step, causing DrVSP switch off and Kv7 current recovery (25 s). Time-dependent current decline following DrVSP activation was quantified as the ratio between the current size measured at the beginning of the second 0 mV step (after DrVSP activation) versus that measured at the end of the first 0 mV step (before DrVSP activation), expressed as a function of the duration of the +100 mV step; these values were then fitted to a Boltzmann function of the following form: y = 1/[1+(x/t½)-Hill slope], where t½ was the duration of the +100 mV step required to inhibit 50% of maximal currents. Kv7 current recovery was instead quantified by normalizing current values recorded during the entire second step at 0 mV to the currents measured at the end of the first 0 mV step. The kinetics of DrVSP-induced Kv7 current inhibition and of Kv7 current recovery after DrVSP switch off were measured by fitting the current traces to a single exponential function of the following form: y = amp×e(−t/τ)+c, where τ was the time constant of the function.
Each data point shown in figures or in the text is the Mean ± SEM of at least 4 determinations, each performed in a single cell or in a separate experiment. Statistically significant differences were evaluated with the Student’s t-test or with the ANOVA followed by the Student-Newman-Keuls test, with the threshold set at p < 0.05.
Both variants herein reported fall at the intron 6-exon 7 boundary of the KCNQ2 gene (Figure 1A). Four in silico tools provided by Alamut software (SpliceSiteFinder-like, MaxEntScan, NNSPLICE, and Gene Splicer; Suppl. Figure 1A) revealed that the SLNE-causing c.Kv7.2 c.928-1G > C intronic variant (affecting the g of the ag acceptor site located at the end of intron 6; Figure 1A) drastically reduced the strength of the affected splice site (Supplementary Figure S1A; Soldovieri et al., 2014); similar results were obtained with the software spliceAI (acceptor loss score: 0.96). In keeping with this prediction, while Kv7.2 mRNAs analysis in lymphoblasts from the unaffected, non-carrier father revealed the presence of only wild-type fragments (295 bp), transcript analysis in lymphoblasts from the proband carrying the c.928-1G > C variant revealed the presence of both wild-type (295 bp) and shorter (265 bp) fragments (Figure 1B), the latter corresponding to the variant-induced use of a cryptic ag site located 30 bp downstream in exon 7, thus causing the removal of encompassed nucleotides (Figure 1A). The intensity of this low molecular weight band was much stronger than that corresponding to the full-length band of the wild-type allele, leading to an intensity ratio higher than the 1:1 expected from the heterozygous state of the patient; this result, possibly explained by the more efficient amplification of shorter amplicons, suggests that the effect of the variant on the splicing is complete. Expression of shorter mRNAs causes a 10 aminoacid in-frame deletion (Kv7.2 p.G310_K319del10, herein referred as Kv7.2 p.G310Δ10), affecting the end of S6 domain and the beginning of the C-terminus of Kv7.2 subunits (Figure 1D).
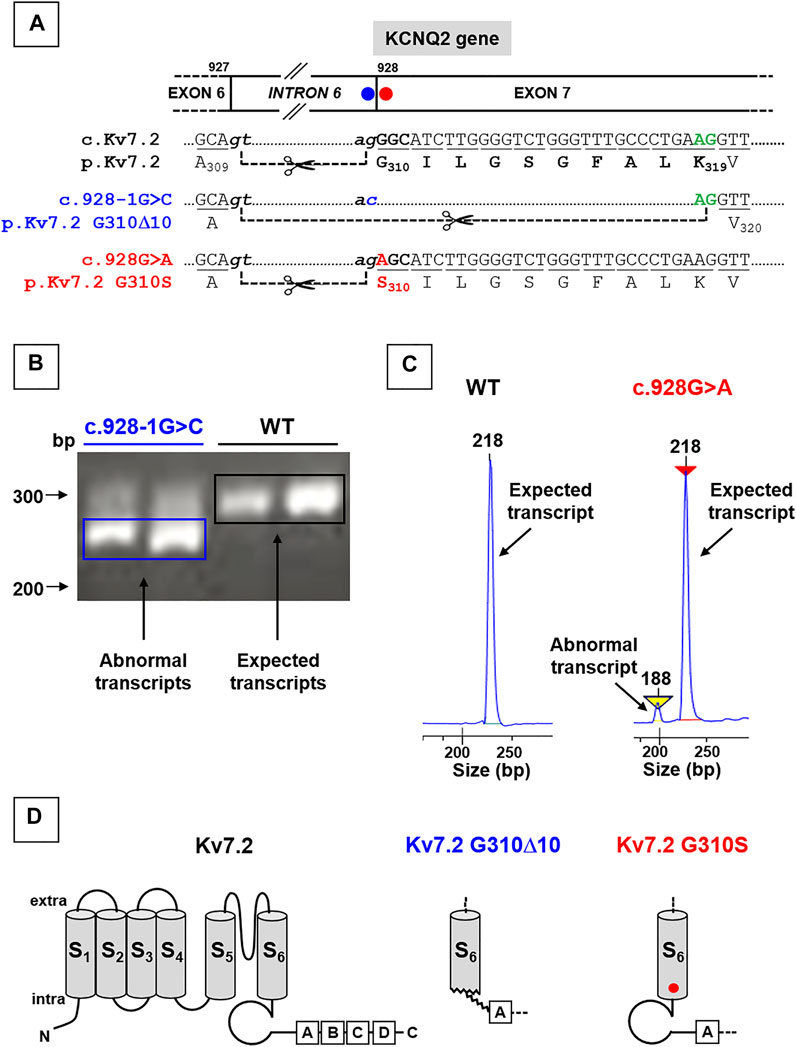
FIGURE 1. Localization of KCNQ2 variants herein investigated and effects on KCNQ2 mRNAs expression. (A) Localization of the two variants herein investigated in the KCNQ2 gene, falling at the last nucleotide of intron 6 (KCNQ2 (C)928-1G > C; blue circle; already reported in Soldovieri et al., 2014) or at the first nucleotide of exon 7 (KCNQ2 (C)928G > (A); already reported in Soldovieri et al., 2020). (B) Representative agarose gel run and (C) electrophoresis profiles of major bands obtained from RT-PCR experiments performed on Kv7.2 mRNAs expressed in father- or probands-deriving lymphoblasts, as indicated; the mRNA fragment of 295 bp (B) or 218 bp (C) are expected when the ag acceptor site at the end of intron 6 is used, while those of 265 bp (B) or 188 bp (C) bands are obtained using the alternative splice site in the exon 7. Panels in C have been obtained cropping the regions of interest from the complete electrophoresis profiles, reported in Supplementary Figure S2 Topology of a Kv7.2 subunit showing the 6 transmembrane segments (called from S1 to S6) and four intracellular domains at the C-terminus (called from A to D), binding many regulatory molecules. Middle and left panels show the structural alterations prompted by the Kv7.2 G310Δ10 in-frame deletion or by the missense Kv7.2 G310S variant, respectively.
By contrast, in the case of the DEE-causing exonic c.928G > A variant, affecting the first nucleotide of exon 7 (Figure 1A), prediction tools provided by Alamut (SpliceSiteFinder-like, MaxEntScan, NNSPLICE, and Gene Splicer) were in favor of only a slight reduction of the strength of the splice site (Supplementary Figure S1A); similar results were obtained with the software spliceAI (acceptor loss score: 0.06). Accordingly, PCR analysis performed in lymphoblasts from the patient carrying this variant revealed that, while only the expected 218 bp-long fragments were detected in samples from the father (used as a control), a shorter (188 bp) Kv7.2 mRNA fragment was also detected in the patient carrying the c.928G > A variant (Figure 1C), with the first being much more abundant than the latter (90:10, respectively) (Figure 1C). The most abundant mRNA incorporates the c.928G > A substitution in exon 7, causing the missense substitution p.G310S (Figures 1A–D).
The functional consequences of p.G310Δ10 or p.G310S Kv7.2 subunits were investigated by electrophysiological recordings upon their expression in CHO cells by transient transfection. Expression of Kv7.2 channels in these cells elicited robust voltage-gated outward K+ currents with slow activation and deactivation kinetics, lack of inactivation and activating at about −40 mV, with a current density of about 30 pA/pF (Figure 2A; Table 1). By contrast, K+ current densities recorded from CHO cells transfected with either mutant plasmids were identical to those of non-transfected cells (Figure 2A; Table 1).
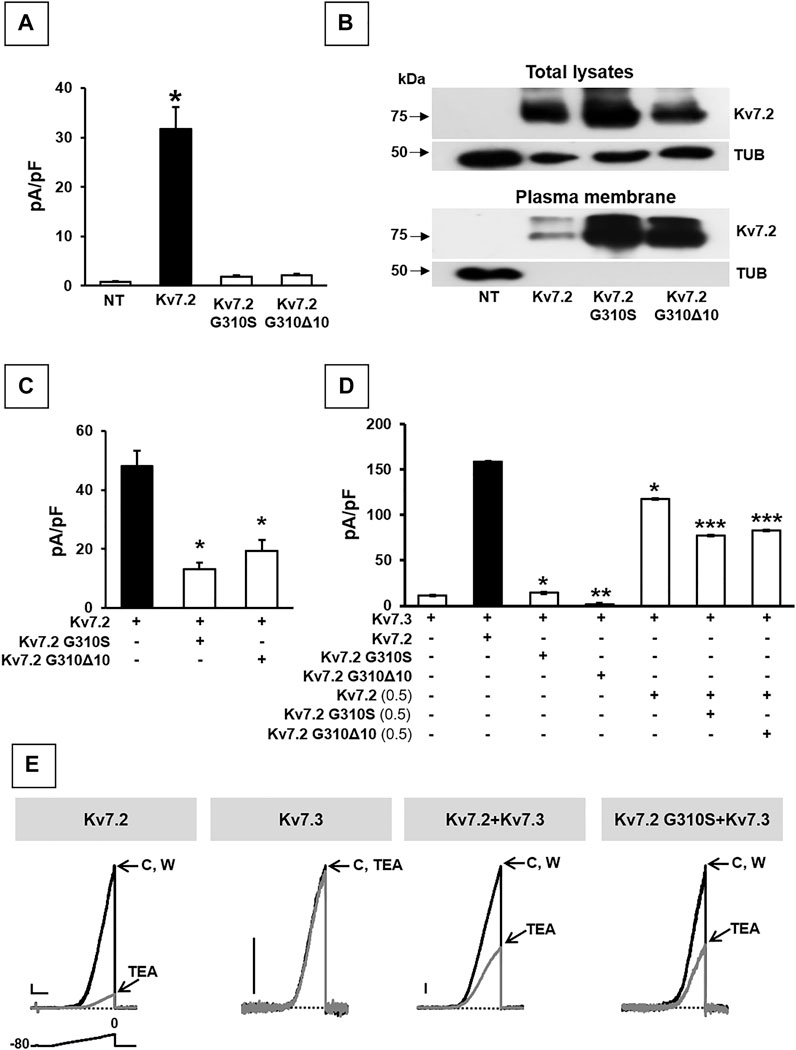
FIGURE 2. Functional and biochemical characterization of Kv7.2 G310S and Kv7.2 G310Δ10 subunits. (A) Maximal current densities measured in non transfected cells (NT) or in cells expressing wild-type or mutant Kv7.2 subunits, as indicated. * = p < 0.05 versus NT cells. (B) Western blot analysis of proteins from total lysates (upper panels) or biotinylated plasma membrane fractions (lower panels) from CHO cells transfected with the indicated constructs. Higher and lower blots were probed with anti-Kv7.2 or anti-α-tubulin antibodies, as indicated. Numbers on the left correspond to the molecular masses of the protein markers. ODKv7.2 TOT/ODTUB ratios were 1.00 ± 0.20, 1.54 ± 0.19, or 0.66 ± 0.25; n = 4; p > 0.05 among each other; ODKv7.2 BIOT/ODTUB ratios were 1.00 ± 0.50, 5.2 ± 1.50, or 3.33 ± 0.45 in Kv7.2-, Kv7.2 p.G310S-, or Kv7.2 p.G310Δ10-transfected cells, respectively; n = 4; p < 0.05 versus Kv7.2. (C,D) Quantification of maximal currents measured in CHO cells expressing either Kv7.2 variant with Kv7.2 (C) and/or Kv7.3 (D) subunits, as indicated. * = p < 0.05 versus the black bar in each panel; ** = p < 0.05 versus Kv7.3; *** = p < 0.05 Kv7.2 + Kv7.3 (0.5:1). (E) Representative current traces recorded upon application of the ramp protocol (shown below the first traces) on CHO cells expressing the indicated constructs in control solution (C), after 2-min 3 mM TEA perfusion, or upon drug washout (W). Current scale: 100 pA; time scale: 1 s.
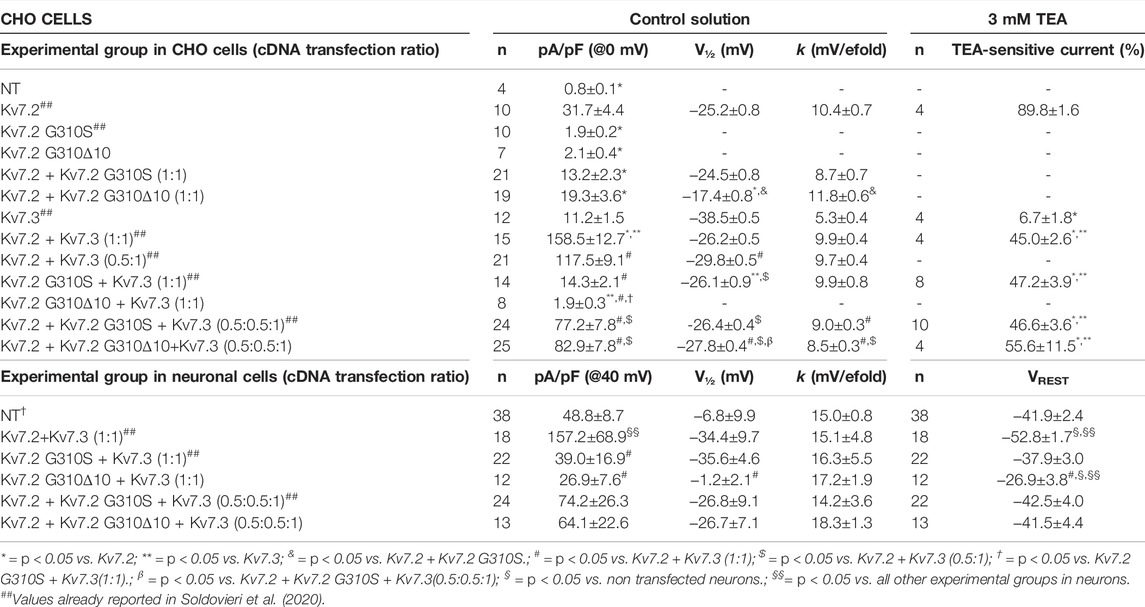
TABLE 1. Functional properties of currents of wild-type or mutant Kv7.2 subunits when expressed in CHO cells or cortical neurons.
To investigate possible variant-induced changes in Kv7.2 subunits trafficking to the plasma membrane, Western-blot experiments were performed in both total lysates and plasma membrane-enriched fractions from CHO cells expressing Kv7.2, Kv7.2 G310S, or Kv7.2 G310Δ10 subunits. The results obtained showed that either substitutions did not interfere with whole-cell expression of Kv7.2 subunits (Figure 2B; p > 0.05 among each other), whilst both significantly increased Kv7.2 subunit expression at the plasma membrane (Figure 2B; p < 0.05 versus Kv7.2). Although this increase in plasma membrane abundance, quantitatively similar for both variants, was not further investigated, these results suggested that the functional impairment observed in electrophysiological experiments upon transfection with plasmids carrying either variant was not due to the inability of Kv7.2 mutant subunits to efficiently traffic to the plasma membrane.
Having detected non-functional mutant subunits at the plasma membrane, we also investigated whether they interfered with the function of wild-type Kv7.2 and/or Kv7.2 subunits, as Kv7.2/3 heteromers underlie most of the M-current in adult neurons (Wang et al., 1998). Notably, co-expression of Kv7.2 subunits carrying either variant with Kv7.2 or Kv7.3 + Kv7.3 subunits significantly reduced maximal currents, thus indicating mutation-induced dominant-negative effects on Kv7.2 (Figure 2C; Table 1) or Kv7.2/Kv7.3 (Figure 2D; Table 1) currents. By contrast, significant dominant-negative effects on Kv7.3 currents only occurred upon co-expression of Kv7.2 p.G310Δ10, but not Kv7.2 p.G310S subunits, whereas currents levels in Kv7.3- or Kv7.2 G310S/Kv7.3-expressing cells were identical (Figure 2D; Table 1). To discern whether currents measured in Kv7.2 G310S/Kv7.3-expressing cells were carried by homomeric Kv7.3 or Kv7.2 G310S/Kv7.3 heteromeric channels, pharmacological experiments using tetraethylammonium (TEA, 3 mM) were performed; in fact, TEA is widely used to discriminate among Kv7 channel of distinct subunit composition since it blocks homomeric Kv7.2, homomeric Kv7.3 or heteromeric Kv7.2/Kv7.3 channels with a high, low, or intermediate potency, respectively (Hadley et al., 2000). In these experiments, currents from cells expressing Kv7.2 G310S/Kv7.3 channels were blocked by TEA to a similar extent as Kv7.2/Kv7.3 channels (Figure 2E; Table 1), thus suggesting that Kv7.2 p.G310S subunits form functional heteromeric channels with Kv7.3 subunits.
Co-expression of Kv7.2 + Kv7.3 subunits in cortical neurons yielded robust outward currents with gating properties very similar to those measured in CHO cells (Figure 3A; Table 1). Notably, the co-expression of Kv7.2 p.G310S or Kv7.2 p.G310Δ10 subunits with wild-type Kv7.3 subunits reduced current densities to background (non-transfected) levels (Table 1). Moreover, cells co-expressing either Kv7.2 variant subunits with Kv7.2 + Kv7.3 subunits channels carried currents whose size was roughly intermediate between that of non-transfected neurons and those expressing Kv7.2 + Kv7.3 channels (Figures 3B,C; Table 1). When compared to non-transfected neurons, neuronal resting membrane potential was hyperpolarized upon Kv7.2 + Kv7.3 co-expression; by contrast, co-expression of either variant failed to cause membrane hyperpolarization (Table 1). Altogether, these results validate the results obtained in CHO cells and suggest that both p.G310Δ10 and p.G310S Kv7.2 variants strongly suppress Kv7.2 and/or Kv7.3 channel function.
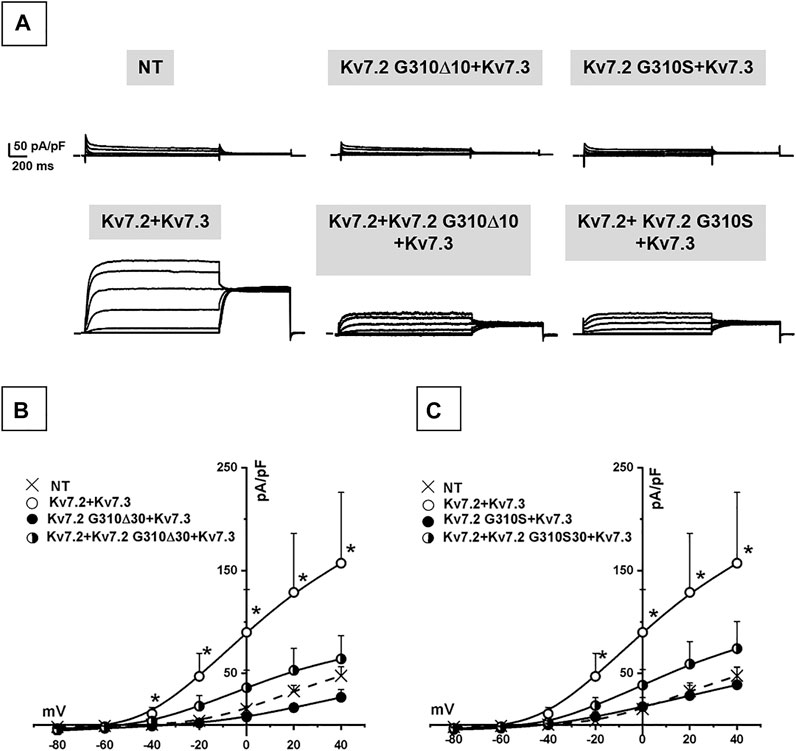
FIGURE 3. Functional characterization of heteromeric Kv7.2 + Kv7.3 channels incorporating Kv7.2 G310S or Kv7.2 G310Δ10 subunits expressed in neurons. (A) Macroscopic currents from neurons expressing wild-type or mutant Kv7.2 + Kv7.3 subunits, as indicated. (B,C) Current density/voltage curves measured in non transfected neurons (NT) or expressing the indicated channels incorporating Kv7.2 G310Δ10 (B) or Kv7.2 G310S (C) subunits, as indicated.
Both Kv7.2 variants herein investigated affect bottom end of the S6 segment in the pore of Kv7.2 subunits (Figure 1C); this region is critical for PIP2-dependent regulation of Kv7.2 subunit function (Zhang et al., 2003; Telezhkin et al., 2013; Zhang et al., 2013; Soldovieri et al., 2016), and is located just prior helix A in the C-terminus (Figure 1C), a major anchoring site for CaM (Yus-Najera et al., 2002; Gamper and Shapiro, 2003).
Both PIP2 and CaM positively affect Kv7.2 subunit function (Telezhkin et al., 2013; Ambrosino et al., 2015). To investigate whether the loss-of-function effects caused by either Kv7.2 variant were dependent on changes in PIP2- and/or CaM-dependent modulation, patch-clamp recordings were performed in CHO cells in which PIP2 levels were either increased by the co-expression of the PIP2-synthesizing enzyme PIP5K (Wenk et al., 2001; Winks et al., 2005), or decreased by a voltage-dependent phosphatase (DrVSP) (Falkenburger et al., 2010); in addition, CaM regulation was tested by measuring the functional consequences of an enhanced CaM availability, achieved by co-expressing wild-type CaM or CaM1234, a CaM variant unable to bind Ca2+ (Geiser et al., 1991).
The results obtained suggest that Kv7.2 currents were significantly increased upon co-expression of PIP5K, CaM, or CaM1234 (Figure 4A; Table 2); notably, potentiation by PIP5K, CaM, or CaM1234 was non-additive in the present experimental conditions. Instead, Kv7.2 p.G310S channels showed strong functional recovery in the presence of PIP5K and CaM1234, but not wild-type CaM (Figure 4A; Table 2); notably, the effects of PIP5K and CaM1234 were synergistic (Figure 4B; Table 1). By contrast, none of the previously mentioned experimental condition (alone or in combination) was able to recover Kv7.2 p.G310Δ10 function (Figures 4A,B; Table 1), possibly suggesting that the G310-K319 region deleted by the mutation is critically involved in Kv7.2 currents functional modulation by PIP2 and/or CaM.
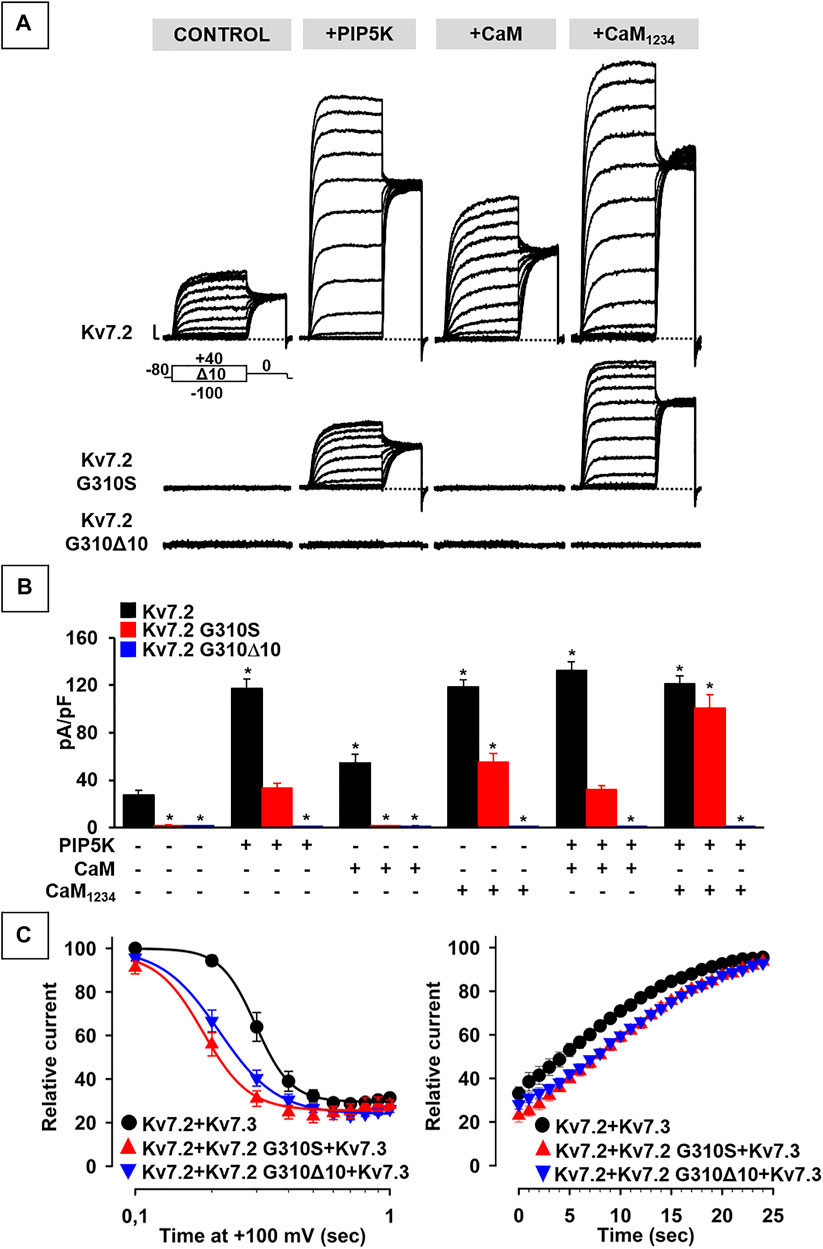
FIGURE 4. Functional study of the modulation of Kv7.2 G310S or Kv7.2 G310Δ10 subunits by PIP2 and calmodulin. (A,B) Representative traces (A) and quantification (B) of currents measured in CHO cells co-expressing homomeric wild-type or mutant Kv7.2 subunits with PIP5K, CaM, and/or CaM1234, as indicated. * = p < 0.05 versus Kv7.2. (C) Time-dependence of current decrease (left panel) and recovery (right panel) of currents measured in CHO cells co-expressing the indicated channels with DrVSP.
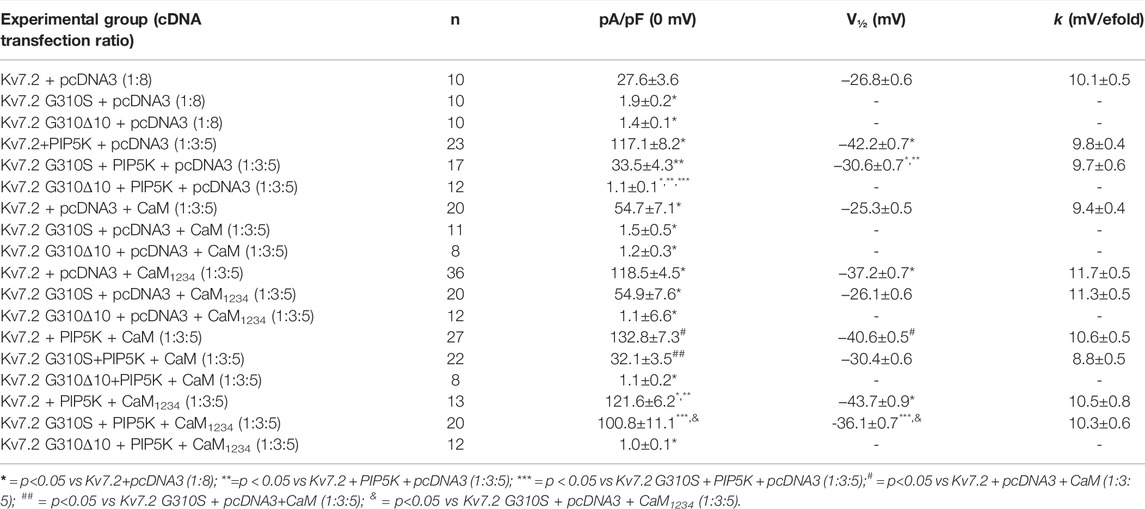
TABLE 2. Functional properties of currents measured in CHO cells expressing wild-type or mutant Kv7.2 subunits upon modulation of PIP2 or CaM levels.
To further investigate variant-induced changes in PIP2-dependent Kv7.2 currents modulation, PIP2 levels were decreased upon co-expression of the voltage-dependent phosphatase DrVSP. As reported in Figure 4C, Kv7.2 + Kv7.3 currents were time-dependently inhibited when +100 mV depolarizing pulses lasting >0.2 s were used to activate DrVSP (t½ was 0.298 ± 0.003 s, n = 16), with a time constant of current inhibition (τ) of 0.24 ± 0.03 s (n = 13). Membrane repolarization at 0 mV, a membrane potential value at which the DrVSP is switched off, led to a complete time-dependent recovery of Kv7.2 + Kv7.3 currents with a time constant of 26.8 ± 3.27 s (n = 15). Notably, currents expressed by Kv7.2 + Kv7.2 G310S + Kv7.3 or Kv7.2 + Kv7.2 G310Δ10 + Kv7.3 channels were more easily inhibited upon DrVSP activation: in fact, t½ were 0.185 ± 0.008 or 0.214 ± 0.003 s, respectively (n = 12–15; p < 0.05 versus control and among each other; Figure 4C); in addition, a faster time constant was measured in cells expressing Kv7.2 + Kv7.2 G310S + Kv7.3 (τ = 0.15 ± 0.02 s; n = 9; p < 0.05 versus wild-type channels), but not Kv7.2 + Kv7.2 G310Δ10 + Kv7.3 subunits (τ = 0.24 ± 0.02 s; n = 10; p > 0.05 versus wild-type channels). No difference in time constant of current recovery was measured for Kv7.2 + Kv7.2 G310S + Kv7.3 or Kv7.2 + Kv7.2 G310Δ10 + Kv7.3 channels when compared to wild-type channels (τ = 26.5 ± 3.85 or 33.1 ± 4.77 s, respectively; n = 9–10; p > 0.05 versus wild-type channels).
Altogether, these results suggest that both variants herein studied prompted significant alterations in CaM- and PIP2-dependent Kv7.2 current modulation, with a greater functional derangement in the presence of the DEE-causing p.G310S variant when compared to the SLNE-causing p.G310Δ10 variant.
Pathogenic variants in Kv7.2 potassium channels have been found in patients affected by widely diverging epileptic phenotypes, ranging from self-limiting (SLFNE) to very severe DEEs (Weckhuysen et al., 2012; Weckhuysen et al., 2013). Given the rarity of KCNQ2-related disorders, genotype-phenotype correlations are difficult to establish; in general, haploinsufficiency caused by the loss of function of a single KCNQ2 allele (by nonsense, splice, or frameshift variants) is the most common cause of familial KCNQ2-SLFNE, while pathogenic variants so far identified in KCNQ2-DEE are all de novo missense variants, and are thought to exert more severe functional defects on potassium current function (Miceli et al., 2013; Orhan et al., 2014). No studies have yet been performed on KCNQ2 variants affecting splice sites (www.rikee.org).
In the present study, we have characterized the genetic, biochemical, and functional consequences prompted by two known variants in the KCNQ2 gene falling at the intron 6-exon 7 boundary: the SLNE-causing c.Kv7.2 c.928-1G > C intronic variant, and the DEE-causing exonic c.928G > A variant, to test whether and how they affected the splicing process and to clarify whether such mechanism might play a pathogenetic role in the probands carrying these two variants.
Kv7.2 mRNAs analysis from patient-derived immortalized lymphoblasts revealed that the SLNE-causing Kv7.2 c.928-1G > C substitution abolished the physiological “ag” acceptor site, and promoted the use of a cryptic site located 30 bp downstream, thus leading to a 10 aminoacid in-frame deletion (p.G310_K319Δ10); in the proband samples, comparison of the intensity of the new, mutation-induced, lower molecular weight band with that corresponding to the wild-type full-length band suggested that the mutation-induced effect of the variant on the splicing process is complete. By contrast, in the case of the c.928G > A substitution, mRNAs analysis revealed more complex effects, with most of the mRNAs (>90%) resulting from the use of the natural “ag” acceptor site localized at the end of intron 6, and leading to Kv7.2 mRNAs of the same length as wild-type, but encoding for the missense substitution p.G310S; in addition, the same splicing alteration described above and leading to the 10 aminoacid in-frame deletion (p.Gly310_Lys319Δ10) was also detected in this patient, although this component only accounted for <10% of total Kv7.2 mRNAs. Although splicing in lymphoblasts might not fully recapitulate that occurring at neuronal level, based on these results we have investigated the functional consequences of Kv7.2 p.G310Δ10 subunits produced by the intronic Kv7.2 c.928-1G > C variant, and those of Kv7.2 p.G310S subunits occurring as a result of the Kv7.2 c.928G > A exonic mutation.
Both Kv7.2 p.G310S and Kv7.2 p.G310Δ10 mutant subunits failed to elicit measurable currents and prompted dominant-negative effects in heteromeric configuration with Kv7.2 and/or Kv7.3 subunits when transiently-transfected in CHO cells. A similar lack of function was also observed when these subunits were co-expressed with Kv7.2/Kv7.3 subunits in primary culture of neonatal rat cortical neurons, a more physiological cellular context.
Kv7.2 currents are regulated by a complex network of mutually-interacting intracellular molecules (Hernandez et al., 2008); mutations herein investigated are located slightly upstream of the C-terminal helix A, a critical anchoring site for CaM (Yus-Najera et al., 2002; Gamper and Shapiro, 2003), and of one of the binding sites for the Kv7 activator PIP2 (Zhang et al., 2003; Li et al., 2005; Gamper and Shapiro, 2007; Telezhkin et al., 2013; Zhang et al., 2013; Soldovieri et al., 2016). Both CaM and PIP2 potentiate Kv7.2 currents; thus, the possibility existed that the observed mutation-induced LoF effects were dependent on alterations in the PIP2/CaM-dependent Kv7.2 current regulation. To test this hypothesis, the intracellular levels of these mediators were modified by overexpressing CaM (either in a wild-type or unable to bind Ca2+ isoform; Geiser et al., 1991), the PIP2-synthesizing enzyme PIP5K (thus increasing PIP2 levels; Wenk et al., 2001) or the PIP2-degrading voltage-dependent phosphatase from Danio rerio (Falkenburger et al., 2010).
In most of these experimental conditions (overexpression of PIP5K and/or CaM1234), currents carried by Kv7.2 subunits carrying the DEE-causing variant p.G310S were recovered to levels identical to those of wild-type Kv7.2 channels, suggesting that the LoF effects prompted by this variant on Kv7.2 currents are dependent to an alteration in the affinity of both mediators. By contrast, none of these experimental conditions (alone or in combination) were able to recover channel function in the case of Kv7.2 subunits incorporating the SLNE-causing variant p.G310Δ10, possibly because of a complete inability of CaM and/or PIP2 to bind Kv7.2 channels as a consequence of the variant-induced in-frame deletion. This could lead to a reduced incorporation of Kv7.2 subunits carrying the SLNE-causing variant in functional heterotetrameric channels and therefore in a reduction of dominant-negative effects prompted on Kv7.2/Kv7.3 currents at high CaM and/or PIP2 levels, thus providing a plausible molecular explanation for the more benign clinical phenotypes observed in the individual carrying this variant. Moreover, both Kv7.2/Kv7.2 G310S/Kv7.3 and Kv7.2/Kv7.2 G310Δ10/Kv7.3 currents were more sensitive than wild-type Kv7.2/Kv7.3 channels to PIP2 depletion; notably, the p.G310S variant prompted slightly stronger functional derangement when compared to the p.G310Δ10 variant, which could further contribute to the divergent clinical phenotypes observed in the two probands.
These results also suggest that a quantitative threshold level exists in terms of tolerance of Kv7.2 to variant-dependent changes in PIP2-dependent current regulation, with smaller effects (such those of the Kv7.2 p.G310Δ10 variant) leading to more benign forms of the clinical spectrum, and stronger derangements (as those caused by the Kv7.2 p.G310S variant) to more severe phenotypes. A similar hypothesis seems to provide a reasonable explanation for the results obtained in a DEE-affected patient carrying two variants in compound heterozygosity in KCNQ3, each inherited by a healthy parent; in this case, Kv7.2/Kv7.3 channels incorporating either KCNQ3 variant showed a smaller alteration in PIP2-dependent current regulation when compared to channels incorporating both KCNQ3 variants (Ambrosino et al., 2018). The fact that, in this latter case, parents were asymptomatic could be explained by the fact that, when compared to KCNQ2, KCNQ3 shows a greater “mutation tolerance,” as also suggested by the higher incidence of KCNQ2-DEE when compared to KCNQ3-DEE cases.
Altogether, the present results suggest that differences in both the alternative splicing process and in PIP2/CaM-dependent Kv7.2 current regulation may contribute to the divergent clinical phenotypes caused by the two KCNQ2 variants herein studied.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The procedures involving animals were conducted according to European (EEC Council Directive 86/609, OJ L 358, 1, December 12 1987) and Italian (D.L. n. 26/2014) laws and policies and approved by the University of Milano-Bicocca Animal Care and Use Ethic Commitee (n.02/2019) and by the Italian Ministry of Health (DGSAF 0017710-P-26/07/2017).
MVS, TG, EF, GL, JCD, and MT designed research; MVS, PA, IM, IR, AL, BC, CG, AB, and MT performed research and analysed data; MVS and MT wrote the paper.
The present study was supported by grants, from the Department of Medicine and Health Science of the University of Molise to MVS, from the Italian Ministry for University and Research (PRIN 2017ALCR7C) to MT and MVS, from the Italian Ministry of Health (Project RF-2019-12370491) to MT and BC, the European Commission H2020 (UNICOM – 875299), and the European Joint Programme on Rare Disease JTC 2020 to MT, and from the Italian Ministry of Health Ricerca Finalizzata Giovani Ricercatori 2016 (Project GR-2016-02363337) to JCD and MVS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are deeply grateful to: Dr. Thomas J. Jentsch, Leibniz-Institut für Molekulare Pharmakologie (FMP), Berlin (Germany) for Kv7.2 and Kv7.3 cDNAs; Dr. Mark S. Shapiro for pcDNA3.1-CaM and pcDNA3.1-CaM1234 plasmids; Dr. Alvaro Villarroel, UPV-CSIC, Leioa (ES) for PIP(4)5Kly; Dr. Yasushi Okamura, Osaka University, Suita, Osaka (JP) for Dr. VSP-IRES-GFP cDNA
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.872645/full#supplementary-material
Ambrosino, P., Alaimo, A., Bartollino, S., Manocchio, L., De Maria, M., Mosca, I., et al. (2015). Epilepsy-Causing Mutations in Kv7.2 C-Terminus Affect Binding and Functional Modulation by Calmodulin. Biochim. Biophys. Acta 1852 (9), 1856–1866. doi:10.1016/j.bbadis.2015.06.012
Ambrosino, P., Freri, E., Castellotti, B., Soldovieri, M. V., Mosca, I., Manocchio, L., et al. (2018). Kv7.3 Compound Heterozygous Variants in Early Onset Encephalopathy Reveal Additive Contribution of C-Terminal Residues to PIP2-Dependent K+ Channel Gating. Mol. Neurobiol. 55 (8), 7009–7024. doi:10.1007/s12035-018-0883-5
Binda, A., Panariti, A., Barbuti, A., Murano, C., Dal Magro, R., Masserini, M., et al. (2018). Modulation of the Intrinsic Neuronal Excitability by Multifunctional Liposomes Tailored for the Treatment of Alzheimer's Disease. Int. J. Nanomedicine 13, 4059–4071. doi:10.2147/IJN.S161563
Falkenburger, B. H., Jensen, J. B., and Hille, B. (2010). Kinetics of PIP2 Metabolism and KCNQ2/3 Channel Regulation Studied with a Voltage-Sensitive Phosphatase in Living Cells. J. Gen. Physiol. 135, 99–114. doi:10.1085/jgp.200910345
Gamper, N., and Shapiro, M. S. (2003). Calmodulin Mediates Ca2+-Dependent Modulation of M-Type K+ Channels. J. Gen. Physiol. 122, 17–31. doi:10.1085/jgp.200208783
Gamper, N., and Shapiro, M. S. (2007). Regulation of Ion Transport Proteins by Membrane Phosphoinositides. Nat. Rev. Neurosci. 8, 921–934. doi:10.1038/nrn2257
Geiser, J. R., van Tuinen, D., Brockerhoff, S. E., Neff, M. M., and Davis, T. N. (1991). Can Calmodulin Function Without Binding Calcium? Cell 65, 949–959. doi:10.1016/0092-8674(91)90547-c
Hadley, J. K., Noda, M., Selyanko, A. A., Wood, I. C., Abogadie, F. C., and Brown, D. A. (2000). Differential Tetraethylammonium Sensitivity of KCNQ1-4 Potassium Channels. Br. J. Pharmacol. 129, 413–415. doi:10.1038/sj.bjp.0703086
Hernandez, C. C., Zaika, O., Tolstykh, G. P., and Shapiro, M. S. (2008). Regulation of Neural KCNQ Channels: Signalling Pathways, Structural Motifs and Functional Implications. J. Physiol. 586 (7), 1811–1821. doi:10.1113/jphysiol.2007.14830410.1113/jphysiol.2007.148304
Kosenko, A., Kang, S., Smith, I. M., Greene, D. L., Langeberg, L. K., Scott, J. D., et al. (2012). Coordinated Signal Integration at the M-Type Potassium Channel Upon Muscarinic Stimulation. EMBO J. 31 (14), 3147–3156. doi:10.1038/emboj.2012.156
Li, Y., Gamper, N., Hilgemann, D. W., and Shapiro, M. S. (2005). Regulation of Kv7 (KCNQ) K+ Channel Open Probability by Phosphatidylinositol 4,5-Bisphosphate. J. Neurosci. 25, 9825–9835. doi:10.1523/JNEUROSCI.2597-05.2005
Manville, R. W., and Abbott, G. W. (2018). Gabapentin is a Potent Activator of KCNQ3 and KCNQ5 Potassium Channels. Mol. Pharmacol. 94 (4), 1155–1163. doi:10.1124/mol.118.112953
Marrion, N. V. (1997). Control of M-Current. Annu. Rev. Physiol. 59, 483–504. doi:10.1146/annurev.physiol.59.1.483
Miceli, F., Soldovieri, M. V., Ambrosino, P., Barrese, V., Migliore, M., Cilio, M. R., et al. (2013). Genotype-Phenotype Correlations in Neonatal Epilepsies Caused by Mutations in the Voltage Sensor of K(v)7.2 Potassium Channel Subunits. Proc. Natl. Acad. Sci. U. S. A. 110 (11), 4386–4391. doi:10.1073/pnas.1216867110
Miceli, F., Soldovieri, M. V., Ambrosino, P., De Maria, M., Migliore, M., Migliore, R., et al. (2015). Early-Onset Epileptic Encephalopathy Caused by Gain-Of-Function Mutations in the Voltage Sensor of Kv7.2 and Kv7.3 Potassium Channel Subunits. J. Neurosci. 35 (9), 3782–3793. doi:10.1523/JNEUROSCI.4423-14.2015
Miceli, F., Vargas, E., Bezanilla, F., and Taglialatela, M. (2012). Gating Currents From Kv7 Channels Carrying Neuronal Hyperexcitability Mutations in the Voltage-Sensing Domain. Biophys. J. 102 (6), 1372–1382. doi:10.1016/j.bpj.2012.02.004
Millichap, J. J., Miceli, F., De Maria, M., Keator, C., Joshi, N., Tran, B., et al. (2017). Infantile Spasms and Encephalopathy Without Preceding Neonatal Seizures Caused by KCNQ2 R198Q, a Gain-Of-Function Variant. Epilepsia 58 (1), e10–e15. doi:10.1111/epi.13601
Millichap, J. J., Park, K. L., Tsuchida, T., Ben-Zeev, B., Carmant, L., Flamini, R., et al. (2016). KCNQ2 Encephalopathy: Features, Mutational Hot Spots, and Ezogabine Treatment of 11 Patients. Neurol. Genet. 2 (5), e96. doi:10.1212/NXG.0000000000000096
Orhan, G., Bock, M., Schepers, D., Ilina, E. I., Reichel, S. N., Löffler, H., et al. (2014). Dominant-Negative Effects of KCNQ2 Mutations are Associated with Epileptic Encephalopathy. Ann. Neurol. 75, 382–394. doi:10.1002/ana.24080
Schwake, M., Pusch, M., Kharkovets, T., and Jentsch, T. J. (2000). Surface Expression and Single Channel Properties of KCNQ2/KCNQ3, M-Type K+ Channels Involved in Epilepsy. J. Biol. Chem. 275 (18), 13343–13348. doi:10.1074/jbc.275.18.13343
Selyanko, A. A., Hadley, J. K., Wood, I. C., Abogadie, F. C., Jentsch, T. J., and Brown, D. A. (2000). Inhibition of KCNQ1-4 Potassium Channels Expressed in Mammalian Cells via M1 Muscarinic Acetylcholine Receptors. J. Physiol. 522 (Pt 3), 349–355. doi:10.1111/j.1469-7793.2000.t01-2-00349.x
Soldovieri, M. V., Ambrosino, P., Mosca, I., De Maria, M., Moretto, E., Miceli, F., et al. (2016). Early-Onset Epileptic Encephalopathy Caused by a Reduced Sensitivity of Kv7.2 Potassium Channels to Phosphatidylinositol 4,5-Bisphosphate. Sci. Rep. 6, 38167. doi:10.1038/srep38167
Soldovieri, M. V., Boutry-Kryza, N., Milh, M., Doummar, D., Heron, B., Bourel, E., et al. (2014). Novel KCNQ2 and KCNQ3 Mutations in a Large Cohort of Families with Benign Neonatal Epilepsy: First Evidence for an Altered Channel Regulation by Syntaxin-1A. Hum. Mutat. 35, 356–367. doi:10.1002/humu.22500
Soldovieri, M. V., Castaldo, P., Iodice, L., Miceli, F., Barrese, V., Bellini, G., et al. (2006). Decreased Subunit Stability as a Novel Mechanism for Potassium Current Impairment by a KCNQ2 C Terminus Mutation Causing Benign Familial Neonatal Convulsions. J. Biol. Chem. 281 (1), 418–428. doi:10.1074/jbc.M510980200
Soldovieri, M. V., Freri, E., Ambrosino, P., Rivolta, I., Mosca, I., Binda, A., et al. (2020). Gabapentin Treatment in a Patient with KCNQ2 Developmental Epileptic Encephalopathy. Pharmacol. Res. 160, 105200. doi:10.1016/j.phrs.2020.105200
Soldovieri, M. V., Miceli, F., and Taglialatela, M. (2011). Driving with No Brakes: Molecular Pathophysiology of Kv7 Potassium Channels. Physiology (Bethesda) 26 (5), 365–376. doi:10.1152/physiol.00009.2011
Telezhkin, V., Thomas, A. M., Harmer, S. C., Tinker, A., and Brown, D. A. (2013). A Basic Residue in the Proximal C-Terminus Is Necessary for Efficient Activation of the M-Channel Subunit Kv7.2 by PI(4,5)P₂. Pflugers Arch. 465 (7), 945–953. doi:10.1007/s00424-012-1199-3
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 Potassium Channel Subunits: Molecular Correlates of the M-Channel. Science 282 (5395), 1890–1893. doi:10.1126/science.282.5395.1890
Weckhuysen, S., Ivanovic, V., Hendrickx, R., Van Coster, R., Hjalgrim, H., Møller, R. S., et al. (2013). Extending the KCNQ2 Encephalopathy Spectrum: Clinical and Neuroimaging Findings in 17 Patients. Neurology 81 (19), 1697–1703. doi:10.1212/01.wnl.0000435296.72400.a1
Weckhuysen, S., Mandelstam, S., Suls, A., Audenaert, D., Deconinck, T., Claes, L. R., et al. (2012). KCNQ2 Encephalopathy: Emerging Phenotype of a Neonatal Epileptic Encephalopathy. Ann. Neurol. 71 (1), 15–25. doi:10.1002/ana.22644
Wenk, M. R., Pellegrini, L., Klenchin, V. A., Di Paolo, G., Chang, S., Daniell, L., et al. (2001). PIP Kinase Igamma is the Major PI(4,5)P(2) Synthesizing Enzyme at the Synapse. Neuron 32, 79–88. doi:10.1016/s0896-6273(01)00456-1
Winks, J. S., Hughes, S., Filippov, A. K., Tatulian, L., Abogadie, F. C., Brown, D. A., et al. (2005). Relationship Between Membrane Phosphatidylinositol-4,5-Bisphosphate and Receptor-Mediated Inhibition of Native Neuronal M Channels. J. Neurosci. 25, 3400–3413. doi:10.1523/JNEUROSCI.3231-04.2005
Yus-Najera, E., Santana-Castro, I., and Villarroel, A. (2002). The Identification and Characterization of a Noncontinuous Calmodulin-Binding Site in Noninactivating Voltage-Dependent KCNQ Potassium Channels. J. Biol. Chem. 277 (32), 28545–28553. doi:10.1074/jbc.M204130200
Zhang, H., Craciun, L. C., Mirshahi, T., Rohács, T., Lopes, C. M., Jin, T., et al. (2003). PIP(2) Activates KCNQ Channels, and its Hydrolysis Underlies Receptor-Mediated Inhibition of M Currents. Neuron 37 (6), 963–975. doi:10.1016/s0896-6273(03)00125-9
Keywords: alternative splicing, Kv7.2 subunits, epileptic encephalopathy, PIP2, calmodulin, KCNQ2
Citation: Mosca I, Rivolta I, Labalme A, Ambrosino P, Castellotti B, Gellera C, Granata T, Freri E, Binda A, Lesca G, DiFrancesco JC, Soldovieri MV and Taglialatela M (2022) Functional Characterization of Two Variants at the Intron 6—Exon 7 Boundary of the KCNQ2 Potassium Channel Gene Causing Distinct Epileptic Phenotypes. Front. Pharmacol. 13:872645. doi: 10.3389/fphar.2022.872645
Received: 09 February 2022; Accepted: 10 May 2022;
Published: 13 June 2022.
Edited by:
Tarek Mohamed Abd El-Aziz, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Marina Trivisano, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2022 Mosca, Rivolta, Labalme, Ambrosino, Castellotti, Gellera, Granata, Freri, Binda, Lesca, DiFrancesco, Soldovieri and Taglialatela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Virginia Soldovieri, bWFyaWF2aXJnaW5pYS5zb2xkb3ZpZXJpQHVuaW1vbC5pdA==; Maurizio Taglialatela, bXRhZ2xpYWxAdW5pbmEuaXQ=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.