 Musadiq A. Bhat
Musadiq A. Bhat Abolghasem Esmaeili
Abolghasem Esmaeili Elena Neumann1‡
Elena Neumann1‡ Dietmar Benke
Dietmar Benke
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 29 March 2022
Sec. Neuropharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.870861
This article is part of the Research TopicRestoring endoplasmic reticulum proteostasis to treat neurological disordersView all 5 articles
GABAB receptors control neuronal excitability via slow and prolonged inhibition in the central nervous system. One important function of GABAB receptors under physiological condition is to prevent neurons from shifting into an overexcitation state which can lead to excitotoxic death. However, under ischemic conditions, GABAB receptors are downregulated, fostering over-excitation and excitotoxicity. One mechanism downregulating GABAB receptors is mediated via the interaction with the endoplasmic reticulum (ER) stress-induced transcription factor CHOP. In this study, we investigated the hypothesis that preventing the interaction of CHOP with GABAB receptors after an ischemic insult restores normal expression of GABAB receptors and reduces neuronal death. For this, we designed an interfering peptide (R2-Pep) that restored the CHOP-induced downregulation of cell surface GABAB receptors in cultured cortical neurons subjected to oxygen and glucose deprivation (OGD). Administration of R2-Pep after OGD restored normal cell surface expression of GABAB receptors as well as GABAB receptor-mediated inhibition. As a result, R2-Pep reduced enhanced neuronal activity and inhibited progressive neuronal death in OGD stressed cultures. Thus, targeting diseases relevant protein-protein interactions might be a promising strategy for developing highly specific novel therapeutics.
Ischemic stroke is one of the leading causes of mortality and disability worldwide (Katan and Luft 2018). During cerebral ischemia reduced or blocked blood circulation deprives neurons from oxygen and glucose, which induces various destructive cellular reactions leading to neuronal death (White et al., 2000). The main cause of progressive neuronal death after an ischemic insult is neuronal overexcitation, resulting from aberrant activation of glutamate receptors and a massive accumulation of intracellular Ca2+ (excitotoxicity). Currently, there is no pharmacological treatment available for clinical use to inhibit progressive neuronal death after an ischemic insult.
Under physiological conditions, neuronal excitability is controlled by GABAB receptors, preventing neurons to reach a state of overexcitation and thereby excitotoxicity (Chalifoux and Carter 2011). GABAB receptors are heterodimeric G protein-coupled receptors consisting of GABAB1 and GABAB2 subunits. They are expressed in virtually all neurons at pre- and postsynaptic locations (Gassmann and Bettler 2012; Terunuma 2018). The binding of the neurotransmitter GABA to the receptor activates Gi/o proteins, which in turn modulate several effector systems inducing long-lasting neuronal inhibition. The most prominent effect of GABAB receptors located at postsynaptic sites is the activation of G protein-coupled inwardly rectifying potassium (GIRK or Kir3) channels, resulting in the hyperpolarization of the neuronal membrane thereby reducing the likelihood of action potential generation (Gähwiler and Brown 1985; Luscher et al., 1997; Chalifoux and Carter 2011). At presynaptic sites the most obvious effect of GABAB receptors is the inhibition of voltage-gated Ca2+ channels, thereby reducing neurotransmitter release (Mintz and Bean 1993; Santos et al., 1995; Chen and van den Pol 1998; Bussieres and El Manira 1999; Chalifoux and Carter 2011).
There is substantial in vitro and in vivo evidence for downregulation of GABAB receptors under conditions leading to neuronal overexcitation and neuronal death (Vollenweider et al., 2006; Guetg et al., 2010; Maier et al., 2010; Terunuma et al., 2010; Kim et al., 2011; Kantamneni et al., 2014; Maier et al., 2014; Huang et al., 2017; Hleihil et al., 2021). This unfortunate situation removes GABAB receptors as “emergency brake” to combat neuronal overexcitation from the system and further fosters excitotoxic neuronal death. One mechanism that downregulates functional GABAB receptors, i. e. receptors expressed at the cell surface, involves the endoplasmic reticulum (ER) stress-induced transcription factor C/EBP homologous protein (CHOP) (Maier et al., 2014). CHOP, also known as C/EBPζ, growth arrest and DNA damage-inducible gene 153 (GADD153) or DNA-damage-inducible transcript 3 (DDIT3), belongs to the CCAAT/enhancer-binding proteins (C/EBP) family and is only marginally expressed under physiological conditions (Oyadomari and Mori 2004; Yang et al., 2017; Hu et al., 2018). However, CHOP is strongly upregulated upon ER stress associated with ischemia and induces apoptotic cell death (Zinszner et al., 1998; Oyadomari and Mori 2004; Morimoto et al., 2007; Oida et al., 2008).
In addition to its pro-apoptotic activity, upregulated CHOP interacts via its C-terminal leucine zipper with the leucine zipper within the C-terminal domain of GABAB2 (Sauter et al., 2005). This interaction blocks the assembly of the GABAB receptor subunits GABAB1 and GABAB2 in the ER and thereby prevent the exit of assembled receptors from ER (Maier et al., 2014). Because the supply of newly formed GABAB receptors is inhibited, ongoing constitutive internalization and lysosomal degradation of cell surface receptors downregulates functional receptors and thereby GABAB receptor-mediated inhibition (Maier et al., 2014).
Enhancing GABAB receptor activity using the agonist baclofen showed beneficial effects in in vivo and in vitro models of cerebral ischemia (Lal et al., 1995; Jackson-Friedman et al., 1997; Kulinskii and Mikhel’son 2000; Babcock et al., 2002; Dave et al., 2005; Zhang et al., 2007; Xu et al., 2008; Cimarosti et al., 2009; Liu et al., 2015; Hleihil et al., 2021). These observations provide strong support that GABAB receptors may constitute valid targets for a neuroprotective strategy. As GABAB receptors are downregulated under ischemic conditions, activation of the receptors by baclofen might have only a limited neuroprotective effect. We, therefore, tested the hypothesis that restoring normal cell surface GABAB receptor expression in neurons subjected to ischemic stress by preventing its interaction with CHOP would reduce ischemia-induced neuronal overexcitation and diminish neuronal death. To inhibit the interaction of GABAB receptors with CHOP, we designed a short interfering peptide (R2-Pep) that comprises a specific sequence corresponding partially to the coiled-coil domain of GABAB2, which is part of the binding site for CHOP (Sauter et al., 2005). R2-Pep indeed restored cell surface expression of GABAB receptors in cultured cortical neurons subjected to OGD (mimicking cerebral ischemia) and inhibited progressive neuronal death.
The use of rat embryos for generating primary neuronal cultures was approved by the Zurich cantonal veterinary office, Zurich, Switzerland (licence ZH011/19). All cell culture media used were from Gibco. The cerebral cortex of 18 days old rat embryos were carefully dissected and washed with 5 ml sterile-filtered PBGA buffer (PBS containing 10 mM glucose, 1 mg/ml bovine serum albumin and antibiotic-antimycotic 1:100 (10,000 units/ml penicillin; 10,000 μg/ml streptomycin; 25 μg/ml amphotericin B)). The cortices were cut into small pieces with a sterile scalpel and digested in 5 ml sterile filtered papain solution for 15 min at 37°C. The supernatant was removed and the tissue was washed twice with complete DMEM/FCS medium (Dulbecco’s Modified Eagle’s Medium containing 10% Fetal Calf Serum and penicillin/streptomycin, 1:100). Then, fresh DMEM/FCS was added, and the tissue was gently triturated and subsequently filtered through a 40 μm cell-strainer. Finally, the neurons were plated at a concentration of 60,000–80,000 cells per well onto the poly-l-lysine (50 μg/ml in PBS) coated coverslips in a 12-well culture dish and incubated overnight at 37°C and 5% CO2. After 24 h of incubation, the DMEM medium was exchanged with freshly prepared NU-medium (Minimum Essential Medium (MEM) with 15% NU serum, 2% B27 supplement, 15 mM HEPES, 0.45% glucose, 1 mM sodium pyruvate, 2 mM GlutaMAX). The cultures were kept for 12–16 days in vitro.
The OGD medium (DMEM lacking glucose, glutamine, sodium pyruvate, HEPES and phenol red) was deprived from oxygen by equilibrating it with nitrogen for 15 min in a water bath at 37°C. 1 ml of equilibrated OGD medium was added into each well of a 12-well culture plate and the coverslips containing the cultured neurons were transferred to the OGD medium. The culture plate was then incubated for 1 hour in a hypoxic incubator at 1% O2, 5% CO2 and 37°C. The coverslips were then transferred back to the culture plate containing the original conditioned culture medium and incubated at 37°C and 5% CO2. Unless otherwise stated, neurons were analyzed after a recovery period of 16 h.
The interfering peptide (R2-Pep) comprises a sequence derived partially from the coiled-coil domain of GABAB2 (LQDTPEKTTYIK). A peptide (Ctrl-Pep) containing the same amino acids but in a random sequence (PETKTDLTQYKI) was used as a control (Ctrl-Pep). To render the peptides cell-permeable, they were tagged at their N-terminus with the protein transduction domain of trans-activator of transcription (TAT) followed by an arginine-rich region (GRKKRRQRRRPQRRRRRRRR). Both peptides were custom synthesized at PEPMIC Co., Ltd., Suzhou, China and labelled at the N-terminus with FITC.
For all experiments, except for the survival assay, the cultures were treated with R2-Pep (10 μg/ml) or with Ctrl-Pep (10 μg/ml) in their original culture medium immediately after the OGD incubation and then incubated for 16 h at 37°C and 5% CO2.
For cell surface staining of GABAB receptors, an antibody directed against the N-terminus of GABAB2 (GABAB2N, custom-made by GeneScript) was used (Benke et al., 2002)). Coverslips containing the cultured neuron/glia cells were washed 3 times with cold buffer A (25 mM HEPES pH 7.4, 119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2 and 30 mM glucose). Then the GABAB2N antibody (1:250 dilution in buffer A containing 10% normal donkey serum (NDS)) was added and incubated on ice for 90 min. The coverslips were then washed 3 times for 5 min with buffer A, followed by incubation with Cy3-labelled donkey anti-rabbit secondary antibody (1:400 in PBS/10% NDS, Jackson ImmunoResearch) for 60 min on ice. Afterwards, the coverslips were washed again 3 times for 5 min with buffer A.
For subsequent staining of intracellular CHOP, the cells were fixed with 4% PFA for 30 min at room temperature. After fixation, the cells were washed with PBS and permeabilized by incubation for 12 min in 0.2% Triton X-100/PBS. Then, CHOP/GADD 153 antibody (1:500, clone B-3, Cat No. sc-7351, Santa Cruz) was added and incubated overnight at 4°C. After incubation, the coverslips were washed 3 times for 5 min with PBS. Then, Cy3-or Cy5-labeled donkey anti-mouse antibody (dilution 1:400 in PBS/10% NDS, Jackson ImmunoResearch) was added and incubated for 1 h at room temperature. Finally, the coverslips were washed again 3 times for 5 min with PBS and mounted in DAKO fluorescence mounting medium onto glass slides for microscopy.
For monitoring Kir3.2 channel expression, the coverslips were washed with PBS, fixed for 30 min in 4% PFA, followed by permeabilization with 0.2% Triton X-100 in PBS for 12 min and incubation with anti-Kir3.2 antibody (1:250, Cat No. APC-006, Alomone Labs). Antibody incubation and further treatment were done as described for staining of CHOP.
In situ PLA was used to investigate the interaction between GABAB1 and GABAB2 subunits as well as CHOP and R2-Pep. The in situ PLA was performed using the Duolink II kit (Sigma Aldrich) according to the instructions of the manufacturer. Briefly, the neurons were washed with PBS and then fixed with 4% PFA for 30 min at room temperature. Then the coverslips were rinsed in PBS for 5 min and permeabilized for 12 min with 0.2% Triton X-100/PBS. After rinsing the coverslips in PBS, they were incubated with the primary antibody solution consisting of a rabbit antibody raised against the C-terminal domain of GABAB2 (1:250, Cat. No. ab75838, Abcam) and a mouse antibody directed against GABAB1 (1:50, Cat No. ab55051, Abcam) or in case of the CHOP/R2-Pep interaction with mouse CHOP antibody (1:40, clone B-3, Cat. No. sc-7351, Santa Cruz) and goat FITC antibody (1:400, Cat. No. NB600-1273, Novus Biologicals; R2-Pep was labeled with FITC at the N-terminus) in a humidity chamber overnight at 4°C. Subsequently, the cultures were washed four times for 5 min with PBS and incubated for 20–30 min at room temperature with the PLA probes (prepared by diluting anti-Mouse MINUS and anti-Rabbit PLUS or anti-Goat PLUS (Duolink II) 1:5 in 5% BSA/PBS). Afterwards, 40 μL of the PLA probe solution were pipetted on top of each coverslip and incubated in a humidity chamber for 1 h at 37°C. The coverslips were then washed two times for 5 min in PBS and incubated for 1 h at 37°C with ligation solution. Subsequently, the cells were washed two times in Duolink II Wash Buffer A and incubated with the amplification solution at 37°C for 100 min. Finally, the coverslips were washed two times for 10 min with Duolink II Wash Buffer B in the dark and mounted onto microscope slides with DAKO fluorescent mounting medium.
To quantify neuronal death, co-cultures of neurons and glia cells subjected to OGD and subsequently treated with R2-Pep or Ctrl-Pep were stained with an antibody directed against the neuron-specific marker protein NeuN (1:400, Cat. No. ABN78, Millipore), followed by staining with Alexa Fluor Plus 488 secondary antibodies (1:2000, Jackson ImmunoResearch). The total number of cells was determined by counting the cell nuclei stained with DAPI included in the fluorescent mounting medium. After microscopy, neuronal loss was quantified by counting the number of neurons and number of DAPI positive nuclei using the ImageJ plugin “Cell Counter” and the ratio of neurons to total cells (DAPI positive nuclei) per image was calculated.
Images were taken with a Zeiss laser scanning confocal microscope (CLSM700 or CLSM710) in sequential mode using the Zeiss 40x (1.3 NA) and 100x (1.45 NA) plan-fluar objective, with a resolution of 1024 × 1024 pixels. The laser intensity and the detector gain were adjusted to values that avoid signal saturation and all images of one experiment was imaged with the same settings in one continuous session. The images were quantitatively analysed using the software ImageJ.
For quantification of cell surface staining, the outer and the inner perimeter of the cell surface were exactly outlined. Then the fluorescence intensity value obtained from the inner border was subtracted from the one of the outer border so that only the fluorescence present at the cell surface was determined and used for statistical evaluation.
For quantification of the total cell staining, only the outer border of the cell was marked and the mean fluorescence intensity was measured.
For quantification of in situ PLA signals, the soma of neurons was surrounded and the fluorescent dots inside these borders were counted using the ImageJ option “Find maxima.” A fixed noise tolerance value was used for the analysis of all images of the same experiment. The PLA signals were normalized to the area analyzed.
Cortical neurons were recorded in the whole-cell voltage-clamp configuration at room temperature using a HEKA EPC10 amplifier and the Patchmaster software. Spontaneous postsynaptic currents (sPSCs) were recorded at a holding potential of -60 mV. Patch electrodes were filled with 120 mM CsCl or KCl, 10 mM EGTA, 10 mM HEPES pH 7.4, 4 mM MgCl2, 0.5 mM GTP and 2 mM ATP. Spontaneous PSCs recordings were performed using intracellular CsCl, whereas the potassium currents were recorded using an intracellular solution containing KCl. The external solution contained 140 mM NaCl, 10 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES pH 7.4 and 10 mM glucose.
Potassium currents were recorded in the presence of tetrodotoxin (0.5 μM), 7-nitro-2,3-dioxo-1,4-dihydroquinoxaline-6-carbonitrile (CNQX, 2 μM), and bicuculline (4 μM). To enhance the amplitude of the baclofen-evoked currents, the potassium concentration of the extracellular solution was increased to 30 mM, and the sodium concentration was reduced to 120 mM (to keep osmolarity constant). GABAB receptor-evoked potassium currents were elicited using a 10 s pulse of 50 μM baclofen at −90 mV.
All synaptic events displaying amplitudes above the background noise (5–12 pA) were identified and analyzed off-line using the MiniAnalysis 6.0.7 software (Synaptosoft). Mean amplitudes and frequency values were obtained from 1 min recording epochs on each experimental condition and normalized to the control condition of the individual neuron.
The statistical evaluation of data was performed using the software GraphPad Prism (version 8.4.3.). All results were given as mean value ±standard error of the mean (SEM). The data were analyzed by one- or two-way ANOVA or Kruskal–Wallis test followed by appropriate post hoc tests as indicated in the figure legends. Data sets were tested for normal or lognormal distributions by the D’Agostino-Pearson test and QQ-plots. In case of significant deviation from homoscedasticity Welch and Brown Forsythe variations of ANOVA was used. A p-value of <0.05 was considered as statistically significant.
We previously showed that CHOP upregulation is maintained in cultured neurons for at least 24 h after being exposed to OGD stress (Maier et al., 2014). However, we did not test for the earliest time point after OGD at which a significant up-regulation of CHOP occurs that could affect GABAB receptor cell surface expression. This information is important for selecting the optimal time point for the electrophysiological experiments, which should as early as possible after the OGD stress for technical reasons. We, therefore, subjected cultured neurons to OGD stress and monitored CHOP expression as well as GABAB receptor cell surface levels either directly after OGD stress or after a recovery period of 30–150 min. Significant up-regulation of CHOP was visible immediately OGD stress and progressively increased thereafter (Figure 1). Conversely, cell surface expression of GABAB receptors concomitantly decreased (Figure 1). These results show a correlated up- and down-regulation of CHOP and GABAB receptors, respectively, directly after 1 h of OGD-induced stress interval.
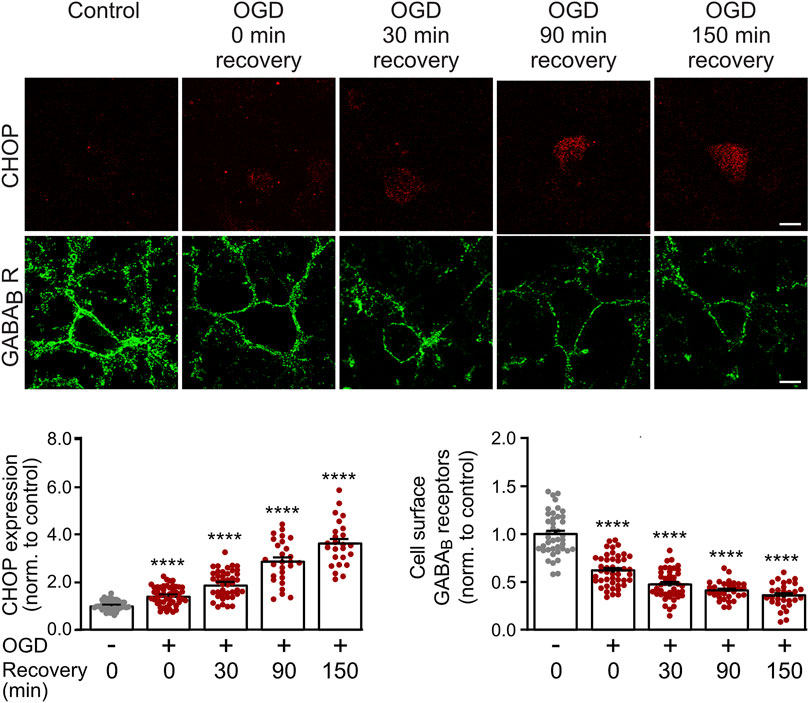
FIGURE 1. Time course of OGD-induced downregulation of GABAB receptor cell surface expression and CHOP upregulation. Neurons were exposed to OGD for 1 h, returned to the normal culture medium and tested for GABAB2 cell surface as well as CHOP expression at different time intervals (0, 30, 90 and 150 min) thereafter. For the 0 min recovery time, the cultures were removed from the OGD medium, washed once in PBS for 2 min and then fixated. Top: representative images, the scale bar corresponds to 10 μm. Bottom: quantification of fluorescence intensities. Neurons not subjected to OGD served as a control. The data represent the mean ± S.E.M. of 31–46 neurons for each experimental condition derived from three independent experiments. ****, p ˂ 0.0001; Brown-Forsythe/Welch one-way ANOVA followed by Dunnett’s T3 multiple comparison test.
CHOP interacts with GABAB receptors via the coiled-coil motif of GABAB2 located in the intracellularly located C-terminal domain (Sauter et al., 2005) and prevents heteromerization of GABAB1 with GABAB2 in the ER (Maier et al., 2014). For the identification of a small peptide interfering with this interaction, we designed a set of synthetic peptides covering the GABAB2 coiled-coil domain and tested them for their ability to restore ER stress-induced (1 μM thapsigargin for 2 h) impairment of heteromerization of GABAB1 and GABAB2 in cultured neurons using in situ PLA. The screening yielded one peptide comprising the GABAB2 sequence 815–826 (LQDTPEKTTYIK) that reliably restored the expression of GABAB1/GABAB2 heteromers (Figures 2A,B). This peptide was rendered cell-permeable by tagging it at the N-terminus with the HIV trans-activator of transcription (TAT) sequence followed by an arginine-rich region (this peptide was named R2-Pep). In addition, the peptide was labelled at the N-terminus with FITC for identification. R2-Pep interacted with upregulated CHOP (cultures treated with 1 μM thapsigargin for 2 h) as indicated by numerous in situ PLA signals (Figure 2C).
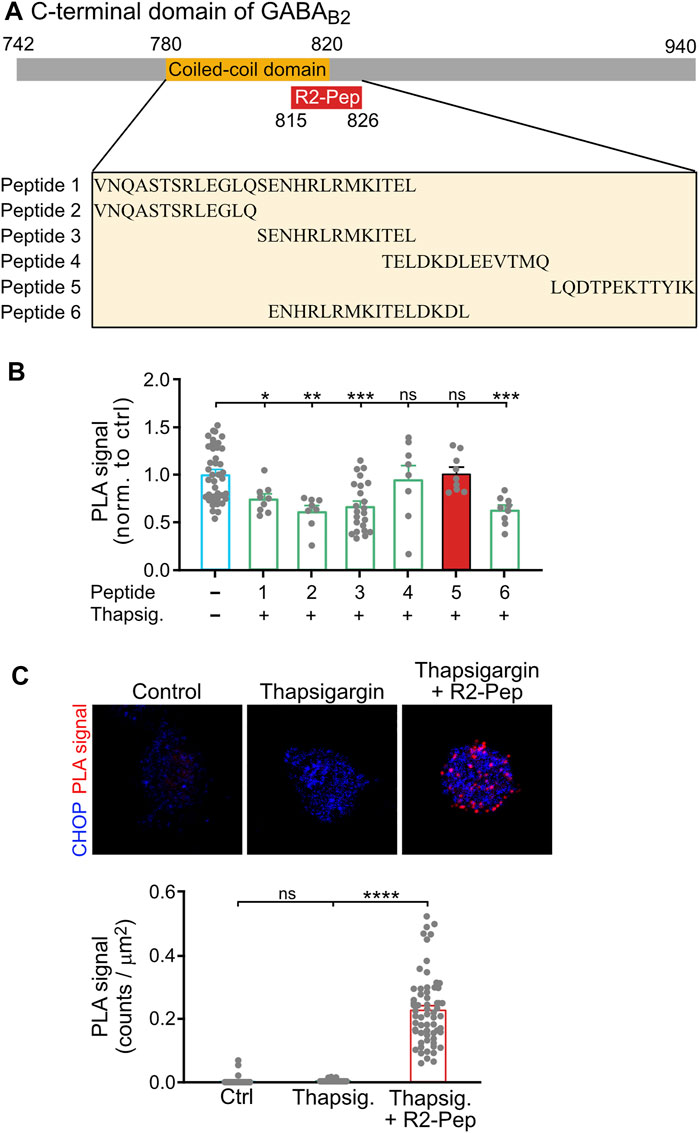
FIGURE 2. Screening for a peptide (R2-Pep) interfering with the interaction of CHOP with GABAB receptors. (A) Scheme of the C-terminal domain of GABAB2 with the coiled-coil domain (orange box), which contributes to the CHOP-GABAB receptor interaction site and location of the interfering peptide R2-Pep sequence (red box). The box below depicts the GABAB2 peptide sequences used for screening. (B) Peptide 5 reliably prevented stress-induced decrease in GABAB1/GABAB2 heteromers. Cultures were stressed for 2 h with thapsigargin (1 μM) and immediately thereafter treated for 30 min with the peptides indicated in (A). Neurons were then tested for the interaction between GABAB1 and GABAB2 by in situ PLA. The in situ PLA signals of the untreated neurons served as control (-, no peptide). The data represent the mean ± S.E.M. of 8–40 neurons derived from two independent experiments (control, n = 4). ns, p > 0.05; *, p < 0.05; **, p < 0.005; ***; p < 0.0005. Brown-Forsythe/Welch one-way ANOVA followed by Games-Howell’s multiple comparison test. (C) R2-Pep (peptide 5 in B) interacted with CHOP. Cultures were stressed for 2 h with thapsigargin (1 μM) and immediately thereafter treated with R2-Pep for 30 min. Neurons were then tested for the interaction of CHOP with R2-Pep by in situ PLA using antibodies directed against CHOP and FITC (R2-Pep was labeled with FITC at the N-terminus). The data represent the mean ± S.E.M. of 47–62 neurons derived from two independent experiments. ns, p > 0.05; ****; p < 0.0001. Kruskal–Wallis test followed by Dunn’s multiple comparison test.
Next, we tested for the ability of R2-Pep to normalize OGD-induced downregulation of GABAB receptors. Treatment of neurons with R2-Pep immediately after OGD restored cell surface expression of GABAB receptors to normal physiological levels (Figure 3A, tested after a recovery period of 16 h). Interestingly, treatment with R2-Pep also reversed the upregulated CHOP expression to normal levels (Figure 3B). As CHOP is constitutively polyubiquitinated and degraded by proteasomes (Hattori et al., 2003), the most likely explanation was the proteasomal degradation of the CHOP/R2-Pep complex. Indeed, inhibition of proteasomal degradation by lactacystin or MG132 completely abolished the downregulation of CHOP induced by R2-Pep, whereas inhibition of lysosomal degradation had no effect (Figure 3C).
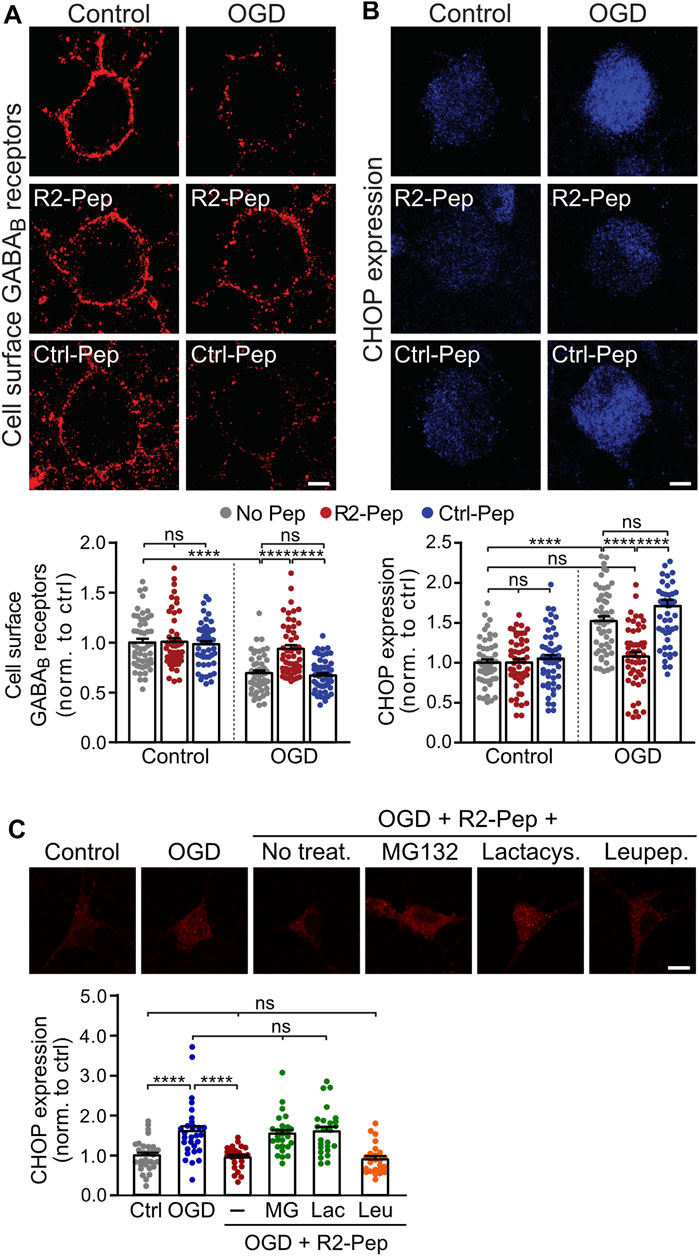
FIGURE 3. CHOP-mediated down-regulation of cell surface GABAB receptors is rescued by R2-Pep. (A) and (B), Cultures were stressed for 1 h with OGD and immediately thereafter treated with R2-Pep, the inactive Ctrl-Pep or remained untreated. The neurons were immunostained for cell surface GABAB2 (A) and CHOP (B) 16 h after OGD stress. Top: representative images (scale bar: 5 μm). Bottom: quantification of fluorescence intensities. The fluorescence intensity of the untreated neurons served as controls. The data represent the mean ± S.E.M. of 49–53 neurons for each experimental condition derived from three independent experiments. ns, p > 0.05; ****; p < 0.0001; two-way ANOVA with Tukey’s multiple comparisons test. (C) R2-Pep induces proteasomal degradation of CHOP upregulated by OGD. Cultures were stressed for 1 h with OGD and immediately thereafter remained untreated or were treated with R2-Pep in the presence of proteasome inhibitors (50 μM lactacystin or 50 μM MG132) or the lysosome inhibitor leupeptin (50 μM). The neurons were immunostained for CHOP expression 16 h after OGD stress. Top: representative images (scale bar: 10 μm). Bottom: quantification of fluorescence intensities. The fluorescence intensity of untreated neurons served as control. The data represent the mean ± S.E.M. of 25–30 neurons for each experimental condition derived from two independent experiments. ****, p < 0.0001; ns, p > 0.05; one-way ANOVA with Tukey’s multiple comparisons test.
Treatment of R2-Pep to neurons not subjected to OGD did neither affect basal CHOP expression nor cell surface GABAB receptor expression. As expected, an inactive control peptide (Ctrl-Pep, same amino acids as R2-Pep but in a random order) did not affect GABAB receptor and CHOP expression under control or OGD conditions (Figures 3A,B).
Up-regulated CHOP interacts with GABAB receptors in the ER, prevents their heterodimerization and thereby inhibits their forward trafficking to the cell surface (Maier et al., 2014). To test whether R2-Pep normalizes the CHOP-induced impaired heterodimerization of GABAB receptors, cultured neurons were subjected to OGD and analyzed for GABAB1-GABAB2 interaction by in situ PLA in the presence or absence of the interfering peptide R2-Pep or the inactive Ctrl-Pep (Figure 4). OGD stress significantly decreased GABAB receptor heteromers in neurons, which was normalized to physiological levels (control) upon administration of R2-Pep. In contrast, treatment of neurons with R2-Pep not subjected to OGD did not affect the level of GABAB receptors heteromers. In addition, the inactive control peptide did not affect the levels of GABAB receptor heteromers under control or OGD conditions, indicating the specificity of R2-Pep (Figure 4).
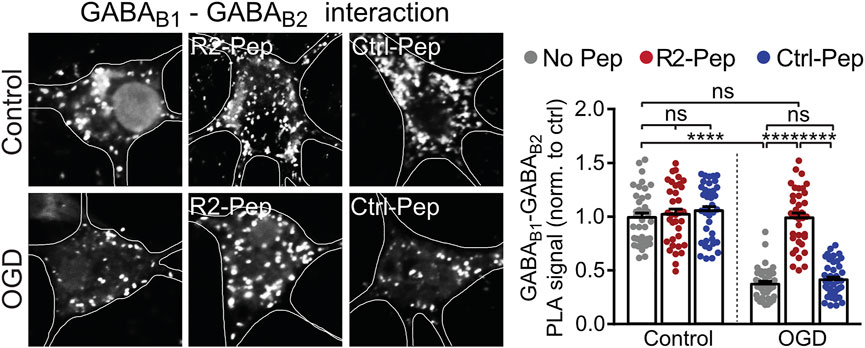
FIGURE 4. CHOP-mediated decrease of GABAB receptor heterodimerization is rescued by R2-Pep. Cultures were stressed for 1 h with OGD and immediately thereafter treated with R2-Pep, the inactive Ctrl-Pep or remained untreated. After 3 h, the interaction between GABAB1 and GABAB2 was tested by in situ PLA. Left: representative images (scale bar: 5 μm). Right: quantification of the GABAB1-GABAB2 interaction in control or OGD treated neurons in the absence or presence of R2-Pep or Rand-Pep. The interaction level observed in untreated neurons served as control. The data represent the mean ± S.E.M. of 37–41 neurons for each experimental condition derived from three independent experiments. ****, p < 0.0001; ns: p > 0.05; two-way ANOVA with Tukey’s multiple comparisons test.
Postsynaptically, GABAB receptor-mediated inhibition is mainly conveyed via activation of inwardly rectifying K+ channels (Kir3.2), leading to inhibitory postsynaptic potentials (Luscher et al., 1997). As GABAB receptors form signalling complexes with their effector proteins (Schwenk et al., 2016), we expected a correlated regulation of the receptors with Kir3.2 channels. Therefore, we analyzed the effect of OGD and R2-Pep on the expression of Kir3.2 channels. In neurons stressed with OGD, Kir3.2 channels were downregulated (Figure 5), comparable to GABAB receptors. Treatment with R2-Pep immediately after OGD restored normal expression levels of Kir3.2, being in line with a co-regulation of GABAB receptors and Kir3.2 in signalling complexes. The inactive Ctrl-Pep did not affect expression of the effectors, documenting the specificity of R2-Pep (Figure 5). In addition, R2-Pep did not affect expression of the effectors under control conditions, i. e. unstressed neurons (Figure 5).
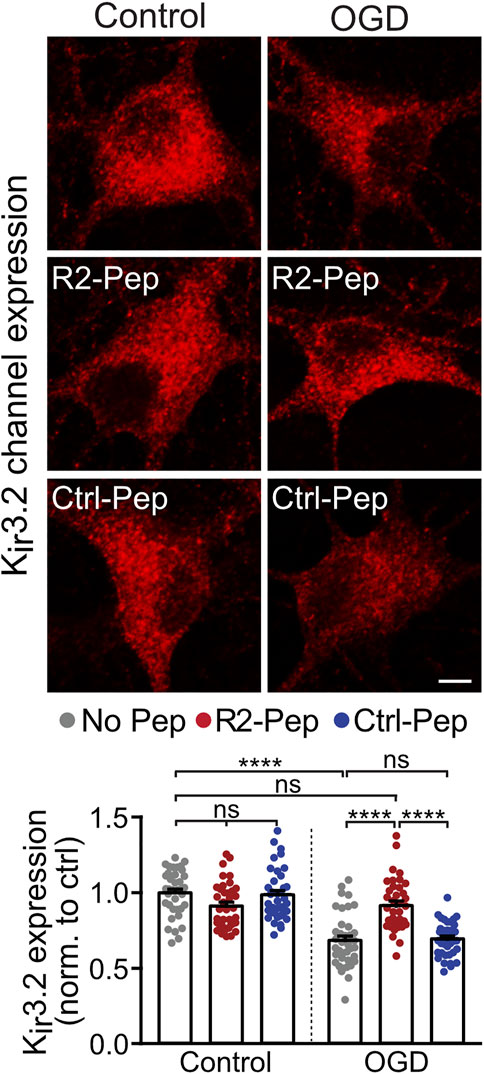
FIGURE 5. R2-Pep normalize OGD-mediated dysregulation of Kir3.2 channels. Cultures were stressed for 1 h with OGD and immediately treated with R2-Pep, Ctrl-Pep or remained untreated. After 16 h neurons were immunostained for Kir3.2 channel expression. Top: representative images (scale bar: 5 μm). Bottom: quantification of fluorescence intensities. The fluorescence intensity of the untreated neurons served as control. The data represent the mean ± S.E.M. of 37–38 neurons from two independent preparations. ns: p > 0.05; ****, p < 0.0001; two-way ANOVA with Tukey’s multiple comparisons test.
Next, we assessed the functional consequences of restored GABAB receptor levels after exposing cultured neurons to OGD in the presence or absence of R2-Pep by measuring baclofen-induced K+ currents using whole-cell patch-clamp recordings. As expected, we observed a strong reduction of baclofen-induced K+ currents amplitudes (i. e. GABAB receptor-mediated currents) in OGD-stressed neurons, which were nearly completely restored to control amplitude values after treatment with R2-Pep (Figure 6A). In contrast, the control peptide (Ctrl-Pep) showed no effect. This finding supports the hypothesis that R2-Pep restores functional GABAB receptors available for neuronal inhibition.
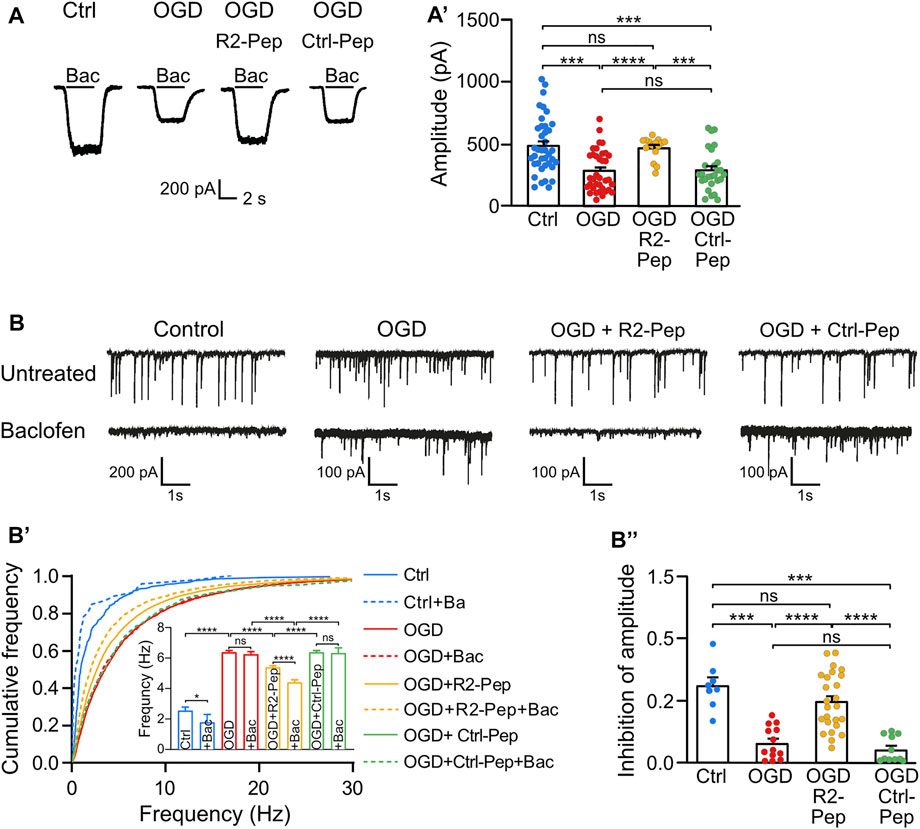
FIGURE 6. Treatment of OGD-stressed neurons with R2-Pep restores GABAB receptor-mediated inhibition. (A) R2-Pep restored OGD-induced downregulated GABAB receptor-mediated K+ currents. Representative traces of baclofen-induced K+ currents were recorded in untreated neurons or OGP-treated neurons in the presence or absence of R2-Pep or the inactive Ctrl-Pep. (A′), Quantitative evaluation of K+ current amplitudes. The data represent the mean ± S.E.M. of Ctrl: n = 39, OGD: n = 36, OGD + R2-Pep: n = 15, OGD + Ctrl-Pep: n = 27. Ns, p > 0.05; ***, p < 0.001 ****, p < 0.0001, Brown-Forsythe/Welch one-way ANOVA followed by Dunnett’s T3 multiple comparison test. (B) Treatment of OGD-stressed neurons with R2-Pep restores baclofen-induced inhibition of the amplitude and frequency of spontaneous postsynaptic currents (sPSCs). Representative current traces depicting sPSCs recorded from untreated neurons and OGD-stressed neurons in the absence or presence of R2-Pep or the inactive Ctrl-Pep. (B′), Cumulative frequency histogram of inter-event-intervals of sPSCs recorded from cells without (solid lines) or with baclofen (dashed lines). Ns, p > 0.05; *, p < 0.05; ****, p < 0.0001; Kruskal–Wallis test followed by Dunn’s multiple comparison test (Ctrl: n = 8, OGD: n = 13, OGD + R2-Pep: n = 25, OGD + Ctrl: n = 11). (B”), Inhibition of sPSC amplitudes by baclofen. Mean amplitudes values were normalized to the control condition of the individual neuron. Ns, p > 0.05; ***, p < 0.001; ****, p < 0.0001; Brown-Forsythe/Welch one-way ANOVA followed by Games-Howell’s multiple comparison test (Ctrl: n = 8, OGD: n = 13, OGD + R2-Pep: n = 25, OGD + Ctrl-Pep: n = 11).
Next, we analyzed whether restored GABAB receptor-mediated inhibition can reduce OGD-induced neuronal overexcitation. OGD treatment strongly increased the frequency of spontaneous postsynaptic currents (sPSCs), indicating enhanced neuronal excitation (Figure 6B). In contrast to untreated neurons, application of baclofen did not reduce the frequency of sPSCs in OGD-treated neurons, suggesting insufficient GABAB receptor-mediated inhibition after ischemic stress. However, the application of R2-Pep to OGD-stressed neurons significantly reduced the frequency of sPSCs (Figure 6B’) and significantly increased baclofen-induced inhibition of current amplitudes (Figure 6B’’). These findings indicate that restoration of GABAB receptor-mediated inhibition in neurons subjected to OGD reduces neuronal overexcitation.
Finally, we tested whether preventing downregulation of GABAB receptors by R2-Pep is neuroprotective. Co-cultures of neurons and glia cells were submitted for 1 h to OGD and treated thereafter with R2-Pep or Ctrl-Pep at different time intervals (see Figure 7A for the experimental outline). To monitor loss of neurons, cultures were stained for the neuronal marker protein NeuN and with DAPI for total cells (the vast majority of cells in the cultures are glia cells, which require much longer exposure to OGD to induce death (Benavides et al., 2005)).
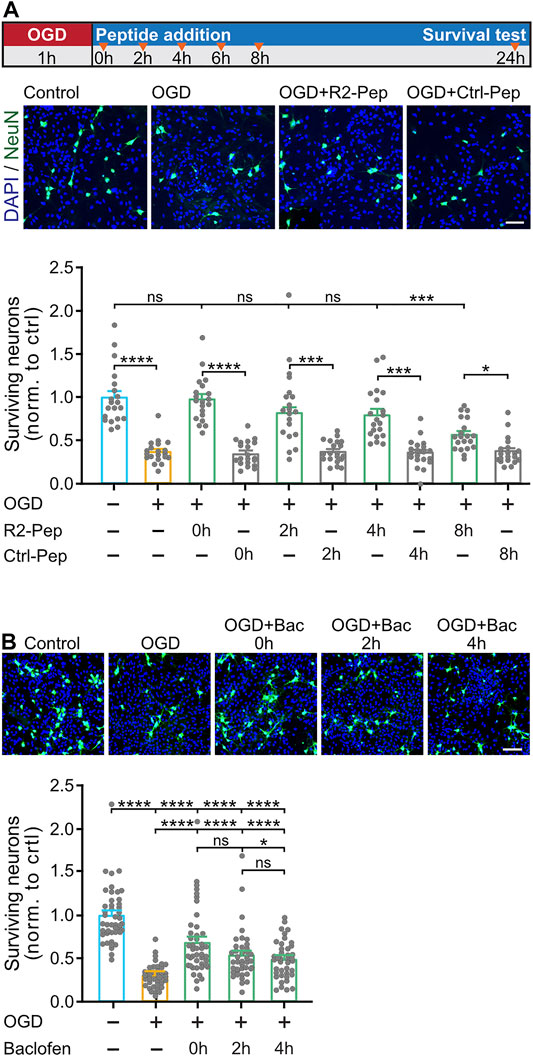
FIGURE 7. R2-Pep limits OGD-induced neuronal death and is more effective than baclofen. A, OGD-stressed cultures were treated with R2-Pep or Crtl-Pep immediately (0 h), 2, 4, 6 or 8 h after the stress. After 24 h the cultures were immunostained for neurons (NeuN, green) and total cells (DAPI, blue). Top: scheme of experimental design. Middle: representative images (scale bar: 100 μm). Bottom: Quantification of surviving neurons. The value of the untreated cultures served as control. The data represent the mean ± S.E.M. of 20 frames (fields of view) for each experimental condition derived from 2 independent experiments. ns: p > 0.05; *, p < 0.05; ***, p < 0.001; ****, p < 0.0001; Brown-Forsythe/Welch one-way ANOVA followed by Dunnett’s T3 multiple comparison test. B, OGD-stressed cultures were treated with baclofen immediately (0 h), 2 h or 4 h after the stress. After 24 h the cultures were immunostained for neurons (NeuN, green) and total cells (DAPI, blue). Top: representative images (scale bar: 100 μm). Bottom: Quantification of surviving neurons. The value of the untreated cultures served as control. The data represent the mean ± S.E.M. of 40–45 frames for each experimental condition derived from 3 independent experiments. ****, p < 0.0001; one-way ANOVA with Tukey’s multiple comparisons test.
In OGD-stressed cultures, a substantial loss of neurons (37 ± 3% of control) was observed after 24 h, indicating considerable neuronal death (Figure 7A). Loss of neurons was completely blocked upon administration of R2-Pep immediately after the stress (98 ± 6% of control) and strongly attenuated (not statistically significant different from control) upon treatment after 2 h (82 ± 7% of control) and 4 h (80 ± 7% of control). The neuroprotective activity dropped to 57 ± 4% of control when cultures were treated with R2-Pep 8 h after the OGD-stress.
Next, we tested whether the neuroprotective activity of R2-Pep is superior to that of the GABAB receptor agonist baclofen. Baclofen exhibits some level of neuroprotection under ischemic conditions (Lal et al., 1995; Babcock et al., 2002; Zhang et al., 2007; Han et al., 2008; Xu et al., 2008; Zhou et al., 2008; Liu et al., 2015; Fu et al., 2016; Lu et al., 2016; Hleihil et al., 2021), but this might be suboptimal in view of the downregulation of the receptors. As expected, compared to R2-Pep, baclofen showed considerably less neuroprotection at all time points tested (0 h: 68 ± 6%, 2 h: 54 ± 5% and 4 h: 50 ± 3% of control) (Figure 7B).
Stressful conditions, such as cerebral ischemia, impair ER function which causes the accumulation of proteins in the ER. This triggers the activation of ER stress response pathways to restore proper function of the ER (Hetz 2012; Kaneko et al., 2017). When the functions of the ER cannot be restored and ER response pathways can no longer limit the damage, apoptosis is induced. One important ER stress-induced apoptotic pathway is triggered by upregulation of the transcription factor CHOP, whose expression level is otherwise marginal under normal physiological conditions (Oyadomari and Mori 2004; Yang et al., 2017; Hu et al., 2018). Apart from functioning as an apoptosis-inducing transcription factor, CHOP has also been shown to interact with GABAB receptors (Sauter et al., 2005). This interaction obstructs the assembly of the GABAB receptor subunits GABAB1 and GABAB2 in the ER, prevents the ER exit of GABAB1/GABAB2 receptor complexes, thereby blocking the supply of new receptors to the cell surface (Maier et al., 2014). This mechanism downregulates GABAB receptors from the cell surface since constitutive internalization and degradation of the receptors continues but the supply of new receptors is blocked. The resulting loss of GABAB receptor-mediated neuronal inhibition is another detrimental effect of CHOP after ischemic stress which fosters neuronal overexcitation and excitotoxic neuronal death.
In this study, we reasoned that blocking the GABA receptor/CHOP interaction after ischemic stress would restore normal GABAB receptor expression and function, which might limit excitotoxic neuronal death. To test this hypothesis, we designed a cell-permeable synthetic peptide (R2-Pep) comprising a short amino acid sequence located within the C-terminal domain of GABAB2, which we formerly identified as part of the CHOP interaction site (Sauter et al., 2005). To mimic ischemic conditions in vitro, we used oxygen and glucose deprivation (OGD) of primary neurons co-cultured with glia cells. Co-cultures of neurons and glia cells were chosen because they closely mimic the brain situation, as glia cells support neurons and make them more resistant to stressful condition like OGD (Gouix et al., 2014; Liu and Chopp 2016). Glia cells have been reported to express GABAB receptors. For instance, in astrocytes, activation of GABAB receptors induce Ca2+ transients resulting in astrocytic glutamate release (Meier et al., 2008). Thus, we cannot rule out a contribution of glial GABAB receptors to the effects of R2-Pep treatment. However, in our neuron/glia cultures, expression of GABAB receptors on glia cells appears to be rare or at a very low levels not detectable with the GABAB receptor antibodies used in this study. In addition, the conditions used (OGD for 1 h) hardly upregulates CHOP in glia cells, which are much more resistant to ischemic stress as compared to neurons. At least 4 h of OGD is required to induce upregulation of CHOP in astrocytes (Benavides et al., 2005). Therefore, it is rather unlikely that R2-Pep affects GABAB receptor expression in glia cells.
We found that exposure of neuron/glia cultures to 1-h OGD upregulated CHOP expression, reduced GABAB1/GABAB2 heteromers and downregulated GABAB receptors from the cell surface. As hypothesized, the interfering peptide R2-Pep restored normal heterodimerization and cell surface expression of the receptors. This was reflected by recovered GABAB receptor function (i.e., baclofen-induced potassium currents) and a reduction of OGD-induced increased neuronal activity (i.e., reduction of enhanced spontaneous postsynaptic currents). The complete restoration of GABAB receptor expression to normal levels was unexpected since the interaction of CHOP with GABAB receptors is only one pathway contributing to the downregulation of GABAB receptors under ischemic/excitotoxic conditions. An additional main pathway is the aberrant sorting of internalized receptors to lysosomal degradation (Guetg et al., 2010; Maier et al., 2010; Terunuma et al., 2010; Kantamneni et al., 2014). However, our results suggest that interfering with the CHOP/GABAB receptor interaction is sufficient to normalize cell surface expression of the receptors by enabling the supply of new receptors from the ER.
Furthermore, treatment with R2-Pep also normalized OGD stress-induced CHOP expression back to control levels. The expression of CHOP is tightly controlled by proteasomal degradation. Monomeric CHOP is constitutively polyubiquitinated and degraded by the proteasome, whereas dimerization via their leucine zippers stabilizes CHOP (Hattori et al., 2003). CHOP bound to R2-Pep is most likely recognized as monomer and rapidly degraded. This view is supported by our finding that blocking proteasomal degradation prevents the R2-Pep induced downregulation of CHOP. Another mechanism downregulating stress-induced CHOP expression by R2-Pep might be directly related to the normalized GABAB receptor expression and function. Activation of GABAB receptors by baclofen has been shown to reduce enhanced CHOP expression induced by hypoxia in retinal ganglion cells (Fu et al., 2016). It is currently unclear whether directly interfering with the GABAB/CHOP interaction or indirectly via the downregulation of CHOP is the primary cause for the R2-Pep-induced normalization of GABAB receptor expression and function. However, the ability of R2-Pep to reduce CHOP expression to normal physiological levels offers the opportunity to prevent CHOP-mediated cell death in pathological conditions primary unrelated to GABAB receptors such as diabetes (Oyadomari et al., 2002), asthma (Wang et al., 2017) or Alzheimer’s (Prasanthi et al., 2011).
GABAB receptors are organized in multiprotein signalling complexes comprising the receptor, G-proteins, KCTD proteins, effector proteins and other associated proteins (Schwenk et al., 2010; Schwenk et al., 2016). We, therefore, assumed that the inwardly rectifying K+ channel Kir3.2, which is one of the main postsynaptic effectors, is co-regulated with the GABAB receptors under ischemic conditions. Indeed, we found that Kir3.2 was downregulated to a similar extent as GABAB receptors after OGD stress and application of R2-Pep normalized their expression. This result agrees with previous findings showing the association and co-regulation of GABAB receptors with inwardly rectifying K+ channels under different physiological or pathological conditions (David et al., 2006; Fernandez-Alacid et al., 2009; Padgett et al., 2012; Hearing et al., 2013).
Since GABAB receptors are involved in the regulation of virtually all main brain functions and can regulate gene expression, it would be very interesting to analyze the effect of R2-Pep after an ischemic insult on gene expression on a genome wide level. In addition to identifying negative off-target effects of R2-Pep, this would also generate valuable information of the pathways affected by the restoration of GABAB receptors.
In line with a neuroprotective function of GABAB receptors in in vivo models of cerebral ischemia (Lal et al., 1995; Babcock et al., 2002; Zhang et al., 2007; Han et al., 2008; Xu et al., 2008; Zhou et al., 2008; Liu et al., 2015; Fu et al., 2016; Lu et al., 2016; Hleihil et al., 2021), we found that treatment of cultures with R2-Pep for up to 4 h after OGD stress largely prevented neuronal death. We also found that R2-Pep has a considerably higher neuroprotective activity than the GABAB receptor agonist baclofen. This was expected because of the downregulation of the receptors after OGD. Based on the observation that activation of GABAB receptors is neuroprotective in in vivo models and the finding that CHOP deficient mice show a reduced level of neuronal death after an ischemic insult (Tajiri et al., 2004), it is very likely that the neuroprotective activity of R2-Pep we observed in our in vitro model also translates to in vivo models. The main challenge for peptide therapeutics targeting the brain is to cross the blood-brain barrier and escape degradation by peptidases and rapid elimination from the circulation. However, there are currently promising nanoparticle-based drug delivery approaches available, that are reported to overcome these problems (Han et al., 2016; Li et al., 2019; Pinheiro et al., 2020). This issue needs to be addressed in future experiments.
Since GABAB receptors are ubiquitously expressed in the brain and are involved in virtually all main brain functions, global activation of GABAB receptors by its orthosteric agonist baclofen is associated with numerous side effects when given systemically (Slonimski et al., 2004; Brennan and Whittle 2008). For this reason, baclofen is largely restricted to the treatment of severe spasticity via intrathecal application (Slonimski et al., 2004). In contrast to global activation of GABAB receptors, targeting a specific disease-relevant aberrant protein-protein interaction, such as the GABAB receptor/CHOP interaction, offers the opportunity to develop highly specific therapeutics that preferentially target the diseased cells and might leave healthy cells largely unaffected. This is expected to result in safe treatments with favourable side effect profiles.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Veterinäramt Zürich, Zollstrasse 20, 8090 Zürich.
MB, AE, and KB performed the experimental work and analyzed data. KB and DB designed the study. KB, EN and DB supervised parts of the experimental work and analyzed data. MB and DB wrote the manuscript. All authors discussed the results and commented on the manuscript.
This work was funded by the Swiss National Science Foundation grants 31003A_156648 and IZSEZO_185519 to DB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Patrick Maier for the initial screening of R2-Pep, Matilda Leardo and Sandra Gobat for contributing to the experimental work and Thomas Grampp for excellent technical assistance.
Babcock, A. M., Everingham, A., Paden, C. M., and Kimura, M. (2002). Baclofen Is Neuroprotective and Prevents Loss of Calcium/Calmodulin-dependent Protein Kinase II Immunoreactivity in the Ischemic Gerbil hippocampus. J. Neurosci. Res. 67 (6), 804–811. doi:10.1002/jnr.10169
Benavides, A., Pastor, D., Santos, P., Tranque, P., and Calvo, S. (2005). CHOP Plays a Pivotal Role in the Astrocyte Death Induced by Oxygen and Glucose Deprivation. Glia 52 (4), 261–275. doi:10.1002/glia.20242
Benke, D., Michel, C., and Mohler, H. (2002). Structure of GABAB Receptors in Rat Retina. J. Recept Signal. Transduct. Res. 22 (1-4), 253–266. doi:10.1081/rrs-120014600
Brennan, P. M., and Whittle, I. R. (2008). Intrathecal Baclofen Therapy for Neurological Disorders: a Sound Knowledge Base but many Challenges Remain. Br. J. Neurosurg. 22 (4), 508–519. doi:10.1080/02688690802233364
Bussieres, N., and El Manira, A. (1999). GABA(B) Receptor Activation Inhibits N- and P/Q-type Calcium Channels in Cultured Lamprey Sensory Neurons. Brain Res. 847 (2), 175–185. doi:10.1016/s0006-8993(99)02002-8
Chalifoux, J. R., and Carter, A. G. (2011). GABAB Receptor Modulation of Synaptic Function. Curr. Opin. Neurobiol. 21, 339–344. doi:10.1016/j.conb.2011.02.004
Chen, G., and van den Pol, A. N. (1998). Presynaptic GABAB Autoreceptor Modulation of P/Q-type Calcium Channels and GABA Release in Rat Suprachiasmatic Nucleus Neurons. J. Neurosci. 18 (5), 1913–1922. doi:10.1523/jneurosci.18-05-01913.1998
Cimarosti, H., Kantamneni, S., and Henley, J. M. (2009). Ischaemia Differentially Regulates GABA(B) Receptor Subunits in Organotypic Hippocampal Slice Cultures. Neuropharmacology 56 (8), 1088–1096. doi:10.1016/j.neuropharm.2009.03.007
Dave, K. R., Lange-Asschenfeldt, C., Raval, A. P., Prado, R., Busto, R., Saul, I., et al. (2005). Ischemic Preconditioning Ameliorates Excitotoxicity by Shifting Glutamate/Gamma-Aminobutyric Acid Release and Biosynthesis. J. Neurosci. Res. 82 (5), 665–673. doi:10.1002/jnr.20674
David, M., Richer, M., Mamarbachi, A. M., Villeneuve, L. R., Dupré, D. J., and Hebert, T. E. (2006). Interactions between GABA-B1 Receptors and Kir 3 Inwardly Rectifying Potassium Channels. Cell Signal 18 (12), 2172–2181. doi:10.1016/j.cellsig.2006.05.014
Fernandez-Alacid, L., Aguado, C., Ciruela, F., Martín, R., Colón, J., Cabañero, M. J., et al. (2009). Subcellular Compartment-specific Molecular Diversity of Pre- and post-synaptic GABA-Activated GIRK Channels in Purkinje Cells. J. Neurochem. 110 (4), 1363–1376. doi:10.1111/j.1471-4159.2009.06229.x
Fu, P., Wu, Q., Hu, J., Li, T., and Gao, F. (2016). Baclofen Protects Primary Rat Retinal Ganglion Cells from Chemical Hypoxia-Induced Apoptosis through the Akt and PERK Pathways. Front. Cel Neurosci. 10, 255. doi:10.3389/fncel.2016.00255
Gähwiler, B. H., and Brown, D. A. (1985). GABAB-receptor-activated K+ Current in Voltage-Clamped CA3 Pyramidal Cells in Hippocampal Cultures. Proc. Natl. Acad. Sci. U S A. 82 (5), 1558–1562. doi:10.1073/pnas.82.5.1558
Gassmann, M., and Bettler, B. (2012). Regulation of Neuronal GABA(B) Receptor Functions by Subunit Composition. Nat. Rev. Neurosci. 13 (6), 380–394. doi:10.1038/nrn3249
Gouix, E., Buisson, A., Nieoullon, A., Kerkerian-Le Goff, L., Tauskela, J. S., Blondeau, N., et al. (2014). Oxygen Glucose Deprivation-Induced Astrocyte Dysfunction Provokes Neuronal Death through Oxidative Stress. Pharmacol. Res. 87, 8–17. doi:10.1016/j.phrs.2014.06.002
Guetg, N., Aziz, S. A., Holbro, N., Turecek, R., Rose, T., Seddik, R., et al. (2010). NMDA Receptor-dependent GABA B Receptor Internalization via CaMKII Phosphorylation of Serine 867 in GABA B1. Proc. Natl. Acad. Sci. U.S.A. 107 (31), 13924–13929. doi:10.1073/pnas.1000909107
Han, D., Zhang, Q. G., Yong-Liu, L., Li, C., Zong, Y. Y., Yu, C. Z., et al. (2008). Co-activation of GABA Receptors Inhibits the JNK3 Apoptotic Pathway via the Disassembly of the GluR6-PSD95-MLK3 Signaling Module in Cerebral Ischemic-Reperfusion. FEBS Lett. 582 (9), 1298–1306. doi:10.1016/j.febslet.2008.02.044
Han, L., Cai, Q., Tian, D., Kong, D. K., Gou, X., Chen, Z., et al. (2016). Targeted Drug Delivery to Ischemic Stroke via Chlorotoxin-Anchored, Lexiscan-Loaded Nanoparticles. Nanomedicine 12 (7), 1833–1842. doi:10.1016/j.nano.2016.03.005
Hattori, T., Ohoka, N., Inoue, Y., Hayashi, H., and Onozaki, K. (2003). C/EBP Family Transcription Factors are Degraded by the Proteasome but Stabilized by Forming Dimer. Oncogene 22, 1273–1280. doi:10.1038/sj.onc.1206204
Hearing, M., Kotecki, L., Marron Fernandez de Velasco, E., Fajardo-Serrano, A., Chung, H. J., Luján, R., et al. (2013). Repeated Cocaine Weakens GABA(B)-Girk Signaling in Layer 5/6 Pyramidal Neurons in the Prelimbic Cortex. Neuron 80 (1), 159–170. doi:10.1016/j.neuron.2013.07.019
Hetz, C. (2012). The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and beyond. Nat. Rev. Mol. Cel Biol. 13 (2), 89–102. doi:10.1038/nrm3270
Hleihil, M., Vaas, M., Bhat, M. A., Balakrishnan, K., and Benke, D. (2021). Sustained Baclofen-Induced Activation of GABA B Receptors after Cerebral Ischemia Restores Receptor Expression and Function and Limits Progressing Loss of Neurons. Front. Mol. Neurosci. 14, 726133. doi:10.3389/fnmol.2021.726133
Hu, H., Tian, M., Ding, C., and Yu, S. (2018). The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 9, 3083. doi:10.3389/fimmu.2018.03083
Huang, L., Li, Q., Wen, R., Yu, Z., Li, N., Ma, L., et al. (2017). Rho-kinase Inhibitor Prevents Acute Injury against Transient Focal Cerebral Ischemia by Enhancing the Expression and Function of GABA Receptors in Rats. Eur. J. Pharmacol. 797, 134–142. doi:10.1016/j.ejphar.2017.01.021
Jackson-Friedman, C., Lyden, P. D., Nunez, S., Jin, A., and Zweifler, R. (1997). High Dose Baclofen Is Neuroprotective but Also Causes Intracerebral Hemorrhage: a Quantal Bioassay Study Using the Intraluminal Suture Occlusion Method. Exp. Neurol. 147 (2), 346–352. doi:10.1006/exnr.1997.6637
Kaneko, M., Imaizumi, K., Saito, A., Kanemoto, S., Asada, R., Matsuhisa, K., et al. (2017). ER Stress and Disease: Toward Prevention and Treatment. Biol. Pharm. Bull. 40 (9), 1337–1343. doi:10.1248/bpb.b17-00342
Kantamneni, S., Gonzàlez-Gonzàlez, I. M., Luo, J., Cimarosti, H., Jacobs, S. C., Jaafari, N., et al. (2014). Differential Regulation of GABAB Receptor Trafficking by Different Modes of N-Methyl-D-Aspartate (NMDA) Receptor Signaling. J. Biol. Chem. 289 (10), 6681–6694. doi:10.1074/jbc.M113.487348
Katan, M., and Luft, A. (2018). Global Burden of Stroke. Semin. Neurol. 38 (2), 208–211. doi:10.1055/s-0038-1649503
Kim, J. Y., Kim, N., Yenari, M. A., and Chang, W. (2011). Mild Hypothermia Suppresses Calcium-Sensing Receptor (CaSR) Induction Following Forebrain Ischemia while Increasing GABA-B Receptor 1 (GABA-B-R1) Expression. Transl. Stroke Res. 2 (2), 195–201. doi:10.1007/s12975-011-0082-4
Kulinskii, V. I., and Mikhel'son, G. V. (2000). Additivity and independence of Neuroprotective Effects of GABAA and GABAB Receptor Agonists in Complete Global Cerebral Ischemia. Bull. Exp. Biol. Med. 130 (8), 772–774. doi:10.1007/BF02766091
Lal, S., Shuaib, A., and Ijaz, S. (1995). Baclofen Is Cytoprotective to Cerebral Ischemia in Gerbils. Neurochem. Res. 20 (2), 115–119. doi:10.1007/BF00970534
Li, R., Huang, Y., Chen, L., Zhou, H., Zhang, M., Chang, L., et al. (2019). Targeted Delivery of Intranasally Administered Nanoparticles-Mediated Neuroprotective Peptide NR2B9c to Brain and Neuron for Treatment of Ischemic Stroke. Nanomedicine 18, 380–390. doi:10.1016/j.nano.2018.10.013
Liu, Z., and Chopp, M. (2016). Astrocytes, Therapeutic Targets for Neuroprotection and Neurorestoration in Ischemic Stroke. Prog. Neurobiol. 144, 103–120. doi:10.1016/j.pneurobio.2015.09.008
Liu, L., Li, C. J., Lu, Y., Zong, X. G., Luo, C., Sun, J., et al. (2015). Baclofen Mediates Neuroprotection on Hippocampal CA1 Pyramidal Cells through the Regulation of Autophagy under Chronic Cerebral Hypoperfusion. Sci. Rep. 5, 14474. doi:10.1038/srep14474
Lu, Y., Li, C. J., Chen, C., Luo, P., Zhou, M., Li, C., et al. (2016). Activation of GABAB2 Subunits Alleviates Chronic Cerebral Hypoperfusion-Induced Anxiety-like Behaviours: A Role for BDNF Signalling and Kir3 Channels. Neuropharmacology 110, 308–321. doi:10.1016/j.neuropharm.2016.08.007
Luscher, C., Jan, L. Y., Stoffel, M., Malenka, R. C., and Nicoll, R. A. (1997). G Protein-Coupled Inwardly Rectifying K+ Channels (GIRKs) Mediate Postsynaptic but Not Presynaptic Transmitter Actions in Hippocampal Neurons. Neuron 19 (3), 687–695. doi:10.1016/s0896-6273(00)80381-5
Maier, P. J., Marin, I., Grampp, T., Sommer, A., and Benke, D. (2010). Sustained Glutamate Receptor Activation Down-Regulates GABAB Receptors by Shifting the Balance from Recycling to Lysosomal Degradation. J. Biol. Chem. 285 (46), 35606–35614. doi:10.1074/jbc.M110.142406
Maier, P. J., Zemoura, K., Acuña, M. A., Yévenes, G. E., Zeilhofer, H. U., and Benke, D. (2014). Ischemia-like Oxygen and Glucose Deprivation Mediates Down-Regulation of Cell Surface γ-aminobutyric acidB Receptors via the Endoplasmic Reticulum (ER) Stress-Induced Transcription Factor CCAAT/enhancer-binding Protein (C/EBP)-homologous Protein (CHOP). J. Biol. Chem. 289 (18), 12896–12907. doi:10.1074/jbc.M114.550517
Meier, S. D., Kafitz, K. W., and Rose, C. R. (2008). Developmental Profile and Mechanisms of GABA-Induced Calcium Signaling in Hippocampal Astrocytes. Glia 56 (10), 1127–1137. doi:10.1002/glia.20684
Mintz, I. M., and Bean, B. P. (1993). GABAB Receptor Inhibition of P-type Ca2+ Channels in central Neurons. Neuron 10 (5), 889–898. doi:10.1016/0896-6273(93)90204-5
Morimoto, N., Oida, Y., Shimazawa, M., Miura, M., Kudo, T., Imaizumi, K., et al. (2007). Involvement of Endoplasmic Reticulum Stress after Middle Cerebral Artery Occlusion in Mice. Neuroscience 147 (4), 957–967. doi:10.1016/j.neuroscience.2007.04.017
Oida, Y., Shimazawa, M., Imaizumi, K., and Hara, H. (2008). Involvement of Endoplasmic Reticulum Stress in the Neuronal Death Induced by Transient Forebrain Ischemia in Gerbil. Neuroscience 151 (1), 111–119. doi:10.1016/j.neuroscience.2007.10.047
Oyadomari, S., and Mori, M. (2004). Roles of CHOP/GADD153 in Endoplasmic Reticulum Stress. Cell Death Differ. 11 (4), 381–389. doi:10.1038/sj.cdd.4401373
Oyadomari, S., Koizumi, A., Takeda, K., Gotoh, T., Akira, S., Araki, E., et al. (2002). Targeted Disruption of the CHOP Gene Delays Endoplasmic Reticulum Stress-Mediated Diabetes. J. Clin. Invest. 109, 525–532. doi:10.1172/JCI14550
Padgett, C. L., Lalive, A. L., Tan, K. R., Terunuma, M., Munoz, M. B., Pangalos, M. N., et al. (2012). Methamphetamine-evoked Depression of GABA(B) Receptor Signaling in GABA Neurons of the VTA. Neuron 73 (5), 978–989. doi:10.1016/j.neuron.2011.12.031
Pinheiro, R. G. R., Granja, A., Loureiro, J. A., Pereira, M. C., Pinheiro, M., Neves, A. R., et al. (2020). RVG29-functionalized Lipid Nanoparticles for Quercetin Brain Delivery and Alzheimer's Disease. Pharm. Res. 37 (7), 139. doi:10.1007/s11095-020-02865-1
Prasanthi, J. R., Larson, T., Schommer, J., and Ghribi, O. (2011). Silencing GADD153/CHOP Gene Expression Protects against Alzheimer's Disease-like Pathology Induced by 27-hydroxycholesterol in Rabbit hippocampus. PLoS One 6 (10), e26420. doi:10.1371/journal.pone.0026420
Santos, A. E., Carvalho, C. M., Macedo, T. A., and Carvalho, A. P. (1995). Regulation of Intracellular [Ca2+] and GABA Release by Presynaptic GABAB Receptors in Rat Cerebrocortical Synaptosomes. Neurochem. Int. 27 (4-5), 397–406. doi:10.1016/0197-0186(95)00021-y
Sauter, K., Grampp, T., Fritschy, J. M., Kaupmann, K., Bettler, B., Mohler, H., et al. (2005). Subtype-selective Interaction with the Transcription Factor CCAAT/enhancer-binding Protein (C/EBP) Homologous Protein (CHOP) Regulates Cell Surface Expression of GABA(B) Receptors. J. Biol. Chem. 280, 33566–33572. doi:10.1074/jbc.M503482200
Schwenk, J., Metz, M., Zolles, G., Turecek, R., Fritzius, T., Bildl, W., et al. (2010). Native GABA(B) Receptors Are Heteromultimers with a Family of Auxiliary Subunits. Nature 465 (7295), 231–235. doi:10.1038/nature08964
Schwenk, J., Pérez-Garci, E., Schneider, A., Kollewe, A., Gauthier-Kemper, A., Fritzius, T., et al. (2016). Modular Composition and Dynamics of Native GABAB Receptors Identified by High-Resolution Proteomics. Nat. Neurosci. 19 (2), 233–242. doi:10.1038/nn.4198
Slonimski, M., Abram, S. E., and Zuniga, R. E. (2004). Intrathecal Baclofen in Pain Management. Reg. Anesth. Pain Med. 29 (3), 269–276. doi:10.1016/j.rapm.2004.01.005
Tajiri, S., Oyadomari, S., Yano, S., Morioka, M., Gotoh, T., Hamada, J. I., et al. (2004). Ischemia-induced Neuronal Cell Death Is Mediated by the Endoplasmic Reticulum Stress Pathway Involving CHOP. Cell Death Differ. 11 (4), 403–415. doi:10.1038/sj.cdd.4401365
Terunuma, M., Vargas, K. J., Wilkins, M. E., Ramírez, O. A., Jaureguiberry-Bravo, M., Pangalos, M. N., et al. (2010). Prolonged Activation of NMDA Receptors Promotes Dephosphorylation and Alters Postendocytic Sorting of GABAB Receptors. Proc. Natl. Acad. Sci. U S A. 107 (31), 13918–13923. doi:10.1073/pnas.1000853107
Terunuma, M. (2018). Diversity of Structure and Function of GABAB Receptors: a Complexity of GABAB-Mediated Signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 94 (10), 390–411. doi:10.2183/pjab.94.026
Vollenweider, F., Bendfeldt, K., Maetzler, W., Otten, U., and Nitsch, C. (2006). GABA(B) Receptor Expression and Cellular Localization in Gerbil hippocampus after Transient Global Ischemia. Neurosci. Lett. 395 (2), 118–123. doi:10.1016/j.neulet.2005.10.079
Wang, Y., Zhu, J., Zhang, L., Zhang, Z., He, L., Mou, Y., et al. (2017). Role of C/EBP Homologous Protein and Endoplasmic Reticulum Stress in Asthma Exacerbation by Regulating the IL-4/signal Transducer and Activator of Transcription 6/transcription Factor EC/IL-4 Receptor α Positive Feedback Loop in M2 Macrophages. J. Allergy Clin. Immunol. 140 (6), 1550. doi:10.1016/j.jaci.2017.01.024
White, B. C., Sullivan, J. M., DeGracia, D. J., O'Neil, B. J., Neumar, R. W., Grossman, L. I., et al. (2000). Brain Ischemia and Reperfusion: Molecular Mechanisms of Neuronal Injury. J. Neurol. Sci. 179 (S 1-2), 1–33. doi:10.1016/s0022-510x(00)00386-5
Xu, J., Li, C., Yin, X. H., and Zhang, G. Y. (2008). Additive Neuroprotection of GABA A and GABA B Receptor Agonists in Cerebral Ischemic Injury via PI-3K/Akt Pathway Inhibiting the ASK1-JNK cascade. Neuropharmacology 54 (7), 1029–1040. doi:10.1016/j.neuropharm.2008.01.014
Yang, Y., Liu, L., Naik, I., Braunstein, Z., Zhong, J., and Ren, B. (2017). Transcription Factor C/EBP Homologous Protein in Health and Diseases. Front. Immunol. 8, 1612. doi:10.3389/fimmu.2017.01612
Zhang, F., Li, C., Wang, R., Han, D., Zhang, Q. G., Zhou, C., et al. (2007). Activation of GABA Receptors Attenuates Neuronal Apoptosis through Inhibiting the Tyrosine Phosphorylation of NR2A by Src after Cerebral Ischemia and Reperfusion. Neuroscience 150 (4), 938–949. doi:10.1016/j.neuroscience.2007.09.070
Zhou, C., Li, C., Yu, H. M., Zhang, F., Han, D., and Zhang, G. Y. (2008). Neuroprotection of Gamma-Aminobutyric Acid Receptor Agonists via Enhancing Neuronal Nitric Oxide Synthase (Ser847) Phosphorylation through Increased Neuronal Nitric Oxide Synthase and PSD95 Interaction and Inhibited Protein Phosphatase Activity in Cerebral Ischemia. J. Neurosci. Res. 86 (13), 2973–2983. doi:10.1002/jnr.21728
Keywords: GABAB receptor, neuroprotection, interfering peptide, CHOP, cerebral ischemia, oxygen and glucose deprivation (OGD)
Citation: Bhat MA, Esmaeili A, Neumann E, Balakrishnan K and Benke D (2022) Targeting the Interaction of GABAB Receptors With CHOP After an Ischemic Insult Restores Receptor Expression and Inhibits Progressive Neuronal Death. Front. Pharmacol. 13:870861. doi: 10.3389/fphar.2022.870861
Received: 07 February 2022; Accepted: 14 March 2022;
Published: 29 March 2022.
Edited by:
Ning Liu, Tulane University, United StatesReviewed by:
Siva Sankara Vara Prasad Sakamuri, Tulane University, United StatesCopyright © 2022 Bhat, Esmaeili, Neumann, Balakrishnan and Benke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dietmar Benke, YmVua2VAcGhhcm1hLnV6aC5jaA==
†These authors share senior authorship
‡Present address: Elena Neumann, Institute of Anesthesiology, University of Zurich, Zurich, SwitzerlandKarthik Balakrishnan, Dewpoint Therapeutics GmbH, Dresden, Germany
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.