
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 12 May 2022
Sec. Neuropharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.863762
 Alan Umfress1
Alan Umfress1 Sarbjit Singh2
Sarbjit Singh2 Kevin J. Ryan3
Kevin J. Ryan3 Ayanabha Chakraborti1
Ayanabha Chakraborti1 Florian Plattner4
Florian Plattner4 Yogesh Sonawane2
Yogesh Sonawane2 Jayapal Reddy Mallareddy2Edward P. Acosta3
Jayapal Reddy Mallareddy2Edward P. Acosta3 Amarnath Natarajan2
Amarnath Natarajan2 James A. Bibb1,5,6*
James A. Bibb1,5,6*Cyclin-dependent kinase 5 (Cdk5) is a crucial regulator of neuronal signal transduction. Cdk5 activity is implicated in various neuropsychiatric and neurodegenerative conditions such as stress, anxiety, depression, addiction, Alzheimer’s disease, and Parkinson’s disease. While constitutive Cdk5 knockout is perinatally lethal, conditional knockout mice display resilience to stress-induction, enhanced cognition, neuroprotection from stroke and head trauma, and ameliorated neurodegeneration. Thus, Cdk5 represents a prime target for treatment in a spectrum of neurological and neuropsychiatric conditions. While intracranial infusions or treatment of acutely dissected brain tissue with compounds that inhibit Cdk5 have allowed the study of kinase function and corroborated conditional knockout findings, potent brain-penetrant systemically deliverable Cdk5 inhibitors are extremely limited, and no Cdk5 inhibitor has been approved to treat any neuropsychiatric or degenerative diseases to date. Here, we screened aminopyrazole-based analogs as potential Cdk5 inhibitors and identified a novel analog, 25–106, as a uniquely brain-penetrant anti-Cdk5 drug. We characterize the pharmacokinetic and dynamic responses of 25–106 in mice and functionally validate the effects of Cdk5 inhibition on open field and tail-suspension behaviors. Altogether, 25–106 represents a promising preclinical Cdk5 inhibitor that can be systemically administered with significant potential as a neurological/neuropsychiatric therapeutic.
Cyclin-dependent kinase 5 (Cdk5) is a proline-directed kinase predominantly expressed in mature neurons (Dhavan and Tsai, 2001). Unlike traditional cyclin kinases, Cdk5 does not require coactivation by binding to a cyclin partner. Instead, Cdk5 is constitutively activated through interactions with its unique coactivators p35 or p39 (Dhavan and Tsai, 2001). Over 3 decades of research on Cdk5 functionality has revealed this kinase to play unique roles in modulating neuronal development, complex behavior, and neurodegeneration. Cdk5 is expressed within excitatory and inhibitory circuitry and regulates glutamatergic, dopaminergic, and GABAergic signaling (Liu et al., 2001; Chergui et al., 2004; Li et al., 2018). The current scenario shows that Cdk5 is a critical regulator of homeostatic neuronal signaling (Dhavan and Tsai, 2001; Barnett and Bibb, 2011; Plattner et al., 2014; Plattner et al., 2015). Cdk5 conditional knockout mice display reduced anxiety-like behaviors and enhanced cocaine locomotor sensitization (Bibb et al., 2001a; Meyer et al., 2008; Plattner et al., 2015). In addition, knockout of Cdk5 within the striatum of adult mice results in impaired long-term plasticity (LTP) and altered motor learning (Benavides and Bibb, 2004; Hernandez et al., 2016). Similarly, mice constitutively lacking the p35 co-factor have dramatically altered cortical layer architecture and display hyperactivity (Chae et al., 1997; Drerup et al., 2010). Within the hippocampus, Cdk5-dependent phosphorylation of NMDA receptors has been linked to decreases in the cell surface expression of NMDA receptors, thereby decreasing cell excitability. Infusion of Cdk5 targeting small-interfering peptides blocks Cdk5 phosphorylation of the NR2B (GluN2B) subunit of NMDA receptors, increases hippocampal LTP, and enhances fear-learning and memory (Hawasli et al., 2007; Plattner et al., 2014). Many other studies have assigned both pre-and post-synaptic functions for Cdk5 in virtually every brain region and associated circuitry. Together, these studies have established a crucial role for homeostatic Cdk5 signaling in normal brain function.
Conversely, Cdk5 activity also serves as a principle feature in excitotoxic signaling and has been implicated in Alzheimer’s disease (AD), Parkinson’s disease (PD), ischemia, and brain injury (Patrick et al., 1999; Meyer et al., 2014; Binukumar et al., 2015; Yousuf et al., 2016). This is because Cdk5 can transform from a constitutively active homeostatic kinase to an aberrantly active neurotoxic enzyme. For example, excitotoxicity or loss of membrane potential can cause loss of Ca2+ homeostasis. Increased intracellular Ca2+ can activate the protease calpain, which then cleaves Cdk5’s physiological coactivator p35 into a truncated aberrant coactivator p25 and a p10 fragment (Kusakawa et al., 2000). The p25 activator lacks a myristoylation site on the N-terminal region of p35. Consequently, the Cdk5/p25 complex is no longer membrane-sequestered (Dhavan and Tsai, 2001). Furthermore, p25 is degraded less quickly than p35 (Patrick et al., 1998). These combined factors lead to mislocalization and hyperactivation of the Cdk5 holoenzyme toward a subset of substrates, resulting in neuronal injury and cellular death (Dhavan and Tsai, 2001). For these reasons, Cdk5/p25 has been implicated in various neurogenerative diseases and neurotoxic insults (Patrick et al., 1999; Cruz et al., 2003; Miao et al., 2012; Sundaram et al., 2012). Elevated levels of the aberrant coactivator p25 induce AD pathologies, including cellular death and hyperphosphorylation of the neurofibrillary tangle protein, Tau (Cruz et al., 2003). Deletion or inhibition of the Cdk5/p25 complex is neuroprotective in various disease models (Sundaram et al., 2013; Yousuf et al., 2016). Thus, Cdk5 serves as a critical regulator of both homeostatic and pathological brain function, and inhibition of Cdk5 may be of significant therapeutic value.
Therapeutic infusion of preclinical inhibitors of Cdk5 into the brain has been successfully shown to ameliorate neurological disease phenotypes in mice. However, none of these compounds has proven effective in clinical trials (Cicenas et al., 2015; Pao and TSAI, 2021). A major barrier for most of these inhibitors has been specificity and tissue penetrance. Most CDK inhibitors target the ATP-binding site of the kinase domain. Unfortunately, cyclin-dependent kinase family members share high sequence homology, particularly in this drug-targeting domain (Sonawane et al., 2016), leading to nonspecific drug interactions and off-target inhibition (Khair et al., 2019). The most prominent Cdk5 inhibitor, roscovitine, has proven to be a useful tool in neuroscience and has also yielded promising results in the treatment of various cancers but has not been approved for use in neurological conditions (Cicenas et al., 2015). More recent work using small-interfering peptides (SiPs) that selectively block Cdk5-p25 but not Cdk5-p35 interactions by both pharmacological and transgenic means has shown promise in preclinical models of neurodegeneration and stroke (Binukumar et al., 2015; Binukumar and Pant, 2016). While these SiPs are proof of concept advances, the strategy of only targeting Cdk5/p25 limits this approach to excitotoxicity-/neuronal injury–linked disease rather than neuropsychiatric conditions mediated through Cdk5/p35 activity. Numerous CDK inhibitors have been developed for the treatment of various cancer types (Asghar et al., 2015; Mangini et al., 2015; Syed, 2017). Development of inhibitors with an aminopyrazole core that forms hydrogen bonds within the hinge region of kinases such as AT7519 has progressed through clinical trials. Derivation of aminopyrazole analogs has shown increased specificity for Cdk2/5 over other CDK family members and effectively halted preclinical models of cancer (Pevarello et al., 2004; Chen et al., 2014; Rana et al., 2018). However, the ability of these aminopyrazole analogs to penetrate the blood–brain barrier and effectively inhibit Cdk5 in the brain remains unknown.
Here, by screening a small panel of aminopyrazole analogs, we identify analog, 25–106, that can be delivered systemically, penetrate the blood–brain barrier, and pharmacologically inhibit Cdk5/p35 activity in the brain. These data demonstrate that 25–106 inhibits Cdk5 activity in vivo and displays high potency for inhibition in vitro. Furthermore, this inhibitor modulates behavioral phenotypes linked to Cdk5 through previous characterizations of conditional knockout mice. These studies further advance our understanding of Cdk5’s role in neuropsychiatric behavior, bring forth a new pharmacological tool to assess Cdk5 activity, and suggest a promising new preclinical drug.
The wild-type group housed male C57BL/6 mice 10–12 weeks of age were used for all in vivo, ex vivo, and behavioral studies. This age range was selected to be consistent with previous studies of conditional Cdk5 knockout mice (Plattner et al., 2015). 25–106 was administered in a 20-fold dose range intravenously (I.V.) at 10, 50, 100, and 200 mg/kg for pharmacokinetic studies. This delivery mode was used as I.V. injections to avoid first-pass drug metabolism observed using other injection methods (Pond and Tozer, 1984). The mice were subsequently euthanized 1, 2, 6, and 24 h post injection to monitor pharmacokinetic distribution and clearance rates from plasma, brain, liver, and kidney. All animal experiments were performed under approved protocols by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committees and in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals.
All reagents were purchased from commercial sources and were used without further purification. Flash chromatography was carried out on silica gel (200–400 mesh). Thin-layer chromatography (TLC) was run on pre-coated ANALTECH uniplate and observed under UV light at 254 nM and with a basic potassium permanganate dip. Column chromatography was performed with silica gel (230–400 mesh, grade 60, Fisher Scientific, United States). 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded in chloroform-d or DMSO-d6 on a Bruker-400 spectrometer. DMSO-d6 was 2.50 ppm for 1H and 39.55 ppm for 13C, and CDCl3 was 7.26 ppm for 1H and 77.23 ppm for 13C. Proton and carbon chemical shifts were reported in ppm relative to the signal from residual solvent proton and carbon. The data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet and/or multiple resonances), coupling constant in hertz (Hz), and integration. The purity of the final compounds was determined by analytical HPLC and was found to be ≥95% pure. HPLC was performed on a Waters Alliance 2,690 system equipped with a Waters 2,996 photodiode array detector and an auto-sampler under the following conditions: column, Phenomenex Luna-2 RP-C18 (5 μM, 4.6 mm × 250 mm, 120 Å, Torrance, CA); solvent A, H2O containing 0.1% formic acid (FA); solvent B, CH3CN containing 0.1% FA; gradient, 60% B to 100% B over 8 min followed by 100% B over 6 min; injection volume, 25 μL; flow rate, 1 ml/min retention times and purity data for each target compound are provided in the Experimental Section. The purified compound was further confirmed by MS analysis using a Quattro Micro triple–quadrupole mass spectrometer using an electron spray ionization (ESI) technique and a TOF mass analyzer.
Synthesis of 2-(3,4,5-trimethoxyphenyl) acetyl chloride (Rana et al., 2018). To a solution of 3,4,5-trimethoxyphenylacetic acid (1 g, 4.42 mmol) in anhydrous dichloromethane at 0°C was added oxalyl chloride (1.7 g, 13.26 mmol), followed by the addition of the catalytic amount of dimethylformamide (DMF) (50 µL). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. The completion of the reaction was monitored by TLC by quenching a small aliquot of the crude mixture in methanol. After completion of the reaction, the reaction mixture was concentrated to dryness, and the residue was used in the next step without further purification.
Synthesis of tert-butyl 3-amino-5-cyclobutyl-1H-pyrazole-1-carboxylate (2) (Rana et al., 2018). 3-amino-5-cyclobutyl-1H-pyrazole (1.86 g, 13.55 mmol) in dichloromethane was placed in a round-bottomed flask, and 4N KOH (6.10 g, 108.4 mmol) was added to the stirring solution. The reaction mixture was allowed to stir at room temperature, followed by the addition of Boc anhydride (3.11 g, 14.23 mmol) in small batches. The reaction mixture was monitored for completion of the reaction by TLC (∼3 h), and the mixture was diluted with CH2Cl2, washed with brine, and dried with MgSO4. The crude product was purified using silica gel column chromatography (2.76 g, 86%).
Synthesis of tert-butyl 5-cyclobutyl-3-[2-(3,4,5-trimethoxyphenyl)acetamido]-1H-pyrazole-1-carboxylate (3) (Rana et al., 2018). Compound 2 (tert-butyl 3-amino-5-cyclobutyl-1H-pyrazole-1-carboxylate, 257 mg, 1.08 mmol) was dissolved in dichloromethane followed by addition of diisopropylethylamine (280 mg, 2.16) under N2. The reaction mixture was cooled to 0°C and freshly prepared 2-(3,4,5-trimethoxyphenyl)acetyl chloride (345 mg, 1.40 mmol) dichloromethane was added dropwise at 0°C. Following addition, the reaction temperature was allowed to rise to room temperature and, the mixture was stirred for an additional 3 h. The crude mixture was washed with brine, extracted with dichloromethane, dried with MgSO4, and concentrated. The crude mixture was purified using silica gel column chromatography (381 mg, 79%); 1H NMR (400 MHz, CDCl3) δ 10.31 (s, 1H), 6.87 (s, 1H), 6.57 (s, 2H), 3.91 (s, 6H), 3.87 (d, J = 3.8 Hz, 3H), 3.72 (s, 2H), 3.55 (m, 1H), 2.41–2.14 (m, 4H), 2.12–1.85 (m, 2H), 1.72–1.56 (m, 9H).
Synthesis of N-(5-cyclobutyl-1H-pyrazol-3-yl)-2-(3,4,5-trimethoxyphenyl)acetamide (25–106). To a stirred solution of tert-butyl 5-cyclobutyl-3-[2-(3,4,5-trimethoxyphenyl)acetamido]-1H-pyrazole-1-carboxylate (3, 60 mg) in dichloromethane (5 ml) at 0°C, trifluoroacetic acid (1 ml) was added dropwise, and the reaction mixture was stirred at room temperature for 3 h. After completion, the reaction mixture was concentrated in vacuo, and the resulting solid was recrystallized to yield a colorless solid (35 mg, 76%). Purity was assessed by HPLC (>98%); 1H NMR (400 MHz, DMSO) δ 12.04 (s, 1H), 10.41 (s, 1H), 6.64 (s, 2H), 6.31 (s, 1H), 3.76 (s, 6H), 3.63 (s, 3H), 3.51 (s, 2H), 3.48–3.37 (m, 1H), 2.35–2.18 (m, 2H), 2.18–2.02 (m, 2H), 1.95 (m, 8.6 Hz, 1H), 1.89–1.73 (m, 1H); 13C NMR (100 MHz, DMSO) δ 168.1, 153.1, 147.8, 147.5, 136.7, 132.2, 107.0, 93.8, 60.4, 56.3, 43.3, 31.6, 29.4, 18.6; MS calculated for C18H23N3O4 m/z 345.16, found mass: 346.14.
A quantitative determination of 25–106 in mouse plasma and tissue homogenate was accomplished by the use of protein precipitation and high-performance liquid chromatography with tandem mass spectrometry detection (LC-MS/MS). 5-(N,N-hexamethylene) amiloride (HMA) was used as the internal standard (IS). 25–106 and IS were typically extracted from 50 µL of mouse plasma or tissue homogenate using protein precipitation with methanol. The extracts were analyzed by reverse-phase chromatography using an ACE Excel 3 C18-PFP column under isocratic conditions at a flow rate of 500 μL/min. The column temperature was maintained at 40°C. The mobile phase A consisted of 0.1% formic acid in water, and the mobile phase B was acetonitrile. A triple–quadrupole mass spectrometer (AB Sciex 5,000) equipped with TurboV IonSpray operating in positive-ion mode was used. Column effluents were analyzed by multiple reaction monitoring (MRM). The precursor/product transitions are 346.1 to 137.1 m/z for 25–106 and 312.37 to 129.5 m/z for IS. The calibration curve was fit using weighted (1/x2) linear regression analysis of the 25–106/IS peak area ratio vs. the 25–106 concentration from 1.0–80.0 ng/ml (ng/g for tissue) for the low curve and 80.0–10,000 ng/ml (ng/g for tissue) for the high curve. The concentrations of incurred and quality control samples were calculated with the same regression analysis, and the results were reported in ng/mL (plasma) or ng/g (tissue) of 25–106.
Molecular modeling was performed using the Schrödinger small-molecule drug discovery suite 2020–1. The CDK/p25 (PDB: 1H4L) crystal structure was retrieved from the Protein Data Bank and analyzed using Maestro version 12.3.013 (Schrödinger Inc.). The protein preparation was performed using Protein Preparation Wizard in which missing hydrogen atoms, side chains, and loops were added, followed by grid generation, ligand preparation, and docking. The protein structures were minimized using the OPLS3e force field to optimize hydrogen bonding networks and converging heavy atoms to an RMSD of 0.3 Å. The prepared protein structure was subjected to SiteMap for binding site analysis as implemented in Schrödinger Suite 2020–1. At least 15 site points per reported site were required. A more restricted definition of hydrophobicity together with a standard grid was used. Site maps at 4 Å or more from the nearest site points were cropped. The structures of 25–106 and roscovitine were subjected to Lig Prep to generate conformers and possible protonation at pH of 7 ± 2, which serves as an input for the docking process. The receptor grid was generated using the receptor grid generation tool in Maestro (Schrödinger Inc.). GLIDE XP was used to perform all the dockings with the van der Waals radii of nonpolar atoms for each of the ligands being scaled by a factor of 0.8 and the partial charge cutoff of 0.15. The docked poses from GLIDE XP were analyzed using Maestro version 12.3.013 (Schrödinger Inc.).
Brain slice pharmacology was performed as described (Nishi et al., 2000). Briefly, the mice were decapitated, and the brains were rapidly dissected and submerged in ice-cold normal Kreb’s solution (124 mM NaCl, 4 mM KCl, 26 mM NaHCO3, 1.25 mM KH2PO4, 1.5 mM MgSO4, 10 mM d-glucose, and 1.5 mM CaCl2). The brains were subsequently sectioned into normal Kreb’s solution at 350 µ thickness. The brain slices were transferred to a 30°C solution of normal Kreb’s containing 10 µM deaminase for a 1-h recovery period. The sections were then incubated in normal Kreb’s solution with and without the indicated compounds across the dose and time ranges indicated. The sections were rapidly snap-frozen in dry ice to terminate treatment regimes.
Immunoblotting was performed as previously described (Umfress et al., 2021). Mouse brains were rapidly dissected in an ice-cold solution of 50 mM NaF/PBS. The regions of interest were further dissected out and rapidly snap-frozen in dry ice. The frozen tissue was homogenized in boiling lysis buffer (1% SDS, 50 mM NaF) and sonicated with several three to four pulses at 40 dB. The samples were subsequently denatured at 90°C for 5 min. Protein concentration was determined by the BCA assay, and homogenates were diluted in 4x sample buffer, separated by SDS-PAGE, and transferred to 0.45-µm nitrocellulose membranes, blocked with 5% milk solution (1 h), and probed with primary antibodies (overnight). The following antibodies were used in the study: phospho-Ser549 synapsin I (Phosphosolutions), total synapsin I (Phosphosolutions), actin (Invitrogen), phospho-Ser1116 (in-house) (Plattner et al., 2014), and total NR2B (Phosphosolutions), phospho-Thr75, and total DARPP-32 (Cell Signaling Technology). Visualization was performed using the Licor CLX membrane scanner and Licor secondary antibodies.
In vitro phosphorylation reactions were conducted as dose-response kinase inhibition assays with three highly homologous CDKs, namely, Cdk2/CyclinE (1.5 nM), CDK5/p25 (0.35 nM), and Cdk9/CyclinT1 (9 nM). Briefly, the abovementioned kinases were incubated with ATP (30 μM), the corresponding substrate (20 μM), and an increasing concentration of 25–106 (0–5 μM) for 30 min. The kinase activity of the abovementioned reactions was graphed against the concentration of 25–106 to determine the IC50 and Ki values for each kinase.
The OFT was performed as described (Chakraborti et al., 2021). With the adaptation duration of the test being 1 h for each mouse, briefly, the mice were placed in a square box (40 cm × 40 cm) and allowed to freely explore. The center square portion of the maze was defined as the “open area.” Video tracking using EthoVision 16 software was used to track animal movement throughout the box and stereotypic and locomotive behaviors such as hopping, sniffing, and rearing. Total distance traveled, entries into the center of the maze, and duration in the center were tracked and analyzed in EthoVision.
The tail suspension test was conducted as previously described (Plattner et al., 2015). The mice were suspended by their tails for a total duration of 6 min and video-recorded. Post-hoc blind experimentalists quantified the duration and latency of immobility for each randomized video. The average quantitative determents of each observer were averaged to generate the final analytic values of motion.
The physicochemical properties of five FDA-approved CNS drugs (droperidol, buspirone, benperidol, amisulpride, and alfentanil) (Table 1) (Ghose et al., 2012), were established as benchmarks for the query of aminopyrazole-based Cdk inhibitors with similar properties from an in-house combinatorial library. This led to the identification of three analogs, 25–143, 25–107, and 25–132, that were previously reported as Cdk2/5 inhibitors (Figure 1A) (Rana et al., 2018). In addition, we designed a fourth analog, 25–106, with an improved predicted brain/blood partition coefficient (QPlogBB), as compared to the reported compounds (Table 1). This trimethoxy aminopyrazole analog was synthesized in three steps from the commercially available starting structure, 3-amino-5-cyclobutyl-1H-pyrazole 1 (Figure 1B). The pyrazole nitrogen (N1) of 1 was protected using Boc-anhydride under strongly basic conditions. Interestingly, Boc protection under less basic conditions produced a mixture of regioisomers (4) and (2) (Figure 1C). The Boc-protected aminopyrazole (2) was reacted with 2-(3,4,5-trimethoxyphenyl) acetyl chloride which was freshly prepared from the corresponding acid. This resulted in compound 3 in 79% yield as a colorless solid. The N-Boc was deprotected on 3 using TFA to produce the title compound, 25–106, in a 76% yield as a colorless solid. Collectively, the three previously reported compounds and the newly synthesized 25–106 were predicted to serve as potent Cdk5 inhibitors, with 25–106 exhibiting the highest predicted brain/blood partition coefficient.
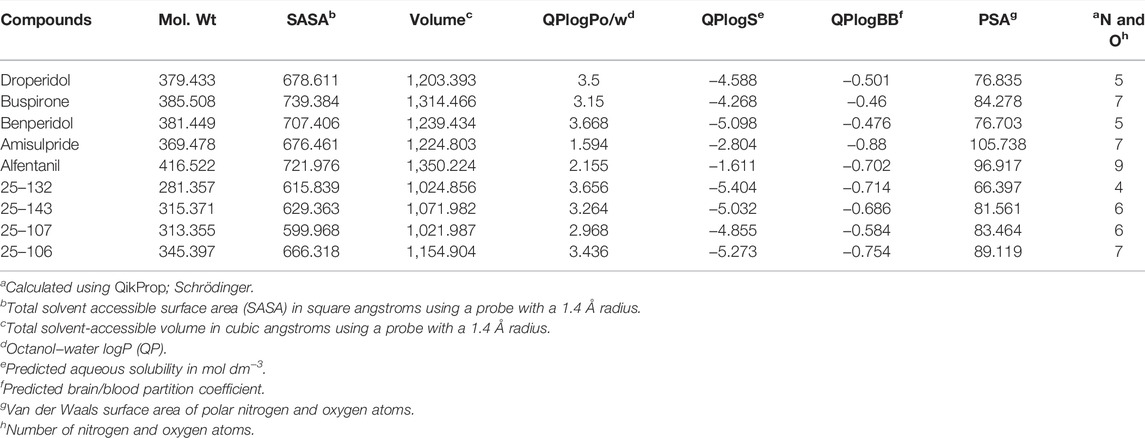
TABLE 1. Physicochemical properties of 25–132, 25–143, 25–107, 25–106, and five approved CNS drugs.a
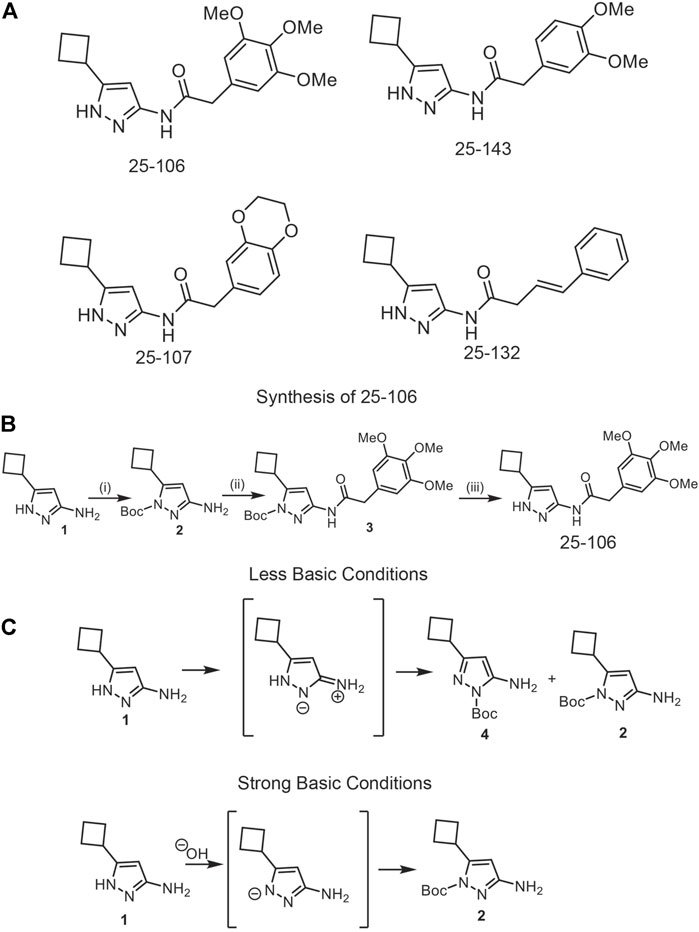
FIGURE 1. Development of new Cdk5 inhibitors. (A) Structures of four potential Cdk5 inhibitors: 25–106, 25–143, 25–107, and 25–132 (B). Synthesis of novel inhibitor, 25–106 (C). Regio-isomers are synthesized under strong or less basic conditions.
In order to assess the functional inhibition of Cdk5 in intact brain tissue by the four compounds summarized in Figure 1, acutely prepared striatal slices were treated in a dose-dependent manner (0, 0.25, 0.5, 1, 5, and 10 µM) with each inhibitor for 1 h, and Cdk5 activity was assessed via immunoblotting the phosphorylation of established Cdk5 sites, phospho-Thr75 DARPP-32 and phospho-Ser1116 NR2B (Bibb et al., 1999; Plattner et al., 2014). All four compounds tested, 25–106 (Figure 2A), 25–107 (Figure 2B), 25–132 (Figure 2C) and 25–143 (Figure 2D), displayed comparable efficacy as ex vivo inhibitors of Cdk5-dependent phosphorylation. All compounds caused significant reductions in Cdk5-dependent phosphorylation of DARPP-32 and NR2B with IC50 values ranging approximately 0.5–1 µm. Thus, each of these newly synthesized compounds functioned in intact brain tissue as potent Cdk5 inhibitors and thus exhibited relatively equal potential to serve as anti-Cdk5 therapeutics.
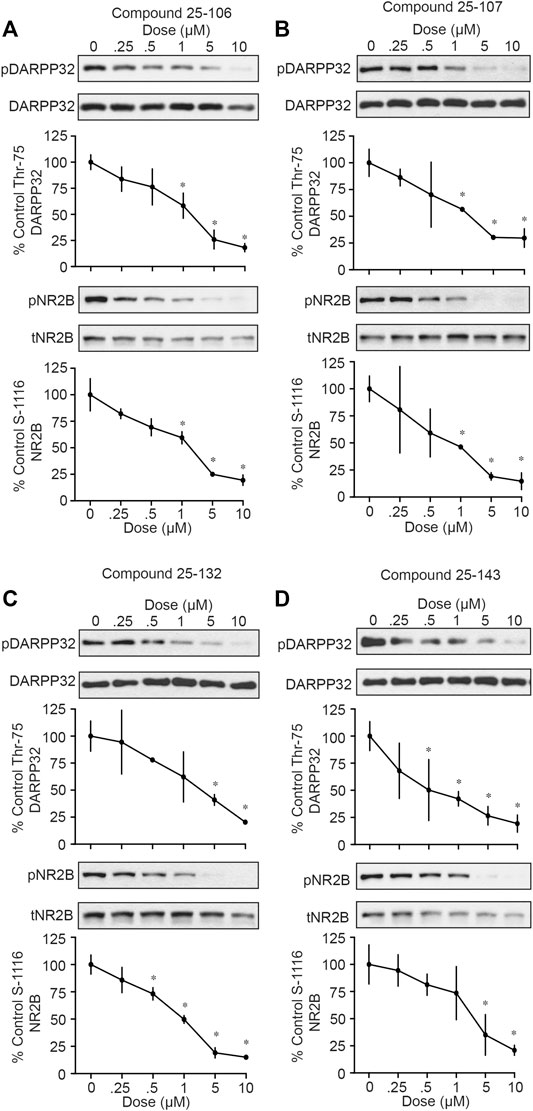
FIGURE 2. In vitro testing of Cdk5 inhibitors. Western blot for phosphor-DARPP32 (T75) and NR2B (S1116) of acute striatal brain slices treated (1 h) with indicated concentrations of (A) 25–106, (B) 25–107, (C) 25–132, and (D) 25–143 (n = four to six slices per treatment) *p <0.05 Student’s t-test.
Each of the four compounds tested in brain slices was further screened for their ability to penetrate the blood–brain barrier (BBB) and inhibit Cdk5-dependent activity in vivo. For these studies, cohorts of mice were treated with each compound via I.V. injection (50 mg/kg), and brain striatal lysates from various time points were subjected to quantitative immunoblot analysis for phosho-Ser549 Synapsin I levels. Three compounds, 20–107, 20–132, and 20–143, had no effect on Cdk5 activity compared to that of vehicle alone (data not shown). In contrast, 25–106 significantly reduced phospho-Ser549 synapsin I levels in the striatum 2 h after I.V. injection (Figure 3A). Moreover, this effect persisted even 24 h after dosing. Interestingly, a dose–response test revealed that 25–106 caused significant reduction in striatal phospho-Ser549 synapsin I levels 24 h post injection across a 20-fold dose range, with approximately equal efficacy at 10, 50, 100, and 200 mg/kg.
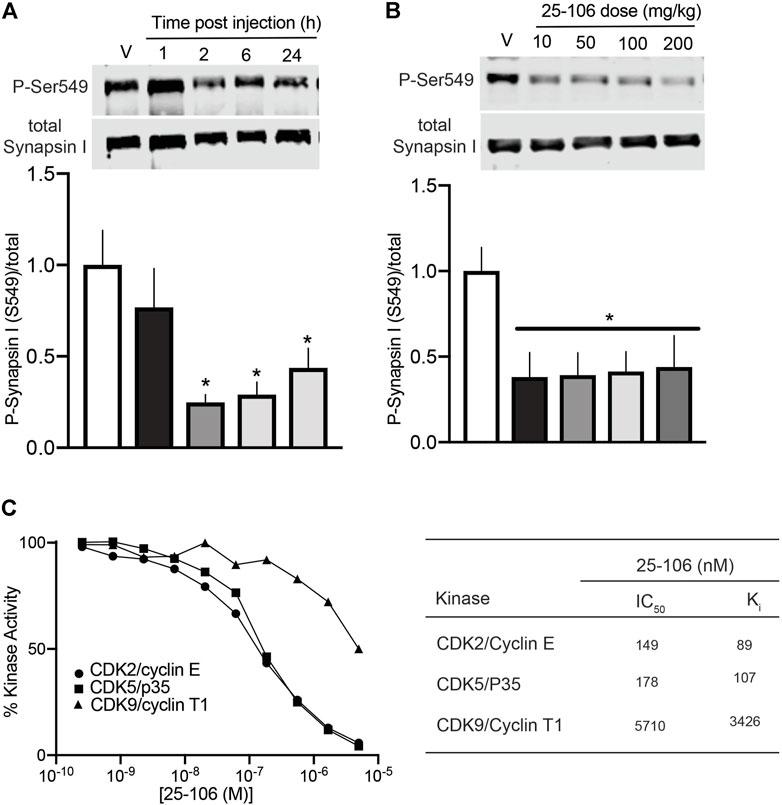
FIGURE 3. In vivo efficacy and in vitro specificity of 25–106 (A). Quantitative immunoblot analysis of striatal brain lysates for phospho-Ser549 synapsin I after dosing 50 mg/kg 25–106 across the times indicated (n = 4 per group) (B). Quantitative immunoblot of phospho-Ser549 synapsin I 24 h post administration across doses indicated (n = 4 per group). (C). In vitro kinase assay (30 min) with Cdk2/Cyclin E, Cdk5/p25, and Cdk9/Cyclin T1 treated with 0–5 µM 25–106 (left) calculated IC50, Ki values of each kinase (right). *p <0.05 student’s t-test.
For assessment of the in vitro selectivity of 25–106, we conducted a dose–response kinase inhibition assay with three highly homologous Cdks, Cdk2/CyclinE, Cdk5/p25, and Cdk9/CyclinT1 (Sonawane et al., 2016). 25–106 exhibited dose-dependent inhibition of all three kinases tested (Figure 3C). However, the compound showed greater than 30-fold selectivity for Cdk2 and Cdk5 than Cdk9.
Due to the prolonged inhibition of Cdk5 by 25–106 observed in (Figure 3), we decided to investigate the specific drug interactions of 25–106 with Cdk5 as compared to the well-established Cdk5 inhibitor, roscovitine. The crystal structure of a complex between CDK5 and p25 was used to elucidate the binding mode of 25–106 and compare it to that of roscovitine (Tarricone et al., 2001). A binding pocket analysis was performed on the CDK5/p25 protein structure (PDB: 1H4L) using the SiteMap tool by Schrödinger Inc. (Halgren, 2009). The ATP binding pocket yielded site and druggability scores of 1.05 and 1.07, respectively, and was selected for docking. The analysis was performed using Glide XP with 25–106 and roscovitine, which revealed that the compounds had similar binding modes with CDK5. The hydrogen bonds formed by the purine core in roscovitine with the hinge region residue Cys83 are mimicked by the aminopyrazole core in 25–106. The cyclobutyl group on the pyrazole ring of 25–106 and the isopropyl group on the imidazole ring of roscovitine occupy the same hydrophobic binding pocket (Figure 4). In addition to the common hydrogen bond interactions with the hinge region residue Cys83, 25–106 forms hydrogen bonds with Glu81 and Lys89 (Figure 4). The extra bonds formed by 25–106 result in a lower predicted required binding energy of −9.122 kcal/mol than −5.960 kcal/mol for roscovitine (Figure 4).
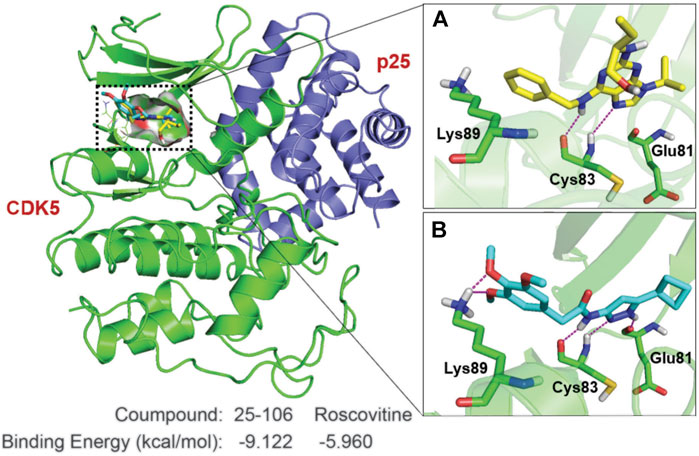
FIGURE 4. Key interactions made by 25–106 and roscovitine with CDK5/p25. CDK5 (green cartoon)/p25 (blue cartoon) (PDB:1H4L). Hydrogen bonds are shown in magenta lines. (A) Roscovitine (represented by yellow sticks), (B) 25–106 (represented by cyan sticks).
In order to derive the pharmacokinetic properties of 25–106 in vivo, we developed a protein precipitation and high-performance liquid chromatography with tandem mass spectrometry (LC-MS/MS) detection method to assess the distribution, concentration, and elimination of 25–106 throughout several organ systems. For these studies, 5-(N,N-hexamethylene) amiloride (HMA) was used as an internal standard. Mass spectra product transitions were observed as 25–106 displayed a prominent peak of the parent compound at 346.6 m/z, with the product transition for 25–106 displaying a larger peak at 137.9 m/z (Figure 5A). Likewise, the internal standard HMA displayed a parent peak of 312.2 m/z with subsequent fragmentation to 129.4 m/z (Figure 5B). Chromatograms displaying the selectivity for each compound’s transition state show distinctly detected peaks for a 100 ng/ml concentration of HMA (0.90 min) and a 125 ng/ml concentration of the extracted calibration standard of 25–106 (1.17 min) (Figure 5C). Altogether, these data display a selective approach in detecting and measuring the concentration of 25–106 that can be applied to various tissue samples.
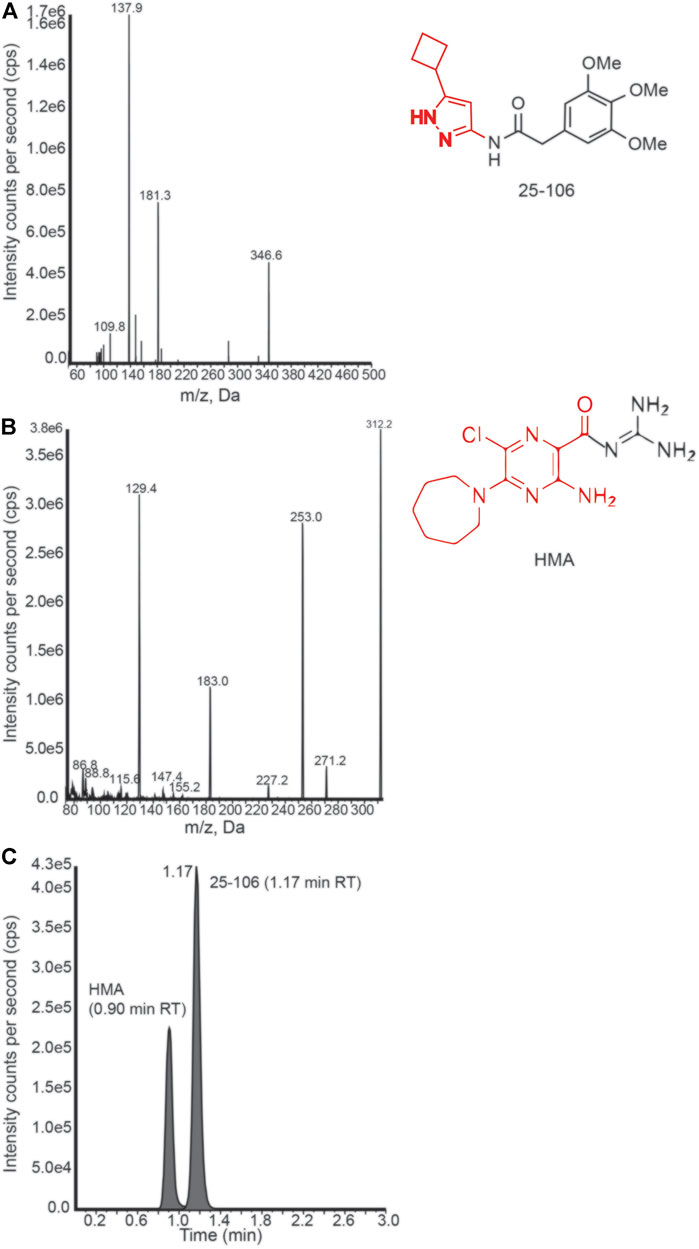
FIGURE 5. LC-MS/MS detection of 25–106 (A). Mass spectrum peaks generated from ion fragmentation of parent compound 25–106 (right). Major peak (137.9 highlighted in red). (B) Mass spectrum peaks generated from the ion fragmentation internal standard (IS) HMA (right). Major peak (129.4 outlined red). (C) Chromatogram of 25–106 (125 ng) and HMA (100 ng) mixture displaying two temporally distinct peaks.
To assess the pharmacokinetic and pharmacodynamic parameters of 25–106 in vivo, we utilized the LC-MS/MS detection method established above (see Figure 5). The mice were administered IV injections of 25–106 doses of 10, 50, 100, or 200 mg/kg. The animals were euthanized, and organs were harvested at 1, 2, 6, and 24 h post injection. Within the same experiment, tissues were used for biochemical analysis of 25–106 effects on Cdk5 activity (see Figures 3A, B). 25–106 showed rapid distribution following 1 h post injection in both the plasma and brain at all four doses tested (Figures 6A,B). 25–106 concentrations steadily decreased over time, remaining detectable in the brain and plasma 24 h after injection. From the concentration-time curves derived for the plasma and brain, several pharmacokinetic parameters, including maximum concentration (Cmax), elimination rate constant (Ke), half-life (T1/2), last measurable concentration (Clast), 24-h area-under-curve (AUC24h), distribution volume (V), and clearance (CL), were calculated (Tables 2, 3). 25–106 showed rapid uptake in the plasma across all doses tested (10, 50, 100, 200 mg/kg), reaching average plasma CMax values of 2567, 1375, 2,577, and 2,322 ng/ml, respectively (Table 2). In the plasma, 25–106 demonstrated linearity between the AUC vs. dose (R2 = 0.983) (Figure 6C). The CL of 25–106 from the plasma displayed varied but linear results as well (R2 = 0.9454) (Figure 6C). Of note, the half-life T1/2 of 25–106 in the plasma appeared to increase with each increasing dose injected (Table 2).
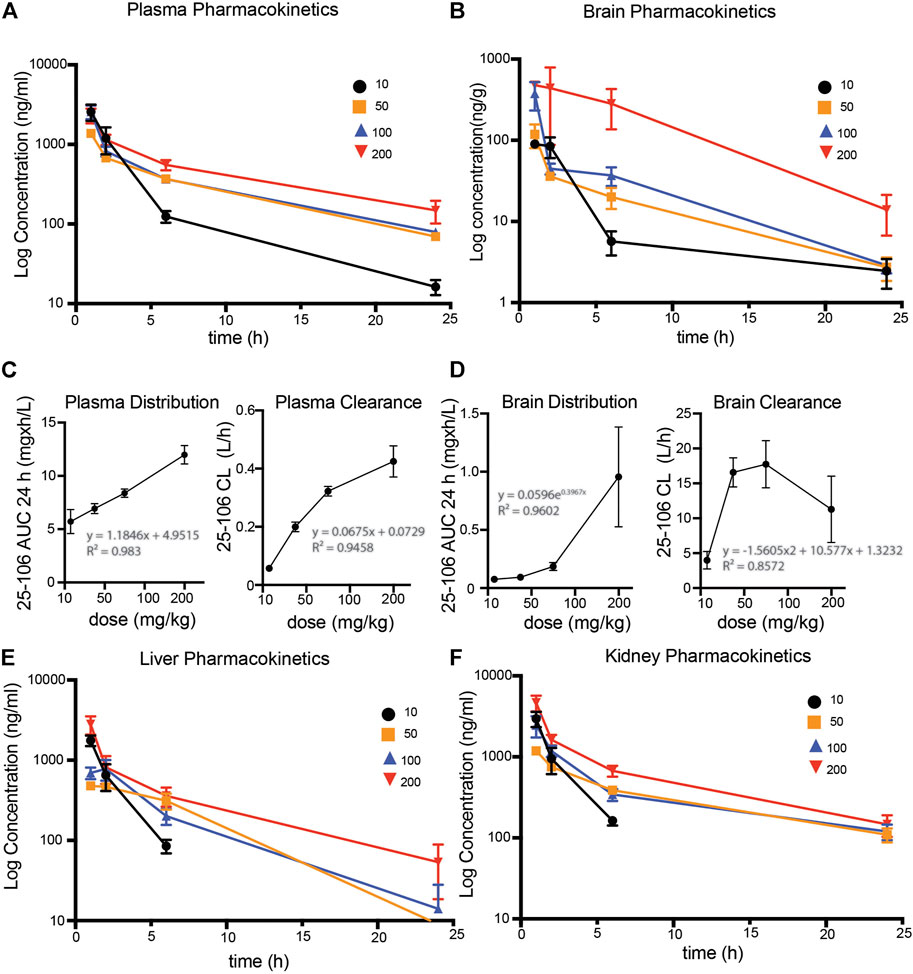
FIGURE 6. Pharmacokinetic profile of 25–106 (A,B). LC-MS/MS detected 25–106 used at four doses (10, 50, 100, and 200 mg/kg) across 1, 2, 6, and 24 h post injection in the plasma (A) and brain (B). (C) distribution and clearance of 25–106 in plasma. (D) Distribution and clearance of 25–106 in the brain. (E,F). LC-MS/MS detected a concentration of 25–106 in the liver (E) and kidney (F) (n = 4 for each treatment).
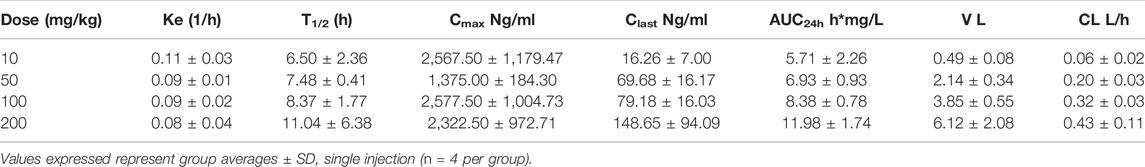
TABLE 2. Pharmacokinetic values of 25–106 in plasma.

TABLE 3. Pharmacokinetic values of 25–106 in the brain.
Similar to the plasma, the highest distribution of 25–106 in the brain was observed 1 h post injection and decreased over time, remaining detectable 24 h after injection. Unlike the plasma, 25–106 distribution in the brain increased exponentially with the dose injected (AUC vs. dose), reaching Cmax results of 101.13, 118.25, 376.88, and 668.75 ng/g for 10, 50, 100, and 200 mg/kg injections, respectively (Figure 6D; Table 3). The T1/2 of 25–106 in the brain remained relatively constant (excluding the lowest dose of 10 mg/kg), with an average half-life of 6.23, 5.03, and 4.39 h for 50, 100, and 200 mg/kg injections, respectively (Table 3). Interestingly, the clearance (CL) of 25–106 from the brain stabilized and decreased at the highest doses tested, with the average CL of 10 mg/kg (3.98 L/h), 50 mg/kg (16.5 L/h), 100 mg/kg (17.7 L/h) and 200 mg/kg (11.29 L/h) (Figure 6D; Table 3). Taken together, these data demonstrate that 25–106 show the properties of a linear drug in the plasma which enters the brain in a dose-dependent exponential manner.
In order to see if there was a 25–106 distribution in other off-target organ systems, in the same experiment, we again utilized the LC-MS/MS detection of liver and kidney samples. Much like the plasma and brain, 25–106 showed dose-dependent increased concentrations in the liver (Figure 6E) and kidney (Figure 6F), with the highest concentrations detected at the 1 h post-injection time point. In the liver, across all doses tested, 25–106 remained undetectable within some samples. Therefore, only the Cmax, Clast, and AUC could be determined for these samples (Table 4). Within the kidney, 25–106 was only undetectable at the lowest dose (10 mg/kg). For each other dose, all values of Cmax, Ke,T1/2, Clast, AUC, V, and CL were calculated (Table 5). Taken together, these results demonstrate 25–106’s temporal and dose-related abilities to distribute into several organ systems, including the brain.
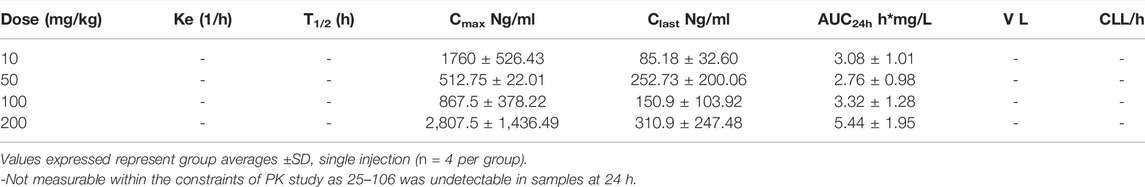
TABLE 4. Pharmacokinetic values of 25–106 in the liver.
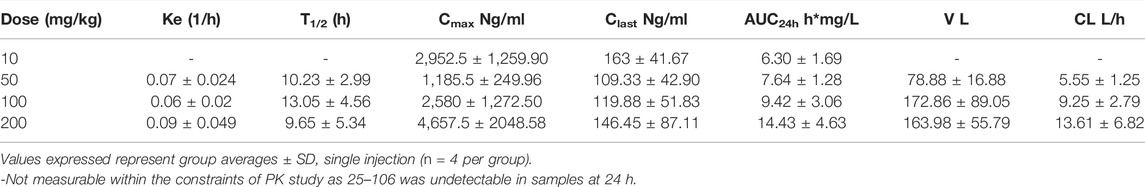
TABLE 5. Pharmacokinetic values of 25–106 in the kidney.
To establish a functional effect of pharmacological Cdk5 inhibition in the brain, we tested mouse performance in two behavioral paradigms previously shown to be altered in Cdk5 knockout mice: the open-field maze and the tail suspension test (Plattner et al., 2015). The open-field maze was used to measure locomotion and anxiety-like behavior (Cacace et al., 2011; Seibenhener and Wooten, 2015). Mice were injected with either vehicle or 50 mg/kg of 25–106 and allowed to freely explore an open-field maze for 1 h with locomotor activity being tracked (Figure 7). The mice injected with 25–106 displayed an increased number of entries into the open portion of the maze and a longer duration of time spent in the open portion of the maze as compared to vehicle-injected mice (Figure 7A). In addition, mice injected with 25–106 displayed increased hopping bouts, increased time sniffing, and increased rearing behaviors (Figure 7B).

FIGURE 7. Neurobehavioral effects of systemic Cdk5 inhibition. (A) Open-field testing (n = 10–12 per group) in mice treated with vehicle or (50 mg/kg) 25–106 1 h after injection. Parameters displayed include entries into the open, duration in the open, and distance traveled. (B) Stereotypic behaviors were assessed during the open-field testing. (C) Effect of systemic 25–106 (50 mg/kg) treatment vs. vehicle on behavioral responses in the tail suspension test (n = 11 per group) in vehicle or 25–106–treated mice 6 h s after injection. *p <0.05 Student’s t-test, **p <0.01 student’s t-test, and ***p <0.001 student’s t-test.
Next, we tested the effects of 25–106 on mouse performance in the tail-suspension test to measure depressive-like phenotypes in mice (Cryan et al., 2005; Can et al., 2012). Mice treated with 50 mg/kg 25–106 displayed an increased latency to suspend immobility and displayed a reduction in total immobility time as compared to vehicle-injected mice (Figure 7C). Thus, 25–106 modulated several neurobehavioral behaviors that have previously been linked to Cdk5.
A multitude of studies have implicated Cdk5 as an important neuronal signal transduction mediator in a variety of diseases and suggest anti-Cdk5 approaches as rational therapeutic strategies (Lee et al., 2000; Hung et al., 2005; Camins et al., 2006; Alvira et al., 2008; Giese, 2014). Initial enthusiasm for anti-Cdk5 treatment for neurodegenerative diseases such as Alzheimer’s disease (AD) led to the generation of a number of lead compounds which showed reasonable potency in vitro. However, the lack of specificity, recognition of potential postmortem confounds in human AD studies, and possibility that chronic inhibition of Cdk5 might lead to neuronal hyperexcitability and epileptiform seizures, as found in aged Cdk5 conditional knockout mice, all contributed to impedance in the pursuit of anti-Cdk5 therapeutics, leaving first- and second-generation compounds, which had poor brain permeable systemic properties, unsuitable for in vivo study (Yoo and Lubec, 2001; Hawasli et al., 2009). For example, pharmacokinetic analysis has demonstrated that roscovitine could distribute into the brain. However, a rapid half-life with no detectable active metabolites was limiting (Vita et al., 2005).
Here, we show 25–106 is a nonselective Cdk2/5 inhibitor that displays low nM IC50 and Ki values. While the shared specificity for these two kinases is consistent with the in vitro profiles for other Cdk5 inhibitors, it should be noted that very low levels of Cdk2 are detected in the brain (Bibb et al., 2001b). Moreover, aminopyrazole-based inhibitors selectively inhibit Cdk5 substrates over Cdk2 substrates in cellular models (Robb et al., 2018). CDK inhibitors, in general, lack selectivity for individual CDKs, given the strong homology within the catalytic domains across kinase family members. While this lack of specificity is most frequently evidenced by in vitro phosphorylation effects, where the drugs can act at their highest potency on purified and often recombinant kinases, within the intracellular milieu, conditions and interactions with other proteins or cell constituents may afford different or even more selective inhibition profiles (Knight and Shokat, 2005; Smyth and Collins, 2009). Indeed, other Cdk5 inhibitors in this class of molecules have been shown to be more selective for Cdk5 than Cdk1/2 in cultured cells (Pozo et al., 2013; Gupta et al., 2022). However, any off-target or toxic effects of systemic inhibition of Cdk2 by 25–106 remain unknown.
Although it has been postulated that pilot inhibitors of Cdk5, such as enantiomers of roscovitine, display improved BBB permeability and functional protection following ischemia, Cdk5 site-directed decreases in phosphorylation stoichiometry have not been clearly shown (Menn et al., 2010). Here, we introduce a new Cdk5 inhibitor and robustly characterize its pharmacokinetic and dynamic responses within several organ systems, including the brain. We observed the highest concentration of 25–106 in the brain at the earliest time point measured (1 h post injection). Therefore, peak levels in the brain are likely achieved rapidly after injection. In order to ascertain the true Tmax of this molecule, future studies of 25–106’s brain distribution across the initial post-injection period will be necessary. Interestingly, although the highest concentrations of 25–106 detected in the brain were at the 1 h post-injection timepoint, Cdk5-dependent phosphorylation remained unchanged 1 h post injection. This likely indicates a time-dependent delay for 25–106 to diffuse into neurons and appropriately bind and inhibit Cdk5-dependent phosphorylation states. No demethylated metabolite structures of 25–106 were detected. The degradation of 25–106 into active or inactive metabolites and their subsequent distribution within the body remain unknown. 25–106 remained detectable in the brain 24 h after injection but was also well-absorbed by peripheral tissues such as the liver and kidney, which could contribute to off-target effects or toxicity arising from prolonged treatment. Therefore, the development of derivatives of 25–106 with greater brain permeability and tissue specificity is a reasonable goal. Molecular modeling of 25–106 as compared to another Cdk5 inhibitor, roscovitine, revealed that 25–106 forms two additional bonds within the hinge region of the ATP binding domain of Cdk5. These extra binding motifs may explain the high activity of 25–106, the decreased binding energy required as compared to Roscovitine, and the prolonged inhibition of Cdk5-dependent substrates.
Previous studies have demonstrated striatal Cdk5 knockout or inhibition results in reduced anxiety-like behaviors in mice via regulation of phosphodiesterase4 (PDE4) activity and protein kinase A (PKA) signaling (Plattner et al., 2015). Impaired PKA activity is associated with major depressive disorder and inhibition of PDE4 and elevated levels of cAMP induce antidepressant effects (Fujita et al., 2012; O’donnell and Zhang, 2004; O’donnell and Xu, 2012). Here, we observed similar behavioral changes in two paradigms with brain-wide pharmacological inhibition of Cdk5. While 25–106 may impart the same mechanistic regulation of PKA activity, this mechanism is not explored here. Other studies have implicated Cdk5 activity in the septum in regulating anxiety-like behaviors in animals (Bignante et al., 2008). Therefore, the exact mechanisms by which 25–106 mediates the phenotypes observed here remain unknown. Alternatively, chronic deletion of Cdk5 in aged mice results in increased acoustic startle responses, a well-defined behavioral paradigm to measure animal responses to an emotional context or stressor (Hawasli et al., 2009; Hantsoo et al., 2018; Pantoni et al., 2020). Since this behavioral adaptation is only observed in aged mice with chronic depletion of Cdk5, we did not assess this paradigm using acute pharmacological inhibition of Cdk5. Chronic treatment and subsequent behavioral changes remain an interesting area of exploration. Additional studies have demonstrated that Cdk5’s cofactor p35 increases following stress exposure and Cdk5 activity increases following restraint stress (Bignante et al., 2010; Papadopoulou et al., 2015). In addition, the reduced anxiety-like behaviors observed here may also be in part due to regulation of various neuropeptides that modulate stress and anxiety-like behaviors (Kormos and Gaszner, 2013; Rana et al., 2022). Cdk5 activity has been shown to increase in mice following corticosterone injections, and this increase in activity correlates with increased phosphorylation of the glucocorticoid receptor following acute stress. However, direct phosphorylation by Cdk5 was not observed. Cdk5’s role in regulation of these neuropeptides implicated in stress remains an understudied area (Papadopoulou et al., 2015).
As perhaps the first robust systemic inhibitor, 25–106 represents an exciting and expandable and translatable pharmacological tool to study the function of Cdk5 activity in wild-type animals. Achieving systemic applicability may be considered a step forward toward the testing of Cdk5 inhibitors to treat neuropsychiatric and neurodegenerative diseases. This provides a promising landscape for future studies to assess the effects of brain-permeable Cdk5 inhibitors to combat stress, anxiety, depression, addiction, cancer, and neurodegeneration.
The raw data supporting the conclusion of this article will be made available by the authors; without undue reservation.
The animal study was reviewed and approved by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committees.
JB, AN, EA, FP, AC, KR, and AU conceived the study and designed the experiments. SS and YS conducted chemical synthesis of drugs. JM conducted drug modeling. AU conducted in vivo studies and in vitro testing. AC conducted behavioral studies. FP conducted in vitro studies. JR conducted molecular modeling. KR and EA conducted LC/MSMS and derived pharmacokinetic data. All authors contributed to the editing and approval of the final manuscript.
This research was facilitated by NIH support MH116896, MH126948 (JB), P50CA127297 (AN), and NINDS T32 Predoctoral Training Fellow (NS061788) (AU). Aspects of this work were also facilitated by an SDHB Pheo Para Coalition Investigator Award, a NETRF Accelerator Award, and pilot grant funding from the Yale Neuroproteomics Center (DA018343) (JB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We give thanks to the UAB behavioral assessment core for assistance with behavioral studies. We thank Daniel Epstein for advice concerning these studies. The Yale Neuroproteomics Center is gratefully acknowledged for providing pilot grant funding.
Alvira, D., Ferrer, I., Gutierrez-Cuesta, J., Garcia-Castro, B., Pallàs, M., and Camins, A. (2008). Activation of the Calpain/cdk5/p25 Pathway in the Girus Cinguli in Parkinson's Disease. Parkinsonism Relat. Disord. 14, 309–313. doi:10.1016/j.parkreldis.2007.09.005
Asghar, U., Witkiewicz, A. K., Turner, N. C., and Knudsen, E. S. (2015). The History and Future of Targeting Cyclin-dependent Kinases in Cancer Therapy. Nat. Rev. Drug Discov. 14, 130–146. doi:10.1038/nrd4504
Barnett, D. G., and Bibb, J. A. (2011). The Role of Cdk5 in Cognition and Neuropsychiatric and Neurological Pathology. Brain Res. Bull. 85, 9–13. doi:10.1016/j.brainresbull.2010.11.016
Benavides, D. R., and Bibb, J. A. (2004). Role of Cdk5 in Drug Abuse and Plasticity. Ann. N. Y Acad. Sci. 1025, 335–344. doi:10.1196/annals.1316.041
Bibb, J. A., Chen, J., Taylor, J. R., Svenningsson, P., Nishi, A., Snyder, G. L., et al. (2001a). Effects of Chronic Exposure to Cocaine Are Regulated by the Neuronal Protein Cdk5. Nature 410, 376–380. doi:10.1038/35066591
Bibb, J. A., Nishi, A., O'callaghan, J. P., Ule, J., Lan, M., Snyder, G. L., et al. (2001b). Phosphorylation of Protein Phosphatase Inhibitor-1 by Cdk5. J. Biol. Chem. 276, 14490–14497. doi:10.1074/jbc.M007197200
Bibb, J. A., Snyder, G. L., Nishi, A., Yan, Z., Meijer, L., Fienberg, A. A., et al. (1999). Phosphorylation of DARPP-32 by Cdk5 Modulates Dopamine Signalling in Neurons. Nature 402, 669–671. doi:10.1038/45251
Bignante, E. A., Paglini, G., and Molina, V. A. (2010). Previous Stress Exposure Enhances Both Anxiety-like Behaviour and P35 Levels in the Basolateral Amygdala Complex: Modulation by Midazolam. Eur. Neuropsychopharmacol. 20, 388–397. doi:10.1016/j.euroneuro.2010.02.007
Bignante, E. A., Rodriguez Manzanares, P. A., Mlewski, E. C., Bertotto, M. E., Bussolino, D. F., Paglini, G., et al. (2008). Involvement of Septal Cdk5 in the Emergence of Excessive Anxiety Induced by Stress. Eur. Neuropsychopharmacol. 18, 578–588. doi:10.1016/j.euroneuro.2008.02.007
Binukumar, B. K., and Pant, H. C. (2016). TFP5/TP5 Peptide Provides Neuroprotection in the MPTP Model of Parkinson's Disease. Neural Regen. Res. 11, 698–701. doi:10.4103/1673-5374.182681
Binukumar, B. K., Shukla, V., Amin, N. D., Grant, P., Bhaskar, M., Skuntz, S., et al. (2015). Peptide TFP5/TP5 Derived from Cdk5 Activator P35 Provides Neuroprotection in the MPTP Model of Parkinson's Disease. Mol. Biol. Cel 26, 4478–4491. doi:10.1091/mbc.E15-06-0415
Cacace, S., Plescia, F., La Barbera, M., and Cannizzaro, C. (2011). Evaluation of Chronic Alcohol Self-Administration by a 3-bottle Choice Paradigm in Adult Male Rats. Effects on Behavioural Reactivity, Spatial Learning and Reference Memory. Behav. Brain Res. 219, 213–220. doi:10.1016/j.bbr.2011.01.004
Camins, A., Verdaguer, E., Folch, J., Canudas, A. M., and Pallàs, M. (2006). The Role of CDK5/P25 Formation/inhibition in Neurodegeneration. Drug News Perspect. 19, 453–460. doi:10.1358/dnp.2006.19.8.1043961
Can, A., Dao, D. T., Terrillion, C. E., Piantadosi, S. C., Bhat, S., and Gould, T. D. (2012). The Tail Suspension Test. J. Vis. Exp. 1, e3769. doi:10.3791/3769
Chae, T., Kwon, Y. T., Bronson, R., Dikkes, P., Li, E., and Tsai, L. H. (1997). Mice Lacking P35, a Neuronal Specific Activator of Cdk5, Display Cortical Lamination Defects, Seizures, and Adult Lethality. Neuron 18, 29–42. doi:10.1016/s0896-6273(01)80044-1
Chakraborti, A., Graham, C., Chehade, S., Vashi, B., Umfress, A., Kurup, P., et al. (2021). High Fructose Corn Syrup-Moderate Fat Diet Potentiates Anxio-Depressive Behavior and Alters Ventral Striatal Neuronal Signaling. Front. Neurosci. 15, 669410. doi:10.3389/fnins.2021.669410
Chen, E. X., Hotte, S., Hirte, H., Siu, L. L., Lyons, J., Squires, M., et al. (2014). A Phase I Study of Cyclin-dependent Kinase Inhibitor, AT7519, in Patients with Advanced Cancer: NCIC Clinical Trials Group IND 177. Br. J. Cancer 111, 2262–2267. doi:10.1038/bjc.2014.565
Chergui, K., Svenningsson, P., and Greengard, P. (2004). Cyclin-dependent Kinase 5 Regulates Dopaminergic and Glutamatergic Transmission in the Striatum. Proc. Natl. Acad. Sci. U S A. 101, 2191–2196. doi:10.1073/pnas.0308652100
Cicenas, J., Kalyan, K., Sorokinas, A., Stankunas, E., Levy, J., Meskinyte, I., et al. (2015). Roscovitine in Cancer and Other Diseases. Ann. Transl Med. 3, 135. doi:10.3978/j.issn.2305-5839.2015.03.61
Cruz, J. C., Tseng, H. C., Goldman, J. A., Shih, H., and Tsai, L. H. (2003). Aberrant Cdk5 Activation by P25 Triggers Pathological Events Leading to Neurodegeneration and Neurofibrillary Tangles. Neuron 40, 471–483. doi:10.1016/s0896-6273(03)00627-5
Cryan, J. F., Mombereau, C., and Vassout, A. (2005). The Tail Suspension Test as a Model for Assessing Antidepressant Activity: Review of Pharmacological and Genetic Studies in Mice. Neurosci. Biobehav Rev. 29, 571–625. doi:10.1016/j.neubiorev.2005.03.009
Dhavan, R., and Tsai, L. H. (2001). A Decade of CDK5. Nat. Rev. Mol. Cel Biol 2, 749–759. doi:10.1038/35096019
Drerup, J. M., Hayashi, K., Cui, H., Mettlach, G. L., Long, M. A., Marvin, M., et al. (2010). Attention-deficit/hyperactivity Phenotype in Mice Lacking the Cyclin-dependent Kinase 5 Cofactor P35. Biol. Psychiatry 68, 1163–1171. doi:10.1016/j.biopsych.2010.07.016
Fujita, M., Hines, C. S., Zoghbi, S. S., Mallinger, A. G., Dickstein, L. P., Liow, J. S., et al. (2012). Downregulation of Brain Phosphodiesterase Type IV Measured with 11C-(R)-rolipram Positron Emission Tomography in Major Depressive Disorder. Biol. Psychiatry 72, 548–554. doi:10.1016/j.biopsych.2012.04.030
Ghose, A. K., Herbertz, T., Hudkins, R. L., Dorsey, B. D., and Mallamo, J. P. (2012). Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 3, 50–68. doi:10.1021/cn200100h
Giese, K. P. (2014). Generation of the Cdk5 Activator P25 Is a Memory Mechanism that Is Affected in Early Alzheimer's Disease. Front. Mol. Neurosci. 7, 36. doi:10.3389/fnmol.2014.00036
Gupta, P., Strange, K., Telange, R., Guo, A., Hatch, H., Sobh, A., et al. (2022). Genetic Impairment of Succinate Metabolism Disrupts Bioenergetic Sensing in Adrenal Neuroendocrine Tumor. doi:10.1101/2022.01.09.475410
Halgren, T. A. (2009). Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 49, 377–389. doi:10.1021/ci800324m
Hantsoo, L., Golden, C. E. M., Kornfield, S., Grillon, C., and Epperson, C. N. (2018). Startling Differences: Using the Acoustic Startle Response to Study Sex Differences and Neurosteroids in Affective Disorders. Curr. Psychiatry Rep. 20, 40. doi:10.1007/s11920-018-0906-y
Hawasli, A. H., Benavides, D. R., Nguyen, C., Kansy, J. W., Hayashi, K., Chambon, P., et al. (2007). Cyclin-dependent Kinase 5 Governs Learning and Synaptic Plasticity via Control of NMDAR Degradation. Nat. Neurosci. 10, 880–886. doi:10.1038/nn1914
Hawasli, A. H., Koovakkattu, D., Hayashi, K., Anderson, A. E., Powell, C. M., Sinton, C. M., et al. (2009). Regulation of Hippocampal and Behavioral Excitability by Cyclin-dependent Kinase 5. PLoS One 4, e5808. doi:10.1371/journal.pone.0005808
Hernandez, A., Tan, C., Mettlach, G., Pozo, K., Plattner, F., and Bibb, J. A. (2016). Cdk5 Modulates Long-Term Synaptic Plasticity and Motor Learning in Dorsolateral Striatum. Sci. Rep. 6, 29812. doi:10.1038/srep29812
Hung, K. S., Hwang, S. L., Liang, C. L., Chen, Y. J., Lee, T. H., Liu, J. K., et al. (2005). Calpain Inhibitor Inhibits P35-P25-Cdk5 Activation, Decreases Tau Hyperphosphorylation, and Improves Neurological Function after Spinal Cord Hemisection in Rats. J. Neuropathol. Exp. Neurol. 64, 15–26. doi:10.1093/jnen/64.1.15
Khair, N. Z., Lenjisa, J. L., Tadesse, S., Kumarasiri, M., Basnet, S. K. C., Mekonnen, L. B., et al. (2019). Discovery of CDK5 Inhibitors through Structure-Guided Approach. ACS Med. Chem. Lett. 10, 786–791. doi:10.1021/acsmedchemlett.9b00029
Knight, Z. A., and Shokat, K. M. (2005). Features of Selective Kinase Inhibitors. Chem. Biol. 12, 621–637. doi:10.1016/j.chembiol.2005.04.011
Kormos, V., and Gaszner, B. (2013). Role of Neuropeptides in Anxiety, Stress, and Depression: from Animals to Humans. Neuropeptides 47, 401–419. doi:10.1016/j.npep.2013.10.014
Kusakawa, G., Saito, T., Onuki, R., Ishiguro, K., Kishimoto, T., and Hisanaga, S. (2000). Calpain-dependent Proteolytic Cleavage of the P35 Cyclin-dependent Kinase 5 Activator to P25. J. Biol. Chem. 275, 17166–17172. doi:10.1074/jbc.M907757199
Lee, M. S., Kwon, Y. T., Li, M., Peng, J., Friedlander, R. M., and Tsai, L. H. (2000). Neurotoxicity Induces Cleavage of P35 to P25 by Calpain. Nature 405, 360–364. doi:10.1038/35012636
Li, Y., Wang, L., Zhang, X., Huang, M., Li, S., Wang, X., et al. (2018). Inhibition of Cdk5 Rejuvenates Inhibitory Circuits and Restores Experience-dependent Plasticity in Adult Visual Cortex. Neuropharmacology 128, 207–220. doi:10.1016/j.neuropharm.2017.10.015
Liu, F., Ma, X. H., Ule, J., Bibb, J. A., Nishi, A., Demaggio, A. J., et al. (2001). Regulation of Cyclin-dependent Kinase 5 and Casein Kinase 1 by Metabotropic Glutamate Receptors. Proc. Natl. Acad. Sci. U S A. 98, 11062–11068. doi:10.1073/pnas.191353898
Mangini, N. S., Wesolowski, R., Ramaswamy, B., Lustberg, M. B., and Berger, M. J., (2015). Palbociclib: A Novel Cyclin-dependent Kinase Inhibitor for Hormone Receptor-Positive Advanced Breast Cancer. Ann. Pharmacother. 49, 1252–1260. doi:10.1177/1060028015602273
Menn, B., Bach, S., Blevins, T. L., Campbell, M., Meijer, L., and Timsit, S. (2010). Delayed Treatment with Systemic (S)-roscovitine Provides Neuroprotection and Inhibits In Vivo CDK5 Activity Increase in Animal Stroke Models. PLoS One 5, e12117. doi:10.1371/journal.pone.0012117
Meyer, D. A., Richer, E., Benkovic, S. A., Hayashi, K., Kansy, J. W., Hale, C. F., et al. (2008). Striatal Dysregulation of Cdk5 Alters Locomotor Responses to Cocaine, Motor Learning, and Dendritic Morphology. Proc. Natl. Acad. Sci. U S A. 105, 18561–18566. doi:10.1073/pnas.0806078105
Meyer, D. A., Torres-Altoro, M. I., Tan, Z., Tozzi, A., Di Filippo, M., Dinapoli, V., et al. (2014). Ischemic Stroke Injury Is Mediated by Aberrant Cdk5. J. Neurosci. 34, 8259–8267. doi:10.1523/JNEUROSCI.4368-13.2014
Miao, Y., Dong, L. D., Chen, J., Hu, X. C., Yang, X. L., and Wang, Z. (2012). Involvement of Calpain/p35-p25/Cdk5/NMDAR Signaling Pathway in Glutamate-Induced Neurotoxicity in Cultured Rat Retinal Neurons. PLoS One 7, e42318. doi:10.1371/journal.pone.0042318
Nishi, A., Bibb, J. A., Snyder, G. L., Higashi, H., Nairn, A. C., and Greengard, P. (2000). Amplification of Dopaminergic Signaling by a Positive Feedback Loop. Proc. Natl. Acad. Sci. U S A. 97, 12840–12845. doi:10.1073/pnas.220410397
o'donnell, J. M., and Xu, Y. (2012). Evidence for Global Reduction in Brain Cyclic Adenosine Monophosphate Signaling in Depression. Biol. Psychiatry 72, 524–525. doi:10.1016/j.biopsych.2012.07.017
O'donnell, J. M., and Zhang, H. T. (2004). Antidepressant Effects of Inhibitors of cAMP Phosphodiesterase (PDE4). Trends Pharmacol. Sci. 25, 158–163. doi:10.1016/j.tips.2004.01.003
Pantoni, M. M., Herrera, G. M., Van alstyne, K. R., and Anagnostaras, S. G. (2020). Quantifying the Acoustic Startle Response in Mice Using Standard Digital Video. Front. Behav. Neurosci. 14, 83. doi:10.3389/fnbeh.2020.00083
Pao, P. C., and Tsai, L. H. (2021). Three Decades of Cdk5. J. Biomed. Sci. 28, 79. doi:10.1186/s12929-021-00774-y
Papadopoulou, A., Siamatras, T., Delgado-Morales, R., Amin, N. D., Shukla, V., Zheng, Y. L., et al. (2015). Acute and Chronic Stress Differentially Regulate Cyclin-dependent Kinase 5 in Mouse Brain: Implications to Glucocorticoid Actions and Major Depression. Transl Psychiatry 5, e578. doi:10.1038/tp.2015.72
Patrick, G. N., Zhou, P., Kwon, Y. T., Howley, P. M., and Tsai, L. H. (1998). p35, the Neuronal-specific Activator of Cyclin-dependent Kinase 5 (Cdk5) Is Degraded by the Ubiquitin-Proteasome Pathway. J. Biol. Chem. 273, 24057–24064. doi:10.1074/jbc.273.37.24057
Patrick, G. N., Zukerberg, L., Nikolic, M., De la monte, S., Dikkes, P., and Tsai, L. H. (1999). Conversion of P35 to P25 Deregulates Cdk5 Activity and Promotes Neurodegeneration. Nature 402, 615–622. doi:10.1038/45159
Pevarello, P., Brasca, M. G., Amici, R., Orsini, P., Traquandi, G., Corti, L., et al. (2004). 3-Aminopyrazole Inhibitors of CDK2/cyclin A as Antitumor Agents. 1. Lead Finding. J. Med. Chem. 47, 3367–3380. doi:10.1021/jm031145u
Plattner, F., Hayashi, K., Hernández, A., Benavides, D. R., Tassin, T. C., Tan, C., et al. (2015). The Role of Ventral Striatal cAMP Signaling in Stress-Induced Behaviors. Nat. Neurosci. 18, 1094–1100. doi:10.1038/nn.4066
Plattner, F., Hernández, A., Kistler, T. M., Pozo, K., Zhong, P., Yuen, E. Y., et al. (2014). Memory Enhancement by Targeting Cdk5 Regulation of NR2B. Neuron 81, 1070–1083. doi:10.1016/j.neuron.2014.01.022
Pond, S. M., and Tozer, T. N. (1984). First-pass Elimination. Basic Concepts and Clinical Consequences. Clin. Pharmacokinet. 9, 1–25. doi:10.2165/00003088-198409010-00001
Pozo, K., Castro-Rivera, E., Tan, C., Plattner, F., Schwach, G., Siegl, V., et al. (2013). The Role of Cdk5 in Neuroendocrine Thyroid Cancer. Cancer Cell 24, 499–511. doi:10.1016/j.ccr.2013.08.027
Rana, S., Sonawane, Y. A., Taylor, M. A., Kizhake, S., Zahid, M., and Natarajan, A. (2018). Synthesis of Aminopyrazole Analogs and Their Evaluation as CDK Inhibitors for Cancer Therapy. Bioorg. Med. Chem. Lett. 28, 3736–3740. doi:10.1016/j.bmcl.2018.10.020
Rana, T., Behl, T., Sehgal, A., Singh, S., Sharma, N., Abdeen, A., et al. (2022). Exploring the Role of Neuropeptides in Depression and Anxiety. Prog. Neuropsychopharmacol. Biol. Psychiatry 114, 110478. doi:10.1016/j.pnpbp.2021.110478
Robb, C. M., Kour, S., Contreras, J. I., Agarwal, E., Barger, C. J., Rana, S., et al. (2018). Characterization of CDK(5) Inhibitor, 20-223 (Aka CP668863) for Colorectal Cancer Therapy. Oncotarget 9, 5216–5232. doi:10.18632/oncotarget.23749
Seibenhener, M. L., and wooten, M. C. (2015). Use of the Open Field Maze to Measure Locomotor and Anxiety-like Behavior in Mice. J. Vis. Exp. 1, e52434. doi:10.3791/52434
Smyth, L. A., and Collins, I. (2009). Measuring and Interpreting the Selectivity of Protein Kinase Inhibitors. J. Chem. Biol. 2, 131–151. doi:10.1007/s12154-009-0023-9
Sonawane, Y. A., Taylor, M. A., Napoleon, J. V., Rana, S., Contreras, J. I., and Natarajan, A. (2016). Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. J. Med. Chem. 59, 8667–8684. doi:10.1021/acs.jmedchem.6b00150
Sundaram, J. R., Chan, E. S., Poore, C. P., Pareek, T. K., Cheong, W. F., Shui, G., et al. (2012). Cdk5/p25-induced Cytosolic PLA2-Mediated Lysophosphatidylcholine Production Regulates Neuroinflammation and Triggers Neurodegeneration. J. Neurosci. 32, 1020–1034. doi:10.1523/JNEUROSCI.5177-11.2012
Sundaram, J. R., Poore, C. P., Sulaimee, N. H., Pareek, T., Asad, A. B., Rajkumar, R., et al. (2013). Specific Inhibition of p25/Cdk5 Activity by the Cdk5 Inhibitory Peptide Reduces Neurodegeneration In Vivo. J. Neurosci. 33, 334–343. doi:10.1523/JNEUROSCI.3593-12.2013
Syed, Y. Y. (2017). Ribociclib: First Global Approval. Drugs 77, 799–807. doi:10.1007/s40265-017-0742-0
Tarricone, C., Dhavan, R., Peng, J., Areces, L. B., Tsai, L. H., and Musacchio, A. (2001). Structure and Regulation of the CDK5-P25(nck5a) Complex. Mol. Cel 8, 657–669. doi:10.1016/s1097-2765(01)00343-4
Umfress, A., Speed, H. E., Tan, C., Ramezani, S., Birnbaum, S., Brekken, R. A., et al. (2021). Neuropathological Effects of Chemotherapeutic Drugs. ACS Chem. Neurosci. 12, 3038–3048. doi:10.1021/acschemneuro.1c00338
Vita, M., Abdel-Rehim, M., Olofsson, S., Hassan, Z., Meurling, L., Sidén, A., et al. (2005). Tissue Distribution, Pharmacokinetics and Identification of Roscovitine Metabolites in Rat. Eur. J. Pharm. Sci. 25, 91–103. doi:10.1016/j.ejps.2005.02.001
Yoo, B. C., and Lubec, G. (2001). p25 Protein in Neurodegeneration. Nature 411, 763–765. discussion 764-5. doi:10.1038/35081146
Keywords: cyclin-dependent kinase 5, pharmacokinetics, inhibitors, behavior, kinase inhibitors
Citation: Umfress A, Singh S, Ryan KJ, Chakraborti A, Plattner F, Sonawane Y, Mallareddy JR, Acosta EP, Natarajan A and Bibb JA (2022) Systemic Administration of a Brain Permeable Cdk5 Inhibitor Alters Neurobehavior. Front. Pharmacol. 13:863762. doi: 10.3389/fphar.2022.863762
Received: 27 January 2022; Accepted: 01 April 2022;
Published: 12 May 2022.
Edited by:
Julia A. Chester, Purdue University, United StatesReviewed by:
Carla Cannizzaro, University of Palermo, ItalyCopyright © 2022 Umfress, Singh, Ryan, Chakraborti, Plattner, Sonawane, Mallareddy, Acosta, Natarajan and Bibb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James A. Bibb, amJpYmJAdWFiLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.