
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 27 April 2022
Sec. Experimental Pharmacology and Drug Discovery
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.841766
This article is part of the Research TopicTherapeutic Potential of the Cannabinoid CB2 ReceptorView all 13 articles
 Samuel Ruiz de Martín Esteban1†
Samuel Ruiz de Martín Esteban1† Irene Benito-Cuesta1†
Irene Benito-Cuesta1† Itziar Terradillos2,3Ana M. Martínez-Relimpio1M. Andrea Arnanz1Gonzalo Ruiz-Pérez1
Itziar Terradillos2,3Ana M. Martínez-Relimpio1M. Andrea Arnanz1Gonzalo Ruiz-Pérez1 Claudia Korn4Catarina Raposo4Roman C. Sarott5Matthias V. Westphal5
Claudia Korn4Catarina Raposo4Roman C. Sarott5Matthias V. Westphal5 Izaskun Elezgarai2,3Erick M. Carreira5
Izaskun Elezgarai2,3Erick M. Carreira5 Cecilia J. Hillard6
Cecilia J. Hillard6 Uwe Grether4
Uwe Grether4 Pedro Grandes2,3M. Teresa Grande1
Pedro Grandes2,3M. Teresa Grande1 Julián Romero1*
Julián Romero1*The distribution and roles of the cannabinoid CB2 receptor in the CNS are still a matter of debate. Recent data suggest that, in addition to its presence in microglial cells, the CB2 receptor may be also expressed at low levels, yet biologically relevant, in other cell types such as neurons. It is accepted that the expression of CB2 receptors in the CNS is low under physiological conditions and is significantly elevated in chronic neuroinflammatory states associated with neurodegenerative diseases such as Alzheimer’s disease. By using a novel mouse model (CB2EGFP/f/f), we studied the distribution of cannabinoid CB2 receptors in the 5xFAD mouse model of Alzheimer’s disease (by generating 5xFAD/CB2EGFP/f/f mice) and explored the roles of CB2 receptors in microglial function. We used a novel selective and brain penetrant CB2 receptor agonist (RO6866945) as well as mice lacking the CB2 receptor (5xFAD/CB2−/−) for these studies. We found that CB2 receptors are expressed in dystrophic neurite-associated microglia and that their modulation modifies the number and activity of microglial cells as well as the metabolism of the insoluble form of the amyloid peptide. These results support microglial CB2 receptors as potential targets for the development of amyloid-modulating therapies.
Cannabinoid receptors include two types of G-protein coupled receptors (GPCRs), CB1 and CB2, that exhibit profound differences in their distribution in the organism of mammals (Pertwee et al., 2010). While the CB1 receptor is one of the most abundant GPCRs in the brain and its expression is constitutive in a wide variety of cells and tissues, the distribution of CB2 receptors is restricted to specific types of cells (B-lymphocytes, natural killer cells, monocytes, etc) and tissues (spleen, Peyer’s patches) and its brain expression is low under physiological conditions (for review, see Mechoulam and Parker, 2013). Importantly, the expression of cannabinoid CB2 receptors is significantly increased under pathological conditions and, specifically, in the context of chronic neuroinflammation (Maresz et al., 2005; Mecha et al., 2016).
The pattern of expression of CB2 receptors and its biological relevance in the CNS is still a matter of debate. It is currently accepted that microglial cells express CB2 receptors under both normal and pathological conditions (Komorowska-Müller and Schmöle, 2020). Interestingly, although its presence in neuronal elements is believed to be low (if any), there are reports that CB2 receptors contribute to functions ascribed to neurons, such as pain or reward (Zhang et al., 2014; Cabañero et al., 2020; He et al., 2021). CB2 receptors also seem to play important roles in neurodegenerative conditions, although their precise contribution has not been elucidated yet due to conflicting results (Mecha et al., 2016; Galán-Ganga et al., 2021; Rodríguez-Cueto et al., 2021).
The amyloid hypothesis of Alzheimer’s disease (AD) is currently the most widely accepted among the scientific community (Hampel et al., 2021). Together with hyperphosphorylated tau-enriched neurofibrillary tangles, neuritic plaques (primarily constituted by amyloid peptides and, specifically, beta amyloid 1-42, Aβ) are the main pathologic features of AD. Multiple deleterious consequences derive from the accumulation of both in the brain, including mitochondrial dysfunction, axonal degeneration, alterations in synaptic transmission and neuroinflammation (Hampel et al., 2021).
There is an urgent need for novel approaches for the treatment of this devastating disease. Efforts have been focused on tackling the neuroinflammatory process triggered by the presence of pathological forms of Aβ as it is presently thought that these peptides possess intrinsic pro-inflammatory properties that play a crucial role in the loss of neurons in specific areas of the AD brain. This process involves several types of cells (microglia, astrocytes) and mediators (cytokines, reactive oxygen species, lipids) that, acting in a concerted and time-dependent manner, expand the damage initiated by neuritic plaques and neurofibrillary tangles (see Hampel et al., 2021, for review).
Microglia seem to play a prominent role in this scenario. In the healthy brain, these cells of myeloid origin are continuously sensing their surrounding environment (Nimmerjahn et al., 2005). When an alteration takes place in the brain parenchyma, these cells become “activated”, and shape their phenotype to cope with this alteration by modifying their structural properties, gene expression profile, ability to produce cytokines and other cell mediators, and phagocytic activity (becoming “damage-associated microglia”, DAM; Deczkowska et al., 2018). Among other adaptations, microglia express cannabinoid CB2 receptors in the context of AD (Mecha et al., 2016), and remarkably, in neuritic plaque-associated microglia (Benito et al., 2003).
In the present study, we analyzed the expression of cannabinoid CB2 receptors in cortical areas of the brain of an AD mouse model (5xFAD/CB2EGFP/f/f) by electron microscopy. In addition, we explored the potential roles of this receptor through its activation with a selective agonist (RO6866945) and through its genetic deletion (5xFAD/CB2−/−).
Mice used in these experiments were described in our previous study (López et al., 2018) and were housed and bred in the animal facilities of Universidad Francisco de Vitoria (Pozuelo de Alarcón, Madrid, Spain). Experimental protocols met the European and Spanish regulations for protection of experimental animals (86/609/EEC and RD 1201/2005 and 53/2013) and were approved by the committee of Ethics for Animal Welfare of the Universidad Francisco de Vitoria and University of the Basque Country (M20/2015/093). Efforts were made to minimize the number and suffering of animals.
Mice co-expressing five familial Alzheimer’s disease mutations (5xFAD) were purchased from Jackson Laboratories (Bar Harbor, ME, United States; Oakley et al., 2006) on the C57BL/6J background and were mated with CB2EGFP/f/f and CB2−/− mice and backcrossed for at least ten generations to generate 5xFAD/CB2EGFP/f/f and 5xFAD/CB2−/− mice.
Prior to the experiment, mice were homogenously distributed per group according to bodyweight. A stock solution of 90 mg/ml RO6866945 (Roche Pharma Research and Early Development, Roche Innovation Center Basel, Basel, Switzerland) in ethanol was conserved at −20°C, and diluted in vehicle solution [5% ethanol, 5% kolliphor (Sigma, C5135), 90% NaCl 0.9% (Braun, 857367)] the day of use. 6 months old 5xFAD/CB2EGFP/f/f and 5xFAD/CB2−/− male mice were treated (i.p.) with RO6866945 10 mg/kg, or vehicle (VEH) daily for 28 days. RO6866945 ((3S)-1-[5-tert-butyl-3-[(4-methyl-1,2,5-oxadiazol-3-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol; CAS Registry Number 1433360-72-5) was synthesized as described in US20130116236 A1 (Example 136) (Adam et al. (2013). Preparation of [1,2,3]triazolo[4,5-d]pyrimidine derivatives useful as cannabinoid receptor 2 agonists, US20130116236 A1). It is a highly potent CB2 agonist across species (human CB2 cAMP EC50 0.2 nM, 104% efficacy; mouse CB2 cAMP EC50 0.2 nM, 101% efficacy) which does neither interact with the CB1 receptor in the cAMP (human CB1 cAMP EC50 > 10′000 nM) nor in the radioligand binding assay (human CB1 Ki > 10′000 nM; Ouali Alami et al., 2018). RO6866945 exhibits an excellent early ADME profile including an oral bioavailability of 44% in mice and penetrates through the blood brain barrier.
Twenty-four hours before the end of the treatment, mice were intraperitoneally injected with 10 mg/kg methoxy-X04 (Tocris, 4920) in 15% DMSO, 15% kolliphor and 70% NaCl 0.9%. Then, mice were anaesthetised with 170 mg/kg ketamine (Richter Pharma, 580393.7) and 10.7 mg/kg xylazine (Calier, 572599.4) in NaCl 0.9%, and transcardially perfused with cold PBS pH 7.4. From each mouse, right cortex, hippocampi and cerebellum were dissected and stored at −80°C. Left cortex and the rest of the brain were immediately processed to isolate microglia for analysis by flow cytometry.
Three male CB2EGFP/f/f and three 5xFAD/CB2EGFP/f/f mice were anaesthetized with ketamine/xylazine (100mg/10 mg/kg body weight, intraperitoneal injection) and subsequently perfused transcardially at room temperature (RT) with 4% formaldehyde (freshly depolymerized from paraformaldehyde), 0.2% picric acid and 0.1% glutaraldehyde in PBS 0.1 M (pH 7.4) for 10–15 min. The brains were then removed from the skull, post-fixed in the fixative solution for 1 week at 4°C and cut into 50 μm thick coronal sections using a vibratome.
Our protocol previously published was used (Puente et al., 2019). Brain sections containing the subiculum were pre-incubated in a blocking solution of 10% bovine serum albumin (BSA), 0.02% saponin and 0.1% sodium azide in Tris-hydrogen chloride buffered saline (TBS 1X), for 30 min on a shaker at RT. Tissue was then incubated for 2 days at 4°C with both a rat monoclonal anti-GFP antibody (1:500, GF090R, Nacalai) and a rabbit polyclonal anti-Iba1 antibody (1:500, 019-19741, FUJIFILM Wako Pure Chemical Corporation) prepared in 10% BSA, 0.1% sodium azide and 0.004% saponin. After washes in 1% BSA/TBS, sections were incubated with 1.4 nm gold-conjugated goat anti-rat IgG antibody (Fab’ fragment, 1:100; Nanoprobes Inc., Yaphank, NY, United States) and with biotinylated anti-rabbit IgG antibody (1:200; Biotin-SP-AffiniPure donkey anti-rabbit IgG) diluted in 1% BSA/TBS with 0.004% saponin on a shaker for 4 h at RT. They were washed in 1% BSA/TBS and then incubated with the avidin-biotin peroxidase complex (1:50; Elite, Vector Laboratories, Burlingame, CA, United States) for 1.5 h at RT. Sections were then washed in 1% BSA/TBS and kept in the same washing solution overnight at 4°C, postfixed with 1% glutaraldehyde in TBS for 12 min at RT and washed in double distilled water. Gold particles were silver-intensified with the HQ Silver kit (Nanoprobes Inc., Yaphank, NY, United States) in the dark for 12 min at RT. The biotinylated antibody was exposed to 0.05% diaminobenzidine (pH 7.4) with 0.01% hydrogen peroxide for 3.5 min at RT. Sections were incubated with 1% osmium tetroxide, pH 7.4, in the dark for 20 min, washed in PB 0.1 M, dehydrated and embedded in Epon 812 resin. 50 nm-thick sections were cut with an ultra-diamond knife (Diatome United States) and collected on nickel mesh grids. They were counterstained with 2.5% lead citrate for 20 min and examined with a transmission electron microscope (JEOL JEM 1400 Plus, Canada). Tissue was photographed using a Hamamatsu FLASH digital camera inserted in the electron microscope. Anatomical landmarks were taken to locate the subiculum region.
To ensure homogeneous labelling between all samples, only the first 1.5 µm from the section surface of each specimen was collected. Random electron micrographs were taken of the subicula. Areas of 3,524 μm2 in CB2EGFP/f/f and 4,078 μm2 in 5xFAD/CB2EGFP/f/f mice were examined to assess CB2 receptors in Iba1-positive microglia. GFP gold particles were counted and differentiated between their localization in membrane (between 0 and 30 nm of the membrane) or cytosol (more than 30 nm). Minor contrast and brightness adjustments were made to the figures using ImageJ software (NIH; RRID: SCR_003070), Adobe Photoshop and Gimp.
Flow cytometry was employed to determine the ability of microglial cells to phagocytize Aβ (stained with methoxy-X04), and the levels of CB2 with RO7246360 probe (compound 3b in Sarott et al., 2020). 6-month-old animals were injected i. p. with Methoxy-X04 (Tocris Bioscience) at 10 mg/kg body weight. 24 h after injection, animals were deeply anesthetized by i. p. administration of a mixture of ketamine (170 mg/kg) and xylazine (10.7 mg/kg) and transcardially perfused with cold PBS 1X, pH 7.4. Brains were dissected and enzymatically digested to facilitate microglia separation. The cell suspension was mechanically dissociated and filtered through a 70 µm-cell strainer. Microglial cells, isolated by percoll gradient (GE Healthcare), were washed with PBS 1X and blocked with 1% BSA/PBS 1X for 20 min. Cells were stained with CD11b-PE and CD45-APC antibodies and with RO7246360 fluorescent probe for 40 min. Samples were read on a MACSQuant Flow Cytometer and analysed with MACS Quantify software (Miltenyi Biotec).
Debris and aggregates were eliminated from analysis by forward and side scatter characteristics. Then microglia were identified as CD11b+ CD45lo. The CB2 receptor expression was determined by the fluorescent signal of RO7246360 probe. Fluorescence signals were corrected by fluorescence minus one (FMO) control. For each hemisphere, approximately ten thousand CD11b + singlets were analysed.
Extracts from frozen brain cortices were obtained by homogenization in magnesium lysis buffer (MLB: 25 mM HEPES, pH 7.5, 150 mM NaCl, 1% Igepal CA-630, 10 mM MgCl2, 1 mM EDTA) containing 10% glycerol, and protease and phosphatase inhibitors (1 mM Na3VO4, 25 mM NaF and protease inhibitor cocktail; Roche) and were maintained at 4°C. Homogenates were centrifuged at 12000 g for 20 min at 4°C and supernatants were collected to determine their protein content by BCA protein assay (Pierce™ BCA protein assay kit, Thermo Scientific). Homogenates were used to measure cAMP levels using an ELISA kit (cat.no. ab65355, Abcam) following the manufacturer’s instructions. Standards and samples were plated in duplicate, and the absorbance was measured at 450 nm using a Varioskan Flash multifunction plate reader (Sunrise, Tecan).
Frozen mouse brain cortices were homogenized in four volumes (weight: volume) of TBS extracting buffer (140 mM NaCl, 3 mM KCl, 25 mM Tris, pH 7.4, 5 mM EDTA and protease inhibitor cocktail; Roche). Homogenates were centrifuged at 16,000 g for 20 min at 4°C. The supernatants were saved to quantify the soluble Aβ1-42 peptide fraction and the pellets were again homogenized in four volumes (weight: volume) of 5M guanidine 50 mM Tris-HCl pH 8. The supernatants obtained after the centrifugation step were collected to quantify the insoluble Aβ1-42 peptide fraction. An equal volume of PBS containing 1 mM serine protease inhibitor AEBSF (Sigma) was added to all samples and their protein content was determined by micro-BCA protein assay (Micro BCA™ protein assay kit, Thermo Scientific). Human Aβ1-42 Ultrasensitive ELISA kit (cat.no. KHB3544 Invitrogen) was used for the quantification of soluble and insoluble fractions of Aβ1-42 peptide following the instructions provided by the manufacturer. Standards and samples were plated in duplicate, and the absorbance was measured at 450 nm using a Varioskan Flash multifunction plate reader (Sunrise, Tecan).
Extracts from frozen brain cortices were obtained following the procedure previously described for cAMP assay. Lysates (60 μg/lane) were separated by SDS-PAGE and transferred onto nitrocellulose membranes (BioRad) and PVDF membranes (BioRad, used for the transference of phosphorylated proteins). After blocking in 5% bovine serum albumin in TTBS (10 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween 20) membranes were incubated overnight at 4°C, as appropriate, with primary antibodies: anti-phospho-p38 MAPK (1:1000; Cell Signaling Technology, 4511T), anti-p38 MAPK (1:1000; Cell Signaling Technology, 8690T), anti-phospho-CREB (1:1000; Cell Signaling Technology, 9198S), anti-CREB (1:1000; Cell Signaling Technology, 9197T), anti-phospho-ERK1/2 MAPK (1:1000; Cell Signaling Technology, 9101S), anti-Erk1/2 MAPK (1:1000; Santa Cruz Biotechnology, sc514302), Anti-Iba1 (1:1000, FUJIFILM Wako Pure Chemical, 016-20001), Anti-APP N-terminus (1:1000, EMD Millipore, MAB348), Anti-APP C-terminus (1:2000, Sigma, A8717), anti-BACE1 (1:500, Abcam, ab 2077) and anti-GAPDH (1:1000; Abcam, ab8245). Membranes were incubated with corresponding horseradish peroxidase (HRP)-conjugated secondary antibody anti-mouse IgG-HRP (1:10000; Abcam, ab97046), anti-rabbit IgG-HRP (1:5000; Cell Signaling Technology, 7074S) and were developed using a chemoluminiscent reagent (Western Lighting ECL Plus, PerkinElmer, NEL103001EA) in the appropriate equipment (ChemiDoc, Bio-Rad). GAPDH was used as an internal control. The relative quantity of protein levels in western blot was measured using ImageJ software (ImageJ; NIH).
All statistical analyses were performed, and graphs were generated using GraphPad Prism v 9.0 (GraphPad). Graphs represent average values ± standard error of the mean. Normality of data distribution was determined with the Shapiro-Wilk or the D’Agostino-Pearson tests. For GFP labeling, data were analyzed by means of the Mann-Whitney U test. For the rest of determinations, data were analysed by means of two-way ANOVA, followed by Tukey’s post-hoc tests. A p-value < 0.05 was considered as statistically significant. Only male animals were used in the experiments. The number of animals used for each experiment is reported in the figure legends.
The GFP/CB2 labelling was localized in Iba-1 immunopositive microglial processes in both CB2EGFP/f/f and 5xFAD/CB2EGFP/f/f mice (Figure 1). GFP-positive microglial processes increased significantly in 5xFAD/CB2EGFP/f/f (0.7126 ± 0.2311) relative to CB2EGFP/f/f (0.1648 ± 0.07686, *p: 0.0176; Figure 2). Likewise, a significant increase in the proportion of GFP-positive microglial ramifications was seen in 5xFAD/CB2EGFP/f/f (16.71 ± 3.664%) with respect to CB2EGFP/f/f (5.430 ± 2.631%; p = 0.0191; Figure 2). Also, the total number of GFP particles per area of microglial ramifications was significantly greater in 5xFAD/CB2EGFP/f/f (1.238 ± 0.2534) than in CB2EGFP/f/f mice (0.6962 ± 0.4138; p = 0.0467; Figure 2), and the number of GFP particles in microglial branches per 100 μm2 was statistically higher in 5xFAD/CB2EGFP/f/f (0.8343 ± 0.2962) than in CB2EGFP/f/f (0.1648 ± 0.07686; p = 0.0176; Figure 2). Noticeably in 5xFAD/CB2EGFP/f/f, the percentage of GFP immunoparticles localized in microglial membranes (77.22 ± 11.40%) was significantly higher than the proportion distributed in the cytosol (22.78 ± 11.40%; p = 0.0106; Figure 2). As to CB2EGFP/f/f, 100% of the GFP particles were found in microglial membranes.
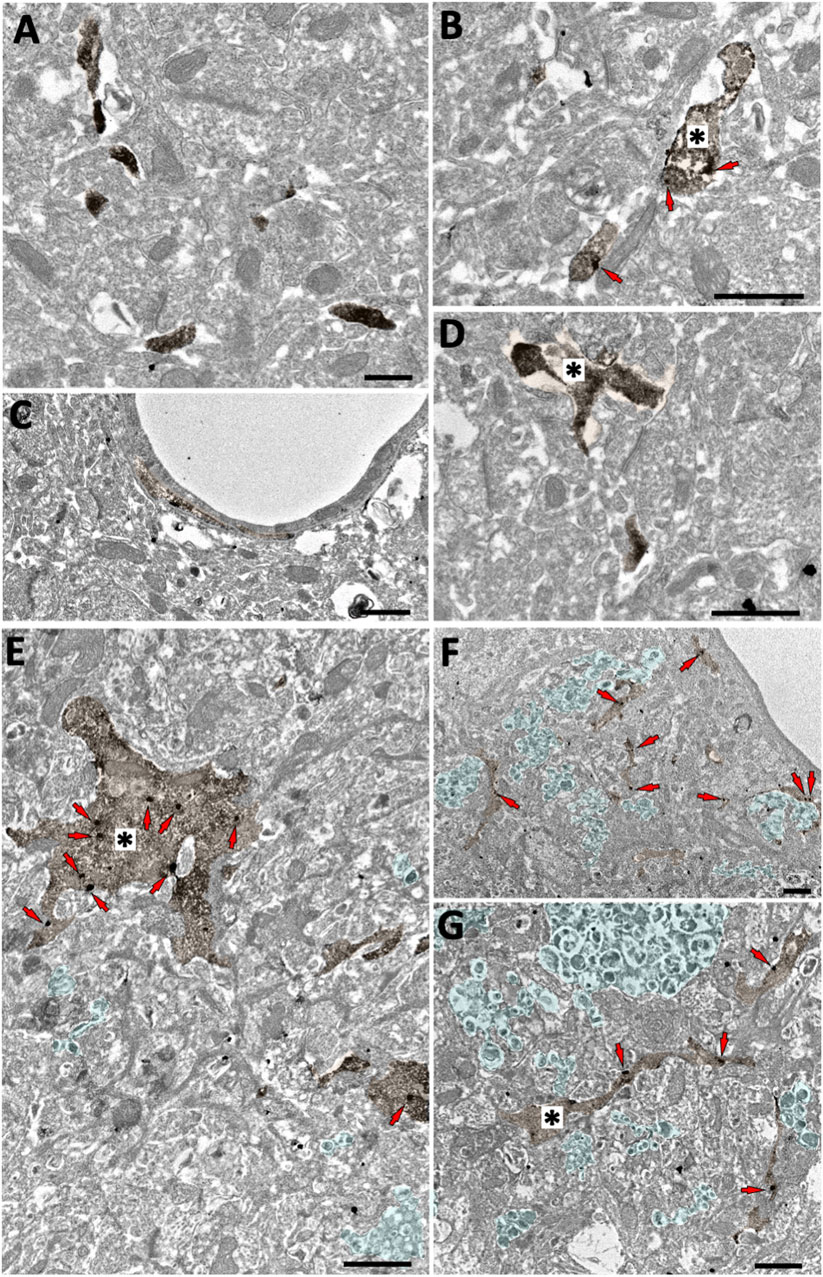
FIGURE 1. Microglial GFP localization in the subiculum of CB2EGFP/f/f and 5xFAD/CB2EGFP/f/f mice. Double pre-embedding immunogold (GFP) and immunoperoxidase (Iba1) method for electron microscopy. GFP particles (red arrows) localize in Iba1-positive microglial elements (DAB immunodeposits, brown, *). In CB2EGFP/f/f (A–D), only GFP membrane localization is observed (arrows, (B). In 5xFAD/CB2EGFP/f/f, GFP particles are found in both membranes and cytosol (E–G). Notice dystrophic neurites (light green areas contoured by white dashed lines) in 5xFAD/CB2EGFP/f/f. Scale bars: 1 µm.
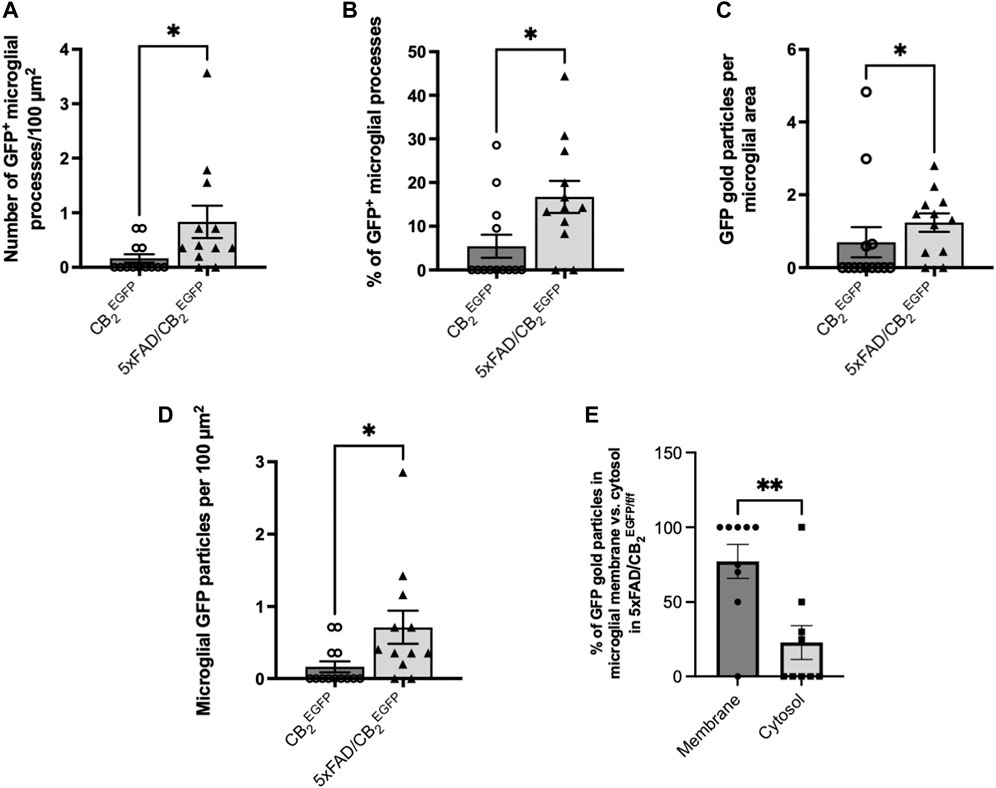
FIGURE 2. Assessment of the microglial GFP/CB2 localization in the subiculum of CB2EGFP/f/f and 5xFAD/CB2EGFP/f/f mice. (A) Number of microglial GFP-positive processes per 100 μm2. (B) Percentage of GFP-positive microglial processes. (C) GFP gold particles per microglial area. (D) Microglial GFP particles per 100 μm2. (E) Percentage of GFP particles in microglial membrane vs. cytosol in 5xFAD/CB2EGFP/f/f mice. Mann-Whitney U test. *<p0.05; **<p0.01; ***<p0.001; ****<p0.0001. N = 3 mice per group. Data represent mean ± SEM.
We then studied whether the chronic treatment with RO6866945 had an impact on the expression levels of cannabinoid CB2 receptors. We used two different approaches: first, RT-PCR revealed no significant effects of the 28-days treatment with the agonist on CB2 mRNA levels (F(1,23) = 0.6509, p = 0.4280) and confirmed the absence of CB2 expression in samples from 5xFAD/CB2-/- mice (Figure 3A; F(1,23) = 437.6, p < 0.0001). Second, we employed flow cytometry to quantify the binding of the selective fluorescent probe RO7246360 to the CB2 receptor; we found no changes induced by the chronic exposure to the agonist [F(1,22) = 0.02066, p = 0.8870] and confirmed the absence of CB2 protein in isolated microglia from 5xFAD/CB2-/- mice (Figure 3B; F(1,22) = 55.62, p < 0.0001).
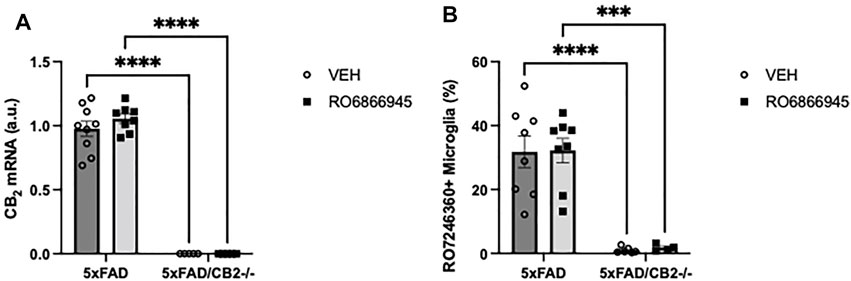
FIGURE 3. The chronic exposure to the CB2 selective agonist, RO6866945, did not modify the expression of cannabinoid CB2 receptors. (A) mRNA levels of the cannabinoid CB2 receptor did not vary after treatment with RO6866945 but were completely absent in 5xFAD/CB2-/- mice. (B) Binding of the fluorescent probe RO7246360 to cannabinoid CB2 receptors was used to quantify protein levels, revealing no changes after treatment with the agonist and the negligible levels of CB2 protein in 5xFAD/CB2-/- mice. Two-way ANOVA followed by Tukey’s post-hoc test. **<p0.01; ****<p0.0001. N = 4–9 mice per group. Data represent mean ± SEM.
We next analyzed the signaling cascades affected by CB2 activation or deletion (Figure 4). We found that the CB2 agonist had a significant impact on cAMP levels [Figure 4A; F(1,19) = 8.851, p = 0.0078]. Post-hoc analysis revealed a decrease in cAMP in 5xFAD/CB2EGFP/f/f mice as a consequence of the treatment (p < 0.0001) that was absent in 5xFAD/CB2-/- mice (p = 0.9802). No differences due to the genotype were observed in vehicle-treated mice (p = 0.2537), although were significant between RO6866945-treated 5xFAD/CB2EGFP/f/f vs. 5xFAD/CB2-/- mice (p < 0.0001).
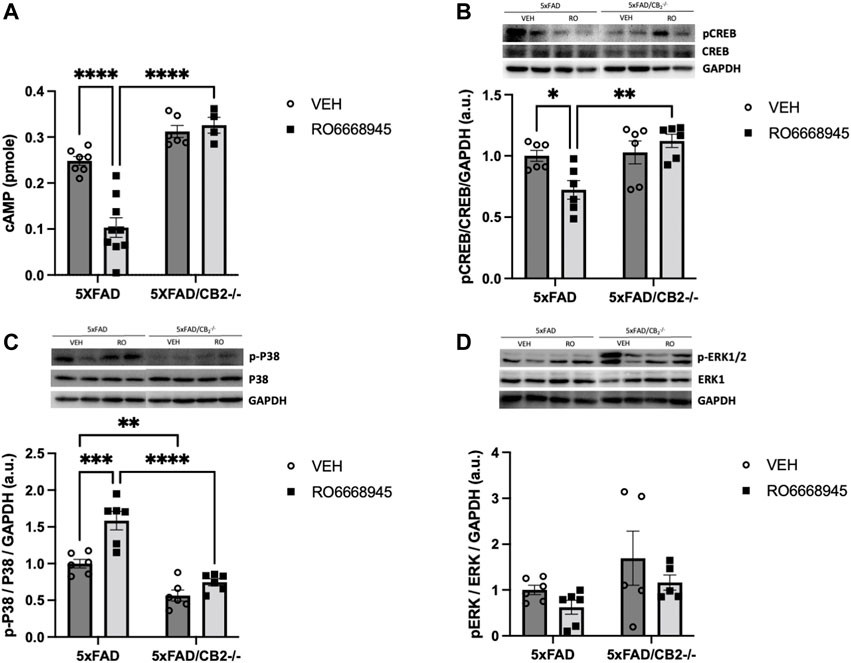
FIGURE 4. Signaling cascades regulated by the activation and deletion of cannabinoid CB2 receptors. (A) cAMP and p-CREB (B) levels were significantly decreased by the treatment with the CB2 agonist, remaining unaltered in samples from CB2-null mice. (C) p-p38MAPK levels were significantly elevated by the exposure to the agonist; in addition, samples from 5xFAD/CB2-/- mice exhibited significantly lower levels. (D) p-ERK levels were not modified by the treatment with the CB2 agonist or the genetic deletion of the receptor. Two-way ANOVA followed by Tukey’s post-hoc test. *<p0.05; **<p0.01; ***<p0.001; ****<p0.0001. N = 4–9 mice per group. Data represent mean ± SEM.
Regarding pCREB levels (Figure 4B), genotype had a significant effect [F(1,20) = 9.370, p = 0.0062]. Post-hoc analysis revealed that the agonist significantly decreased pCREB levels in 5xFAD/CB2EGFP/f/f mice (p = 0.0492) but not in 5xFAD/CB2-/- mice (p = 0.7765). No differences due to the genotype were observed in vehicle-treated mice (p = 0.9917), although were significant between RO6866945-treated 5xFAD/CB2EGFP/f/f vs. 5xFAD/CB2−/− mice (p = 0.0033).
p-p38MAPK levels (Figure 4C) were modified by the treatment with RO6866945 [F(1,20) = 21.77, p = 0.0001], by genotype [F(1,20) = 60.28, p < 0.0001] and by the interaction of both factors [F(1,20) = 6.088, p = 0.0228]. p-p38MAPK was increased in 5xFAD/CB2EGFP/f/f mice as a consequence of CB2 activation by the agonist (p = 0.0003) and exhibited significantly lower levels in samples from both vehicle- and RO6866945-treated CB2-lacking mice (p = 0.0064 and p < 0.0001, respectively). These observations highlight the selectivity of RO6866945 as a CB2-selective agonist and suggest a putative constitutive activation of p-38MAPK signaling cascade by CB2 receptors in the context of AD.
Finally, p-ERK levels remained unaltered after treatment with the agonist [F(1,18) = 2.377, p = 0.1405] as well as in 5xFAD/CB2−/− mice (Figure 4D; F(1,18) = 4.339, p = 0.0518).
As microglia are the main source of cannabinoid CB2 receptors in the brain of 5xFAD/CB2EGFP/f/f mice, we analyzed the putative changes triggered in these cells by the activation of the receptor and by its genetic deletion (Figure 5). We found no changes in Iba1+ microglia (Figure 5A; F(1,20) = 0,7931, p = 0.3837) nor in its phagocytic activity (measured by its ability to internalize methoxy-X04-stained amyloid; Figures 5B,C; F(1,22) = 3.602, p = 0.0709) derived from CB2 activation by the agonist. However, significant differences were evident between 5xFAD/CB2EGFP/f/f and 5xFAD/CB2-/- microglia; thus, we found a decrease in Iba1+ microglia abundance [Figure 5A; F(1,20) = 34.95, p < 0.0001] as well as an impairment in its phagocytic activity (Figure 5B; F(1,22) = 69.96, p < 0.0001).
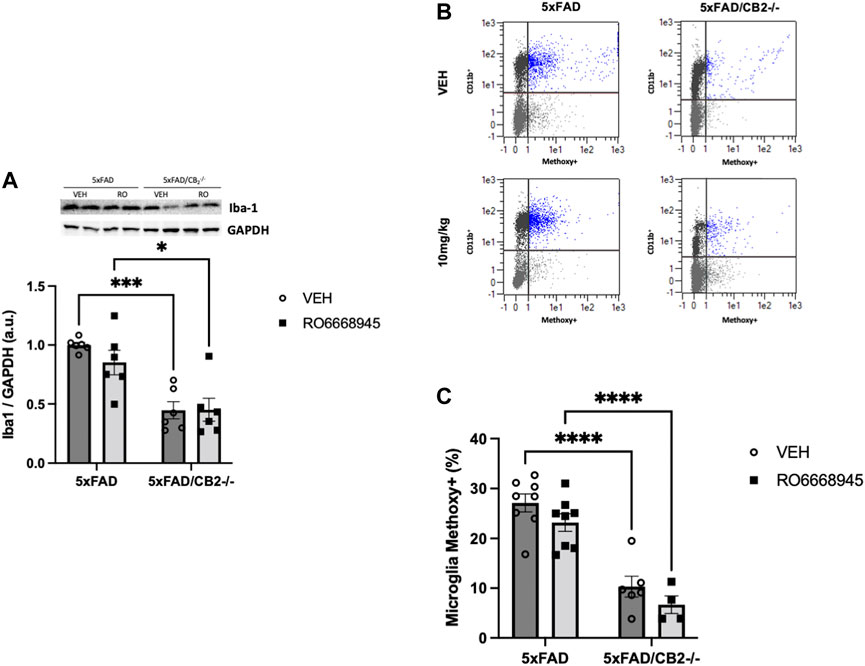
FIGURE 5. Iba1+ microglia and phagocytic activity is decreased after genetic deletion of the cannabinoid CB2 receptor. Analysis of cortices by western blot (A) and flow cytometry (B,C) revealed no changes associated to the treatment with the agonist together with a significant decrease in Iba1+ microglia (A). Phagocytic activity (B,C) of microglia was significantly impaired in 5xFAD/CB2-/- mice. Scatter plots of CD11b isolated microglia after intraperitoneal administration of methoxy-X04 are shown (B). Phagocytic capacity was calculated as percentage of methoxy-X04+/CD11b+/CD45lo cells to CD11b+/CD45lo cells (C). Two-way ANOVA followed by Tukey’s post-hoc test. *<p0.05. **<p0.01; ***<p0.001; ****<p0.0001. N = 5–6 mice per group. Data represent mean ± SEM.
We next measured the impact of CB2 modulation on Aβ levels. To that end, we quantified several amyloid-related peptides (APP, C83 and BACE1) as well as the soluble and insoluble forms of Aβ1-42, the main component of neuritic plaques (Figure 6). Our data showed no changes in APP [F(1,20) = 0.08911, p = 0.7684], C83 [F(1,20) = 0.1794, p = 0.6764] or BACE1 [F(1,20) = 3.026, p = 0.0973] after treatment with RO6866945. CB2 deletion induced significant differences in protein levels of BACE1 [F(1,20) = 10.34, p = 0.0043], but not in C83 (F(1,20) = 1.705, p = 0.2065) and APP [F(1,20) = 2.468, p = 0.1319].
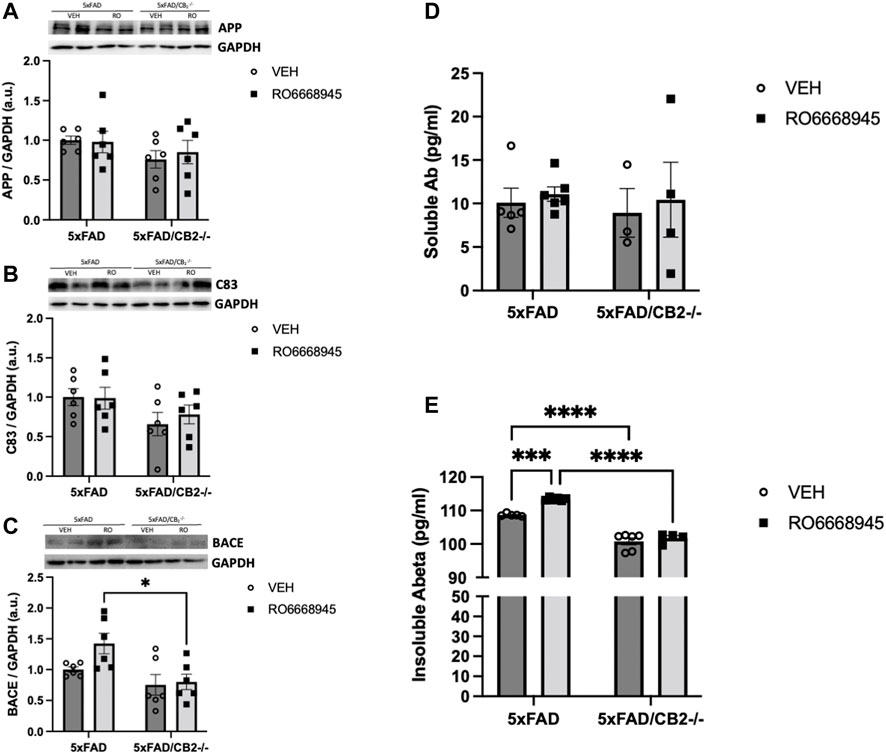
FIGURE 6. Cannabinoid CB2 receptors modulate amyloid dynamics in vivo. No changes were evident after treatment in APP (A), C83 (B) or BACE1 (C), as measured by western blot. Soluble amyloid levels (D) remained also unaltered, while those of insoluble amyloid (E) were significantly increased by the exposure to the CB2 agonist and decreased in 5xFAD/CB2-/- mice. Two-way ANOVA followed by Tukey’s post-hoc test. *<p0.05; ****<p0.0001. N = 4-6 mice per group. Data represent mean ± SEM.
Levels of soluble amyloid were unaltered after treatment with the CB2 agonist [F(1,14) = 0.2681, p = 0.6127] or genetic inactivation of the receptor [F(1,14) = 0.1408, p = 0.1731; Figure 6D]. Cortical amounts of insoluble amyloid, however, were significantly modified by both (Figure 6E). Thus, the treatment with RO6866945 led to a significant increase in insoluble amyloid levels [F(1,17) = 17.88, p = 0.0006] in 5xFAD/CB2EGFP/f/f mice, while 5xFAD/CB2−/− mice exhibited decreased levels of this peptide [F(1,17) = 209.3, p < 0.0001].
In the present manuscript we report a significant role of cannabinoid CB2 receptors in microglial functions and in the metabolism of Aβ in an animal model of Alzheimer’s disease (5xFAD). Specifically, we found that the absence of CB2 receptors decrease the total number of microglial cells as well as their ability to phagocytose Aβ and have a modulatory role in the accumulation of the insoluble form of this pathogenic peptide. Furthermore, our data suggest that microglial CB2 receptors may be constitutively activated in the context of AD, as indicated by p38 phosphorylation state.
Our present data confirm the increased expression of cannabinoid CB2 receptors in plaque-associated microglia (Benito et al., 2007a). By using electronic microscopy, we have observed that the presence of EGFP (expressed under the control of the Cnr2 promoter region) was enhanced specifically in these cells, in 5xFAD/CB2EGFP/f/f mice, while in controls remained low or undetectable in microglial cells as well as in other cell types (such as neurons or astrocytes). These observations match with previous data obtained from our group (Benito et al., 2007b; López et al., 2018) and from others (Savonenko et al., 2015; Spangenberg et al., 2019) and confirm the selective expression of CB2 receptors in activated microglial cells in the context of the chronic neuroinflammation triggered by amyloid accumulation.
These findings allow us to assume that the changes in signal transduction cascades observed after treatment with the CB2 agonist, RO6866945, or after the genetic deletion of the receptor are mostly derived from the modulation of microglial cells, although the contribution of other receptor’s populations located in different types of cells cannot be completely ruled out. It might be possible that, even when expressed at very low levels, CB2 receptors could modulate the activity of neurons and/or astrocytes, as has been reported by other authors (Onaivi et al., 2008; Espejo-Porras et al., 2019). In our hands, however, microglial CB2 receptors must play a major role in the observed changes in cAMP, CREB and p38MAPK signaling cascades. Interestingly, p38MAPK regulation might be under the tonic influence of CB2 receptors, as its activity was significantly reduced in CB2-lacking mice.
These data match with those recently reported by Reusch et al. (Reusch et al., 2022) regarding microglial phagocytosis and signaling cascade (p38MAPK) profiles. By using cultures of BV-2 and primary microglia cells, these authors found that CB2 receptors are necessary for TLR-mediated activation, as shown by gene transcription, morphological and functional (LPS/IFN-γ, CpG and Polyl:C stimulation) analysis and that p38MPK signaling was directly involved in the CB2-mediated regulation of TLR function. Thus, primary neonatal microglia from CB2-/- exhibited a dysregulation of this intracellular route at the transcriptional level that was especially evident after challenge with LPS/IFN-γ and Polyl:C, with a significant reduction in the phosphorylation level of p38. Furthermore, the significant decrease in the phagocytic activity of CB2-lacking microglia we herein report may be also associated to the loss of TLR function, as these receptors are well-known for their critical role in the uptake and clearance of amyloid by these cells (Tahara et al., 2006). Finally, our present observations match well with our previously published study in which a decrease in methoxy-X04+ plaques in 5xFAD/CB2−/− mice was found (López et al., 2018).
As a limitation of the present studies, only male mice were employed. The question on putative sex differences in the 5xFAD model has been recently addressed by Forner et al. (Forner et al., 2021). These authors performed a comprehensive analysis of pathology-associated changes in male and female 5xFAD mice and found that female mice develop the disease at an earlier age, exhibit more significant weight loss, higher levels of insoluble Aβ and improved motor performance in the rotarod test than their males counterparts. A trend to increased microgliosis was also observed.
The current view on the pathogenesis of AD indicates that the accumulation of Aβ is one of the main hallmarks of this disease, together with the formation of tau-enriched neurofibrillary tangles (Querfurth and LaFerla, 2010). Both factors contribute to a significant loss of active synaptic connections in the cortex and hippocampus, triggering the well-known symptoms of this disease, such as memory loss, cognitive decline, etc. The animal model that we have employed in the present studies exhibits an enhanced amyloidogenic status, leading to the production of increased amounts of Aβ at early stages of the mouse’s lifespan and to the formation of neuritic plaques as early as 3 months of age (Oakley et al., 2006).
Though still controversial, the role of microglia in the formation and accumulation of amyloid-enriched neuritic plaques seems very relevant (Song and Colonna, 2018). It is thought that, in the context of AD and as the presence of increased species of Aβ extends in time, activated microglia become a relevant contributor to neuronal damage mainly by secreting elevated amounts of cytokines, ROS, and other mediators, and by losing their ability to phagocytose and degrade these pathological peptides (Song and Colonna, 2018). Importantly, microglia are thought to perform a “shielding” task by effectively surrounding neuritic plaques and thus preventing the expansion of the damage in the brain parenchyma (Condello et al., 2015). The complex role of microglia has been recently highlighted by recent reports showing that in vivo depletion of microglia (for instance, by the administration of antagonists of colony stimulating factor receptor-1, CSFR1, to mice) significantly alters plaque dynamics in the mouse brain. Spangenberg et al. (2019) and, very recently, Casali et al. (2020) have shown that microglia depletion prevents the formation of Aβ-enriched neuritic plaques and that microglia restoration favors its compact structure (Spangenberg et al., 2019; Casali et al., 2020). Our observations that CB2-deficient AD mice exhibit a decreased phagocytic activity combined with a decrease in cortical insoluble amyloid levels are suggestive of a role of CB2 receptors in plaque dynamics, in which their activation could contribute to a compaction of amyloid plaques while their deletion could lead to a more diffuse appearance. A similar effect has been described in TREM2-lacking mice, suggesting an impairment in microglial function (Wang et al., 2016).
Finally, it is important to note that other authors have reported conflicting data associated to the genetic deletion of cannabinoid CB2 receptors, different to those reported here. Koppel et al. (2014) used J20 APP mice to study the effects of CB2 genetic inactivation and found increased levels of soluble amyloid and plaques as well as enhanced plaque-associated microgliosis. In line with these data, Aso et al. (2016) also found significant increases in Aβ1-40 as well as in amyloid deposition in the APP/PS1 mouse model of AD. Wu et al. (2017) reported a stimulatory effect of a CB2 agonist on amyloid clearance combined with decreased microgliosis in the hippocampus of APP/PS1 mice. Conversely, Schmöle et al. (2015); Schmöle et al. (2018) found decreased microgliosis and amyloid levels as a consequence of CB2 deletion in APP/PS1 mice. Most of these studies also revealed no CB2-mediated effects on spatial memory. This variability regarding the effects of cannabinoid CB2 receptors may be partially explained by the variety of AD mouse models employed in these studies but may be also suggestive of the subtle and limited effects of modulating the activity of these receptors, as well as may reflect putative adaptive responses in constitutive knock-out models.
We have confirmed (by immunoelectron microscopy) microglia as the main source of cannabinoid CB2 receptors in the 5xFAD/CB2EGFP/f/f mouse model of AD. In addition, we have found that these receptors regulate the ability of these cells to phagocytose amyloid peptides in vivo and, probably in direct relation with this, in the composition of amyloid species in the brain. These data thus suggest a role for microglial cannabinoid CB2 receptors in the initiation, maintenance and removal of plaques and open new venues for the microglia-based therapeutic approaches in AD.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Universidad Francisco de Vitoria.
PG, MTG, and JR designed the experiments; SRdME, IB-C, IT, AMM-R, MAA, GR-P, and IE carried out the experiments; CK, CJH, CR, RCS, MVW, EMC, and UG provided materials. PG, UG, and JR wrote the manuscript. SRdME and IB-C contributed equally to this work. All authors revised the final version of the manuscript.
The present work has been supported by a grant from Ministerio de Ciencia e Innovación (ref PID2019-108992RB-I00 and ref PID2019-107548RB-I00) to JR and PG, respectively, by the Basque Government (ref IT1230-19) to PG, and the Research and Education Component of the Advancing a Healthier Wisconsin Endowment at the Medical College of Wisconsin to CJH.
Authors CK, CR and UG were employed by the company F. Hoffmann-La Roche Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aβ, Amyloid beta; AD, Alzheimer’s Disease; ALS, Amyotrophic Lateral Sclerosis; APP, Amyloid Precursor Protein; BACE1, Beta-site Amyloid Precursor Protein Cleaving Enzyme 1; cAMP, Cyclic adenosine monophosphate; CNS, Central Nervous System; CREB, cAMP Response Element-Binding; CSFR1, Colony Stimulating Factor Receptor-1; EGFP, Enhanced Green Fluorescent Protein; ERK1/2, Extracellular Signal-Regulated Kinases ½; GABA-A, Gamma-Amino Butyric Acid Receptor Subunit Alpha-1; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; GPCRs, G-protein coupled receptors; HRP, Horseradish Peroxidase; Iba1, Ionized Calcium Binding Adapter Molecule 1; MAPK, Mitogen-Activated Protein Kinases; TREM2, Triggering Receptor Expressed On Myeloid Cells 2.
Aso, E., Andrés-Benito, P., Carmona, M., Maldonado, R., and Ferrer, I. (2016). Cannabinoid Receptor 2 Participates in Amyloid-β Processing in a Mouse Model of Alzheimer's Disease but Plays a Minor Role in the Therapeutic Properties of a Cannabis-Based Medicine. J. Alzheimers Dis. 51, 489–500. doi:10.3233/JAD-150913
Benito, C., Núñez, E., Pazos, M. R., Tolón, R. M., and Romero, J. (2007a). The Endocannabinoid System and Alzheimer's Disease. Mol. Neurobiol. 36, 75–81. doi:10.1007/s12035-007-8006-8
Benito, C., Núñez, E., Tolón, R. M., Carrier, E. J., Rábano, A., Hillard, C. J., et al. (2003). Cannabinoid CB2 Receptors and Fatty Acid Amide Hydrolase Are Selectively Overexpressed in Neuritic Plaque-Associated Glia in Alzheimer's Disease Brains. J. Neurosci. 23, 11136–11141. doi:10.1523/jneurosci.23-35-11136.2003
Benito, C., Romero, J. P., Tolón, R. M., Clemente, D., Docagne, F., Hillard, C. J., et al. (2007b). Cannabinoid CB1 and CB2 Receptors and Fatty Acid Amide Hydrolase are Specific Markers of Plaque Cell Subtypes in Human Multiple Sclerosis. J. Neurosci. 27, 2396–2402. doi:10.1523/JNEUROSCI.4814-06.2007
Cabañero, D., Ramírez-López, A., Drews, E., Schmöle, A., Otte, D. M., Wawrzczak-Bargiela, A., et al. (2020). Protective Role of Neuronal and Lymphoid Cannabinoid CB2 Receptors in Neuropathic Pain. Elife 9, e55582. doi:10.7554/eLife.55582
Casali, B. T., MacPherson, K. P., Reed-Geaghan, E. G., and Landreth, G. E. (2020). Microglia Depletion Rapidly and Reversibly Alters Amyloid Pathology by Modification of Plaque Compaction and Morphologies. Neurobiol. Dis. 142, 104956. doi:10.1016/j.nbd.2020.104956
Condello, C., Yuan, P., Schain, A., and Grutzendler, J. (2015). Microglia Constitute a Barrier that Prevents Neurotoxic Protofibrillar Aβ42 Hotspots Around Plaques. Nat. Commun. 6, 6176. doi:10.1038/ncomms7176
Deczkowska, A., Keren-Shaul, H., Weiner, A., Colonna, M., Schwartz, M., and Amit, I. (2018). Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173, 1073–1081. doi:10.1016/j.cell.2018.05.003
Espejo-Porras, F., García-Toscano, L., Rodríguez-Cueto, C., Santos-García, I., de Lago, E., and Fernandez-Ruiz, J. (2019). Targeting Glial Cannabinoid CB2 Receptors to Delay the Progression of the Pathological Phenotype in TDP-43 (A315T) Transgenic Mice, a Model of Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 176, 1585–1600. doi:10.1111/bph.14216
Forner, S., Kawauchi, S., Balderrama-Gutierrez, G., Kramár, E. A., Matheos, D. P., Phan, J., et al. (2021). Systematic Phenotyping and Characterization of the 5xFAD Mouse Model of Alzheimer's Disease. Sci. Data 8, 270. doi:10.1038/s41597-021-01054-y
Galán-Ganga, M., Rodríguez-Cueto, C., Merchán-Rubira, J., Hernández, F., Ávila, J., Posada-Ayala, M., et al. (2021). Cannabinoid Receptor CB2 Ablation Protects against TAU Induced Neurodegeneration. Acta Neuropathol. Commun. 9, 90. doi:10.1186/s40478-021-01196-5
Hampel, H., Hardy, J., Blennow, K., Chen, C., Perry, G., Kim, S. H., et al. (2021). The Amyloid-β Pathway in Alzheimer's Disease. Mol. Psychiatry 26, 5481–5503. doi:10.1038/s41380-021-01249-0
He, X. H., Galaj, E., Bi, G. H., He, Y., Hempel, B., Wang, Y. L., et al. (2021). β-Caryophyllene, an FDA-Approved Food Additive, Inhibits Methamphetamine-Taking and Methamphetamine-Seeking Behaviors Possibly via CB2 and Non-CB2 Receptor Mechanisms. Front. Pharmacol. 12, 722476. doi:10.3389/fphar.2021.722476
Komorowska-Müller, J. A., and Schmöle, A.-C. (2020). CB2 Receptor in Microglia: The Guardian of Self-Control. Int. J. Mol. Sci. 22, 19. doi:10.3390/ijms22010019
Koppel, J., Vingtdeux, V., Marambaud, P., d'Abramo, C., Jimenez, H., Stauber, M., et al. (2014). CB2 Receptor Deficiency Increases Amyloid Pathology and Alters Tau Processing in a Transgenic Mouse Model of Alzheimer's Disease. Mol. Med. 20, 29–36. doi:10.2119/molmed.2013.00140.revised
López, A., Aparicio, N., Pazos, M. R., Grande, M. T., Barreda-Manso, M. A., Benito-Cuesta, I., et al. (2018). Cannabinoid CB2 Receptors in the Mouse Brain: Relevance for Alzheimer's Disease. J. Neuroinflammation 15, 158. doi:10.1186/s12974-018-1174-9
Maresz, K., Carrier, E. J., Ponomarev, E. D., Hillard, C. J., and Dittel, B. N. (2005). Modulation of the Cannabinoid CB2 Receptor in Microglial Cells in Response to Inflammatory Stimuli. J. Neurochem. 95, 437–445. doi:10.1111/j.1471-4159.2005.03380.x
Mecha, M., Carrillo-Salinas, F. J., Feliú, A., Mestre, L., and Guaza, C. (2016). Microglia Activation States and Cannabinoid System: Therapeutic Implications. Pharmacol. Ther. 166, 40–55. doi:10.1016/j.pharmthera.2016.06.011
Mechoulam, R., and Parker, L. A. (2013). The Endocannabinoid System and the Brain. Annu. Rev. Psychol. 64, 21–47. doi:10.1146/annurev-psych-113011-143739
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science 308, 1314–1318. doi:10.1126/science.1110647
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal Beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer's Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 26, 10129–10140. doi:10.1523/JNEUROSCI.1202-06.2006
Onaivi, E. S., Ishiguro, H., Gong, J. P., Patel, S., Meozzi, P. A., Myers, L., et al. (2008). Functional Expression of Brain Neuronal CB2 Cannabinoid Receptors are Involved in the Effects of Drugs of Abuse and in Depression. Ann. N. Y. Acad. Sci. 1139, 434–449. doi:10.1196/annals.1432.036
Ouali Alami, N., Schurr, C., Olde Heuvel, F., Tang, L., Li, Q., Tasdogan, A., et al. (2018). NF-κB Activation in Astrocytes Drives a Stage-Specific Beneficial Neuroimmunological Response in ALS. EMBO J. 37 (16), e98697. doi:10.15252/embj.201798697
Pertwee, R. G., Howlett, A. C., Abood, M. E., Alexander, S. P., Di Marzo, V., Elphick, M. R., et al. (2010). International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: beyond CB₁ and CB₂. Pharmacol. Rev. 62, 588–631. doi:10.1124/pr.110.003004
Puente, N., Río, I. B., Achicallende, S., Nahirney, P. C., and Grandes, P. (2019). High-Resolution Immunoelectron Microscopy Techniques for Revealing Distinct Subcellular Type 1 Cannabinoid Receptor Domains in Brain. Bio Protoc. 9, e3145. doi:10.21769/BioProtoc.3145
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer's Disease. N. Engl. J. Med. 362, 329–344. doi:10.1056/NEJMra0909142
Reusch, N., Ravichandran, K. A., Olabiyi, B. F., Komorowska-Müller, J. A., Hansen, J. N., Ulas, T., et al. (2022). Cannabinoid Receptor 2 Is Necessary to Induce Toll-like Receptor-Mediated Microglial Activation. Glia 70, 71–88. doi:10.1002/glia.24089
Rodríguez-Cueto, C., Gómez-Almería, M., García Toscano, L., Romero, J., Hillard, C. J., Lago, E., et al. (2021). Inactivation of the CB 2 Receptor Accelerated the Neuropathological Deterioration in TDP‐43 Transgenic Mice, a Model of Amyotrophic Lateral Sclerosis. Brain Pathol. 31, e12972. doi:10.1111/bpa.12972
Sarott, R. C., Westphal, M. V., Pfaff, P., Korn, C., Sykes, D. A., Gazzi, T., et al. (2020). Development of High-Specificity Fluorescent Probes to Enable Cannabinoid Type 2 Receptor Studies in Living Cells. J. Am. Chem. Soc. 142, 16953–16964. doi:10.1021/jacs.0c05587
Savonenko, A. V., Melnikova, T., Wang, Y., Ravert, H., Gao, Y., Koppel, J., et al. (2015). Cannabinoid CB2 Receptors in a Mouse Model of Aβ Amyloidosis: Immunohistochemical Analysis and Suitability as a PET Biomarker of Neuroinflammation. PLoS One 10, e0129618. doi:10.1371/journal.pone.0129618
Schmöle, A. C., Lundt, R., Ternes, S., Albayram, Ö., Ulas, T., Schultze, J. L., et al. (2015). Cannabinoid Receptor 2 Deficiency Results in Reduced Neuroinflammation in an Alzheimer's Disease Mouse Model. Neurobiol. Aging 36, 710–719. doi:10.1016/j.neurobiolaging.2014.09.019
Schmöle, A. C., Lundt, R., Toporowski, G., Hansen, J. N., Beins, E., Halle, A., et al. (2018). Cannabinoid Receptor 2-Deficiency Ameliorates Disease Symptoms in a Mouse Model with Alzheimer's Disease-Like Pathology. J. Alzheimers. Dis. 64, 379–392. doi:10.3233/JAD-180230
Song, W. M., and Colonna, M. (2018). The Identity and Function of Microglia in Neurodegeneration. Nat. Immunol. 19, 1048–1058. doi:10.1038/s41590-018-0212-1
Spangenberg, E., Severson, P. L., Hohsfield, L. A., Crapser, J., Zhang, J., Burton, E. A., et al. (2019). Sustained Microglial Depletion with CSF1R Inhibitor Impairs Parenchymal Plaque Development in an Alzheimer's Disease Model. Nat. Commun. 10, 3758. doi:10.1038/s41467-019-11674-z
Tahara, K., Kim, H. D., Jin, J. J., Maxwell, J. A., Li, L., and Fukuchi, K. (2006). Role of Toll-Like Receptor Signalling in Abeta Uptake and Clearance. Brain 129, 3006–3019. doi:10.1093/brain/awl249
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-Mediated Early Microglial Response Limits Diffusion and Toxicity of Amyloid Plaques. J. Exp. Med. 213, 667–675. doi:10.1084/jem.20151948
Wu, J., Hocevar, M., Foss, J. F., Bie, B., and Naguib, M. (2017). Activation of CB2 Receptor System Restores Cognitive Capacity and Hippocampal Sox2 Expression in a Transgenic Mouse Model of Alzheimer's Disease. Eur. J. Pharmacol. 811, 12–20. doi:10.1016/j.ejphar.2017.05.044
Keywords: cannabinoids, CB2 receptor, amyloid, Alzheimer’s disease, microglia
Citation: Ruiz de Martín Esteban S, Benito-Cuesta I, Terradillos I, Martínez-Relimpio AM, Arnanz MA, Ruiz-Pérez G, Korn C, Raposo C, Sarott RC, Westphal MV, Elezgarai I, Carreira EM, Hillard CJ, Grether U, Grandes P, Grande MT and Romero J (2022) Cannabinoid CB2 Receptors Modulate Microglia Function and Amyloid Dynamics in a Mouse Model of Alzheimer’s Disease. Front. Pharmacol. 13:841766. doi: 10.3389/fphar.2022.841766
Received: 22 December 2021; Accepted: 30 March 2022;
Published: 27 April 2022.
Edited by:
Maria Grazia Morgese, University of Foggia, ItalyReviewed by:
Agnes Nadjar, Université de Bordeaux, FranceCopyright © 2022 Ruiz de Martín Esteban, Benito-Cuesta, Terradillos, Martínez-Relimpio, Arnanz, Ruiz-Pérez, Korn, Raposo, Sarott, Westphal, Elezgarai, Carreira, Hillard, Grether, Grandes, Grande and Romero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julián Romero, Si5yb21lcm8ucHJvZkB1ZnYuZXM=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.