 Angela V. Berry
Angela V. Berry Joseph L. Kuti
Joseph L. Kuti
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 18 March 2022
Sec. Translational Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.833189
This article is part of the Research TopicPharmacokinetics, Pharmacodynamics (PK/PD) of Antibiotics: A Reality CheckView all 9 articles
Beta-lactams remain a critical member of our antibiotic armamentarium and are among the most commonly prescribed antibiotic classes in the inpatient setting. For these agents, the percentage of time that the free concentration remains above the minimum inhibitory concentration (%fT > MIC) of the pathogen has been shown to be the best predictor of antibacterial killing effects. However, debate remains about the quantity of fT > MIC exposure needed for successful clinical response. While pre-clinical animal based studies, such as the neutropenic thigh infection model, have been widely used to support dosing regimen selection for clinical development and susceptibility breakpoint evaluation, pharmacodynamic based studies in human patients are used validate exposures needed in the clinic and for guidance during therapeutic drug monitoring (TDM). For the majority of studied beta-lactams, pre-clinical animal studies routinely demonstrated the fT > MIC should exceed approximately 40–70% fT > MIC to achieve 1 log reductions in colony forming units. In contrast, clinical studies tend to suggest higher exposures may be needed, but tremendous variability exists study to study. Herein, we will review and critique pre-clinical versus human-based pharmacodynamic studies aimed at determining beta-lactam exposure thresholds, so as to determine which targets may be best suited for optimal dosage selection, TDM, and for susceptibility breakpoint determination. Based on our review of murine and clinical literature on beta-lactam pharmacodynamic thresholds, murine based targets specific to each antibiotic are most useful during dosage regimen development and susceptibility breakpoint assessment, while a range of exposures between 50 and 100% fT > MIC are reasonable to define the beta-lactam TDM therapeutic window for most infections.
Beta-lactams are among the most prescribed antibiotic classes in the hospital setting, as these drugs are frequently used as prophylaxis and as empiric therapy for pneumonia, bloodstream infections, intra-abdominal infections, and infections of the urinary tract (Holten and Onusko, 2000; Bush and Bradford, 2016). For instance, penicillin-based antibiotics are the most frequently used antibiotic class in the ICU and accounted for 36% of all antibiotic use (Vincent et al., 2020). In support of their robust utilization, beta-lactams are frequently recommended by national and international treatment guidelines, and have been prescribed for many indications that correspond with their utilization (Holten and Onusko, 2000; Guilhaumou et al., 2019).
Despite their robust indications and frequent utilization, therapeutic failures among beta-lactam antibiotics, like any antibiotic, are not completely unavoidable. Reasons for clinical failure are often related to host issues (e.g., source control, underlying co-morbidities, and severity of infection) or lack of optimal exposure due to lower than anticipated concentrations, bacterial resistance, or both. Moreover, the variability in beta-lactam pharmacokinetics that can be observed across certain patient populations suggests additional efforts are sometimes required to ensure optimal exposure, even against susceptible organisms. Therapeutic drug monitoring (TDM) is one approach used clinically to optimize drug exposure while minimizing toxicity (Reeves et al., 2016). Currently, beta-lactam TDM is relatively uncommon, but there is gaining interest in its role and implementation given the diversity of indications and patient types for which these drugs are utilized (Roberts et al., 2010). TDM is also performed for other antimicrobials, including aminoglycosides, voriconazole, and vancomycin (Abdul-Aziz et al., 2020).
Well before a beta-lactam is ever available for prescription to patients, however, pre-clinical models are conducted to understand in vivo efficacy and establish exposure response relationships (Waack et al., 2020). These preclinical models, including the classic neutropenic murine thigh infection model, demonstrate the killing profile of the antibiotic versus a pathogen (concentration-dependent versus concentration-independent (i.e., time-dependent) killing, the pharmacodynamic index that best predicts killing [e.g., time that free drug concentrations remain above the minimum inhibitory concentration (fT > MIC)], and quantify the exposure thresholds needed for specific microbiological endpoints. These data are paramount to successful establishment of an optimal dosing regimen for further clinical development, as well as support provisional susceptibility breakpoints (Bulitta et al., 2019). Dosing regimens developed based on supportive pre-clinical animal models increases the likelihood that drug candidates are successful in phase 3 clinical trials. Once approved and available for clinical use, beta-lactam antibiotics are then often prescribed to treat infections in patient populations not included in original phase 3 studies, as well as for organisms resistant to prior standard of care. As such, these more challenging indications for use of beta-lactams beg the question of whether approved dosing regimens remain sufficient and if similar pharmacodynamic thresholds apply. Therefore, human pharmacodynamic studies are requisite to confirm pre-clinical observations and to further guide management of serious infections in critically ill patients via TDM. Herein, we will summarize and critique pre-clinical and human-based pharmacodynamic studies of beta-lactams developed after year 2000 with the aim of comparing beta-lactam exposure thresholds required to achieve defined endpoints. We searched Pubmed/Medline with search terms (beta-lactams, pharmacodynamics, murine, thigh-infection, pneumonia, and time above the MIC). We identified all beta-lactams approved after 2000 and restricted studies to these agents for our murine review. We included all human studies for any beta-lactam published after year 2000. The citation list of included papers was also reviewed to identify studies missed during initial search.
In vitro and in vivo animal studies consistently demonstrate that fT > MIC is the pharmacodynamic parameter that best describes the killing profile of beta-lactam antibiotics (Vogelman and Craig, 1986; Reed, 2000; Lodise et al., 2006). In fact this has been established since the first beta-lactam was discovered. Eagle and others first demonstrated that longer “continuous” infusions of penicillin G led to improved microbiologic success against Streptococcus pyogenes in a mouse infection model in 1953 (Eagle et al., 1953). But it wasn’t until the 1980s that Craig and Ebert (Leggett et al., 1989) introduced the neutropenic murine thigh and lung infection models tools to identify thresholds required for specific antibacterial endpoints. Exposures generated from these experiments were analyzed using a non-linear concentration (or dose) response model, referred to as the maximal effect (Emax) and sigmoidal Emax model (Holford, 2017). Fitting a model to the exposure effect data leads to reasonable estimates of effect of increasing exposure (Figure 1).
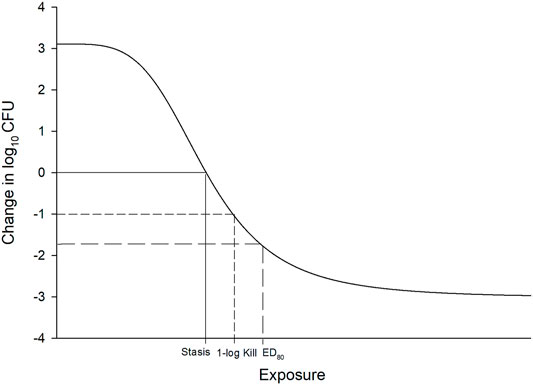
FIGURE 1. Example of sigmoidal Emax model used to define exposure response relationship in pre-clinical and clinical studies. The x-axis is typically drug exposure (e.g., dose, concentration, T > MIC) and the y-axis is effect (i.e., change in log10 CFU from baseline). Stasis, 1 log10 CFU reduction, and ED80 requirements are demonstrated.
The most common endpoints utilized in these pharmacodynamic studies include the exposure required for stasis, 1-log10 reductions in colony forming units (CFU), and 2-log10 CFU reductions after 24 h on the Emax curve. Alternative endpoints such as the effective dose (ED) or exposure (EE) that reduces the bacterial population by a certain percentage (e.g., 50, 80, and 90%) from baseline have also been proposed. However, based on guidance by the European Medicine’s Agency (EMA) and Food and Drug Administration (FDA), most antibiotic developers seek to develop a dosage that provides the most rapid and greatest amount of killing in the model, but this must be balanced with risks of toxicity and cost of goods (Bulitta et al., 2019). Notably, many antibiotics are incapable of achieving ≥2 log10 reductions over 24 h if they are “slower killing” agents or have higher MICs against a specific targeted pathogen. As a result, static and 1 log10 CFU reductions are the most referenced thresholds used during pre-clinical development, with stasis generally reserved for less serious infections such as complicated urinary tract infections and acute bacterial skin and skin structure infection, while ≥1-log10 CFU reductions are targeted for more serious infections including pneumonia and bloodstream infections. The application of these pharmacodynamic thresholds combined with Monte Carlo simulation to identify optimal dosing of antibiotics for human studies has led to successful application to the FDA for respiratory tract infections (Bulik et al., 2013).
The earliest pre-clinical studies of beta-lactam antibiotics partitioned these drugs by their ring structure class. Against Gram-negative and many Gram-positive bacteria, cephalosporin based beta-lactams typically required 60–70% fT > MIC for 1–2 log10 CFU reductions and 30–40% fT > MIC for stasis (e.g., cefotaxime against Klebsiella pneumoniae in the neutropenic murine lung infection model) (Craig, 1998). In contrast, penicillin based beta-lactams generally required less (i.e., 50–60% fT > MIC) for maximal effects, while carbapenems required approximately 40% fT > MIC due to a longer post antibiotic effect (Turnidge, 1998). Similar reductions in static exposure requirements were observed.
Studies over the last 20 years have validated these observations in the neutropenic murine infection models. The efficacy and exposure-response of ceftaroline, the first approved cephalosporin with activity against methicillin-resistant Staphylococcus aureus (MRSA), was first evaluated in the murine thigh and lung infection models against S. aureus, Streptococcus pneumoniae, Escherichia coli, and K. pneumoniae (Andes and Craig, 2006). Stasis, 1-log10, and 2-log10 CFU reductions were observed at ceftaroline exposures of 26 ± 8%, 33 ± 9%, and 45 ± 13% fT > MIC against the S. aureus isolates, respectively. These values were 39 ± 9%, 43 ± 9%, and 50 ± 10% for S. pneumoniae. Finally against the Gram-negative organisms combined, 28 ± 9% fT > MIC was required for stasis, and 41 ± 11% was required for 1 log10 CFU reductions. Notably, 2 log10 reductions were not achieved for all Gram-negative isolates, although a mean of 54% was reported for the isolates that did. Later, ceftaroline was evaluated as a human simulated regimen (equivalent to 600 mg q12h) in the lung infection model. Seventeen S. aureus isolates (2 MSSA, 15 MRSA) with ceftaroline MICs between 0.5 and 4 mg/L were studied. The composite Emax curve identified 16% fT > MIC as the threshold required for stasis and 41% fT > MIC for
Studies conducted of a second anti-MRSA cephalosporin, ceftobiprole, which is approved in Europe and Canada, but not yet the United States, demonstrated bacteriostatic effects when the fT > MIC was 36–45%, 14–28%, and 15–22% against Enterobacterales, S. aureus, and S. pneumoniae, respectively, in the neutropenic thigh infection model (Craig and Andes, 2008). Although thresholds for 1 log10 reductions were not provided, the mean fT > MIC required for 2 log10 CFU reductions were 64.5, 29.3, and 25.8%, respectively. For isolates that also grew in the pneumonia model, plasma exposure requirements were identical between infection sites. Notably, this was because of very high penetration of ceftobiprole into the epithelial lining fluid of mice, a trait that did not translate to humans, thereby leading to poor pulmonary exposures during phase 3 studies and inferior clinical outcomes (Rodvold et al., 2009).
The pharmacodynamics of the potent anti-Pseudomonas cephalosporin, ceftolozane, was determined using the neutropenic thing infection model (Craig and Andes, 2013). Against wild-type E. coli and K. pneumoniae, stasis and 1-log CFU reductions were achieved at 26.3 ± 2.1% and 31.6 ± 1.65 T > MIC, respectively. These exposure requirements were 25.5 ± 2.8% and 31.5 ± 2.8% for Pseudomonas aeruginosa. While total drug exposures were reported in the study, protein binding was less than 5% in the mice. Clinically, ceftolozane is only available combined with tazobactam to add stability against hydrolysis from extended spectrum beta-lactamases (ESBL). Tazobactam was added in 2:1, 4:1, and 8:1 ratios to the ceftolozane doses and all data were analyzed together against ESBL producing Enterobacterales. Stasis and 1 log10 CFU reductions were observed at 31.1 and 34.8% T > MIC, respectively. Notably, the reported 1 log thresholds for ceftolozane were numerically lower than required for previous anti-Pseudomonas cephalosporins, so a follow up study including 14 P. aeruginosa with ceftolozane MICs ranging from 2–16 mg/L was conducted (Lepak et al., 2014). Stasis and 1 log thresholds were increased to 31.2 ± 6.9% and 39.4 ± 7.5% T > MIC, values still lower than historical cephalosporins that require 60–70% fT > MIC for similar activity. Although 2 log thresholds were reported to be ∼42% T > MIC, this was only achieved (i.e., Emax) against five of the 14 isolates, and is therefore not representable of all isolates included in the study.
Cefiderocol is the first approved siderophore cephalosporin antibiotic. Its pharmacodynamics were studied in a number of murine thigh and lung infection models against various multi-drug resistant Gram-negative pathogens of interest (Katsube et al., 2019). Against a wild-type P. aeruginosa (cefiderocol MIC: 0.25 mg/L, cefepime MIC: 1 mg/L), stasis and 1 log10 CFU reductions were achieved at 47.5 and 57.6% fT > MIC in the murine thigh infection model (Nakamura et al., 2019). For comparison, cefepime required 61.7 and 87.7% fT > MIC for similar activity against the isolate. Pharmacodynamic analyses of cefiderocol against additional Enterobacterales and P. aeruginosa, including carbapenem resistant strains, resulted in stasis requirements of 62.5 ± 27.4% and 63 ± 15.5% fT > MIC, respectively; 1 log10 CFU reductions were observed with fT > MIC of 73.3 ± 23.3% and 72.2 ± 21.4%. T > MIC requirements were greater for carbapenem resistant isolates (85.2%) compared with carbapenem susceptible isolates (61.3%). In the lung infection model, 1 log10 CFU reductions were achieved with thresholds of 64.4 ± 22.5%, 70.3 ± 9.0%, 88.1 ± 3.4% and 53.9 ± 18.1% fT > MIC against Enterobacterales, P. aeruginosa, Acinetobacter baumannii, and Stenotrophomonas maltophilia, respectively. A similar cefiderocol fT > MIC target (∼81%) was documented in a separate murine thigh infection model using against eight P. aeruginosa (Ghazi et al., 2018).
Carbapenems have historically been reported to require lower T > MIC thresholds compared with other beta-lactams due to a prolonged post-antibiotic effect. Over the last 20 years, two carbapenems were developed and successfully approved. Ertapenem pharmacodynamics in the murine thigh infection model observed stasis requirements of 19% (range: 2–38) fT > MIC against E. coli and K. pneumoniae, including ESBL producing strains (Maglio et al., 2005). Although 1 log10 reductions were not reported, the ED80 exposure requirement was 33% (range: 13–65) fT > MIC. Notably, exposure requirements were similar between ESBL producing and non-ESBL producing isolates. Doripenem was studied in the neutropenic murine thigh infection model against 24 clinical P. aeruginosa isolates with MICs ranging from 0.125 to 16 mg/L (Kim et al., 2008). Using an Emax curve comprised of data for all 24 P. aeruginosa isolates included, stasis, 1 log10, and 2 log10 CFU reductions were observed at ∼20, 30, and 40% fT > MIC, respectively. Similar exposure requirements have been reported for carbapenems against Acinetobacter spp. The pharmacodynamics of meropenem, imipenem, and doripenem were studied against 14 A. baumannii; stasis, 1 log10, and 2 log10 CFU reductions were achieved at 24, 33, and 48% fT > MIC (Macvane et al., 2014a). A separate thigh infection model identified meropenem fT > MIC requirements of 16.7% (range: 7.3–24.2) for stasis and 26.8% (14.5–36.9%) for 1 log10 CFU reductions against six A. baumannii (Sabet et al., 2020). Two log reductions were achieved in five of the six isolates at 41.6% (range 26–53.2%) fT > MIC.
Many beta-lactams approved in the last decade, as well as the majority of those in pre-clinical development, have aimed to thwart beta-lactamase mediated resistance by combining an older beta-lactam backbone with a novel beta-lactamase inhibitor drug. Ceftazidime/avibactam, meropenem/vaborbactam, and imipenem/cilastatin/relebactam were developed to target multidrug resistant gram-negatives that had developed resistance to their beta-lactam backbone components. With their development, the science of beta-lactamase inhibitor pharmacodynamics has made significant advances (Monogue and Nicolau, 2019).
During development of these agents, the common assumption is that the pharmacodynamics of the backbone beta-lactam remains consistent with the agent when administered alone. Instead, the focus is on what inhibitor exposures are required to potentiate the MIC of the backbone. Meropenem T > MIC requirements for stasis, 1 log10 and 2 log10 reductions were 30, 35, and 45% fT > MIC against Gram-negatives in the neutropenic thigh infection model; however, vaborbactam required a free area under the curve to MIC (fAUC/MIC) of 38 to enable meropenem to achieve 1 log10 kill at 35% fT > MIC against K. pneumoniae producing the KPC-carbapenemase (Food and Drug Administration, 2021). For ceftazidime/avibactam, the values used during development were 50% fT > MIC for ceftazidime and free avibactam concentrations that remained above a critical concentration threshold of 1 mg/L for at least 50% of the dosing interval (50% fT > CT 1 mg/L) (Das et al., 2019; Das et al., 2020). Indeed, studies in the neutropenic thigh infection model of humanized ceftazidime/avibactam (2g/0.5 g every 8 h as 2 h infusion) in lung epithelial lining fluid (ELF) demonstrated ≥1 log10 CFU reductions against P. aeruginosa with MICs up to 32/4 mg/L, which corresponded with ceftazidime exposures ≥19% fT > MIC in ELF (Housman et al., 2014). A subsequent study in the murine thigh and lung infection model identified ceftazidime exposures of 0–29% fT > MIC required for stasis, noting that the ceftazidime MICs of the study isolates were 32–128 mg/L. However, avibactam exposures of 46.9% fT > CT 1 mg/L, when combined with a fixed ceftazidime dosage that resulted in 50% fT > ceftazidime/avibactam MIC, achieved 1 log kill against P. aeruginosa (Berkhout et al., 2016). Finally, for imipenem/cilastatin/relebactam, 40% fT > MIC was assumed to be the imipenem threshold used for 1 log10 CFU reductions, while relebactam exposure targets were fAUC/MIC of 3.3, 4.3, and 7.0 for stasis, 1 log10, and 2 log10 reductions against P. aeruginosa in the murine thigh infection model (Patel et al., 2020).
For the beta-lactams developed and approved since year 2000, neutropenic murine thigh and lung infection models have consistently identified fT > MIC as the pharmacodynamic index associated with CFU reduction at 24 h; furthermore, required exposures have been similar to studies conducted since the 1980s. For the two cephalosporins targeted at Gram-positive bacteria (ceftaroline and ceftobiprole), fT > MIC of 25–40% was required for 1 log10 CFU reductions. For Gram-negative targeted cephalosporins (ceftolozane and cefiderocol), fT > MIC requirements for 1 log10 reductions encompassed a wider range of 30–40% for ceftolozane, 61% for cefiderocol against wild-type gram-negatives, and 85% for cefiderocol against carbapenemase producing Gram-negatives. The carbapenems (ertapenem and doripenem) achieved approximately 1 log10 CFU reductions with exposures of 30–40% fT > MIC. Finally, when combined with novel beta-lactamase inhibitors targeting enzyme mediated resistance, the pharmacodynamic threshold requirements for ceftazidime/avibactam, meropenem/vaborbactam, and imipenem/cilastatin/relebactam were consistent with the beta-lactam backbones alone. However, the pharmacodynamic index of each beta-lactamase inhibitor will vary by drug as well as which beta-lactam backbone it is combined with. With few exceptions, a range of 40–70% fT > MIC was observed for beta-lactams to achieve 1 log10 CFU reductions in the mouse infection models. These data were very useful in defining optimal dosing regimens during drug development and for support of susceptibility breakpoints.
Although mean exposure requirements are similar to those previously reported, strain to strain variability should be better appreciated. Even in these contemporary pharmacodynamic studies, coefficient of variation (CVs) around threshold requirements were routinely 20–30%. It should be noted, however, that exposure requirements exceeding 100% fT > MIC in specific isolates were absent from the majority of these studies. While pharmacokinetic variability among mice in these studies can account for some of this variability, differences within individual isolates still contribute to varying pharmacodynamic thresholds, as demonstrated in in vitro chemostat experiments where drug exposure can be quantified in each individual model (Grupper et al., 2019). As a result, some have suggested that the variability in thresholds observed should be accounted for alongside pharmacokinetic variability and natural MIC variation when conducting Monte Carlo simulations to guide optimal dosing and susceptibility breakpoints (Soon et al., 2013).
While original dosing for most antibiotics is primarily guided by pre-clinical studies, the use of these drugs in patient populations who were excluded from original regulatory clinical trials may complicate confidence in the dosing regimen. These patients may have different pharmacokinetics due to underlying illness, co-morbidities, or life-saving treatment modalities (Roberts et al., 2014a). They may have different infection sources or be infected with difficult to treat/more resistant pathogens. Collectively, these challenges are responsible for endorsement of beta-lactam TDM and individualized dosing (Roberts et al., 2010; Fratoni et al., 2021). The question then remains, if one is to pursue TDM, what exposures should be targeted to increase the likelihood of success? The pre-clinical exposure thresholds that generated many of the dosing strategies to begin with and have a part in influencing susceptibility breakpoints are a reasonable start. But clinical studies are valuable to validate or further guide exposure requirements aimed at achieving successful clinical response.
Critically ill patients receiving “standard” beta-lactam doses appear to be less likely to achieve pharmacodynamic thresholds; however, many of these studies use high exposure targets such as 100% T > MIC or even 100% T > 4x MIC (i.e., a minimum concentration that is four fold higher than the MIC for the entire duration of the dosing interval). The DALI (Defining Antibiotic Levels in Intensive Care Patients) study was a prospective, multicenter, pharmacokinetic point-prevalence study that sought to define beta-lactam exposure variability in 248 critically-ill patients (Roberts et al., 2014b). As this was among the first studies to quantify exposure attainment in critically ill patients, several thresholds were defined a priori, including 50 and 100% fT > MIC, as well as more aggressive thresholds of 50 and 100% fT>4xMIC. The investigators observed that 16% of infected patients did not achieve at least 50% fT > MIC, and 39.6% of all patients had exposures less than 100% fT > MIC. Patients who did not achieve at least 50% fT > MIC exposures were 32% less likely to have a positive clinical outcome. Although the authors preferred utilization of the more aggressive thresholds, the receiver operator curves (ROC) suggested no differences in predicting clinical failures between 50 and 100% fT > MIC.
The EXPAT trial, a prospective, observational study that included 147 ICU patients receiving various beta-lactams, found that 63.3% of patients achieved 100% fT > MIC while only 36.7% attained 100% fT>4xMIC (Abdulla et al., 2020). Lower exposure targets were not assessed. Male gender, a larger glomerular filtration rate (i.e., >90 ml/min/1.73m2), and larger body mass increased the risk of non-target attainment. However, neither threshold was significantly associated with 30 days survival. A number of additional point-prevalence type studies have demonstrated low rates of achieving beta-lactam exposure targets but none of these studies have attempted to link clinical outcomes with exposure attainment (Carlier et al., 2013; Fournier et al., 2015; Ehmann et al., 2017; Cies et al., 2018). A common theme in these studies is the absence of identified pathogens and their antibiotic MICs. When unavailable, investigators reasonably assumed worse-case scenario by substituting the susceptibly breakpoint or epidemiological cut-off (ECOFF). However, this potentially leads to underestimation of exposure, which may explain why high exposures were not associated with clinical response (i.e., actual MICs were much lower than the breakpoint and sufficient drug exposure was already achieved).
A number of studies specifically aimed at evaluating beta-lactam exposure response relationships in patient populations have been conducted. Most of these studies use covariate based population pharmacokinetic models (e.g., clearance equal to a proportion of creatinine clearance and volume of distribution calculated by body weight) to estimate pharmacokinetic parameters that are then used to calculate T > MIC. While a reasonable estimate, this is a limitation in contrast to determining observed concentration and pharmacokinetic profiles in individuals. Many studies also routinely include various infection types, use limited MICs derived from automated susceptibility testing (AST) systems, and varying clinical endpoints. These limitations must be taken into consideration when evaluating identified pharmacodynamic thresholds.
Crandon et al. (2010) evaluated cefepime pharmacodynamics in 56 patients with pneumonia (66%), skin and skin structure infections (25%), and bacteremia (9%). Using a covariate based model to estimate pharmacokinetics, but actual isolate MICs determined by Etest, they identified exposures >60% fT > MIC to be significantly associated with a positive microbiological response. In 180 patients with Gram-negative bloodstream infections (most commonly E. coli, P. aeruginosa, and K. pneumoniae), cefepime exposures of 68–74% fT > MIC were associated with a higher odds of survival (Rhodes et al., 2015). The study is strengthened by its larger numbers, survival endpoint, and the use of two different covariate based models to estimate cefepime pharmacokinetics independently; however, MICs were derived from a VITEK AST system with limited range between 1 and 64 mg/L. T > MIC thresholds of 43 and 53% were identified in two additional studies evaluating cefepime and ceftazidime in patients with nosocomial pneumonia; both studies also utilized covariate based pharmacokinetic models to estimate exposures (Muller et al., 2013; Macvane et al., 2014b). Finally, piperacillin/tazobactam pharmacodynamics was assessed in a retrospective study of 78 patients with P. aeruginosa bacteremia (Tannous et al., 2020). The investigators employed a covariate based population pharmacokinetic model and identified fT > MIC of >60.9% to be associated with in-hospital survival.
A few studies with cephalosporins suggest higher exposure thresholds. Tam and others evaluated 20 patients treated with cefepime plus an aminoglycoside who were infected with various Gram-negatives from various sources (Tam et al., 2002). The investigators used observed cefepime concentrations to calculate exposure, and MICs were conducted via Etest. Microbiological success was 89% when T > MIC was at least 100%, and 0% when it was less than 100% (p = 0.032); however, the range of exposures was not described. When Classification and Regression Tree (CART) was employed, a minimum concentration (Cmin)/MIC ratio of 4.3 was significantly associated with microbiological success. The final logistic regression model predicted 80 and 90% success when T > 4.3 x MIC was 83 and 95% of the dosing interval. A follow up study in 33 patients with pneumonia found similar exposure requirements for cefepime (Aitken et al., 2015). In contrast to the original study, the latter used a covariate population model to estimate cefepime pharmacokinetics and MICs from VITEK. The majority of patients had P. aeruginosa (n = 17) followed by Acinetobacter baumannii complex (n = 5), but the highest observed MIC was 8 mg/L thus 32/33 patients were reported to have achieved 100% fT > MIC. As a result, clinical failure (12/33) was most significantly correlated with a fCmin/MIC ratio <2.1. Gatti and others reported a case series of 13 patients treated with cefiderocol for extensively drug resistant A. baumannii bloodstream infections or ventilator associated pneumonia (Gatti et al., 2021). Cefiderocol total drug concentrations were determined only at the end of the dosing interval and corrected for protein binding of 58%. The authors arbitrarily defined exposures as sub-optimal (fCmin/MIC <1), quasi-optimal (fCmin/MIC 1- <4), or optimal (fCmin/MIC ≥4). Microbiological failure occurred in 80% of patients with suboptimal fCmin/MIC compared with 29% of those achieving optimal or quasi-optimal fCmin/MIC ratio. The majority of failures occurred in patients with ventilator associated pneumonia.
Meropenem pharmacodynamics were evaluated in patients participating in clinical trials of lower respiratory tract infections (Li et al., 2007). The investigators used a covariate based population model to estimate meropenem pharmacokinetics and identified 54% fT > MIC to be associated with microbiological eradication. Notably, a fCmin/MIC >5 was the only pharmacodynamic parameter significantly associated with clinical response, which was defined as cure or improvement of all signs and symptoms caused by the infection without additional antibiotic therapy. A population-based predictive model that evaluated the use of meropenem in adult patients with febrile neutropenia found an 80% clinical response rate when the fT > MIC was >75% (Ariano et al., 2005). Out of the 60 patients in the study, 42 clinical responders achieved an average exposure of 83% fT > MIC, whereas the average exposure for the 18 non-responders was 59% fT > MIC (p = 0.04). In 28 patients treated with meropenem, imipenem, or doripenem for P. aeruginosa ventilator-associated pneumonia, clinical response was 75% when the fT > MIC was 19.2%, and survival was 80% when >47.9% fT > MIC was achieved (Crandon et al., 2016). Kuti et al. (2018) analyzed the meropenem exposure in 15 children with cystic fibrosis (CF) acute pulmonary exacerbation to determine the pharmacodynamic threshold required for relative increased in forced expiratory volume (FEV1). Observed concentration profiles were observed for all participants and MICs were conducted by Etest. The majority of participants were infected P. aeruginosa. The meropenem fT > MIC was significantly associated
A study that prospectively evaluated the fCmin/MIC ratios of meropenem, piperacillin/tazobactam, ceftazidime, aztreonam, cefepime, and ceftriaxone in 98 patients with Gram-negative bloodstream infections identified a fCmin/MIC >1.3 to be associated with clinical response (Wong et al., 2020). This exposure threshold was fCmin/MIC >4.95 for meropenem, aztreonam, and ceftriaxone. While the investigators collected observed trough concentrations for all patients, MICs were conducted via a mix of Etest and VITEK AST, and the majority (71%) of patients had fCmin/MIC ratios >5.
To our knowledge, the results of three randomized controlled trials studying TDM of beta-lactam antibiotics are available at the time of writing. These studies may be useful to determine if the exposure thresholds sought by employing TDM achieved significant improvements in clinical outcomes. In one study, TDM was used to guide meropenem and piperacillin/tazobactam extended infusions in 41 critically ill patients (De Waele et al., 2014). The TDM target was 100% fT>4xMIC. Notably, very few patients achieved this aggressive exposure threshold with standard dosing. By 72 h, TDM patients were more likely to achieve the threshold (58% versus 16%, p = 0.007). However, no outcomes were reported in the study. A second clinical trial in 32 patients with neutropenic fever and hematologic malignancies aimed to achieve 100% fT > MIC via TDM for piperacillin/tazobactam (Sime et al., 2015). Like the first study, a greater percentage of patients in the TDM intervention arm achieved 100% fT > MIC (69% vs. 19%, p = 0.012). A greater number of patients also achieved 50% fT > MIC in the TDM arm (94 vs. 38%, p = 0.0001). However, no differences in clinical outcomes of fever or days to recovery from neutropenia were observed.
Finally, the TARGET study evaluated the role of TDM guided dosing for continuous infusion piperacillin/tazobactam in a multicenter, open-label randomized controlled trail of 249 adult patients with sepsis or septic shock (Hagel et al., 2021). Patients were randomized to TDM with goal exposure of 100% fT>4xMIC or f100% T > 64 if the pathogen or MIC were unavailable (i.e., 4 × 16 mg/L). Attainment of the pharmacodynamic threshold was more often achieved in patients with TDM; however, when adjusted for disease severity using baseline Sequential Organ Failure Assessment (SOFA) scores, the TDM-guided strategy for continuous infusion piperacillin/tazobactam did not result in greater resolution of organ dysfunction. Importantly, patients with steady state-concentrations >96 mg/L, as all well as those with concentrations <32 mg/L, had greater mortality compared with patients with concentrations absolute concentrations of 32–96 mg/L, which suggests there may be an upper limit to therapeutic range. Indeed, beta-lactams are known to have concentration dependent toxicities, particularly neurotoxicity, which is important to consider but outside the scope of this paper (Barreto et al., 2021).
The phase 3 randomized controlled registrational trials for beta-lactams are an ideal opportunity to confirm exposure response relationships and demonstrate adequacy of the dosage selected because clinical outcomes are rigorously defined, sparse pharmacokinetic sampling are routinely collected, and complete microbiology data are often available. Notably, some of these studies were unable to identify an exposure-response relationship. While this may be viewed as “unfortunate,” this is actually a positive observation, as it suggests the dosage developed from pre-clinical models was sufficient to achieve the murine based thresholds and result in successful clinical responses. Such studies are exemplified by ceftaroline in the treatment of community acquired pneumonia (Bhavnani et al., 2013) and cefiderocol (Kawaguchi et al., 2021) in the treatment of nosocomial pneumonia, blood stream infections, and complicated urinary tract infections. For these studies, the large majority of patients had fT > MIC exposures above the thresholds identified by pre-clinical murine studies and therefore no relationships with clinical study endpoints were identified. Additionally, during the cefiderocol analyses, no relationship with any of the study endpoints was observed even for 100% fT>4xMIC.
Ceftaroline exposure response relationships were identified during the phase 3 clinical studies of acute bacterial skin and skin structure infections (ABSSSI) (Bhavnani et al., 2015). Although failure rates were low in these studies, there was a robust spread of fT > MIC across the participants (range: 0–100%). As a result, classification and regression tree analyses identified 54.2 and 55% fT > MIC as thresholds predictive of microbiological success in all patients and those specifically with S. aureus infections, respectively. Microbiological success was 95.3% (486/510) for patients with fT > MIC of at least 54.2 compared with 68.8% (11/16) for those who did not achieve this threshold.
In contrast, ceftibiprole did not achieve pre-defined non-inferiority margins when studied for nosocomial pneumonia (Muller et al., 2014). Approximately 15% of patients (n = 52) contributed concentration data, while the remaining participants (n = 312) had their exposures estimated via a covariate based population model. Multiple logistic regression found fT > MIC to be associated with successful response. For patients with pathogens identified, 51% fT > MIC was identified as the threshold associated with eradication of the baseline pathogen by the end of treatment. Additionally, 62.2% fT > MIC was identified as threshold associated with eradication of any pathogen by end of therapy.
The majority of clinical studies (Table 1) suggest a pharmacodynamic threshold predictive of response that is congruent with pre-clinical murine studies (i.e., 40–70% fT > MIC). A few report higher threshold requirements (i.e., 100% fT>4xMIC or fCmin/MIC >4); however, this may have been a result of too few patients achieving low fT > MIC in the trial or only evaluating Cmin/MIC exposures. Collectively, most clinical pharmacodynamic studies suffer from a number of limitations. Most notable include the use of covariate based population models to estimate the exposure instead of observed concentrations, small sample sizes, lack of full range MICs, mixed organisms and infection types, as well as inter-study variation in endpoint utilized (clinical success, microbiological eradication, mortality, etc), thus making systematic review/meta-analyses difficult. Future studies aimed at identifying pharmacodynamic thresholds for the beta-lactams should consider solutions to these limitations during study design.
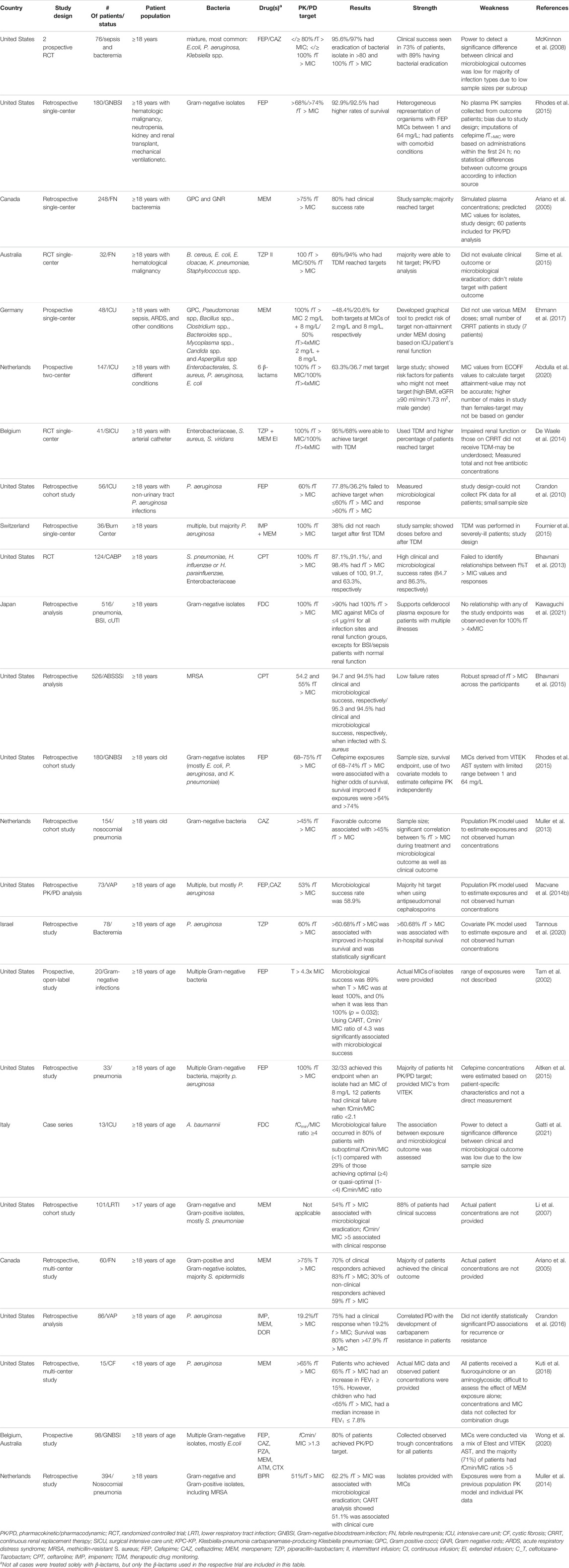
TABLE 1. Summary of details for clinical pharmacodynamic studies of beta-lactams.
It should also be noted that, by design, clinical pharmacodynamic studies are unable to parse pharmacodynamic parameters due to fixed dosages used during treatment. Because dose fractionation is not feasible, multiple pharmacodynamic indices often demonstrate statistical significance (Li et al., 2007; McKinnon et al., 2008; Muller et al., 2014; Kuti et al., 2018). It is our opinion that the pre-clinical models should set precedent on the index most predictive of effect, and the aforementioned clinical data are not sufficient to counter the robustness of dose ranging and dose fractionation study design utilized in pre-clinical models. Clinical studies are also not able to assess threshold variability by strain. Finally, the MIC itself is limited by routine two dilution testing and permitted variability of at least a dilution in each direction (i.e., 100% variability on each side of the MIC). This is further complicated in the clinical setting when the MIC is derived by automated susceptibility testing instead of a reference method and only once. These limitations on the MIC are in part why some have suggested much higher PD thresholds for TDM than observed during pre-clinical studies; however, once again, our review has not identified evidence of needing such high exposures for response. In contrast, such high exposure could lead to avoidable adverse events.
Herein, we reviewed pre-clinical and clinical studies of beta-lactams with the aim of determining optimal exposure thresholds. These studies collectively support fT > MIC as the pharmacodynamic parameter associated with beta-lactam effect. Pre-clinical murine infection studies continue to report thresholds in the range of 40–70% fT > MIC for 1 log10 CFU reductions, with Gram-negatives typically requiring greater exposure compared with Gram-positives. In only a few murine studies were exposure thresholds greater than 70% fT > MIC. Furthermore, these thresholds have been successfully applied to optimize dosage development for use in clinical registrational studies, as well as for decision support in setting accurate susceptibility breakpoints.
While limitations in study design exist, the majority of clinical pharmacodynamic studies, including those conducted post-hoc on registrational trials, are supportive of pre-clinical thresholds. However, a small number of studies do report higher thresholds, and these studies are routinely referenced as justification for requiring excessive exposure (e.g., 100% fT>4x MIC). As a result, TDM guidelines and review articles continue to recommend high exposures for beta-lactams, while randomized controlled trials of TDM requiring such exposures have yet to demonstrate superiority.
In conclusion, our review of pre-clinical and clinical pharmacodynamic studies of the beta-lactams support exposure thresholds derived from drug specific pre-clinical murine studies to support optimal dosage selection and to guide susceptibility breakpoint assessments. Until further evaluated in antibiotic specific clinical studies, exposures of 50–100% fT > MIC are reasonable to define the beta-lactam therapeutic window for most infections.
JK developed the concept and structure of the manuscript. AB and JK researched the topic, wrote the manuscript, and reviewed the final draft.
This review was funded internally by the Center for Anti-Infective Research and Development, Hartford Hospital.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdul-Aziz, M. H., Alffenaar, J. C., Bassetti, M., Bracht, H., Dimopoulos, G., Marriott, D., et al. (2020). Antimicrobial Therapeutic Drug Monitoring in Critically Ill Adult Patients: a Position Paper. Intensive Care Med. 46 (6), 1127–1153. doi:10.1007/s00134-020-06050-1
Abdulla, A., Dijkstra, A., Hunfeld, N. G. M., Endeman, H., Bahmany, S., Ewoldt, T. M. J., et al. (2020). Failure of Target Attainment of Beta-Lactam Antibiotics in Critically Ill Patients and Associated Risk Factors: a Two-center Prospective Study (EXPAT). Crit. Care 24 (1), 558. doi:10.1186/s13054-020-03272-z
Aitken, S. L., Altshuler, J., Guervil, D. J., Hirsch, E. B., Ostrosky-Zeichner, L. L., Ericsson, C. D., et al. (2015). Cefepime Free Minimum Concentration to Minimum Inhibitory Concentration (fCmin/MIC) Ratio Predicts Clinical Failure in Patients with Gram-Negative Bacterial Pneumonia. Int. J. Antimicrob. Agents 45 (5), 541–544. doi:10.1016/j.ijantimicag.2014.12.018
Andes, D., and Craig, W. A. (2006). Pharmacodynamics of a New Cephalosporin, PPI-0903 (TAK-599), Active against Methicillin-Resistant Staphylococcus aureus in Murine Thigh and Lung Infection Models: Identification of an In Vivo Pharmacokinetic-Pharmacodynamic Target. Antimicrob. Agents Chemother. 50 (4), 1376–1383. doi:10.1128/AAC.50.4.1376-1383.2006
Ariano, R. E., Nyhlén, A., Donnelly, J. P., Sitar, D. S., Harding, G. K., and Zelenitsky, S. A. (2005). Pharmacokinetics and Pharmacodynamics of Meropenem in Febrile Neutropenic Patients with Bacteremia. Ann. Pharmacother. 39 (1), 32–38. doi:10.1345/aph.1E271
Barreto, E. F., Webb, A. J., Pais, G. M., Rule, A. D., Jannetto, P. J., and Scheetz, M. H. (2021). Setting the Beta-Lactam Therapeutic Range for Critically Ill Patients: Is There a Floor or Even a Ceiling? Crit. Care Explor 3 (6), e0446. doi:10.1097/CCE.0000000000000446
Berkhout, J., Melchers, M. J., van Mil, A. C., Seyedmousavi, S., Lagarde, C. M., Schuck, V. J., et al. (2016). Pharmacodynamics of Ceftazidime and Avibactam in Neutropenic Mice with Thigh or Lung Infection. Antimicrob. Agents Chemother. 60 (1), 368–375. doi:10.1128/AAC.01269-15
Bhalodi, A. A., Crandon, J. L., Biek, D., and Nicolau, D. P. (2012). Efficacy of Ceftaroline Fosamil in a Staphylococcal Murine Pneumonia Model. Antimicrob. Agents Chemother. 56 (12), 6160–6165. doi:10.1128/AAC.01078-12
Bhavnani, S. M., Hammel, J. P., Van Wart, S. A., Rubino, C. M., Reynolds, D. K., Forrest, A., et al. (2013). Pharmacokinetic-pharmacodynamic Analyses for Efficacy of Ceftaroline Fosamil in Patients with Community-Acquired Bacterial Pneumonia. Antimicrob. Agents Chemother. 57 (12), 6348–6350. doi:10.1128/AAC.01748-13
Bhavnani, S. M., Hammel, J. P., Van Wart, S. A., Rubino, C. M., Reynolds, D. K., Forrest, A., et al. (2015). Pharmacokinetic-pharmacodynamic Analysis for Efficacy of Ceftaroline Fosamil in Patients with Acute Bacterial Skin and Skin Structure Infections. Antimicrob. Agents Chemother. 59 (1), 372–380. doi:10.1128/AAC.02531-14
Bulik, C. C., Bhavnani, S. M., Hammel, J. P., Forrest, A., Dudley, M. N., Ellis-Grosse, E. J., et al. (2013). “Evaluation of the Probability of Regulatory Approval Based on Pre-clinical PK-PD Target Attainment for Community-Acquired and Hospital-Acquired Pneumonia,” in Proceeding of the 53rd InterScience Conference on Antimicrobial Agents and Chemotherapy, September 2013 (Washington, DC: American Society of Microbiology). A-295.
Bulitta, J. B., Hope, W. W., Eakin, A. E., Guina, T., Tam, V. H., Louie, A., et al. (2019). Generating Robust and Informative Nonclinical In Vitro and In Vivo Bacterial Infection Model Efficacy Data to Support Translation to Humans. Antimicrob. Agents Chemother. 63 (5), e02307–18. doi:10.1128/AAC.02307-18
Bush, K., and Bradford, P. A. (2016). β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb Perspect. Med. 6 (8), a025247. doi:10.1101/cshperspect.a025247
Carlier, M., Carrette, S., Roberts, J. A., Stove, V., Verstraete, A., Hoste, E., et al. (2013). Meropenem and Piperacillin/tazobactam Prescribing in Critically Ill Patients: Does Augmented Renal Clearance Affect Pharmacokinetic/pharmacodynamic Target Attainment when Extended Infusions Are Used. Crit. Care 17 (3), R84. doi:10.1186/cc12705
Cies, J. J., Moore, W. S., Enache, A., and Chopra, A. (2018). β-Lactam Therapeutic Drug Management in the PICU. Crit. Care Med. 46 (2), 272–279. doi:10.1097/CCM.0000000000002817
Craig, W. A., and Andes, D. R. (2008). In Vivo pharmacodynamics of Ceftobiprole against Multiple Bacterial Pathogens in Murine Thigh and Lung Infection Models. Antimicrob. Agents Chemother. 52 (10), 3492–3496. doi:10.1128/AAC.01273-07
Craig, W. A., and Andes, D. R. (2013). In Vivo activities of Ceftolozane, a New Cephalosporin, with and without Tazobactam against Pseudomonas aeruginosa and Enterobacteriaceae, Including Strains with Extended-Spectrum β-lactamases, in the Thighs of Neutropenic Mice. Antimicrob. Agents Chemother. 57 (4), 1577–1582. doi:10.1128/AAC.01590-12
Craig, W. A. (1998). Pharmacokinetic/pharmacodynamic Parameters: Rationale for Antibacterial Dosing of Mice and Men. Clin. Infect. Dis. 26 (1), 1–2. quiz 1-2. doi:10.1086/516284
Crandon, J. L., Bulik, C. C., Kuti, J. L., and Nicolau, D. P. (2010). Clinical Pharmacodynamics of Cefepime in Patients Infected with Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 54 (3), 1111–1116. doi:10.1128/AAC.01183-09
Crandon, J. L., Luyt, C. E., Aubry, A., Chastre, J., and Nicolau, D. P. (2016). Pharmacodynamics of Carbapenems for the Treatment of Pseudomonas aeruginosa Ventilator-Associated Pneumonia: Associations with Clinical Outcome and Recurrence. J. Antimicrob. Chemother. 71 (9), 2534–2537. doi:10.1093/jac/dkw200
Das, S., Li, J., Riccobene, T., Carrothers, T. J., Newell, P., Melnick, D., et al. (2019). Dose Selection and Validation for Ceftazidime-Avibactam in Adults with Complicated Intra-abdominal Infections, Complicated Urinary Tract Infections, and Nosocomial Pneumonia. Antimicrob. Agents Chemother. 63 (4), e02187–18. doi:10.1128/AAC.02187-18
Das, S., Zhou, D., Nichols, W. W., Townsend, A., Newell, P., and Li, J. (2020). Selecting the Dosage of Ceftazidime-Avibactam in the Perfect Storm of Nosocomial Pneumonia. Eur. J. Clin. Pharmacol. 76 (3), 349–361. doi:10.1007/s00228-019-02804-z
De Waele, J. J., Carrette, S., Carlier, M., Stove, V., Boelens, J., Claeys, G., et al. (2014). Therapeutic Drug Monitoring-Based Dose Optimisation of Piperacillin and Meropenem: a Randomised Controlled Trial. Intensive Care Med. 40 (3), 380–387. doi:10.1007/s00134-013-3187-2
Eagle, H., Fleischman, R., and Levy, M. (1953). Continuous vs. Discontinuous Therapy with Penicillin. N. Engl. J. Med. 248 (12), 481–488. doi:10.1056/nejm195303192481201
Ehmann, L., Zoller, M., Minichmayr, I. K., Scharf, C., Maier, B., Schmitt, M. V., et al. (2017). Role of Renal Function in Risk Assessment of Target Non-attainment after Standard Dosing of Meropenem in Critically Ill Patients: a Prospective Observational Study. Crit. Care 21 (1), 263. doi:10.1186/s13054-017-1829-4
Food and Drug Administration, (2021). Vabormere (Meropenem and Vaborbactam for Injection) Clinical Pharmacology and Biopharmacuetics Review. Accessed at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209776Orig1s000ClinPharmR.pdf.
Fournier, A., Eggimann, P., Pagani, J. L., Revelly, J. P., Decosterd, L. A., Marchetti, O., et al. (2015). Impact of the Introduction of Real-Time Therapeutic Drug Monitoring on Empirical Doses of Carbapenems in Critically Ill Burn Patients. Burns 41 (5), 956–968. doi:10.1016/j.burns.2015.01.001
Fratoni, A. J., Nicolau, D. P., and Kuti, J. L. (2021). A Guide to Therapeutic Drug Monitoring of β-lactam Antibiotics. Pharmacotherapy 41 (2), 220–233. doi:10.1002/phar.2505
Gatti, M., Bartoletti, M., Cojutti, P. G., Gaibani, P., Conti, M., Giannella, M., et al. (2021). A Descriptive Case Series of Pharmacokinetic/pharmacodynamic Target Attainment and Microbiological Outcome in Critically Ill Patients with Documented Severe Extensively Drug-Resistant Acinetobacter Baumannii Bloodstream Infection And/or Ventilator-Associated Pneumonia Treated with Cefiderocol. J. Glob. Antimicrob. Resist. 27, 294–298. doi:10.1016/j.jgar.2021.10.014
Ghazi, I. M., Monogue, M. L., Tsuji, M., and Nicolau, D. P. (2018). Pharmacodynamics of Cefiderocol, a Novel Siderophore Cephalosporin, in a Pseudomonas aeruginosa Neutropenic Murine Thigh Model. Int. J. Antimicrob. Agents 51 (2), 206–212. doi:10.1016/j.ijantimicag.2017.10.008
Grupper, M., Stainton, S. M., Nicolau, D. P., and Kuti, J. L. (2019). In Vitro Pharmacodynamics of a Novel Ceftibuten-Clavulanate Combination Antibiotic against Enterobacteriaceae. Antimicrob. Agents Chemother. 63 (7), e00144–19. doi:10.1128/AAC.00144-19
Guilhaumou, R., Benaboud, S., Bennis, Y., Dahyot-Fizelier, C., Dailly, E., Gandia, P., et al. (2019). Optimization of the treatment with beta-lactam antibiotics in critically ill patients-guidelines from the French Society of Pharmacology and Therapeutics (Société Française de Pharmacologie et Thérapeutique-SFPT) and the French Society of Anaesthesia and Intensive Care Medicine (Société Française d'Anesthésie et Réanimation-SFAR). Crit. Care 23 (1), 104. doi:10.1186/s13054-019-2378-9
Hagel, S., Brinkman, A., Bach, F., Bauer, M., Brenner, T., Witzke, D., et al. (2021). “Effect of Therapeutic Drug Monitoring-Based Dose Optimization of Piperacillin-Tazobactam on Sepsis-Related Organ Dysfunction in Patients with Sepsis: a Randomized Clinical Trial (Abstract 00843),” in Proceeding of the 31st ECCMID, July 2021.
Holford, N. (2017). Pharmacodynamic Principles and the Time Course of Immediate Drug Effects. Transl Clin. Pharmacol. 25 (4), 157–161. doi:10.12793/tcp.2017.25.4.157
Holten, K. B., and Onusko, E. M. (2000). Appropriate Prescribing of Oral Beta-Lactam Antibiotics. Am. Fam. Physician 62 (3), 611–620.
Housman, S. T., Crandon, J. L., Nichols, W. W., and Nicolau, D. P. (2014). Efficacies of Ceftazidime-Avibactam and Ceftazidime against Pseudomonas aeruginosa in a Murine Lung Infection Model. Antimicrob. Agents Chemother. 58 (3), 1365–1371. doi:10.1128/AAC.02161-13
Katsube, T., Echols, R., and Wajima, T. (2019). Pharmacokinetic and Pharmacodynamic Profiles of Cefiderocol, a Novel Siderophore Cephalosporin. Clin. Infect. Dis. 69 (Suppl. 7), S552–S8. doi:10.1093/cid/ciz828
Kawaguchi, N., Katsube, T., Echols, R., and Wajima, T. (2021). Population Pharmacokinetic and Pharmacokinetic/Pharmacodynamic Analyses of Cefiderocol, a Parenteral Siderophore Cephalosporin, in Patients with Pneumonia, Bloodstream Infection/Sepsis, or Complicated Urinary Tract Infection. Antimicrob. Agents Chemother. 65 (3), e01437–20. doi:10.1128/AAC.01437-20
Keel, R. A., Crandon, J. L., and Nicolau, D. P. (2011). Efficacy of Human Simulated Exposures of Ceftaroline Administered at 600 Milligrams Every 12 hours against Phenotypically Diverse Staphylococcus aureus Isolates. Antimicrob. Agents Chemother. 55 (9), 4028–4032. doi:10.1128/AAC.00372-11
Kim, A., Banevicius, M. A., and Nicolau, D. P. (2008). In Vivo pharmacodynamic Profiling of Doripenem against Pseudomonas aeruginosa by Simulating Human Exposures. Antimicrob. Agents Chemother. 52 (7), 2497–2502. doi:10.1128/AAC.01252-07
Kuti, J. L., Pettit, R. S., Neu, N., Cies, J. J., Lapin, C., Muhlebach, M. S., et al. (2018). Meropenem Time above the MIC Exposure Is Predictive of Response in Cystic Fibrosis Children with Acute Pulmonary Exacerbations. Diagn. Microbiol. Infect. Dis. 91 (3), 294–297. doi:10.1016/j.diagmicrobio.2018.01.020
Leggett, J. E., Fantin, B., Ebert, S., Totsuka, K., Vogelman, B., Calame, W., et al. (1989). Comparative Antibiotic Dose-Effect Relations at Several Dosing Intervals in Murine Pneumonitis and Thigh-Infection Models. J. Infect. Dis. 159 (2), 281–292. doi:10.1093/infdis/159.2.281
Lepak, A. J., Reda, A., Marchillo, K., Van Hecker, J., Craig, W. A., and Andes, D. (2014). Impact of MIC Range for Pseudomonas aeruginosa and Streptococcus Pneumoniae on the Ceftolozane In Vivo Pharmacokinetic/pharmacodynamic Target. Antimicrob. Agents Chemother. 58 (10), 6311–6314. doi:10.1128/AAC.03572-14
Li, C., Du, X., Kuti, J. L., and Nicolau, D. P. (2007). Clinical Pharmacodynamics of Meropenem in Patients with Lower Respiratory Tract Infections. Antimicrob. Agents Chemother. 51 (5), 1725–1730. doi:10.1128/AAC.00294-06
Lodise, T. P., Lomaestro, B. M., Drusano, G. L., and Society of Infectious Diseases, P. (2006). Application of Antimicrobial Pharmacodynamic Concepts into Clinical Practice: Focus on Beta-Lactam Antibiotics: Insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy 26 (9), 1320–1332. doi:10.1592/phco.26.9.1320
Macvane, S. H., Crandon, J. L., and Nicolau, D. P. (2014a). Characterizing In Vivo Pharmacodynamics of Carbapenems against Acinetobacter Baumannii in a Murine Thigh Infection Model to Support Breakpoint Determinations. Antimicrob. Agents Chemother. 58 (1), 599–601. doi:10.1128/AAC.02029-13
Macvane, S. H., Kuti, J. L., and Nicolau, D. P. (2014b). Clinical Pharmacodynamics of Antipseudomonal Cephalosporins in Patients with Ventilator-Associated Pneumonia. Antimicrob. Agents Chemother. 58 (3), 1359–1364. doi:10.1128/AAC.01463-13
Maglio, D., Banevicius, M. A., Sutherland, C., Babalola, C., Nightingale, C. H., and Nicolau, D. P. (2005). Pharmacodynamic Profile of Ertapenem against Klebsiella pneumoniae and Escherichia coli in a Murine Thigh Model. Antimicrob. Agents Chemother. 49 (1), 276–280. doi:10.1128/AAC.49.1.276-280.2005
McKinnon, P. S., Paladino, J. A., and Schentag, J. J. (2008). Evaluation of Area under the Inhibitory Curve (AUIC) and Time above the Minimum Inhibitory Concentration (T>MIC) as Predictors of Outcome for Cefepime and Ceftazidime in Serious Bacterial Infections. Int. J. Antimicrob. Agents 31 (4), 345–351. doi:10.1016/j.ijantimicag.2007.12.009
Monogue, M. L., and Nicolau, D. P. (2019). Pharmacokinetics-pharmacodynamics of β-lactamase Inhibitors: Are We Missing the Target? Expert Rev. Anti Infect. Ther. 17 (8), 571–582. doi:10.1080/14787210.2019.1647781
Muller, A. E., Punt, N., and Mouton, J. W. (2013). Optimal Exposures of Ceftazidime Predict the Probability of Microbiological and Clinical Outcome in the Treatment of Nosocomial Pneumonia. J. Antimicrob. Chemother. 68 (4), 900–906. doi:10.1093/jac/dks468
Muller, A. E., Punt, N., and Mouton, J. W. (2014). Exposure to Ceftobiprole Is Associated with Microbiological Eradication and Clinical Cure in Patients with Nosocomial Pneumonia. Antimicrob. Agents Chemother. 58 (5), 2512–2519. doi:10.1128/AAC.02611-13
Nakamura, R., Ito-Horiyama, T., Takemura, M., Toba, S., Matsumoto, S., Ikehara, T., et al. (2019). In Vivo Pharmacodynamic Study of Cefiderocol, a Novel Parenteral Siderophore Cephalosporin, in Murine Thigh and Lung Infection Models. Antimicrob. Agents Chemother. 63 (9), e02031–18. doi:10.1128/AAC.02031-18
Patel, M., Daryani, N. M., Fen, H. P., Hilbert, D. W., Melchers, M. J., Mavridou, E., et al. (2020). “Imipenem/Relebactam Pharmacokinetic/Pharmacodynamic Analyses from an In Vivo Neutropenic Murine Thigh Infection Model (Abstract 1693),” in Proceeding of the 30th ECCMID, Paris, France, April 2020.
Reed, M. D. (2000). Optimal Antibiotic Dosing. The Pharmacokinetic-Pharmacodynamic Interface. Postgrad. Med. 108 (7 Suppl. Contemporaty), 17–24. doi:10.3810/pgm.12.2000.suppl10.52
Reeves, D., Lovering, A., and Thomson, A. (2016). Therapeutic Drug Monitoring in the Past 40 Years of the Journal of Antimicrobial Chemotherapy. J. Antimicrob. Chemother. 71 (12), 3330–3332. doi:10.1093/jac/dkw408
Rhodes, N. J., Kuti, J. L., Nicolau, D. P., Van Wart, S., Nicasio, A. M., Liu, J., et al. (2015). Defining Clinical Exposures of Cefepime for Gram-Negative Bloodstream Infections that Are Associated with Improved Survival. Antimicrob. Agents Chemother. 60 (3), 1401–1410. doi:10.1128/AAC.01956-15
Roberts, J. A., Hope, W. W., and Lipman, J. (2010). Therapeutic Drug Monitoring of Beta-Lactams for Critically Ill Patients: Unwarranted or Essential. Int. J. Antimicrob. Agents 35 (5), 419–420. doi:10.1016/j.ijantimicag.2010.01.022
Roberts, J. A., Abdul-Aziz, M. H., Lipman, J., Mouton, J. W., Vinks, A. A., Felton, T. W., et al. (2014). Individualised Antibiotic Dosing for Patients Who Are Critically Ill: Challenges and Potential Solutions. Lancet Infect. Dis. 14 (6), 498–509. doi:10.1016/S1473-3099(14)70036-2
Roberts, J. A., Paul, S. K., Akova, M., Bassetti, M., De Waele, J. J., Dimopoulos, G., et al. (2014). DALI: Defining Antibiotic Levels in Intensive Care Unit Patients: Are Current β-lactam Antibiotic Doses Sufficient for Critically Ill Patients. Clin. Infect. Dis. 58 (8), 1072–1083. doi:10.1093/cid/ciu027
Rodvold, K. A., Nicolau, D. P., Lodise, T. P., Khashab, M., Noel, G. J., Kahn, J. B., et al. (2009). Identifying Exposure Targets for Treatment of Staphylococcal Pneumonia with Ceftobiprole. Antimicrob. Agents Chemother. 53 (8), 3294–3301. doi:10.1128/AAC.00144-09
Sabet, M., Tarazi, Z., and Griffith, D. C. (2020). Pharmacodynamics of Meropenem against Acinetobacter Baumannii in a Neutropenic Mouse Thigh Infection Model. Antimicrob. Agents Chemother. 64 (4), e02388–19. doi:10.1128/AAC.02388-19
Sime, F. B., Roberts, M. S., Tiong, I. S., Gardner, J. H., Lehman, S., Peake, S. L., et al. (2015). Can Therapeutic Drug Monitoring Optimize Exposure to Piperacillin in Febrile Neutropenic Patients with Haematological Malignancies? A Randomized Controlled Trial. J. Antimicrob. Chemother. 70 (8), 2369–2375. doi:10.1093/jac/dkv123
Soon, R. L., Ly, N. S., Rao, G., Wollenberg, L., Yang, K., Tsuji, B., et al. (2013). Pharmacodynamic Variability beyond that Explained by MICs. Antimicrob. Agents Chemother. 57 (4), 1730–1735. doi:10.1128/AAC.01224-12
Tam, V. H., McKinnon, P. S., Akins, R. L., Rybak, M. J., and Drusano, G. L. (2002). Pharmacodynamics of Cefepime in Patients with Gram-Negative Infections. J. Antimicrob. Chemother. 50 (3), 425–428. doi:10.1093/jac/dkf130
Tannous, E., Lipman, S., Tonna, A., Hector, E., Hussein, Z., Stein, M., et al. (2020). Time above the MIC of Piperacillin-Tazobactam as a Predictor of Outcome in Pseudomonas aeruginosa Bacteremia. Antimicrob. Agents Chemother. 64 (8), e02571–19. doi:10.1128/AAC.02571-19
Turnidge, J. D. (1998). The Pharmacodynamics of Beta-Lactams. Clin. Infect. Dis. 27 (1), 10–22. doi:10.1086/514622
Vincent, J. L., Sakr, Y., Singer, M., Martin-Loeches, I., Machado, F. R., Marshall, J. C., et al. (2020). Prevalence and Outcomes of Infection Among Patients in Intensive Care Units in 2017. JAMA 323 (15), 1478–1487. doi:10.1001/jama.2020.2717
Vogelman, B., and Craig, W. A. (1986). Kinetics of Antimicrobial Activity. J. Pediatr. 108 (5 Pt 2), 835–840. doi:10.1016/s0022-3476(86)80754-5
Waack, U., Weinstein, E. A., and Farley, J. J. (2020). Assessing Animal Models of Bacterial Pneumonia Used in Investigational New Drug Applications for the Treatment of Bacterial Pneumonia. Antimicrob. Agents Chemother. 64 (5), e02242–19. doi:10.1128/AAC.02242-19
Keywords: beta-lactam, pharmacodynamics, pre-clinical models, therapeutic drug monitoring, susceptibility breakpoints
Citation: Berry AV and Kuti JL (2022) Pharmacodynamic Thresholds for Beta-Lactam Antibiotics: A Story of Mouse Versus Man. Front. Pharmacol. 13:833189. doi: 10.3389/fphar.2022.833189
Received: 10 December 2021; Accepted: 24 February 2022;
Published: 18 March 2022.
Edited by:
Markus Zeitlinger, Medical University of Vienna, AustriaReviewed by:
Alexander (a.k.a. Sander) A. Vinks, Cincinnati Children’s Hospital Medical Center, United StatesCopyright © 2022 Berry and Kuti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph L. Kuti, am9zZXBoLmt1dGlAaGhjaGVhbHRoLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.