 Genri Numata1,2
Genri Numata1,2 Eiki Takimoto1,3*
Eiki Takimoto1,3*- 1Department of Cardiovascular Medicine, The University of Tokyo Hospital, Tokyo, Japan
- 2Department of Advanced Translational Research and Medicine in Management of Pulmonary Hypertension, The University of Tokyo Hospital, Tokyo, Japan
- 3Division of Cardiology, Department of Medicine, The Johns Hopkins Medical Institutions, Baltimore, MD, United States
Cyclic guanosine monophosphate (cGMP), produced by guanylate cyclase (GC), activates protein kinase G (PKG) and regulates cardiac remodeling. cGMP/PKG signal is activated by two intrinsic pathways: nitric oxide (NO)-soluble GC and natriuretic peptide (NP)-particulate GC (pGC) pathways. Activation of these pathways has emerged as a potent therapeutic strategy to treat patients with heart failure, given cGMP-PKG signaling is impaired in heart failure with reduced ejection fraction (HFrEF) and preserved ejection fraction (HFpEF). Large scale clinical trials in patients with HFrEF have shown positive results with agents that activate cGMP-PKG pathways. In patients with HFpEF, however, benefits were observed only in a subgroup of patients. Further investigation for cGMP-PKG pathway is needed to develop better targeting strategies for HFpEF. This review outlines cGMP-PKG pathway and its modulation in heart failure.
Introduction
Heart failure is a major health problem, and its prevalence is increasing worldwide. The traditional guideline directed therapies target the renin-angiotensin-aldosterone system and the sympathetic nervous system, but recently, cyclic guanosine 3′,5′-monophosphate (cGMP) and its downstream protein kinase G (PKG) signaling has attracted attention as a novel therapeutic target (Tsai and Kass, 2009). cGMP-PKG pathway regulates diverse cellular mechanisms to maintain cellular homeostasis and is activated by two different pathways. One is natriuretic peptide (NP)-NP receptor (NPR)-particulate guanylate cyclase (pGC) pathway, and the other is NO-soluble GC (sGC) pathway. cGMP-PKG pathway has been suggested to be blunted or dysregulated in patients with HFrEF or HFpEF (Paulus et al., 2013; Redfield et al., 2013). Increased plasma levels of inflammatory cytokines including TNF-α and IL-6 in HF are related to endothelial dysfunction with low NO-sGC-cGMP signaling in the heart and vasculature (Torre-Amione et al., 1996; Lommi et al., 1997), where its degradation by cGMP-PDEs might be enhanced. In HFpEF patients, myocardial homogenates from biopsy samples revealed low PKG activity and cGMP concentration compared with HFrEF and aortic stenosis patients (Van Heerebeek et al., 2012). Thus, the therapeutic strategy to recover blunted cGMP/PKG signaling in heart failure is very reasonable. Sacubitril/valsartan is the first agent in this class that has been approved for use in heart failure. It consists of the neprilysin (NEP) inhibitor and the angiotensin receptor blocker, and is described as an angiotensin receptor-neprilysin inhibitor (ARNi). NEP hydrolyzes several peptide hormones including NPs (ANP, BNP, CNP), adrenomedullin, glucagon, enkephalins, substance P, neurotensin, oxytocin, and bradykinin. Thus, its inhibition enhances NPs-pGC-cGMP. ARNi improved clinical outcomes in patients with HFrEF (McMurray et al., 2014; Velazquez et al., 2018) and also exhibited favorable outcomes in a particular sub-group (female) in HFpEF (Solomon et al., 2019; Pieske et al., 2021). Vericiguat, an sCG stimulator that enhances (NO)-sGC-cGMP pathway independently of NO, was approved for the treatment of heart failure. Vericugat is effective in patients with HFrEF (Armstrong et al., 2020a), but it failed to reveal clinical improvement in HFpEF (Armstrong et al., 2020b; Udelson et al., 2020) (Table 1). The clinical importance of cGMP-PKG pathway is clear; however, a better understanding of underlying mechanisms is necessary for the optimal therapeutic strategy with enhancement of cGMP-PKG signaling pathway. This review focuses on the regulatory mechanisms of cGMP-PKG pathway in heart failure.
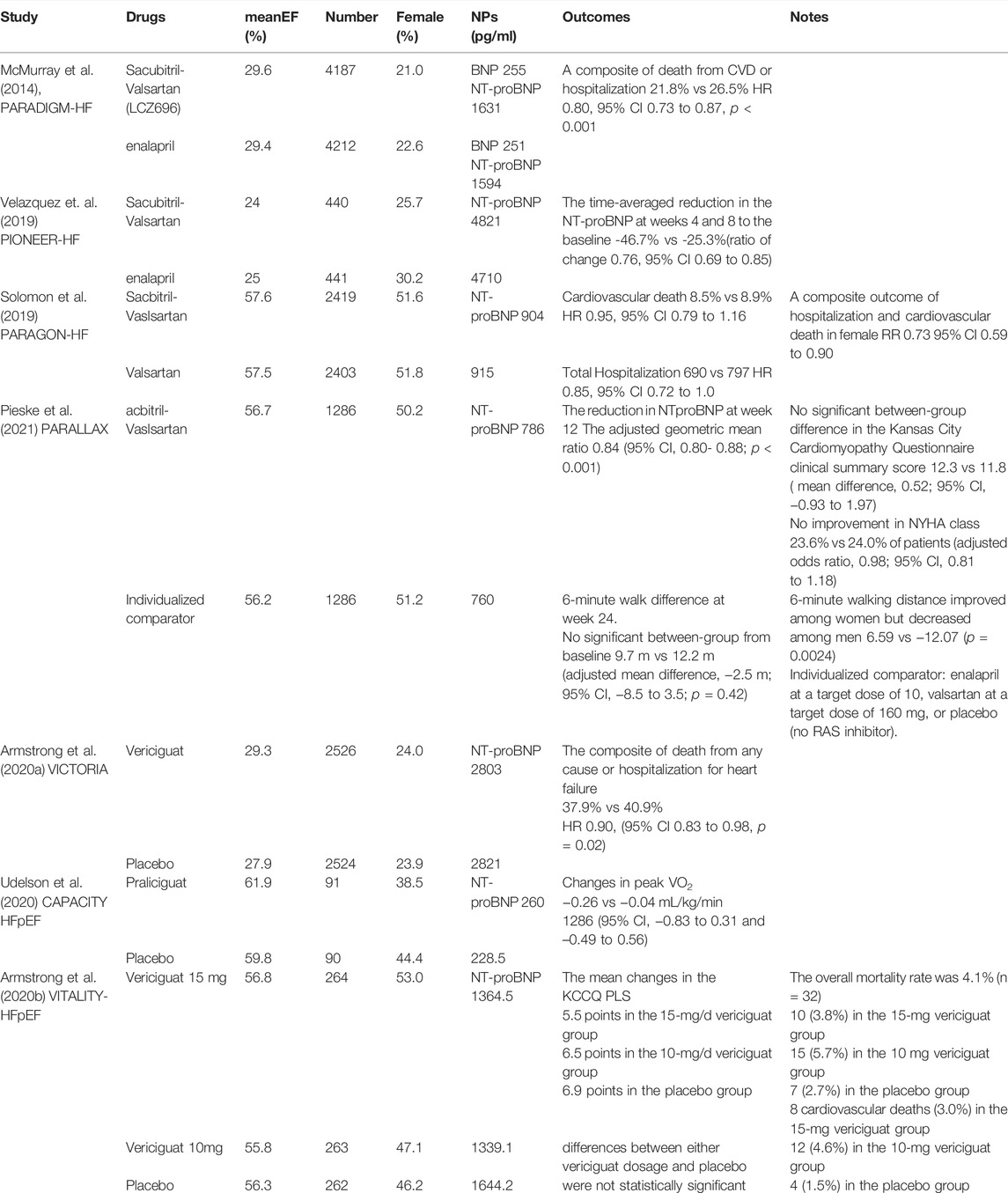
TABLE 1. Clinical trials associated with sGC inhibitors and neprilysin inhibitors.
Phosphodiesterase and cGMP/PKG Signaling
Phosphodiesterase (PDE) has 11 superfamilies and more than 100 isoform variants that hydrolyze cAMP or cGMP to their inactive respective 5′-monophosphate form. Seven PDEs (PDE1, 2, 3, 4, 5, 8, and 9) are currently known to be expressed in myocardium. PDE1, 2, and 3 hydrolyze both cAMP and cGMP, while PDE5 and 9 are selective for cGMP and PDE4 and 8 are selective for cAMP (Kim and Kass, 2017). PDEs are differentially localized within the cells, contributing to the compartmentalized regulation of cGMP and cAMP signaling in both space and time.
Inhibition of PDE1, a dual substrate esterase, demonstrates acute inotropic and lusitropic effects largely via cAMP pathway (Hashimoto et al., 2018), demonstrated in large animal models. PDE1A, one the three isoforms of PDE1, modulates pathological hypertrophy via cGMP-PKG in rodent and cell models, while PDE1C, coupled with adenosine A2A receptor and TRPC3, hydrolyzes cAMP and regulates apoptosis in cardiac myocytes (Miller et al., 2009; Zhang et al., 2018).
PDE2 is also a dual substrate esterase and involved in the regulation of cardiac hypertrophy via cGMP. PDE2 specifically plays an important role in the crosstalk between cGMP and cAMP pathways because its activity is stimulated by cGMP (Baliga et al., 2018). PDE2 has three splice variants (PDE2A1, 2A2, 2A3), which are differently localized: PDE2A1 in cytoplasm, PDE2A2 in mitochondrial matrix, and PDE2A3 at membrane (mostly PDE2A3) (Geoffroy et al., 1999; Lugnier et al., 1999; Mongillo et al., 2006; Weber et al., 2017). In the heart, in particular, PDE2A might be localized in both cytosolic and particulate fractions of cardiac ventricle, though it differs from species to species (Le Trong et al., 1990; Bode et al., 1991; Muller et al., 1992; Sugioka et al., 1994; Geoffroy et al., 1999; Mongillo et al., 2006). In humans, PDE2A3 is expressed in cardiomyocytes and vascular endothelial cells (Sadhu et al., 1999). Under the normal conditions, PDE2 is less abundant in cardiomyocytes than in fibroblasts and endothelial cells (Stephenson et al., 2009; Vettel et al., 2014), but under the pathological conditions, PDE2 expressions and cAMP-hydrolyzing activity significantly increase (Levy, 2013; Mehel et al., 2013). (Chen et al., 2016). Cardiac PDE2A expressions increase in rat cardiac hypertrophy and also in human ischemic or non-ischemic heart failure (Mehel et al., 2013). PDE2 can hydrolyze cGMP produced by either pGC (Stangherlin et al., 2011) and sGC (Mongillo et al., 2006) with the allosteric hydrolyzing ability activated by cGMP, but this might depend on the stress conditions and cGMP concentrations (Terasaki and Appleman, 1975; Prigent et al., 1988; Mery et al., 1993; Dittrich et al., 2001; Herring et al., 2001; Weber et al., 2017). PDE2A overexpression blunts norepinephrine-induced cellular hypertrophy with marked decrease in cAMP levels (Mehel et al., 2013). On the other hand, PDE2A inhibition suppresses cardiac hypertrophy induced by norepinephrine in rats (Zoccarato et al., 2015). These apparently opposite results might be attributable to the cAMP and cGMP regulation levels which might depend on the contexts. In the computer modeling, Zhao et al. reported that PDE2A hydrolyzed increasing amount of cAMP with increasing levels of β adrenergic stimulation, and hydrolyzed increasing amounts of cGMP with decreasing levels of NO stimulation (Zhao et al., 2016).
We elaborate on cGMP-specific PDEs (PDE5 and PDE9) in the next section, reviewing their effects on cardiac remodeling. PDE5 hydrolyzes cGMP derived from NO-sGC pathway and PDE9 degrades cGMP from NP-pGC pathway, modulating various signaling related to cardiac remodeling (Figure 1).
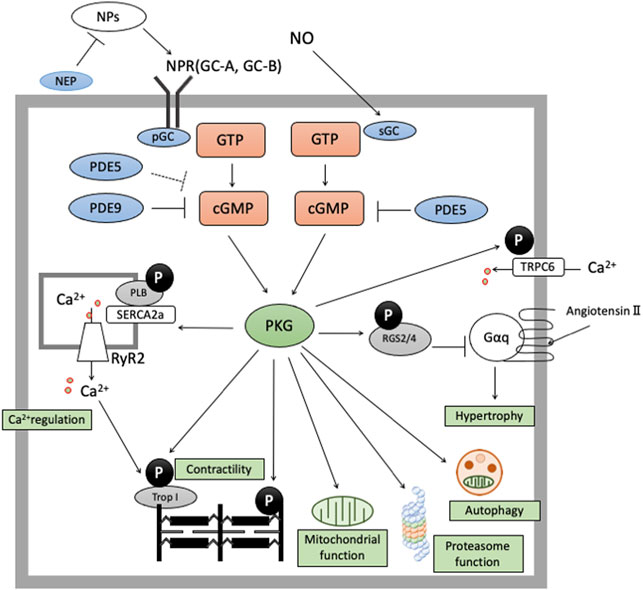
FIGURE 1. cGMP/PKG signaling in cardiomyocyte cGMP-PKG signaling is enhanced by two pathways. The former is NO-sGC-cGMP pathway and the latter is NP-NPR-pGC pathway. cGMP derived from NO-sGC pathway takes hydrolyzation by PDE5, and cGMP from NP-pGC pathway by PDE9 and PDE5 (especially in stressed conditions). cGMP/PKG signaling exerts protective effects in cardiomyocyte by phosphorylate various proteins like RGS2/4, Troponin I, TSC2, cMyBP-C, or Titin.
NO-sGC Pathway (PDE5)
Nitric oxide (NO) stimulates sGC to produce cGMP, which is hydrolyzed specifically by PDE5. PDE5A is localized at Z-disks in cardiac myocytes under physiological conditions but it is diffusely distributed under diseased conditions (Takimoto et al., 2005; Zhang et al., 2008). The expression of PDE5A is up-regulated in failing hearts (Shan et al., 2012), though it is very low under physiological conditions. In experimental animal models, PDE5 inhibition (PDE5i) provides cardiac protection against pressure-overload, ischemia-reperfusion injury, and doxorubicin-toxicity (Takimoto et al., 2005; Burley et al., 2007; Kukreja et al., 2012; Jin et al., 2013), with multiple myocardial signaling pathways altered (Takimoto, 2012). A regulator of G-protein signaling (RGS), 2/4 is phospho-activated to inhibit Gq-signaling (Takimoto et al., 2009) and transient receptor potential canonical Ca2+ channel-type6 (TRPC6) coupled with calcinurin (Cn) signaling (Koitabashi et al., 2010; Seo et al., 2014) is deactivated by PDE5i-PKG-phosphorylation. Mechanisms related to proteostasis are also regulated. PDE5i-activated PKG enhances proteasome function, blocking the accumulation of misfolded proteins via posttranslational modifications of proteasome subunits (Ranek et al., 2013). PDE5i also phosphorylates tuberin (TSC2), an intrinsic regulator of the mechanistic target of rapamycin complex-1 (mTORC1), and enhances autophagy. In a model of ischemia re-perfusion injury, PDE5i-cGMP-PKG exerts cardio-protective effects against necrosis and apoptosis through modulating mitochondrial functions (RAMZI et al., 2002; Salloum et al., 2008; Li et al., 2016; Patel et al., 2019) Additionally, PDE5i alone or in combination with natriuretic peptide, phosphorylates sarcomeric proteins including titin (Bishu et al., 2011a), troponin-I (Layland et al., 2005; Wijnker et al., 2014), and cardiac myosin-binding protein C (Thoonen et al., 2015), which improves systolic and diastolic function. Some may still have debate on PDE5A or PKG effects on cardiomyocyte. Straubinger et al. reported that sildenafil failed to limit the progressive cardiomyocyte growth, fibrosis, or cardiac dysfunctions in the cardiomyocyte-specific overexpression of the AT1 receptor mice (Straubinger et al., 2015). Lukowski et al. showed that the deletion of cardiomyocyte-specific PKG had no effect on cardiac hypertrophy caused by pressure overload and isoproterenol administration (Lukowski et al., 2010). Patrucco et al. reported that the lack of PKG in cardiomyocyte, endothelial cells, or cardiac fibroblast did not augment hypertrophic response and sildenafil had modest effects on angiotensin II-induced cardiac hypertrophy (Patrucco et al., 2014). On the other hand, however, cardiomyocyte-specific overexpression of PDE5A recovered impaired cardiac functions from pressure overload (Zhang et al., 2010) and myocardial infarction (Pokreisz et al., 2009). Frantz and Kuhn et al. generated animals with cardiomyocyte-restricted deletion of PKG, and demonstrated the animals developed severe hypertrophy by chronic angiotensin II infusion or pressure overload (Frantz et al., 2013). Recently, we have consistently reported that sildenafil exhibited protective effects against cardiac hypertrophy via proliferator activated receptor γ co-activator-1α-PKG cascade (Zhu et al., 2021). Together it would be reasonable to conclude that cGMP-PKG signaling in cardiomyocyte would be important in cardiac hypertrophy and remodeling. With regard to the cardiac-specific role and regulation of PDE5, a tissue-specific conditional deletion model would be awaited.
sGC stimulators and sGC activators are direct modulators of sGC, increasing the production of cGMP: the former stimulates NO-sensitive (unoxidized) sGC and the latter can activate NO-insensitive (oxidized) sGC. sGC stimulators have shown cardiac benefits in an HFpEF model (Wilck et al., 2018) as well as in an HFrEF model. Double-transgenic rats (dTGR) harboring the renin and angiotensinogen genes exhibit an HFpEF phenotype of diastolic dysfunction, preserved EF, systemic hypertension, cardiac hypertrophy, fibrosis, inflammation, and endothelial dysfunction, and dies between 7 and 8 weeks from severe heart failure (Damage et al., 1999; Mervaala et al., 2001; Wellner et al., 2005; Fischer et al., 2008; Finckenberg et al., 2012; Haase et al., 2014). Treatment with an sGC stimulator improved cardiac function, cardiac fibrosis, and inflammation, with minimal effects on cardiac hypertrophy (Wilck et al., 2018). sGC activators have also shown cardio-protective effects in another HFpEF model (Dahl salt-sensitive model: DSS) (Kolijn et al., 2020), where an sGC activator (cinaciguat) phosphorylates titin and improves passive stiffness. In human cardiomyocytes from HFpEF patients, cinaciguat phosphorylates titin and improves passive tension, associated with a reduction in proinflammatory cytokines and oxidative stress markers (Kolijn et al., 2020). sGC-bound cofactor heme (Fe2+) is oxidized to Fe3+ under oxidative conditions, leading to the inactive Apo form that no longer is responsive to NO. sGC stimulators stimulate only Fe2+-sGC, while sGC activators act on oxidated sGC(Fe3+-sGC or Apo-sGC) (Krishnan et al., 2018) to produce cGMP. In oxidated conditions such as HFpEF, sGC activator might have an advantage.
Although preclinical studies have revealed cardio-protective and anti-remodeling effects from NO-sGC-cGMP activation in either type of heart failure (HFrEF or HFpEF), clinical studies have yielded mixed results. Two meta-analyses of controlled clinical trials (928 patients in 14 studies (De Vecchis et al., 2017), 555 patients in 13 studies (De Vecchis et al., 2018)) demonstrate that PDE5 inhibitors improve clinical outcomes, exercise capacity, and pulmonary hemodynamics in patients with HFrEF, but not HFpEF. The negative results in HFpEF might be partially attributable to the female-specific response of PDE5i depending on estrogen levels, given the prevalence of HFpEF in older women: nearly half of the patients were older women (average age 67) in the negative RELAX trial. Epidemiological studies have demonstrated that women are likely to develop HFpEF. In clinical trials of HFpEF women account for around 50–60% of the trial cohorts (Forman and Gaziano, 2009; Savill, 2014), whereas they account for 20–25% of those of HFrEF (Pablo, 2017; Zannad et al., 2018; Pieske et al., 2019). In a recent multicenter, observational study, female sex was reported to be independently associated with the presence of diastolic dysfunction and worse clinical outcomes (Sotomi et al., 2021). Sex-hormone estrogen plays a pivotal role in cGMP-PKG signal coupled with NO via estrogen receptor (ERɑ)-mediated non-nuclear signaling, also known as rapid signaling or membrane-initiated steroid signaling (Adlanmerini et al., 2014; Arnal et al., 2017). In a female mouse model of heart failure, PDE5i fails to provide heart-protective effects in the absence of estrogen. We previously demonstrated that sildenafil treatment failed to exert anti-remodeling effects in female pathological hypertrophy heart in from Gαq-overexpressing or pressure-overloaded mice after ovary removal; on the contrary, estrogen replacement recovered the protective effects of sildenafil (Sasaki et al., 2014). Rüdebusch et al. (2020) also demonstrated that sGC stimulation has protective effects associated with improved gene expressions in mice heart failure model induced by pressure overload (Rüdebusch et al., 2020) and interestingly we have recently reported that this sGC protective effects are independent of estrogen status in rodent pressure-overload model (Nobuaki et al., 2020).
Despite promising preclinical results, however, a clinical study testing vericiguat in patients with HFpEF turned out negative (Vitality HFpEF). Although the reason for the negative results remains an open question, the redox status related to HFpEF might be speculated to be involved. NO–sGC–cGMP signaling can be compromised either by reducing the bioavailability of NO or by altering the redox state of sGC itself (Costell et al., 2012). Several groups reported that redox conditions altered cysteine residues (Cys) on sGC, affecting its catalytic or regulatory functions (Craven and DeRubertis, 1978a; Craven and DeRubertis, 1978b; Braughler, 1983). The redox status also alters the heme conditions within sGC. Heme iron in the reduced status (Fe2+) is necessary for NO binding, and sGC stimulator can stimulate only the reduced form of sGC, while the sGC activator can activate both reduced sGC and oxidized sGC (containing Fe3+) (Evgenov et al., 2006). In rat external iliac arteries without endothelium, peroxynitrite was reported to alter the redox state of sGC. Under the exposure of peroxynitrite, vascular relaxation induced by an sGC stimulator was impaired, whereas that by an sGC activator was enhanced. Additionally, this response correlated well with tissue levels of cGMP (Tawa et al., 2014). In Sprague Dawley rats fed with high salt/fat diet, an sGC activator, but not an sGC stimulator, attenuated the development of cardiac hypertrophy in a blood pressure-independent manner (Evgenov et al., 2006). Although there are no data about sGC redox status in patients with heart failure, inflammation and oxidative stress conditions in HFpEF might critically affect the efficacy of cGMP-modifying drugs (Tawa et al., 2014).
Thus, an sGC activator might serve as a potential novel treatment of HFpEF. So far, cinaciguat, an sGC activator, has been tested only in acute heart failure, with increased hypotensive events but no clear benefits, and sGC activators have not yet been explored in patients with chronic heart failure.
NP-pGC Pathway (PDE9 and PDE5)
Natriuretic peptides stimulate transmembrane receptor guanylate cyclase to produce cGMP. Atrial and B-type natriuretic peptides (ANP, BNP) bind to receptor particulate guanylyl cyclase A (pGC-A or NPRA), while C-type natriuretic peptide (CNP) binds to particulate guanylyl cyclase B (pGC-B or NPRB). pGC-A is localized at T-tubules and pGC-B is distributed throughout the sarcolemma. This spatial difference renders compartmentalized ANP/NPRA/cGMP signaling vs. CNP/NPRB/cGMP: the former have little impact on contractility and the latter have positive-lusitropic effects (Kuhn, 2016; Subramanian et al., 2018; Michel et al., 2020). cGMP from NP-pGC axis is degraded by PDE9 (Volpe et al., 2016; Goetze et al., 2020), which is expressed prominently in the brain and less in the heart (GraceKim et al., 2017). Similar to PDE5, myocardial PDE9 expression is low under physiological conditions but is upregulated under disease conditions such as HFpEF and aortic stenosis (Lee et al., 2015). PDE9 inhibition, either with a pharmacological or a genetic approach, suppressed cardiac hypertrophy in rodent pressure-overload (PO) model (Lee et al., 2015; Kokkonen-Simon et al., 2018; Richards et al., 2021). Importantly, both PDE5i and PDE9i similarly improve diastolic distensibility and ameliorate cardiac remodeling, associated with better profiles of hypertrophic/fibrosis-related gene expression (Lee et al., 2015), (Bishu et al., 2011a); however, comprehensive analyses of RNA-sequence data of myocardium reveals significant differences between PDE5i and PDE9i (Kokkonen-Simon et al., 2018), particularly in miRNA profiles related to hypertrophy and fibrosis: marked down-regulation of pro-hypertrophic and pro-fibrotic miRs by PDE5i vs. virtually no effect by PDE9i.
As previously described, ARNI exhibited favorable outcome in female patients with HFpEF (Solomon et al., 2019; Pieske et al., 2021). There has been no explanation provided for this observation of female-only benefit. We would speculate that this might be possibly related to difference of plasma NPs levels. Female patients with HFpEF are reported to exhibit lower plasma NPs levels as follows. ARNI might compensate lower levels of NPs in female patients with HFpEF. In HFpEF patients, plasma BNP levels are reported to be lower than in HFrEF (Harada et al., 2017); interestingly, women with HFpEF had lower BNP levels than men [43.9 vs. 76.1 pmol/L, p = 0.0193 (Tasevska-Dinevska et al., 2011), 104 vs 133, p < 0.001 (Savarese and D’Amario, 2018)] while in HFrEF the levels of NPs were inconsistent. One group reported that the plasma levels of ANP and BNP were similar in both genders (ANP: 114.9 vs. 141.2 pg/ml, p = 0.2606, BNP: 252.0 vs. 381.9 pg/ml, p = 0.1577). Another group reported that the plasma levels of NT-proBNP were higher in female HFrEF (8481 vs. 7543 pg/ml, p < 0.001) (Kim et al., 2017) and there is another group reporting that plasma NT-proBNP levels were similar in both genders (2532 vs. 2677 pg/ml, p = 0.978) (Sobhani et al., 2018). Another possible reason why ARNI is effective in female HFpEF might be related to CNP regulation. CNP exerts biological effects by binding to two types of natriuretic receptors; cGMP-coupled NPR-B and NPR-C (Chauhan et al., 2003; Villar et al., 2007). Endothelial deletion of CNP or global deletion of NPR-C revealed hypertensive phenotype only in female mice (Moyes et al., 2014), while the absence of eNOS and COX-1 in endothelial cells had no effect on mean blood pressure in female mice, but resulted in significantly high blood pressure in male animals (Scotland et al., 2005).
These suggest the pivotal contribution of CNP to female blood pressure maintenance. It is thus tempting to speculate that cardiac protection from ARNI therapy might depend more on the regulation of CNP in females than in males, although the contribution of cGMP might be unclear.
Although PDE5 hydrolyzes cGMP coupled with NO under normal conditions. PDE5 could become interactive with NPs-derived cGMP under stressed conditions (Zhang et al., 2012). Cardiomyocyte PDE5 is normally localized at Z-bands of sarcomeres, but becomes diffusely localized when exposed to pathological stress such as TAC or NOS inhibition (Nagayama et al., 2008; Zhang et al., 2012). In a dog hypertension model produced by bilateral renal wrapping, sildenafil treatment with concomitant BNP administration enhances plasma cGMP concentration, and recovers left ventricular diastolic capacitance in association with titin phosphorylation compared with sildenafil treatment alone (Bishu et al., 2011b). The beneficial synergistic effects of the combined PDE5 and NPs were also reported in pulmonary hypertension (PH). In a mouse model of hypoxia-induced PH, global deletion of NPRA blunts the beneficial effects of sildenafil on right ventricular systolic pressure (Zhao et al., 2003). Also, in hypoxia-induced PH rats, ANP and sildenafil show synergistic effects on decreasing right ventricular systolic pressure and on increasing plasma cGMP levels (Preston et al., 2004). Furthermore, a recent clinical trial of pulmonary arterial hypertension also demonstrated that the combined inhibition of neprilysin and PDE5 increase both plasma NP and cGMP levels and decreased pulmonary vascular resistance without affecting systemic blood pressure (Hobbs et al., 2019), which makes contrast to the concomitant use of PDE5 inhibitor (sildenafil) with sGC stimulator (riociguat) having been reported to be associated with hypotension but without beneficial effects on hemodynamics or exercise capacity (Galiè et al., 2015). The combination of pGC-related pathway and PDE5 might be a potential therapeutic option also in heart failure.
PKG Oxidation in Failing Heart
cGMP activated PKG targets various molecules to regulate cellular function in cardiomyocytes (Takimoto, 2012), including RGS2/4, TRPC6, proteasome systems, mitochondria, and sarcomere components. Two PKG genes, prkg1 and prkg2, encode PKG1 and PKG2, respectively, and PKG1 is the primary isotype in cardiomyocyte. PKG1 is activated classically by cGMP, but also by oxidation (Figure 2): When oxidized, a cysteine residue C43(C42 in mice) forms a disulfide bond to form a homodimer of PKG1 (Burgoyne et al., 2007). Oxidized PKG1 is increased in failing hearts, though it accounts for only a small portion of PKG1 in normal hearts (Paulus et al., 2013; Nakamura et al., 2015; Prysyazhna et al., 2016). Oxidative PKG1 resides only at cytosol but not at the plasma membrane, while unoxidized PKG1 resides in both (Nakamura et al., 2015). Therefore, oxidized PKGI is no longer able to exert beneficial effects by the mechanisms mediated by membrane-localized PKGI, including inhibition of TRPC6-Cn-NFAT hypertrophy signaling and TSC2-mTORC1 metabolic/autophagy signaling (Oeing et al., 2020). Interestingly, PKG1 oxidation is required for the anti-remodeling effects from PDE5i as cytosol-localized PDE5 needs cGMP-activation via its GAF domain, while sGC stimulation exerts anti-remodeling effects independent of redox status of PKG1 (Nakamura et al., 2018). PDE5 inhibitor could be effective only under the sufficient myocardial stress to oxidate PKG1α, whereas an sGC stimulator provides benefits independent of redox conditions.
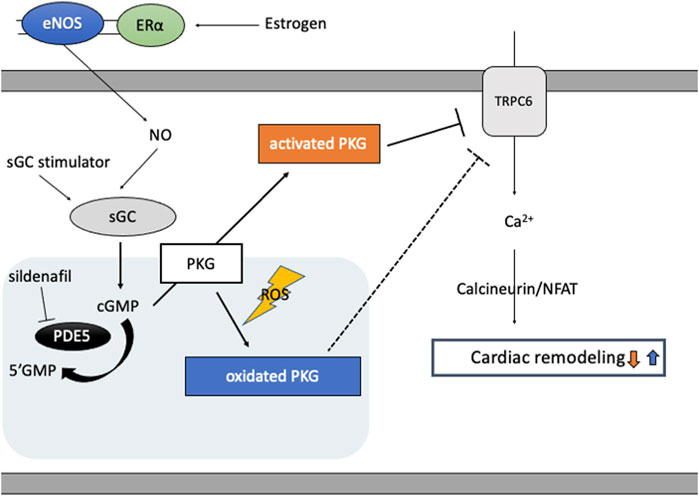
FIGURE 2. cGMP/PKG signaling and PKG oxidation. Estrogen plays a pivotal role in cGMP-PKG signaling coupled with eNOS via estrogen receptor (ERɑ)-mediated non-nuclear signaling. In diseased conditions like heart failure, eNOS activity is impaired and PKG undertakes oxidation and localizes in cytosol, inhibiting protective effects of PKG signaling independent of cGMP and enhancing cardiac remodeling via target proteins like TRPC6. PDE5 inhibitor, sildenafil, reveals protective effects only under the condition of sufficient oxidated PKG1α, while sGC stimulator improves cardiac remodeling independent of PKG redox status.
Conclusion
cGMP/PKG signaling can be augmented by stimulation of either NO-sGC pathway or NP-pGC pathway. Although activation of either provides anti-remodeling benefits, they do not necessarily share the same molecular mechanisms in common. Furthermore, benefits might be also affected by the PKG redox status. Although ample preclinical evidence shows the benefits of cGMP/PKG augmentation in HFrEF or HFpEF models, clinical studies thus far provide consistent efficacy of cGMP/PKG augmentation in patients with HFrEF and limited efficacy in patients with HFpEF. Further studies would be helpful to better understand the pathophysiology of HFpEF and the development of novel treatments.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the research grants from Fukuda Foundation for Medical Technology and JSPS KAKENHI (Grant-in-Aid for Scientific Research #21K08047).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adlanmerini, M., Solinhac, R., Abot, A., Fabre, A., Raymond-Letron, I., Guihot, A.-L., et al. (2014). Mutation of the Palmitoylation Site of Estrogen Receptor α In Vivo Reveals Tissue-specific Roles for Membrane versus Nuclear Actions. Proc. Natl. Acad. Sci. 111, E283 LP–E290. doi:10.1073/pnas.1322057111
Armstrong, P. W., Lam, C. S. P., Anstrom, K. J., Ezekowitz, J., Hernandez, A. F., O’Connor, C. M., et al. (2020). Effect of Vericiguat vs Placebo on Quality of Life in Patients with Heart Failure and Preserved Ejection Fraction: The VITALITY-HFpEF Randomized Clinical Trial. JAMA - J. Am. Med. Assoc. 324, 1512–1521. doi:10.1001/jama.2020.15922
Armstrong, P. W., Pieske, B., Anstrom, K. J., Ezekowitz, J., Hernandez, A. F., Butler, J., et al. (2020). Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 382, 1883–1893. doi:10.1056/NEJMoa1915928
Arnal, J.-F., Lenfant, F., Metivier, R., Flouriot, G., Henrion, D., Adlanmerini, M., et al. (2017). Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 97, 1045–1087. doi:10.1152/physrev.00024.2016
Baliga, R. S., Preedy, M. E. J., Dukinfield, M. S., Chu, S. M., Aubdool, A. A., Bubb, K. J., et al. (2018). Phosphodiesterase 2 Inhibition Preferentially Promotes NO/guanylyl cyclase/cGMP Signaling to Reverse the Development of Heart Failure. Proc. Natl. Acad. Sci. U. S. A. 115, E7428–E7437. doi:10.1073/pnas.1800996115
Bishu, K., Hamdani, N., Mohammed, S. F., Kruger, M., Ohtani, T., Ogut, O., et al. (2011). Sildenafil and B-type Natriuretic Peptide Acutely Phosphorylate Titin and Improve Diastolic Distensibility In Vivo. Circulation 124, 2882–2891. doi:10.1161/CIRCULATIONAHA.111.048520
Bishu, K., Hamdani, N., Mohammed, S. F., Kruger, M., Ohtani, T., Ogut, O., et al. (2011). Sildenafil and B-type Natriuretic Peptide Acutely Phosphorylate Titin and Improve Diastolic Distensibility In Vivo. Circulation 124, 2882–2891. doi:10.1161/CIRCULATIONAHA.111.048520
Bode, D. C., Kanter, J. R., and Brunton, L. L. (1991). Cellular Distribution of Phosphodiesterase Isoforms in Rat Cardiac Tissue. Circ. Res. 68, 1070–1079. doi:10.1161/01.res.68.4.1070
Braughler, J. M. (1983). Soluble Guanylate Cyclase Activation by Nitric Oxide and its Reversal. Involvement of Sulfhydryl Group Oxidation and Reduction. Biochem. Pharmacol. 32, 811–818. doi:10.1016/0006-2952(83)90581-6
Burgoyne, J. R., Madhani, M., Cuello, F., Charles, R. L., Brennan, J. P., Schröder, E., et al. (2007). Cysteine Redox Sensor in PKGIa Enables Oxidant-Induced Activation. Science 317, 13931397. doi:10.1126/science.1144318
Burley, D. S., Ferdinandy, P., and Baxter, G. F. (2007). Cyclic GMP and Protein Kinase-G in Myocardial Ischaemia-Reperfusion: Opportunities and Obstacles for Survival Signaling. Br. J. Pharmacol. 152, 855–869. doi:10.1038/sj.bjp.0707409
Chauhan, S. D., Nilsson, H., Ahluwalia, A., and Hobbs, A. J. (2003). Release of C-type Natriuretic Peptide Accounts for the Biological Activity of Endothelium-Derived Hyperpolarizing Factor. Proc. Natl. Acad. Sci. U. S. A. 100, 1426–1431. doi:10.1073/pnas.0336365100
Chen, W., Spitzl, A., Mathes, D., Nikolaev, V. O., Werner, F., Weirather, J., et al. (2016). Endothelial Actions of ANP Enhance Myocardial Inflammatory Infiltration in the Early Phase after Acute Infarction. Circ. Res. 119, 237–248. doi:10.1161/CIRCRESAHA.115.307196
Costell, M. H., Ancellin, N., Bernard, R. E., Zhao, S. F., Upson, J. J., Morgan, L. A., et al. (2012). Comparison of Soluble Guanylate Cyclase Stimulators and Activators in Models of Cardiovascular Disease Associated with Oxidative Stress. Front. Pharmacol. 3 (JUL), 1–14. doi:10.3389/fphar.2012.00128
Craven, P. A., and DeRubertis, F. R. (1978). Effects of Thiol Inhibitors on Hepatic Guanylate Cyclase Activity Evidence for the Involvement of Vicinal Dithiols in the Expression of Basal and Agonist-Stimulated Activity. BBA - Enzymol. 524, 231–244. doi:10.1016/0005-2744(78)90121-3
Craven, P. A., and DeRubertis, F. R. (1978). Restoration of the Responsiveness of Purified Guanylate Cyclase to Nitrosoguanidine, Nitric Oxide, and Related Activators by Heme and Hemeproteins. Evidence for Involvement of the Paramagnetic nitrosyl.Heme Complex in Enzyme Activation. J. Biol. Chem. 253, 8433–8443. doi:10.1016/s0021-9258(17)34310-7
Damage, H. E., Luft, F. C., Mervaala, E., Mu, D. N., Gross, V., Schmidt, F., et al. (1999). Influence of Exercise Training on Neurogenic Control of Blood Pressure in Spontaneously Hypertensive Rats. State-of-the-Art Lecture 34, 212–218. doi:10.1161/01.HYP.34.4.720
De Vecchis, R., Cesaro, A., and Ariano, C. (2018). Differential Effects of the Phosphodiesterase Inhibition in Chronic Heart Failure Depending on the Echocardiographic Phenotype (HFREF or HFpEF): a Meta-Analysis. Minerva Cardioangiol 66, 659–670. doi:10.23736/S0026-4725.17.04382-1
De Vecchis, R., Cesaro, A., Ariano, C., Giasi, A., and Cioppa, C. (2017). Phosphodiesterase-5 Inhibitors Improve Clinical Outcomes, Exercise Capacity and Pulmonary Hemodynamics in Patients with Heart Failure with Reduced Left Ventricular Ejection Fraction: A Meta-Analysis. J. Clin. Med. Res. 9, 488–498. doi:10.14740/jocmr3008w
Dittrich, M., Jurevicius, J., Georget, M., Rochais, F., Fleischmann, B. K., Hescheler, J., et al. (2001). Local Response of L-type Ca2+ Current to Nitric Oxide in Frog Ventricular Myocytes. J. Physiol. 534, 109–121. doi:10.1111/j.1469-7793.2001.00109.x
Evgenov, O. V., Pacher, P., Schmidt, P. M., Haskó, G., Schmidt, H. H. H. W., and Stasch, J. P. (2006). NO-independent Stimulators and Activators of Soluble Guanylate Cyclase: Discovery and Therapeutic Potential. Nat. Rev. Drug Discov. 5, 755–768. doi:10.1038/nrd2038
Finckenberg, P., Eriksson, O., Baumann, M., Merasto, S., Lalowski, M. M., Levijoki, J., et al. (2012). Caloric Restriction Ameliorates Angiotensin II-Induced Mitochondrial Remodeling and Cardiac Hypertrophy. Hypertension 59, 76–84. doi:10.1161/HYPERTENSIONAHA.111.179457
Fischer, R., Dechend, R., Qadri, F., Markovic, M., Feldt, S., Herse, F., et al. (2008). Dietary N-3 Polyunsaturated Fatty Acids and Direct Renin Inhibition Improve Electrical Remodeling in a Model of High Human Renin Hypertension. Hypertension 51, 540–546. doi:10.1161/HYPERTENSIONAHA.107.103143
Forman, D., and Gaziano, J. M. (2009). Irbesartan in Patients with Heart Failure and Preserved Ejection Fraction. Curr. Cardiovasc. Risk Rep. 3, 311–312. doi:10.1007/s12170-009-0056-1
Frantz, S., Klaiber, M., Baba, H. A., Oberwinkler, H., Völker, K., Gaßner, B., et al. (2013). Stress-dependent Dilated Cardiomyopathy in Mice with Cardiomyocyte- Restricted Inactivation of Cyclic GMP-dependent Protein Kinase I. Eur. Heart J. 34, 1233–1244. doi:10.1093/eurheartj/ehr445
Galiè, N., Müller, K., Scalise, A. V., and Grünig, E. (2015). PATENT PLUS: A Blinded, Randomised and Extension Study of Riociguat Plus Sildenafil in Pulmonary Arterial Hypertension. Eur. Respir. J. 45, 1314–1322. doi:10.1183/09031936.00105914
Geoffroy, V., Fouque, F., Nivet, V., Clot, J. P., Lugnier, C., Desbuquois, B., et al. (1999). Activation of a cGMP-Stimulated cAMP Phosphodiesterase by Protein Kinase C in a Liver Golgi-Endosomal Fraction. Eur. J. Biochem. 259, 892–900. doi:10.1046/j.1432-1327.1999.00123.x
Goetze, J. P., Bruneau, B. G., Ramos, H. R., Ogawa, T., de Bold, M. K., and de Bold, A. J. (2020). Cardiac Natriuretic Peptides. Nat. Rev. Cardiol. 17, 698–717. doi:10.1038/s41569-020-0381-0
GraceKim, E., PhDa, , David, A., and Kass, M. (2017). Cardiac Phosphodiesterases and Their Modulation for Treating Heart Disease Grace. Handb. Exp. Pharmacol. 243, 249–269. doi:10.1007/164_2016_82
Haase, N., Rugor, J., Przybyl, L., Qadri, F., Müller, D. N., and Dechend, R. (2014). Relaxin Does Not Improve Angiotensin II-Induced Target-Organ Damage. PLoS One 9, 1–7. doi:10.1371/journal.pone.0093743
Harada, E., Mizuno, Y., Kugimiya, F., Shono, M., Maeda, H., Yano, N., et al. (2017). B-type Natriuretic Peptide in Heart Failure with Preserved Ejection Fraction: Relevance to Age-Related Left Ventricular Modeling in Japanese. Circ. J. 81, 1006–1013. doi:10.1253/circj.CJ-16-1282
Hashimoto, T., Kim, G. E., Tunin, R. S., Adesiyun, T., Hsu, S., Nakagawa, R., et al. (2018). Acute Enhancement of Cardiac Function by Phosphodiesterase Type 1 Inhibition Translational Study in the Dog and Rabbit. Circulation 138, 1974–1987. doi:10.1161/CIRCULATIONAHA.117.030490
Herring, N., Rigg, L., Terrar, D. A., and Paterson, D. J. (2001). NO-cGMP Pathway Increases the Hyperpolarisation-Activated Current, if, and Heart Rate during Adrenergic Stimulation. Cardiovasc. Res. 52, 446–453. doi:10.1016/s0008-6363(01)00425-4
Hobbs, A. J., Moyes, A. J., Baliga, R. S., Ghedia, D., Ochiel, R., Sylvestre, Y., et al. (2019). Neprilysin Inhibition for Pulmonary Arterial Hypertension: a Randomized, Double-Blind, Placebo-Controlled, Proof-Of-Concept Trial. Br. J. Pharmacol. 176, 1251–1267. doi:10.1111/bph.14621
Jin, Z., Zhang, J., Zhi, H., Hong, B., Zhang, S., Guo, H., et al. (2013). The Beneficial Effects of Tadalafil on Left Ventricular Dysfunction in Doxorubicin-Induced Cardiomyopathy. J. Cardiol. 62, 110–116. doi:10.1016/j.jjcc.2013.03.018
Kim, G. E., and Kass, D. A. (2017). Cardiac Phosphodiesterases and Their Modulation for Treating Heart Disease. Handb. Exp. Pharmacol. 243, 249–269. doi:10.1007/164_2016_82
Kim, H. L., Kim, M. A., Choi, D. J., Han, S., Jeon, E. S., Cho, M. C., et al. (2017). Gender Difference in the Prognostic Value of N-Terminal Pro-B Type Natriuretic Peptide in Patients with Heart Failure ― a Report from the Korean Heart Failure Registry (KorHF). Circ. J. 81, 1329–1336. doi:10.1253/circj.CJ-16-1345
Koitabashi, N., Aiba, T., Hesketh, G. G., Rowell, J., Zhang, M., Takimoto, E., et al. (2010). Cyclic GMP/PKG-dependent Inhibition of TRPC6 Channel Activity and Expression Negatively Regulates Cardiomyocyte NFAT Activation: Novel Mechanism of Cardiac Stress Modulation by PDE5 Inhibition. J. Mol. Cell. Cardiol. 48, 713–724. doi:10.1016/j.yjmcc.2009.11.015
Kokkonen-Simon, K. M., Saberi, A., Nakamura, T., Ranek, M. J., Zhu, G., Bedja, D., et al. (2018). Marked Disparity of microRNA Modulation by cGMP-Selective PDE5 versus PDE9 Inhibitors in Heart Disease. JCI Insight 3, e121739. doi:10.1172/jci.insight.121739
Kolijn, D., Kovács, Á., Herwig, M., Lódi, M., Sieme, M., Alhaj, A., et al. (2020). Enhanced Cardiomyocyte Function in Hypertensive Rats with Diastolic Dysfunction and Human Heart Failure Patients after Acute Treatment with Soluble Guanylyl Cyclase (sGC) Activator. Front. Physiol. 11, 1–21. doi:10.3389/fphys.2020.00345
Krishnan, S. M., Kraehling, J. R., Eitner, F., Bénardeau, A., and Sandner, P. (2018). The Impact of the Nitric Oxide (No)/soluble Guanylyl Cyclase (sGC) Signaling cascade on Kidney Health and Disease: A Preclinical Perspective. Int. J. Mol. Sci. 19. doi:10.3390/ijms19061712
Kuhn, M. (2016). Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 96, 751–804. doi:10.1152/physrev.00022.2015
Kukreja, R. C., Salloum, F. N., and Das, A. (2012). Cyclic Guanosine Monophosphate Signaling and Phosphodiesterase-5 Inhibitors in Cardioprotection. J. Am. Coll. Cardiol. 59, 1921–1927. doi:10.1016/j.jacc.2011.09.086
Layland, J., Solaro, R. J., and Shah, A. M. (2005). Regulation of Cardiac Contractile Function by Troponin I Phosphorylation. Cardiovasc. Res. 66, 12–21. doi:10.1016/j.cardiores.2004.12.022
Le Trong, H., Walsh, K. A., Charbonneau, H., Beier, N., Sonnenburg, W. K., Stroop, S. D., et al. (1990). Amino Acid Sequence of the Cyclic GMP Stimulated Cyclic Nucleotide Phosphodiesterase from Bovine Heart. Biochemistry 29, 10280–10288. doi:10.1021/bi00496a018
Lee, D. I., Zhu, G., Sasaki, T., Cho, G.-S., Hamdani, N., Holewinski, R., et al. (2015). Phosphodiesterase 9A Controls Nitric-oxide-independent cGMP and Hypertrophic Heart Disease. Nature 519, 472–476. doi:10.1038/nature14332
Levy, F. O. (2013). Cardiac PDEs and Crosstalk between cAMP and cGMP Signalling Pathways in the Regulation of Contractility. Naunyn. Schmiedebergs. Arch. Pharmacol. 386, 665–670. doi:10.1007/s00210-013-0874-z
Li, N., Yuan, Y., Li, S., Zeng, C., Yu, W., Shen, M., et al. (2016). Pde5 Inhibitors Protect against post-infarction Heart Failure. Front. Biosci. - Landmark 21, 1194–1210. doi:10.2741/4450
Lommi, J., Pulkki, K., Koskinen, P., Naveri, H., Leinonen, H., Harkonen, M., et al. (1997). Haemodynamic, Neuroendocrine and Metabolic Correlates of Circulating Cytokine Concentrations in Congestive Heart Failure. Eur. Heart J. 18, 1620–1625. doi:10.1093/oxfordjournals.eurheartj.a015142
Lugnier, C., Keravis, T., Le Bec, A., Pauvert, O., Proteau, S., and Rousseau, E. (1999). Characterization of Cyclic Nucleotide Phosphodiesterase Isoforms Associated to Isolated Cardiac Nuclei. Biochim. Biophys. Acta - Gen. Subj. 1472, 431–446. doi:10.1016/s0304-4165(99)00145-2
Lukowski, R., Rybalkin, S. D., Loga, F., Leiss, V., Beavo, J. A., and Hofmann, F. (2010). Cardiac Hypertrophy Is Not Amplified by Deletion of cGMP-dependent Protein Kinase I in Cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 107, 5646–5651. doi:10.1073/pnas.1001360107
McMurray, J. J. V., Packer, M., Desai, A. S., Gong, J., Lefkowitz, M. P., Rizkala, A. R., et al. (2014). Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. N. Engl. J. Med. 371, 993–1004. doi:10.1056/NEJMoa1409077
Mehel, H., Emons, J., Vettel, C., Wittköpper, K., Seppelt, D., Dewenter, M., et al. (2013). Phosphodiesterase-2 Is Up-Regulated in Human Failing Hearts and Blunts β-adrenergic Responses in Cardiomyocytes. J. Am. Coll. Cardiol. 62, 1596–1606. doi:10.1016/j.jacc.2013.05.057
Mervaala, E. M. A., Cheng, Z. J., Tikkanen, I., Lapatto, R., Nurminen, K., Vapaatalo, H., et al. (2001). Endothelial Dysfunction and Xanthine Oxidoreductase Activity in Rats with Human Renin and Angiotensinogen Genes. Hypertension 37, 414–418. doi:10.1161/01.hyp.37.2.414
Mery, P., Pavoines, C., Belhassen, L., and Peckers, F. (1993). Nitric Oxide Regulates Cardiac Ca2+ Current. J. Biol. Chem. 268, 26286–26295. doi:10.1016/s0021-9258(19)74313-0
Michel, K., Herwig, M., Werner, F., Spes, K. Š., Abeßer, M., Schuh, K., et al. (2020). C-type Natriuretic Peptide Moderates Titin-Based Cardiomyocyte Stiffness. JCI Insight 5. doi:10.1172/jci.insight.139910
Miller, C. L., Oikawa, M., Cai, Y., Wojtovich, A. P., Nagel, D. J., Xu, X., et al. (2009). Role of Ca2+/calmodulin-Stimulated Cyclic Nucleotide Phosphodiesterase 1 in Mediating Cardiomyocyte Hypertrophy. Circ. Res. 105, 956–964. doi:10.1161/CIRCRESAHA.109.198515
Mongillo, M., Tocchetti, C. G., Terrin, A., Lissandron, V., Cheung, Y. F., Dostmann, W. R., et al. (2006). Compartmentalized Phosphodiesterase-2 Activity Blunts β-adrenergic Cardiac Inotropy via an NO/cGMP-dependent Pathway. Circ. Res. 98, 226–234. doi:10.1161/01.RES.0000200178.34179.93
Moyes, A. J., Khambata, R. S., Villar, I., Bubb, K. J., Baliga, R. S., Lumsden, N. G., et al. (2014). Endothelial C-type Natriuretic Peptide Maintains Vascular Homeostasis. J. Clin. Invest. 124, 4039–4051. doi:10.1172/JCI74281
Muller, B., Stoclet, J. C., and Lugnier, C. (1992). Cytosolic and Membrane-Bound Cyclic Nucleotide Phosphodiesterases from guinea Pig Cardiac Ventricles. Eur. J. Pharmacol. Mol. Pharmacol. 225, 263–272. doi:10.1016/0922-4106(92)90028-t
Nagayama, T., Zhang, M., Hsu, S., Takimoto, E., and Kass, D. A. (2008). Sustained Soluble Guanylate Cyclase Stimulation Offsets Nitric-Oxide Synthase Inhibition to Restore Acute Cardiac Modulation by Sildenafil. J. Pharmacol. Exp. Ther. 326, 380–387. doi:10.1124/jpet.108.137422
Nakamura, T., Ranek, M. J., Lee, D. I., Hahn, V. S., Kim, C., Eaton, P., et al. (2015). Prevention of PKG1a Oxidation Augments Cardioprotection in the Stressed Heart. J. Clin. Invest. 125, 2468–2472. doi:10.1172/JCI80275
Nakamura, T., Zhu, G., Ranek, M. J., Kokkonen-Simon, K., Zhang, M., Kim, G. E., et al. (2018). Prevention of PKG-1α Oxidation Suppresses Antihypertrophic/Antifibrotic Effects from PDE5 Inhibition but Not sGC Stimulation. Circ. Hear. Fail. 11, e004740. doi:10.1161/CIRCHEARTFAILURE.117.004740
Nobuaki, F., Eiki, T., Kazutaka, U., Pangyen, L., Miyu, T., Yu, O., et al. (2020). Estrogen Receptor-α Non-nuclear Signaling Confers Cardioprotection and Is Essential to cGMP-PDE5 Inhibition Efficacy. JACC Basic Transl. Sci. 5, 282–295.
Oeing, C. U., Nakamura, T., Pan, S., Mishra, S., Dunkerly-Eyring, B. L., Kokkonen-Simon, K. M., et al. (2020). PKG1a Cysteine-42 Redox State Controls mTORC1 Activation in Pathological Cardiac Hypertrophy. Circ. Res. 127, 522–533. doi:10.1161/CIRCRESAHA.119.315714
Pablo, A. (2017). Olavegogeascoechea. De la evidencia a la práctica en la insuficiencia cardíaca. Rev. Argentina Med. 5, 132–133.
Patel, C. H., Nakamura, T., Zhu, G., Bedja, D., Sasaki, M., Holewinski, R. J., et al. (2019). PKG1-modified TSC2 Regulates mTORC1 Activity to Counter Adverse Cardiac Stress. Nature 566, 264–269.
Patrucco, E., Domes, K., Sbroggió, M., Blaich, A., Schlossmann, J., Desch, M., et al. (2014). Roles of cGMP-dependent Protein Kinase I (cGKI) and PDE5 in the Regulation of Ang II-Induced Cardiac Hypertrophy and Fibrosis. Proc. Natl. Acad. Sci. U. S. A. 111, 12925–12929. doi:10.1073/pnas.1414364111
Paulus, W. J., Tschöpe, C., and D, P. H. (2013). A Novel Paradigm for Heart Failure with Preserved Ejection Fraction: Comorbidities Drive Myocardial Dysfunction and Remodeling through Coronary Microvascular Endothelial Inflammation. J. Am. Coll. Cardiol. 62, 263–271. doi:10.1016/j.jacc.2013.02.092
Pieske, B., Patel, M. J., Westerhout, C. M., Anstrom, K. J., Butler, J., Ezekowitz, J., et al. (2019). Baseline Features of the VICTORIA (Vericiguat Global Study in Subjects with Heart Failure with Reduced Ejection Fraction) Trial. Eur. J. Heart Fail. 21, 1596–1604. doi:10.1002/ejhf.1664
Pieske, B., Wachter, R., Shah, S. J., Baldridge, A., Szeczoedy, P., Ibram, G., et al. (2021). Effect of Sacubitril/Valsartan vs Standard Medical Therapies on Plasma NT-proBNP Concentration and Submaximal Exercise Capacity in Patients with Heart Failure and Preserved Ejection Fraction: The PARALLAX Randomized Clinical Trial. JAMA - J. Am. Med. Assoc. 326, 1919–1929.
Pokreisz, P., Vandenwijngaert, S., Bito, V., Van Bergh, A. Den., Lenaerts, I., Busch, C., et al. (2009). Ventricular Phosphodiesterase-5 Expression Is Increased in Patients with Advanced Heart Failure and Contributes to Adverse Ventricular Remodeling after Myocardial Infarction in Mice. Circulation 119, 408–416. doi:10.1161/CIRCULATIONAHA.108.822072
Preston, I. R., Hill, N. S., Gambardella, L. S., Warburton, R. R., and Klinger, J. R. (2004). Synergistic Effects of ANP and Sildenafil on cGMP Levels and Amelioration of Acute Hypoxic Pulmonary Hypertension. Exp. Biol. Med. 229, 920–925. doi:10.1177/153537020422900908
Prigent, A. F., Fougier, S., Nemoz, G., Anker, G., Pacheco, H., Lugnier, C., et al. (1988). Comparison of Cyclic Nucleotide Phosphodiesterase Isoforms from Rat Heart and Bovine Aorta. Separation and Inhibition by Selective Reference Phosphodiesterase Inhibitors. Biochem. Pharmacol. 37, 3671–3681. doi:10.1016/0006-2952(88)90400-5
Prysyazhna, O., Burgoyne, J. R., Scotcher, J., Grover, S., Kass, D., and Eaton, P. (2016). Phosphodiesterase 5 Inhibition Limits Doxorubicin-Induced Heart Failure by Attenuating Protein Kinase G Iα Oxidation. J. Biol. Chem. 291, 17427–17436. doi:10.1074/jbc.M116.724070
Ramzi, O., Salloum, F., Hawkins, J., and Kukreja, R. C. (2002). Sildenafil (Viagra) Induces Powerful Cardioprotective Effect via Opening of Mitochondrial KATP Channels in Rabbits. Am. J. Physiol. Hear. Circ. Physiol. 283, H1263–H1269. doi:10.1152/ajpheart.00324.2002
Ranek, M. J., Terpstra, E. J. M., Li, J., Kass, D. A., and Wang, X. (2013). Protein Kinase G Positively Regulates Proteasome-Mediated Degradation of Misfolded Proteins. Circulation 128, 365–376. doi:10.1161/CIRCULATIONAHA.113.001971
Redfield, M. M., Chen, H. H., Borlaug, B. A., Semigran, M. J., Lee, K. L., Lewis, G., et al. (2013). Effect of Phosphodiesterase-5 Inhibition on Exercise Capacity and Clinical Status in Heart Failure with Preserved Ejection Fraction: A Randomized Clinical Trial. JAMA - J. Am. Med. Assoc. 309, 1268–1277. doi:10.1001/jama.2013.2024
Richards, D. A., Aronovitz, M. J., Liu, P., Martin, G. L., Tam, K., Pande, S., et al. (2021). CRD-733, a Novel PDE9 (Phosphodiesterase 9) Inhibitor, Reverses Pressure Overload-Induced Heart Failure. Circ. Hear. Fail. 14, e007300. doi:10.1161/circheartfailure.120.007300
Rüdebusch, J., Benkner, A., Nath, N., Fleuch, L., Kaderali, L., Grube, K., et al. (2020). Stimulation of Soluble Guanylyl Cyclase (sGC) by Riociguat Attenuates Heart Failure and Pathological Cardiac Remodelling. Br. J. Pharmacol. 1, 13. doi:10.1111/bph.15333
Sadhu, K., Hensley, K., Florio, V. A., and Wolda, S. L. (1999). Differential Expression of the Cyclic GMP-Stimulated Phosphodiesterase PDE2A in Human Venous and Capillary Endothelial Cells. J. Histochem. Cytochem. 47, 895–905. doi:10.1177/002215549904700707
Salloum, F. N., Abbate, A., Das, A., Houser, J. E., Mudrick, C. A., Qureshi, I. Z., et al. (2008). Sildenafil (Viagra) Attenuates Ischemic Cardiomyopathy and Improves Left Ventricular Function in Mice. Am. J. Physiol. - Hear. Circ. Physiol. 294, 1398–1406. doi:10.1152/ajpheart.91438.2007
Sasaki, H., Nagayama, T., Blanton, R. M., Seo, K., Zhang, M., Zhu, G., et al. (2014). PDE5 Inhibitor Efficacy Is Estrogen Dependent in Female Heart Disease. J. Clin. Invest. 124, 2464–2471. doi:10.1172/JCI70731
Savarese, G., and D’Amario, D. (2018). Sex Differences in Heart Failure. Adv. Exp. Med. Biol. 1065, 529–544. doi:10.1007/978-3-319-77932-4_32
Savill, P. (2014). Spironolactone in Heart Failure with Preserved Ejection Fraction. Practitioner 258, 10.
Scotland, R. S., Madhani, M., Chauhan, S., Moncada, S., Andresen, J., Nilsson, H., et al. (2005). Investigation of Vascular Responses in Endothelial Nitric Oxide Synthase/cyclooxygenase-1 Double-Knockout Mice: Key Role for Endothelium-Derived Hyperpolarizing Factor in the Regulation of Blood Pressure In Vivo. Circulation 111, 796–803. doi:10.1161/01.CIR.0000155238.70797.4E
Seo, K., Rainer, P. P., Lee, D. I., Hao, S., Bedja, D., Birnbaumer, L., et al. (2014). Hyperactive Adverse Mechanical Stress Responses in Dystrophic Heart Are Coupled to Transient Receptor Potential Canonical 6 and Blocked by Cgmp-Protein Kinase G Modulation. Circ. Res. 114, 823–832. doi:10.1161/CIRCRESAHA.114.302614
Shan, X., Quaile, M. P., Monk, J. K., French, B., Cappola, T. P., and Margulies, K. B. (2012). Differential Expression of Pde5 in Failing and Nonfailing Human Myocardium. Circ. Hear. Fail. 5, 79–86. doi:10.1161/CIRCHEARTFAILURE.111.961706
Sobhani, K., Nieves Castro, D. K., Fu, Q., Gottlieb, R. A., Van Eyk, J. E., and Noel Bairey Merz, C. (2018). Sex Differences in Ischemic Heart Disease and Heart Failure Biomarkers. Biol. Sex. Differ. 9, 1–13. doi:10.1186/s13293-018-0201-y
Solomon, S. D., McMurray, J. J. V., Anand, I. S., Ge, J., Lam, C. S. P., Maggioni, A. P., et al. (2019). Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 381, 1609–1620. doi:10.1056/NEJMoa1908655
Sotomi, Y., Hikoso, S., Nakatani, D., Mizuno, H., Okada, K., Dohi, T., et al. (2021). Sex Differences in Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 10, 1–20. doi:10.1161/jaha.120.018574
Stangherlin, A., Gesellchen, F., Zoccarato, A., Terrin, A., Fields, L. A., Berrera, M., et al. (2011). CGMP Signals Modulate Camp Levels in a Compartment-specific Manner to Regulate Catecholamine-dependent Signaling in Cardiac Myocytes. Circ. Res. 108, 929–939. doi:10.1161/CIRCRESAHA.110.230698
Stephenson, D. T., Coskran, T. M., Wilhelms, M. B., Adamowicz, W. O., O’Donnell, M. M., Muravnick, K. B., et al. (2009). Immunohistochemical Localization of Phosphodiesterase 2A in Multiple Mammalian Species. J. Histochem. Cytochem. 57, 933–949. doi:10.1369/jhc.2009.953471
Straubinger, J., Schöttle, V., Bork, N., Subramanian, H., Dünnes, S., Russwurm, M., et al. (2015). Sildenafil Does Not Prevent Heart Hypertrophy and Fibrosis Induced by Cardiomyocyte Angiotensin II Type 1 Receptor Signalings. J. Pharmacol. Exp. Ther. 354, 406–416. doi:10.1124/jpet.115.226092
Subramanian, H., Froese, A., Jönsson, P., Schmidt, H., Gorelik, J., and Nikolaev, V. O. (2018). Distinct Submembrane Localisation Compartmentalises Cardiac NPR1 and NPR2 Signalling to cGMP. Nat. Commun. 9, 1–9. doi:10.1038/s41467-018-04891-5
Sugioka, M., Ito, M., Masuoka, H., Ichikawa, K., Konishi, T., Tanaka, T., et al. (1994). Identification and Characterization of Isoenzymes of Cyclic Nucleotide Phosphodiesterase in Human Kidney and Heart, and the Effects of New Cardiotonic Agents on These Isoenzymes. Naunyn. Schmiedebergs. Arch. Pharmacol. 350, 284–293. doi:10.1007/BF00175034
Takimoto, E. (2012). Cyclic GMP-dependent Signaling in Cardiac Myocytes. Circ. J. 76, 1819–1825. doi:10.1253/circj.cj-12-0664
Takimoto, E., Koitabashi, N., Hsu, S., Ketner, E. A., Zhang, M., Nagayama, T., et al. (2009). The. Regulator of G Protein Signaling 2 Mediates Cardiac Compensation to Pressure Overload and Antihypertrophic Effects of PDE5 Inhibition in Mice. J. Clin. Invest. 119, 408–420. doi:10.1172/JCI35620
Takimoto, E., Champion, H. C., Li, M., Belardi, D., Ren, S., Rodriguez, E. R., et al. (2005). Chronic Inhibition of Cyclic GMP Phosphodiesterase 5A Prevents and Reverses Cardiac Hypertrophy. Nat. Med. 11, 214–222. doi:10.1038/nm1175
Tasevska-Dinevska, G., Kennedy, L. M., Cline-Iwarson, A., Cline, C., Erhardt, L., and Willenheimer, R. (2011). Gender Differences in Variables Related to B-Natriuretic Peptide, Left Ventricular Ejection Fraction and Mass, and Peak Oxygen Consumption, in Patients with Heart Failure. Int. J. Cardiol. 149, 364–371. doi:10.1016/j.ijcard.2010.02.018
Tawa, M., Shimosato, T., Iwasaki, H., Imamura, T., and Okamura, T. (2014). Effects of Peroxynitrite on Relaxation through the NO/sGC/cGMP Pathway in Isolated Rat Iliac Arteries. J. Vasc. Res. 51, 439–446. doi:10.1159/000371491
Terasaki, W. L., and Appleman, M. M. (1975). The Role of Cyclic GMP in the Regulation of Cyclic AMP Hydrolysis. Metabolism 24, 311–319. doi:10.1016/0026-0495(75)90112-2
Thoonen, R., Giovanni, S., Govindan, S., Lee, D. I., Wang, G.-R., Calamaras, T. D., et al. (2015). Molecular Screen Identifies Cardiac Myosin-Binding Protein-C as a Protein Kinase G-Iα Substrate. Circ. Heart Fail. 8, 1115–1122. doi:10.1161/CIRCHEARTFAILURE.115.002308
Torre-Amione, G., Kapadia, S., Benedict, C., Oral, H., Young, J. B., and Mann, D. L. (1996). Proinflammatory Cytokine Levels in Patients with Depressed Left Ventricular Ejection Fraction: A Report from the Studies of Left Ventricular Dysfunction (SOLVD). J. Am. Coll. Cardiol. 27, 1201–1206. doi:10.1016/0735-1097(95)00589-7
Tsai, E. J., and Kass, D. A. (2009). Cyclic GMP Signaling in Cardiovascular Pathophysiology and Therapeutics. Pharmacol. Ther. 122, 216–238. doi:10.1016/j.pharmthera.2009.02.009
Udelson, J. E., Lewis, G. D., Shah, S. J., Zile, M. R., Redfield, M. M., Burnett, J., et al. (2020). Effect of Praliciguat on Peak Rate of Oxygen Consumption in Patients with Heart Failure with Preserved Ejection Fraction: The CAPACITY HFpEF Randomized Clinical Trial. JAMA - J. Am. Med. Assoc. 324, 1522–1531. doi:10.1001/jama.2020.16641
Van Heerebeek, L., Hamdani, N., Falcão-Pires, I., Leite-Moreira, A. F., Begieneman, M. P. V., Bronzwaer, J. G. F., et al. (2012). Low Myocardial Protein Kinase G Activity in Heart Failure with Preserved Ejection Fraction. Circulation 126, 830–839. doi:10.1161/CIRCULATIONAHA.111.076075
Velazquez, E. J., Morrow, D. A., DeVore, A. D., Duffy, C. I., Ambrosy, A. P., McCague, K., et al. (2018). Angiotensin–Neprilysin Inhibition in Acute Decompensated Heart Failure. N. Engl. J. Med. 380, 539–548. doi:10.1056/NEJMoa1812851
Vettel, C., Lämmle, S., Ewens, S., Cervirgen, C., Emons, J., Ongherth, A., et al. (2014). PDE2-mediated cAMP Hydrolysis Accelerates Cardiac Fibroblast to Myofibroblast Conversion and Is Antagonized by Exogenous Activation of cGMP Signaling Pathways. Am. J. Physiol. - Hear. Circ. Physiol. 306, 1246–1252. doi:10.1152/ajpheart.00852.2013
Villar, I. C., Panayiotou, C. M., Sheraz, A., Madhani, M., Scotland, R. S., Nobles, M., et al. (2007). Definitive Role for Natriuretic Peptide Receptor-C in Mediating the Vasorelaxant Activity of C-type Natriuretic Peptide and Endothelium-Derived Hyperpolarising Factor. Cardiovasc. Res. 74, 515–525. doi:10.1016/j.cardiores.2007.02.032
Volpe, M., Carnovali, M., and Mastromarino, V. (2016). The Natriuretic Peptides System in the Pathophysiology of Heart Failure: From Molecular Basis to Treatment. Clin. Sci. 130, 57–77. doi:10.1042/CS20150469
Weber, S., Zeller, M., Guan, K., Wunder, F., Wagner, M., and El-Armouche, A. (2017). PDE2 at the Crossway between cAMP and cGMP Signalling in the Heart. Cell. Signal. 38, 76–84. doi:10.1016/j.cellsig.2017.06.020
Wellner, M., Dechend, R., Park, J. K., Shagdarsuren, E., Al-Saadi, N., Kirsch, T., et al. (2005). Cardiac Gene Expression Profile in Rats with Terminal Heart Failure and Cachexia. Physiol. Genomics 20, 256–267. doi:10.1152/physiolgenomics.00165.2004
Wijnker, P. J. M., Murphy, A. M., Stienen, G. J. M., and van der Velden, (2014). J. Troponin I Phosphorylation in Human Myocardium in Health and Disease. Neth. Heart J. 22, 463–469. doi:10.1007/s12471-014-0590-4
Wilck, N., Markó, L., Balogh, A., Kräker, K., Herse, F., Bartolomaeus, H., et al. (2018). Nitric Oxide-Sensitive Guanylyl Cyclase Stimulation Improves Experimental Heart Failure with Preserved Ejection Fraction. JCI insight 3. doi:10.1172/jci.insight.96006
Zannad, F., Anker, S. D., Byra, W. M., Cleland, J. G. F., Fu, M., Gheorghiade, M., et al. (2018). Rivaroxaban in Patients with Heart Failure, Sinus Rhythm, and Coronary Disease. N. Engl. J. Med. 379, 1332–1342. doi:10.1056/NEJMoa1808848
Zhang, M., Koitabashi, N., Nagayama, T., Rambaran, R., Feng, N., Takimoto, E., et al. (2008). Expression, Activity, and Pro-hypertrophic Effects of PDE5A in Cardiac Myocytes. Cell. Signal. 20, 2231–2236. doi:10.1016/j.cellsig.2008.08.012
Zhang, M., Takimoto, E., Hsu, S., Lee, D. I., Nagayama, T., Danner, T., et al. (2010). Myocardial Remodeling Is Controlled by Myocyte-Targeted Gene Regulation of Phosphodiesterase Type 5. J. Am. Coll. Cardiol. 56, 2021–2030. doi:10.1016/j.jacc.2010.08.612
Zhang, M., Takimoto, E., Lee, D. I., Santos, C. X. C., Nakamura, T., Hsu, S., et al. (2012). Pathological Cardiac Hypertrophy Alters Intracellular Targeting of Phosphodiesterase Type 5 from Nitric Oxide Synthase-3 to Natriuretic Peptide Signaling. Circulation 126, 942–951. doi:10.1161/CIRCULATIONAHA.112.090977
Zhang, Y., Knight, W., Chen, S., Mohan, A., and Yan, C. (2018). Multiprotein Complex with TRPC (Transient Receptor Potential-Canonical) Channel, PDE1c (Phosphodiesterase 1C), and A2R (Adenosine A2 Receptor) Plays a Critical Role in Regulating Cardiomyocyte cAMP and Survival. Circulation 138, 1988–2002. doi:10.1161/CIRCULATIONAHA.118.034189
Zhao, C. Y., Greenstein, J. L., and Winslow, R. L. (2016). Roles of Phosphodiesterases in the Regulation of the Cardiac Cyclic Nucleotide Cross-Talk Signaling Network. J. Mol. Cell. Cardiol. 91, 215–227. doi:10.1016/j.yjmcc.2016.01.004
Zhao, L., Mason, N. A., Strange, J. W., Walker, H., and Wilkins, M. R. (2003). Beneficial Effects of Phosphodiesterase 5 Inhibition in Pulmonary Hypertension Are Influenced by Natriuretic Peptide Activity. Circulation 107, 234–237. doi:10.1161/01.cir.0000050653.10758.6b
Zhu, G., Ueda, K., Hashimoto, M., Zhang, M., Sasaki, M., Kariya, T., et al. (2021). The Mitochondrial Regulator PGC1α Is Induced by cGMP–PKG Signaling and Mediates the Protective Effects of Phosphodiesterase 5 Inhibition in Heart Failure. FEBS Lett. 596, 17–28. doi:10.1002/1873-3468.14228
Keywords: NO, SGC, NPR, PGC, cGMP, PKG
Citation: Numata G and Takimoto E (2022) Cyclic GMP and PKG Signaling in Heart Failure. Front. Pharmacol. 13:792798. doi: 10.3389/fphar.2022.792798
Received: 11 October 2021; Accepted: 09 February 2022;
Published: 11 April 2022.
Edited by:
Antonio Lax, University of Murcia, SpainReviewed by:
Kristen Bubb, Monash University, AustraliaRobert Lukowski, University of Tübingen, Germany
Copyright © 2022 Numata and Takimoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eiki Takimoto, ZWlraXRha2ltb3RvQGdtYWlsLmNvbQ==