 Fa-Da Wang
Fa-Da Wang Jing Zhou
Jing Zhou En-Qiang Chen
En-Qiang Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 11 February 2022
Sec. Gastrointestinal and Hepatic Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.787748
This article is part of the Research TopicFibrosis in the Respiratory and Digestive SystemsView all 50 articles
Liver fibrosis is the pathological process of excessive extracellular matrix deposition after liver injury and is a precursor to cirrhosis, hepatocellular carcinoma (HCC). It is essentially a wound healing response to liver tissue damage. Numerous studies have shown that hepatic stellate cells play a critical role in this process, with various cells, cytokines, and signaling pathways engaged. Currently, the treatment targeting etiology is considered the most effective measure to prevent and treat liver fibrosis, but reversal fibrosis by elimination of the causative agent often occurs too slowly or too rarely to avoid life-threatening complications, especially in advanced fibrosis. Liver transplantation is the only treatment option in the end-stage, leaving us with an urgent need for new therapies. An in-depth understanding of the mechanisms of liver fibrosis could identify new targets for the treatment. Most of the drugs targeting critical cells and cytokines in the pathogenesis of liver fibrosis are still in pre-clinical trials and there are hardly any definitive anti-fibrotic chemical or biological drugs available for clinical use. In this review, we will summarize the pathogenesis of liver fibrosis, focusing on the role of key cells, associated mechanisms, and signaling pathways, and summarize various therapeutic measures or drugs that have been trialed in clinical practice or are in the research stage.
Hepatic fibrosis is a universal pathological process that occurs in various types of chronic liver disease, including viral hepatitis, alcoholic hepatitis, fatty liver disease, nonalcoholic fatty liver disease (NAFLD), wilson’s disease, and cholangitis. When hepatocytes are damaged, the release of signals such as reactive oxygen species (ROS) and intercellular interactions lead to the differentiation of HSCs towards myofibroblasts, and the latter is the primary source of the extracellular matrix (ECM) (Casini et al., 1997; Novo et al., 2009; Ghatak et al., 2011; Mederacke et al., 2013). Damaged hepatocytes also activate inflammatory cells such as macrophages and lymphocytes to generate multiple types of cytokines, including transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF). These cytokines would result in dysregulation of ECM degradation and synthesis, leading to the development of liver fibrosis (Luedde et al., 2014; Seki and Schwabe, 2015). Suppose the injury persists and therapeutic interventions are not taken in time, the liver parenchyma will gradually be replaced by scar tissue formed by excessive ECM, leading to the loss of standard structure and the formation of cirrhosis. Additionally, the risk of hepatocellular carcinoma (HCC) and serious complications such as gastrointestinal bleeding increased.
Over the past decades, we have made some important progress in the mechanism study of liver fibrosis, but the complex pathogenesis of liver fibrosis poses certain difficulties for the development of anti-hepatic fibrosis drugs. Many therapeutic interventions are effective in experimental models, but their efficacy and safety in humans are unknown and cannot be applied in the clinic for the time being. Though still lack of specific anti-fibrosis agents in clinical, numerous studies have shown that the etiological treatment of primary liver disease is effective and even partially reversible for liver fibrosis (Dienstag et al., 2003; Czaja and Carpenter, 2004; Schiff et al., 2008; Chang et al., 2010). In addition, it is worth mentioning that traditional Chinese medicine (TCM) has a beneficial effect on anti-fibrosis (Chen et al., 2015). In this review, we will summarize the various therapeutic measures or drugs that have been trialed in clinical practice or are in the research stage.
Liver fibrosis is caused by an excessive accumulation of scar tissue, accompanied by angiogenesis (Lin et al., 2021), which ultimately leads to changes in the architecture of the liver. The mechanisms of liver fibrosis can be generalized as follows, multiple stimuli (such as toxins, viruses, cholestasis, hypoxia, and insulin resistance, etc.) attack the liver cells and induce the formation of reactive oxygen species (such as hydrogen peroxide, hydroxyl radicals, and aldehyde end products, etc.), which in turn cause hepatocyte damage, apoptosis, steatosis, and immune cell infiltration, especially kupffer cells (KCs) (Wehr et al., 2013; Pradere et al., 2013). At the same time, sinusoidal endothelial cells experience the loss of fenestrae, known as capillarization of the sinusoids (Marrone et al., 2016). Chronic damage to hepatocytes is the initiator of the fibrotic cascade, it induces the production of pro-fibrotic cytokines/growth factors (e.g., TNF-a, IL-6, TGF-β, and PDGF) indirectly through interactions with hepatic macrophages and natural killer (NK) cells. Meanwhile, it directly activates primary response cells (e.g., hepatic stellate cells) through the release of cellular contents, ultimately leading to the activation of HSCs and the fibrotic network and excessive deposition of ECM (Figure 1) (Canbay et al., 2002; Elpek, 2014). Based on the pathogenesis, we can regress liver fibrosis by protecting hepatocytes, inhibiting the activation of hepatic stellate cells, and fibrotic scar evolution.
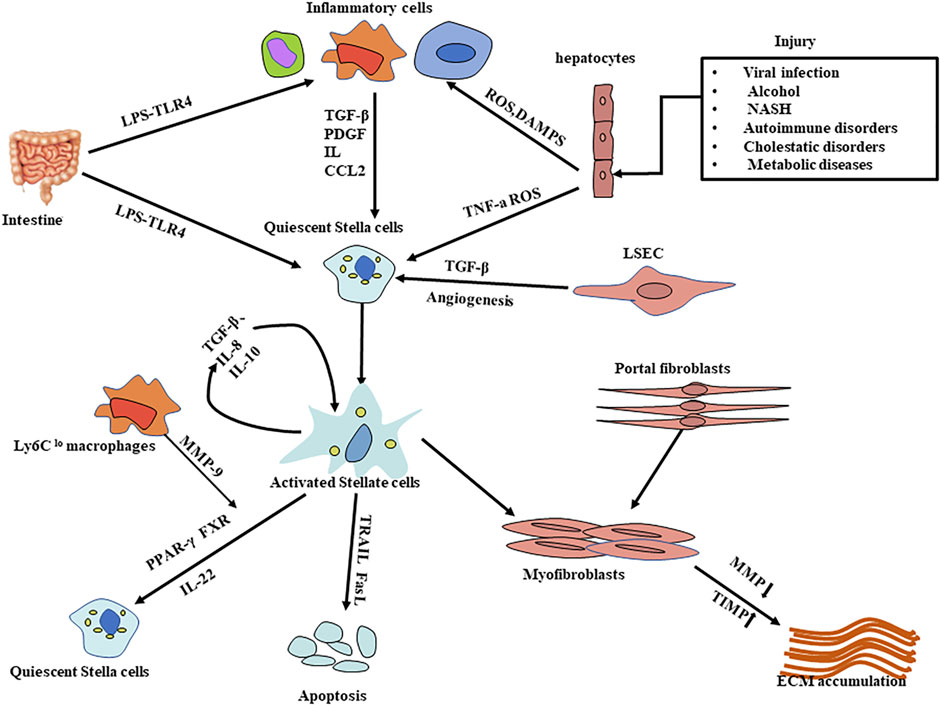
FIGURE 1. Mechanisms of liver fibrosis. Liver injury is caused by a variety of stimuli that result in hepatocyte damage and the release of substances such as ROS; in response to persistent hepatocyte injury, HSCs and macrophages (including Kupffer cells) are activated, activated myofibroblasts increase and excessive ECM is produced, leading to the progression of liver fibrosis. The activation of hepatic stellate cells is a key step in the process of liver fibrosis. Many influential factors regulating HSC activation, proliferation, function, and survival have emerged as important therapeutic targets; likewise, protection of hepatocytes from damage and degradation of excessive ECM deposition provide therapeutic options. HSCs: Hepatic stellate cells CCL2:C-C chemokine ligands types 2; LPS: Lipopolysaccharide LSEC: Liver sinusoidal endothelial cells TIMP: inhibitors of matrix metalloproteinase; MMP: matrix metalloproteinase; DAMPS:damage-associated molecular patterns; ECM: extracellular matrix; ROS: reactive oxygen species.
The ECM is a complex network of macromolecular substances that can regulate various physiological functions such as cell growth, proliferation, migration, differentiation, adhesion, metabolism, damage repair, and tissue remodeling through various signaling systems. In the normal liver, it is a highly dynamic substrate that maintains an exact balance between synthesis and degradation (Theocharis et al., 2016; Villesen et al., 2020). However, in chronic liver disease, the balance is disturbed due to the involvement of multiple cells and cytokines, leading to a greater synthesis than degradation. But most of these changes can be reversed if the liver injury is transient (Hernandez-Gea and Friedman, 2011). The process of liver fibrosis is complicated, involving both hepatic parenchymal and non-parenchymal cells as well as immune cells, and the main functions of different cells and cytokines in liver fibrosis are described in detail below.
In normal liver, HSCs exhibit a quiescent state, whose physiological functions are related to fat storage and the metabolism of vitamin A. Another function of the quiescent HSC is to secrete adequate amounts of ECM proteins such as type III collagen, type IV collagen, and laminin. Besides, HSC secretes a variety of degradative enzymes called matrix metalloproteinases (MMPs), such as MMP-1, which promote the degradation of ECM. HSC also produces tissue inhibitors of matrix metalloproteinases (TIMPs), such as the TIMP-1 and TIMP-2. The TIMP1 can prevent ECM degradation by blocking MMPs and can inhibit HSC apoptosis (Carloni et al., 1996; Roeb et al., 1997; Benyon and Arthur, 2001; Geerts, 2001; Yoneda et al., 2016). The highly regulated interaction between MMPs and TIMPs is responsible for the renewal of the liver matrix and the maintenance of homeostasis and healthy liver architectures in vivo (Murphy et al., 2002). When the liver injury occurs, numerous key cells and inflammatory mediators are involved, including inflammatory stimuli, fibrogenic cytokines TGF-β, ROS, produced by activating macrophages, platelets, and products of damaged hepatocytes drive HSC activation. Quiescent HSCs become activated and TIMP-1 expression is upregulated, which is an essential and central step of liver fibrogenesis. The activated HSCs can not only transform into myofibroblasts and secrete enough ECM, but also secret cytokines such as TGF-β to maintain a constant state of activation, ultimately resulting in the deposition of mature collagen fibers in the space of the Disse and leading to the formation of scars (Tsuchida and Friedman, 2017).
In past, our understanding of HSCs has been dominated by their crucial role in liver fibrosis, thus generating anti-fibrotic strategies that target this cell. As research has progressed and understanding of the role of HSCs in disease has increased, we have found that HSCs have a role in promoting liver cell regeneration (Yang et al., 2008), which may be achieved mainly through the following mechanisms, secretion of cytokines that promote liver cell proliferation, promote the migration of stem cells to the liver, and promote the epithelial transformation of mesenchymal cells into hepatocytes. Therefore, we need to consider their role in liver regeneration when targeting HSCs in liver fibrosis (Ge et al., 2020).
Myofibroblasts (MFs) are key cells in fibrotic diseases, including lung, kidney, and liver disease (Friedman et al., 2013). It is the major cell that produces ECM in the process of liver fibrosis, such as collagen I and III. The origin of myofibroblasts has been controversial, but experiments and data now demonstrate that the main sources are HSCs and portal myofibroblasts. Following an injury to liver tissue, myofibroblasts are transformed from activated HSCs in response to a large number of cytokines and inflammatory cells. The overproduced cytokines can continue to act on myofibroblasts to keep them activated, which in turn produces large aggregates of ECM. In biliary disease, the main source of myofibroblasts is portal myofibroblasts (Iwaisako et al., 2014; Wells et al., 2015). Besides, animal experiments have shown that HSCs and myofibroblasts can be converted from mesothelial cells via mesothelial-mesenchymal transition after liver injury (Li et al., 2013).
Hepatocytes make up 80% of the total cell population and volume of the human liver, and under physiological conditions perform a variety of functions such as detoxification, secretion of bile, proteins, and lipids (Schulze et al., 2019). It is also a primary target for toxic substances that attack the liver. Hepatocyte death is an important initial event in all liver diseases. Dead hepatocytes release intracellular compounds called damage-associated molecular patterns (DAMPs) that signal to surround hepatic stellate cells and Kuffer’s cells and therefore play an important role in the development of fibrosis and inflammation (An et al., 2020; Gaul et al., 2021). Therefore, protecting hepatocytes from damage is an important therapeutic intervention.
Inflammation is a fundamental characteristic of chronic liver disease, cell death is typically the precipitating event. The release of signals such as reactive oxygen species (ROS) from the damaged cells can activate the inflammatory cells, including macrophages, lymphocytes, and NK cells etc (Jaeschke, 2011). Among them, hepatic macrophages (Kupffer cells) play major roles and are known as regulators in the process of liver fibrosis (Wynn and Barron, 2010). KCs are an essential component of the innate immune mononuclear phagocytic system and play critical functions in homeostasis, and act as first responders following liver injury. In response to tissue damage, numerous Ly-6Chi macrophages are recruited to the liver, releasing cytokines and attracting NK cells and other immune cells (Karlmark et al., 2009; Reid et al., 2016). These macrophages could induce the transdifferentiation of HSCs into collagen-producing myofibroblasts by secreting TGF-β1 and PDGF. Dendritic cells (DCs) increase fibrosis regression, mainly through the production of MMP-9 (Jiao et al., 2012). NK cells directly kill target cells and are capable of producing a variety of cytokines that play various roles in liver injury, fibrosis, and hepatocarcinogenesis, activated NKT cells have a role in killing activated HSCs (Radaeva et al., 2006). However, in chronic liver disease, NKT cells have a pro-inflammatory function, recruit neutrophils and myeloid cells, and promote the activation of hepatic stellate, leading to hepatocyte necrosis, fibrosis, and even HCC (Jin et al., 2011; Wolf et al., 2014).
Activated inflammatory cells are the primary source of cytokines, such as C-C chemokine ligands types 2 and 5 (CCL2 and CCL5), IL, TGF-β1, PDGF, etc. The role of inflammatory cells is double-sided, which could promote the regression of liver fibrosis and accelerate the deterioration of fibrosis. For example, hepatic macrophages can not only relieve inflammation and fibrosis by degrading the ECM and releasing anti-inflammatory cytokines but also promote liver fibrosis by activating HSCs (Duffield et al., 2005; Tacke and Zimmermann, 2014). The Ly6Chi macrophages can differentiate into restorative Ly6Clo macrophages to engulf cell debris and secrete MMP-9 and MMP-1/MMP-2 to promote scar regression (Ramachandran et al., 2012).
In normal liver tissue, liver sinusoidal endothelial cells (LSECs) have characteristics of vasodilatory, anti-inflammatory, anti-thrombotic, anti-angiogenic, anti-fibrotic, and regeneration-promoting effects (Ding et al., 2010), so LSECs are considered to be the gatekeepers of hepatic homeostasis. At the same time, LSECs are the main source of endothelium-derived nitric oxide (NO), which keeps HSCs in a resting state. In the presence of liver injury, LSECs become capillarized, which can not only reduce the production of vasodilators (such as NO, cyclooxygenase, and prostaglandin I2 [PGI2]) but also increase the production of vasoconstrictors (endothelin 1, thromboxane A2, angiotensin II). This imbalance not only alters the phenotype of LSECs but also contributes to the activation of HSCs and promotes inflammation and liver fibrosis (Deleve et al., 2008; Xie et al., 2012; Poisson et al., 2017). It also secretes TGF-β, PDGF or activates signaling pathways such as Wntβcatenin, which can activate HSCs in a paracrine and autocrine manner. Due to the unique properties of LSECs, selective LSEC-targeted therapy appears to be an attractive strategy for the treatment of liver fibrosis (Gracia-Sancho et al., 2021).
Transforming growth factors (TGF) have the function of regulating the growth and development of various cells, which are essential for the homeostasis of tissues and organs. In the liver, they are mainly produced by HSCs, LSECs, KCs, and DCs as well as NKT cells, and can act on themselves or other cells through autocrine or paracrine secretion (Schon and Weiskirchen, 2014). The functions of TGF-β vary between different types and stages of liver disease. During liver fibrosis, TGF-β is up-regulated, the main function of TGF-β is to activate HSCs, which are considered to be the main pro-fibrotic factor in the process of liver fibrosis. It also enhances the expression of TIMPs and directly promotes the synthesis of interstitial fibrillar collagens (García-Trevijano et al., 1999; Hellerbrand et al., 1999; Dewidar et al., 2019). Due to its important function in liver fibrosis, blocking the signaling pathway of TGF-β is now a potential target for the treatment of liver fibrosis (Guo et al., 2012).
The platelet-derived growth factor (PDGF) is a member of the family of growth factors whose biological functions include angiogenesis, regulation of cell proliferation and survival, cell migration, and stimulation of the synthesis of major components of the connective tissue matrix (Heldin and Westermark, 1999). In the context of liver disease, the expression of PDGF and its receptors have now been shown to be significantly upregulated (Pinzani et al., 1996). It has been regarded as the most effective growth factor for HSC proliferation in hepatic fibrosis, and the receptors of PDGF have become a new promising direction in the treatment of liver fibrosis (Pinzani et al., 1989; Borkham-Kamphorst et al., 2007).
The progression and regression of liver fibrosis are regulated by a complex signaling pathway consisting of cytokines, growth factors, and chemokines. IL-6, TNF-a, Interleukins, PDGF, and TGF-β are the key pro-inflammatory and profibrogenic cytokines that drive liver fibrosis. Interleukins (ILs) are important immunomodulatory cytokines. During liver injury, it is produced by various cell types and exerts pro-inflammatory (such as IL-13, IL-17, and IL-33) as well as anti-inflammatory effects (such as IL-22 and IL-10) in hepatic cells (Hu et al., 2016; Xu et al., 2016; Liu et al., 2019). For instance, An animal study has shown that the IL-6/gp130 pathway plays a protective role for non-parenchymal hepatocytes in the progression of fibrosis (Streetz et al., 2003). But a recent study has demonstrated that IL-6 can induce differentiation of HSCs towards myofibroblast via MAPK and JAK/STAT signaling pathways (Kagan et al., 2017). In addition, IL-22 has been shown to attenuate liver fibrosis by binding to cell receptors, attenuating the activation of HSCs and down-regulating levels of inflammatory cytokines (Lu et al., 2015). This suggests that a strategy using blocking pro-inflammatory interleukins or inducing anti-inflammatory interleukin production to treat liver fibrosis can be effective.
Tumour necrosis factor (TNF) and related receptor pathways can activate apoptosis in hepatocytes via the caspases pathway and exert anti-apoptotic effects via the NF-κB pathway (Osawa et al., 2018). TNF also plays a vital role in the activation of HSCs and the synthesis of ECM (Osawa et al., 2013). TNF reduces apoptosis of activated rat HSCs through upregulation of the anti-apoptotic factor NF-κB. However, the effects of TNF-α on HSCs and fibrosis are multiple. In animal experiments, it has shown an anti-fibrotic effect by reducing glutathione and inhibiting the secretion of pro-collagen α1 (Hernandez-Munoz et al., 1997; Varela-Rey et al., 2007).
The liver is exposed to venous blood from the small and large intestines through the portal vein. Due to this unique blood supply system, the liver is easily exposed to bacterial products that are transferred from the lumen of the intestine via the portal vein. The small and large intestines are rich in flora. In healthy organisms, due to the barrier effect of the intestinal mucosa, only a small amount of translocated bacterial products reach the liver, and the liver immune system tolerates these bacterial products to avoid harmful reactions. After the injury to the liver or the intestinal mucosa, the flora becomes disturbed, and intestinal bacteria can translocate to the liver. Its metabolites such as lipopolysaccharide (LPS) can conjugate with functional toll-like receptor 4 (TLR4) to activate reactive cells such as HSCs and Kupffer cells, and also enhance the activity of transforming growth factors thus leading to the development of liver fibrosis (Seki et al., 2007; Schnabl and Brenner, 2014). In the liver, both hepatocytes and non-parenchymal cells (NPCs) have expressed TLR4. Compared to other organs, healthy livers have a low level of TLR4. However, damaged livers increase the expression of TLR4 and its co-receptors, thus making TLR4 signaling-mediated inflammatory responses more sensitive (Kitazawa et al., 2008; Guo and Friedman, 2010).
Recently, it has been shown that fibrosis can reverse after the removal of pathogenic conditions. Although no drugs are currently approved for the treatment of liver fibrosis, some treatment modalities have shown effectiveness in patients, such as antiviral therapy for patients with viral hepatitis, zinc for wilson’s disease, phlebotomy for hemochromatosis, alcohol withdrawal for alcoholic liver disease and ursodeoxycholic acid (UDCA) in the treatment of primary biliary cholangitis (Powell and Klatskin, 1968; Marcellini et al., 2005; Takahashi et al., 2014; Bardou-Jacquet et al., 2020; Ye et al., 2020). glucocorticoids, vitamin E, and angiotensin receptor antagonists have gradually been shown to have antifibrotic effects as well. In addition, TCM appears to have an increasingly prominent role in the treatment of liver fibrosis and its efficacy is promising (Fujiwara et al., 2010; He et al., 2013), but more clinical trials are needed to confirm its effectiveness. In the following, we will describe these potential treatments in detail below (Table 1).
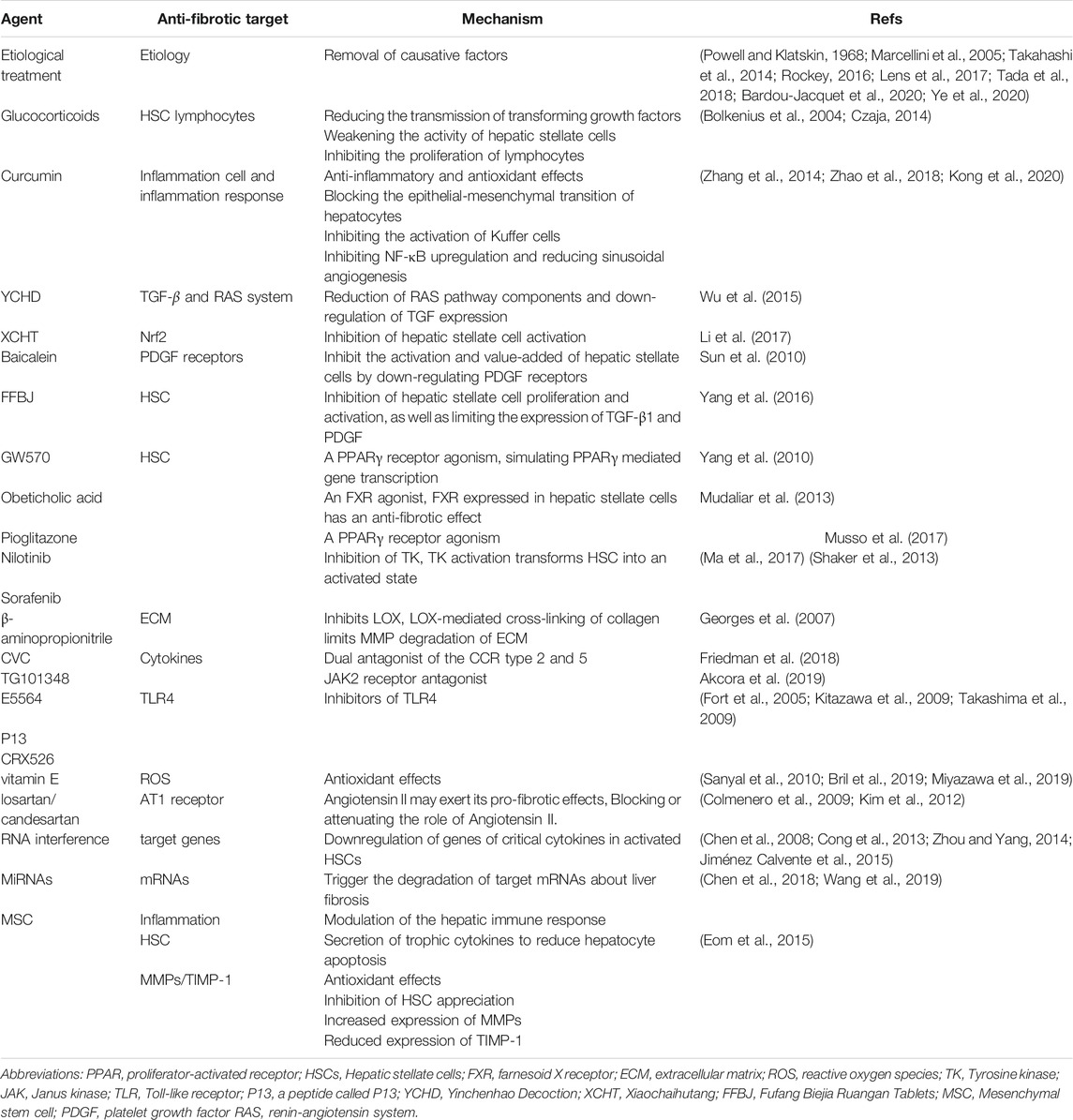
TABLE 1. Targets and main mechanisms of some of existing anti-fibrotic drugs and novel therapeutic approaches.
Among all the factors that contribute to chronic liver disease, hepatitis virus infection is the most common, primarily hepatitis B and C. Chronic hepatitis B virus infection is a worldwide public health problem, with approximately 250 million people chronically infected and at high risk of developing cirrhosis and liver cancer. When liver cells are infected with the virus, cellular damage induces an inflammatory response, at the same time the virus itself can directly induce activation of the immune system, leading to activation of HSCs and progress to liver fibrosis. Clearing hepatitis viruses or inhibiting hepatitis virus replication is the most effective way to reduce liver cell damage. With the advent of antiviral drugs, we have now made considerable progress in the fight against the hepatitis B and C virus. Through effective antiviral therapy, most liver fibrosis can be reversed, and liver cirrhosis and its related complications can be reduced (Rockey, 2016; Lens et al., 2017; Tada et al., 2018).
Glucocorticoids have immunomodulatory and anti-inflammatory effects. As we mentioned earlier, the inflammatory response and immune cells play critical roles in the process of liver fibrosis. Thus glucocorticoids may have some therapeutic effects in liver fibrosis. It has been shown that glucocorticoids can reduce liver fibrosis by reducing the transmission of transforming growth factors, weakening the activity of HSCs, and inhibiting the proliferate of lymphocytes, but the efficacy of glucocorticoids differently in different diseases (Bolkenius et al., 2004; Czaja, 2014). Glucocorticoids or immunosuppressive agents are the most significant treatment options for chronic autoimmune liver disease, and liver fibrosis can be reversed with adequate management (Valera et al., 2011). Early glucocorticoid treatment is effective for prognosis in hepatitis and liver failure due to viral hepatitis B (Fujiwara et al., 2010; He et al., 2013). Nevertheless, the use of corticosteroids for alcohol-related acute liver failure or slow-onset acute liver failure is still controversial in clinical practice, although the AASLD and EASL guidelines recommend treatment with corticosteroids. Studies have shown that the use of glucocorticoids improves short-term survival but does not significantly improve long-term prognosis and carries risks such as infection (Thursz et al., 2015; Sersté et al., 2018; Gustot and Jalan, 2019). Similarly, the use of glucocorticoids to treat a drug-induced liver injury is also in dispute (Andrade et al., 2019). Consequently, there is still a need for extensive trials and data to evaluate the safety and efficacy of glucocorticoids in liver disease.
Notably, there is growing evidence that TCM is effective in the prevention and treatment of liver fibrosis (Pan et al., 2020). TCM can suppress liver fibrosis activity through different mechanisms, including inhibition of cytokine production and suppression of HSCs activation, as well as regulating the progression of liver fibrosis through other molecular mechanisms (Shan et al., 2019). Turmeric is an herb that grows in Asia and has been widely used as a spice in food and for therapeutic applications. In China, it is also an ingredient in TCM and recently its extract curcumin has received much attention. Curcumin has been widely used in anti-fibrotic models due to its anti-inflammatory and antioxidant effects. It has been shown to alleviate liver fibrosis by blocking the epithelial-mesenchymal transition of hepatocytes through the regulation of oxidative stress and autophagy (Cai et al., 2018; Cai et al., 2019), and to weaken the role of Ly6Chi cells in liver fibrosis by inhibiting the activation of Kuffer cells, thereby reducing the secretion of chemokines (Cai et al., 2018; Cai et al., 2019). In addition, it also has the effect of inhibiting NF-κB upregulation and reducing sinusoidal angiogenesis (Cai et al., 2018; Cai et al., 2019). More research is underway on the mechanism of curcumin against liver fibrosis.
YCHD (Yinchenhao Decoction) is a traditional Chinese herbal formulation used to treat liver fibrosis and has been experimentally shown to have multiple active ingredients targeting various targets of liver fibrosis (Cai et al., 2018; Cai et al., 2019). Recent studies have indicated RAS system in liver fibrosis/cirrhosis may exert pro-fibrotic effects, and the antifibrotic effects of the YCHD fibrosis effect may be related to the reduction of RAS pathway components and down-regulation of TGF expression (Bataller et al., 2000; Yoshiji et al., 2007). XCHT (Xiaochaihutang) is a water decoction traditionally used in china for the treatment of liver diseases, and the mechanism for its anti-fibrosis is not completely clear. Nrf2 is an important redox-sensitive transcription factor in vivo, which can promote cell survival, as well as maintain the redox state of cells. Animal experiments have shown that XCHT is an effective drug for the treatment of liver fibrosis, and its therapeutic effect is mainly through upregulation of the Nrf2 pathway thus resulting in the inhibition of HSCs activation (Bataller et al., 2000; Yoshiji et al., 2007). In addition, baicalein, the main component of XCHT, can inhibit the activation and proliferation of HSCs by down-regulating PDGF receptors, thus exerting an anti-fibrotic effect (Bataller et al., 2000; Yoshiji et al., 2007). It has been reported that ETV combined with FFBJ (Fufang Biejia Ruangan Tablets) showed significant anti-fibrotic effects in CHB patients. The mechanism of action may be related to the inhibition of HSCs proliferation and activation, as well as limiting the expression of TGF-β1 and PDGF (Bataller et al., 2000; Yoshiji et al., 2007).
Similar TCMs also include Huangqi Decoction Dahuang Zhechong Pills, Fuzheng Huayu Formula, Anluo Huaxian Pills (Li, 2020), etc. Although TCM has been used for thousands of years, its clinical effectiveness in liver fibrosis needs further evaluation due to the lack of rigorous randomized controlled trials.
Vitamin E, angiotensin-converting enzyme inhibitors (ACE-I), and angiotensin II type 1 (AT1) receptor blockers have recently drawn attention to the treatment of liver fibrosis. Vitamin E is an important nutrient with antioxidant effects, it can inhibit the production of singlet free radicals, oxygen, lipid hydroperoxides, and lipid radicals. Since products such as free radicals play an important role in the development of liver fibrosis, it may be a potential option for the treatment of liver fibrosis. However, in patients with non-alcoholic liver disease, vitamin E could not significantly reduce the severity of fibrosis (Sanyal et al., 2010; Bril et al., 2019; Miyazawa et al., 2019).
Recent studies have also shown that the production of angiotensin II type 1 receptor is increased in activated HSCs and enhanced the activity of the renin-angiotensin system (RAS) in liver fibrosis/cirrhosis. Angiotensin II may exert its pro-fibrotic effects through increased oxidative stress, activation and proliferation of HSCs, upregulation of TGF-β and TIMP1, and accelerated deposition of collagen (Bataller et al., 2000; Yoshiji et al., 2007). Based on these mechanisms, ACE-I and AT1 receptor blockers are potential treatment options for liver fibrosis. A clinical study evaluating the efficacy of angiotensin II receptor blocker (ARB) losartan in patients with HCV showed that it can reduce inflammation and decrease fibrosis gene expression (Colmenero et al., 2009).In patients with chronic alcoholic liver diseases, the combination of UDCA and ARB candesartan improved patients’ fibrosis scores compared to UDCA treatment alone (Kim et al., 2012). However, as no clear effects have been shown in other clinical trials, further studies are needed to demonstrate the benefit of RAS antagonists in liver fibrosis.
There are no directly effective anti-fibrotic drugs in clinical practice and most of them are still in clinical trials and in the research stage. Although etiological treatment has proven to be effective, some etiologies cannot be eliminated. In addition, even with effective etiological treatment, reversal of advanced liver fibrosis cannot completely avoid complications such as gastrointestinal bleeding and HCC, so we urgently need direct anti-fibrotic drugs.
The main pathogenesis of liver fibrosis can be summarized as follows: Following the chronic injury to hepatocytes, the multitude of cellular and cytokine interactions lead to the activation of key cells such as HSCs and MFs, which in turn leads to the overproduction of ECM and the development of liver fibrosis (Parola and Pinzani, 2019; Kisseleva and Brenner, 2021). Based on the pathogenesis of liver fibrosis, the drugs we are exploring focus on the following aspects: protecting hepatocytes from damage, inhibiting cytokine activity and cell proliferation, promoting apoptosis of key cells, reducing ECM synthesis, and promoting its degradation. Many relevant drugs are already in clinical trials, including FXR agonists, PPAR agonists, and TAK agonists, which inhibit hepatic stellate cell activation and promote ECM degradation, TLR4 receptor antagonists, and novel therapeutic approaches such as miRNA and mesenchymal stem cell therapy. Below we describe in detail the novel treatments and drugs according to the different targets (Table 1).
Because of its important role in liver fibrosis, HSCs become a major target for anti-fibrotic drugs (Elpek, 2014). Reversing liver fibrosis by converting activated HSCs to a quiescent state or promoting their apoptosis is our main goal. Experimental models of fibrosis consistently demonstrate that elimination of activated HSCs by apoptosis or other pathways can lead to regression of fibrosis (Iredale et al., 1998; Troeger et al., 2012). Recent studies have shown that activated HSCs can be transformed into non-fibrotic cells by transcriptional reprogramming, such as ectopic expression of GATA4, FOXA3, HNF1a, and HNF4a in vivo (Song et al., 2016). Besides, cellular senescence may be an anti-fibrotic strategy. Expression of nuclear receptors PPAR and FXR in HSCs suppress HSCs activation, as studies have shown that HSC senescence can be invoked by PPARγ, FXR agonist, such as GW570 (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015), pioglitazone (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015), obeticholic acid (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015), thereby alleviating liver fibrosis degree. TK (Tyrosine kinase) is expressed in HSCs and its activation transforms them into an activated state, thus inhibition of TK may be a potential target for the treatment of liver fibrosis. Sorafenib has been used as a treatment for patients with HCC, where complications of cirrhosis (such as portal hypertension) have been reduced, and its anti-fibrotic activity has been confirmed in numerous trials (Ma et al., 2017). Nilotinib triggers apoptosis and autophagic cell death via inhibition of histone deacetylase in activated HSC cells (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015). Unfortunately, most of these drugs are in animal studies and are not yet available for clinical use, their safety and efficacy deserve to be evaluated.
The removal of the excess ECM is one of our goals for treating liver fibrosis. Collagen is the most abundant ECM protein in liver fibrosis. Specific inhibition of type 1 collagen fibrils synthesis has now been achieved in animals by miRNA, and this miRNA leads to a significant reduction in collagen 1 synthesis in fibrosis models (Jiménez Calvente et al., 2015). Besides, LOX is a copper-dependent amine oxidase (Perepelyuk et al., 2013), and LOX-mediated cross-linking of collagen limits MMP degradation of ECM. The β-aminopropionitrile inhibits LOX, reduces liver stiffness, decreases the number of fibroblasts, and attenuates cell injury-induced liver fibrosis (Georges et al., 2007). However, the clinical trial did not demonstrate significant efficacy (Loomba et al., 2018). Similar to LOX, transglutaminase (TGs) forms a covalent isopeptide bond by covalently linking a glutamine residue of one protein chain to a lysine residue of another protein chain. Intercross-linking of TGs can promote liver fibrosis, and therefore invoking specific inhibitors of TGs could be a potential target for the treatment of liver fibrosis (Van Herck et al., 2010).
Cytokines are involved in the entire process of liver fibrosis, blocking their signaling pathways, and receptors may inhibit the production of the ECM and accelerate its degradation. Cenicriviroc (CVC), an oral dual antagonist of the CCR type 2 and 5, has been shown to have antifibrotic effects in animal studies. A clinical trial has shown amelioration of liver fibrosis in patients with nonalcoholic steatohepatitis (NASH) after 1 year of treatment with CVC (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015), and further clinical trials on CVC are currently underway, and we hope that it will become an anti-fibrotic option in the future. The Janus kinases (JAK) signaling pathway plays an important role in the pathogenesis of hepatic fibrosis and can be activated by a variety of cytokines such as IL. Studies have shown that the use of the JAK2 receptor antagonist TG101348 can reduce hepatic fibrosis in animal models (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015). However, cytokine function is also important for maintaining the immune response, tissue repair, etc. Long-term targeting of these cytokines is challenging due to the severe adverse effects. A great deal of research has been done on cytokine antagonism (Gressner and Weiskirchen, 2006).
As we mentioned earlier that intestinal microbiota is closely associated with the development of liver fibrosis. the main mechanism by which liver fibrosis occurs, in this case, is the combination of the bacterial metabolites LPS and TLR4, further activating key cells in the liver fibrosis process. Therefore, inhibition of TLR4-related intracellular signaling may be effective in reducing TLR4-mediated inflammation and inhibiting liver fibrosis (Beutler, 2004). It was shown that a peptide called P13, which was previously shown to be a potent inhibitor of TLR signaling in vitro. Using this peptide to treat mice effectively inhibited LPS-induced inflammatory mediator production and significantly limited liver damage, enhancing survival in a mouse model of inflammation (Tsung et al., 2007). Several small-molecule inhibitors of TLR4 are currently being tested, including lipid A mimetics, e.g., E5564 and CRX526 (Fort et al., 2005; Kitazawa et al., 2009; Takashima et al., 2009), soluble fusion proteins with extracellular structural domains. However, these are still in animal studies and may become targets for anti-fibrotic drugs in the future.
Liver fibrosis is highly related to activated HSCs, and the activation of HSCs is regulated by a variety of cytokines. Downregulation of these cytokines in activated HSCs using RNA interference (RNAi) is a promising strategy for reversing liver fibrosis. RNA interference is a new technique that uses small interfering RNAs (siRNAs) of 21–23 nucleotides to specifically knock out target genes, and this new technique is based on the high specificity of siRNAs and their ability to downregulate genes associated with liver fibrosis (Kim and Rossi, 2007). Many therapies for siRNA are currently in clinical trials to translocate siRNA into HSC or other hepatic parenchymal cells, for example, lipid nanoparticles containing HSP47 siRNA for the treatment of liver fibrosis (Kulkarni et al., 2018). The main mechanism of siRNA action is to cause homologous degradation of the targeted mRNA (Aagaard and Rossi, 2007). It has been shown that siRNA can address liver fibrosis by regulating collagen expression in HSC (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015). Meanwhile, it has been reported that direct knockdown of TGF-β expression using siRNA can exert antifibrotic effects in a rat model (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015). Similarly, the use of PDGF siRNA suppressed the advancement of liver fibrosis in mice (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015). Besides, MMP2-specific siRNA and TIMP-specific siRNAs also exert an anti-fibrotic effect on the liver (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015).
MiRNAs are endogenous small non-coding RNAs that can post-transcriptionally regulate the expression of mRNAs and ultimately trigger the degradation of target mRNAs. miRNAs are associated with a variety of liver diseases, including liver fibrosis, and therefore miRNAs are an alternative treatment for liver fibrosis. It has been found that miRNAs can be both up and down-regulated during liver fibrosis. Up-regulated miRNA can be reverted by anti-miRNA oligonucleotides, and miRNA masking (Krützfeldt et al., 2005; Janssen et al., 2013; Thakral and Ghoshal, 2015), unlike the upregulated miRNA, some downregulation of miRNA inhibiting liver fibrosis has been found (Chen et al., 2018; Wang et al., 2019), down-regulated MiRNA can be restored by MiRNA mimics or plasmids expressing miRNA (Cheng and Mahato, 2011). Similar to siRNAs, the biggest challenge for miRNAs is to overcome degradation and targeted transport in the blood. To date, there are no clinical trials on MiRNA about the treatment of liver fibrosis. A lot of effort has been spent on siRNA with MiRNA and viral and non-viral transport systems have been developed, which also still face significant challenges. There are already anti-fibrotic siRNAs in clinical trials, and in the future siRNA with miRNA may become a novel treatment for liver disease.
Recently, MSC therapy has been regarded as an effective alternative for the treatment of liver disease. MSCs possess the ability to self-renew and differentiate into many types of cells, and differentiation of MSCs into hepatocytes is the prospect of liver regeneration (Hu et al., 2020). The main mechanisms of the anti-fibrotic effects of MSCs can be generalized as follows, modulation of the hepatic immune response, secretion of trophic cytokines to reduce hepatocyte apoptosis, antioxidant effects, inhibition of HSC proliferation, and increased expression of MMPs or reduced expression of TIMP-1 (Hernandez-Munoz et al., 1997; Varela-Rey et al., 2007). Mesenchymal stem cells are now widely used in clinical and preclinical studies of liver fibrosis, Jang and others showed the beneficial effects of autologous bone marrow MSC transplantation for the treatment of alcoholic cirrhosis (Jang et al., 2014), Kharaziha et al. (2009) showed that liver function improved in patients with cirrhosis after injected autologous mesenchymal stem cells. However, due to their multi-differentiation potential, MSCs can differentiate into myofibroblasts rather than hepatocytes (Baertschiger et al., 2009; di Bonzo et al., 2008). Besides, another risk of MSC transplantation is that they are susceptible to malignant transformation and promote the growth of existing tumors (Zhu et al., 2006). MSCs have the potential to differentiate into hepatocytes, immunomodulatory properties, and the ability to secrete trophic cytokines, making them a potential treatment for liver disease. however, with both their fibrotic potential and their ability to promote the growth of pre-existing tumor cells, MSC therapy needs to be evaluated further.
Recently, with our greater understanding of the mechanisms of liver fibrosis, a plethora of therapeutic strategies have been generated. but the treatment of liver fibrosis remains a difficult clinical problem that we face today and etiological treatment is currently recognized as the most effective anti-fibrotic approach. Multiple interactions between ECM, hepatic stellate, endothelial cells, and immune cells have been demonstrated during liver fibrosis, but the central event in fibrosis is the activation of HSCs. Due to multiple cells and cytokines being involved in the progression of liver fibrosis, it is crucial for us to fully understand the biology of critical cells such as HSCs, myofibroblasts, and macrophages, including their activation and inactivation, to facilitate the development of specific targeted drugs. In addition, the inflammatory response is one of the fundamental features of liver fibrosis, so controlling liver inflammation and inflammatory cells is also a viable strategy for treating liver fibrosis. Besides, novel therapies targeting intestinal microecology, mRNA, and mesenchymal stem cells are also becoming available for clinical trials, and several drugs have been successful in regressing liver fibrosis in experimental models.
A growing number of potential drugs are in phase II and III trials, and we expect that some of these drugs may soon be approved for use in patients. These new drugs target multiple pathways in the pathogenesis of chronic liver disease, but the mechanisms of liver fibrosis are complex. With certain cells having a dual role in the development and regression of liver fibrosis, and targeted therapies may have some side effects. Therefore we must understand the mechanisms more clearly so that we can establish scientific treatments that are safe and effective in achieving long-term results. In the future, a better understanding of the molecular mechanisms involved in the regression of liver fibrosis may provide new preventive and therapeutic strategies for patients with fibrosis and even cirrhosis.
F-DW, JZ conceived this review and collected the literature, E-QC conducted the study supervision and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was partially supported by key R&D projects of Sichuan Province (No. 2019YFS0209).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aagaard, L., and Rossi, J. J. (2007). RNAi Therapeutics: Principles, Prospects and Challenges. Adv. Drug Deliv. Rev. 59 (2-3), 75–86. doi:10.1016/j.addr.2007.03.005
Akcora, B. Ö., Dathathri, E., Ortiz-Perez, A., Gabriël, A. V., Storm, G., Prakash, J., et al. (2019). TG101348, a Selective JAK2 Antagonist, Ameliorates Hepatic Fibrogenesis In Vivo. Faseb j 33 (8), 9466–9475. doi:10.1096/fj.201900215RR
An, P., Wei, L. L., Zhao, S., Sverdlov, D. Y., Vaid, K. A., Miyamoto, M., et al. (2020). Hepatocyte Mitochondria-Derived Danger Signals Directly Activate Hepatic Stellate Cells and Drive Progression of Liver Fibrosis. Nat. Commun. 11 (1), 2362. doi:10.1038/s41467-020-16092-0
Andrade, R. J., Chalasani, N., Björnsson, E. S., Suzuki, A., Kullak-Ublick, G. A., Watkins, P. B., et al. (2019). Drug-induced Liver Injury. Nat. Rev. Dis. Primers 5 (1), 58. doi:10.1038/s41572-019-0105-0
Baertschiger, R. M., Serre-Beinier, V., Morel, P., Bosco, D., Peyrou, M., Clément, S., et al. (2009). Fibrogenic Potential of Human Multipotent Mesenchymal Stromal Cells in Injured Liver. PLoS One 4 (8), e6657. doi:10.1371/journal.pone.0006657
Bardou-Jacquet, E., Morandeau, E., Anderson, G. J., Ramm, G. A., Ramm, L. E., Morcet, J., et al. (2020). Regression of Fibrosis Stage with Treatment Reduces Long-Term Risk of Liver Cancer in Patients with Hemochromatosis Caused by Mutation in HFE. Clin. Gastroenterol. Hepatol. 18 (8), 1851–1857. doi:10.1016/j.cgh.2019.10.010
Bataller, R., Ginès, P., Nicolás, J. M., Görbig, M. N., Garcia-Ramallo, E., Gasull, X., et al. (2000). Angiotensin II Induces Contraction and Proliferation of Human Hepatic Stellate Cells. Gastroenterology 118 (6), 1149–1156. doi:10.1016/s0016-5085(00)70368-4
Benyon, R. C., and Arthur, M. J. (2001). Extracellular Matrix Degradation and the Role of Hepatic Stellate Cells. Semin. Liver Dis. 21 (3), 373–384. doi:10.1055/s-2001-17552
Beutler, B. (2004). Inferences, Questions and Possibilities in Toll-like Receptor Signalling. Nature 430 (6996), 257–263. doi:10.1038/nature02761
Bolkenius, U., Hahn, D., Gressner, A. M., Breitkopf, K., Dooley, S., and Wickert, L. (2004). Glucocorticoids Decrease the Bioavailability of TGF-Beta Which Leads to a Reduced TGF-Beta Signaling in Hepatic Stellate Cells. Biochem. Biophys. Res. Commun. 325 (4), 1264–1270. doi:10.1016/j.bbrc.2004.10.164
Borkham-Kamphorst, E., van Roeyen, C. R., Ostendorf, T., Floege, J., Gressner, A. M., and Weiskirchen, R. (2007). Pro-fibrogenic Potential of PDGF-D in Liver Fibrosis. J. Hepatol. 46 (6), 1064–1074. doi:10.1016/j.jhep.2007.01.029
Bril, F., Biernacki, D. M., Kalavalapalli, S., Lomonaco, R., Subbarayan, S. K., Lai, J., et al. (2019). Role of Vitamin E for Nonalcoholic Steatohepatitis in Patients with Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Care 42 (8), 1481–1488. doi:10.2337/dc19-0167
Cai, F. F., Bian, Y. Q., Wu, R., Sun, Y., Chen, X. L., Yang, M. D., et al. (2019). Yinchenhao Decoction Suppresses Rat Liver Fibrosis Involved in an Apoptosis Regulation Mechanism Based on Network Pharmacology and Transcriptomic Analysis. Biomed. Pharmacother. 114, 108863. doi:10.1016/j.biopha.2019.108863
Cai, F. F., Wu, R., Song, Y. N., Xiong, A. Z., Chen, X. L., Yang, M. D., et al. (2018). Yinchenhao Decoction Alleviates Liver Fibrosis by Regulating Bile Acid Metabolism and TGF-β/Smad/ERK Signalling Pathway. Sci. Rep. 8 (1), 15367. doi:10.1038/s41598-018-33669-4
Canbay, A., Higuchi, H., Bronk, S. F., Taniai, M., Sebo, T. J., and Gores, G. J. (2002). Fas Enhances Fibrogenesis in the Bile Duct Ligated Mouse: a Link between Apoptosis and Fibrosis. Gastroenterology 123 (4), 1323–1330. doi:10.1053/gast.2002.35953
Carloni, V., Romanelli, R. G., Pinzani, M., Laffi, G., and Gentilini, P. (1996). Expression and Function of Integrin Receptors for Collagen and Laminin in Cultured Human Hepatic Stellate Cells. Gastroenterology 110 (4), 1127–1136. doi:10.1053/gast.1996.v110.pm8613002
Casini, A., Ceni, E., Salzano, R., Biondi, P., Parola, M., Galli, A., et al. (1997). Neutrophil-derived Superoxide Anion Induces Lipid Peroxidation and Stimulates Collagen Synthesis in Human Hepatic Stellate Cells: Role of Nitric Oxide. Hepatology 25 (2), 361–367. doi:10.1053/jhep.1997.v25.pm0009021948
Chang, T. T., Liaw, Y. F., Wu, S. S., Schiff, E., Han, K. H., Lai, C. L., et al. (2010). Long-term Entecavir Therapy Results in the Reversal of Fibrosis/cirrhosis and Continued Histological Improvement in Patients with Chronic Hepatitis B. Hepatology 52 (3), 886–893. doi:10.1002/hep.23785
Chen, S. R., Chen, X. P., Lu, J. J., Wang, Y., and Wang, Y. T. (2015). Potent Natural Products and Herbal Medicines for Treating Liver Fibrosis. Chin. Med. 10, 7. doi:10.1186/s13020-015-0036-y
Chen, S. W., Zhang, X. R., Wang, C. Z., Chen, W. Z., Xie, W. F., and Chen, Y. X. (2008). RNA Interference Targeting the Platelet-Derived Growth Factor Receptor Beta Subunit Ameliorates Experimental Hepatic Fibrosis in Rats. Liver Int. 28 (10), 1446–1457. doi:10.1111/j.1478-3231.2008.01759.x
Chen, Y., Ou, Y., Dong, J., Yang, G., Zeng, Z., Liu, Y., et al. (2018). Osteopontin Promotes Collagen I Synthesis in Hepatic Stellate Cells by miRNA-129-5p Inhibition. Exp. Cel Res 362 (2), 343–348. doi:10.1016/j.yexcr.2017.11.035
Cheng, K., and Mahato, R. I. (2011). Biological and Therapeutic Applications of Small RNAs. Pharm. Res. 28 (12), 2961–2965. doi:10.1007/s11095-011-0609-0
Colmenero, J., Bataller, R., Sancho-Bru, P., Domínguez, M., Moreno, M., Forns, X., et al. (2009). Effects of Losartan on Hepatic Expression of Nonphagocytic NADPH Oxidase and Fibrogenic Genes in Patients with Chronic Hepatitis C. Am. J. Physiol. Gastrointest. Liver Physiol. 297 (4), G726–G734. doi:10.1152/ajpgi.00162.2009
Cong, M., Liu, T., Wang, P., Fan, X., Yang, A., Bai, Y., et al. (2013). Antifibrotic Effects of a Recombinant Adeno-Associated Virus Carrying Small Interfering RNA Targeting TIMP-1 in Rat Liver Fibrosis. Am. J. Pathol. 182 (5), 1607–1616. doi:10.1016/j.ajpath.2013.01.036
Czaja, A. J., and Carpenter, H. A. (2004). Decreased Fibrosis during Corticosteroid Therapy of Autoimmune Hepatitis. J. Hepatol. 40 (4), 646–652. doi:10.1016/j.jhep.2004.01.009
Czaja, A. J. (2014). Hepatic Inflammation and Progressive Liver Fibrosis in Chronic Liver Disease. World J. Gastroenterol. 20 (10), 2515–2532. doi:10.3748/wjg.v20.i10.2515
Deleve, L. D., Wang, X., and Guo, Y. (2008). Sinusoidal Endothelial Cells Prevent Rat Stellate Cell Activation and Promote Reversion to Quiescence. Hepatology 48 (3), 920–930. doi:10.1002/hep.22351
Dewidar, B., Meyer, C., Dooley, S., and Meindl-Beinker, A. N. (2019). TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 8 (11). doi:10.3390/cells8111419
di Bonzo, L. V., Ferrero, I., Cravanzola, C., Mareschi, K., Rustichell, D., Novo, E., et al. (2008). Human Mesenchymal Stem Cells as a Two-Edged Sword in Hepatic Regenerative Medicine: Engraftment and Hepatocyte Differentiation versus Profibrogenic Potential. Gut 57 (2), 223–231. doi:10.1136/gut.2006.111617
Dienstag, J. L., Goldin, R. D., Heathcote, E. J., Hann, H. W., Woessner, M., Stephenson, S. L., et al. (2003). Histological Outcome during Long-Term Lamivudine Therapy. Gastroenterology 124 (1), 105–117. doi:10.1053/gast.2003.50013
Ding, B. S., Nolan, D. J., Butler, J. M., James, D., Babazadeh, A. O., Rosenwaks, Z., et al. (2010). Inductive Angiocrine Signals from Sinusoidal Endothelium Are Required for Liver Regeneration. Nature 468 (7321), 310–315. doi:10.1038/nature09493
Duffield, J. S., Forbes, S. J., Constandinou, C. M., Clay, S., Partolina, M., Vuthoori, S., et al. (2005). Selective Depletion of Macrophages Reveals Distinct, Opposing Roles during Liver Injury and Repair. J. Clin. Invest. 115 (1), 56–65. doi:10.1172/jci22675
Elpek, G. Ö. (2014). Cellular and Molecular Mechanisms in the Pathogenesis of Liver Fibrosis: An Update. World J. Gastroenterol. 20 (23), 7260–7276. doi:10.3748/wjg.v20.i23.7260
Eom, Y. W., Shim, K. Y., and Baik, S. K. (2015). Mesenchymal Stem Cell Therapy for Liver Fibrosis. Korean J. Intern. Med. 30 (5), 580–589. doi:10.3904/kjim.2015.30.5.580
Fort, M. M., Mozaffarian, A., Stöver, A. G., Correia, Jda. S., Johnson, D. A., Crane, R. T., et al. (2005). A Synthetic TLR4 Antagonist Has Anti-inflammatory Effects in Two Murine Models of Inflammatory Bowel Disease. J. Immunol. 174 (10), 6416–6423. doi:10.4049/jimmunol.174.10.6416
Friedman, S. L., Ratziu, V., Harrison, S. A., Abdelmalek, M. F., Aithal, G. P., Caballeria, J., et al. (2018). A Randomized, Placebo-Controlled Trial of Cenicriviroc for Treatment of Nonalcoholic Steatohepatitis with Fibrosis. Hepatology 67 (5), 1754–1767. doi:10.1002/hep.29477
Friedman, S. L., Sheppard, D., Duffield, J. S., and Violette, S. (2013). Therapy for Fibrotic Diseases: Nearing the Starting Line. Sci. Transl Med. 5 (167), 167sr1. doi:10.1126/scitranslmed.3004700
Fujiwara, K., Yasui, S., Okitsu, K., Yonemitsu, Y., Oda, S., and Yokosuka, O. (2010). The Requirement for a Sufficient Period of Corticosteroid Treatment in Combination with Nucleoside Analogue for Severe Acute Exacerbation of Chronic Hepatitis B. J. Gastroenterol. 45 (12), 1255–1262. doi:10.1007/s00535-010-0280-y
García-Trevijano, E. R., Iraburu, M. J., Fontana, L., Domínguez-Rosales, J. A., Auster, A., Covarrubias-Pinedo, A., et al. (1999). Transforming Growth Factor β1induces the Expression of α1(i) Procollagen mRNA by a Hydrogen Peroxide-c/ebpβ-dependent Mechanism in Rat Hepatic Stellate Cells. Hepatology 29 (3), 960–970. doi:10.1002/hep.510290346
Gaul, S., Leszczynska, A., Alegre, F., Kaufmann, B., Johnson, C. D., Adams, L. A., et al. (2021). Hepatocyte Pyroptosis and Release of Inflammasome Particles Induce Stellate Cell Activation and Liver Fibrosis. J. Hepatol. 74 (1), 156–167. doi:10.1016/j.jhep.2020.07.041
Ge, J. Y., Zheng, Y. W., Tsuchida, T., Furuya, K., Isoda, H., Taniguchi, H., et al. (2020). Hepatic Stellate Cells Contribute to Liver Regeneration through Galectins in Hepatic Stem Cell Niche. Stem Cel Res Ther 11 (1), 425. doi:10.1186/s13287-020-01942-x
Geerts, A. (2001). History, Heterogeneity, Developmental Biology, and Functions of Quiescent Hepatic Stellate Cells. Semin. Liver Dis. 21 (3), 311–335. doi:10.1055/s-2001-17550
Georges, P. C., Hui, J. J., Gombos, Z., McCormick, M. E., Wang, A. Y., Uemura, M., et al. (2007). Increased Stiffness of the Rat Liver Precedes Matrix Deposition: Implications for Fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 293 (6), G1147–G1154. doi:10.1152/ajpgi.00032.2007
Ghatak, S., Biswas, A., Dhali, G. K., Chowdhury, A., Boyer, J. L., and Santra, A. (2011). Oxidative Stress and Hepatic Stellate Cell Activation Are Key Events in Arsenic Induced Liver Fibrosis in Mice. Toxicol. Appl. Pharmacol. 251 (1), 59–69. doi:10.1016/j.taap.2010.11.016
Gracia-Sancho, J., Caparrós, E., Fernández-Iglesias, A., and Francés, R. (2021). Role of Liver Sinusoidal Endothelial Cells in Liver Diseases. Nat. Rev. Gastroenterol. Hepatol. 18 (6), 411–431. doi:10.1038/s41575-020-00411-3
Gressner, A. M., and Weiskirchen, R. (2006). Modern Pathogenetic Concepts of Liver Fibrosis Suggest Stellate Cells and TGF-Beta as Major Players and Therapeutic Targets. J. Cel Mol Med 10 (1), 76–99. doi:10.1111/j.1582-4934.2006.tb00292.x
Guo, J., and Friedman, S. L. (2010). Toll-like Receptor 4 Signaling in Liver Injury and Hepatic Fibrogenesis. Fibrogenesis Tissue Repair 3, 21. doi:10.1186/1755-1536-3-21
Guo, Y., Xiao, L., Sun, L., and Liu, F. (2012). Wnt/beta-catenin Signaling: a Promising New Target for Fibrosis Diseases. Physiol. Res. 61 (4), 337–346. doi:10.33549/physiolres.932289
Gustot, T., and Jalan, R. (2019). Acute-on-chronic Liver Failure in Patients with Alcohol-Related Liver Disease. J. Hepatol. 70 (2), 319–327. doi:10.1016/j.jhep.2018.12.008
He, B., Zhang, Y., Lü, M. H., Cao, Y. L., Fan, Y. H., Deng, J. Q., et al. (2013). Glucocorticoids Can Increase the Survival Rate of Patients with Severe Viral Hepatitis B: a Meta-Analysis. Eur. J. Gastroenterol. Hepatol. 25 (8), 926–934. doi:10.1097/MEG.0b013e32835f4cbd
Heldin, C. H., and Westermark, B. (1999). Mechanism of Action and In Vivo Role of Platelet-Derived Growth Factor. Physiol. Rev. 79 (4), 1283–1316. doi:10.1152/physrev.1999.79.4.1283
Hellerbrand, C., Stefanovic, B., Giordano, F., Burchardt, E. R., and Brenner, D. A. (1999). The Role of TGFbeta1 in Initiating Hepatic Stellate Cell Activation In Vivo. J. Hepatol. 30 (1), 77–87. doi:10.1016/s0168-8278(99)80010-5
Hernandez-Gea, V., and Friedman, S. L. (2011). Pathogenesis of Liver Fibrosis. Annu. Rev. Pathol. 6, 425–456. doi:10.1146/annurev-pathol-011110-130246
Hernandez-Munoz, I., de la Torre, P., Sanchez-Alcazar, J., Garcia, I., Santiago, E., Munoz-Yague, M., et al. (1997). Tumor Necrosis Factor Alpha Inhibits Collagen Alpha 1(I) Gene Expression in Rat Hepatic Stellate Cells through a G Protein. Gastroenterology 113 (2), 625–640. doi:10.1053/gast.1997.v113.pm9247485
Hu, B. L., Shi, C., Lei, R. E., Lu, D. H., Luo, W., Qin, S. Y., et al. (2016). Interleukin-22 Ameliorates Liver Fibrosis through miR-200a/beta-Catenin. Sci. Rep. 6, 36436. doi:10.1038/srep36436
Hu, C., Zhao, L., Zhang, L., Bao, Q., and Li, L. (2020). Mesenchymal Stem Cell-Based Cell-free Strategies: Safe and Effective Treatments for Liver Injury. Stem Cel Res Ther 11 (1), 377. doi:10.1186/s13287-020-01895-1
Iredale, J. P., Benyon, R. C., Pickering, J., McCullen, M., Northrop, M., Pawley, S., et al. (1998). Mechanisms of Spontaneous Resolution of Rat Liver Fibrosis. Hepatic Stellate Cell Apoptosis and Reduced Hepatic Expression of Metalloproteinase Inhibitors. J. Clin. Invest. 102 (3), 538–549. doi:10.1172/jci1018
Iwaisako, K., Jiang, C., Zhang, M., Cong, M., Moore-Morris, T. J., Park, T. J., et al. (2014). Origin of Myofibroblasts in the Fibrotic Liver in Mice. Proc. Natl. Acad. Sci. U S A. 111 (32), E3297–E3305. doi:10.1073/pnas.1400062111
Jaeschke, H. (2011). Reactive Oxygen and Mechanisms of Inflammatory Liver Injury: Present Concepts. J. Gastroenterol. Hepatol. 26(Suppl. 1), 173–179. doi:10.1111/j.1440-1746.2010.06592.x
Jang, Y. O., Kim, Y. J., Baik, S. K., Kim, M. Y., Eom, Y. W., Cho, M. Y., et al. (2014). Histological Improvement Following Administration of Autologous Bone Marrow-Derived Mesenchymal Stem Cells for Alcoholic Cirrhosis: a Pilot Study. Liver Int. 34 (1), 33–41. doi:10.1111/liv.12218
Janssen, H. L., Reesink, H. W., Lawitz, E. J., Zeuzem, S., Rodriguez-Torres, M., Patel, K., et al. (2013). Treatment of HCV Infection by Targeting microRNA. N. Engl. J. Med. 368 (18), 1685–1694. doi:10.1056/NEJMoa1209026
Jiao, J., Sastre, D., Fiel, M. I., Lee, U. E., Ghiassi-Nejad, Z., Ginhoux, F., et al. (2012). Dendritic Cell Regulation of Carbon Tetrachloride-Induced Murine Liver Fibrosis Regression. Hepatology 55 (1), 244–255. doi:10.1002/hep.24621
Jiménez Calvente, C., Sehgal, A., Popov, Y., Kim, Y. O., Zevallos, V., Sahin, U., et al. (2015). Specific Hepatic Delivery of Procollagen α1(I) Small Interfering RNA in Lipid-like Nanoparticles Resolves Liver Fibrosis. Hepatology 62 (4), 1285–1297. doi:10.1002/hep.27936
Jin, Z., Sun, R., Wei, H., Gao, X., Chen, Y., and Tian, Z. (2011). Accelerated Liver Fibrosis in Hepatitis B Virus Transgenic Mice: Involvement of Natural Killer T Cells. Hepatology 53 (1), 219–229. doi:10.1002/hep.23983
Kagan, P., Sultan, M., Tachlytski, I., Safran, M., and Ben-Ari, Z. (2017). Both MAPK and STAT3 Signal Transduction Pathways Are Necessary for IL-6-dependent Hepatic Stellate Cells Activation. PLoS One 12 (5), e0176173. doi:10.1371/journal.pone.0176173
Karlmark, K. R., Weiskirchen, R., Zimmermann, H. W., Gassler, N., Ginhoux, F., Weber, C., et al. (2009). Hepatic Recruitment of the Inflammatory Gr1+ Monocyte Subset upon Liver Injury Promotes Hepatic Fibrosis. Hepatology 50 (1), 261–274. doi:10.1002/hep.22950
Kharaziha, P., Hellström, P. M., Noorinayer, B., Farzaneh, F., Aghajani, K., Jafari, F., et al. (2009). Improvement of Liver Function in Liver Cirrhosis Patients after Autologous Mesenchymal Stem Cell Injection: a Phase I-II Clinical Trial. Eur. J. Gastroenterol. Hepatol. 21 (10), 1199–1205. doi:10.1097/MEG.0b013e32832a1f6c
Kim, D. H., and Rossi, J. J. (2007). Strategies for Silencing Human Disease Using RNA Interference. Nat. Rev. Genet. 8 (3), 173–184. doi:10.1038/nrg2006
Kim, M. Y., Cho, M. Y., Baik, S. K., Jeong, P. H., Suk, K. T., Jang, Y. O., et al. (2012). Beneficial Effects of Candesartan, an Angiotensin-Blocking Agent, on Compensated Alcoholic Liver Fibrosis - a Randomized Open-Label Controlled Study. Liver Int. 32 (6), 977–987. doi:10.1111/j.1478-3231.2012.02774.x
Kisseleva, T., and Brenner, D. (2021). Molecular and Cellular Mechanisms of Liver Fibrosis and its Regression. Nat. Rev. Gastroenterol. Hepatol. 18 (3), 151–166. doi:10.1038/s41575-020-00372-7
Kitazawa, T., Tsujimoto, T., Kawaratani, H., Fujimoto, M., and Fukui, H. (2008). Expression of Toll-like Receptor 4 in Various Organs in Rats with D-Galactosamine-Induced Acute Hepatic Failure. J. Gastroenterol. Hepatol. 23 (8 Pt 2), e494–8. doi:10.1111/j.1440-1746.2007.05246.x
Kitazawa, T., Tsujimoto, T., Kawaratani, H., and Fukui, H. (2009). Therapeutic Approach to Regulate Innate Immune Response by Toll-like Receptor 4 Antagonist E5564 in Rats with D-Galactosamine-Induced Acute Severe Liver Injury. J. Gastroenterol. Hepatol. 24 (6), 1089–1094. doi:10.1111/j.1440-1746.2008.05770.x
Kong, D., Zhang, Z., Chen, L., Huang, W., Zhang, F., Wang, L., et al. (2020). Curcumin Blunts Epithelial-Mesenchymal Transition of Hepatocytes to Alleviate Hepatic Fibrosis through Regulating Oxidative Stress and Autophagy. Redox Biol. 36, 101600. doi:10.1016/j.redox.2020.101600
Krützfeldt, J., Rajewsky, N., Braich, R., Rajeev, K. G., Tuschl, T., Manoharan, M., et al. (2005). Silencing of microRNAs In Vivo with 'antagomirs'. Nature 438 (7068), 685–689. doi:10.1038/nature04303
Kulkarni, J. A., Cullis, P. R., and van der Meel, R. (2018). Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 28 (3), 146–157. doi:10.1089/nat.2018.0721
Lens, S., Alvarado-Tapias, E., Mariño, Z., Londoño, M. C., Martinez, J., LLop, E., et al. (2017). Effects of All-Oral Anti-viral Therapy on HVPG and Systemic Hemodynamics in Patients with Hepatitis C Virus-Associated Cirrhosis. Gastroenterology 153 (5), 1273–e1. doi:10.1053/j.gastro.2017.07.016
Li, H. (2020). Advances in Anti Hepatic Fibrotic Therapy with Traditional Chinese Medicine Herbal Formula. J. Ethnopharmacol 251, 112442. doi:10.1016/j.jep.2019.112442
Li, J., Hu, R., Xu, S., Li, Y., Qin, Y., Wu, Q., et al. (2017). Xiaochaihutang Attenuates Liver Fibrosis by Activation of Nrf2 Pathway in Rats. Biomed. Pharmacother. 96, 847–853. doi:10.1016/j.biopha.2017.10.065
Li, Y., Wang, J., and Asahina, K. (2013). Mesothelial Cells Give Rise to Hepatic Stellate Cells and Myofibroblasts via Mesothelial-Mesenchymal Transition in Liver Injury. Proc. Natl. Acad. Sci. U S A. 110 (6), 2324–2329. doi:10.1073/pnas.1214136110
Lin, Y., Dong, M. Q., Liu, Z. M., Xu, M., Huang, Z. H., Liu, H. J., et al. (2021). A Strategy of Vascular‐targeted Therapy for Liver Fibrosis. Hepatology. doi:10.1002/hep.32299
Liu, J., Yang, Y., Zheng, C., Chen, G., Shen, Z., Zheng, S., et al. (2019). Correlation of Interleukin-33/ST2 Receptor and Liver Fibrosis Progression in Biliary Atresia Patients. Front. Pediatr. 7, 403. doi:10.3389/fped.2019.00403
Loomba, R., Lawitz, E., Mantry, P. S., Jayakumar, S., Caldwell, S. H., Arnold, H., et al. (2018). The ASK1 Inhibitor Selonsertib in Patients with Nonalcoholic Steatohepatitis: A Randomized, Phase 2 Trial. Hepatology 67 (2), 549–559. doi:10.1002/hep.29514
Lu, D. H., Guo, X. Y., Qin, S. Y., Luo, W., Huang, X. L., Chen, M., et al. (2015). Interleukin-22 Ameliorates Liver Fibrogenesis by Attenuating Hepatic Stellate Cell Activation and Downregulating the Levels of Inflammatory Cytokines. World J. Gastroenterol. 21 (5), 1531–1545. doi:10.3748/wjg.v21.i5.1531
Luedde, T., Kaplowitz, N., and Schwabe, R. F. (2014). Cell Death and Cell Death Responses in Liver Disease: Mechanisms and Clinical Relevance. Gastroenterology 147 (4), 765–e4. doi:10.1053/j.gastro.2014.07.018
Ma, R., Chen, J., Liang, Y., Lin, S., Zhu, L., Liang, X., et al. (2017). Sorafenib: A Potential Therapeutic Drug for Hepatic Fibrosis and its Outcomes. Biomed. Pharmacother. 88, 459–468. doi:10.1016/j.biopha.2017.01.107
Marcellini, M., Di Ciommo, V., Callea, F., Devito, R., Comparcola, D., Sartorelli, M. R., et al. (2005). Treatment of Wilson's Disease with Zinc from the Time of Diagnosis in Pediatric Patients: a Single-Hospital, 10-year Follow-Up Study. J. Lab. Clin. Med. 145 (3), 139–143. doi:10.1016/j.lab.2005.01.007
Marrone, G., Shah, V. H., and Gracia-Sancho, J. (2016). Sinusoidal Communication in Liver Fibrosis and Regeneration. J. Hepatol. 65 (3), 608–617. doi:10.1016/j.jhep.2016.04.018
Mederacke, I., Hsu, C. C., Troeger, J. S., Huebener, P., Mu, X., Dapito, D. H., et al. (2013). Fate Tracing Reveals Hepatic Stellate Cells as Dominant Contributors to Liver Fibrosis Independent of its Aetiology. Nat. Commun. 4, 2823. doi:10.1038/ncomms3823
Miyazawa, T., Burdeos, G. C., Itaya, M., Nakagawa, K., and Miyazawa, T. (2019). Vitamin E: Regulatory Redox Interactions. IUBMB Life 71 (4), 430–441. doi:10.1002/iub.2008
Mudaliar, S., Henry, R. R., Sanyal, A. J., Morrow, L., Marschall, H. U., Kipnes, M., et al. (2013). Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 145 (3), 574–e1. doi:10.1053/j.gastro.2013.05.042
Murphy, F. R., Issa, R., Zhou, X., Ratnarajah, S., Nagase, H., Arthur, M. J., et al. (2002). Inhibition of Apoptosis of Activated Hepatic Stellate Cells by Tissue Inhibitor of Metalloproteinase-1 Is Mediated via Effects on Matrix Metalloproteinase Inhibition: Implications for Reversibility of Liver Fibrosis. J. Biol. Chem. 277 (13), 11069–11076. doi:10.1074/jbc.M111490200
Musso, G., Cassader, M., Paschetta, E., and Gambino, R. (2017). Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-Analysis. JAMA Intern. Med. 177 (5), 633–640. doi:10.1001/jamainternmed.2016.9607
Novo, E., di Bonzo, L. V., Cannito, S., Colombatto, S., and Parola, M. (2009). Hepatic Myofibroblasts: a Heterogeneous Population of Multifunctional Cells in Liver Fibrogenesis. Int. J. Biochem. Cel Biol 41 (11), 2089–2093. doi:10.1016/j.biocel.2009.03.010
Osawa, Y., Hoshi, M., Yasuda, I., Saibara, T., Moriwaki, H., and Kozawa, O. (2013). Tumor Necrosis Factor-α Promotes Cholestasis-Induced Liver Fibrosis in the Mouse through Tissue Inhibitor of Metalloproteinase-1 Production in Hepatic Stellate Cells. PLoS One 8 (6), e65251. doi:10.1371/journal.pone.0065251
Osawa, Y., Kojika, E., Hayashi, Y., Kimura, M., Nishikawa, K., Yoshio, S., et al. (2018). Tumor Necrosis Factor-α-Mediated Hepatocyte Apoptosis Stimulates Fibrosis in the Steatotic Liver in Mice. Hepatol. Commun. 2 (4), 407–420. doi:10.1002/hep4.1158
Pan, X., Ma, X., Jiang, Y., Wen, J., Yang, L., Chen, D., et al. (2020). A Comprehensive Review of Natural Products against Liver Fibrosis: Flavonoids, Quinones, Lignans, Phenols, and Acids. Evid. Based Complement. Alternat Med. 2020, 7171498. doi:10.1155/2020/7171498
Parola, M., and Pinzani, M. (2019). Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol. Aspects Med. 65, 37–55. doi:10.1016/j.mam.2018.09.002
Perepelyuk, M., Terajima, M., Wang, A. Y., Georges, P. C., Janmey, P. A., Yamauchi, M., et al. (2013). Hepatic Stellate Cells and portal Fibroblasts Are the Major Cellular Sources of Collagens and Lysyl Oxidases in normal Liver and Early after Injury. Am. J. Physiol. Gastrointest. Liver Physiol. 304 (6), G605–G614. doi:10.1152/ajpgi.00222.2012
Pinzani, M., Gesualdo, L., Sabbah, G. M., and Abboud, H. E. (1989). Effects of Platelet-Derived Growth Factor and Other Polypeptide Mitogens on DNA Synthesis and Growth of Cultured Rat Liver Fat-Storing Cells. J. Clin. Invest. 84 (6), 1786–1793. doi:10.1172/jci114363
Pinzani, M., Milani, S., Herbst, H., DeFranco, R., Grappone, C., Gentilini, A., et al. (1996). Expression of Platelet-Derived Growth Factor and its Receptors in normal Human Liver and during Active Hepatic Fibrogenesis. Am. J. Pathol. 148 (3), 785–800.
Poisson, J., Lemoinne, S., Boulanger, C., Durand, F., Moreau, R., Valla, D., et al. (2017). Liver Sinusoidal Endothelial Cells: Physiology and Role in Liver Diseases. J. Hepatol. 66 (1), 212–227. doi:10.1016/j.jhep.2016.07.009
Powell, W. J., and Klatskin, G. (1968). Duration of Survival in Patients with Laennec's Cirrhosis. Influence of Alcohol Withdrawal, and Possible Effects of Recent Changes in General Management of the Disease. Am. J. Med. 44 (3), 406–420. doi:10.1016/0002-9343(68)90111-3
Pradere, J. P., Kluwe, J., De Minicis, S., Jiao, J. J., Gwak, G. Y., Dapito, D. H., et al. (2013). Hepatic Macrophages but Not Dendritic Cells Contribute to Liver Fibrosis by Promoting the Survival of Activated Hepatic Stellate Cells in Mice. Hepatology 58 (4), 1461–1473. doi:10.1002/hep.26429
Radaeva, S., Sun, R., Jaruga, B., Nguyen, V. T., Tian, Z., and Gao, B. (2006). Natural Killer Cells Ameliorate Liver Fibrosis by Killing Activated Stellate Cells in NKG2D-dependent and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-dependent Manners. Gastroenterology 130 (2), 435–452. doi:10.1053/j.gastro.2005.10.055
Ramachandran, P., Pellicoro, A., Vernon, M. A., Boulter, L., Aucott, R. L., Ali, A., et al. (2012). Differential Ly-6C Expression Identifies the Recruited Macrophage Phenotype, Which Orchestrates the Regression of Murine Liver Fibrosis. Proc. Natl. Acad. Sci. U S A. 109 (46), E3186–E3195. doi:10.1073/pnas.1119964109
Reid, D. T., Reyes, J. L., McDonald, B. A., Vo, T., Reimer, R. A., and Eksteen, B. (2016). Kupffer Cells Undergo Fundamental Changes during the Development of Experimental NASH and Are Critical in Initiating Liver Damage and Inflammation. PLoS One 11 (7), e0159524. doi:10.1371/journal.pone.0159524
Rockey, D. C. (2016). Liver Fibrosis Reversion after Suppression of Hepatitis B Virus. Clin. Liver Dis. 20 (4), 667–679. doi:10.1016/j.cld.2016.06.003
Roeb, E., Purucker, E., Breuer, B., Nguyen, H., Heinrich, P. C., Rose-John, S., et al. (1997). TIMP Expression in Toxic and Cholestatic Liver Injury in Rat. J. Hepatol. 27 (3), 535–544. doi:10.1016/s0168-8278(97)80359-5
Sanyal, A. J., Chalasani, N., Kowdley, K. V., McCullough, A., Diehl, A. M., Bass, N. M., et al. (2010). Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 362 (18), 1675–1685. doi:10.1056/NEJMoa0907929
Schiff, E., Simsek, H., Lee, W. M., Chao, Y. C., Sette, H., Janssen, H. L., et al. (2008). Efficacy and Safety of Entecavir in Patients with Chronic Hepatitis B and Advanced Hepatic Fibrosis or Cirrhosis. Am. J. Gastroenterol. 103 (11), 2776–2783. doi:10.1111/j.1572-0241.2008.02086.x
Schnabl, B., and Brenner, D. A. (2014). Interactions between the Intestinal Microbiome and Liver Diseases. Gastroenterology 146 (6), 1513–1524. doi:10.1053/j.gastro.2014.01.020
Schon, H. T., and Weiskirchen, R. (2014). Immunomodulatory Effects of Transforming Growth Factor-β in the Liver. Hepatobiliary Surg. Nutr. 3 (6), 386–406. doi:10.3978/j.issn.2304-3881.2014.11.06
Schulze, R. J., Schott, M. B., Casey, C. A., Tuma, P. L., and McNiven, M. A. (2019). The Cell Biology of the Hepatocyte: A Membrane Trafficking Machine. J. Cel Biol 218 (7), 2096–2112. doi:10.1083/jcb.201903090
Seki, E., De Minicis, S., Osterreicher, C. H., Kluwe, J., Osawa, Y., Brenner, D. A., et al. (2007). TLR4 Enhances TGF-Beta Signaling and Hepatic Fibrosis. Nat. Med. 13 (11), 1324–1332. doi:10.1038/nm1663
Seki, E., and Schwabe, R. F. (2015). Hepatic Inflammation and Fibrosis: Functional Links and Key Pathways. Hepatology 61 (3), 1066–1079. doi:10.1002/hep.27332
Sersté, T., Cornillie, A., Njimi, H., Pavesi, M., Arroyo, V., Putignano, A., et al. (2018). The Prognostic Value of Acute-On-Chronic Liver Failure during the Course of Severe Alcoholic Hepatitis. J. Hepatol. 69 (2), 318–324. doi:10.1016/j.jhep.2018.02.022
Shaker, M. E., Ghani, A., Shiha, G. E., Ibrahim, T. M., and Mehal, W. Z. (2013). Nilotinib Induces Apoptosis and Autophagic Cell Death of Activated Hepatic Stellate Cells via Inhibition of Histone Deacetylases. Biochim. Biophys. Acta 1833 (8), 1992–2003. doi:10.1016/j.bbamcr.2013.02.033
Shan, L., Liu, Z., Ci, L., Shuai, C., Lv, X., and Li, J. (2019). Research Progress on the Anti-hepatic Fibrosis Action and Mechanism of Natural Products. Int. Immunopharmacol 75, 105765. doi:10.1016/j.intimp.2019.105765
Song, G., Pacher, M., Balakrishnan, A., Yuan, Q., Tsay, H. C., Yang, D., et al. (2016). Direct Reprogramming of Hepatic Myofibroblasts into Hepatocytes In Vivo Attenuates Liver Fibrosis. Cell Stem Cell 18 (6), 797–808. doi:10.1016/j.stem.2016.01.010
Streetz, K. L., Tacke, F., Leifeld, L., Wüstefeld, T., Graw, A., Klein, C., et al. (2003). Interleukin 6/gp130-dependent Pathways Are Protective during Chronic Liver Diseases. Hepatology 38 (1), 218–229. doi:10.1053/jhep.2003.50268
Sun, H., Che, Q. M., Zhao, X., and Pu, X. P. (2010). Antifibrotic Effects of Chronic Baicalein Administration in a CCl4 Liver Fibrosis Model in Rats. Eur. J. Pharmacol. 631 (1-3), 53–60. doi:10.1016/j.ejphar.2010.01.002
Tacke, F., and Zimmermann, H. W. (2014). Macrophage Heterogeneity in Liver Injury and Fibrosis. J. Hepatol. 60 (5), 1090–1096. doi:10.1016/j.jhep.2013.12.025
Tada, T., Kumada, T., Toyoda, H., Sone, Y., Takeshima, K., Ogawa, S., et al. (2018). Viral Eradication Reduces Both Liver Stiffness and Steatosis in Patients with Chronic Hepatitis C Virus Infection Who Received Direct-Acting Anti-viral Therapy. Aliment. Pharmacol. Ther. 47 (7), 1012–1022. doi:10.1111/apt.14554
Takahashi, H., Shigefuku, R., Maeyama, S., and Suzuki, M. (2014). Cirrhosis Improvement to Alcoholic Liver Fibrosis after Passive Abstinence. BMJ Case Rep. 2014. doi:10.1136/bcr-2013-201618
Takashima, K., Matsunaga, N., Yoshimatsu, M., Hazeki, K., Kaisho, T., Uekata, M., et al. (2009). Analysis of Binding Site for the Novel Small-Molecule TLR4 Signal Transduction Inhibitor TAK-242 and its Therapeutic Effect on Mouse Sepsis Model. Br. J. Pharmacol. 157 (7), 1250–1262. doi:10.1111/j.1476-5381.2009.00297.x
Thakral, S., and Ghoshal, K. (2015). miR-122 Is a Unique Molecule with Great Potential in Diagnosis, Prognosis of Liver Disease, and Therapy Both as miRNA Mimic and Antimir. Curr. Gene Ther. 15 (2), 142–150. doi:10.2174/1566523214666141224095610
Theocharis, A. D., Skandalis, S. S., Gialeli, C., and Karamanos, N. K. (2016). Extracellular Matrix Structure. Adv. Drug Deliv. Rev. 97, 4–27. doi:10.1016/j.addr.2015.11.001
Thursz, M. R., Richardson, P., Allison, M., Austin, A., Bowers, M., Day, C. P., et al. (2015). Prednisolone or Pentoxifylline for Alcoholic Hepatitis. N. Engl. J. Med. 372 (17), 1619–1628. doi:10.1056/NEJMoa1412278
Troeger, J. S., Mederacke, I., Gwak, G. Y., Dapito, D. H., Mu, X., Hsu, C. C., et al. (2012). Deactivation of Hepatic Stellate Cells during Liver Fibrosis Resolution in Mice. Gastroenterology 143 (4), 1073–e22. doi:10.1053/j.gastro.2012.06.036
Tsuchida, T., and Friedman, S. L. (2017). Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 14 (7), 397–411. doi:10.1038/nrgastro.2017.38
Tsung, A., McCoy, S. L., Klune, J. R., Geller, D. A., Billiar, T. R., and Hefeneider, S. H. (2007). A Novel Inhibitory Peptide of Toll-like Receptor Signaling Limits Lipopolysaccharide-Induced Production of Inflammatory Mediators and Enhances Survival in Mice. Shock 27 (4), 364–369. doi:10.1097/01.shk.0000239773.95280.2c
Valera, J. M., Smok, G., Márquez, S., Poniachik, J., and Brahm, J. (2011). Histological Regression of Liver Fibrosis with Immunosuppressive Therapy in Autoimmune Hepatitis. Gastroenterol. Hepatol. 34 (1), 10–15. doi:10.1016/j.gastrohep.2010.10.003
Van Herck, J. L., Schrijvers, D. M., De Meyer, G. R., Martinet, W., Van Hove, C. E., Bult, H., et al. (2010). Transglutaminase 2 Deficiency Decreases Plaque Fibrosis and Increases Plaque Inflammation in Apolipoprotein-E-Deficient Mice. J. Vasc. Res. 47 (3), 231–240. doi:10.1159/000255966
Varela-Rey, M., Fontán-Gabás, L., Blanco, P., López-Zabalza, M. J., and Iraburu, M. J. (2007). Glutathione Depletion Is Involved in the Inhibition of Procollagen alpha1(I) mRNA Levels Caused by TNF-Alpha on Hepatic Stellate Cells. Cytokine 37 (3), 212–217. doi:10.1016/j.cyto.2007.03.013
Villesen, I. F., Daniels, S. J., Leeming, D. J., Karsdal, M. A., and Nielsen, M. J. (2020). Review Article: the Signalling and Functional Role of the Extracellular Matrix in the Development of Liver Fibrosis. Aliment. Pharmacol. Ther. 52 (1), 85–97. doi:10.1111/apt.15773
Wang, Y. Z., Zhang, W., Wang, Y. H., Fu, X. L., and Xue, C. Q. (2019). Repression of Liver Cirrhosis Achieved by Inhibitory Effect of miR-454 on Hepatic Stellate Cells Activation and Proliferation via Wnt10a. J. Biochem. 165 (4), 361–367. doi:10.1093/jb/mvy111
Wehr, A., Baeck, C., Heymann, F., Niemietz, P. M., Hammerich, L., Martin, C., et al. (2013). Chemokine Receptor CXCR6-dependent Hepatic NK T Cell Accumulation Promotes Inflammation and Liver Fibrosis. J. Immunol. 190 (10), 5226–5236. doi:10.4049/jimmunol.1202909
Wells, R. G., Schwabe, R. F., and Schwabe, R. (2015). Origin and Function of Myofibroblasts in the Liver. Semin. Liver Dis. 35 (2), 97–106. doi:10.1055/s-0035-1550061
Wolf, M. J., Adili, A., Piotrowitz, K., Abdullah, Z., Boege, Y., Stemmer, K., et al. (2014). Metabolic Activation of Intrahepatic CD8+ T Cells and NKT Cells Causes Nonalcoholic Steatohepatitis and Liver Cancer via Cross-Talk with Hepatocytes. Cancer Cell 26 (4), 549–564. doi:10.1016/j.ccell.2014.09.003
Wu, L., Zhou, P. Q., Xie, J. W., Zhu, R., Zhou, S. C., Wang, G., et al. (2015). Effects of Yinchenhao Decoction on Self-Regulation of Renin-Angiotensin System by Targeting Angiotensin Converting Enzyme 2 in Bile Duct-Ligated Rat Liver. J. Huazhong Univ. Sci. Technolog Med. Sci. 35 (4), 519–524. doi:10.1007/s11596-015-1463-9
Wynn, T. A., and Barron, L. (2010). Macrophages: Master Regulators of Inflammation and Fibrosis. Semin. Liver Dis. 30 (3), 245–257. doi:10.1055/s-0030-1255354
Xie, G., Wang, X., Wang, L., Wang, L., Atkinson, R. D., Kanel, G. C., et al. (2012). Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 142 (4), 918–e6. doi:10.1053/j.gastro.2011.12.017
Xu, Y., Liang, P., Bian, M., Chen, W., Wang, X., Lin, J., et al. (2016). Interleukin-13 Is Involved in the Formation of Liver Fibrosis in Clonorchis Sinensis-Infected Mice. Parasitol. Res. 115 (7), 2653–2660. doi:10.1007/s00436-016-5012-7
Yang, L., Jung, Y., Omenetti, A., Witek, R. P., Choi, S., Vandongen, H. M., et al. (2008). Fate-mapping Evidence that Hepatic Stellate Cells Are Epithelial Progenitors in Adult Mouse Livers. Stem Cells 26 (8), 2104–2113. doi:10.1634/stemcells.2008-0115
Yang, L., Stimpson, S. A., Chen, L., Wallace Harrington, W., and Rockey, D. C. (2010). Effectiveness of the PPARγ Agonist, GW570, in Liver Fibrosis. Inflamm. Res. 59 (12), 1061–1071. doi:10.1007/s00011-010-0226-0
Yang, N. H., Yuan, G. S., Zhou, Y. C., Liu, J. W., Huang, H. P., Hu, C. G., et al. (2016). Entecavir Combined with Fufang Biejia Ruangan Tablet in Treatment of Chronic Hepatitis B Patients with Liver Fibrosis: 96-week Efficacy Analyses. Nan Fang Yi Ke Da Xue Xue Bao 36 (6), 775–779.
Ye, H. L., Zhang, J. W., Chen, X. Z., Wu, P. B., Chen, L., and Zhang, G. (2020). Ursodeoxycholic Acid Alleviates Experimental Liver Fibrosis Involving Inhibition of Autophagy. Life Sci. 242, 117175. doi:10.1016/j.lfs.2019.117175
Yoneda, A., Sakai-Sawada, K., Niitsu, Y., and Tamura, Y. (2016). Vitamin A and Insulin Are Required for the Maintenance of Hepatic Stellate Cell Quiescence. Exp. Cel Res 341 (1), 8–17. doi:10.1016/j.yexcr.2016.01.012
Yoshiji, H., Kuriyama, S., and Fukui, H. (2007). Blockade of Renin-Angiotensin System in Antifibrotic Therapy. J. Gastroenterol. Hepatol. 22 (Suppl. 1), S93–S95. doi:10.1111/j.1440-1746.2006.04663.x
Zhang, F., Zhang, Z., Chen, L., Kong, D., Zhang, X., Lu, C., et al. (2014). Curcumin Attenuates Angiogenesis in Liver Fibrosis and Inhibits Angiogenic Properties of Hepatic Stellate Cells. J. Cel Mol Med 18 (7), 1392–1406. doi:10.1111/jcmm.12286
Zhao, X. A., Chen, G., Liu, Y., Chen, Y., Wu, H., Xiong, Y., et al. (2018). Curcumin Reduces Ly6Chi Monocyte Infiltration to Protect against Liver Fibrosis by Inhibiting Kupffer Cells Activation to Reduce Chemokines Secretion. Biomed. Pharmacother. 106, 868–878. doi:10.1016/j.biopha.2018.07.028
Zhou, X., and Yang, X. F. (2014). Progress of Targeting Transforming Growth Factor-Β1 Small Interfering RNA in Liver Fibrosis. Chin. Med. Sci. J. 29 (4), 231–235. doi:10.1016/s1001-9294(14)60076-6
Keywords: liver fibrosis, hepatic stellate cells, cytokines, extracellular matrix, traditional Chinese medicine
Citation: Wang F-D, Zhou J and Chen E-Q (2022) Molecular Mechanisms and Potential New Therapeutic Drugs for Liver Fibrosis. Front. Pharmacol. 13:787748. doi: 10.3389/fphar.2022.787748
Received: 01 October 2021; Accepted: 17 January 2022;
Published: 11 February 2022.
Edited by:
Jing Qu, Huazhong University of Science and Technology, ChinaReviewed by:
V. Nathan Subramaniam, Queensland University of Technology, AustraliaCopyright © 2022 Wang, Zhou and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: En-Qiang Chen, Y2hlbmVucWlhbmcxOTgzQGhvdG1haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.