 Sandra Grañana-Castillo
Sandra Grañana-Castillo Maiara Camotti Montanha
Maiara Camotti Montanha Rachel Bearon
Rachel Bearon Saye Khoo1
Saye Khoo1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 15 December 2022
Sec. Drug Metabolism and Transport
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1076266
This article is part of the Research TopicDrug-Drug Interactions in PharmacologyView all 14 articles
Tuberculosis remains the leading cause of death among people living with HIV. Rifapentine is increasingly used to treat active disease or prevent reactivation, in both cases given either as weekly or daily therapy. However, rifapentine is an inducer of CYP3A4, potentially interacting with antiretrovirals like rilpivirine. This in silico study investigates the drug-drug interaction (DDI) magnitude between daily oral rilpivirine 25 mg with either daily 600 mg or weekly 900 mg rifapentine. A physiologically based pharmacokinetic (PBPK) model was built in Simbiology (Matlab R2018a) to simulate the drug-drug interaction. The simulated PK parameters from the PBPK model were verified against reported clinical data for rilpivirine and rifapentine separately, daily rifapentine with midazolam, and weekly rifapentine with doravirine. The simulations of concomitant administration of rifapentine with rilpivirine at steady-state lead to a maximum decrease on AUC0-24 and Ctrough by 83% and 92% on day 5 for the daily rifapentine regimen and 68% and 92% for the weekly regimen on day 3. In the weekly regimen, prior to the following dose, AUC0-24 and Ctrough were still reduced by 47% and 53%. In both simulations, the induction effect ceased 2 weeks after the interruption of rifapentine’s treatment. A daily double dose of rilpivirine after initiating rifapentine 900 mg weekly was simulated but failed to compensate the drug-drug interaction. The drug-drug interaction model suggested a significant decrease on rilpivirine exposure which is unlikely to be corrected by dose increment, thus coadministration should be avoided.
Tuberculosis affects one-fourth of the world-wide population (WHO, 2021c). People with tuberculosis have 5–10% life-time risk of falling ill and without treatment there is a 45% risk of death (WHO, 2021c). Among people living with HIV, tuberculosis remains a primary cause of death as without adequate treatment nearly all die (WHO, 2021a,c).
Rilpivirine is a NNRTI drug to treat HIV infection, and alongside other antiretrovirals it increases the life expectancy of people living with HIV to similar levels of their HIV negative peers. Rilpivirine is commercially available combined with emtricitabine and tenofovir, or combined with dolutegravir (Drugbank Rilpivirine, 2022). Additionally, it is approved as the first long-acting intramuscular HIV treatment when given together with cabotegravir; oral cabotegravir and rilpivirine are given during lead-in therapy, or for bridging specific clinical scenarios (FDA, 2022).
Rifapentine can be given daily as substitution for rifampicin, to treat active tuberculosis (WHO, 2021b; Dorman et al., 2021; CDC, 2022) and as prophylaxis in combination with isoniazid weekly for 3 months (3HP) or daily for a month (1HP) (FDA, 2010; WHO, 2020). 3HP and 1HP shortens the treatment of latent tuberculosis and decreases pill burden compared to the six or 9 months of daily isoniazid (6H/9H) or 3-month daily isoniazid with rifampicin (3HR).
Rifamycins (rifampicin, rifapentine, rifabutin) are antibiotics to treat tuberculosis. However, they are metabolic inducers that can potentially interact with some antiretrovirals. Rifapentine promotes the synthesis of enzymes, namely CYP3A4, which is the primary metabolising enzyme of rilpivirine, leading to a potential drug-drug interaction (DDI) if taken together (FDA, 2011a; Williamson et al., 2013).
Physiologically based pharmacokinetic (PBPK) modelling is a mathematical approach that aims to predict the potential magnitude of DDIs prior or, in some occasions, as replacement of clinical trials, among other applications (FDA, 2018). PBPK modelling mechanistically describes a drug’s pharmacokinetic behaviour by combining physiology, population, and drug properties (FDA, 2020). It comprises of multiple compartments each representing a primary body organ which are then interconnected by the arterial and venous systems, mimicking the physiological composition (Nestorov, 2003). This technique permits exploring potential scenarios that otherwise would not be feasible due to ethical or logistical constraints (Zhuang and Lu, 2016).
In this in silico study, we aimed to investigate the DDI magnitude between daily oral rilpivirine 25 mg with either daily 600 mg or weekly 900 mg rifapentine.
A cohort of 100 adults (50% female) was generated between the age of 18 and 60. No ethic approval was required as this study was based on virtual patients. The PBPK model was designed in Simbiology (Matlab version 2018a). The following assumptions were made: 1) well-stirred compartments with instant distribution of the drug; 2) no drug absorption from the colon; and 3) the model drug transport into tissues was blood-flow limited.
Anatomical properties were randomised following a normal distribution using the height from CDC (2020) and BMI from de la Grandmaison et al. (2001). From these predefined characteristics, weight was the product of BMI divided by height square and body surface area was calculated with the Du Bois formula. Organ volumes were the product of organ density (Brown et al., 1997) and organ weight (Bosgra et al., 2012). Blood flows connected the organ compartments and they were derived from the total cardiac output (Brown et al., 1997).
A previously defined compartmental absorption and transit model were used to represent the oral absorption (Yu and Amidon, 1999). The drug absorption rate constant (Ka) was either derived from clinical data, retrograde calculation from effective permeability (Peff) or using Caco-2 cells. The parameters are described in Table 1. Additionally, a tablet disintegration rate (Kdisin) was included in rifapentine’s absorption.
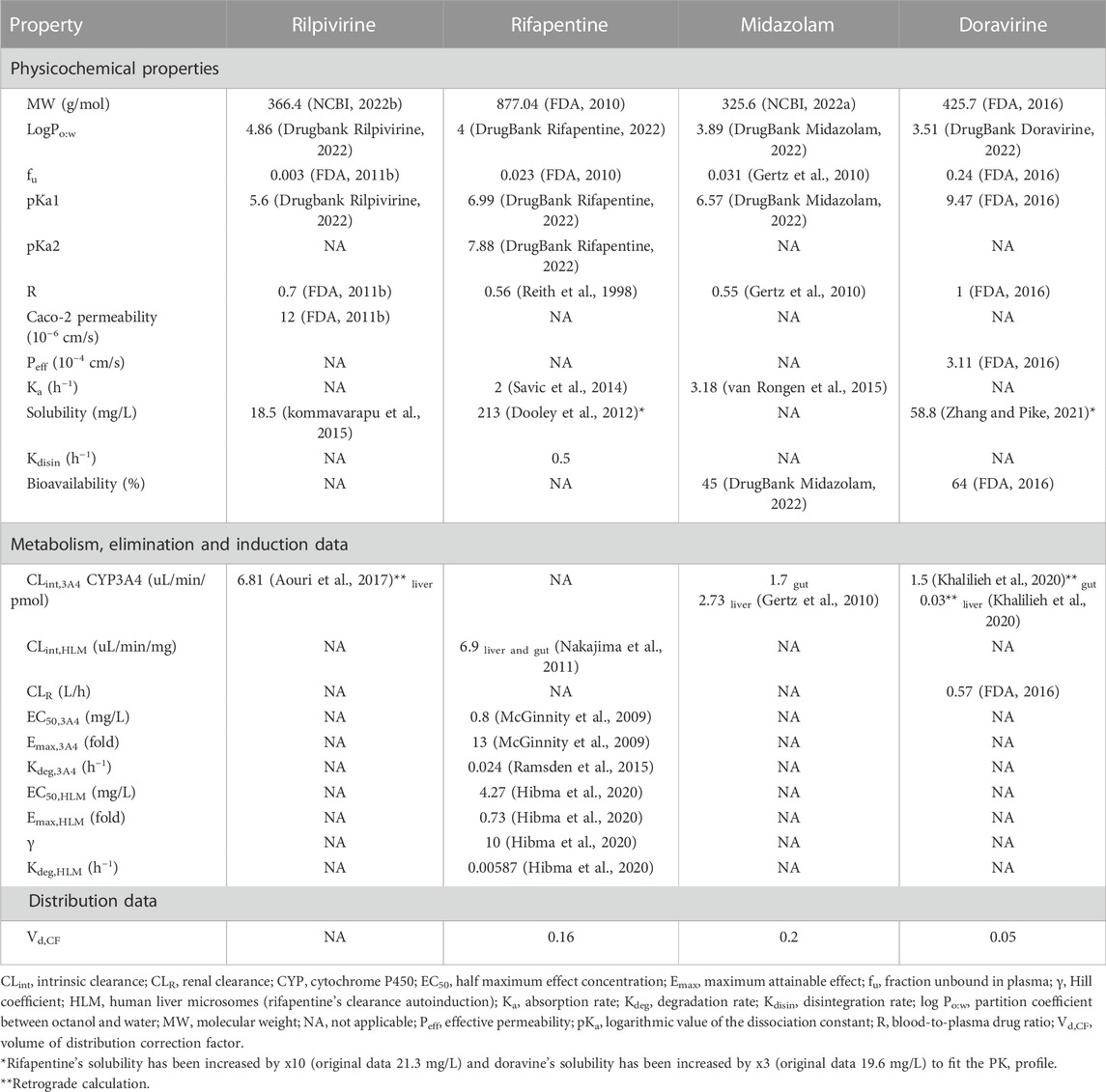
TABLE 1. Summary of physicochemical and in vitro data for rilpivirine, rifapentine, midazolam and doravirine.
Intestinal clearance (L/h) was implemented using Eq. 1 for rilpivirine, midazolam and doravirine, the latter two drugs are used for the model qualification, and using Eq. 2 for rifapentine.
Where CLint,3A4 is the intrinsic clearance for CYP3A4, and CLint,HLM is the intrinsic clearance for rifapentine. Ab3A4,intestines is the abundance of CYP3A4 in the intestine (43 ± 8.6 pmol enzyme/mg microsome) (Paine et al., 1997), MPPGI is the microsomal protein per Gram of intestine (2.7 ± 0.49 mg microsome/g protein) (Paine et al., 1997), WTintestines is the intestine weight in kilograms, and EnzAct3A4 and AutoIndHLM are the relative increases in enzyme activity.
CYP3A4 contributed to rilpivirine, midazolam and doravirine metabolism. Hepatic metabolism (CLhep) was calculated considering the CYP3A4 in vitro intrinsic clearance (CLint,3A4) (Eq. 3) and was scaled to the whole liver. When CLint,3A4 data was unavailable, the hepatic metabolism was estimated using a retrograde calculation from the systemic clearance and absolute bioavailability.
Where Ab3A4,liver is the abundance of CYP3A4 in the liver (138.92 ± 27.78 pmol/mg microsomes) (Barter et al., 2006), MPPGL is the microsomal protein per gram of liver, and WTliver is the liver weight in kilograms. MPPGL was normally distributed using Eq. 4 and a standard deviation of ± 4 mg microsome/g protein (Barter et al., 2008), where age is expressed in years.
The CYP3A4 induction was represented as a relative increase of enzyme activity (EnzAct3A4), considering the differential rate of synthesis and degradation of CYP3A4 (Kdeg,3A4) using Eq. 5:
Where Kdeg,3A4 is 0.024 h−1 (Ramsden et al., 2015), and INDSYN3A4 is the induction of CYP3A4 enzyme synthesis (Eq. 6):
Where Emax is the maximum fold effect, EC50 is the rifapentine’s concentration that elicits half of the maximum effect (McGinnity et al., 2009), Cp is the plasma concentration and fu is the fraction unbound in plasma.
The Arylacetamide Deacetylase (AADAC) was the primary metabolising enzyme for rifapentine. Hepatic metabolism (CLhep) was calculated considering the deacetylase activity in human liver microsomes (CLint,HLM) using Eq. 7 (Nakajima et al., 2011).
Where AutoIndHLM is the relative increase in the acetylase enzyme according to Hibma et al. (2020) The model consisted of an indirect response semi-mechanistic enzyme-turnover model, where the synthesis deacetylase rate was affected by INDSYNHLM (Eq. 8).
Where Kdeg is the enzyme degradation rate (Kdeg 0.00587 h−1) and INDSYNHLM is the induction of rifapentine’s metabolism (Eq. 9) and γ is the Hill coefficient:
The total systemic clearance was the sum of the hepatic metabolism (CLhep) limited by the hepatic blood flow and renal clearance (CLR).
The total volume of distribution (Vss) was calculated following Poulin and Theil (2002) work. However, rifapentine, midazolam and doravirine needed a correction factor applied to the volume of distribution (Vd,CF) to match the clinical parameter; Vd,CF was identified via fitting the observed PK profile.
The PK profiles of rilpivirine and rifapentine were simulated to verify the performance of the PBPK models. Clinically relevant doses for each drug were simulated: once daily oral rilpivirine 25 mg, once daily oral rifapentine 600 mg and once weekly oral rifapentine 900 mg. The predicted PK values of the PBPK model were compared to the typical population estimates from clinical studies (Aouri et al., 2017; Hibma et al., 2020). If the PK parameters were not reported, these were extracted from PK graphs using the Plot Digitizer Tool (plotdigitizer.sourceforge.net). The model performance was successfully verified if the simulated values were within 2-fold range of the reported clinical values and the absolute average-fold error (AAFE) was below 2 (Eq. 10).
4The rifapentine model was verified against clinical DDI data with CYP3A4 sensitive substrates, midazolam and doravirine. The DDI model was validated using two studies: one using daily rifapentine at ascending doses (5, 10, 15, and 20 mg/kg) with a single dose of midazolam 15 mg and the other using weekly rifapentine 900 mg and isoniazid 900 mg with doravirine 100 mg twice daily (Dooley et al., 2012; Lam et al., 2020). To verify the simulations, the dose and schedule were matched to the DDI study design, with the exception of the daily rifapentine, which was simulated as a 600 mg fixed-dose instead of multiple doses dependant on weight. This is because the daily rifapentine study showed a similar decrease of AUC and Cmax of midazolam across the four escalating doses of rifapentine which ranged from 91 to 93% and 82–87%, respectively (Dooley et al., 2012). In addition, the dose was fixed because most studies with rifapentine do not show a body weight dependency (Savic et al., 2017; Hibma et al., 2020; Pham et al., 2022). Therefore, to verify the midazolam-rifapentine DDI, a fixed dose of 600 mg was used for simplicity. Isoniazid was omitted in the validation of the doravirine-rifapentine DDI model (vide discussion). The DDI model performance was evaluated by comparing the observed and simulated PK parameter values of the substrate with the CYP3A4 modulator, and the observed and simulated percentage decrease resulting from the PK fold of rilpivirine with and without rifapentine. Similarly, to the PK simulations, the DDI simulations were successfully validated if the values were within 2-fold range of the reported clinical values and if the AAFE was equal or below 2.
The verified model was used to predict the effect of either daily or weekly rifapentine on daily rilpivirine in a virtual population. The protocol consisted of 14 days daily dose of rilpivirine alone to reach steady state concentrations, followed by 14 days of daily dose of rilpivirine with either daily or weekly dose of rifapentine and 14 days more of daily dose of rilpivirine alone. To further evaluate treatment options, an additional 25 mg daily dose adjustment on rilpivirine was simulated right after the initiation of the weekly rifapentine’s regimen to identify if it could be an alternative to circumvent the potential DDI (Figure 1).
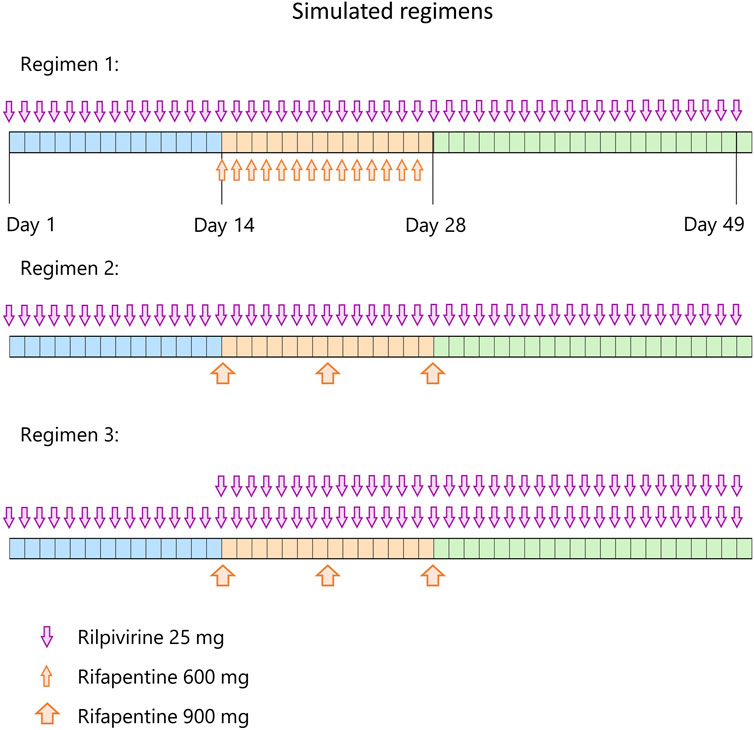
FIGURE 1. Simulated regimens. Each regimen starts with 14 days of rilpivirine 25 mg once daily (blue period), followed by 14 days (orange period) of rilpivirine 25 mg once daily and 600 mg rifapentine once daily on regimen 1, rilpivirine 25 mg once daily and once weekly 900 mg rifapentine on regimen 2, and rilpivirine 50 mg once daily and once weekly 900 mg rifapentine on regimen 3. On the third period (green), rifapentine’s administration is ceased but rilpivirine is maintained with the same dose strategy as the previous period during 21 days.
The PBPK model was qualified successfully against clinical data for rifapentine 600 mg daily and 900 mg weekly with an average (min-max) AAFE of 1.20 (1.00–1.59) for AUC, Cmax, Ctrough, CL/F, t1/2, and Vss/F (Hibma et al., 2020). The rifapentine model included the autoinduction of its own metabolism. In line with the literature, the simulations showed a clearance increase of 71% in the daily regimen, where it reached its maximum potential, and minimal increase of clearance (30%) in the weekly regimen (Hibma et al., 2020).
Rilpivirine was qualified against clinical data for rilpivirine 25 mg daily at steady state (Aouri et al., 2017). All parameters were within the acceptance criteria with an AAFE of 1.12 (1.04–1.22).
To qualify the induction of CYP3A4 by rifapentine, midazolam was used as a single 15 mg dose with and without rifapentine daily 600 mg steady state and doravirine 100 mg twice daily alone and also after co-administration of weekly 900 mg rifapentine. The midazolam-rifapentine yielded an AAFE of 1.25 (1.00–1.74) and the doravirine-rifapentine of 1.27 (1.04–1.89), all in agreement with the acceptance criteria.
The validation is outlined in Supplementary Tables S1–S9 and Supplementary Figures S1–S7, available as Supplementary data.
Results are summarized in Figure 2 and Table 2.
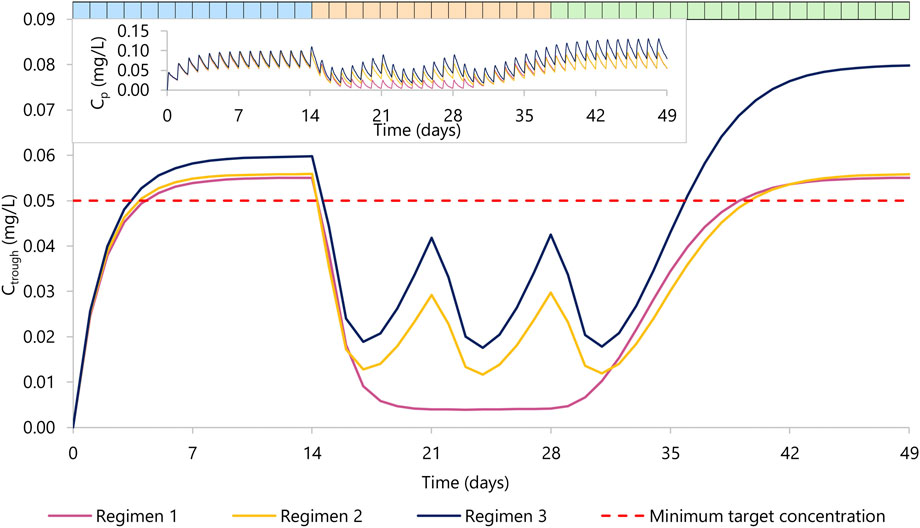
FIGURE 2. Rilpivirine Ctrough concentration over time. Regimen 1 (violet) represents 14 days of rilpivirine 25 mg daily dose (period one in blue), 14 days of rilpivirine 25 mg daily dose with rifapentine 600 mg daily dose (period two in orange), and 21 days of rilpivirine 25 mg daily dose (period three in green). Regimen 2 (yellow) represents 14 days of rilpivirine 25 mg daily dose (period 1), 14 days of rilpivirine 25 mg daily dose with rifapentine 900 mg weekly dose, a total of three doses) (period 2), and 21 days of rilpivirine 25 mg daily dose (period 3). Regimen 3 (dark blue) represents 14 days of rilpivirine 25 mg daily dose (period 1), 14 days of rilpivirine 50 mg daily dose with rifapentine 900 mg weekly dose a total of three doses (period 2), and 21 days of rilpivirine 50 mg daily dose (period 3). The red dashed line represents the minimum target concentration for rilpivirine. The top left corner contains the mean plasma concentration PK profile for rilpivirine for each regimen.
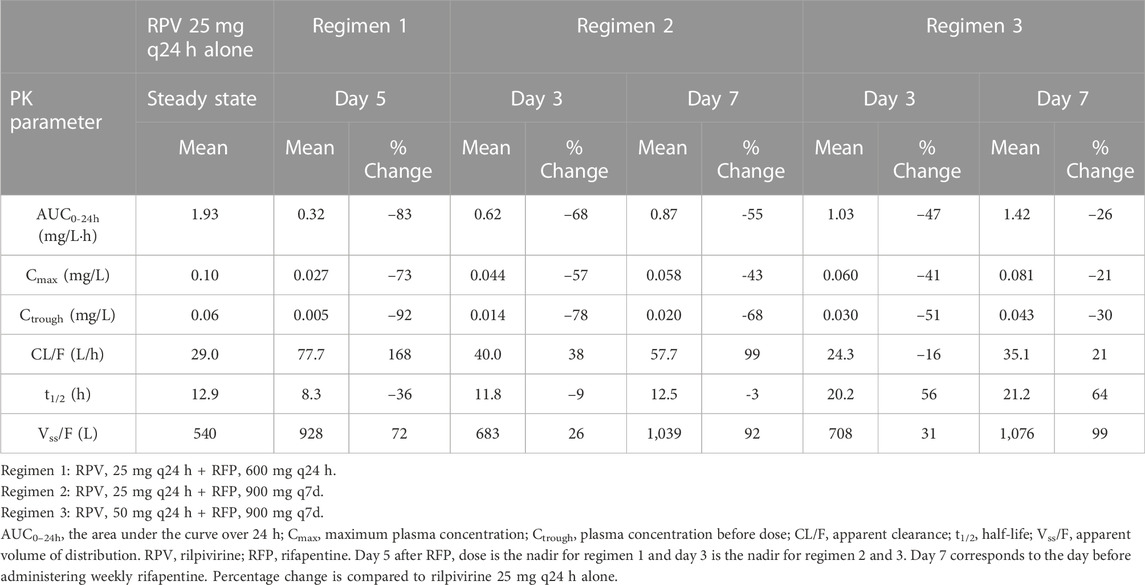
Table 2. Predicted PK parameters of orally administered rilpivirine with and without rifapentine in multiple dosing regimens.
In the regimen 1, we simulated the administration of rilpivirine 25 mg daily for 14 days (period 1), followed by the concomitant administration of daily rifapentine 600 mg for 14 days (period 2), and on day 28, we ceased the administration of rifapentine and continued with rilpivirine 25 mg once daily alone for 21 additional days (period 3). Within the first 3 days of concomitant administration of daily rifapentine 600 mg, rilpivirine’s plasma concentration (Cp) showed a linear but steep decrease. After the first dose of rifapentine, the values of AUC0-24 and Ctrough for rilpivirine were reduced by 18% and 37%, by day 2 they were reduced by 51% and 71%, and by day 3 72% and 85%, respectively. On the fifth day, the plateau maximum effect was observed, with reductions in AUC0-24 and Ctrough of 83% and 92% (Ctrough = 0.005 mg/L), respectively and remained similar until the cease of rifapentine at the start of period 3. In period 3, the plasma concentrations of rilpivirine slowly recovered over time, with more than 95% restored after 14 days of the last rifapentine dose.
In the regimen 2, a similar decrease was observed on the simulated weekly rifapentine 900 mg with rilpivirine. Period one consisted of once daily 25 mg rilpivirine, period two of once daily 25 mg rilpivirine with once weekly 900 mg rifapentine, a total of three doses, and period three of once daily 25 mg rilpivirine. The maximum effect was achieved on day 3 with reductions on the AUC0-24 and Ctrough by 68% and 92% (Ctrough = 0.013 mg/L). Shortly after, as rifapentine’s Cp declined, rilpivirine’s Cp slowly recovered; but prior to the following weekly dose, AUC0-24 and Ctrough were still reduced by 47% and 53% (Ctrough = 0.029 mg/L). After the second dose, rilpivirine reached its maximum induction at day 3 after rifapentine’s dose. Analogously to regimen 1, rilpivirine Cp was restored to more than 95% its original Cp after the last rifapentine dose.
Regimen three evaluated a double dose administration of rilpivirine; identical to regimen two except for an additional 25 mg rilpivirine daily dose on period two and 3. The PK profile was comparable to regimen 2, with AUC0-24 and Ctrough slightly higher due to the increase of dose. Maximum induction was also reached on day 3 after rifapentine’s first dose, with a simulated AUC0-24 and Ctrough reduced by 66% and 69% (Ctrough = 0.019 mg/L) compared to the standard regimen. Considering that in period three rilpivirine is still administered as a double dose, minimum target concentration is reached earlier on day 8 after rifapentine’s cessation.
The DDI between rilpivirine and rifapentine has not yet been clinically studied, although it has been with rifampicin and rifabutin, two other rifamycins (FDA, 2011a). Rifapentine is a stronger CYP3A4 inducer than rifabutin but weaker than rifampicin; for example, rifapentine decreases indinavir’s AUC by 70%, rifampicin by 92% while rifabutin by 34% (Burman et al., 2001). This PBPK model suggests an AUC and Ctrough decrease of rilpivirine by 83 and 92% at its maximum induction when administered with daily oral rifapentine. These findings are comparable to the rifampicin-rilpivirine interaction, where the AUC and Ctrough decreased by 80% and 89%, respectively (Ford et al., 2011). Considering the clinical data provided by the FDA, rifampicin and rifabutin coadministration with rilpivirine is contraindicated (FDA, 2011a). However, a double dose of rilpivirine might be sufficient to overcome the DDI with rifabutin, with rilpivirine doubled at least 2 weeks after rifabutin’s cessation (Liverpool HIV Drug Interactions, 2022b).
In this PBPK study we aimed to identify the DDI magnitude between rilpivirine and rifapentine. The PBPK model showed a strong DDI between rilpivirine and daily rifapentine and less prominent DDI when rifapentine was administered weekly. However, the decrease on key PK parameters were very significant in both scenarios, including when rilpivirine dose was virtually doubled. In this case, the PBPK model suggested that the coadministration of rilpivirine and rifapentine, either daily or weekly, is contraindicated.
There has been an increased interest in studying DDIs via PBPK modelling as it allows the simulation of multiple scenarios with the ultimate goal of informing regulatory agencies, prioritise and design clinical trials, as well as informing healthcare professionals on how to manage DDIs. However, some parameters are not yet fully described, making it difficult to mathematically characterise them. In this model, we considered that all rilpivirine was cleared due to CYP3A4 metabolism without inclusion of renal clearance, as there is limited information on drug metabolism. In the same way, rilpivirine bioavailability was assumed to be 100% due to lack of clinical data. Because some physiological processes are not well understood, in vitro data occasionally did not match the PK profile accurately, and data had to be fitted or calculated in retrograde. This model captured the DDI mechanistically and in a time dependant manner, including the synthesis and degradation of CYP3A4 as well as the autoinduction of rifapentine’s clearance although it did not account for potential interaction with transporters. An in vitro study showed a 3-fold increase in ABCB1 relative gene expression at the highest rifapentine concentration (10 uM), which encodes for p-glycoprotein, while no change for OATP1B1 and OATP1B3 transporters (Williamson et al., 2013). A DDI between moxifloxacin and three times weekly rifapentine 900 mg showed a decrease on the moxifloxacin’s AUC by 17.2%, the authors suggested that transporters could have played a role as moxifloxacin is not metabolised by CYP P450 isoforms but that was not assessed (Dooley et al., 2008). There is little evidence that transporters play an important role on rifapentine’s DDI. Genetic polymorphisms were not assessed in this PBPK model, although Aouri et al. (2017) demonstrated that CYP3A4*22 polymorphism among others did not affect rilpivirine’s pharmacokinetics.
While 1HP and 3HP treatments include isoniazid and rifapentine, the DDI model omitted isoniazid PK. Isoniazid presents an in vitro inhibition constant (Ki) of 51.8–75.9 μmol/L (7.1–10.4 mg/L) (Desta et al., 2001; Wen et al., 2002) and desirable Cp levels range between 3 and 6 mg/L (Huerta-García et al., 2020). At therapeutic concentrations, theoretically 40% of the CYP3A activity is inhibited (Desta et al., 2001). Considering that rifapentine is a moderate to strong CYP3A inducer with a Cmax more than ten times higher than its CYP3A EC50, the overall DDI between isoniazid, rifapentine and a substrate is driven by the rifapentine induction effect.
Antiretroviral therapy (ART) including rilpivirine is highly advantageous as the fixed-dose combined pills are relatively small compared to the alternatives as well as the oral-lead in or substitute for missed doses of long-acting injectables antiretrovirals (Bennet, 2020). Nonetheless, rilpivirine presents higher rates of virologic failure in patients with high viral load (>100,000 copies/mL) or ≤95% adherence, relative to patients taking efavirenz (Bennet, 2020). Maintenance of adequate plasma concentrations is essential for optimal antiretroviral therapy. Current target trough concentration (Ctrough) for rilpivirine is 0.05 mg/L (Néant et al., 2020), which is suggested as four times the concentration required for 90% inhibition (IC90 0.012 mg/L) (Margolis et al., 2015). However, a recent study with rilpivirine based regimen have highlighted that the current Ctrough target might need to be reassessed (Néant et al., 2019) and an optimal target Ctrough of 0.07 mg/L is required to achieve virologic response, especially in pre-treated patients (Néant et al., 2020). This is further complicated considering that 11% of a population in Aouri et al. study did not reach a Ctrough of 0.05 mg/L (Aouri et al., 2017). Considering the repurposed target concentration, many more would fall in subtherapeutic concentrations. Dose increase is usually done by 25 mg, as rilpivirine is only available in this dose (EMC, 2021). A practical example is the management of the DDIs with rifabutin where rilpivirine dose can be doubled to overcome the DDI (EMC, 2021). However, increasing the dose even further (x3-12 times) increases the risk of QTc prolongation as this phenomenon is dose dependant and should be avoided (Aouri et al., 2017).
This in silico study, suggests that co-administration of rifapentine with rilpivirine is contraindicated and replacement or inclusion of an additional antiretroviral therapy is recommended. Currently, 3HP is only recommended with raltegravir 400 mg twice daily or efavirenz 600 mg once daily for treatment of latent tuberculosis infection in people living with HIV (WHO, 2020). These drugs are good substitute candidates as raltegravir is primarily metabolised by UGT1A1 and efavirenz by CYP2B6 with marginal contribution of CYP3A4 (Ogburn et al., 2010). A study in 2014 showed a 71% AUC0-12 increase of raltegravir after 900 mg weekly rifapentine, which was tolerated and safe (Weiner et al., 2014). Differently, daily rifapentine treatment decreased raltegravir’s Cmin by 41%, which requires more clinical investigation (Weiner et al., 2014; Liverpool HIV Drug Interactions, 2022a). Podany et al. (2015) observed that 88% of participants taking efavirenz with 1HP maintained the minimum target concentration ≥1 mg/L and viral suppression. Recently, dolutegravir twice daily has proven safe and well tolerated with 3HP and 1HP. Dolutegravir’s AUC decreased by 26% when co-administered with 3HP, suggesting that dolutegravir could be administered without dose adjustments but a double dose is recommended in individuals at risk of treatment failure or blips (Dooley et al., 2020). A double dose of dolutegravir with 1HP showed concentrations higher than dolutegravir once daily alone and was suggested to be safe (Imperial et al., 2022). As seen in the validation study of weekly rifapentine, doravirine 100 mg twice daily could potentially be used with 3HP (Lam et al., 2020). On the other hand, bictegravir is contraindicated (Arora et al., 2021; Liou et al., 2021) and there is no data with protease inhibitors or darunavir yet.
This modelling approach provides a potential tool to study the magnitude of DDIs of daily and weekly regimens which can help designing clinical trials when necessary or avoid them when the interaction is unmanageable. This PBPK study suggests that rilpivirine antiretroviral therapy does not reach sufficient exposure to be managed with 3HP or 1HP on its own and potentially an additional antiretroviral regimen should be included. Alternatively, some antiretrovirals are manageable both with 3HP and 1HP, and others can only be managed with 3HP as the DDI is less marked. 3HP is preferred if it does not require switching therapies and 1HP if it does and it is a suitable regimen. This PBPK model is characterised by some limitations including in vitro data availability and description of processes involved in drug disposition such as drug transport that can hinder the ability to accurately predict complex scenarios. Non-etheless, it is based on a detailed description of the human physiology, drug metabolism, and PK processes representing a powerful tool to explore different scenarios and aid clinicians on how to manage DDIs.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
All authors contributed to the overall concept and design. SG-C performed the modelling analysis. SG-C wrote the manuscript with support from MM, RB, SK, and MS. All authors reviewed and contributed to the final manuscript.
This project is funded by Engineering and Physical Sciences Research Council (EPSRC).
This work has been presented at the American Conference of Pharmacometrics 13, Denver, CO, United States, 2022 (Poster T-058).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2022.1076266/full#supplementary-material
Aouri, M., Barcelo, C., Guidi, M., Rotger, M., Cavassini, M., Hizrel, C., et al. (2017). Population pharmacokinetics and Pharmacogenetics analysis of rilpivirine in HIV-1-Infected individuals. Antimicrob. Agents Chemother. 61. doi:10.1128/aac.00899-16
Arora, P., Collins, S., Martin, H., Zhang, X., Mak, L., Ling, J., et al. (2021). “Drug interactions with once-daily B F/TAF in combination with once-weekly rifapentine,” in Virtual conference on retroviruses and opportunistic infections.
Barter, Z. E., Chowdry, J. E., Harlow, J. R., Snawder, J. E., Lipscomb, J. C., and Rostami-Hodjegan, A. (2008). Covariation of human microsomal protein per gram of liver with age: Absence of influence of operator and sample storage may justify interlaboratory data pooling. Drug Metab. Dispos. 36 (12), 2405–2409. doi:10.1124/dmd.108.021311
Barter, Z. E., Rowland Yeo, K., Tucker, G. T., and Rostami-Hodjegan, A. (2006). A meta-analysis of CYP3A4 abudance in human liver: Use of different standards contributes to apparent heterogeneity in reported values. [Online]. Available: https://www.certara.com/app/uploads/Resources/Posters/Barter_2006_ACCP_CYP3A4.pdf (Accessed 05 13, 2022).
Bennet, J. E. (2020). Principles and practice of infectious diseases, chapter 128: Antiretroviral therapy for human immunodeficiency virus infection. Ninth Edition ed. Elsevier, 1739–1760.
Bosgra, S., van Eijkeren, J., Bos, P., Zeilmaker, M., and Slob, W. (2012). An improved model to predict physiologically based model parameters and their inter-individual variability from anthropometry. Crit. Rev. Toxicol. 42 (9), 751–767. doi:10.3109/10408444.2012.709225
Brown, R. P., Delp, M. D., Lindstedt, S. L., Rhomberg, L. R., and Beliles, R. P. (1997). Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13 (4), 407–484. doi:10.1177/074823379701300401
Burman, W. J., Gallicano, K., and Peloquin, C. (2001). Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin. Pharmacokinet. 40 (5), 327–341. doi:10.2165/00003088-200140050-00002
CDC (2020). Anthropometric reference data for children and adults: United States, 2015–2018. [Online]. Available: https://www.cdc.gov/nchs/fastats/body-measurements.htm (Accessed 05 11, 2022).
CDC (2022). Treatment for TB disease. [Online]. Available: https://www.cdc.gov/tb/topic/treatment/tbdisease.htm (Accessed 11 16, 2022).
de la Grandmaison, G. L., Clairand, I., and Durigon, M. (2001). Organ weight in 684 adult autopsies: New tables for a caucasoid population. Forensic Sci. Int. 119 (2), 149–154. doi:10.1016/s0379-0738(00)00401-1
Desta, Z., Soukhova, N. V., and Flockhart, D. A. (2001). Inhibition of cytochrome P450 (CYP450) isoforms by isoniazid: Potent inhibition of CYP2C19 and CYP3A. Antimicrob. Agents Chemother. 45 (2), 382–392. doi:10.1128/aac.45.2.382-392.2001
Dooley, K., Flexner, C., Hackman, J., Peloquin, C. A., Nuermberger, E., Chaisson, R. E., et al. (2008). Repeated administration of high-dose intermittent rifapentine reduces rifapentine and moxifloxacin plasma concentrations. Antimicrob. Agents Chemother. 52 (11), 4037–4042. doi:10.1128/aac.00554-08
Dooley, K. E., Bliven-Sizemore, E. E., Weiner, M., Lu, Y., Nuermberger, E. L., Hubbard, W. C., et al. (2012). Safety and pharmacokinetics of escalating daily doses of the antituberculosis drug rifapentine in healthy volunteers. Clin. Pharmacol. Ther. 91 (5), 881–888. doi:10.1038/clpt.2011.323
Dooley, K. E., Savic, R., Gupte, A., Marzinke, M. A., Zhang, N., Edward, V. A., et al. (2020). Once-weekly rifapentine and isoniazid for tuberculosis prevention in patients with HIV taking dolutegravir-based antiretroviral therapy: A phase 1/2 trial. Lancet. HIV 7 (6), e401–e409. doi:10.1016/s2352-3018(20)30032-1
Dorman, S. E., Nahid, P., Kurbatova, E. V., Phillips, P. P. J., Bryant, K., Dooley, K. E., et al. (2021). Four-month rifapentine regimens with or without moxifloxacin for tuberculosis. N. Engl. J. Med. 384 (18), 1705–1718. doi:10.1056/NEJMoa2033400
DrugBank Doravirine (2022). DrugBank doravirine. [Online]. Available: https://go.drugbank.com/drugs/DB12301 (Accessed September 12, 2022).
DrugBank Midazolam (2022). DrugBank midazolam. [Online]. Available: https://go.drugbank.com/drugs/DB00683 (Accessed September 12, 2022).
DrugBank Rifapentine (2022). DrugBank rifapentine. [Online]. Available: https://go.drugbank.com/drugs/DB01201 (Accessed May 19, 2022).
Drugbank Rilpivirine (2022). Rilpivirine. [Online]. Available: https://go.drugbank.com/drugs/DB08864 (Accessed October 19, 2022).
FDA (2010). Priftin (rifapentine) Tablets label. [Online]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021024s009lbl.pdf (Accessed 05 19, 2022).
FDA (2011a). Edurant (rilpivirine) tablets label. [Online]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202022s000lbl.pdf (Accessed 07 25, 2022).
FDA (2011b). Rilpivirine: Clinical pharmacology and biopharmaceutics review(s). [Online]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202022Orig1s000ClinPharmR.pdf (Accessed 05 19, 2022).
FDA (2016). NDA multi-disciplinary review and evaluation – NDA 210806 and NDA 210807 doravirine (DOR) and DOR/3TC/DOR. [Online]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210806Orig1s000,%20210807Orig1s000MultidisciplineR.pdf (Accessed 09 15, 2022).
FDA (2018). Program of physiologically-based pharmacokinetic and pharmacodynamic modeling (PBPK Program). [Online]. Available: https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/program-physiologically-based-pharmacokinetic-and-pharmacodynamic-modeling-pbpk-program#:∼:text=On%20August%2031%2C%202018%2C%20FDA%20issued%20the%20%E2%80%9C,applications%20%28BLAs%29%2C%20or%20abbreviated%20new%20drug%20applications%20%28ANDAs%29 (Accessed 06 16, 2022).
FDA (2020). The use of physiologically based pharmacokinetic analyses - biopharmaceutics applications for oral drug product development, manufacturig changes, and controls guidance for industry. C.f.D.E.a.R. (CDER).
FDA (2022). CABENUVA (cabotegravir extended-release injectable suspension; rilpivirine extended-release injectable suspension), co-packaged for intramuscular use. [Online]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/212888s003lbl.pdf (Accessed 06 22, 2022).
Ford, N., Lee, J., Andrieux-Meyer, I., and Calmy, A. (2011). Safety, efficacy, and pharmacokinetics of rilpivirine: Systematic review with an emphasis on resource-limited settings. HIV AIDS (Auckl) 3, 35–44. doi:10.2147/hiv.S14559
Gertz, M., Harrison, A., Houston, J. B., and Galetin, A. (2010). Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab. Dispos. 38 (7), 1147–1158. doi:10.1124/dmd.110.032649
Hibma, J. E., Radtke, K. K., Dorman, S. E., Jindani, A., Dooley, K. E., Weiner, M., et al. (2020). Rifapentine population pharmacokinetics and dosing recommendations for latent tuberculosis infection. Am. J. Respir. Crit. Care Med. 202 (6), 866–877. doi:10.1164/rccm.201912-2489OC
Huerta-García, A. P., Medellín-Garibay, S. E., Ortiz-Álvarez, A., Magaña-Aquino, M., Rodríguez-Pinal, C. J., Portales-Pérez, D. P., et al. (2020). Population pharmacokinetics of isoniazid and dose recommendations in Mexican patients with tuberculosis. Int. J. Clin. Pharm. 42 (4), 1217–1226. doi:10.1007/s11096-020-01086-1
Imperial, M. Z., Luetkemeyer, A., Dawson, R., Cramer, Y., Rosenkranz, S., Swindells, S., et al. (2022). “Dolutegravir pharmacokinetics in people with HIV receiving daily 1HP for latent tuberculosis treatment (ACTG A5372) - abstract 78,” in Conference on retroviruses and opportunistic infections.
Khalilieh, S., Yee, K. L., Sanchez, R., Stoch, S. A., Wenning, L., and Iwamoto, M. (2020). Clinical pharmacokinetics of the novel HIV-1 non-nucleoside reverse transcriptase inhibitor doravirine: An assessment of the effect of patient characteristics and drug-drug interactions. Clin. Drug Investig. 40 (10), 927–946. doi:10.1007/s40261-020-00934-2
kommavarapu, P., Maruthapillai, A., Palanisamy, K., and Sunkara, M. (2015). Preparation and characterization of rilpivirine solid dispersions with the application of enhanced solubility and dissolution rate. Beni-Suef Univ. J. Basic Appl. Sci. 4 (1), 71–79. doi:10.1016/j.bjbas.2015.02.010
Lam, E., Schaefer, J., Zheng, R., Zhan, T., and Kraft, W. K. (2020). Twice-daily doravirine overcomes the interaction effect from once-weekly rifapentine and isoniazid in healthy volunteers. Clin. Transl. Sci. 13 (6), 1244–1250. doi:10.1111/cts.12810
Liou, B. H., Cheng, C. N., Lin, Y. T., Lin, Y. J., Chuang, Y. C., Lin, K. Y., et al. (2021). Short-course daily isoniazid and rifapentine for latent tuberculosis infection in people living with HIV who received coformulated bictegravir/emtricitabine/tenofovir alafenamide. J. Int. AIDS Soc. 24 (11), e25844. doi:10.1002/jia2.25844
Liverpool HIV Drug Interactions (2022a). Raltegravir with rifapentine interaction. [Online]. Available: https://www.hiv-druginteractions.org/checker (Accessed 06 23, 2022).
Liverpool HIV Drug Interactions (2022b). Rilpivirine with rifapentine. [Online]. Available: https://www.hiv-druginteractions.org/checker (Accessed 06 24, 2022).
Margolis, D. A., Brinson, C. C., Smith, G. H. R., de Vente, J., Hagins, D. P., Eron, J. J., et al. (2015). Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): A randomised, phase 2b, dose-ranging trial. Lancet. Infect. Dis. 15 (10), 1145–1155. doi:10.1016/s1473-3099(15)00152-8
McGinnity, D. F., Zhang, G., Kenny, J. R., Hamilton, G. A., Otmani, S., Stams, K. R., et al. (2009). Evaluation of multiple in vitro systems for assessment of CYP3A4 induction in drug discovery: Human hepatocytes, pregnane X receptor reporter gene, and Fa2N-4 and HepaRG cells. Drug Metab. Dispos. 37 (6), 1259–1268. doi:10.1124/dmd.109.026526
Nakajima, A., Fukami, T., Kobayashi, Y., Watanabe, A., Nakajima, M., and Yokoi, T. (2011). Human arylacetamide deacetylase is responsible for deacetylation of rifamycins: Rifampicin, rifabutin, and rifapentine. Biochem. Pharmacol. 82 (11), 1747–1756. doi:10.1016/j.bcp.2011.08.003
NCBI (2022a). PubChem database, midazolam, CID = 4192 [Online]. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/4192 (Accessed October 20, 2022).
NCBI (2022b). PubChem database, rilpivirine, CID = 6451164 [Online]. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/rilpivirine (Accessed October 20, 2022).
Néant, N., Solas, C., Bouazza, N., Lê, M. P., Yazdanpanah, Y., Dhiver, C., et al. (2019). Concentration-response model of rilpivirine in a cohort of HIV-1-infected naive and pre-treated patients. J. Antimicrob. Chemother. 74 (7), 1992–2002. doi:10.1093/jac/dkz141
Néant, N., Lê, M. P., Bouazza, N., Gattacceca, F., Yazdanpanah, Y., Dhiver, C., et al. (2020). Usefulness of therapeutic drug monitoring of rilpivirine and its relationship with virologic response and resistance in a cohort of naive and pretreated HIV-infected patients. Br. J. Clin. Pharmacol. 86 (12), 2404–2413. doi:10.1111/bcp.14344
Nestorov, I. (2003). Whole body pharmacokinetic models. Clin. Pharmacokinet. 42 (10), 883–908. doi:10.2165/00003088-200342100-00002
Ogburn, E. T., Jones, D. R., Masters, A. R., Xu, C., Guo, Y., and Desta, Z. (2010). Efavirenz primary and secondary metabolism in vitro and in vivo: Identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab. Dispos. 38 (7), 1218–1229. doi:10.1124/dmd.109.031393
Paine, M. F., Khalighi, M., Fisher, J. M., Shen, D. D., Kunze, K. L., Marsh, C. L., et al. (1997). Characterization of interintestinal and intraintestinal variations in human CYP3A-dependent metabolism. J. Pharmacol. Exp. Ther. 283 (3), 1552–1562.
Pham, M. M., Podany, A. T., Mwelase, N., Supparatpinyo, K., Mohapi, L., Gupta, A., et al. (2022). Population pharmacokinetic modeling and simulation of rifapentine supports concomitant antiretroviral therapy with efavirenz and non-weight based dosing. Antimicrob. Agents Chemother. 66 (9), e0238521. doi:10.1128/aac.02385-21
Podany, A. T., Bao, Y., Swindells, S., Chaisson, R. E., Andersen, J. W., Mwelase, T., et al. (2015). Efavirenz pharmacokinetics and pharmacodynamics in HIV-infected persons receiving rifapentine and isoniazid for tuberculosis prevention. Clin. Infect. Dis. 61 (8), 1322–1327. doi:10.1093/cid/civ464
Poulin, P., and Theil, F. P. (2002). Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J. Pharm. Sci. 91 (1), 129–156. doi:10.1002/jps.10005
Ramsden, D., Zhou, J., and Tweedie, D. J. (2015). Determination of a degradation constant for CYP3A4 by direct suppression of mRNA in a novel human hepatocyte model, HepatoPac. Drug Metab. Dispos. 43 (9), 1307–1315. doi:10.1124/dmd.115.065326
Reith, K., Keung, A., Toren, P. C., Cheng, L., Eller, M. G., and Weir, S. J. (1998). Disposition and metabolism of 14C-rifapentine in healthy volunteers. Drug Metab. Dispos. 26 (8), 732–738.
Savic, R. M., Lu, Y., Bliven-Sizemore, E., Weiner, M., Nuermberger, E., Burman, W., et al. (2014). Population pharmacokinetics of rifapentine and desacetyl rifapentine in healthy volunteers: Nonlinearities in clearance and bioavailability. Antimicrob. Agents Chemother. 58 (6), 3035–3042. doi:10.1128/aac.01918-13
Savic, R. M., Weiner, M., MacKenzie, W. R., Engle, M., Whitworth, W. C., Johnson, J. L., et al. (2017). Defining the optimal dose of rifapentine for pulmonary tuberculosis: Exposure-response relations from two phase II clinical trials. Clin. Pharmacol. Ther. 102 (2), 321–331. doi:10.1002/cpt.634
van Rongen, A., Kervezee, L., Brill, M., van Meir, H., den Hartigh, J., Guchelaar, H. J., et al. (2015). Population pharmacokinetic model characterizing 24-hour variation in the pharmacokinetics of oral and intravenous midazolam in healthy volunteers. CPT. Pharmacometrics Syst. Pharmacol. 4 (8), 454–464. doi:10.1002/psp4.12007
Weiner, M., Egelund, E. F., Engle, M., Kiser, M., Prihoda, T. J., Gelfond, J. A., et al. (2014). Pharmacokinetic interaction of rifapentine and raltegravir in healthy volunteers. J. Antimicrob. Chemother. 69 (4), 1079–1085. doi:10.1093/jac/dkt483
Wen, X., Wang, J. S., Neuvonen, P. J., and Backman, J. T. (2002). Isoniazid is a mechanism-based inhibitor of cytochrome P450 1A2, 2A6, 2C19 and 3A4 isoforms in human liver microsomes. Eur. J. Clin. Pharmacol. 57 (11), 799–804. doi:10.1007/s00228-001-0396-3
WHO (2020). WHO operational handbook on tuberculosis (Module 1 – prevention): Tuberculosis preventive treatment. [Online]. Available: https://www.who.int/publications/i/item/9789240001503 (Accessed 06 23, 2022).
WHO (2021a). HIV and tuberculosis. [Online]. Available: https://www.who.int/westernpacific/health-topics/hiv-aids/hiv-and-tuberculosis (Accessed 09 17, 2022).
WHO (2021b). Treatment of drug-susceptible tuberculosis: Rapid communication [Online]. Available at: https://www.who.int/publications/i/item/9789240028678 (Accessed November 16, 2022).
WHO (2021c). Tuberculosis [Online]. Available at: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (Accessed June 16, 2022).
Williamson, B., Dooley, K. E., Zhang, Y., Back, D. J., and Owen, A. (2013). Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob. Agents Chemother. 57 (12), 6366–6369. doi:10.1128/aac.01124-13
Yu, L. X., and Amidon, G. L. (1999). A compartmental absorption and transit model for estimating oral drug absorption. Int. J. Pharm. 186 (2), 119–125. doi:10.1016/s0378-5173(99)00147-7
Zhang, Y., and Pike, A. (2021). Pyridones in drug discovery: Recent advances. Bioorg. Med. Chem. Lett. 38, 127849. doi:10.1016/j.bmcl.2021.127849
Keywords: drug interaction (DI), PBPK, antiretroviral therapy, tuberculosis, rilpivirine, rifapentine
Citation: Grañana-Castillo S, Montanha MC, Bearon R, Khoo S and Siccardi M (2022) Evaluation of drug-drug interaction between rilpivirine and rifapentine using PBPK modelling. Front. Pharmacol. 13:1076266. doi: 10.3389/fphar.2022.1076266
Received: 21 October 2022; Accepted: 28 November 2022;
Published: 15 December 2022.
Edited by:
Simona Pichini, National Institute of Health (ISS), ItalyReviewed by:
Eric F. Egelund, University of Florida, United StatesCopyright © 2022 Grañana-Castillo, Montanha, Bearon, Khoo and Siccardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Grañana-Castillo, Uy5HcmFuYW5hLUNhc3RpbGxvQGxpdmVycG9vbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.