 Yen-Chieh Chuang
Yen-Chieh Chuang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 13 December 2022
Sec. Inflammation Pharmacology
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1074986
Dysregulation of macrophages in the pro-inflammatory (M1) and anti-inflammatory (M2) sub-phenotypes is a crucial element in several inflammation-related diseases and injuries. We investigated the role of aquaporin (AQP) in macrophage polarization using AQP pan-inhibitor mercury chloride (HgCl2). Lipopolysaccharides (LPSs) induced the expression of AQP-1 and AQP-9 which increased the cell size of bone marrow-derived macrophages. The inhibition of AQPs by HgCl2 abolished cell size changes and significantly suppressed M1 polarization. HgCl2 significantly reduced the activation of the nuclear factor kappa B (NF-κB) and p38 mitogen-activated protein kinase (MAPK) pathways and inhibited the production of IL-1β. HgCl2 attenuated LPS-induced activation of mitochondria and reactive oxygen species production and autophagy was promoted by HgCl2. The increase in the light chain three II/light chain three I ratio and the reduction in PTEN-induced kinase one expression suggests the recycling of damaged mitochondria and the restoration of mitochondrial activity by HgCl2. In summary, the present study demonstrates a possible mechanism of the AQP inhibitor HgCl2 in macrophage M1 polarization through the restriction of cell volume change, suppression of the p38 MAPK/NFκB pathway, and promotion of autophagy.
Macrophages are highly plastic immune cells involved in both infection defense and tissue homeostasis. The dysregulation of macrophage activation is the leading cause of inflammation-related tissue injury (Laskin et al., 2011). They are classified by their response to environmental stimuli and macrophages can be polarized into a pro-inflammatory classically activated phenotype (M1) or an anti-inflammatory alternatively activated phenotype (M2) (Aggarwal et al., 2014). Induction of M1 polarization exaggerates alveolar inflammation and tissue damage (Ying et al., 2015), whereas promotion of M2 polarization mitigates pulmonary and systemic inflammation and aids in repair (Vergadi et al., 2014; Mishra et al., 2021). Macrophage polarization is mainly regulated by cytokines and chemokines and is modulated by tissue structures and physical factors in the extracellular environment (Lomas-Neira et al., 2006). The lipopolysaccharide (LPS)-induced inflammatory response and phagocytic activity of RAW264.7 cells can be modulated by altering the cell volume in various osmotic microenvironments (Hung et al., 2018). Macrophages cultured on scaffolds with smaller pore sizes promote M1 polarization (Yin et al., 2020) and mechanical elongation of macrophages leads to M2 polarization (McWhorter et al., 2015). Research on drugs that modulate physical stress in macrophages may help to develop novel therapeutic strategies against sepsis-related tissue damage.
Aquaporins (AQPs) are water channel-forming proteins that facilitate the transport of water and small non-charged molecules across the plasma membrane (Agre et al., 2002) and mediate the mechanisms of water homeostasis and osmotic regulation (Verkman et al., 2000; King et al., 2004). AQPs are also involved in shape and volume changes of various immune cells (Moon et al., 2004) and play crucial roles in the migration and phagocytosis of inflammatory cells (Meli et al., 2018). AQP has emerged as a potential target for new drug development against inflammation-related tissue damage (da Silva and Soveral, 2021). However, whether AQPs play a role in regulating macrophage polarization remains unknown. A recent study showed that knockout ablation of AQP-1 is functionally equivalent to IL-4-induced M2 polarization (Tyteca et al., 2015). In contrast, Liu et al. (Liu et al., 2020a) reported that AQP-1 protects against LPS-induced kidney injury by promoting macrophage M2 polarization.
Mercury ion (Hg2+) is an established water channel inhibitor as it effectively occludes the water transportation pore of AQPs by covalent modification of their cysteine residue (Preston et al., 1993; Zhang et al., 1993). Despite been reported as a highly toxic metal (Rice et al., 2014), recent study has shown the therapeutic potential with relatively low dose of mercury compounds against glioma (Pires et al., 2022). In immune cells, research showed that mercury chloride (HgCl2) inhibits the nitric oxide production of macrophage through modulate NFκB and MAPK signaling pathway (Kim et al., 2002), and attenuate caspase-1 activity, and IL-1β secretion (Ahn et al., 2018). Therefore, in the present study, we investigated the effect of HgCl2 on macrophage polarization.
Bone marrow-derived macrophages (BMDMs) were isolated from the bone marrow of C57B/6 mice aged 10–12 weeks old. All procedures were approved by the Institutional Animal Care and Use Committee of the National Defense Medical Center, Taiwan and followed the National Institutes of Health guidelines for animal care. After euthanasia, the legs of the mice were sprayed with 70% ethanol, and the femurs and tibias were dissected, separated, and transferred to a sterile flow hood. After removing the skin and muscle tissue, the bone was sprayed with 70% ethanol and the ends of the femurs and tibias were cut with sterile scissors. The bone marrow was flushed with ice-cold PBS and filtered through a 70 μm cell strainer (BD Biosciences, CA, United States) to remove solid debris. The recovered cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco, ThermoFisher Scientific, Waltham, MA, United States) supplemented with 10% heat-inactivated fetal bovine serum (FBS; HyClone, Cytiva, Logan, UT, United States), 1% penicillin–streptomycin (Biological Industries, CT, United States), and 20 ng/ml recombinant monocytic colony-stimulating factor (M-CSF; Peprotech, Rocky Hill, NJ, United States) for 7 days to fully differentiate into macrophages.
BMDMs were seeded into 6-well culture plates at a density of 1 × 106 cells/well and subjected to the respective experiments. BMDMs were treated with 1 μg/ml LPS (Escherichia coli serotype 026:B6, Sigma-Aldrich, St Louis, MO, United States) or 20 μg/ml IL-4 (Peprotech) to induce macrophage polarization. To modulate cell size, BMDMs were pretreated with 20 mM NaCl (final osmolarity of 400 mOsmol) for 30 min before LPS or IL-4 stimulation. In some experiments, the cells were pretreated with 1 μM HgCl2 (Sigma-Aldrich). HgCl2 was dissolved in PBS buffer. M1/M2 polarization, expression of AQPs, cell size, production of pro-inflammatory mediators, signaling pathways, cell autophagy, and mitochondrial stress were analyzed at 8 and 24 h.
Cells were scraped and stained with the following antibodies: APC-eFluor 780 anti-mouse F4/80 (clone BM8, eBioscience, San Diego, CA, United States), Alexa Fluor 488 anti-mouse CD11b (clone M1/70, BD Biosciences), PE anti-mouse iNOS (clone CXNFT, eBioscience), and eFluor450 anti-mouse CD38 (clone 90, eBioscience). The isotype controls were rat IgG2bκ (eBR2a) and rat IgG2bκ (DA/HA) from BD Biosciences (San Jose, CA, United States). The cells were washed and stained with Flow Cytometry Staining Buffer (eBioscience) for 30 min at 4°C. For intracellular staining, cells were fixed in IC Fixation Buffer (eBioscience) for 30 min at 4°C, permeabilized, and stained in permeabilization buffer (eBioscience). Results were obtained using the FACSVerse (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR, United States). The F4/80+ and CD11b+ cells were gated as fully differentiated macrophages.
Total RNA was isolated from cells using the commercial kit RNA Miniprep Plus (Zymo Research Corp., Irvine, CA, United States), and cDNA was prepared using 20–100 ng/μL total RNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, United States) following the manufacturer’s instructions. Real-time quantitative PCR (qPCR) was performed on the cDNA using TaqMan probes for nos2, tnf, ccl17 (Applied Biosystems). qPCR was performed on a QuantStudio™ five System (Applied Biosystems) using the TaqMan® universal Master Mix II (Applied Biosystems). Fold changes in expression were calculated by the 2−ΔΔCt method using mouse gapdh (glyceraldehyde-3-phosphate dehydrogenase) as an endogenous control for mRNA expression.
Cells were cultivated in a 3.5 cm glass bottom confocal dish (SPL Life Sciences, Pocheon, Korea) at a density of 1.2 × 106 cells per dish. After incubating for 24 h, the cells were fixed with 4% paraformaldehyde for 20 min and blocked with 10% bovine serum albumin (BSA) for 20 min. The cells were incubated with conjugated antibodies eFluor 660 anti-mouse F4/80 (, clone BM8, eBioscience) (1:100), PE anti-mouse iNOS (clone CXNFT, eBioscience) (1:300), and DAPI (4′,6-diamidino-2-phenylindole, Invitrogen, Carlsbad, CA, United States) for 1 h. Images were captured using a laser scanning confocal microscope (LSM 880, Carl Zeiss, United States) with a ×40 objective (original magnification).
Cells in 6-well culture plates were lysed in a radioimmunoprecipitation assay (RIPA) cell lysis buffer containing protease and phosphatase inhibitor cocktail (Halt, ThermoFisher Scientific). To collect the nuclear fraction, cells were seeded and cultivated in 10 cm culture dishes and isolated with Nuclear and Cytoplasmic Extraction Reagent kits (NE-PER, ThermoFisher Scientific) following the manufacturer’s instructions. Protein concentrations were measured using a bicinchoninic acid protein assay kit (Pierce, ThermoFisher Scientific). The lysate (30 μg/lane) was resolved on a 10% SDS-polyacrylamide gel using a Hoefer electrophoresis blotting system (Hoefer, United States) and transferred to a 0.45 μm polyvinylidene fluoride membrane (Merck Millipore, United States). The membrane was probed with antibodies against JNK, p-JNK (Thr183/Tyr185), p38, p-p38 (Thr180/Tyr182), NF-κB (p65), p-NF-κB (p65) (Ser536), cleaved Caspase-3, LC3A/B, P62 (SQSTM1) purchased from Cell Signaling Technology (Danvers, MA, United States), PINK1 from Abcam, AQP1, and AQP9 from Biorbyt (Berkeley, CA, United States) (1:1000 dilution). β-actin (Sigma-Aldrich) and PCNA (OriGene Technologies, Rockville, MD, United States) were incubated at 1:10000 dilutions and used as internal controls. Densitometry was analyzed semi-quantitatively using ImageJ software (National Institutes of Health, Bethesda, MD, United States).
The culture medium was collected from the 6-well culture plates. The concentration of IL-1β was quantified using ELISA kits (R&D Systems, Minneapolis, MN, United States) following the manufacturer’s instructions.
LA-4 murine lung epithelium (CCL-196) purchased from American Type Culture Collection was seeded into 24-well culture plates at 80–90% confluency. Epithelial cells were then treated with 1 μg/ml LPS for 24 h. LPS- or HgCl2-treated BMDMs labeled with green, fluorescent dye (Invitrogen) were seeded on top of the activated LA-4 monolayer and incubated for 1 h. Non-adherent cells were washed off with warm PBS. The remaining adhered BMDMs were counted from images captured at ×50 magnification using a fluorescence microscope (Leica DM 2500, Wetzlar, Germany). The experiment was repeated at least three times.
Mitochondrial stress was assessed using a Seahorse XFp Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, United States) following the manufacturer’s instructions. BMDMs were seeded in XFp cell culture miniplates (Agilent Technologies) at a density of 3.2 × 105 cells/well. After 8 h of incubation, the medium was replaced with the XF medium. Oligomycin (1 µM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 4 µM), and antimycin A (0.5 µM) were added to the wells through a Seahorse cartridge. Real-time readings of the oxygen consumption rate (OCR) were calculated and recorded using Seahorse XFp software.
Mitochondrial reactive oxygen species (ROS) levels were measured using MitoSox-Red (Molecular Probes, Invitrogen). BMDMs were seeded in 12-well culture plates at a density of 4 × 105 cells/well. The cells were double-stained with 5 μM MitoSox-Red and 50 nM MitoTracker Green FM (Molecular Probes) in HBSS for 30 min. Images were captured at ×200 and ×400 magnifications using a fluorescence microscope (Leica). The percentage of MitoSOX-positive cells was calculated at magnification of ×200.
All results are expressed as mean ± standard error of the mean (SEM). One-way analysis of covariance (ANOVA) and Student’s t-test were used to compare differences between the study groups. Tukey’s correction was used for post-hoc comparisons. Statistical significance was set at p < 0.05.
LPS is commonly used to polarize macrophages into the M1 phenotype. LPS (1 μg/ml) significantly promoted M1 polarization in BMDM, as characterized by the increase of iNOS+/CD38+ population (Figures 1C–E). Although hypertonicity (+20 mM NaCl) restricted cell swelling (Figures 1A,B), it further enhanced LPS-induced M1 polarization (Figures 1C–E). In contrast, hypertonicity did not affect M2 polarization in BMDM (Figures 1F–H).
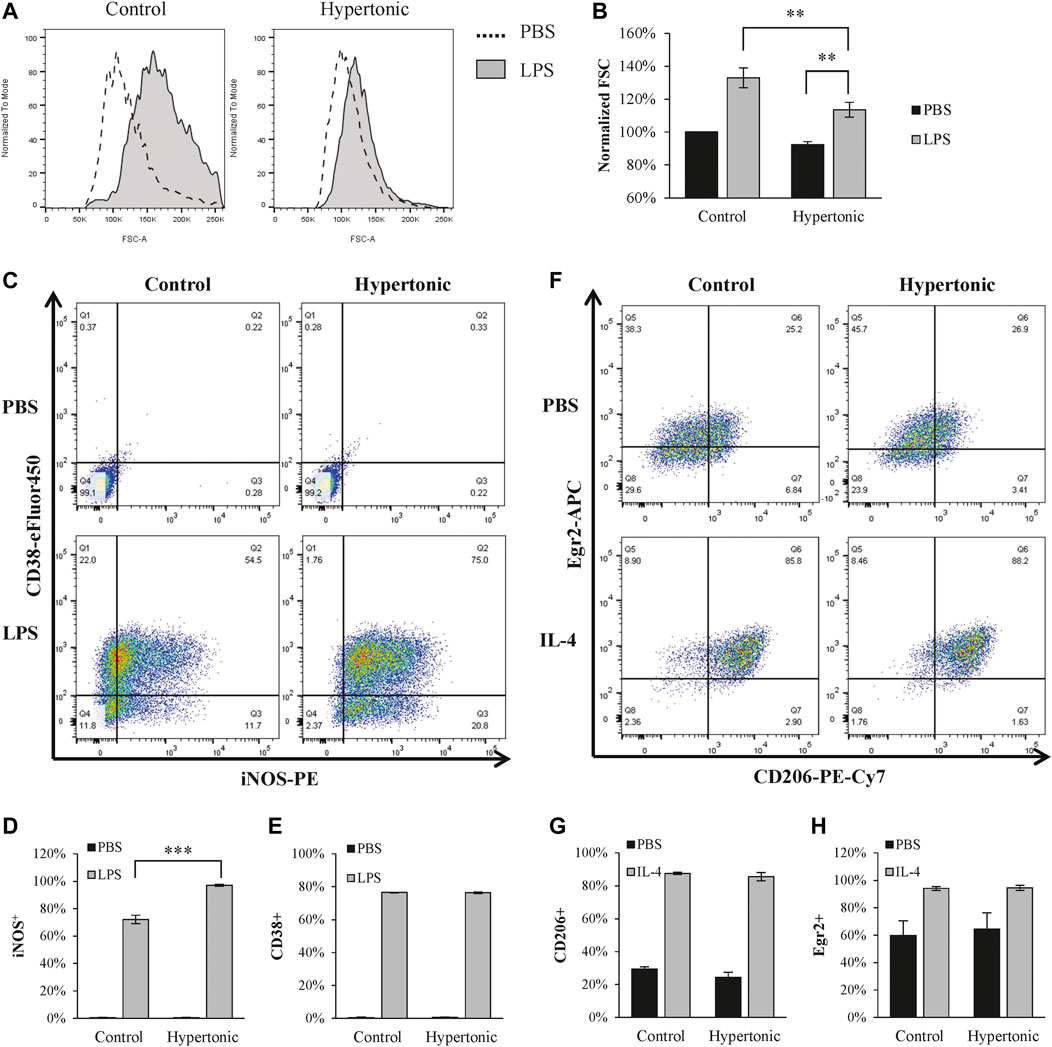
FIGURE 1. Hypertonicity induces promotes M1 polarization. (A,B) Normalized forward scatter (FCS) in flow cytometry was used to determine the relative cell size 24 h after tonicity (+20 mM NaCl) and LPS (1 μg/ml) treatment. (n = 5–7). (C–E) M1 polarization of BMDMs (F4/80+ and CD11b+) were estimated by iNOS and CD38 in flow cytometry (n = 3). (F–H) M2 polarization of BMDMs were estimated by CD206 and Egr2 in flow cytometry 24 h after tonicity (+20 mM NaCl) and IL-4 (20 ng/ml) treatment (n = 3). Values are mean ± SEM. **p < 0.01 vs. control. ***p < 0.001 vs. control.
Inhibition of AQPs by HgCl2 (1 μM) restricted the increase in BMDM cell size caused by LPS and also abolished cell size changes caused by hypertonicity (Figures 2A,B). On the effects to polarization, HgCl2 significantly inhibited LPS-induced M1 polarization (Figures 2E–I). Immunofluorescence staining showed that HgCl2 significantly inhibited iNOS and CD38 expression in BMDM (Figures 2C,D), the percentage of macrophage presenting high amount of iNOS (iNOShi) and CD38 (CD38hi) signals were reduced almost by half with HgCl2 (Figures 2E,F). The effect of HgCl2 on M1 polarization was also assessed by qPCR, which showed that the expression of M1-related genes nos2, tnf, and ccl17 was reduced by 76%, 21%, and 72%, respectively (Figures 2G–I).
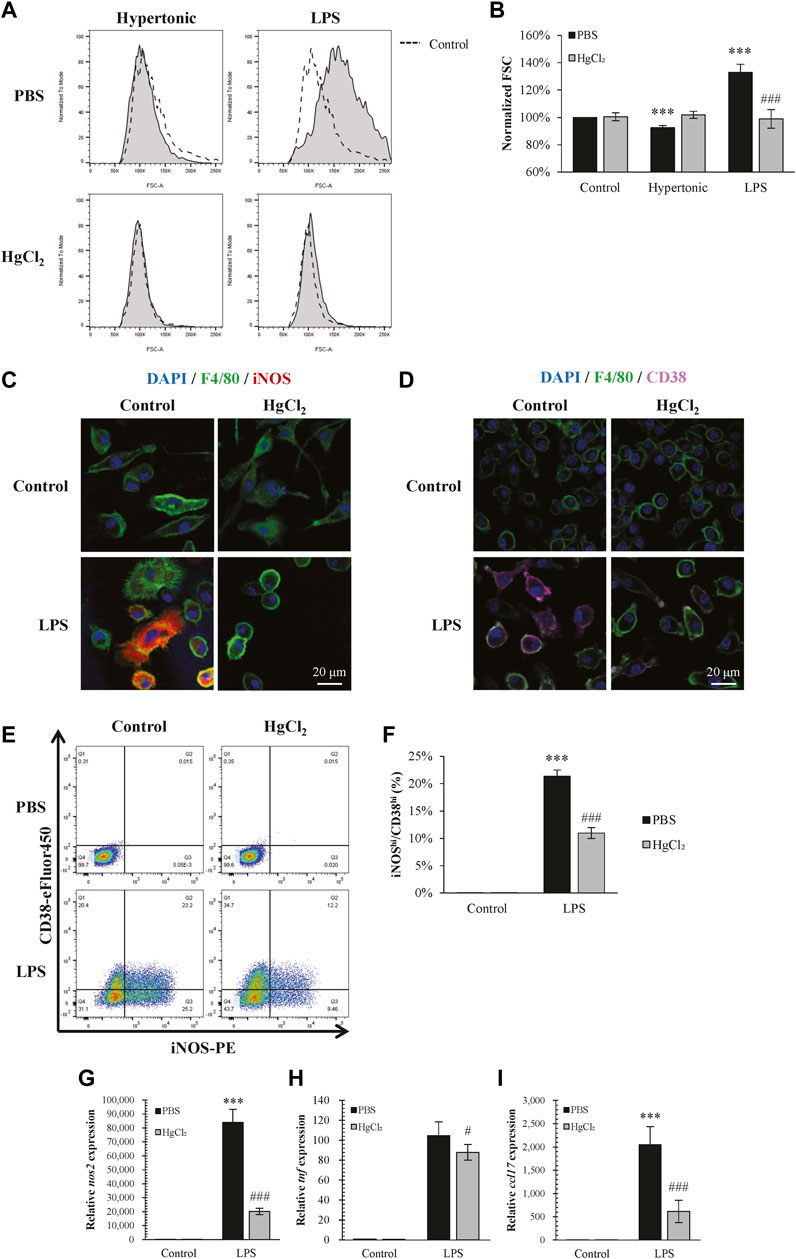
FIGURE 2. HgCl2 restricts lipopolysaccharides (LPS)-induced cell swelling and inhibits M1 polarization. (A,B) Normalized forward scatter (FCS) in flow cytometry was used to determine the relative cell size 24 h after tonicity (+20 mM NaCl) and LPS (1 μg/ml) treatment and HgCl2 (1 μM) pre-treatment. (n = 5–7). (C,D) Immunofluorescence image showing iNOS and CD38 staining 24 h after LPS (1 μg/ml) treatment and HgCl2 (1 μM) pre-treatment. (E,F) M1 polarization of bone marrow-derived macrophages (F4/80+ and CD11b+) were estimated by iNOS and CD38 in flow cytometry (n = 4–7). (G–I) M1-related gene expression were evaluated by TaqMan ™ qPCR assay 4 h after LPS treatment (n = 3). Values are mean ± SEM. ***p < 0.001 vs. control. #p < 0.05 vs. LPS. ###p < 0.001 vs. LPS.
Activation of nuclear factor kappa B (NF-κB) pathway and increased adhesion ability are two notably features for M1 macrophage. LPS increased the cytoplasmic abundance of phosphorylated p38 (Figures 3A–C) and the nuclear translocation of p-p65 NF-κB (Figures 3D–G) in BMDM, and produced large amounts of IL-1β (Figure 3H). LPS also significantly increased the adhesion ability of BMDM (Figures 3I–K). HgCl2 effectively suppressed the activation of the NF-κB and p38 MAPK pathways and inhibited the production of IL-1β (Figures 3A–H). The presence of HgCl2 also significantly reduced the adhesion ability (Figures 3I–K).

FIGURE 3. HgCl2 suppress inflammatory pathway and adherence function. (A,D,F) Inflammatory cascade 30 min after lipopolysaccharides (LPS) (1 μg/ml) and HgCl2 (1 μM) treatment was determined by western blot analysis. and (B–G) interpreted by semi quantitative densitometric analysis (n = 3–4). (H) The level of IL-1β production 24 h after LPS (1 μg/ml) and HgCl2 (1 μM) treatment. (I) Images of adhered BMDMs (green) on LA-4 murine lung epithelium (DIC) in different magnification (×100, 200×, and 400×). (J,K) Adhered BMDMs (green) in adherence assay were imaged and counted by fluorescence microscope (50×) and imageJ (n = 7–11). Values are mean ± SEM. **p = 0.001 vs. control. ***p < 0.001 vs. control. ###p < 0.001 vs. LPS.
Mitochondrial ROS production plays an important role in macrophage immunity; however, over-activated mitochondria are associated with cellular stress and dysfunction. Mitochondrial ROS production was estimated using MitoSOX staining. LPS induced substantial mitochondrial ROS production in BMDM (Figure 4A) and significantly increased the percentage of MitoSOX-positive cells (Figure 4B). The LPS-induced mitochondrial ROS production was significantly suppressed by HgCl2 treatment (Figure 4).
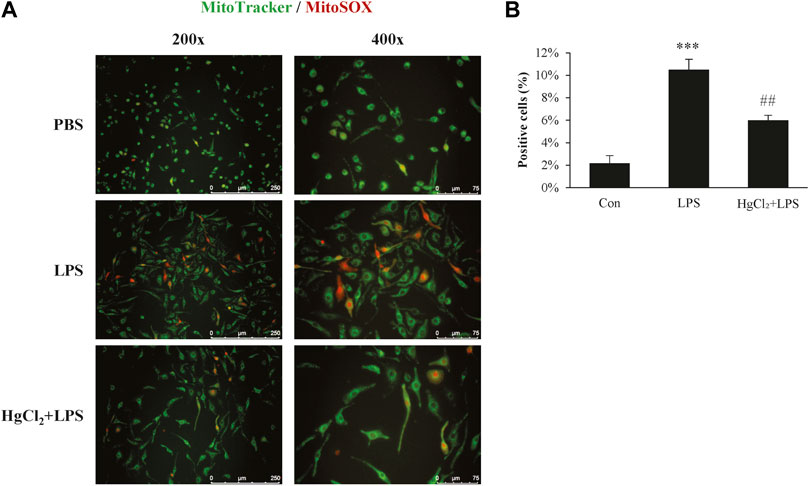
FIGURE 4. HgCl2 reduces mitochondrial reactive oxygen species production. (A) Immunofluorescence image of MitoSOX staining 8 h after lipopolysaccharides (LPS) (1 μg/ml) and HgCl2 (1 μM) treatment. (B) The percentage of MitoSOX positive cells per field in ×200 magnification was analyzed with imageJ (n = 7–11). Values are mean ± SEM. ***p < 0.001 vs. control. ##p < 0.01 vs. LPS.
The Seahorse XF Cell Mito Stress Test was used to calculate the fundamental parameters of mitochondrial function (Figure 5A). LPS significantly suppressed oxygen consumption in BMDM under basal conditions (Figure 5C) and during maximal respiration (Figure 5D). LPS also reduced the ATP-linked OCR and spare respiratory capacity (Figures 5E,F). HgCl2 significantly restored mitochondrial function (Figures 5C–F).
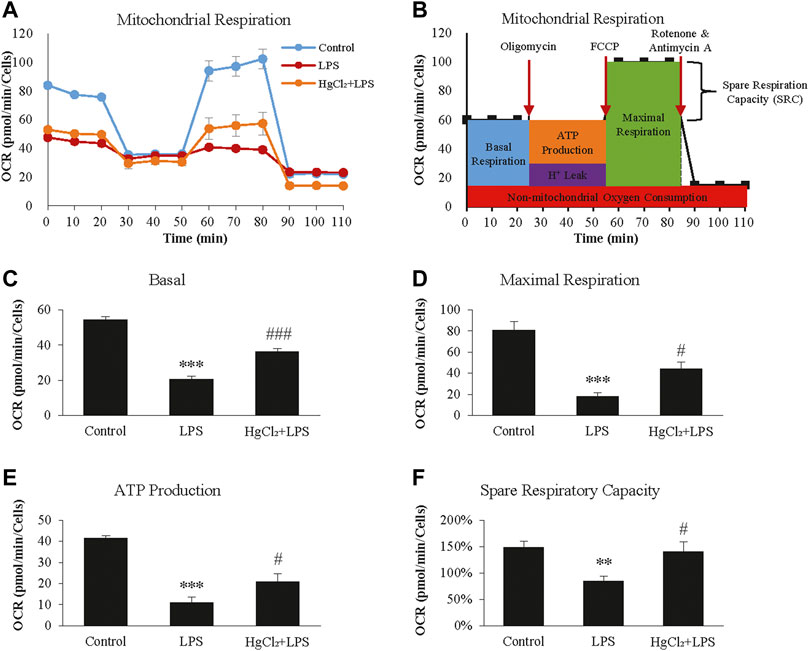
FIGURE 5. HgCl2 reduces lipopolysaccharides (LPS)-induced mitochondrial stress of bone marrow-derived macrophages (BMDM). (A) Oxygen consumption rates (OCR, in pmol/min) profile of BMDMs after 8 h of LPS (1 μg/ml) and HgCl2 (1 μM) treatment. (B) Schematic diagram of how the fundamental parameters of mitochondrial function measured by XF Cell Mito Stress Test Kit. Calculated basal (C), maximal respiration (D), ATP production (E), and spare respiratory capacity (F). All measurements were normalized for cell count (n = 4). Values are mean ± SEM. **p < 0.01 vs. control. ***p < 0.001 vs. control. #p < 0.05 vs. LPS. ###p < 0.001 vs. LPS.
Autophagy is a recycling mechanism of cellular homeostasis responsible for maintaining healthy mitochondria. LPS increased light chain three II/light chain three I level in the presence of bafilomycin A1 (Figures 6A,B) and caused substantial p62/SQSTM1 abundance in BMDM (Figures 6A,C). HgCl2 further augmented the LPS-induced increase in autophagic flux (Figures 6A–C). LPS also stimulated the expression of mitophagy marker PTEN-induced kinase 1 (PINK1), indicating mitochondrial stress. Pre-treatment of BMDM with HgCl2 reduced LPS-induced PINK1 abundance, suggesting that the present of AQP inhibitors alleviated mitochondrial stress (Figure 6D).
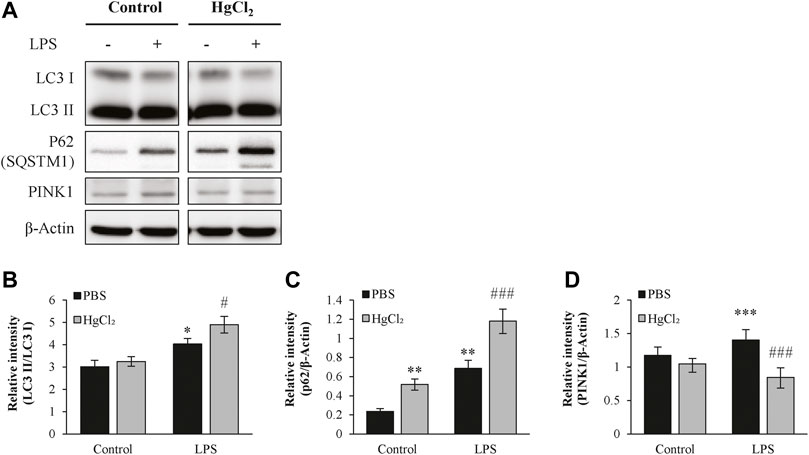
FIGURE 6. HgCl2 upregulates the autophagy in M1 bone marrow-derived macrophages (BMDMs). (A) Autophagy of BMDMs 8 h after LPS (1 μg/ml) and HgCl2 (1 μM) treatment was determined by western blot analysis and (B–D) interpreted by semi quantitative densitometric analysis (n = 4–6). Bafilomycin A1 (100 nM) were cotreated with LPS to normalize autophagy flux. Values are mean ± SEM. *p < 0.05 vs. control. **p < 0.01 vs. control. ***p < 0.001 vs. control. #p < 0.05 vs. LPS. ###p < 0.001 vs. LPS.
Our previous studies in primary alveolar macrophages and RAW 264.7 cells, have found that cell size is one of the critical parameters determining the inflammatory response to LPS treatment (Hung et al., 2018; Hung et al., 2020). Shrinking the size of macrophages with osmolarity differences is associated with a lower activated IKK-NfκB cascade and less TNF-α and IL-6 secretion following LPS treatment. In contrast, further enlargement of macrophage cell size with a hypoosmotic medium enhances its inflammatory responses. However, in the present study with BMDMs, M1 polarization induced by LPS seems to be augmented by hypertonicity. This may contribute to the differences in the macrophage origin. Studies have shown that inflammasome activity is boosted with high osmolarity in BMDM (Ip and Medzhitov, 2015), but significantly suppressed in alveolar or peritoneal macrophages (Rabolli et al., 2014).
HgCl2 is well established as an AQP inhibitor, and mercury physically blocks water transportation pores by thiol binding to its pore-localized cysteine (Savage and Stroud, 2007; Yool et al., 2010). Although mercury is considered a highly toxic metal, studies have shown that low-dose treatment with HgCl2 (up to 10 μM) is not toxic to macrophages (Rabolli et al., 2014) and could even be beneficial against inflammation. In immune cells, studies have shown that the introduction of HgCl2 alters the inflammatory response of macrophages by modulating the NF-κB and MAPK signaling pathways (Kim et al., 2002), attenuate NLRP3 related inflammasome activation and caspase-1 and IL-1β secretion (Ahn et al., 2018). In the present study, the viability of BMDMs showed no significant reduction up to 5 μM HgCl2, and the real-time cell metabolism assay revealed that mitochondrial activity was restored with 1 μM HgCl2, further suggesting that metabolic integrity was intact.
Autophagy is a highly conserved mechanism in eukaryotic cells that maintains cell homeostasis and organelle quality and can be stimulated by nutrient starvation, genotoxic agents, cytokines, or oxidative stress. Studies have revealed that exposure to mercury promotes autophagy in hepatocytes and stem cells (Chang et al., 2013; Chatterjee et al., 2014), and in macrophages, autophagy is crucial for defense against infection (Gutierrez et al., 2004; Nakagawa et al., 2004). A decrease in autophagy activity correlates positively with the inflammatory response. A recent study found that macrophage autophagy might play an important role in macrophage differentiation and polarization. Liu et al. demonstrated that autophagy deficiency in macrophages promotes M1 polarization and suppresses the M2 polarization potential (Liu et al., 2015). Our results confirm this finding using the opposite approach. The initiation of autophagy is a common response of macrophages to LPS stimulation (Xu et al., 2007; Delgado et al., 2008), but further boosting autophagy activity with HgCl2 is associated with reduced M1 polarization.
NF-κB activity is essential for LPS-mediated polarization to the M1 phenotype and is widely used to simulate pulmonary infection in acute lung injury (ALI) models. NF-κB is central to many inflammatory macrophage functions, such as the expression and secretion of iNOS, TNF-α, IL-1, IL-6, and bacterial phagocytosis (Xie et al., 1994; Goldring et al., 1995; Sha et al., 1995). The MAPK pathways, JNK and p38, are crucial in regulating the activation of NF-κB, and the phosphorylation of JNK and p38 dissociates the bonding of cytosolic NF-κB and IκB, resulting in the nuclear translocation of NF-κB, thus promoting the correlated gene expression (Liu et al., 2020b). Moreover, both p38 and JNK are associated with the regulation of apoptosis and autophagy. However, the roles of p38 and JNK in regulating apoptosis and autophagy are still unclear, both of which play a dual role in regulating autophagy as positive and negative regulators (Sui et al., 2014). Our results demonstrated that although the translocation of NF-κB was suppressed by HgCl2 treatment, it reduced only the phosphorylation of p38, but not JNK. Similar results were found in a study on microglia, in which the inhibition of p38 restored the kinase activity of ULK1, an initiation complex of the autophagic cascade in microglia, thus increasing autophagy activity (He et al., 2018).
Macrophages are the frontline of innate and adaptive immune defense against pathogen infection, and regulate immune responses. However, during inflammation, macrophage mitochondria are major targets of oxidative stress. Mitochondrial damage in macrophages is associated with apoptosis, metabolic switching, and systemic inflammatory responses (Ramond et al., 2019). In ALI, the level of mitochondrial dysfunction and ROS production in macrophages correlates positively with acute lung injury outcomes (Deng et al., 2017; Dong and Yuan, 2018). Recent studies have shown that artificially transferring healthy mitochondria from mesenchymal stem cells to damaged tissue protects against mitochondria-related injuries such as ALI (Islam et al., 2012), cardiac ischemia/reperfusion injury (Han et al., 2016) and cortical neurons after stroke (Babenko et al., 2015). These findings further emphasize the importance of mitochondrial integrity in inflammatory diseases. Our results show that treatment with HgCl2 significantly reduces the generation of mitochondrial ROS following LPS stimulation. Similar results were found in the study by H. Ahn et al. in an inflammasome model. They demonstrated that the introduction of HgCl2 prevents mitochondrial ROS production and cytosolic release of mitochondria DNA (Ahn et al., 2018). However, our study further evaluated mitochondrial health using a real-time cell metabolism assay and showed that HgCl2 also restored mitochondrial function during respiration.
In summary, the present study demonstrates a possible mechanism of action for the AQP inhibitor HgCl2 in macrophage polarization. The LPS-induced M1 activation of macrophages was attenuated by the inhibitory effect of HgCl2. It reduces cytokine production, adhesion, apoptosis, and mitochondrial ROS production. NF-κB inhibition is mainly mediated by the downregulation of p38 MAPK signaling. The evoked autophagy level can accelerate the recycling of damaged mitochondria, regain mitochondrial activity, and prevent mitochondrial damage (Figure 7). However, additional research is needed to clarify the inhibition specificity of HgCl2 on the AQP family and the molecular interactions between AQPs and MAPKs to provide a more complete view of this mechanism.
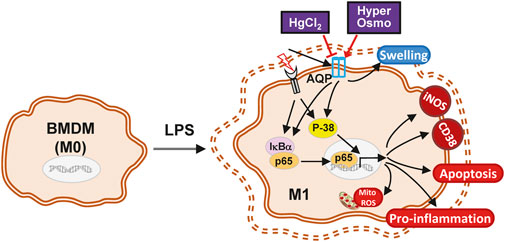
FIGURE 7. The schematic diagram of the effect of HgCl2 on bone marrow-derived macrophages in inflammatory response. The stimulation from LPS triggers the M1 polarization of BMDMs, activating the inflammatory cascade and causing the swelling of the cell. Although hypertonicity and HgCl2 induction are both able to restrict cell enlargement, the AQP inhibition effect of HgCl2 can significantly reduce the M1 phenotype of BMDMs. HgCl2 suppress the NF-κB activation mainly through the downregulation of p38/MAPK pathway, reducing the translocation of NF-κB into nucleus. Elevated autophagy levels may also help to recycle over-activated mitochondria, preventing mitochondrial ROS stress, and maintaining cell metabolic stability.
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of National Defense Medical Center.
The authors confirm contribution to the paper as follows: study conception and design: Y-CC, S-YW, C-KP, S-ET, and K-LH; data collection: Y-CC; statistical analysis: Y-CC, S-YW, and K-LH; interpretation of results: Y-CC, S-YW, Y-CH, S-ET, and K-LH; drafting manuscript: Y-CC; project administration: S-YW and K-LH. All authors reviewed the results and approved the final version of the manuscript.
This study was financially supported by grants from the Ministry of Science and Technology, Taiwan (MOST 109-2314-B-016-029-MY3), Tri-Service General Hospital (TSGH-C108-103, TSGH-D-109102, and TSGH-E-111233), and National Defense Medical Center, Taiwan (MAB-109-018).
We would like to acknowledge the Instrument Center of the National Defense Medical Center and the Department of Medical Research of Tri-Service General Hospital for technical support, especially. Y-CH for her helpful advice and technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aggarwal, N. R., King, L. S., and D'Alessio, F. R. (2014). Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell. Mol. Physiol. 306 (8), L709–L725. doi:10.1152/ajplung.00341.2013
Agre, P., King, L. S., Yasui, M., Guggino, W. B., Ottersen, O. P., Fujiyoshi, Y., et al. (2002). Aquaporin water channels-from atomic structure to clinical medicine. J. Physiol. 542 (1), 3–16. doi:10.1113/jphysiol.2002.020818
Ahn, H., Kim, J., Kang, S. G., Yoon, S. I., Ko, H. J., Kim, P. H., et al. (2018). Mercury and arsenic attenuate canonical and non-canonical NLRP3 inflammasome activation. Sci. Rep. 8 (1), 13659. doi:10.1038/s41598-018-31717-7
Babenko, V. A., Silachev, D. N., Zorova, L. D., Pevzner, I. B., Khutornenko, A. A., Plotnikov, E. Y., et al. (2015). Improving the post-stroke therapeutic potency of mesenchymal multipotent stromal cells by cocultivation with cortical neurons: The role of crosstalk between cells. Stem Cells Transl. Med. 4 (9), 1011–1020. doi:10.5966/sctm.2015-0010
Chang, S. H., Lee, H. J., Kang, B., Yu, K. N., Minai-Tehrani, A., Lee, S., et al. (2013). Methylmercury induces caspase-dependent apoptosis and autophagy in human neural stem cells. J. Toxicol. Sci. 38 (6), 823–831. doi:10.2131/jts.38.823
Chatterjee, S., Ray, A., Mukherjee, S., Agarwal, S., Kundu, R., and Bhattacharya, S. (2014). Low concentration of mercury induces autophagic cell death in rat hepatocytes. Toxicol. Ind. Health 30 (7), 611–620. doi:10.1177/0748233712462442
da Silva, I. V., and Soveral, G. (2021). Aquaporins in immune cells and inflammation: New targets for drug development. Int. J. Mol. Sci. 22 (4), 1845. doi:10.3390/ijms22041845
Delgado, M. A., Elmaoued, R. A., Davis, A. S., Kyei, G., and Deretic, V. (2008). Toll-like receptors control autophagy. EMBO J. 27 (7), 1110–1121. doi:10.1038/emboj.2008.31
Deng, S. Y., Zhang, L. M., Ai, Y. H., Pan, P. H., Zhao, S. P., Su, X. L., et al. (2017). Role of interferon regulatory factor-1 in lipopolysaccharide-induced mitochondrial damage and oxidative stress responses in macrophages. Int. J. Mol. Med. 40 (4), 1261–1269. doi:10.3892/ijmm.2017.3110
Dong, Z., and Yuan, Y. (2018). Accelerated inflammation and oxidative stress induced by LPS in acute lung injury: Ιnhibition by ST1926. Int. J. Mol. Med. 41 (6), 3405–3421. doi:10.3892/ijmm.2018.3574
Goldring, C. E., Narayanan, R., Lagadec, P., and Jeannin, J. F. (1995). Transcriptional inhibition of the inducible nitric oxide synthase gene by competitive binding of NF-kappa B/Rel proteins. Biochem. Biophys. Res. Commun. 209 (1), 73–79. doi:10.1006/bbrc.1995.1472
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119 (6), 753–766. doi:10.1016/j.cell.2004.11.038
Han, H., Hu, J., Yan, Q., Zhu, J., Zhu, Z., Chen, Y., et al. (2016). Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 13 (2), 1517–1524. doi:10.3892/mmr.2015.4726
He, Y., She, H., Zhang, T., Xu, H., Cheng, L., Yepes, M., et al. (2018). p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 217 (1), 315–328. doi:10.1083/jcb.201701049
Hung, C. M., Peng, C. K., Wu, C. P., and Huang, K. L. (2018). Bumetanide attenuates acute lung injury by suppressing macrophage activation. Biochem. Pharmacol. 156, 60–67. doi:10.1016/j.bcp.2018.08.013
Hung, C. M., Peng, C. K., Yang, S. S., Shui, H. A., and Huang, K. L. (2020). WNK4-SPAK modulates lipopolysaccharide-induced macrophage activation. Biochem. Pharmacol. 171, 113738. doi:10.1016/j.bcp.2019.113738
Ip, W. K., and Medzhitov, R. (2015). Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 6, 6931. doi:10.1038/ncomms7931
Islam, M. N., Das, S. R., Emin, M. T., Wei, M., Sun, L., Westphalen, K., et al. (2012). Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18 (5), 759–765. doi:10.1038/nm.2736
Kim, S. H., Johnson, V. J., and Sharma, R. P. (2002). Mercury inhibits nitric oxide production but activates proinflammatory cytokine expression in murine macrophage: differential modulation of NF-kappaB and p38 MAPK signaling pathways. Nitric Oxide 7 (1), 67–74. doi:10.1016/s1089-8603(02)00008-3
King, L. S., Kozono, D., and Agre, P. (2004). From structure to disease: the evolving tale of aquaporin biology. Nat. Rev. Mol. Cell Biol. 5 (9), 687–698. doi:10.1038/nrm1469
Laskin, D. L., Sunil, V. R., Gardner, C. R., and Laskin, J. D. (2011). Macrophages and tissue injury: agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 51, 267–288. doi:10.1146/annurev.pharmtox.010909.105812
Liu, K., Zhao, E., Ilyas, G., Lalazar, G., Lin, Y., Haseeb, M., et al. (2015). Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 11 (2), 271–284. doi:10.1080/15548627.2015.1009787
Liu, C., Li, B., Tang, K., Dong, X., Xue, L., Su, G., et al. (2020). Aquaporin 1 alleviates acute kidney injury via PI3K-mediated macrophage M2 polarization. Inflamm. Res. 69 (5), 509–521. doi:10.1007/s00011-020-01334-0
Liu, L., Guo, H., Song, A., Huang, J., Zhang, Y., Jin, S., et al. (2020). Progranulin inhibits LPS-induced macrophage M1 polarization via NF-кB and MAPK pathways. BMC Immunol. 21 (1), 32. doi:10.1186/s12865-020-00355-y
Lomas-Neira, J., Chung, C. S., Perl, M., Gregory, S., Biffl, W., and Ayala, A. (2006). Role of alveolar macrophage and migrating neutrophils in hemorrhage-induced priming for ALI subsequent to septic challenge. Am. J. Physiol. Lung Cell. Mol. Physiol. 290 (1), L51–L58. doi:10.1152/ajplung.00028.2005
McWhorter, F. Y., Davis, C. T., and Liu, W. F. (2015). Physical and mechanical regulation of macrophage phenotype and function. Cell. Mol. Life Sci. 72 (7), 1303–1316. doi:10.1007/s00018-014-1796-8
Meli, R., Pirozzi, C., and Pelagalli, A. (2018). New perspectives on the potential role of aquaporins (AQPs) in the physiology of inflammation. Front. Physiol. 9, 101. doi:10.3389/fphys.2018.00101
Mishra, P., Pandey, N., Pandey, R., and Tripathi, Y. B. (2021). Role of macrophage polarization in acute respiratory distress syndrome. JoR. 1 (4), 260–272. doi:10.3390/jor1040024
Moon, C., Rousseau, R., Soria, J. C., Hoque, M. O., Lee, J., Jang, S. J., et al. (2004). Aquaporin expression in human lymphocytes and dendritic cells. Am. J. Hematol. 75 (3), 128–133. doi:10.1002/ajh.10476
Nakagawa, I., Amano, A., Mizushima, N., Yamamoto, A., Yamaguchi, H., Kamimoto, T., et al. (2004). Autophagy defends cells against invading group A Streptococcus. Science 306 (5698), 1037–1040. doi:10.1126/science.1103966
Pires, V., Bramatti, I., Aschner, M., Branco, V., and Carvalho, C. (2022). Thioredoxin reductase inhibitors as potential antitumors: Mercury compounds efficacy in glioma cells. Front. Mol. Biosci. 9, 889971. doi:10.3389/fmolb.2022.889971
Preston, G. M., Jung, J. S., Guggino, W. B., and Agre, P. (1993). The mercury-sensitive residue at cysteine 189 in the CHIP28 water channel. J. Biol. Chem. 268 (1), 17–20. doi:10.1016/s0021-9258(18)54108-9
Rabolli, V., Wallemme, L., Lo Re, S., Uwambayinema, F., Palmai-Pallag, M., Thomassen, L., et al. (2014). Critical role of aquaporins in interleukin 1β (IL-1β)-induced inflammation. J. Biol. Chem. 289 (20), 13937–13947. doi:10.1074/jbc.M113.534594
Ramond, E., Jamet, A., Coureuil, M., and Charbit, A. (2019). Pivotal role of mitochondria in macrophage response to bacterial pathogens. Front. Immunol. 10, 2461. doi:10.3389/fimmu.2019.02461
Rice, K. M., Walker, E. M., Wu, M., Gillette, C., and Blough, E. R. (2014). Environmental mercury and its toxic effects. J. Prev. Med. Public Health 47 (2), 74–83. doi:10.3961/jpmph.2014.47.2.74
Savage, D. F., and Stroud, R. M. (2007). Structural basis of aquaporin inhibition by mercury. J. Mol. Biol. 368 (3), 607–617. doi:10.1016/j.jmb.2007.02.070
Sha, W. C., Liou, H. C., Tuomanen, E. I., and Baltimore, D. (1995). Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 80 (2), 321–330. doi:10.1016/0092-8674(95)90415-8
Sui, X., Kong, N., Ye, L., Han, W., Zhou, J., Zhang, Q., et al. (2014). p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 344 (2), 174–179. doi:10.1016/j.canlet.2013.11.019
Tyteca, D., Nishino, T., Debaix, H., Van Der Smissen, P., N'Kuli, F., Hoffmann, D., et al. (2015). Regulation of macrophage motility by the water channel aquaporin-1: crucial role of M0/M2 phenotype switch. PLoS One 10 (2), e0117398. doi:10.1371/journal.pone.0117398
Vergadi, E., Vaporidi, K., Theodorakis, E. E., Doxaki, C., Lagoudaki, E., Ieronymaki, E., et al. (2014). Akt2 deficiency protects from acute lung injury via alternative macrophage activation and miR-146a induction in mice. J. Immunol. 192 (1), 394–406. doi:10.4049/jimmunol.1300959
Verkman, A. S., Matthay, M. A., and Song, Y. (2000). Aquaporin water channels and lung physiology. Am. J. Physiol. Lung Cell. Mol. Physiol. 278 (5), L867–L879. doi:10.1152/ajplung.2000.278.5.L867
Xie, Q. W., Kashiwabara, Y., and Nathan, C. (1994). Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J. Biol. Chem. 269 (7), 4705–4708. doi:10.1016/s0021-9258(17)37600-7
Xu, Y., Jagannath, C., Liu, X. D., Sharafkhaneh, A., Kolodziejska, K. E., and Eissa, N. T. (2007). Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27 (1), 135–144. doi:10.1016/j.immuni.2007.05.022
Yin, Y., He, X.-T., Wang, J., Wu, R.-X., Xu, X.-Y., Hong, Y.-L., et al. (2020). Pore size-mediated macrophage M1-to-M2 transition influences new vessel formation within the compartment of a scaffold. Appl. Mater. Today 18, 100466. doi:10.1016/j.apmt.2019.100466
Ying, H., Kang, Y., Zhang, H., Zhao, D., Xia, J., Lu, Z., et al. (2015). MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J. Immunol. 194 (3), 1239–1251. doi:10.4049/jimmunol.1402088
Yool, A. J., Brown, E. A., and Flynn, G. A. (2010). Roles for novel pharmacological blockers of aquaporins in the treatment of brain oedema and cancer. Clin. Exp. Pharmacol. Physiol. 37 (4), 403–409. doi:10.1111/j.1440-1681.2009.05244.x
Keywords: aquaporin, bone marrow-derived macrophages, mercury chloride, macrophage polarization, autophagy
Citation: Chuang Y-C, Wu S-Y, Huang Y-C, Peng C-K, Tang S-E and Huang K-L (2022) Cell volume restriction by mercury chloride reduces M1-like inflammatory response of bone marrow-derived macrophages. Front. Pharmacol. 13:1074986. doi: 10.3389/fphar.2022.1074986
Received: 20 October 2022; Accepted: 23 November 2022;
Published: 13 December 2022.
Edited by:
Galina Sud’ina, Lomonosov Moscow State University, RussiaReviewed by:
Monika Sharma, Brigham and Women’s Hospital, United StatesCopyright © 2022 Chuang, Wu, Huang, Peng, Tang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun-Lun Huang, kun@mail.ndmctsgh.edu.tw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.