
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 16 December 2022
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2022 | https://doi.org/10.3389/fphar.2022.1072651
This article is part of the Research TopicEpigenetic Regulation and Therapy Resistance in CancerView all 5 articles
 Zhixiong Zhang1,2†
Zhixiong Zhang1,2† Guan Wang1†
Guan Wang1† Yuyan Li1,2†
Yuyan Li1,2† Dongsheng Lei3Jin Xiang1
Dongsheng Lei3Jin Xiang1 Liang Ouyang1,4Yanyan Wang1,4*Jinliang Yang1*
Liang Ouyang1,4Yanyan Wang1,4*Jinliang Yang1*DNA methylation mediated by DNA methyltransferase is an important epigenetic process that regulates gene expression in mammals, which plays a key role in silencing certain genes, such as tumor suppressor genes, in cancer, and it has become a promising therapeutic target for cancer treatment. Similar to other epigenetic targets, DNA methyltransferase can also be modulated by chemical agents. Four agents have already been approved to treat hematological cancers. In order to promote the development of a DNA methyltransferase inhibitor as an anti-tumor agent, in the current review, we discuss the relationship between DNA methylation and tumor, the anti-tumor mechanism, the research progress and pharmacological properties of DNA methyltransferase inhibitors, and the future research trend of DNA methyltransferase inhibitors.
DNA methylation is a major covalent modification occurring at the C-5 position of cytosine in CpG dinucleotides, with cofactor S-adenosyl-l-methionine (SAM) as a methyl donor, which suppresses the transcription of the methylated gene (Costa et al., 2017). DNA methylation plays an important role in maintaining normal cell function and embryonic development. Once the modification is abnormal, it leads to the occurrence of tumor. Abnormal DNA methylation in a tumor cell is manifested in the following three aspects: 1) DNA methyltransferase (DNMT) is overexpressed and its catalytic activity is enhanced in the tumor cell. 2) The DNA methylation level near highly repetitive sequences in the tumor cell is significantly reduced, resulting in chromosomal instability and reactivation of transposon elements. 3) The tumor suppressor gene (TSG) cannot be normally expressed due to hyper methylation in the promoter (Pechalrieu et al., 2017).
DNA methylation is mediated by DNMT in mammals. Currently, five types of DNMT have been reported, among which DNMT1 and DNMT3A/3B are related to DNA methylation (Masaki et al., 1999; Goffin and Eisenhauer, 2002). Inhibition of DNMT by the DNA methyltransferase inhibitor (DNMTi) is of great significance for tumor immunotherapy. On one hand, the DNMTi can promote tumor antigen presentation by upregulating the expression of tumor-associated antigen (TAA). On the other hand, the DNMTi can enhance the function of cytotoxic T lymphocyte (CTL) by maintaining or upregulating the expression of IL-2, IFN-γ, and other anti-tumor cytokines through demethylation. The DNMTi can also promote immune response by promoting NK cell-mediated cytotoxic effect and modulating immunosuppressive cells. In addition, the DNMTi can remodel the chromatin of tumor cells by inhibiting DNA methylation and histone modification.
Up to now, there are nearly 70 pipelines of DNMTi development worldwide, among which azacitidine, decitabine, clofarabine, and arsenic trioxide have been approved for the treatment of hematological tumors. A few DNMTis have entered the clinical stage for anti-tumor research. However, most of them are still in the pre-clinical stage. In the current review, we discussed the relationship between DNA methylation and tumor, the research progress, mechanism, and pharmacological properties of DNMTis with the purpose of promoting the development of DNMTis as an anti-tumor agent.
In a broad sense, DNA methylation refers to the chemical modification of methyl binding to a specific base on DNA through a covalent bond, which may occur at the C-5 position of cytosine, N-6 position of adenine, and N-7 position of guanine (Ahmad and Rao, 1996). Strictly speaking, DNA methylation in common research refers to the process in which the methyl group of SAM is transferred to the C-5 position of cytosine in the 5′-CpG-3′ dinucleotides (CpGs) under the function of DNMTs to form 5-methyl cytosine (Figure 1A). DNA methylation is mediated by DNMT and five DNMTs have been found so far. DNMT1 is mainly involved in the maintenance of the methylation state and the de novo methylation of the non-CpG site. DNMT3 comprises DNMT3A, DNMT3B, and DNMT3L, in which DNMT3A and 3B are de novo DNA methylation enzymes, while DNMT3L is the regulator of DNMT3A/3B. DNMT2 has no catalytic activity and its function in DNA methylation is unclear (Margot et al., 2001).
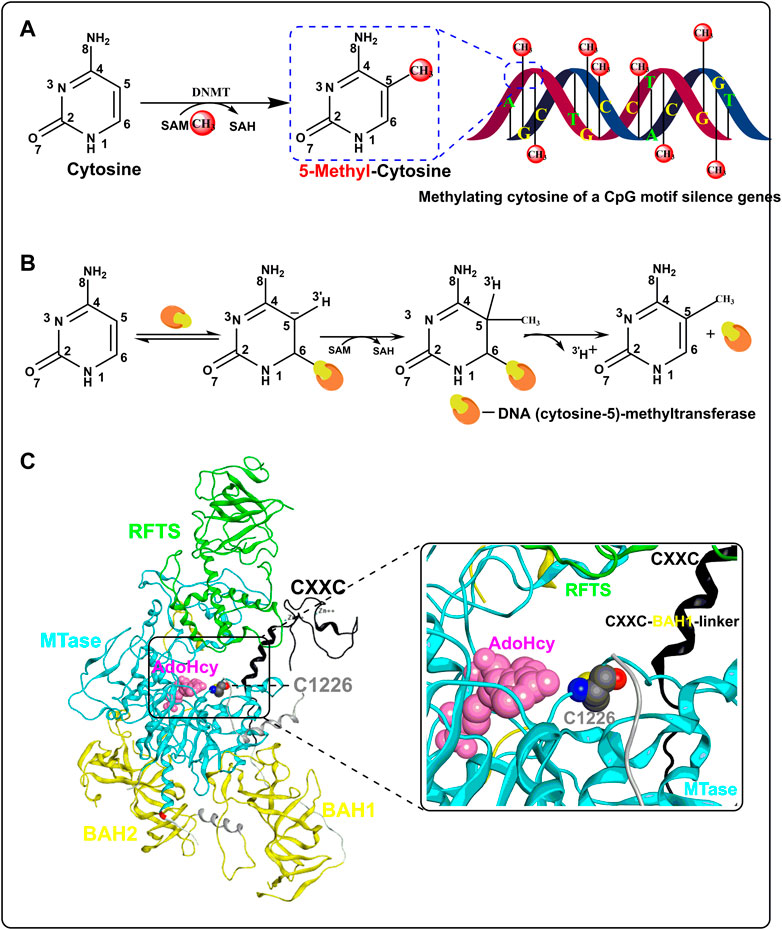
FIGURE 1. Process and mechanism of DNA methylation. (A) The overall process of CpG methylation by DNMT. (B) Mechanism of DNA methylation in DNA (cytosine-5)-methyltransferase. (C) Crystal structure of human DNA methyltransferase 1 with AdoHcy (PBD code: 4WXX). The RFTS, CXXC, MTase, and BAH domains are colored in green, black, light blue, and yellow, respectively. SAH is colored in pink and residue C1226 is colored in gray.
The mechanism of DNA methylation was first reported in prokaryotic DNA (cytosine-5)-methyltransferase (Wu and Santi, 1987). First, the C atom of cytosine is flipped out of the DNA double helix and embedded in the catalytic domain when DNA (cytosine-5)-methyltransferase binds to DNA. Second, mercaptan of cysteine at the active site of DNA (cytosine-5)-methyltransferase forms a covalent bond with cytosine C6 through nucleophilic action and simultaneously activates C5. Third, SAM is extremely unstable for methyl binding to the S atom. With the action of DNA (cytosine-5)-methyltransferase, methyl is transferred to activated C5 from SAM, which becomes S-adenosyl-Homocysteine (SAH) later. Finally, with the release of protons at C5 and the breaking of the covalent bond at C6, the process of DNA methylation is completed (Figure 1B). In addition, Zhang et al. (2015) resolved the crystal structure of human DNMT1 in complex with SAH (PDB code: 4WXX) in 2015, which helped us understand the mechanism of DNA methylation catalyzed by DNMT in eukaryotes. Human DNMT1 mainly contains the RFTS domain, the CXXC domain, the MTase domain, and two BAH domains (Figure 1C). SAH is mainly bound in the cavity enclosed by the RFTS, CXXC, and MTase domains, which are also the pockets where DNA binds. Structural analysis showed that when human DNMT1 was not bound to DNA, the RFTS domain could bind to the MTase domain directly through a hydrogen bond and bind indirectly through a CXXC–BAH1 linker, thus forming an auto-inhibitory conformation. However, when DNMT1 binds to unmethylated DNA, the activity of human DNMT1 is inhibited by the CXXC–BAH1 linker. The inhibitory activity of the CXXC–BAH1 linker on human DNMT1 mainly showed two aspects. On one hand, the CXXC–BAH1 linker occupies the DNA binding site and pushes the residue C1226, which is important for the activity of human DNMT1, away from the DNA binding pocket through steric hindrance. On the other hand, the CXXC–BAH1 linker undergoes a helix-to-loop conformational transition and is positioned along the catalytic cleft of DNMT1 to inhibit the interaction between DNMT1 and DNA. Upon binding to the hemi-methylated DNA and SAM, the auto-inhibitory state of DNMT1 was released with the conformation change of the RFTS domain and CXXC–BAH1 linker Zhang et al.,2015). At that moment, the thiol of C1226 in the MTase domain flips over to cytosine and initiates a nucleophilic addition reaction to the C-6 atom of cytosine to form a covalent bond (Figure 1C). The unstable methyl group on SAM was transferred to the C-5 atom of cytosine and then the proton and DNMT are shed from cytosine to form 5-methylcytosine.
Hypomethylation plays an important role in the initiation, progression, invasion, and metastasis of different types of cancer. Normal cells ensure genomic stability by silencing satellite sequences through DNA hypermethylation, which is reversed in tumor cells. The loss of DNA methylation of satellite-2 and satellite-α is associated with early breast cancer development, while in ovarian cancer, they promote tumor progression (Widschwendter et al., 2004; Costa et al., 2006). The melanoma-associated cancer-testis (CT) antigen (MAGE) is reactivated due to DNA hypomethylation in melanoma and colorectal cancer, which promotes the development of melanoma and colorectal cancer (Kim et al., 2006). Loss of DNA methylation in the CDH3 gene promoter results in the overexpression of P-cadherin in colorectal and breast carcinomas, which decreased cell polarity and promotes the invasion and migration of colorectal and breast cancer cells (Paredes et al., 2005; Milicic et al., 2008). Overexpression of P-glycoproteins involved in drug resistance in breast cancer patients due to DNA hypomethylation is associated with the advanced tumor stage (Chekhun et al., 2006).
Hypermethylation also plays an important role in the initiation, progression, invasion, and metastasis of different types of cancer. For example, cell adhesion genes CDH1 in gastric cancer cells and CDH13 in primary non-small cell lung cancer cells are silenced due to hypermethylation, which promotes the invasion and metastasis of both types of tumor cells (Seuk Kim et al., 2005; Huang et al., 2015). Various genes associated with DNA repair processes are also hypermethylated in tumor tissues; hypermethylation of the MLH1 gene in gastric cancer leads to abnormal DNA mismatch repair and promotes the progression of gastric cancer (Fleisher et al., 2001). In addition, the apoptosis pathway mediated by death-associated protein kinase 1 (DAPK1)-TMS1 promotes tumor cell apoptosis. The silencing of TMS1 due to hypermethylation promotes the proliferation of breast cancer cells, thus promoting the progression of breast cancer (Gordian et al., 2009).
Taken together, the abnormal methylation of tumor cells is mainly characterized by genome-wide hypomethylation and local hypermethylation. A low level of DNA methylation is associated with the activation of proto-oncogenes. DNA hypomethylation is commonly found in solid tumors and the degree of DNA methylation increases with the increase in the malignant degree. A high level of DNA methylation is associated with the silence of TSG (Akhavan-Niaki and Samadani, 2013). In addition, the ways of DNA hypermethylation in tumor cells are varied. Hypermethylation of multiple genes can occur simultaneously in a tumor, such as APC, DKPK1, MGMT, P16, and RASSF1 in lung cancer, while others, like RASSF1 and P16, could be hypermethylated in a variety of tumors (Castro et al., 2010).
DNMTis can enhance the immunogenicity of tumor cells (Figure 2). For example, DNMTis can upregulate the major histocompatibility complex I (MHC-I) by inhibiting DNA hypermethylation in the promoter of MHC-I, thus improving the presentation of TAA and enhancing immunogenicity (Gallagher et al., 2017). In addition, DNMTis can promote the expression of the cancer testis antigen, which can help host CTL to distinguish tumor cells from healthy cells (Gallagher et al., 2017).
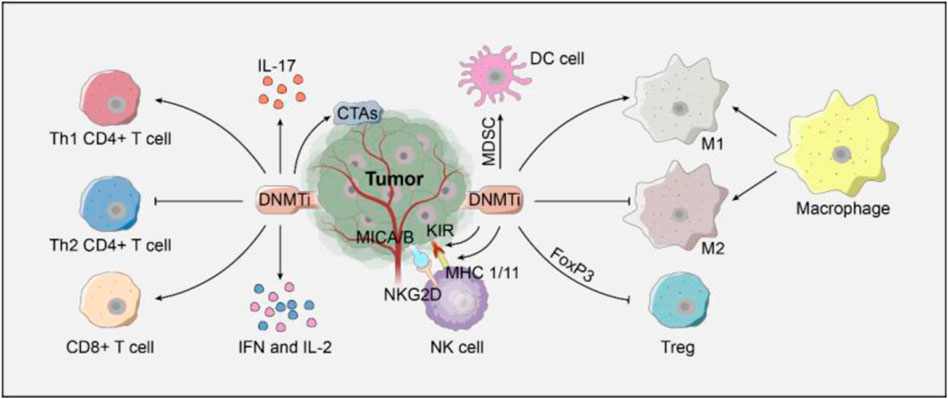
FIGURE 2. Regulation of DNMTis on immune cells.
DNMTis can enhance the cytotoxic effect of CD8+ T cells and help CD4+ T cells and NK cells by inducing the expression of immune-stimulatory cytokines (Figure 2). When CD8+ T cells were exposed to TAA, DNMTis enhanced the cytotoxic activity of CTL by inhibiting methylation and promoting IL-2 expression (Qiu et al., 2017). When immature CD8+ T cells were transformed into effector T cells, DNMTis also induced the demethylation of the three hypermethylated CpG islands in the upstream of IFN-γ, thus enhancing the bioactivity of CTL (Lucarini et al., 2017). The DNMTi promotes CD4+ T cells to differentiate into Th1 CD4+ T cells by promoting the secretion of IL-17. The killer cell immunoglobulin-like receptor (KIR) on the surface of NK cells can express normally under the demethylation of the DNMTi, which promotes the recognition of abnormal tumor cells after binding to MHC-I specifically (Küçük et al., 2016).
DNMTis can also promote the immune response by regulating the bioactivity of immunosuppressive cells (Figure 2). On one hand, the DNMTi inhibits the differentiation of T lymphocytes into regulatory T cells by regulating the expression of Foxp3 (Costantini et al., 2013). The DNMTi can also inhibit the immunosuppressive effect of myeloid-derived suppressor cells and induce their differentiation into dendritic cells (Zhou et al., 2017). M1 macrophages display anti-tumor phagocytic properties, whereas M2 macrophages have pro-tumorigenic properties. On the other hand, DNMTis can promote the differentiation of macrophages into M1 rather than M2 macrophages by regulating the inflammatory response.
Currently, there are nearly 70 DNMTi pipelines under development worldwide. In total, 16 pipelines have already been in the clinical research stage and some of them have already entered the phase III clinical study (Table 1). Azacitidine, decitabine, clofarabine, and arsenic trioxide have been approved for the treatment of myelodysplastic syndrome (MDS) and hematological cancer.

TABLE 1. Progress of DNMTis in clinical anti-tumor research.
5-Azacitidine (1) is a cytarabine derivative synthesized in 1964. It was not clear until 1980 that it exerts anti-tumor activity by inhibiting DNA methylation (Jones and Taylor, 1980). 5-Azacitidine is modified by nucleotide reductase and then phosphorylated by uridine cytidine kinase to form a triphosphate, 5-azacitidine triphosphate, which is integrated into the DNA sequence as a cytosine analog during DNA replication, thereby inhibiting DNA cytosine methylation (Rius et al., 2009). It can also cause the degradation of DNMT1 by binding to the sulfhydryl of DNMT1 with a covalent bond (Ghoshal et al., 2005). 5-Azacitidine was approved by the Food and Drug Administration (FDA) for the treatment of MDS in 2004 and it was the first DNMTi in clinical practice with a trade name of Vidaza (Kaminskas et al., 2005). Studies showed that 5-azacytidine could inhibit the L1210 cell with ID50 and ID90 of 0.019 and 0.15 μg/ml in vitro, respectively (Li et al., 1970). In addition, a study showed that 5-azacitidine can significantly increase the tri-methylation of lysine-4 on histone H3 in mouse primary spermatocytes, which indicated that 5-azacitidine could modulate histone methylation (Burlibaşa et al., 2021). It could also reduce tumor growth in two intracerebral glioblastoma models in vivo.
5-Azacytidine is mainly used for the treatment of MDS and hematologic tumors clinically. In the clinical trial “CALGB 8921,” 72 patients with refractory anemia with excessive blasts, chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML), were subcutaneously administered 75 mg/m2 of azacitidine every 4 weeks for 7 consecutive days. The overall response (OR) was 13.9%. The mean and median durations of partial response were 810 and 430 days, respectively; 80% of patients were still in partial response (PR). Severe side effects, mainly including thrombocytopenia, febrile neutropenia, and fever, were observed. As shown in Figure 3, the pharmacokinetics (PK) of azacitidine was studied in six MDS patients. A single dose of 75 mg/m2 of azacitidine was intramuscularly administered on day 1 (Marcucci et al., 2013). The absolute bioavailability of azacitidine for subcutaneous administration is 89%. The dose of 75 mg/m2 is well-tolerated and it could be rapidly absorbed and widely distributed. It is easy for hydrolysis and deamination in plasma, which results in a short half-life. The median half-lives of azacitidine were 0.36 ± 0.02 h and 0.69 ± 0.14 h for subcutaneous and intravenous administration, respectively. Urinary excretion is the primary route of elimination of 5-azacitidine and its metabolites. Vidaza is currently administered subcutaneously for high-risk MDS, CMML, and AML. The recommended dose for the first treatment cycle is 75 mg/m2, administered subcutaneously daily for 7 consecutive days.
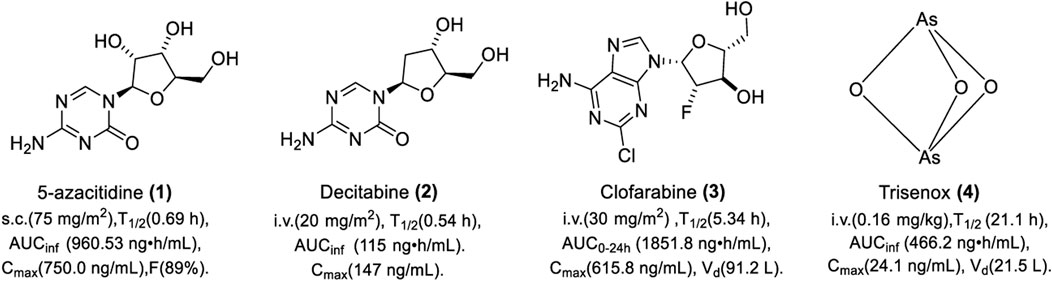
FIGURE 3. Chemical structure of DNMTis approved by the FDA.
Decitabine (2) was approved by the FDA in 2006 for the treatment of MDS with a trade name of Dacogen (Center for Drug Evaluation and Research). Because it is deoxy, decitabine can be converted directly to a cytosine analog (5-azabine triphosphate) by deoxy-cytidine kinase without prior deoxidation. The inhibitory activity of DNMT is 30 times more than that of azacitidine (Rius et al., 2009). High concentrations of decitabine can induce cell death by forming non-functional DNA during DNA replication, while low concentrations of decitabine can bind DNMT covalently, which leads to DNMT inactivation without causing cell death (Momparler, 2005). Studies showed that decitabine could inhibit the proliferation of SNU719, NCC24, and KATOIII cells in vitro (Nakamura et al., 2016). In addition, a study showed that decitabine could enhance apoptosis induced by histone deacetyltransferase inhibitor (HDACi) depsipeptide in human thoracic cancer cells. Further study showed that decitabine increased the depsipeptide-induced acetylation of histone H3 and H4 (Zhu and Otterson, 2003). Decitabine administered once daily at 1 mg/kg for 5 days could result in the regression of EL4 tumor growth in C57BL/6 mice in vivo (Wang et al., 2013). Decitabine is prone to deamination and causes granulocytopenia, which limits its clinical application.
Decitabine was mainly used for the treatment of MDS and cancer in clinical practice. Patients with MDS intravenously receive 20 mg/m2 of decitabine for 1 h daily and continuously receive the same dose for 5 days; the complete response (CR) and OR was 20% and 61%, respectively. The overall survival (OS) was 20 months. The main side effects are neutropenia, nausea, and fatigue. The PK was evaluated in 11 patients, who intravenously received 20 mg/m2 for over 1 h, and plasma concentration–time profiles after discontinuation of infusion showed a biexponential decline. As shown in Figure 3, decitabine can be absorbed rapidly. Upon repeat doses, there was no systemic accumulation or any changes in PK parameters. The deamination of cytidine deaminase in liver, intestinal epithelial cells, granulocytes, and whole blood is the main metabolic pathway of decitabine. The recommended dose of decitabine for its indication of MDS is 15 mg/m2 with continuous intravenous infusion for at least 3 h, once every 8 h for 3 consecutive days.
Clofarabine (3) is a purine nucleoside DNMTi, which was approved by the FDA in 2004 for the treatment of refractory or relapsed AML in children with the trade name of clofarabine. A study showed that clofarabine inhibited the proliferation of tumor cells by downregulating DNMT1 and inhibiting the methylation of TSG, such as PTEN, APC, and RARβ2 (Majda et al., 2010). It can also promote the expression of p21, which competitively binds to the binding site of DNMT1 on the proliferating cell nuclear antigen (PCNA) protein, thus blocking the activity of the PCNA-promoting DNMT1 on hemimethylated DNA (Iida et al., 2010). In another study, clofarabine and fludarabine showed a synergistic effect against AML cells by increasing the methylation of lysine 4/9/27/36/79 on histone H3 (Valdez et al., 2011).
Clofarabine (20, 30, and 40 mg/m2/day) was administered intravenously for 5 days in patients with relapsed AML or elderly patients with newly diagnosed AML (Tatsuya et al., 2013). The OR was 43% with four (29%) patients achieving complete remission, and two (14%) patients achieved incomplete remission. The maximum tolerated dose (MTD) was 30 mg/m2/day and the most common adverse events were thrombocytopenia and anemia. As shown in Figure 3, clofarabine (30 mg/m2/day by i.v. infusion) was absorbed rapidly with a narrow distribution. The Cmax and exposure in 24 h increased with increasing dose. Kidney excretion is the main way for clofarabine elimination. For patients with relapsed or resistant AML who received at least two previous treatments, the recommended therapy for clofarabine is 52 mg/m2/d over 2 h for 5 consecutive days, repeated every 2–6 weeks after organ function has recovered to baseline levels.
Arsenic trioxide (4) was approved by the European Commission in 2002 for the treatment of relapsed or persistent acute promyelocytic leukemia (APL) with the trade name of Trisenox (Cohen et al., 2001). Studies showed that Trisenox could promote the apoptosis of leukemia cells by inhibiting DNA methylation (Badarkhe et al., 2016). However, the mechanism of DNA methylation inhibition is unclear. Interestingly, studies showed that arsenic trioxide could reduce global acetylation of lysine 16 on histone H4 by binding to histone acetyltransferase hMOF in HeLa cells and HEK293T cells, while also increasing deacetyltransferase HDAC4 expression (Liu et al., 2015). These data provide a new theoretical basis for elucidating the mechanism of As toxicity.
Five patients with newly diagnosed APL were intravenously administered at a dose of 0.16 mg/kg for 40 min infusion on the first day followed by 18–20 h daily at a very slow rate with an infusion speed of 8 drips/min (Gao et al., 2018). All patients achieved hematologic complete remission after induction therapy, but also experienced side effects such as abnormal coagulation and anemia. As shown in Figure 3, Trisenox was absorbed slowly with a low clearance rate and a long half-life, which results in wide distribution in tissues. When the drug was stopped, the arsenic content in tissues was detected from high to low in the order of skin, ovary, liver, and kidney. During the treatment, the amount of arsenic excreted in urine in 24 h was 1%–8% of the dosing dose. Concentrations of total arsenic were constant throughout the administration period at a steady state. The PK remained the same during continuous administration. The clearance rate of continuous slow infusion is slower than that of rapid infusion, which facilitates to maintain the effective concentration at a low level and avoid the toxicity caused by a high concentration. It seems that hepatic toxicity is the main reason leading to the termination of Trisenox. For patients with newly diagnosed or relapsed APL, the recommended dose is 0.15 mg/kg intravenously daily in combination with tretinoin until bone marrow remission and not to exceed 60 days in the induction cycle.
Guadecitabine (SGI-110, 5) is a dinucleotide derivative of decitabine, which inhibits DNA methylation by the same mechanism of decitabine, but its PK and stability are superior to those of decitabine due to the phosphodiester bond of SGI-110, which can resist the transient degradation of cytidine deaminase. Studies showed that SGI-110 reduced the methylation of T24 and HCT116 cells and caused the depletion of DNMT1 in vitro (Yoo et al., 2007). The study also showed that treatment with SGI-110 or in combination with HDAC inhibitor MS275 could reverse the epithelial mesenchymal transition (EMT) by inducing tri-methylation of lysine-27 on histone H3 (H3K27me3) in triple-negative breast cancer (TNBC) cells. The level of H3K27me3 in breast cancer patients was positively correlated with their survival. These data suggested that SGI-110 alone or in combination with the HDAC inhibitor could improve the survival of breast cancer patients (Su et al., 2018). In addition, SGI-110 could reduce DNA methylation in the promoter of the p16 gene in a human xenotransplantation tumor model in vivo.
SGI-110 is used for the treatment of MDS and AML in clinics. In total, 74 patients with AML and 20 patients with MDS were randomly assigned to subcutaneous SGI-110 with a dose of 3–125 mg/m2, either once daily for 5 consecutive days (daily×5), or once weekly for 3 weeks. The median OS for patients with AML and MDS was 140 days and 282 days, respectively (Issa et al., 2015). No dose-limiting toxicity (DLT) was noted up to 90 mg/m2. The most common grade 3 adverse events were febrile neutropenia, pneumonia, and thrombocytopenia. There were no differences in the PK between the regimens or between days in a particular regimen. As shown in Figure 4, SGI-110 was absorbed rapidly after subcutaneous injection. The half-life of SGI-110 was longer than that of decitabine. SGI-110 can be present in plasma for up to 8 h or more, resulting from the continuous appearance of decitabine, which creates an exposure window of 11–12 h. In summary, SGI-110, as a prodrug for decitabine, could release decitabine slowly and has a longer exposure window (Issa et al., 2015).
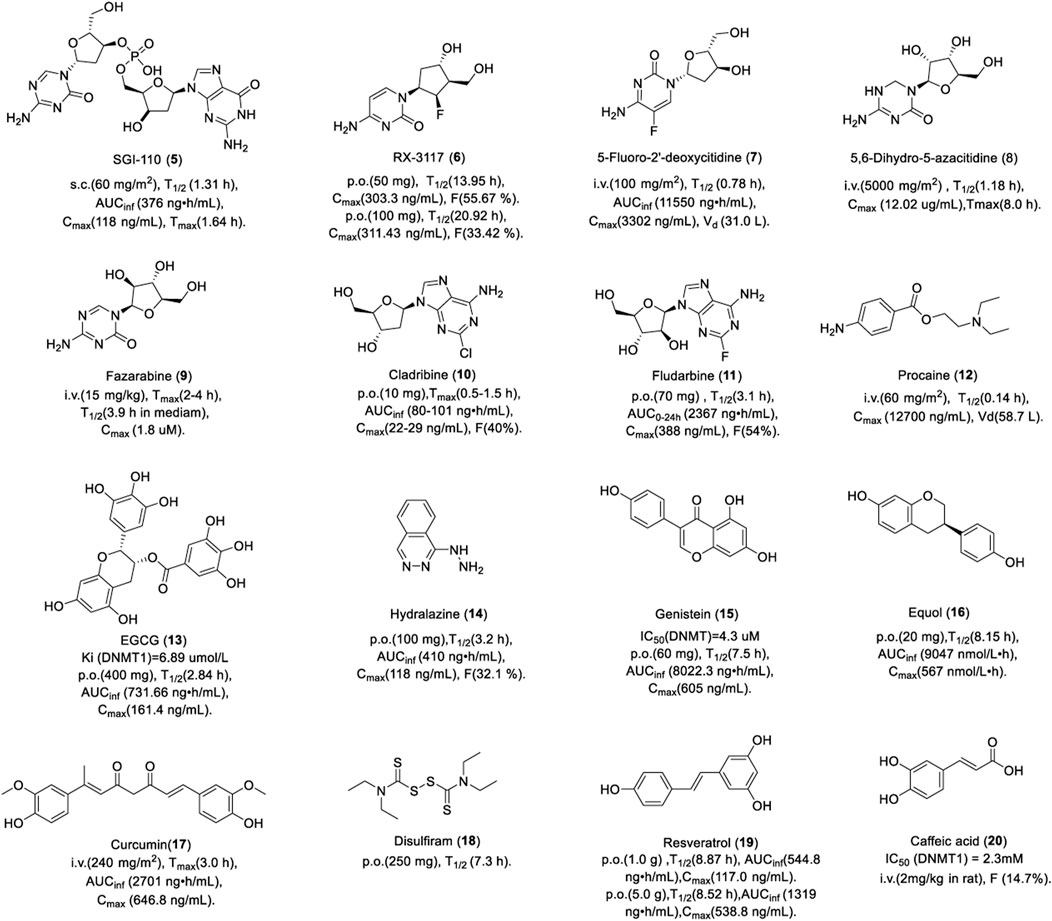
FIGURE 4. Chemical structure of DNMTis in clinical.
RX-3117 (TV-1360, 6) is a potent DNA synthesis inhibitor. A study showed that RX-3117 could inhibit the proliferation of breast, lung, and colon cancer cells with an IC50 of 0.18, 0.25, and 0.28 μM in vitro, respectively (Choi et al., 2012). RX-3117 induced 100%, 78%, 62%, and 66% tumor growth inhibition in human Colo 205, H460, H69, and CaSki tumor xenograft models in vivo, respectively. Further studies showed that RX-3117 could be phosphorylated to diphosphate (RX-DP) and triphosphate (RX-TP) by uridine cytidine kinase. RX-DP is reduced by ribonucleotide reductase to DRX-DP and further phosphorylated to DRX-TP, which can incorporate into a DNA sequence to inhibit DNA synthesis (Peters et al., 2013), while RX-TP can incorporate into RNA to inhibit RNA synthesis.
In a phase II clinical trial, patients with advanced urothelial cancer received a dose of 700 mg of RX-3117 orally once-a-day for 5 consecutive days followed by 2 days off per week for 3 weeks. Among the 45 patients with refractory metastatic pancreatic cancer, progression-free survival (PFS) increased by more than 2 months in 13 patients, disease stability increased by more than 4 months in 5 patients, and partial remission in 1 patient. The adverse effects were anemia, fatigue, and diarrhea. Patients with solid tumors were treated with 50 mg or 100 mg RX-3117 orally or 20 mg intravenously. As shown in Figure 4, RX-3117 has high oral bioavailability (F) and the absolute F was 55.67% and 33.42% for the 50 and 100 mg doses, respectively, which suggests that the F of RX-3117 in plasma may not be dose-proportional. However, the half-life of RX-3117 is proportional to the dose. The plasma PK of intravenous RX-3117 differed from that of oral RX-3117. The 20-mg dose of intravenous RX-3117 had a mean Cmax of 1,143.63 ng/ml, which was approximately a fourfold increase over the peak concentrations of the oral doses. Therefore, a low dose of RX-3117 administered intravenously may be a good choice for the treatment of solid tumors (Udvaros et al., 2015).
In order to solve the instability of decitabine, one pyrimidine analog, 5-fluoro-2-deoxycytidine (FdCyd, 7), was designed by adding fluorine to the pyrimidine ring (Frank and Robert, 2005). FdCyd inhibits DNA methylation by the same mechanism as decitabine. It can inhibit DNA methylation by integrating into DNA sequences in the form of triphosphate or binding to DNMT covalently.
FdCyd is currently in phase II clinical trials for the treatment of solid tumors. A total of 54 patients with advanced malignant tumor received intravenous infusion of FdCyd and tetrahydrouridine (THU, inhibiting the deamination of FdCyd) for 3 h for 5 days every 3 weeks (Newman et al., 2015). The dose of FdCyd was 2.5–180 mg/m2/day and the fixed dose of THU was 350 mg/m2/day. A total of 20 patients with advanced solid tumor had a stable disease plateau between 1.4 and 13.3 months and their CT results showed that the tumor volume reduction was more than 90%. Significant clinical improvement was noted after two cycles of treatment, the PR was obtained after four cycles, and continued shrinkage of the tumor was noted after six cycles. One of six patients at a dose of 134 mg/m2/day experienced a first-cycle DLT, grade 3 colitis. The FdCyd volume of distribution of 30–40 L/m2 suggests the distribution of FdCyd in body water, consistent with its hydrophilic character.
5, 6-Dihydro-5-azacytidine (DHAC, 8) was designed by opening the imine bond at the C5-C6 position of azacitidine. However, DHAC did not show good efficacy in the treatment of mesothelioma either as a single agent or in combination with cisplatin and the response in solid tumors was limited; therefore, the clinical trial had to be terminated (Pechalrieu et al., 2017).
A total of 13 patients with advanced cancer were administered by infusion over 24 h with a dose of 1,000–7,000 mg/m2 (Curt et al., 1985). Dramatic reductions in palpable adenopathy were observed in two patients. The limiting toxicity was pleuritic chest pain. As shown in Figure 4, DHAC was absorbed slowly with a Tmax up to 8 h. A total body clearance of 311 ± 76 ml/min/m2 and post-infusion half-life during 1 and 2 hours were observed. 40% DHAC was excreted unchanged in urine. The slower clearance rate and the large quantity recoverable in the urine are most likely attributable to the chemical stability. It is worth noting that DHAC is stable in aqueous solution and may be administered intravenously, which potentially avoids the acute toxicities associated with the bolus administration of 5-azacytidine.
Fazarabine (9) was synthesized in the 1970s as a molecule with the arabinose of ara-C and the triazine ring of azacitidine (Beisler et al., 1979). Like ara-C, fazarabine requires intracellular activation ofthe nucleotide triphosphate by deoxycytidine kinase before integrating into a DNA sequence. It is reported to be cytotoxic against the Molt-4 human lymphoblastic leukemia cell in vitro (Townsend et al., 1985). In addition, fazarabine has a better activity against both murine and human solid tumor in contrast to its parent compounds (ara-C and 5-AC), including Lewis lung carcinoma and TE-671 medulloblastoma (Dalal et al., 1986).
Fazarabine has undergone several phase I/II clinical trials for the treatment of cancer, which has been discontinued due to low response rates. A total of 16 children with refractory malignancies were administered fazarabine as a 24-hour continuous infusion with a dose of 15 or 20 mg/m2/h (Heideman et al., 1989). No objective tumor responses were noted. Two patients had stable disease for a period of 6 weeks. DLT was granulocytopenia. As shown in Figure 4, there was a rapid increase in plasma concentrations of fazarabine after infusion. Fazarabine is widely distributed and it can cross over the blood–brain barrier into the cerebrospinal fluid. However, these experiments failed to correct the considerable spontaneous degradation of fazarabine in aqueous media (half-life, 3.9 ± 0.45 h in RPMI 1640 medium). Thus, it is possible that the plasma concentrations maintained during continuous infusion may be even more cytotoxic than that was observed.
Adenosine analogs DNMTi cladribine (10) and fludarabine (11) are mainly used for the treatment of hematologic tumors in clinics. They can inhibit DNA methylation through various mechanisms. First, they can be integrated into DNA sequences in the form of triphosphate deoxynucleotide to inhibit DNA synthesis like 5-azacytidine. Second, similar to clofarabine, they blocked the PCNA protein to promote the activity of DNMT1 on semi-methylated DNA by upregulating p21 (Iida et al., 2010). Third, they can also inhibit the activity of SAH hydrolase, a key enzyme for methyl donor recycling, thus decreasing the level of both DNA and histone methylation (Wyczechowska and Fabianowska-Majewska, 2003). It is worth noting that fludarabine as a single drug has no effect on the methylation of histone H3 in AML cells, but can promote the methylation of multiple sites of histone H3 when combined with clofarabine, which is a very interesting question and deserves further study (Valdez et al., 2011).
Cladribine is a nucleoside analog of deoxyadenosine. A chlorine substitution in the purine ring protects cladribine against degradation by adenosine deaminase. Cladribine is currently used for the treatment of multiple sclerosis (MS). The recommended dose is 3.5 mg/kg, consisting of two annual courses of treatment, each comprising two treatment weeks every other month. There were 1,326 patients with MS who received either placebo or a cumulative dose of cladribine of 3.5 or 5.25 mg/kg over the 2-year study period for two courses of treatment. Results showed 86% relative reduction in the mean number of T1 Gd + lesions, 73% relative reduction in the mean number of active T2 lesions, and 74% relative reduction in the mean number of combined unique lesions per patient per scan. As shown in Figure 4, patients with MS were treated with a single dose of 10 mg cladribine tablets by month. Cladribine was rapidly absorbed. Cladribine has good oral bioavailability of about 40%, and it has the potential to penetrate the blood–brain barrier. Cladribine is phosphorylated to cladribine monophosphate (Cd-AMP) by deoxycytidine kinase in cells. Cd-AMP is further phosphorylated to chlorodeoxyadenosine diphosphate (Cd-ADP), which is in turn phosphorylated by 5-nucleotidase to Cd-ATP. Cladribine elimination is equally dependent on renal and non-renal routes (Merck Serono Europe Limited, 2017).
Fludarabine is a fluorinated derivative of adenosine, which can prevent the deamination of adenosine deaminase. Patients with B-cell CML received 10 mg tablets of fludarabine to a dose of 40 mg/m2/d for 5 days. The OR was 71.6% (CR, 37.0%; PR, 34.6%). The median time to progression was 841 days. The grade 3/4 toxicity was myelosuppression (Rossi et al., 2004). In another study, 27 patients with non-Hodgkin’s lymphoma and B-cell CML received fludarabine orally and intravenously. As shown in Figure 4, fludarabine was rapidly absorbed within 1 h of oral administration. AUC0–24h increased in a linear proportion to the oral dose, which is the same as intravenous administration. Bioavailability and time to Cmax were dose-independent. There was no significant difference in the PK between oral fludarabine with or without food. Taken together, oral doses of fludarabine can achieve an AUC0–24h of 2-fluoro-arabinofuranosyl-adenine similar to intravenous administration with dose-independent bioavailability. Fludarabine is largely eliminated by renal excretion.
Procaine (12) has been used as an anesthetic for years, and it was not known that procainamide could inhibit DNA methylation until 1988 (Cornacchia et al., 1988). Inspired by this, researchers turned to procaine, which has a similar structure to procainamide. Procaine (10 nM) inhibited the proliferation of MCF-7 breast cancer cells and reduced the methylation of TSG RARβ by 40% (Villar-Garea et al., 2003). Studies showed that procaine reduced the affinity between DNMT1 and semi-methylated DNA or DNMT1 and SAM by binding to the CpG of DNA at a high concentration of 100–500 μM (Lee et al., 2005). Procaine is easily absorbed from the injection site and distributed to systemic tissues. It can pass the blood–brain barrier and placenta and also can be hydrolyzed by esterase in tissues and plasma. Most of the hydrolyzed products are excreted through urine and a small amount is metabolized by the liver. These data indicate that procaine is a drug with limited distribution and tissue uptake, as well as a short duration of action. As an infiltration anesthetic, 0.25%–0.5% solution can be used in the department of stomatology, no more than 1 g at a time.
Epgallocatechin gallate (EGCG, 13) is a polyphenolic compound extracted from green tea. Studies showed that EGCG inhibited the methylation of WIF-1 in lung cancer cells (Gao et al., 2009) and GSTP1 in prostate cancer cells (Pandey et al., 2010). In addition, EGCG could induce a decrease in the global DNA methylation level in the human epidermoid carcinoma A431 cell by reducing the mRNA and protein levels of DNMT1, DNMT3a, and DNMT3b, especially for TSGs, p16INK4a, and Cip1/p21 (Nandakumar et al., 2011). The mechanism of DNA methylation inhibition is manifested in the following two aspects. First, EGCG inhibited DNMT1 by binding to the catalytic site of DNMT1 non-covalently with a Ki of 6.89 μM (Fang et al., 2007). Second, catechol-O-methyltransferase introduces the methyl of SAM to the catecholamine of EGCG, which leads to depletion of SAM available as a methyl donor, thereby causing an indirect reduction in DNMT-mediated DNA methylation (Hong et al., 2003). In addition to inhibiting DNA methylation, EGCG can also exert anti-tumor activity by regulating the methylation and acetylation of histones. For example, EGCG could promote the transcription of human telomerase reverse-transcriptase (hTERT) by decreasing the acetylation of lysine on histone H3 and H4 in breast cancer cells (Meeran et al., 2011), while increasing the acetylation of lysine 9/14 on histone H3 and lysine 5/12/16 on histone H4 in skin cancer A431 cells (Nandakumar et al., 2011). In addition, EGCG could also inhibit the tri-methylation of lysine 27 on histone H3 and increase the acetylation of lysine 9/18 on histone H3 at the TIMP-3 promoter in prostate cancer cells (Deb et al., 2019).
A total of 40 volunteers received a formulation of 800 mg EGCG once daily, a formulation of 400 mg EGCG twice daily, or placebo once daily. There was no significant difference in the PK between the two doses, while Cmax and AUC increased in a dose-dependent pattern. As shown in Figure 4, EGCG was absorbed slowly and distributed widely in the body, which was first absorbed in the intestine. However, the bioavailability is limited due to its oxidation and metabolism (Chow et al., 2003).
Hydralazine (14) is a smooth muscle relaxant used to treat hypertension. Inspired by the ability to induce reduced DNA methylation in systemic lupus erythematous cells, hydralazine was identified as a DNMTi. A study showed that hydralazine induced demethylation and re-expression of TSG p16 and RAR2 in human bladder cancer and APC in human cervical cancer HeLa and Caski cells (Leung et al., 2011). The mechanism of hypomethylation of hydralazine was controversial. First, docking results showed that hydralazine could bind to DNMT1 as a weak DNMTi. Second, hydralazine decreased the expression of human DNMT1 and DNMT3A. In addition, hydralazine may not only affect DNA methylation but can also be involved in the negative regulation of methylation of lysine 9 on histone H3. Studies have shown that hydralazine could inhibit the dimethylation of lysine 9 on histone H3 by inhibiting the activity of histone methyltransferase G9A in gemcitabine-resistance cervical cancer cells, thereby upregulating the expression of the human equilibrative nucleoside transporter 1 (hENT1) and reversing drug resistance. This has also been demonstrated in enzyme inhibition experiments; hydralazine at 2 μM and 10 µM strongly reduced the methylation of lysine 9 on histone H3 (Candelaria et al., 2012).
In a phase II clinical trial, 14 patients with progressive cutaneous T-cell lymphoma received hydralazine plus magnesium valproate. The OR was 71% (CR, 50%; PR, 21%). The median time to response was 2 months, the median duration of response was 28 months, and the median PFS was 36 months (Candelaria et al., 2007). Healthy volunteers were given an oral administration of 100 mg hydrazine. The oral bioavailability is 25%–55% and half-life is about 2–8 h. An obvious effect was noted 1 hour after oral administration and lasted for 24–30 h. It is mainly metabolized in the liver, mostly excreted in the form of acetylated and hydroxylated metabolites, of which 3%–14% is excreted through urine in its original form (Ludden, 1982).
Genistein (15) and equol (16) are flavonoid compounds extracted from legumes. Equol induced the promoter hypomethylation of TSGs BRCA1 and BRCA2 in breast cancer cells (Bosviel et al., 2012). Genistein can reverse the hypermethylation and re-expression of the P16, RARβ2, MGMT, PTEN, and CYLD genes in esophageal and prostate cancer cells (Fang et al., 2005). Enzyme activity results showed that genistein could inhibit the activity of DNMT1 and DNMT3 (Majid et al., 2010). In addition, genistein could also regulate the epigenetics of histones. A study showed that genistein could significantly reduce the methylation of lysine 9 on histone H3 and decrease both the transcriptional and translational levels of HDAC2 in a patient-derived xenograft (PDX) orthotopic mouse TNBC model (Sharma et al., 2021). Equol, like genistein, could significantly reduce the methylation of lysine 4/27 on histone H3 in MCF-7 and MDA-MB 231 breast cancer cells, while significantly increasing the acetylation of lysine 4/8 on histone H3 (Dagdemir et al., 2013).
In a phase I/II study, 13 patients with metastatic colorectal cancer were treated with genistein plus FOLFOX. The adverse events related to genistein alone were headaches, nausea, and hot flashes. The OR and PFS were 61.5% and 11.5 months, respectively. As shown in Figure 4, 40 volunteers received a dose of 30, 60, 150, or 300 mg genistein orally. Genistein was rapidly absorbed and showed nearly dose-linear PK characteristics. The plasma concentration–time profiles showed a rapid one-peak course until Tmax, followed by a multiphasic decrease, which probably reflected a distribution and elimination phase. Genistein was safe and well-tolerated in the dose range of 30–300 mg. ADME studies revealed that intestinal, biliary, and renal excretions are the excretion pathways for genistein (Ullmann et al., 2005).
Equol has been shown to reduce the risk of prostate cancer in men and breast cancer in women. The incidence of breast cancer in 159 patients was reduced by 20% after intervention with soy isoflavones. The PK of equol was assessed in 14 volunteers by oral administration of 20 mg. The plasma concentration reached the maximum within 1–2 h and decreased rapidly in the first 6 h, showing a unimodal distribution. The half-life is 7–8 h. 61% of the dose was excreted through urine in 72 h. The high bioavailability of equol suggests that low doses of equol taken twice daily may be sufficient (Setchell et al., 2009).
Curcumin (17) is a kind of diketone compound extracted from turmeric, which has anti-inflammatory, anti-oxidant, anti-tumor, anti-angiogenesis, anti-bacterial, anti-viral, anti-fibrotic, anti-depressant, anti-ischemic, immunomodulatory, and neuroprotective properties. The anti-tumor activity of curcumin is mainly attributed to its anti-inflammatory and anti-oxidant properties. Curcumin has also been reported to exert anti-tumor activity by modulating growth factors, enzymes, transcription factors, and kinase- and apoptosis-related proteins (Giordano and Tommonaro, 2019). In addition, the anti-tumor activity of curcumin is also associated with epigenetic modifications, primarily DNA methylation. Curcumin was identified as a DNMTi through virtual screening. Molecular docking results suggested that curcumin blocked the catalytic thiolate of C1226 of DNMT1 covalently, which was validated by the fact that curcumin inhibited the activity of DNMT (M. SssI) with an IC50 of 30 nM (Liu et al., 2009a). Curcumin can inhibit the DNA methylation of RAR2 and FANCF in cervical cancer cells in vitro (Parashar et al., 2012). A 70% decrease in tumor growth in a xenografted MV4-11 model was achieved when the mice were treated with 100 mg/kg curcumin in vivo. In addition to inhibiting DNA methylation, curcumin could also inhibit the methylation of histones. For example, curcumin inhibits the methylation of lysine 4/9/27 on histone H3 in leukemia cells by inhibiting histone lysine methyltransferase (HKMT) and enhancing the activity of histone lysine demethylase (HKDMT) with consequent changes in gene expression that could contribute to its antitumor effect. Interestingly, curcumin also reduced the methylation of arginine 42 and glutamine 19/55 on histone H3 (Sawesi et al., 2022).
In a phase I clinical trial, 32 patients with metastatic cancer were treated with liposomal curcumin infusions at doses between 100 and 300 mg/m2. Of 15 patients under tumor assessment, 14 showed progressive disease, one showed stable disease, and two had significant tumor marker responses. No DLT was observed in 26 patients at doses between 100 and 300 mg/m2 over 8 h. The adverse events were facial edema, anemia, and hemolysis. PK analyses showed that there is a stable plasma concentration of total curcumin during infusion and a rapid decline after the infusion as a consequence of redistribution and extensive metabolism. Plasma concentrations of curcumin showed an apparent linear dependence on the infusion rate. Curcumin has low oral bioavailability in humans and it may undergo intestinal metabolism (Sharma et al., 2001).
Disulfiram (18), a clinical drug for the treatment of alcohol abuse, was found to be a DNMTi by chance. Disulfiram has a sulfhydryl group, which can attack the sulfhydryl of DMNT cysteine to inhibit the activity of DMNT. A study showed disulfiram could inhibit DMNT1 and induce the expression of TSG, such as, p16, and RARβ2 (Lin et al., 2011a). In addition, disulfiram could also decrease the dimethylation and trimethylation of lysine 4 on histone H3 by inducing the degradation of histone methyltransferases MLL1 and MLL2 in pediatric glioma cells (Meier et al., 2021).
Disulfiram is currently in phase II clinical trials for cancer treatment. The efficacy in the treatment of prostate cancer was tested in 19 patients; two cohorts received disulfiram 250 mg and 500 mg daily, respectively (Schweizer et al., 2013). Changes in the global 5meC content were observed in 2 of 9 patients (22.2%) in cohort 1 and 3 of 10 (30.0%) in cohort 2. Six patients developed grade 3 AEs. As shown in Figure 4, the half-life was 7.3 h and the average peak time was 8–10 h when the dose was 250 mg. However, the plasma concentrations varied among individuals, which might be caused by the strong lipid solubility and the hepatointestinal circulation. Disulfiram is absorbed primarily from the gastrointestinal tract and is quickly reduced by glutathione in red blood cells to its monomer. Most of the metabolites are excreted through urine, while carbon disulfide is excreted through the respiratory system.
Resveratrol (19) is an anthraquinone terpenoid found in peanuts with cardiovascular protection effect. The demethylation activity of resveratrol has been shown to be lower than that of EGCG, while it has synergistic effects in combination with cladribine or fludarabine. The mechanism of demethylation lies in the fact that it can reduce the expression of DNMT1 and inhibit the acetylation of STAT3, which can destroy the interaction between acetylated STAT3 and DNMT, thus inhibiting CpG methylation (Lee et al., 2012). Resveratrol can also catalyze post-translational modifications of histones. A study showed that resveratrol decreased the dimethylation of arginine 3 on histone H4 and the trimethylation of lysine 27 on histone H3 by decreasing the expression of arginine methyltransferase 5 in breast cancer cells. It could also enhance the acetylation of lysine 9/27 on histone H3 by inhibiting the activity of lysine deacetylase. These epigenetic modifications lead the expression of genes BRCA1, p53, and p21 and inhibit the growth of breast cancer cells (Chatterjee et al., 2019).
A total of 24 patients with relapsed or refractory multiple myeloma were enrolled in a phase II clinical trial. Nine patients had SD following a minimum of two cycles, and nine patients received a resveratrol and bortezomib combination, achieving an ORR (≥MR) of 22% by a modified intention-to-treat (ITT) population, or an ORR of 8% by ITT analysis. The median time to progression was 2.8 months and overall survival was not reached. Adverse events were nausea, diarrhea, and vomiting. A total of 10 volunteers received a dose of 0.5, 1, 2.5, or 5 g resveratrol orally (Boocock et al., 2007). As shown in Figure 4, resveratrol was rapidly absorbed, reaching a maximum plasma concentration of 0.83–1.5 h, followed by a sharp decline, which was distributed in the organs with abundant blood perfusion. Plasma concentrations showed a “bimodal shape” after oral administration, possibly due to enterohepatic recirculation. AUC and Cmax for resveratrol increased with dose. Resveratrol has low bioavailability, which was consistent with the whole body clearance (2,235–4,930 L/h) and volume of distribution (9,198–22,226 L). Resveratrol was metabolized to glucuronylated and sulfated resveratrol rapidly.
Caffeic acid (20) is a phenolic acid compound derived from honeysuckle, which is often used in the prevention and treatment of bleeding. As a weak DNMTi, it could inhibit DNMT from Spiroplasma sp. strain MQ1 (M Sssl) and human DNMT1 with an IC50 of 3.0 mM and 2.3 mM, respectively. Further study showed that caffeic acid inhibited DNA methylation in a non-competitive way. Caffeic acid, as a substrate of catechol-O-methyltransferase, is methylated to SAH, which further inhibits DNMT1 (Zhu and Zhu, 2006). Dilshara et al. (2016) showed that 50 μM of caffeic acid increased the apoptosis of hepatocarcinoma Hep3B cells in vitro. Caffeic acid (100 mg/kg) can reduce the changes in markers of liver injury in hepatocarcinoma in rats. In addition, caffeic acid could also inhibit the activity of HDAC with an IC50 of 2.54 mM. Its activity is not as potent as we expected, whereas its derivatives such as curcumin and chlorogenic acid have potent activity for HDAC inhibition, which suggests that caffeic acid may be considered a promising lead structure for HDAC inhibitors that have the potential to provide new therapeutics for cancer (Bora-Tatar et al., 2009).
Caffeic acid is in phase II clinical trials for cancer treatment. As shown in Figure 4, the maximum plasma concentration of caffeic acid was observed 1 h after ingestion of food and then rapidly decreased. Only a small amount of caffeic acid is absorbed in the stomach, while 95% absorbed in the colon by the intestinal mucosa (de Oliveira and Markowicz Bastos, 2011). The absorbed caffeic acid converts to its metabolites through methylation, sulfation, and gluconic acid, which are excreted through urine finally.
Although three nucleoside DNMTis azacitidine, decitabine, and clofarabine are already on the market, they are chemically and metabolically unstable with low bioavailability. Thus, a large number of stable second-generation nucleoside DNMTis were developed, which include azacitidine derivatives CP-4200 and zebularine, decitabine derivatives NPEOC-DAC, and thio-cytidine derivatives T-dCyd and 5-aza-t-dCyd.
CP-4200 (21) is an elaidic acid ester of azacitidine that was synthesized with the intention of rendering drug uptake less dependent on the conventional nucleoside transport systems and improving the cellular uptake. As the trans-octadecenoate of azacitidine, the cellular uptake was substantially less dependent on the nucleoside transporters, and it has better membrane permeability. CP-4200 (Figure 5) has better anti-tumor activity than azacitidine in an orthotropic mouse AML model in vivo (Brueckner et al., 2010). As a prodrug of azacitidine, CP-4200 has the advantage of delayed delivery of azacitidine. It could inhibit the methylation of the whole genome and specific genes in human colon cancer HCT116 cell and breast cancer MCF7 cell effectively, which is dependent on the activity of DNMT1 degradation (Brueckner et al., 2010). CP-4200 is currently in the preclinical stage for cancer treatment.
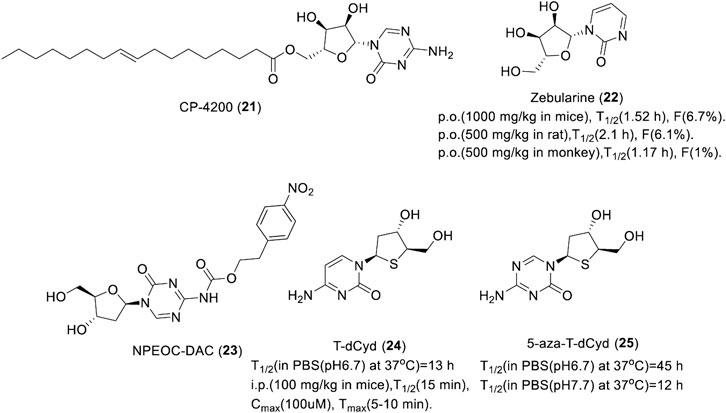
FIGURE 5. Chemical structure of nucleoside DNMTis in preclinical.
Zebularine (22), an azacitidine derivative lacking an amino at the 4th position of the pyrimidine ring, exerts demethylation activity by stabilizing the binding of DNMT to DNA. A study showed that it could induce hypomethylation in the promoter of TSG p57 and p15 in myeloid leukemia cells in vitro (Nakamura et al., 2015). DNMT1 was almost completely suppressed in tumor cells by zebularine, indicating it has high selectivity toward DNMT1. Zebularine could lead the promoter demethylation of p15INK4B and induce p15INK4B expression in AML194 cells by maintaining the high acetylation level of histone H4 and increased the trimethylation of lysine 4 on histone H3 when combined with trans-retinoic acid or phenyl sodium butyrate but had no effect on histone modification, especially histone methylation, when used as a single drug (Scott et al., 2007). As show in Figure 5, Zebularine could be continuously administered to maintain the demethylated state due to its low toxicity. It also works when given orally. However, due to saturation of the absorption process and primary metabolism of the liver, zebularine has limited oral bioavailability. Plasma concentrations declined with a short half-life in mice, rats, and monkeys, which indicates that frequent dosing or continuous i.v. infusion may be necessary to maintain the prolonged inhibition of DNMT.
NPEOC-DAC (23, Figure 5) is a decitabine derivative formed with the introduction of a 2′-deoxy-N4-[2-(4-nitrophenyl)ethoxycarbonyl] (NPEOC) group in the 4-amino of decitabine, which could be cleaved by carboxyl esterase to release decitabine (University of Southern California, 2008). NPEOC-DAC inhibits global DNA methylation by the induction of hypomethylation of the LINE-1 repetitive element and it reverses the hypermethylation of TSG ID4 in tumor cells. The ability to inhibit the DNA methylation of NPEOC-DAC is specific to HepG2 and Hep3B2 liver cancer cell lines, which is dependent on the activity of carboxyl esterase 1 (Byun et al., 2008). NPEOC-DAC was 23-fold less efficient with a 3-day delay at inhibiting DNA methylation than decitabine, which suggested that NPEOC-DAC is capable of releasing decitabine slowly to maintain DNA hypomethylation by continuous administration. In addition, NPEOC-DAC has very low solubility in water, which could potentially lead to very favorable human PK compared to decitabine.
4′-Thio-2′-deoxycytidine (T-dCyd, 24) and 5-aza-4′-thio-2′-deoxycytidine (aza-T-dCyd, 25) are thiocytidine derivatives developed by the Southern Research Institute of the United States (Southern Research Institute, 2009). Studies showed that T-dCyd and aza-T-dCyd could deplete DNMT1 in cancer cells resulting from the re-expression of TSG p15. Both compounds depleted DNMT1 and inhibited tumor growth in human tumor xenograft models. Specifically, CCRF-CEM and KG1a leukemia cells were sensitive to both of them with an IC50 below 1 μM, while aza-T-dCyd was 3–15-fold more potent than T-dCyd. Aza-T-dCyd showed potential cytotoxicity toward NCI-H23 lung cancer cells, HCT-116 colon cancer cells, and IGROV-1 ovarian cancer cells with an IC50 of 4.5, 58, and 36 μM, respectively, while T-dCyd inhibited the proliferation of these cells at 100 μM by less than 50% (Thottassery et al., 2014). The selectivity of aza-T-dCyd was at least 10-fold more potent than that of decitabine. In addition, T-dCyd can incorporate into a DNA sequence with a high level and the presence of the thio-deoxyribose appears to increase the chemical stability of 5-aza-T-dCyd when incubated in PBS solution. The half-life of T-dCyd and aza-T-dCyd was 13 ± 1 h and 45 ± 6 h in PBS (pH 6.7) at 37°C (Figure 5), respectively.
RG108 (26) is the first DNMT1 inhibitor discovered by rational drug design, which inhibits DNMT1 by blocking the active site of DNMT1 with an IC50 of 118 nM (Stresemann and Lyko, 2008). In a patent, 22 of 66 RG108 derivatives could inhibit DNA methylation with an IC50 of 50 μM or less (Cancer Research Technology Ltd, 2007). The modification of histones in tumor cells by RG108 is the same as that of zebularine.
As show in Figure 6, RG108 was slowly absorbed within 30 min of subcutaneous injection in rats and was widely distributed in various tissues, such as the liver, heart, and muscle. It takes about 4 h after absorption for the plasma concentration to decline by half, which indicates that RG108 has a long plasma half-life. In another study, researchers synthesized a large number of indole derivatives and evaluated the inhibitory activity against DNMT1 using tritium-labeled AdoMet (3H-SAM) as a methyl donor. Results showed that 17 of 27 indole derivatives could inhibit DNMT1 with an IC50 of 50 μM or less. Compound 1 (27) had the most potent inhibitory activity with an IC50 of 3.53 μM. Interestingly, compound 2 (28) exerted a more potent inhibition activity in CCFR-CEM hematologic cells and Jurkat over other cancer cell lines with an EC50 of 6.5 μM and 8.5 μM, respectively. Compound 3 (29) displayed a promising inhibitory effect in various cancer cells, representing a potential lead for further optimization (Ikerchem S L, 2014).
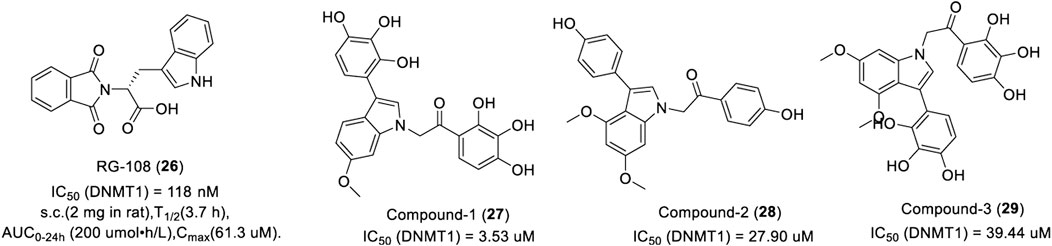
FIGURE 6. Chemical structure of indole-based DNMTis in preclinical.
Based on the crystal structure of mouse DNMT1 in complex with SAH and cytosine (PDB: 4DA4), a new carbazole DNMTi DC-05 (30) was found by a structure-based drug design. DC-05 is a selective DNMT1 inhibitor with an IC50 and Kd of 10.3 μM and 1.09 μM (Figure 7), respectively, which showed poor activities against DNMT3A/3B (IC50 > 200 μM). Due to the high IC50 and good selectivity of compound DC-05 to DNMT1, many DC-05 analogs were searched based on the structure of DC-05. DC-501 (IC50 = 2.5 µM, 31) and DC-517 (IC50 = 1.7 µM, 32) displayed 4.1- and 6.0-fold higher inhibitory activities against DNMT1 than that of DC-05 (Figure 7). The Kd of DC-517 is 0.91 μM, which suggested that DC-517 had a higher affinity for DNMT1. Further studies showed that DC-05, DC-501, and DC-517 can inhibit the proliferation of HCT116 and Capan-1 at a low micromolar concentration (Chen et al., 2014). The SAR analysis and predicted binding model study showed a better occupation of the cytosine and SAM pockets, and the formation of ionic interactions or hydrogen bonds linking the two pockets is necessary for the activity of carbazole DNMTi.

FIGURE 7. Chemical structure of carbazole-based DNMTis in pre-clinical.
SGI-1027 (33) is a quinazoline DNMTi identified by SuperGen from 4-aniline quinazoline derivants, which showed potent activities against DNMT1, DNMT3A, and DNMT3B by a SAM-competitive mechanism with an IC50 of 12.5 μM, 8.0 μM, and 7.5 μM, respectively (Figure 8) (Datta et al., 2009). In addition, SGI-1027 interacted with DNA and resulted in SAM-noncompetitive but DNA-competitive inhibition. Treatment of human colon cancer RKO cells with SGI-1027 led to the re-expression of silenced TSG and the degradation of DNMT1. Furthermore, researchers prepared a series of regioisomers of SGI-1027 by shifting each fragment linkage from the para to the meta or ortho position, of which a novel quinolone DNMTi compound 4 (34) was identified (Figure 8). It was more potent than SGI-1027 and more selective toward other SAM-dependent methyltransferases with less toxicity (Valente et al., 2014).
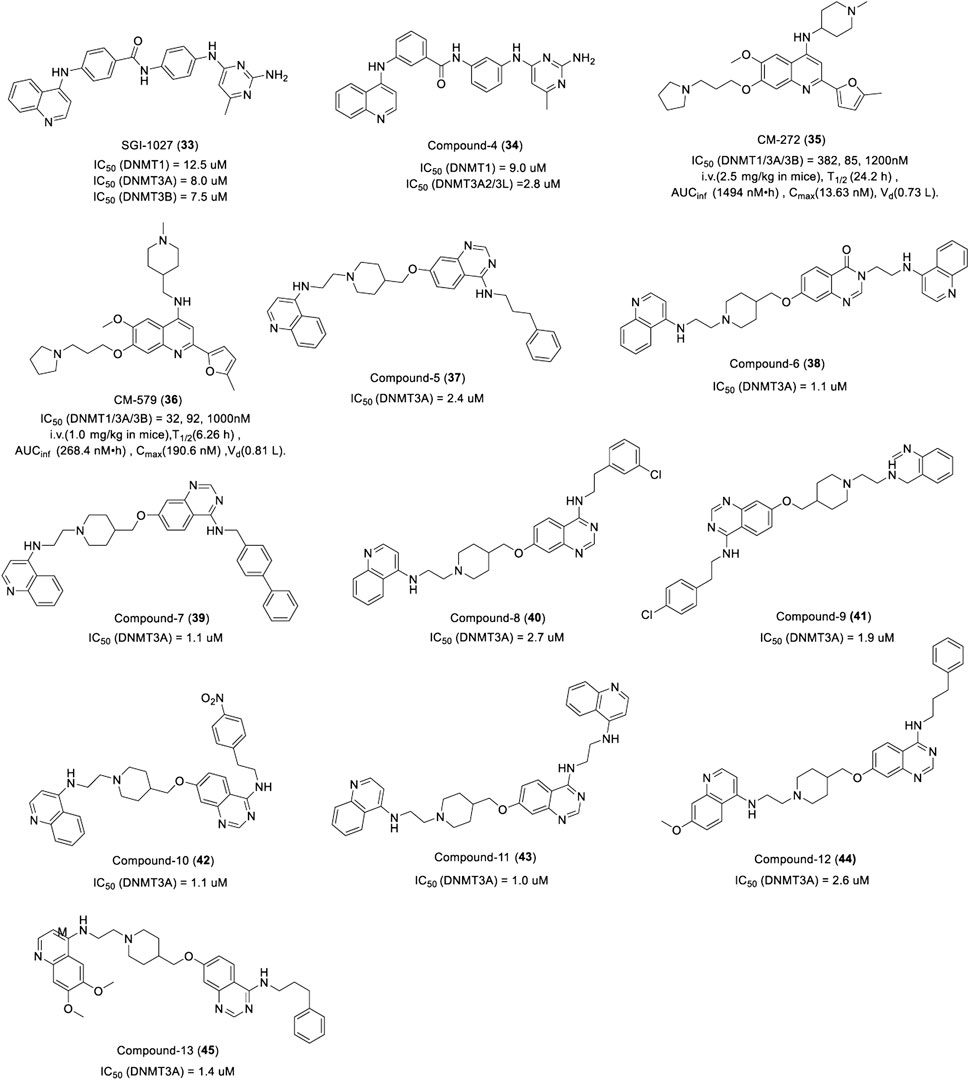
FIGURE 8. Chemical structure of quinolone- and quinazoline-based DNMTis in pre-clinical.
CM-272 (35) and CM-579 (36) are other dual-target quinolone inhibitors targeting G9a and DNMT based on a rational drug design. CM-272 inhibits G9a, DNMT1, DNMT3A, and DNMT3B with an IC50 of 8 nM, 382 nM, 85 nM, and 1,200 nM, respectively (Figure 8), and promotes the apoptosis of various tumor cells in vitro. As an inhibitor of histone methyltransferase G9a, studies have shown that CM-272 can effectively reduce the di-methylation level of lysine 9 on histone H3 in hepatoma cells by inhibiting G9a and downregulating G9a expression (Bárcena-Varela et al., 2019). CM-272 prolongs the survival of CEMO-1 cell xenogeneic models at a dose of 2.5 mg/kg. CM-579 showed more potent inhibitory activity against G9a, DNMT1, DNMT3A, and DNMT3B with an IC50 of 16 nM, 32 nM, 92 nM, and 1,000 nM (Figure 8), respectively, which has a high affinity for DNMT1 with a Kd of 1.5 nM. CM-272 and CM-579 have long plasma half-lives, among which the plasma half-life of CM-272 is four times than that of CM-579 (San José-Enériz et al., 2017). As show in Figure 8, Unfortunately, the distribution of both becomes narrow after absorption. Further studies found that the two compounds did not compete with SAM but inhibited the binding of G9a and DNMTs to their substrates reversibly, which may explain their selectivity and therapeutic potential in different tumors.
Quinazoline DNMTi was found in a series of quinazoline derivatives synthesized by Pierre Fabre Medicament. Among the 36 quinazoline derivatives, compounds 5, 6, 7, 8,9,10, 11, 12, and 13 (37–45) showed potent inhibitory activity against DNMT3A with an IC50 of 2.4, 1.1, 1.1, 2.7, 1.9, 1.1, 1.0, 2.6, and 1.4 μM, respectively (Figure 8). It is worth noting that compounds 6, 9, and 11 also had potent inhibitory activity against DNMT1 with an inhibitory rate of 100%, 75%, and 99% at 32 μM, respectively. Further study showed that compound 6 could inhibit the proliferation of human leukemia cell KG-1, lymphoma cell Karmas 299 with an EC50 of 0.5 and 1.8 μM (Figure 10), respectively. Compounds 6 and 7 could inhibit the proliferation of KG-1 with an EC50 of 1.3 and 0.4 μM, respectively (Pierre Fabre Medicament; Centre National de la Recherche Scientifique (CNRS), 2015).
Psammaplin A (PsA, 46) is a marine natural bromotyrosine compound extracted from the sponge Psammaplinaplysilla, which has inhibitory activity against HDAC and DNMT1 with an IC50 of 4.2 and 18.6 nM, respectively (Figure 9) (Piña et al., 2003). After intravenous administration of PsA in mice, the plasma concentration of PsA decreased rapidly with a plasma half-life of about 10 min. PsA was found to be highly distributed in lung tissues. The high and specific lung-targeting characteristics indicate that PsA has the potential to be developed as a lung cancer treatment agent. Psammaplin G (47), another bromotyrosine compound isolated from Pseudoceratina purpurea, exhibited inhibitory activity against DNMT1 with an IC50 of 12.8 nM (Figure 9) (Piña et al., 2003). UVI5008 (48), a derivative of Psammaplin A, could inhibit the DNA methylation of TSG p16INK4a and RARβ2 (García et al., 2011), which showed strong anti-tumor activity with an IC50 from 0.2 to 3.1 µM. UVI5008 (Figure 9), a direct inhibitor of DNMT3a, has poor activity against DNMT1. To the best of our knowledge, UVI5008 is the first DNMT3a-selective inhibitor. In conclusion, UVI5008 was an epigenetic modifier that inhibited HDAC and DNMT, which efficiently induced the selective death of cancer cells and exerted its activity in a genetic mouse breast cancer model and several human tumor xenografts. As potent DNMT and HDAC inhibitors, Psammaplin A/G and UVI5008 can effectively inhibit DNA methylation and induce hyperacetylation of histone H3. Isofistularin-3 (49), a brominated alkaloid derived from aplysina aerophoba, was found to be a DNMTi due to its structural resemblance to psammaplin A. Isofistularin-3 could inhibit the activity of DNMT1 with an IC50 of 13.5 µM in a DNA-competitive manner (Figure 9). In addition, isofistularin-3 could modify the methylation of the AHR promoter in Raji cells and induce growth arrest of cancer cells in G0/G1 concomitant with increased p21 and p27 expression and reduced cycling E1, PCNA, and c-myc levels (Florean et al., 2016). These results suggest that bromotyrosine derivatives are potential DNMTis. However, a recent study found that Psammaplin A had no inhibitory activity against DNMT (García et al., 2011), and it was further confirmed that histone deacetylases are the main target of Psammaplin and not DNMT (Baud et al., 2012). The inhibitory activity and mechanism of these compounds on DNMT need to be further studied.
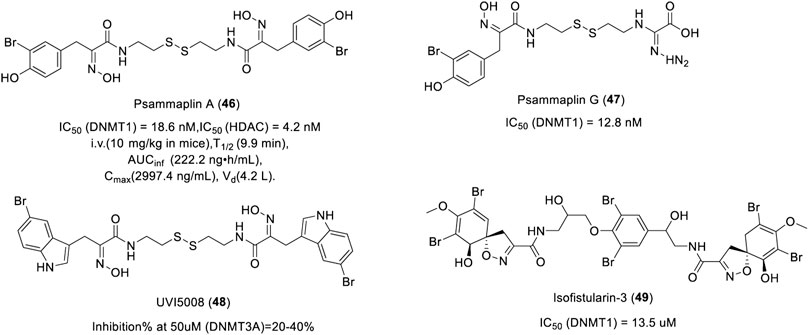
FIGURE 9. Chemical structure of bromotyrosine-based DNMTis in pre-clinical.
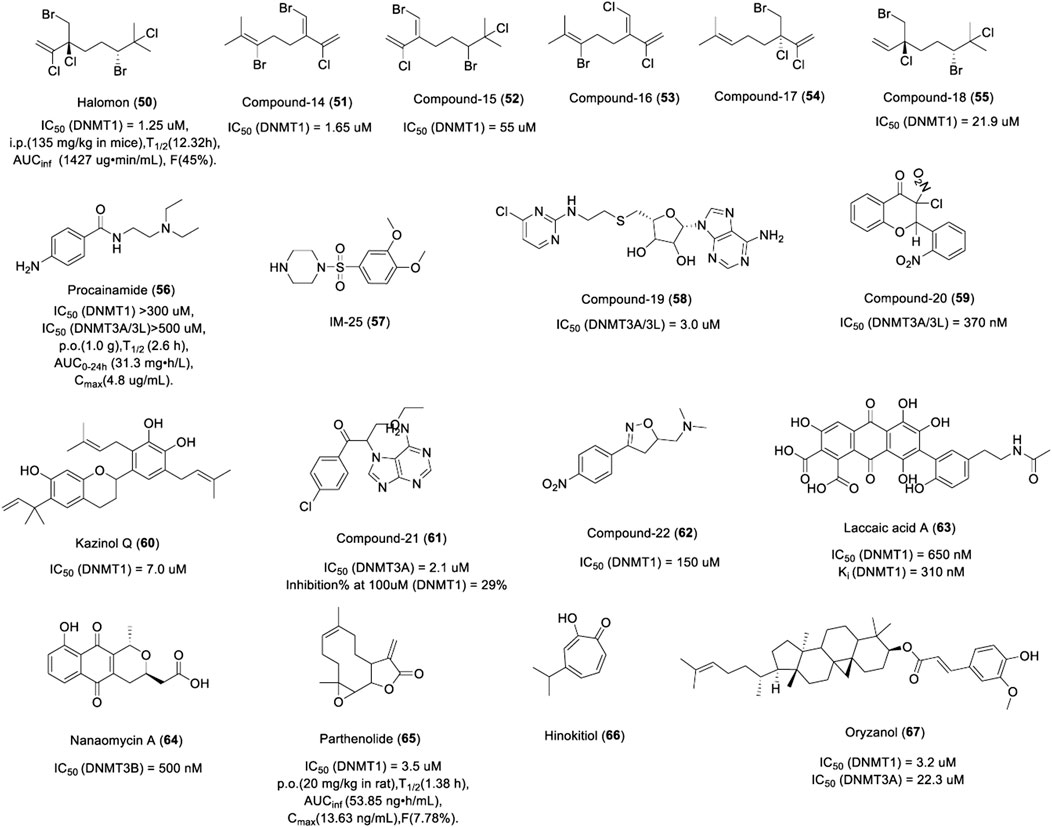
FIGURE 10. Chemical structure of other DNMTis in pre-clinical.
Halogenated monoterpenes, a class of marine products isolated from Madagascar red marine alga Portieria hornemannii, have been described as potential DNMTis (Andrianasolo et al., 2006). Previous studies have shown that halomon (50) and compound 14 (51) have comparable activities against DNMT1 (1.25 µM and 1.65 µM, respectively), while compounds 15 (52) and 18 (55) showed weaker inhibitory activity against DNMT1 with an IC50 of 55 µM and 21.9 µM (Figure 10), respectively. Compounds 16 (53) and 17 (54) were not tested due to the small quantities isolated. Halomon has a long half-life in mice. As show in Figure 10, The bioavailability of halomon was 45% and 47% after i.p and s.c administration, respectively. Urinary excretion of the parent compound was minimal. Halomon was distributed widely in all tissues.
Procaine, procainamide, and its derivant IM25 are aminobenzoic acid DNMTis (Figure 10). Procaine has entered the clinical stage of anti-tumor research while procainamide and IM25 are still in the preclinical research stage for tumor treatment. Procainamide (56) is a common antiarrhythmic drug, but studies have found that it also has the activity of inhibiting DNMT (Scheinbart et al., 1991). Studies showed that procainamide demethylated and re-expressed the RARβ2 and p16 genes in urinary bladder carcinoma and breast cancer cells (Cancer Research Technology Ltd, 2007) and the WIF-1 gene in lung cancer cells (Procaine et al., 2009). Further study showed that procainamide inhibited the activity of DNMT1 but not DNMT3A/3B (Chatterjee et al., 2019), suggesting that procainamide may be a selective inhibitor of DNMT1. Procainamide is almost completely absorbed after oral administration and has a long half-life; 50% of procainamide is metabolized to N-acetyl procainamide and excreted through the kidneys. IM-25 (57) is a novel amino benzoic acid DNMTi screened from 169 procainamide derivants by the Ohio State University, which caused the reversal of promoter hyper methylation of GSTP1 and TRIP10 genes in MCF7 breast cancer cells and induced global hypomethylation and hypomethylated more loci than procainamide and 5-aza-2′-deoxycytidine (Lin et al., 2011b). Procainamide and IM25 inhibited the activity of DNMT1 by reducing the affinity for its two substrates: SAM and hemi-methylated DNA, which is consistent with the inhibition mechanism of procaine. Although amino benzoic acids have been shown to be demethylating in a variety of tumor cells, their side effects limit their application in anti-tumor therapy.
Heteroaryl compounds are a new type of DNMTis produced by GlaxoSmithKline by imitating the structure of methyl donor SAM or SAH. These heteroaryl compounds contain a functional group, which could coordinate easily into the DNA binding region, particularly the cytosine binding pocket. Such small molecules can inhibit DNMT activity via binding in a covalent or non-covalent manner and, in turn, inhibit the enzymes. Of the five heteroaryl compounds synthesized in the first batch, compound 19 (58) has the most potent inhibitory activity against DNMT3A/3L with an IC50 of 3 μM (Figure 10) (Isakovic et al., 2009). A total of 120 heteroaryl compounds, synthesized in the second batch, exhibited an IC50 value in a range of 19.1–300 μM against DNMT1 and seven of them exhibited an IC50 value of less than 100 μM against DNMT1. The IC50 of these compounds for DNMT3B/3L was as low as 0.025 μM and as high as over 300 μM. Also, 24 compounds exhibited an IC50 value of less than 1 μM against DNMT3B/3L (Glaxosmithkline Intellectual Property Development Limited, 2013).
Based on the scaffold of genistein, 36 of 114 flavones and flavanone compounds were found as DNMT3 inhibitors, which could inhibit the murine DNMT3A/3L complex with IC50 values ranging from 0.37–315 μM. The most potent compound 20 (59) possesses a chlorinator motif at the C3 position of the flavanone ring with an IC50 of 370 nM (Figure 10). The docking results showed that it exerted inhibitory activity in a SAM-competitive manner (INSERM, 2011). In another study, researchers screened out another flavonoid DNMTi “Kazinol Q” (60) from 12 natural products, which exhibited potent inhibitory activity against DNMT1 by the same mechanism as EGCG with an IC50 of 7 μM (Figure 10) (Identification of Kazinol et al., 2014).
Propiophenone is a novel DNMT3A inhibitor, which was obtained by high-throughput screening in France. The best inhibitor was compound 21 (61), which showed potent inhibitory activity against DNMT3A with an IC50 of 2.1 μM, but showed weak activity against DNMT1 (29% at 100 μM) (Figure 10). The mechanism of the compound was investigated as it forms a reactive Michael acceptor group in situ. Another study showed that propiophenone derivatives are possibly transformed into Michael acceptors, which most likely inhibit DNMT3A by forming a covalent bond with the catalytic cysteine of the enzyme (Erdmann et al., 2015). Isoxazoline and oxazoline DNMTis are constrained analogs of procaine/procainamide by constraining the N-alkyl amide moiety into a 4-substituted- or 5-substituted-oxazoline ring. Results showed that compound 22 (62), an oxazoline DNMTi, had potent inhibitory activity against DNMT1 (IC50 = 150 μM) (Figure 10) and also exhibited a dose-dependent anti-proliferative effect against HCT116 human colon carcinoma cells. The docking results showed that compound 7b had the same binding site as SAH, suggesting that it inhibited the activity of DNMT in a SAH-competitive manner (Castellano et al., 2011).
Laccaic acid A (63) is a natural product from the red scales of the insect Kerria lacca, which was found to be a new class of anthraquinone-based DNMTi. Laccaic acid A could bind to DNMT1 directly and exhibit 2.5-fold higher potency at DNMT1 inhibition activity than SGI-1027 with a Ki and IC50 of 310 and 650 nM (Figure 10), respectively. Notably, it also inhibited the activity of DNMT3. Further research showed that it is competitive with the DNA substrate in methylation assays and alters the expression of methylated genes in MCF-7 breast cancer cells synergistically with 5-aza-2-deoxycytidine. In conclusion, laccaic acid A is a DNMT1 inhibitor with a DNA-competitive manner, which suggests that specifically substituted anthraquinones may be a useful scaffold for DNMTis (Fagan et al., 2013).
Nanaomycin A (64) is an antibiotic that was recently reported to selectively inhibit DNMT3B with an IC50 of 500 nM, while the activity of DNMT1 was not affected (Figure 10). Nanaomycin A showed anti-proliferation activity in HCT116, A549, and HL60 tumor cell lines with an IC50 of 400, 4,100, and 800 nM by reducing the global methylation levels of the RASSF1A gene. Nanaomycin A does not degrade DNMT1 or DNMT3b, indicating that it is a non-covalent inhibitor different from the nucleoside DNMTi (Kuck et al., 2010). However, it is debatable whether the anthracycline group is a good candidate for clinical drug testing due to some cardiotoxicity issues (Horenstein et al., 2000). Nanaomycin A, the first DNMT3B-selective compound, provides a valuable biochemical tool and benchmark for future studies. The antibiotic mithramycin A (MMA) is a potential DNMT1 inhibitor, which could reduce the CpG island methylation of SLIT2 and TIMP-3 genes and could cause the degradation of DNMT1 in lung cancer cells. MMA is recognized as a CG-rich DNA-binding agent, implying that it might also have a similar inhibitory effect against DNMT as procaine (Lin et al., 2007).
Parthenolide (65), a sesquiterpene lactone of feverfew, could inhibit the activity of DNMT1 with an IC50 of 3.5 μM (Figure 10). The inhibitory activity of parthenolide against DNMT1 may be due to two reasons. Firstly, the electrophilic α-methylene γ-lactone of parthenolide could cause cysteine alkylation of DNMT1. Secondly, it could also down-regulate the expression of DNMT1 by blocking the binding of transcription factor Sp1 to the DNMT1 promoter. A study has shown that parthenolide reversed the methylation of the HIN1 gene and induced re-expression in MCF-7 breast cancer cells. It also induced global DNA hypomethylation in MV4-11 cells (Liu et al., 2009b). Parthenolide could be rapidly absorbed by rats and the plasma concentration decreased by half in about 1 h. The bioavailability was limited and was 7.78% in rats.
Ubiquitin-like plant homeodomain and RING finger domain 1 (UHRF1) is involved in the methylation of DNMTs (Liu et al., 2013). Hinokitiol (66), a component of essential oils extracted from Chymacyparis obtusa, induces DNA demethylation via downregulation of the expression of DNMT1 and UHRF1 as a novel DNMTi (Seo et al., 2017) (Figure 10). A study has shown that hinokitiol altered the methylation of 10 hypermethylated genes in colon cancer cells and reactivated the mRNA expression of O6-methylguanine DNMT, carbohydrate sulfotransferase 10, and B-cell translocation gene 4 (Hu et al., 2015). Interestingly, MGMT is a DNA repair enzyme responsible for demethylating O6-methylguanine in normal cells, which repairs the O6-alkylguanine lesions in DNA by transferring the alkyl groups from the O6-position of guanine to its own active center Cys145 residue and restores normal DNA. It can specifically repair the damage of O6 alkyl guanine in DNA sequences and maintain the accuracy and integrity of the genome (Pegg, 2011). However, for tumor cells, abnormally expressed MGMT specifically removes the lesions at the guanine O6 position, resulting in resistance of tumor cells to guanine O6-alkylating agents like TMZ (Gerson, 2002). In conclusion, the anti-tumor activity of hinokitiol is more like a double-edged sword. On one hand, it can exert anti-tumor activity by inhibiting abnormal DNA methylation; on the other hand, it can cause abnormal MGMT expression and lead to drug resistance toward alkylating agents in tumor cells. This suggests that hinokitiol should not be combined with alkylating agents for antitumor therapy in clinical practice, but should be combined with MGMT inhibitors, which could transfer the alkyl group to the active site Cys145 residue of MGMT and lead the inactivation of MGMT (Sun et al., 2018a). Unfortunately, only two MGMT inhibitors have entered clinical trials. It is urgent to develop new MGMT inhibitors for combination with DNMTis. In a study, researchers predicted the MGMT inhibitory activity of 134 base analogs with QSAR and found some purine analogs, such as O6-benzyl-8-bromo-guanine, O6-thenyl-guanine, and O6-(3-aminomethyl) benzyl-guanine which were potent MGMT inhibitors. In addition, the study also found that there are some substructures, such as 2-bromoprop-1-ene, 2-bromobuta-1,3-diene, thiophene, p-tolylmethanol, ≥2 saturated or aromatic heteroatom-containing ring size 6, E-2-nitroethenamine, ≥3 hetero-aromatic rings, p-xylene, and m-xylene, which are essential for the inhibitory activity of the MGMT inhibitor, which may play an important role in guiding the development of new MGMT inhibitors in the future (Sun et al., 2018b).
Rice-specific component γ-oryzanol (67), a mixture of ferulic acid ester and several phytosterols, is a potent DNMTi in the striatum of mice, which significantly inhibited the activity of DNMT1 (IC50 = 3.2 μM), DNMT 3A (IC50 = 22.3 μM), and DNMT 3B (maximum inhibition 57%) at least partly in an SAH-competitive manner (Figure 10). Furthermore, γ-oryzanol decreased the activity of DNMT1 as an antagonist against ERRγ, which regulates the production of DNMT. Oral administration of γ-oryzanol significantly decreased striatal DNA methylation in the promoter of the D2Rs in HFD-fed mice in vivo (Kozuka et al., 2017).
DNA methylation is an important epigenetic process that regulates gene expression. Abnormal DNA methylation leads to transcription inactivation and silent expression of tumor suppressor genes, which results in the occurrence and development of cancer. DNMTis can reverse the abnormal DNA methylation process to achieve the purpose of tumor treatment.
Currently, many DNMTis are under development, primarily including nucleoside and non-nucleoside DNMTis according to the chemical structure. Nucleoside DNMTi is a cytosine nucleoside analog that either directly integrates into DNA sequences in the form of triphosphate to inhibit DNA replication or binds to DNMT covalently to degrade DNMT. Since 2004, the representative first-generation nucleosides DNMTi, 5-azacitidine, clofarabine, and decitabine have been approved by the FDA for the treatment of MDS, CMML, and AML. However, the structure of 5-azacitidine is very unstable and easily hydrolyzed and deaminated. In order to improve the stability of 5-azacitidine and decitabine, a large number of second-generation cytidine analogs have been developed. Zebularine, obtained by removal of 4-amino from the pyrimidine of decitabine, was stable in neutral aqueous solution with a longer half-life. NPEOC-DAC, obtained by introducing the amino-protective group NPEOC, can infiltrate DNA and inhibit DNMTs after the hydrolysis of carboxyl esterase 1 in human hepatoma cells. SGI-110 can resist the deamination of cytidine deaminase with better PK and metabolic stability because of the introduction of a phosphodiester bond. The thiocytidine derivatives T-dCyd and 5-aza-T-dCyd are more stable due to the presence of thiodeoxyribose. 5-Fluoro-2′-deoxycytidine, obtained by the introduction of fluorine in the 5-position of the pyrimidine ring with increasing stability, has entered phase II clinical anti-tumor research. CP-4200, an oleic acid derivative of azacitidine, has better membrane permeability. In general, the chemical stability, metabolic stability, and water solubility of the second-generation nucleoside DNMTis have been greatly improved and some of them can even be taken orally. However, both the first and second generation nucleoside DNMTis are characterized by high toxicity, poor selectivity, and low bioavailability because most of them need to be integrated into DNA sequences to play a role.
In contrast, non-nucleoside DNMTi with various chemical scaffolds has attracted great attention (Medina-Franco et al., 2015). Many non-nucleoside DNMTis have been developed through various strategies. RG108 is the first DNMTi discovered through rational drug design, which can inhibit DNA methylation at low concentrations by blocking the active site of DNMT. Local anesthetic procaine and antiarrhythmic drug procainamide are amino benzoic acid DNMTis found based on drug reposition, which can block DNA methylation by binding the CpG island of DNA. Carbazole DNMTi DC-05 and DC-517 are obtained by virtual screening based on the crystal structure of the DNMT–DNMTi complex, which are the first selective inhibitors of DNMT1. In addition, a large number of DNMTis have been found in natural products. EGCG in green tea, genistein and equol in beans, curcumin in turmeric, resveratrol in peanuts, and caffeic acid in honeysuckle have entered clinical research for cancer treatment. Non-nucleoside DNMTis have a variety of chemical structures and different mechanisms to inhibit DNA methylation. Since they do not need to be integrated into DNA sequences, they have lower toxicity than nucleoside DNMTi. However, the non-nucleoside DNMTi has lower efficacy and poor selectivity than nucleoside DNMTis.
There is an urgent need to develop a new generation of DNMTi with high efficacy, selectivity, and bioavailability. However, unclear issues about DNMT and DNMTi hinder the development of selective DNMT1. First, it remains controversial about which DNMT isoform is the best therapeutic target in cancer. Second, the mechanism of different isomers of DNMT in DNA methylation inhibition and whether a compensation mechanism for DNMT with different isoforms existed are not clear. Third, a DNMTi derived from natural compounds is difficult to be modified and optimized. Forth, there are rare reports on the crystal structure of the DNMT–DNMTi complex, which limits the development of DNMTis through structure-based drug discovery. Fifth, the re-expression of genes following hypomethylation of DNA requires the eviction of nucleosomes from gene promoters. Thus, another complicating factor in the development of DNMTi is the contribution of the nucleosome in the expression of genes (Esteller et al., 2010). Sixth, high structural conservation in the catalytic domains of DNMTs poses a big challenge to design selective DNMTis for a specific DNMT isoforms.
The high toxicity of nucleoside DNMTis is determined by their own properties and mechanism of DNA demethylation. Therefore, it is expected that non-nucleoside-selective DNMTis will be the focus of DNMTi development in the future. First, it is an important way to discover novel DNMTis by exploring the structure–activity relationship of non-nucleoside DNMTis reported so far. Second, we can fuse a nucleoside and a non-nucleoside DNMTi together using a fusion strategy based on structural biology. Similarly, one general approach, called multisubstrate adduct, can be applied in DNMTi design, in which the two substrates SAM and cytosine analog are combined into one single molecule. These adjunctive inhibitors are likely to result in targeting both catalytic and cofactor binding sites, which will increase the binding affinity and selectivity of different DNMT isoforms. Third, chimeric DNMTi contains DNA sequence-specific recognition and effect regions; thus, it can inhibit DNA methylation specifically. Forth, DNMTis with a new structure and mechanism can be found by large-scale computer virtual screening. Fifth, fragment-based drug discovery is another critical way for selective DNMTi discovery. Selective DNMTis can be obtained by growing or linking the new scaffolds that could bind to different isoforms of DNMT. Finally, drug reposition has the benefit to reduce the enormous cost in time and resources devoted to novel drugs.
In conclusion, with the identification of DNMT and inhibitor complex structures and the enrichment of DNMTs’ chemical space, specific and highly effective inhibitors targeting DNMTs will emerge in the coming years. Concerning the high complexity of epigenetic regulation mechanisms, DNMTi may not only be identified as potential therapeutic agents targeting DNA methylation but also could be served as chemical probes that contribute to the better understanding of DNA methylation in normal and cancer cells.
ZZ, GW, LO, YW, and JL contributed to the conception and design of the study. ZZ wrote the first draft of the manuscript and drew the figures. YL, DL, and YW were responsible for checking the spelling and grammar of the manuscript. All authors contributed to manuscript revision and read and approved the submitted version.
This work was supported by grants from the Technological Special Project for “Significant New Drugs Development” (Grant Nos. 2018ZX09201018-021 and 2018ZX09201018-020), the National Natural Science Foundation of China (Grant No. 82073318), the National Major Scientific and Sichuan Science and Technology Program (Grant No. 2019YFS0003), the National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Grant No. Z20201004), the Post-Doctor Research Project, West China Hospital, Sichuan University (2020HXBH020), the Science and Technology Project of the Health Planning Committee of Sichuan (20PJ022), and the Ministry of Science and Technology Project of the People’s Republic of China (2018YFC2001802-1).
Thanks to Yuxi Wang, GW, YW, and JY for writing, assisting, and proofreading the article. Thanks to Chengyong Wu for his guidance on the figures of the review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahmad, I., and Rao, D. N. (1996). Chemistry and biology of DNA methyltransferases. Crit. Rev. Biochem. Mol. Biol. 31 (5-6), 361–380. doi:10.3109/10409239609108722
Akhavan-Niaki, H., and Samadani, A. (2013). DNA methylation and cancer development: Molecular mechanism [J]. Cell. Biochem. Biophysics 67 (2), 501–513. doi:10.1007/s12013-013-9555-2
Andrianasolo, E. H., France, D., Cornell-Kennon, S., and Gerwick, W. H. (2006). DNA methyl transferase inhibiting halogenated monoterpenes from the Madagascar red marine alga Portieria hornemannii. J. Nat. Prod. 69 (4), 576–579. doi:10.1021/np0503956
Badarkhe, G., Sil, A., Bhattacharya, S., Nath, U. K., and Das, N. K. (2016). Erythema multiforme due to arsenic trioxide in a case of acute promyelocytic leukemia: A diagnostic challenge. Indian J. Pharmacol. 48 (2), 216–218. doi:10.4103/0253-7613.178827
Bárcena-Varela, M., Caruso, S., Llerena, S., Alvarez-Sola, G., Uriarte, I., Latasa, M. U., et al. (2019). Dual targeting of histone methyltransferase G9a and DNA-methyltransferase 1 for the treatment of experimental hepatocellular carcinoma. Hepatology 69 (2), 587–603. doi:10.1002/hep.30168
Baud, M., Leiser, T., Haus, P., Samlal, S., Wong, A. C., Wood, R. J., et al. (2012). Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J. Med. Chem. 55 (4), 1731–1750. doi:10.1021/jm2016182
Beisler, J. A., Abbasi, M., and Driscoll, J. S. (1979). Synthesis and antitumor activity of 5-azacytosine arabinoside. J. Med. Chem. 22 (10), 1230–1234. doi:10.1021/jm00196a015
Boocock, D. J., Faust, G., Patel, K. R., Schinas, A. M., Brown, V. A., Ducharme, M. P., et al. (2007). Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol. Biomarkers Prev. 16 (6), 1246–1252. doi:10.1158/1055-9965.EPI-07-0022
Bora-Tatar, G., Dayangac-Erden, D., Demir, A. S., Dalkara, S., Yelekci, K., and Erdem-Yurter, H. (2009). Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorg. Med. Chem. 17 (14), 5219–5228. doi:10.1016/j.bmc.2009.05.042
Bosviel, R., Durif, J., Déchelotte, P., Bignon, Y. J., and Bernard-Gallon, D. (2012). Epigenetic modulation of BRCA1 and BRCA2 gene expression by equol in breast cancer cell lines. Br. J. Nutr. 108 (7), 1187–1193. doi:10.1017/S000711451100657X
Brueckner, B., Rius, M., Markelova, M. R., Fichtner, I., Hals, P. A., Sandvold, M. L., et al. (2010). Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol. Cancer Ther. 9 (5), 1256–1264. doi:10.1158/1535-7163.MCT-09-1202
Burlibaşa, L., Nicu, A. T., and Domnariu, C. (2021). DNA methyltransferase inhibitors modulate histone methylation: Epigenetic crosstalk between H3K4me3 and DNA methylation during sperm differentiation. Zygote 29 (3), 239–244. doi:10.1017/S0967199420000684
Byun, H. M., Si, H. C., Laird, P. W., Trinh, B., Siddiqui, M. A., Marquez, V. E., et al. (2008). 2'-Deoxy-N4-[2-(4-nitrophenyl)ethoxycarbonyl]-5-azacytidine: A novel inhibitor of DNA methyltransferase that requires activation by human carboxylesterase 1. Cancer Lett. 266 (2), 238–248. doi:10.1016/j.canlet.2008.02.069
Cancer Research Technology Ltd (2007). Phthalimides, succinimides and related compounds and their use as pharmaceuticals. WO2007007054.
Candelaria, M., de La Cruz-Hernandez, E., Taja-Chayeb, L., Perez-Cardenas, E., Trejo-Becerril, C., Gonzalez-Fierro, A., et al. (2012). DNA methylation-independent reversion of gemcitabine resistance by hydralazine in cervical cancer cells. PLoS One 7 (3), e29181. doi:10.1371/journal.pone.0029181
Candelaria, M., Gallardo-Rincon, D., Arce, C., Cetina, L., Aguilar-Ponce, J., Arrieta, O., et al. (2007). A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann. Oncol. 7 (9), 1529–1538. doi:10.1093/annonc/mdm204
Castellano, S., Kuck, D., Viviano, M., Yoo, J., Lopez-Vallejo, F., Conti, P., et al. (2011). Synthesis and biochemical evaluation of δ(2)-isoxazoline derivatives as DNA methyltransferase 1 inhibitors. J. Med. Chem. 54, 7663–7677. doi:10.1021/jm2010404
Castro, M., Grau, L., Puerta, P., Gimenez, L., Venditti, J., Quadrelli, S., et al. (2010). Multiplexed methylation profiles of tumor suppressor genes and clinical outcome in lung cancer. J. Transl. Med. 8 (1), 86. doi:10.1186/1479-5876-8-86
Center for Drug Evaluation and Research (2006). About the center for drug evaluation and research - FDA approves decitabine for injection (dacogen) to treat myelodysplastic syndromes [J]. Center for Drug Evaluation and Research.
Chatterjee, B., Ghosh, K., and Kanade, S. R. (2019). Resveratrol modulates epigenetic regulators of promoter histone methylation and acetylation that restores BRCA1, p53, p21CIP1 in human breast cancer cell lines. BioFactors 45 (5), 818–829. doi:10.1002/biof.1544
Chekhun, V. F., Kulik, G. I., Yurchenko, O. V., Tryndyak, V. P., Todor, I. N., Luniv, L. S., et al. (2006). Role of DNA hypomethylation in the development of the resistance to doxorubicin in human MCF-7 breast adenocarcinoma cells. Cancer Lett. 231 (1), 87–93. doi:10.1016/j.canlet.2005.01.038
Chen, S., Wang, Y., Zhou, W., Li, S., Peng, J., Shi, Z., et al. (2014). Identifying novel selective non-nucleoside DNA methyltransferase 1 inhibitors through docking-based virtual screening. J. Med. Chem. 57 (21), 9028–9041. doi:10.1021/jm501134e
Choi, W. J., Chung, H. J., Chandra, G., Alexander, V., Zhao, L. X., Lee, H. W., et al. (2012). Fluorocyclopentenyl-cytosine with broad spectrum and potent antitumor activity. J. Med. Chem. 55 (9), 4521–4525. doi:10.1021/jm3004009
Chow, H. H., Cai, Y., Hakim, I. A., Crowell, J. A., Shahi, F., Brooks, C. A., et al. (2003). Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals[J]. Clin. Cancer Res. 9 (9), 3312–3319.
Cohen, M. H., Hirschfeld, S., Honig, S. F., Ibrahim, A., Johnson, J. R., O'Leary, J. J., et al. (2001). Drug approval summaries: Arsenic trioxide, tamoxifen citrate, anastrazole, paclitaxel, bexarotene. Oncologist 6 (1), 4–11. doi:10.1634/theoncologist.6-1-4
Cornacchia, E., Golbus, J., Maybaum, J., Strahler, J., HanaSh, S., and Richardson, B. (1988). Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J. Immunol. 140 (7), 2197–2200.
Costa, E. M. D., Mcinnes, G., Beaudry, A., and Raynal, N. J. M. (2017). DNA methylation-targeted drugs. Cancer J. 23 (5), 270–276. doi:10.1097/PPO.0000000000000278
Costa, F., Paixão, V. A., Cavalher, F. P., Ribeiro, K. B., Cunha, I. W., Rinck, J. A., et al. (2006). SATR-1 hypomethylation is a common and early event in breast cancer. Cancer Genet. cytogenet. 165 (2), 135–143. doi:10.1016/j.cancergencyto.2005.07.023
Costantini, B., Kordasti, S. Y., Kulasekararaj, A. G., Jiang, J., Seidl, T., Abellan, P. P., et al. (2013). The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica 98 (8), 1196–1205. doi:10.3324/haematol.2012.074823
Curt, G. A., Kelley, J. A., Fine, R. L., Huguenin, P. N., Roth, J. S., Batist, G., et al. (1985). A phase I and pharmacokinetic study of dihydro-5-azacytidine (NSC 264880). Cancer Res. 45 (7), 3359–3363.
Dagdemir, A., Durif, J., Ngollo, M., Bignon, Y. J., and Bernard-Gallon, D. (2013). Histone lysine trimethylation or acetylation can be modulated by phytoestrogen, estrogen or anti-HDAC in breast cancer cell lines. Epigenomics 5 (1), 51–63. doi:10.2217/epi.12.74
Dalal, M., Plowman, J., Breitman, T. R., Schuller, H. M., del Campo, A. A., Vistica, D. T., et al. (1986). Arabinofuranosyl-5-azacytosine: Antitumor and cytotoxic properties. Cancer Res. 46 (2), 831–838.
Datta, J., Ghoshal, K., Denny, W. A., Gamage, S. A., Brooke, D. G., Phiasivongsa, P., et al. (2009). A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 69 (10), 4277–4285. doi:10.1158/0008-5472.CAN-08-3669
de Oliveira, D. M., and Markowicz Bastos, D. H. (2011). Biodisponibilidade de ácidos fenólicos [J]. Quím. Nova 34 (6), 17. doi:10.1590/S0100-40422011000600023
Deb, G., Shankar, E., Thakur, V. S., Ponsky, L. E., Bodner, D. R., Fu, P., et al. (2019). Green tea-induced epigenetic reactivation of tissue inhibitor of matrix metalloproteinase-3 suppresses prostate cancer progression through histone-modifying enzymes. Mol. Carcinog. 58 (7), 1194–1207. doi:10.1002/mc.23003
Dilshara, M. G., Jayasooriya, R. G., Park, S. R., Choi, Y. H., Choi, I. W., and Kim, G. Y. (2016). Caffeic acid phenethyl ester enhances TRAIL-mediated apoptosis via CHOP-induced death receptor 5 upregulation in hepatocarcinoma Hep3B cells[J]. Mol. Cell. Biochem. 418 (1-2), 13–20. doi:10.1007/s11010-016-2726-x
Erdmann, A., Menon, Y., Gros, C., Molinier, N., Novosad, N., Samson, A., et al. (2015). Design and synthesis of new non nucleoside inhibitors of DNMT3A. Bioorg. Med. Chem. 23 (17), 5946–5953. doi:10.1016/j.bmc.2015.06.066
Esteller, M., Kelly, T. K., and Jones, P. A. (2010). Epigenetics in cancer. Carcinogenesis 31 (1), 27–36. doi:10.1093/carcin/bgp220
Fagan, R. L., Cryderman, D. E., Kopelovich, L., Wallrath, L. L., and Brenner, C. (2013). Laccaic acid A is a direct, DNA-competitive inhibitor of DNA methyltransferase 1. J. Biol. Chem. 288 (33), 23858–23867. doi:10.1074/jbc.M113.480517
Fang, M., Chen, D., and Yang, C. S. (2007). Dietary polyphenols may affect DNA methylation. J. Nutr. 137, 223S–228S. doi:10.1093/jn/137.1.223S
Fang, M. Z., Chen, D., Sun, Y., Jin, Z., Christman, J. K., and Yang, C. S. (2005). Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 11 (19), 7033–7041. doi:10.1158/1078-0432.CCR-05-0406
Fleisher, A. S., Esteller, M., Tamura, G., Rashid, A., Stine, O. C., Yin, J., et al. (2001). Hypermethylation of the hMLH1 gene promoter is associated with microsatellite instability in early human gastric neoplasia. Oncogene 20 (3), 329–335. doi:10.1038/sj.onc.1204104
Florean, C., Schnekenburger, M., Lee, J. Y., Kim, K. R., Mazumder, A., Song, S., et al. (2016). Discovery and characterization of Isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to TRAIL in cancer cells. Oncotarget 7 (17), 24027–24049. doi:10.18632/oncotarget.8210
Frank, L., and Robert, B. (2005). DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl. Cancer Inst. 97 (20), 1498–1506. doi:10.1093/jnci/dji311
Gallagher, S. J., Shklovskaya, E., and Hersey, P. (2017). Epigenetic modulation in cancer immunotherapy. Curr. Opin. Pharmacol. 35, 48–56. doi:10.1016/j.coph.2017.05.006
Gao, C., Shuang, H., Guo, M., Hai, X., and Zhou, J. (2018). Clinical pharmacokinetics and safety profile of single agent arsenic trioxide by continuous slow-rate infusion in patients with newly diagnosed acute promyelocytic leukemia [J]. Cancer Chemother. Pharmacol. 82, 229–236. doi:10.1007/s00280-018-3606-8
Gao, Z., Xu, Z., Hung, M. S., Lin, Y. C., Wang, T., Gong, M., et al. (2009). Promoter demethylation of WIF-1 by epigallocatechin-3-gallate in lung cancer cells. Anticancer Res. 29 (6), 2025–2030.
García, J., Franci, G., Pereira, R., Benedetti, R., Nebbioso, A., Rodriguez-Barrios, F., et al. (2011). Epigenetic profiling of the antitumor natural product psammaplin A and its analogues. Bioorg. Med. Chem. 19 (12), 3637–3649. doi:10.1016/j.bmc.2010.12.026
Gerson, S. L. (2002). Clinical relevance of MGMT in the treatment of cancer. J. Clin. Oncol. 20 (9), 2388–2399. doi:10.1200/JCO.2002.06.110
Ghoshal, K., Ghoshal, J., Majumder, S., Bai, S., Kutay, H., Motiwala, T., et al. (2005). 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell. Biol. 25 (11), 4727–4741. doi:10.1128/MCB.25.11.4727-4741.2005
Giordano, A., and Tommonaro, G. (2019). Curcumin and cancer. Nutrients 11 (10), 2376. doi:10.3390/nu11102376
Goffin, J., and Eisenhauer, E. (2002). DNA methyltransferase inhibitors-state of the art. Ann. Oncol. 13 (11), 1699–1716. doi:10.1093/annonc/mdf314
Gordian, E., Ramachandran, K., and Singal, R. (2009). Methylation mediated silencing of TMS1 in breast cancer and its potential contribution to docetaxel cytotoxicity. Anticancer Res. 29 (8), 3207–3210.
Heideman, R. L., Gillespie, A., Ford, H., Balis, F. M., Reaman, G. H., Tan, C., et al. (1989). Phase I trial and pharmacokinetic evaluation of fazarabine in children. Cancer Res. 49 (18), 5213–5216.
Hong, L., Meng, X., Li, C., Sang, S., Patten, C., Sheng, S., et al. (2003). Glucuronides of tea catechins: Enzymology of biosynthesis and biological activities [J]. Drug Metab. Dispos. 31 (4), 452–461. doi:10.1124/dmd.31.4.452
Horenstein, S., Vander Heide, R. S., and L'Ecuyer, T. J. (2000). Molecular basis of anthracycline-induced cardiotoxicity and its prevention. Mol. Genet. Metab. 71 (1-2), 436–444. doi:10.1006/mgme.2000.3043
Hu, J., Chen, S., Kong, X., Zhu, K., Cheng, S., Zheng, M., et al. (2015). Interaction between DNA/histone methyltransferases and their inhibitors. Curr. Med. Chem. 22 (3), 360–372. doi:10.2174/0929867321666141106114538
Huang, L., Wu, R. L., and Xu, A. M. (2015). Epithelial-mesenchymal transition in gastric cancer. Am. J. Transl. Res. 7 (11), 2141–2158.
Identification of Kazinol, Q., Lai, I. L., Yang, H. C., Lin, C. N., and Bai, L. Y. (2014). Identification of Kazinol Q, a natural product from formosan plants, as an inhibitor of DNA methyltransferase. Phytother. Res. 28 (1), 49–54. doi:10.1002/ptr.4955
Iida, T., Suetake, I., Tajima, S., Morioka, H., Ohta, S., Obuse, C., et al. (2010). PCNA clamp facilitates action of DNA cytosine methyltransferase 1 on hemimethylated DNA. Genes. 7 (10), 997–1007. doi:10.1046/j.1365-2443.2002.00584.x
Ikerchem S L (2014). Indole derivatives, pharmaceutical compositions containing such indoles and their use as a DNA methylation modulators. WO2014072371.
INSERM (2011). Flavones and flavanones derivatives as DNA methyltransferases inhibitors. WO2011029956.
Isakovic, L., Saavedra, O. M., Llewellyn, D. B., Claridge, S., Zhan, L., Bernstein, N., et al. (2009). Constrained (l-)-S-adenosyl-l-homocysteine (SAH) analogues as DNA methyltransferase inhibitors. Bioorg. Med. Chem. Lett. 19 (10), 2742–2746. doi:10.1016/j.bmcl.2009.03.132
Issa, J. J., Roboz, G., Rizzieri, D., Jabbour, E., Stock, W., O'Connell, C., et al. (2015). Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet. Oncol. 16, 1099–1110. doi:10.1016/S1470-2045(15)00038-8
Jones, P. A., and Taylor, S. M. (1980). Cellular differentiation, cytidine analogs and DNA methylation. Cell. 20 (1), 85–93. doi:10.1016/0092-8674(80)90237-8
Kaminskas, E., Farrell, A. T., Wang, Y. C., Sridhara, R., and Pazdur, R. (2005). FDA drug approval summary: Azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist 10 (3), 176–182. doi:10.1634/theoncologist.10-3-176
Kim, K. H., Choi, J. S., Kim, I. J., Ku, J. L., and Park, J. G. (2006). Promoter hypomethylation and reactivation of MAGE-A1 and MAGE-A3 genes in colorectal cancer cell lines and cancer tissues. World J. Gastroenterol. 12 (35), 5651–5657. doi:10.3748/wjg.v12.i35.5651
Kozuka, C., Kaname, T., Shimizu-Okabe, C., Takayama, C., Tsutsui, M., Matsushita, M., et al. (2017). Impact of Brown rice-specific γ-oryzanol on epigenetic modulation of dopamine D2 receptors in brain striatum in high-fat-diet-induced obesity in mice. Diabetologia 60 (8), 1502–1511. doi:10.1007/s00125-017-4305-4
Kuck, D., Caulfield, T., Lyko, F., and Medina-Franco, J. L. (2010). Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 9 (11), 3015–3023. doi:10.1158/1535-7163.MCT-10-0609
Küçük, C., Hu, X., Gong, Q., Jiang, B., Cornish, A., Gaulard, P., et al. (2016). Diagnostic and biological significance of KIR expression profile determined by RNA-seq in natural killer/T-cell lymphoma [J]. Am. J. Pathology 186, 1435–1441. doi:10.1016/j.ajpath.2016.02.011
Lee, B. H., Yegnasubramanian, S., Lin, X., and Nelson, W. G. (2005). Procainamide is a specific inhibitor of DNA methyltransferase 1. J. Biol. Chem. 280 (49), 40749–40756. doi:10.1074/jbc.M505593200
Lee, H., Zhang, P., Herrmann, A., Yang, C., Xin, H., Wang, Z., et al. (2012). Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. U. S. A. 109 (20), 7765–7769. doi:10.1073/pnas.1205132109
Leung, G., Sun, W., Zheng, L., BrookeS, S., TullyM., , and Shi, R. (2011). Anti-acrolein treatment improves behavioral outcome and alleviates myelin damage in experimental autoimmune encephalomyelitis mouse. Neuroscience 173 (2), 150–155. doi:10.1016/j.neuroscience.2010.11.018
Li, L. H., Olin, E. J., Buskirk, H. H., and Reineke, L. M. (1970). Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 30 (11), 2760–2769.
Lin, J., Haffner, M. C., Zhang, Y., Lee, B. H., Brennen, W. N., Britton, J., et al. (2011). Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate 71 (4), 333–343. doi:10.1002/pros.21247
Lin, R. K., Hsu, C. H., and Wang, Y. C. (2007). Mithramycin A inhibits DNA methyltransferase and metastasis potential of lung cancer cells. Anticancer. Drugs 18 (10), 1157–1164. doi:10.1097/CAD.0b013e3282a215e9
Lin, Y. S., Shaw, A. Y., Wang, S. G., Hsu, C. C., Teng, I. W., Tseng, M. J., et al. (2011). Identification of novel DNA methylation inhibitors via a two-component reporter gene system[J]. J. Biomed. Sci. 18 (1), 1–8. doi:10.1186/1423-0127-18-3
Liu, D., Wu, D., Zhao, L., Yang, Y., Ding, J., Dong, L., et al. (2015). Arsenic trioxide reduces global histone H4 acetylation at lysine 16 through direct binding to histone acetyltransferase hMOF in human cells. PLoS One 10 (10), e0141014. doi:10.1371/journal.pone.0141014
Liu, X., Gao, Q., Li, P., Zhao, Q., Zhang, J., Li, J., et al. (2013). UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun. 4, 1563. doi:10.1038/ncomms2562
Liu, Z., Liu, S., Xie, Z., Pavlovicz, R. E., Wu, J., Chen, P., et al. (2009). Modulation of DNA methylation by a sesquiterpene lactone parthenolide. J. Pharmacol. Exp. Ther. 329 (2), 505–514. doi:10.1124/jpet.108.147934
Liu, Z., Xie, Z., Jones, W., Pavlovicz, R. E., Liu, S., Yu, J., et al. (2009). Curcumin is a potent DNA hypomethylation agent. Bioorg. Med. Chem. Lett. 19 (3), 706–709. doi:10.1016/j.bmcl.2008.12.041
Lucarini, V., Buccione, C., Ziccheddu, G., Peschiaroli, F., Sestili, P., Puglisi, R., et al. (2017). Combining type I interferons and 5-aza-2'-deoxycitidine to improve anti-tumor response against melanoma. J. Invest. Dermatol. 137 (1), 159–169. doi:10.1016/j.jid.2016.08.024
Ludden, T. M. (1982). Clinical pharmacokinetics of hydrallazine. Clin. Pharmacokinet. 2 (3), 317–329. doi:10.2165/00003088-197702050-00001
Majda, K., Kaufman-Szymczyk, A., Lubecka-Pietruszewska, K., Bednarek, A., and Fabianowska-Majewska, K. (2010). Influence of clofarabine on transcriptional activity of PTEN, APC, RARB2, ZAP70 genes in K562 cells. Anticancer Res. 30 (11), 4601–4606.
Majid, S., Dar, A. A., Shahryari, V., Hirata, H., Ahmad, A., Saini, S., et al. (2010). Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-Cell translocation gene 3 in prostate cancer. Cancer 116, 66–76. doi:10.1002/cncr.24662
Marcucci, G., Silverman, L., Eller, M., Lintz, L., and Beach, C. L. (2013). Bioavailability of azacitidine subcutaneous versus intravenous in patients with the myelodysplastic syndromes. J. Clin. Pharmacol. 45 (5), 597–602. doi:10.1177/0091270004271947
Margot, J. B., Cardoso, M. C., and Leonhardt, H. (2001). Mammalian DNA methyltransferases show different subnuclear distributions. J. Cell. Biochem. 83 (3), 373–379. doi:10.1002/jcb.1236
Masaki, O., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 99 (3), 247–257. doi:10.1016/s0092-8674(00)81656-6
Medina-Franco, J. L., Méndez-Lucio, O., Duenas-Gonzalez, A., and Yoo, J. (2015). Discovery and development of DNA methyltransferase inhibitors using in silico approaches. Drug Discov. Today 20 (5), 569–577. doi:10.1016/j.drudis.2014.12.007
Meeran, S. M., Patel, S. N., Chan, T. H., and Tollefsbol, T. O. (2011). A novel prodrug of epigallocatechin-3-gallate: Differential epigenetic hTERT repression in human breast cancer cells. Cancer Prev. Res. 4 (8), 1243–1254. doi:10.1158/1940-6207.CAPR-11-0009
Meier, S., Cantilena, S., Niklison Chirou, M. V., Anderson, J., Hargrave, D., Salomoni, P., et al. (2021). Alcohol-abuse drug disulfiram targets pediatric glioma via MLL degradation [J]. Cell. Death Dis. 12 (8), 785. doi:10.1038/s41419-021-04078-9
Milicic, A., Harrison, L. A., Goodlad, R. A., Hardy, R. G., Nicholson, A. M., Presz, M., et al. (2008). Ectopic expression of P-cadherin correlates with promoter hypomethylation early in colorectal carcinogenesis and enhanced intestinal crypt fission in vivo. Cancer Res. 68 (19), 7760–7768. doi:10.1158/0008-5472.CAN-08-0020
Momparler, R. L. (2005). Pharmacology of 5-Aza-2'-deoxycytidine (decitabine). Semin. Hematol. 42, S9–S16. doi:10.1053/j.seminhematol.2005.05.002
Nakamura, K., Nakabayashi, K., Htet Aung, K., Aizawa, K., Hori, N., Yamauchi, J., et al. (2015). DNA methyltransferase inhibitor zebularine induces human cholangiocarcinoma cell death through alteration of DNA methylation status[J]. PLOS ONE 10 (3), 1–21. doi:10.1371/journal.pone.0120545
Nakamura, M., Nishikawa, J., Saito, M., Sakai, K., Sasaki, S., Hashimoto, S., et al. (2016). Decitabine inhibits tumor cell proliferation and up-regulates e-cadherin expression in Epstein-Barr virus-associated gastric cancer. J. Med. Virol. 89 (3), 508–517. doi:10.1002/jmv.24634
Nandakumar, V., Vaid, M., and Katiyar, S. K. (2011). (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 32 (4), 537–544. doi:10.1093/carcin/bgq285
Newman, E. M., Morgan, R. J., Kummar, S., Beumer, J. H., Blanchard, M. S., Ruel, C., et al. (2015). A phase I, pharmacokinetic, and pharmacodynamic evaluation of the DNA methyltransferase inhibitor 5-fluoro-2'-deoxycytidine, administered with tetrahydrouridine. Cancer Chemother. Pharmacol. 75 (3), 537–546. doi:10.1007/s00280-014-2674-7
Pandey, M., Shukla, S., and Gupta, S. (2010). Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells [J]. Int. J. Cancer 126 (11), 2520–2533. doi:10.1002/ijc.24988
Parashar, G., Parashar, N. C., and Capalash, N. (2012). Curcumin causes promoter hypomethylation and increased expression of FANCF gene in SiHa cell line. Mol. Cell. Biochem. 365 (1-2), 29–35. doi:10.1007/s11010-012-1240-z
Paredes, J., Albergaria, A., Oliveira, J. T., Jeronimo, C., Milanezi, F., and Schmitt, F. C. (2005). P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin. Cancer Res. 11 (16), 5869–5877. doi:10.1158/1078-0432.CCR-05-0059
Pechalrieu, D., Etievant, C., and Arimondo, P. B. (2017). DNA methyltransferase inhibitors in cancer: From pharmacology to translational studies [J]. Biochem. Pharmacol. 129, 1–13. doi:10.1016/j.bcp.2016.12.004
Pegg, A. E. (2011). Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 24 (5), 618–639. doi:10.1021/tx200031q
Peters, G. J., Smid, K., Vecchi, L., Kathmann, I., Sarkisjan, D., Honeywell, R. J., et al. (2013). Metabolism, mechanism of action and sensitivity profile of fluorocyclopentenylcytosine (RX-3117; TV-1360). Invest. New Drugs 31 (6), 1444–1457. doi:10.1007/s10637-013-0025-x
Pierre Fabre Medicament; Centre National de la Recherche Scientifique (CNRS) (2015). Quinazoline derivatives and their use as DNA methyltransferase inhibitors. WO2015040169.
Piña, I. C., Gautschi, J. T., Wang, G., Sanders, M. L., Schmitz, F. J., France, D., et al. (2003). Psammaplins from the sponge pseudoceratina purpurea: Inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 68 (10), 3866–3873. doi:10.1021/jo034248t
Procaine, J., Xu, Z., Hung, M. S., Lin, Y. C., Wang, T., Gong, M., et al. (2009). Procaine and procainamide inhibit the Wnt canonical pathway by promoter demethylation of WIF-1 in lung cancer cells. Oncol. Rep. 22 (06), 1479–1484. doi:10.3892/or_00000590
Qiu, H., Hu, X., Gao, L., Chen, L., Chen, J., Yuan, J., et al. (2017). Interleukin 10 enhanced CD8+ T cell activity and reduced CD8+ T cell apoptosis in patients with diffuse large B cell lymphoma. Exp. Cell. Res. 360 (2), 146–152. doi:10.1016/j.yexcr.2017.08.036
Rius, M., Stresemann, C., Keller, D., Brom, M., Schirrmacher, E., Keppler, D., et al. (2009). Human concentrative nucleoside transporter 1-mediated uptake of 5-azacytidine enhances DNA demethylation. Mol. Cancer Ther. 8 (1), 225–231. doi:10.1158/1535-7163.MCT-08-0743
Rossi, J. F., van Hoof, A., de Boeck, K., Johnson, S. A., Bron, D., Foussard, C., et al. (2004). Efficacy and safety of oral fludarabine phosphate in previously untreated patients with chronic lymphocytic leukemia. J. Clin. Oncol. 22 (7), 1260–1267. doi:10.1200/JCO.2004.05.012
San José-Enériz, E., Agirre, X., Rabal, O., Vilas-Zornoza, A., Sanchez-Arias, J. A., Miranda, E., et al. (2017). Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 8, 15424. doi:10.1038/ncomms15424
Sawesi, S., Malkaram, S. A., Abd Elmageed, Z. Y., and Fandy, T. E. (2022). Modulation of the activity of histone lysine methyltransferases and demethylases by curcumin analog in leukaemia cells. J. Cell. Mol. Med. 26, 5624–5633. doi:10.1111/jcmm.17589
Scheinbart, L. S., Johnson, M. A., Gross, L. A., Edelstein, S. R., and Richardson, B. C. (1991). Procainamide inhibits DNA methyltransferase in a human T cell line [J]. J. Rheumatology 18 (4), 530.
Schweizer, M. T., Lin, J., Blackford, A., Bardia, A., King, S., Armstrong, A. J., et al. (2013). Pharmacodynamic study of disulfiram in men with non-metastatic recurrent prostate cancer. Prostate Cancer Prostatic Dis. 16 (4), 357–361. doi:10.1038/pcan.2013.28
Scott, S. A., Lakshimikuttysamma, A., Sheridan, D. P., Sanche, S. E., Geyer, C. R., and DeCoteau, J. F. (2007). Zebularine inhibits human acute myeloid leukemia cell growth in vitro in association with p15INK4B demethylation and reexpression. Exp. Hematol. 35 (2), 263–273. doi:10.1016/j.exphem.2006.10.005
Seo, J. S., Choi, Y. H., Moon, J. W., Kim, H. S., and Park, S. H. (2017). Hinokitiol induces DNA demethylation via DNMT1 and UHRF1 inhibition in colon cancer cells. BMC Cell. Biol. 18 (1), 14–11. doi:10.1186/s12860-017-0130-3
Setchell, K. D., Zhao, X., Jha, P., Heubi, J. E., and Brown, N. M. (2009). The pharmacokinetic behavior of the soy isoflavone metabolite S-(-) equol and its diastereoisomer R-(+) equol in healthy adults determined by using stable-isotope-labeled tracers [J]. Am. J. Clin. Nutr. 90 (4), 1029–1037. doi:10.3945/ajcn.2009.27981
Seuk Kim, J., Han, J., Shim, Y. M., Park, J., and Kim, D. H. (2005). Aberrant methylation of H-cadherin (CDH13) promoter is associated with tumor progression in primary nonsmall cell lung carcinoma. Cancer 104 (9), 1825–1833. doi:10.1002/cncr.21409
Sharma, M., Arora, I., Chen, M., Wu, H., Crowley, M. R., Tollefsbol, T. O., et al. (2021). Therapeutic effects of dietary soybean genistein on triple-negative breast cancer via regulation of epigenetic mechanisms. Nutrients 13 (11), 3944. doi:10.3390/nu13113944
Sharma, R. A., Mclelland, H. R., Hill, K. A., Ireson, C. R., Euden, S. A., Manson, M. M., et al. (2001). Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res. 7 (7), 1894–1900.
Southern Research Institute (2009). The use of 2’-deoxy-4’-thiocytidine and its analogues as DNA hypomethylating anticancer agents. WO2011109012.
Stresemann, C., and Lyko, F. (2008). Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 123 (1), 8–13. doi:10.1002/ijc.23607
Su, Y., Hopfinger, N. R., Nguyen, T. D., Pogash, T. J., Santucci-Pereira, J., and Russo, J. (2018). Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. J. Exp. Clin. Cancer Res. 37 (1), 314–318. doi:10.1186/s13046-018-0988-8
Sun, G., Fan, T., Sun, X., Hao, Y., Cui, X., Zhao, L., et al. (2018). In silico prediction of O6-methylguanine-DNA methyltransferase inhibitory potency of base analogs with QSAR and machine learning methods. Molecules 23 (11), 2892. doi:10.3390/molecules23112892
Sun, G., Zhao, L., Zhong, R., and Peng, Y. (2018). The specific role of O6-methylguanine-DNA methyltransferase inhibitors in cancer chemotherapy. Future Med. Chem. 10 (16), 1971–1996. doi:10.4155/fmc-2018-0069
Tatsuya, S., Yamauchi, T., Ando, K., Nagai, T., Kakihana, K., Miyata, Y., et al. (2013). Phase I study of clofarabine in adult patients with acute myeloid leukemia in Japan. Jpn. J. Clin. Oncol. 43 (12), 1177–1183. doi:10.1093/jjco/hyt155
Thottassery, J. V., Sambandam, V., Allan, P. W., Maddry, J. A., Maxuitenko, Y. Y., Tiwari, K., et al. (2014). Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4'-thio-2'-deoxycytidine and 5-aza-4'-thio-2'-deoxycytidine. Cancer Chemother. Pharmacol. 74 (2), 291–302. doi:10.1007/s00280-014-2503-z
Townsend, A., Leclerc, J. M., Dutschman, G., and Cheng, Y. C. (1985). Metabolism of 1-beta-D-arabinofuranosyl-5-azacytosine and incorporation into DNA of human T-lymphoblastic cells (Molt-4). Cancer Res. 45 (8), 3522–3528.
Udvaros, I., Rethy, A., Lang, I., Hitre, E., and Peterson, C. (2015). A phase 1 exploratory study of RX-3117 to determine oral bioavailability in cancer subjects with solid tumors. J. Clin. Oncol. 33, e13545. doi:10.1200/jco.2015.33.15_suppl.e13545
Ullmann, U., Metzner, J., Frank, T., Cohn, W., and Riegger, C. (2005). Safety, tolerability, and pharmacokinetics of single ascending doses of synthetic genistein (Bonistein) in healthy volunteers. Adv. Ther. 22 (1), 65–78. doi:10.1007/BF02850186
University of Southern California (2008). N4 modifications of pyrimidine analogs and uses thereof. WO2008054767.
Valdez, B. C., Li, Y., Murray, D., Champlin, R. E., and Andersson, B. S. (2011). The synergistic cytotoxicity of clofarabine, fludarabine and busulfan in AML cells involves ATM pathway activation and chromatin remodeling. Biochem. Pharmacol. 81 (2), 222–232. doi:10.1016/j.bcp.2010.09.027
Valente, S., Liu, Y., Schnekenburger, M., Zwergel, C., Cosconati, S., Gros, C., et al. (2014). Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. J. Med. Chem. 57 (3), 701–713. doi:10.1021/jm4012627
Villar-Garea, A., Fraga, M. F., Espada, J., and Esteller, M. (2003). Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 63 (16), 4984–4989.
Wang, L. X., Mei, Z. Y., Zhou, J. H., Yao, Y. S., Li, Y. H., Xu, Y. H., et al. (2013). Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. Plos One 8 (5), e62924. doi:10.1371/journal.pone.0062924
Widschwendter, M., Siegmund, K. D., Müller, H. M., Fiegl, H., Marth, C., Muller-Holzner, E., et al. (2004). Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 64 (11), 3807–3813. doi:10.1158/0008-5472.CAN-03-3852
Wu, J. C., and Santi, D. V. (1987). Kinetic and catalytic mechanism of HhaI methyltransferase. J. Biol. Chem. 262 (10), 4778–4786. doi:10.1016/s0021-9258(18)61263-3
Wyczechowska, D., and Fabianowska-Majewska, K. (2003). The effects of cladribine and fludarabine on DNA methylation in K562 cells. Biochem. Pharmacol. 65 (2), 219–225. doi:10.1016/s0006-2952(02)01486-7
Yoo, C. B., Jeong, S., Egger, G., Liang, G., Phiasivongsa, P., Tang, C., et al. (2007). Delivery of 5-aza-2'-deoxycytidine to cells using oligodeoxynucleotides [J]. Cancer Res. 67 (13), 6400–6408. doi:10.1158/0008-5472.CAN-07-0251
Zhang, Z. M., Liu, S., Lin, K., Luo, Y., Perry, J. J., Wang, Y., et al. (2015). Crystal structure of human DNA methyltransferase 1. J. Mol. Biol. 427 (15), 2520–2531. doi:10.1016/j.jmb.2015.06.001
Zhou, J., Yao, Y., Shen, Q., Li, G., Hu, L., and Zhang, X. (2017). Demethylating agent decitabine disrupts tumor-induced immune tolerance by depleting myeloid-derived suppressor cells. J. Cancer Res. Clin. Oncol. 143 (8), 1371–1380. doi:10.1007/s00432-017-2394-6
Zhu, L. B. T., and Zhu, B. T. (2006). Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis 27 (2), 269–277. doi:10.1093/carcin/bgi206
Zhu, W. G., and Otterson, G. A. (2003). The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr. Med. Chem. Anticancer. Agents 3 (3), 187–199. doi:10.2174/1568011033482440
AML acute myeloid leukemia
APL acute promyelocytic leukemia
DNMT DNA methyltransferase
DNMTi DNA methyltransferase inhibitor
CTL cytotoxic T lymphocyte
MHC-I major histocompatibility complex I
KIR killer cell immunoglobulin-like receptor
MDS myelodysplastic syndrome
MS multiple sclerosis
CMML chronic myelomonocytic leukemia
OR overall response
CR complete response
OS overall survival
PR partial response
PK pharmacokinetics
PD pharmacodynamic
PFS progression-free survival
PCNA proliferating cell nuclear antigen
DLT dose-limited toxicity
EGCG epgallocatechin gallate
SAH S-adenosyl-homocysteine
TSG tumor suppressor gene;
SAM S-adenosyl-L-methionine
TAAs tumor-associated antigens
MTD maximum tolerated dose
Cmax peak concentration
Tmax time to maximum plasma concentration
T1/2 half-life
AUC the area under the curve
Vd volume of distribution
NPEOC 2'-deoxy-N4-[2-(4-nitrophenyl)ethoxycarbonyl]
MAGE melanoma-associated CT antigen
DAPK1 death-associated protein kinase 1
TNBC triple-negative breast cancer
EMT epithelial mesenchymal transition
hTERT human telomerase reverse-transcriptase
TIMP3 tissue inhibitor of matrix metalloproteinase 3
hENT1 human equilibrative nucleoside transporter 1
HDAC histone deacetyltransferase
PDX patient-derived xenograft
HKMT histone lysine methyltransferase
HKDMT histone lysine demethylase
Keywords: epigenetic, DNA methylation, DNA methyltransferase, DNA methyltransferase inhibitor, tumor immunotherapy
Citation: Zhang Z, Wang G, Li Y, Lei D, Xiang J, Ouyang L, Wang Y and Yang J (2022) Recent progress in DNA methyltransferase inhibitors as anticancer agents. Front. Pharmacol. 13:1072651. doi: 10.3389/fphar.2022.1072651
Received: 17 October 2022; Accepted: 28 November 2022;
Published: 16 December 2022.
Edited by:
Yongxia Zhu, Sichuan Cancer Hospital, ChinaReviewed by:
Guohui Sun, Beijing University of Technology, ChinaCopyright © 2022 Zhang, Wang, Li, Lei, Xiang, Ouyang, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanyan Wang, YW15d3l5QDEyNi5jb20=; Jinliang Yang, amx5YW5nMDFAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.